3.2. Chemistry
Infrared spectra were recorded on an IRAffinity-1S FTIR-spectrometer (Shimadzu Europa GmbH, Hannover, Germany). NMR spectra were recorded on an Avance III 300 spectrometer, tempered at 298 K:
1H (300 MHz),
13C-NMR (75 MHz) (Bruker Daltonik GmbH, Bremen, Germany). The data is reported as follows: chemical shift in ppm from tetramethylsilane (TMS) as external standard, multiplicity and coupling constant
J (Hz). Spectra were either referenced to TMS or internal DMSO-
d6 (
1H-NMR δ 2.50) and internal DMSO-
d6 (
13C-NMR δ 39.52) or internal CHCl
3 (
1H-NMR δ 7.26) and internal CDCl
3 (
13C-NMR δ 77.00). The following NMR abbreviations have been used: b (broad), s (singlet), d (doublet), t (triplet), m (unresolved multiplet). Several target compounds show spectra of at least two isomers in DMSO-
d6 with a maximal ratio of 1:3. These are due to atropisomers and tautomers as already reported for similar 4,5-diaryl-imidazoles [
32]. Although such effects have not been observed whenever CDCl
3 was used, DMSO-
d6 was the most frequented solvent with respect to its favorable solubility-mediating properties. For reasons of clarity, signals are only given for the main isomer. The labelling scheme of structures to correlate NMR signals can be found in Supporting Information. LC-MS was performed with an 1100 HPLC system (Agilent Technologies, Santa Clara, CA, USA) over an Agilent Eclipse XDB-C8 column (150 × 4.6 mm, 5 µm) using a 0.1% acetic acid/acetonitrile gradient for mobile phase (flow rate = 1 mL∙min
−1). Mass spectra with nominal solution were recorded on a Bruker Esquire ~LC ion trap mass spectrometer with electron spray ionization (ESI) operating in the positive ion mode, with the following parameters: drying gas nitrogen 8 L∙min
−1, nebulizer 35 psi, drying temperature 350 °C). HRMS spectra were recorded on an AccuTOF™ GCv 4G electron ionization (EI)/field desorption (FD) mass spectrometer (JEOL Germany, Freising, Germany). For clarity, only the highest measured peak is given for mass spectra. Melting points/decomposition temperatures were determined on a SMP3 Melting Point Apparatus (Stuart Scientific, Keison Products, Chelmsford, Essex, UK) and are uncorrected. Column chromatography was performed using a LaFlash system (VWR International GmbH, Darmstadt, Germany). The crude product was loaded on silica gel 60 (63–200 µm) (Macherey-Nagel, Düren, Germany) or PuriFlash IR-50 C18 modified silica gel (50 µm) (Interchim Deutschland GmbH, Mannheim, Germany) and packed in Interchim PuriFlash-DLE/12G dry-load precolumns. Pre-packed Interchim PuriFlash-30SIHP silica gel columns (30 µm, 40 g) and Interchim PuriFlash-15C18HP modified silica gel columns (15 µm, 55 g) were used for separation with flow rates usually adjusted to 30 mL∙min
−1 or 20.5 mL∙min
−1. Progress of reactions was monitored by thin-layer chromatography (TLC) performed with Macherey-Nagel 0.2 mm Polygram
® SIL G/UV
254 pre-coated silica gel polyester sheets and Silicagel 60 RP-18 F
254 modified silica gel aluminum plates (Merck Millipore, Darmstadt, Germany). Where necessary, reactions were carried out in a nitrogen atmosphere using 4 Å molecular sieves. All reagents and solvents were obtained from commercial sources (abcr GmbH, Karlsruhe, Germany; Sigma-Aldrich Chemie GmbH, Munich, Germany; Merck Group, Munich, Germany; Merck Millipore; Acros Organics Thermo Fisher Scientific, Geel, Belgium; VWR International GmbH, Hannover, Germany and used as received: THF was used after distillation over Na/benzophenone. HPLC analysis was performed on a Hewlett Packard HP 1050 Series using either a ZORBAX
® Eclipse XDB-C8 (150 mm × 4.6 mm, 5 µm) or a Kinetex
® C8 (150 × 4.6 mm, 5 µm) column (mobile phase flow 1.5 mL·min
−1, gradient KH
2PO
4 buffer 10 mM, pH 2.3/methanol, UV-detection 254 nm). All key compounds submitted to biological assays were proven by this method to show ≥98% purity. Syntheses under elevated pressure were performed in Berghof high
preactor™ BR-25 with corresponding heating block on a MR Hei-Standard laboratory heating plate (Heidolph, Schwabach, Germany). Microwave syntheses were performed in a CEM Discover Microwave Synthesizer (CEM GmbH, Kamp-Lintfort, Germany) under air cooling and high stirring with a maximal power of 100 W.
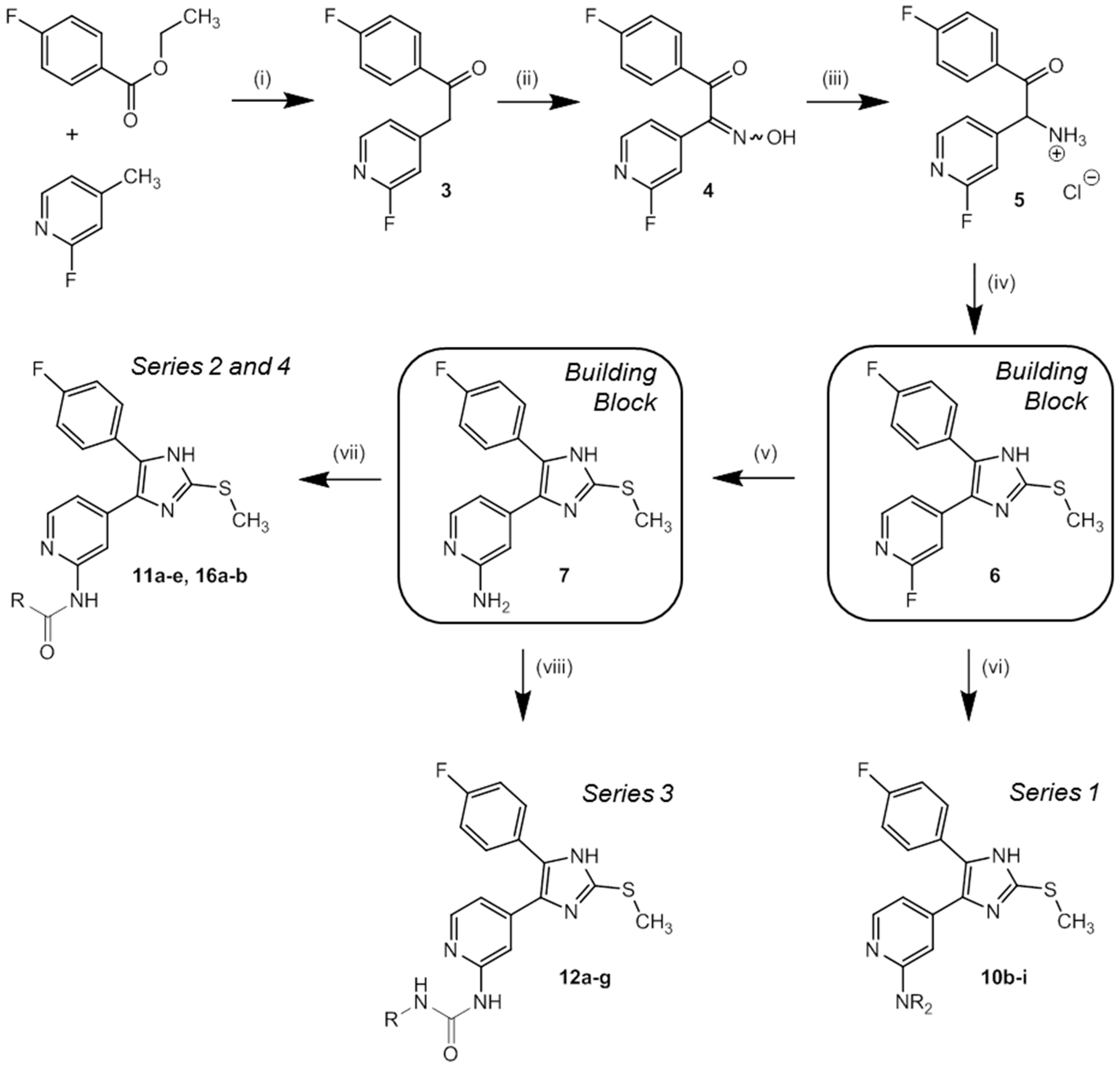
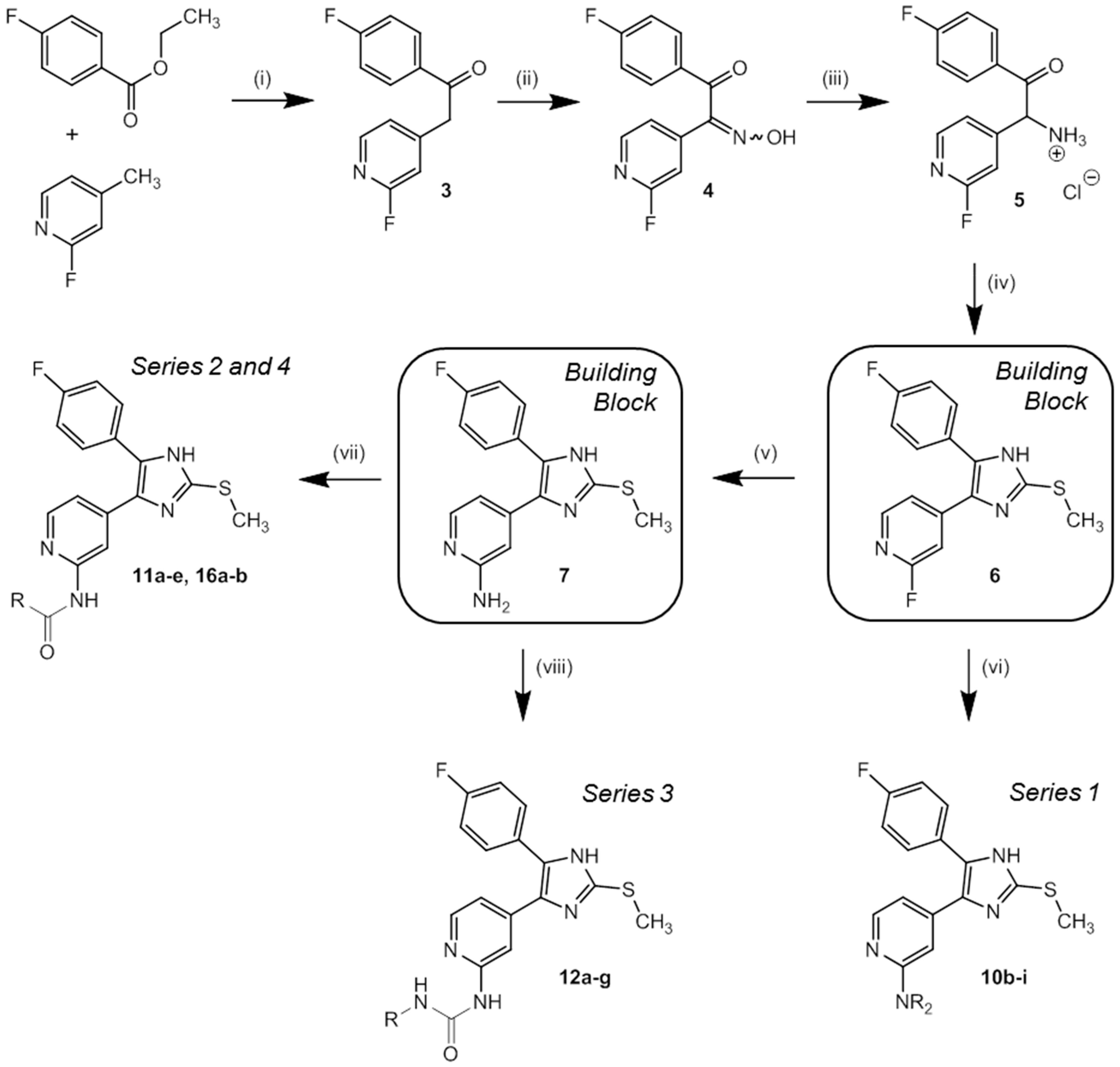
3.2.1. Syntheses of Key Building Blocks 3–7
1-(4-Fluorophenyl)-2-(2-fluoropyridin-4-yl)-ethan-1-one (3). NaHMDS (66.7 mL 2 M solution in THF, 133 mmol) was slowly added to a stirred solution of 2-fluoro-4-methylpyridine (10.6 mL, 103 mmol) and ethyl 4-fluorobenzoate (18.1 mL, 123 mmol) in 40 mL anhyd. THF at 0 °C under a nitrogen atmosphere. After stirring at 0 °C for 2 h the reaction was allowed to reach rt and stirring continued for 1 h. The mixture was diluted with ethyl acetate and washed twice with 10% aq. HCl. The organic layer was dried over anhyd. Na2SO4 and solvent was removed under reduced pressure. Recrystallization from ethyl acetate afforded 3 as colorless solid. Yield 23.9 g (quant.); C13H9F2NO (Mr 233.22); m.p. 102 °C; 1H-NMR (DMSO-d6): δ = 4.59 (s, 2H, CH2), 7.10 (s, 1H, C3H, Pyr), 7.25–7.27 (m, 1H, C5H, Pyr), 7.37–7.43 (m, 2H, C3/5H, F-Phe), 8.11–8.19 (m, 3H, C2/6H, F-Phe, C6H, Pyr) ppm; 13C-NMR (DMSO-d6): δ = 43.6 (d, 4JCF = 2.8 Hz, CH2), 110.8 (d, 2JCF = 37.6 Hz, C3H, Pyr), 115.8 (d, 2JCF = 22.0 Hz, C3/5H, F-Phe), 123.8 (d, 4JCF = 3.8 Hz, C5H, Pyr), 131.3 (d, 3JCF = 9.6 Hz, C2/6H, F-Phe), 132.9 (d, 4JCF = 2.7 Hz, C1, F-Phe), 147.0 (d, 5JCF = 15.5 Hz, C6H, Pyr), 150.7 (d, 3JCF = 8.5 Hz, C1, Pyr), 163.1 (d, 1JCF = 234.3 Hz, C2F, Pyr), 165.2 (d, 1JCF = 252.0 Hz, C4F, F-Phe), 194.6 (CO) ppm; MS (ESI, 70 eV) m/z 234 [MH]+.
1-(4-Fluorophenyl)-2-(2-fluoropyridin-4-yl)-2-(hydroximino)ethan-1-one (4). NaNO2 (2.06 g, 29.8 mmol) in 12 mL H2O was slowly added to a stirred solution of 3 (2.30 g, 9.86 mmol) in 17 mL glacial acetic acid at 10 °C. After stirring at r.t. for 1 h, 30 mL H2O were added and stirring continued for 3.5 h. The suspension was cooled to 8 °C, filtered, and the residue was washed with H2O and dried under reduced pressure to afford 4 as colorless solid. Yield 2.46 g (95%); C13H8F2N2O2 (Mr 262.22); m.p. 185 °C; 1H-NMR (DMSO-d6): δ = 7.19 (bs, 1H, C3H, Pyr), 7.37–7.46 (m, 3H, C5H, Pyr and C3/5H, F-Phe), 7.92–7.98 (m, 2H, C2/6H, F-Phe), 8.30 (d, 3J = 5.3 Hz, 1H, C6H, Pyr), 12.67 (s, 1H, OH) ppm; 13C-NMR (DMSO-d6): δ = 105.4 (d, 2JCF = 38.9 Hz, C3H, Pyr), 116.7 (d, 2JCF = 22.5 Hz, C3/5H, F-Phe), 118.2 (d, 4JCF = 4.2 Hz, C5H, Pyr), 130.8 (d, 4JCF = 2.7 Hz, C1, F-Phe), 132.2 (d, 3JCF = 10.1 Hz, C2/6H, F-Phe), 144.3 (d, 3JCF = 8.4 Hz, C4, Pyr), 148.8 (d, 5JCF = 15.6 Hz, C6H, Pyr), 151.9 (d, 4JCF = 4.0 Hz, CNOH), 163.5 (d, 1JCF = 235.6 Hz, C2F, Pyr), 166.0 (d, 1JCF = 254.8 Hz, C4F, F-Phe), 192.04 (CO) ppm; MS (ESI, 70 eV) m/z = 263 [MH]+.
2-(4-Fluorophenyl)-1-(2-fluoropyridin-4-yl)-2-oxoethan-1-aminium chloride (5). Pd/C 10% (279 mg) was added in one portion under a nitrogen atmosphere to an intensely stirred solution of 4 (1.60 g, 6.10 mmol) in 15 mL 2-propanol and 20 mL HCl sat. 2-propanol and stirring continued for 12 h at r.t. The crude product was obtained by filtration, the residue was resuspended in methanol, filtered again, and the solvent was removed under reduced pressure. Recrystallization from methanol/diethyl ether afforded 5 as beige solid. Yield 1.43 g (82%); C13H11ClF2N2O (Mr 284.69); m.p. 216 °C; 1H-NMR (DMSO-d6): δ = 6.56 (s, 1H, CH), 7.35–7.41 (m, 2H, C3/5H, F-Phe), 7.52–7.54 (m, 2H, C3/5H, Pyr), 8.21 (dd, 3J = 8.4 Hz, 4J = 5.6 Hz, 2H, C2/6H, F-Phe), 8.31 (d, 3J = 5.4 Hz, 1H, C6H, Pyr), 9.36 (bs, 3H, NH3+) ppm; 13C-NMR (DMSO-d6): δ = 56.8 (CNH3+), 110.5 (d, 2JCF = 39.3 Hz, C3H, Pyr), 116.9 (d, 2JCF = 22.1 Hz, C3/5H, F-Phe), 122.3 (d, 4JCF = 4.2 Hz, C5H, Pyr), 130.0 (d, 4JCF = 2.7 Hz, C1, F-Phe), 132.9 (d, 3JCF = 9.8 Hz, C2/6H, F-Phe), 147.4 (d, 3JCF = 8.0 Hz, C4, Pyr), 149.4 (d, 5JCF = 15.0 Hz, C6H, Pyr), 163.5 (d, 1JCF = 236.7 Hz, C2F, Pyr), 166.3 (d, 1JCF = 255.1 Hz, C4F, F-Phe), 191.4 (CO) ppm; MS (ESI, 70 eV) m/z 249 [MCl]+.
2-Fluoro-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridine (6). A mixture of 5 (4.41 g, 15.5 mmol) and methyl thiocyanate (3.18 mL, 46.5 mmol) was refluxed under a nitrogen atmosphere for 45 min in 140 mL anhyd. DMF and stirred 45 min at r.t. before ice-cold H2O (400 mL) was added. The suspension was cooled to 8 °C, filtered, the residue was washed with H2O, and dried under reduced pressure to afford 6 as fine yellow solid. Yield 3.59 g (76%); C15H11F2N3S (Mr 303.33); m.p. 205 °C; 1H-NMR (DMSO-d6): δ = 2.63 (s, 3H, SCH3), 7.09 (bs, 1H, C3H, Pyr), 7.26–7.34 (m, 3H, C5H, Pyr and C3/5H, F-Phe), 7.49–7.54 (m, 2H, C2/6H, F-Phe), 8.09 (d, 3J = 5.3 Hz, 1H, C6H, Pyr), 12.84 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 14.9 (SCH3), 105.0 (d, 2JCF = 39.5 Hz, C3H, Pyr), 115.9 (d, 2JCF = 21.6 Hz, C3/5H, F-Phe), 118.5 (d, 4JCF = 2.5 Hz, C5H, Pyr), 126.5 (C1, F-Phe), 130.9 (C2/6H, F Phe), 132.9 (d, 3JCF = 8.6 Hz, C4, Pyr), 143.0 (C2, Imdz), 147.6 (d, 3JCF = 16.1 Hz, C6H, Pyr), 162.1 (d, 1JCF = 246.4 Hz, CF, F-Phe), 163.7 (d, 1JCF = 233.3 Hz, CF, Pyr) ppm; MS (ESI, 70 eV) m/z 304 [MH]+.
4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-amine (7). A solution of 6 (1.00 g, 3.30 mmol) in 15 mL 32% aq. ammonia solution was heated in a high pressure reactor at 180 °C for 18 h. The reactor was allowed to reach r.t. and H2O was added. The crude product was obtained by filtration, washed with H2O and diisopropyl ether and purified by flash chromatography (SiO2, 50%–100% ethyl acetate/petrol ether) to afford 7 as beige solid. Yield 852 mg (86%); C15H13FN4S (Mr 300.36); 1H-NMR (DMSO-d6): δ = 2.60 (s, 1H, SCH3), 5.79–5.95 (m, 2H, NH2), 6.42–6.67 (m, 2H, C3/5H, Pyr), 7.17–7.27 (m, 2H, C3/5H, F-Phe), 7.48 (bs, 2H, C2/6H, F-Phe), 7.74–7.86 (m, 1H, C6H, Pyr), 12.58 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 105.1 (C5H, Pyr), 110.0 (C3H, Pyr), 115.7 (d, 3JCF = 21.1 Hz, C3/5H, F-Phe), 127.0 (s, C4/C5, Imdz), 129.2 (s, C1, F-Phe), 130.5 (d, 4JCF = 6.6 Hz, C2/6H, F-Phe), 147.5 (C6H, Pyr), 160.1 (CF) ppm; MS (ESI, 70 eV) m/z 301 [MH]+.
3.2.2. Synthesis of 4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-amines and -piperazines 8, 9, 10a–i
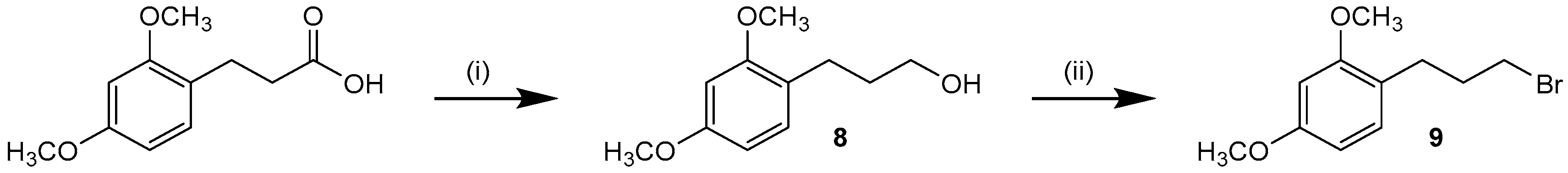
3-(2,4-Dimethoxyphenyl)propan-1-ol (8). Under a nitrogen atmosphere a solution of LiAlH4 (120 mg, 3.16 mmol) in 3 mL anhyd. THF was slowly added to 3-(2,4-dimethoxyphenyl)propionic acid (508 mg, 2.42 mmol) in 6 mL anhyd. THF at r.t. with intense stirring that continued for 1 h. After completion, the reaction was cooled down in an ice-bath and quenched by successive addition of H2O (120 µL), 15% aq. NaOH solution (120 µL), and H2O (360 µL). The precipitate was filtered off, washed with THF, and the filtrate was concentrated under reduced pressure. The crude product was purified by flash chromatography (SiO2, 20%–30% ethyl acetate/petrol ether) to afford 8 as colorless oil. Yield 457 mg (96%); C11H16O3 (Mr 196.25); 1H-NMR (CDCl3): δ = 1.78–1.83 (m, 2H, CH2CH2CH2OH), 2.65 (t, 3J = 7.3 Hz, 2H, CH2CH2CH2OH), 3.59 (t, 3J = 6.3 Hz, 1H, CH2CH2CH2OH), 3.78 (s, 1H, OH), 3.79 (s, 3H, C4OCH3), 3.81 (s, 3H, C2OCH3), 6.42–6.46 (m, 2H, C3/5H, (OCH3)2-Phe), 7.03 (dd, 3J = 7.7 Hz, 5J = 0.6 Hz, 1H, C6H, (OCH3)2-Phe) ppm; 13C-NMR (CDCl3): δ = 25.4 (CH2CH2CH2OH), 33.2 (CH2CH2CH2OH), 55.5 (C2/4OCH3), 62.1 (CH2CH2CH2OH), 98.6 (C3H, (OCH3)2-Phe), 104.3 (C5H, (OCH3)2-Phe), 122.4 (C1, (OCH3)2-Phe), 130.4 (C6H, (OCH3)2-Phe), 158.3 (C2OCH3), 159.3 (C4OCH3) ppm.
1-(3-Bromopropyl)-2,4-dimethoxybenzene (9). To an ice-cold solution of 8 (260 mg, 1.33 mmol) in 4 mL anhyd. DCM, triphenylphosphine (412 mg, 1.57 mmol) and N-bromosuccinimide (263 mg, 1.48 mmol) were added under a nitrogen atmosphere and the reaction was stirred for 2 h. The mixture was washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. Diethyl ether was added, the precipitate filtered off, and the filtrate was concentrated and purified by flash chromatography (SiO2, 2%–10% ethyl acetate/petrol ether) to afford 9 as colorless oil. Yield 205 mg (60%); C11H15BrO (Mr 259.14); 1H-NMR (CDCl3): δ = 2.11 (quint, 3J = 5.6 Hz, 2H, CH2CH2CH2Br), 2.70 (t, 3J = 7.2 Hz, 2H, CH2CH2CH2Br), 3.39 (t, 3J = 6.8 Hz, 2H, CH2CH2CH2Br), 3.80 (s, 6H, C2/4OCH3), 6.42 (dd, 3J = 8.0 Hz, 4J = 2.5 Hz, 1H, C5H, (OCH3)2-Phe), 6.45 (d, 4J = 2.3 Hz, 1H, C3H, (OCH3)2-Phe), 7.05 (dd, 3J = 8.0 Hz, 5J = 0.5 Hz, 1H, C6H, (OCH3)2-Phe) ppm; 13C-NMR (CDCl3): δ = 28.4 (CH2CH2CH2Br), 33.0 (CH2CH2CH2Br), 33.9 (CH2CH2CH2Br), 55.3 (C4OCH3), 55.5 (C2OCH3), 98.7 (C3H, (OCH3)2-Phe), 103.9 (C5H, (OCH3)2-Phe), 121.4 (C1, (OCH3)2-Phe), 130.5 (C6H, (OCH3)2-Phe), 158.5 (C2OCH3), 159.6 (C4OCH3) ppm.
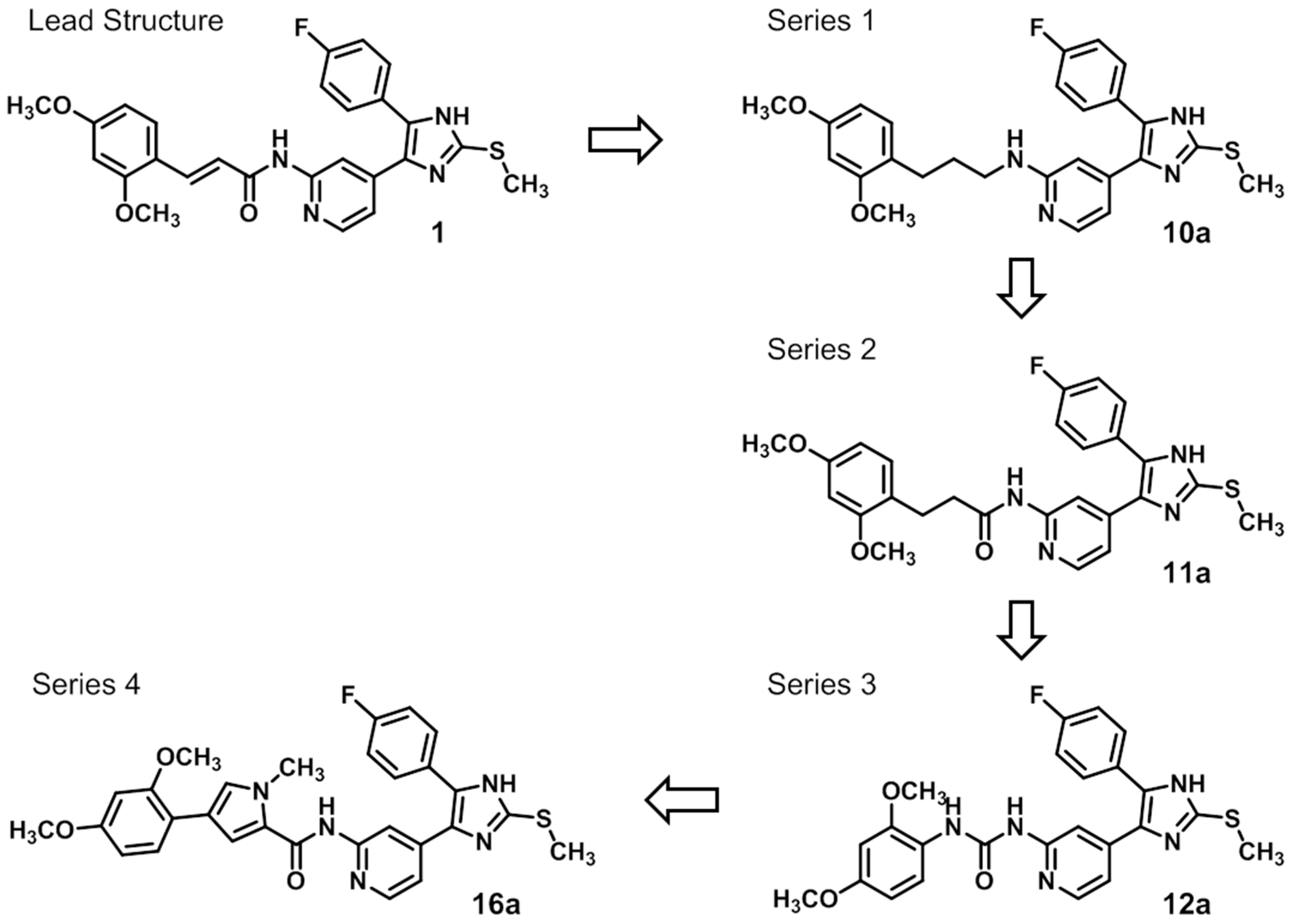
N-(3-(2,4-Dimethoxyphenyl)propyl)-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-amine (10a). The synthetic protocol starting from Boc-protected 2-amino-4-methylpyridine and 9 predominantly matches the procedure described for 7 and can be found in the Supporting Information. 1H-NMR (DMSO-d6): δ = 1.67–1.72 (m, 2H, CH2CH2CH2NH), 2.48–2.52 (m, 2H, CH2CH2CH2NH), 2.60 (s, 3H, SCH3), 3.13–3.17 (m, 2H, CH2CH2CH2NH), 3.72 (s, 3H, C4OCH3), 3.74 (s, 3H, C2OCH3), 6.41–6.50 (m, 5H, C3/5H, Pyr and C3/5H, (OCH3)2-Phe and CH2CH2CH2NH), 7.01 (d, 3J = 8.2 Hz, 1H, C6H, (OCH3)2-Phe), 7.22 (m, 2H, C3/5H, F-Phe), 7.48 (m, 2H, C2/6H, F-Phe), 7.85 (bs, C6H, Pyr), 12.58 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 26.6 (CH2CH2CH2NH), 29.3 (CH2CH2CH2NH), 40.7 (CH2CH2CH2NH), 55.1 (C4OCH3), 55.2 (C2OCH3), 98.3 (C3H, (OCH3)2-Phe), 104.3 (C5H, (OCH3)2-Phe), 104.9 (C3H, Pyr), 109.7 (C5H, Pyr), 115.4 (C3/5H, F-Phe), 121.8 (C1, (OCH3)2-Phe), 126.5 (C4, Imdz), 127.0 (C5, Imdz), 129.7 (C1, F-Phe), 129.7 (C6H, (OCH3)2-Phe), 130.1 (C2/6H, F-Phe), 142.1 (C2, Imdz), 147.5 (C6H, Pyr), 157.9 (C2OCH3), 158.8 (C4OCH3), 159.3 (C2, Pyr) ppm; MS (ESI, 70 eV) m/z 479 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C26H27FN4O2S, 478.1839; found, 478.1839.
General Procedure for the Preparation Compounds 10b–i
Compound 6 (1.0 equiv) was suspended in the appropriate amine/piperazine (4.0 to 6.0 equiv) and the intensely stirred mixture was heated to 160 °C. Progress of the reaction was monitored by HPLC control. After complete conversion the mixture was diluted with ethyl acetate, washed with sat. aq. NaHCO3 solution and H2O, dried over anhyd. Na2SO4, and concentrated under reduced pressure. The crude product was purified by flash chromatography (stationary phase, eluent, and mixing ratio given for each compound, respectively) to afford the particular compound.
N-(2,4-Dimethoxyphenyl)-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-amine (10b). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and 2,4-dimethoxyaniline (808 mg, 5.27 mmol). Purification was achieved by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether) to afford 10b as greyish crystals. Yield 312 mg (54%); C23H21FN4O2S (Mr 436.51); m.p. 206 °C; 1H-NMR (DMSO-d6): δ = 2.60 (s, 3H, SCH3), 3.75 (s, 6H, 2 OCH3), 6.39–6.44 (m, 1H, C5H, (OCH3)2-Phe), 6.56–6.58 (m, 1H, C3H, (OCH3)2-Phe), 6.63 (dd, 3J = 5.3 Hz, 4J = 1.2 Hz, 1H, C5H, Pyr), 6.82 (s, 1H, C3H, Pyr), 7.18–7.28 (m, 2H, C3/5H, F-Phe), 7.41–7.46 (m, 2H, C2/6H, F-Phe), 7.52 (d, 3J = 8.7 Hz, 1H, C6H, (OCH3)2-Phe), 7.71 (s, 1H, NH), 7.95 (d, 3J = 5.3 Hz, 1H, C6H, Pyr), 12.60 (s, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 55.2 (OCH3), 55.2 (OCH3), 99.0 (C3H, (OCH3)2-Phe), 104.0 (C5H, (OCH3)2-Phe), 105.4 (C3H, Pyr), 111.5 (C5H, Pyr), 115.4 (dd, 2JCF = 21.8 Hz, C3/5H, F-Phe), 122.5 (C, Imdz), 123.6 (C6H, (OCH3)2-Phe), 126.8 (d, 4JCF = 3.1 Hz, C1, F-Phe), 130.0 (d, 3JCF = 8.1 Hz, C2/6H, F-Phe), 134.9 (C1, (OCH3)2-Phe), 138.8 (C, Imdz), 141.7 (C2OCH3), 142.7 (C2, Imdz), 147.6 (C6H, Pyr), 152.2 (C4OCH3), 155.9 (C4, Pyr), 157.4 (C2, Pyr), 161.8 (d, 1JCF = 245.0 Hz, CF) ppm; MS (ESI, 70 eV) m/z 437 [MH]+.
N-(2-Ethoxyphenyl)-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-amine (10c). Synthesis was performed according to the general procedure from 6 (1.50 g, 4.95 mmol) and 2-ethoxyaniline (3.88 mL, 29.7 mmol). Purification was achieved by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether) to afford 10c as dark red crystals. Yield 1.10 g (52%); C23H21FN4OS (Mr 420.51); m.p. 179 °C; 1H-NMR (CDCl3): δ = 1.39 (t, 3J = 7.0 Hz, 3H, CH2CH3), 2.63 (s, 3H, SCH3), 4.03 (q, 3J = 7.0 Hz, 2H, CH2CH3), 6.76 (t, 3J = 7.7 Hz, 1H, C5H, EtO-Phe), 6.81–6.84 (m, 2H, C5H, Pyr and C3H, EtO-Phe), 6.91 (t, 3J = 7.7 Hz, 1H, C4H, EtO-Phe), 6.97 (bs, 1H, C3H, Pyr), 7.00–7.05 (t, 3J = 8.7 Hz, 2H, C3/5H, F-Phe), 7.21 (bs, 1H, NH), 7.37–7.41 (m, 2H, C2/6H, F-Phe), 7.49 (d, 3J = 7.7 Hz, 1H, C6H, EtO-Phe), 8.00 (d, 3J = 5.5 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 15.0 (CH3CH2), 16.6 (SCH3), 64.3 (CH2CH3), 106.5 (C3H, Pyr), 111.7 (C3H, EtO-Phe), 112.9 (C5H, Pyr), 116.0 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 120.0 (C6H, EtO-Phe), 120.7 (C5H, EtO-Phe), 122.6 (C4H, EtO-Phe), 129.4 (C1, EtO-Phe), 130.5 (d, 3JCF = 8.1 Hz, C2/6H, F-Phe), 143.2 (C2, Imdz), 146.8 (C6H, Pyr), 148.7 (COEt), 155.5 (C2, Pyr), 162.5 (d, 1JCF = 249.0 Hz, CF) ppm; MS (ESI, 70 eV) m/z = 421 [MH]+.
N-(3,4-Dimethoxyphenethyl)-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-amine (10d). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and 3,4-dimethoxyphenethylamine (900 µL, 5.31 mmol). Purification was achieved by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether) to afford 10d as yellow crystals. Yield 550 mg (90%); C25H25FN4O2S (Mr 464.56); m.p. 87 °C; 1H-NMR (CDCl3): δ = 2.55 (s, 3H, SCH3), 2.69 (t, 3J = 6.9 Hz, 2H, CH2CH2NH), 3.29 (m, 2H, CH2CH2NH), 3.74 (s, 3H, C3OCH3), 3.75 (s, 3H, C4OCH3), 4.86 (bs, 1H, CH2CH2NH), 6.51 (d, 3J = 5.2 Hz, 1H, C5H, Pyr), 6.57 (d, 4J = 2.0 Hz, 1H, C3H, Pyr), 6.57–6.70 (m, 3H, C2/5/6H, (OCH3)2-Phe), 6.91–6.96 (m, 2H, C3/5H, F-Phe), 7.32–7.36 (m, 2H, C2/6H, F-Phe), 7.74 (d, 3J = 5.8 Hz, 1H, C6H, Pyr), 10.77 (bs, 1H, NH) ppm; 13C-NMR (CDCl3): δ = 16.4 (SCH3), 35.1 (CH2CH2NH), 43.5 (CH2CH2NH), 55.9 (C3OCH3), 55.9 (C4OCH3), 104.1 (C3H, Pyr), 111.0 (C5H, Pyr), 111.4 (C5H, (OCH3)2-Phe), 112.1 (C2H, (OCH3)2-Phe), 115.7 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 120.7 (C6H, (OCH3)2-Phe), 130.3 (d, 3JCF = 8.2 Hz, C2/6H, F-Phe), 131.4 (C4/5, Imdz), 142.9 (C4, Pyr), 146.7 (C6H, Pyr, 147.7 (C4OCH3), 149.0 (C3OCH3), 158.3 (C2, Pyr), 162.6 (d, 1JCF = 248.5 Hz, CF) ppm; MS (ESI, 70 eV) m/z 465 [MH]+.
N-(2-(1H-Indol-3-yl)-ethyl)-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-amine (10e). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and tryptamine (845 mg, 5.27 mmol). Purification was achieved by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether) to afford 10e as yellowish-brown crystals. Yield 427 mg (73%); C25H22FN5S (Mr 443.54); m.p. 122 °C; 1H-NMR (CDCl3): δ = 2.61 (s, 3H, SCH3), 2.95 (t, 3J = 6.6 Hz, 2H, CH2CH2NH), 3.39 (t, 3J = 6.6 Hz, 2H, CH2CH2NH), 6.52 (d, 3J = 5.7 Hz, 1H, C5H, Pyr), 6.52 (bs, 1H, C2H, Indole), 6.87 (d, 4J = 2.1 Hz, 1H, C3H, Pyr), 6.95–7.01 (m, 2H, C3/5H, F-Phe), 7.08 (t, 3J = 7.8 Hz, 1H, C6H, Indole), 7.16 (t, 3J = 7.9 Hz, 1H, C5H, Indole), 7.30 (d, 3J = 7.9 Hz, 1H, C4H, Indole), 7.35–7.39 (m, 2H, C2/6H, F-Phe), 7.52 (d, 3J = 7.7 Hz, 1H, C7H, Indole), 7.71 (d, 3J = 5.7 Hz, 1H, C6H, Pyr), 8.38 (s, 1H, NH, Indole) ppm; 13C-NMR (CDCl3): δ = 16.4 (SCH3), 25.1 (CH2CH2NH), 42.2 (CH2CH2NH), 104.0 (C3H, Pyr), 110.6 (C5H, Pyr), 111.3 (C4H, Indole), 112.6 (C3, Indole), 115.8 (d, 2JCF = 21.6 Hz, C3/5H, F-Phe), 118.6 (C7H, Indole), 119.4 (C6H, Indole), 122.1 (C5H, Indole), 122.5 (C2H, Indole), 127.2 (C8a, Indole), 130.3 (3JCF = 8.3 Hz, C2/6H, F-Phe), 136.4 (C3a, Indole), 143.3 (C2, Imdz), 145.1 (C6H, Pyr), 158.0 (C2, Pyr) ppm; MS (ESI, 70 eV) m/z 444 [MH]+.
N-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-N′,N′-dimethyl-propan-1,3-diamine (10f). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and N1,N1-dimethylpropane-1,3-diamine (1.00 mL, 7.93 mmol). Purification was achieved by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether, then methanol) and subsequent filtration, to afford 10f as yellow solid. Yield 175 mg (35%); C20H24FN5S (Mr 385.51); m.p. 129 °C; 1H-NMR (CDCl3): δ = 1.61 (t, 3J = 6.8 Hz, 2H, CH2CH2CH2), 2.13 (s, 6H, N(CH3)2), 2.29 (t, 3J = 6.9 Hz, 2H, CH2N(CH3)2), 2.58 (s, 3H, SCH3), 3.11 (t, 3J = 6.7 Hz, 2H, CH2NH), 5.22 (bs, 1H, CH2NH), 6.42–6.46 (m, 2H, C3/5H, Pyr), 6.95–7.01 (m, 2H, C3/5H, F-Phe), 7.35–7.39 (m, 2H, C2/6H, F-Phe), 7.78 (dd, 3J = 5.4 Hz, 5J = 0.6 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 16.5 (SCH3), 26.7 (CH2CH2CH2), 45.2 (N(CH3)2), 57.6 (CH2N(CH3)2), 104.0 (C3H, Pyr), 111.1 (C5H, Pyr), 115.5 (d, 2JCF = 21.5 Hz, C3/5H, F-Phe), 130.2 (d, 3JCF = 8.1 Hz, C2/6H, F-Phe), 142.7 (C2, Imdz), 147.9 (C6H, Pyr), 159.2 (C2, Pyr), 162.4 (d, 1JCF = 247.7 Hz, CF) ppm; MS (ESI, 70 eV) m/z 386 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-piperazine (10g). Synthesis was performed according to the general procedure from 6 (300 mg, 989 µmol) and piperazine (341 mg, 3.96 mmol). Purification was achieved by flash chromatography (bas. Al2O3, 100% methanol) to afford 10g as yellow solid. Yield 63.0 mg (17%); C19H20FN5S (Mr 369.46); m.p. 146 °C; 1H-NMR (DMSO-d6): δ = 2.60 (bs, 3H, SCH3), 2.73 (t, 3J = 4.5 Hz, 4H, C2/6H2, Piperazine), 3.31 (t, 3J = 4.6 Hz, 4H, C3/5H2, Piperazine), 6.56 (d, 3J = 5.1 Hz, 1H, C5H, Pyr), 6.80 (bs, 1H, C3H, Pyr), 7.20–7.26 (m, 2H, C3/5H, F-Phe), 7.47–7.51 (m, 2H, C2/6H, F-Phe), 7.97 (d, 3J = 5.1 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 45.3 (C2/6H2, Piperazine), 45.8 (C3/5H2, Piperazine), 103.8 (C3H, Pyr), 110.6 (C5H, Pyr), 115.4 (d, 2JCF = 21.6 Hz, C3/5H, F-Phe), 130.3 (d, 3JCF = 7.6 Hz, C2/6H, F-Phe), 142.3 (C2, Imdz), 147.6 (C6H, Pyr), 159.6 (C2, Pyr), 161.6 (d, 1JCF = 245.1 Hz, CF) ppm; MS (ESI, 70 eV) m/z 370 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-4-methyl-piperazine (10h). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and N-methylpiperazine (600 µL, 5.39 mmol). Purification was achieved by flash chromatography (RP-18, 20%–90% methanol/H2O) to afford 10h as pale yellow solid. Yield 115 mg (23%); C20H22FN5S (Mr 383.49); m.p. 111 °C; 1H-NMR (CDCl3): δ = 2.28 (s, 3H, CH3), 2.45 (t, 3J = 4.8 Hz, 4H, C3/5H2, Piperazine), 2.57 (s, 3H, SCH3), 3.40 (t, 3J = 4.8 Hz, 4H, C2/6H2, Piperazine), 6.54 (dd, 3J = 5.3 Hz, 4J = 1.1 Hz, 1H, C5H, Pyr), 6.73 (bs, 1H, C3H, Pyr), 6.91–6.97 (m, 2H, C3/5H, F-Phe), 7.32–7.37 (m, 2H, C2/6H, F-Phe), 7.94 (d, 3J = 5.3 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 16.5 (SCH3), 44.7 (C2/6H2, Piperazine), 45.6 (CH3), 54.4 (C3/5H2, Piperazine), 104.9 (C3H, Pyr), 111.9 (C5H, Pyr), 115.7 (d, 2JCF = 21.6 Hz, C3/5H, F-Phe), 128.2 (d, 4JCF = 4.1 Hz, CC1, F-Phe), 130.2 (d, 3JCF = 8.1 Hz, C2/6H, F-Phe), 142.8 (C2, Imdz), 147.9 (C6H, Pyr), 159.6 (C2, Pyr), 162.5 (d, 1JCF = 248.3 Hz, CF) ppm; MS (ESI, 70 eV) m/z 384 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-4-phenyl-piperazine (10i). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and N-phenylpiperazine (800 µL, 5.23 mmol). The combined organic phases were extracted with 2 M aq. HCl solution, the aq. layer was neutralized with 2 M aq. KOH solution, and the pale yellow precipitate was collected by filtration, and purified by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether) to afford 10i as yellow crystals. Yield 291 mg (50%); C20H22FN5S (Mr 445.56); m.p. 101 °C; 1H- NMR (CDCl3): δ = 2.64 (s, 3H, SCH3), 3.19 (t, 3J = 5.1 Hz, 4H, C2/6H2, Piperazine), 3.58 (t, 3J = 5.1 Hz, 4H, C3/5H2, Piperazine), 6.64 (dd, 3J = 5.4 Hz, 4J = 1.2 Hz, 1H, C5H, Pyr), 6.93–6.85 (m, 4H, C3H, Pyr and C2/4/6H, Phe), 6.99–7.05 (m, 2H, C3/5H, F-Phe), 7.26 (t, 3J = 8.0 Hz, 2H, C3/5H, Phe), 7.39–7.44 (m, 2H, C2/6H, F-Phe), 8.00 (d, 3J = 5.5 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 16.5 (SCH3), 45.4 (C2/6H2, Piperazine), 49.0 (C3/5H2, Piperazine), 105.1 (C3H, Pyr), 111.6 (C5H, Pyr), 115.8 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 116.3 (C2/6H, Phe), 120.2 (C4H, Phe), 129.2 (C3/5H, Phe), 130.3 (d, 3JCF = 8.1 Hz, C2/6H, F-Phe), 143.0 (C2, Imdz), 146.9 (C6H, Pyr), 151.1 (C1, Phe), 159.0 (C2, Pyr), 162.6 (d, 1JCF = 248.5 Hz, CF) ppm; MS (ESI, 70 eV) m/z 446 [MH]+.
3.2.3. Synthesis of N-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-phenyl-propanamides 11a–e
The appropriate methoxy-substituted 3-phenylpropionic acid and CDI (1.1 equiv) were stirred in 3–4 mL anhyd. DMF until formation of CO2 was undetectable. 7 was added to the reaction and the mixture was heated at 110 °C under a nitrogen atmosphere for 12 h. The reaction was cooled to rt and ethyl acetate was added. The resulting mixture was washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (SiO2 and RP-18, eluent and mixing ratio given for each compound, respectively) to afford the particular compound.
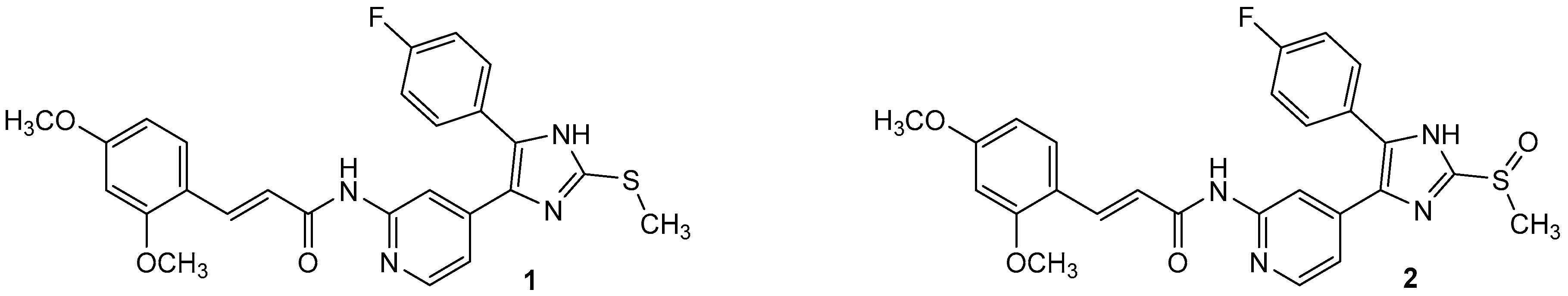
3-(2,4-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)propanamide (11a). Synthesis was performed according to the general procedure from 3-(2,4-dimethoxyphenyl)propionic acid (560 mg, 2.66 mmol) and 7 (400 mg, 1.33 mmol). Purification was achieved by flash chromatography (SiO2, 30%–50% ethyl acetate/petrol ether and RP-18, 30%–80% methanol/H2O) to afford 11a as beige solid. Yield 177 mg (27%); C26H25FN4O3S (Mr 492.57); m.p. 90 °C; 1H-NMR (DMSO-d6): δ = 2.57 (t, 3J = 7.4 Hz, 2H, COCH2CH2), 2.62 (s, 3H, SCH3), 2.75 (t, 3J = 7.5 Hz, 2H, COCH2CH2), 3.72 (s, 3H, C2OH3), 3.77 (s, 3H, C4OCH3), 6.42 (dd, 3J = 8.3 Hz, 4J = 2.4 Hz, 1H, C5H, (OCH3)2-Phe), 6.51 (d, 4J = 2.4 Hz, 1H, C3H, (OCH3)2-Phe), 6.99 (dd, 3J = 5.3 Hz, 1H, C5H, Pyr), 7.03 (d, 3J = 8.3 Hz, 1H, C6H, (OCH3)2-Phe), 7.28 (bs, 2H, C3/5H, F-Phe), 7.46–7.51 (m, 2H, C2/6H, F-Phe), 8.12 (bs, 1H, C6H, Pyr), 8.32 (bs, 1H, C3H, Pyr), 10.29 (bs, 1H, CONH), 12.72 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 24.7 (COCH2CH2), 36.3 (COCH2CH2), 55.1 (C2OCH3), 55.3 (C4OCH3), 98.3 (C3H, (OCH3)2-Phe), 104.3 (C5H, (OCH3)2-Phe), 110.6 (C3H, Pyr), 115.8 (d, 2JCF = 24.1 Hz, C3/5H, F-Phe), 116.4 (C5H, Pyr), 120.9 (C1, (OCH3)2-Phe), 126.7 (C1, F-Phe), 129.8 (C6H, (OCH3)2-Phe), 130.7 (C5, Imdz and C2/6H, F-Phe), 134.5 (C4, Imdz), 143.7 (C2, Imdz), 147.6 (C6H, Pyr), 148.0 (C4, Pyr), 152.5 (C2, Pyr), 157.9 (C4, (OCH3)2-Phe), 159.0 (C2, (OCH3)2-Phe), 161.9 (d, 1JCF = 246.7 Hz, CF), 171.4 (CO) ppm; IR (ATR): ν = 2935, 1670, 1609, 1547, 1505, 1414, 1289, 1262, 1221, 1207, 1153, 1121, 1036, 835 cm−1; MS (ESI, 70 eV) m/z 493 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C26H25FN4O3S, 492.1631; found, 492.1631.
3-(2,5-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-propanamide (
11b). Synthesis was performed according to the general procedure from 3-(2,5-dimethoxyphenyl)propionic acid (560 mg, 2.66 mmol) and
7 (400 mg, 1.33 mmol). Purification was achieved by flash chromatography (SiO
2, 20%–100% ethyl acetate/petrol ether and RP-18, 20%–100% methanol/H
2O) to afford
11b as colorless solid. Yield 255 mg (39%); C
26H
25FN
4O
3S (M
r 492.57); m.p. 91 °C;
1H-NMR (DMSO-
d6): δ = 2.59–2.64 (m, 5H, SC
H3 and COC
H2CH
2), 2.80 (t,
3J = 7.5 Hz, 2H, COCH
2C
H2), 3.66 (s, 3H, C
5OC
H3), 3.73 (s, 3H, C
2OC
H3), 6.72 (dd,
3J = 8.8 Hz,
4J = 3.1 Hz, 1H, C
4H, (OCH
3)
2-Phe), 6.78 (d,
4J = 3.0 Hz, 1H, C
6H, (OCH
3)
2-Phe), 6.86 (d,
3J = 8.8 Hz, 1H, C
3H, (OCH
3)
2-Phe), 6.99 (dd,
3J = 5.2 Hz,
4J = 1.6 Hz, 1H, C
5H, Pyr), 7.26 (bs, 2H, C
3/5H, F-Phe), 7.46–7.51 (m, 2H, C
2/6H, F-Phe), 8.13 (bs, 1H, C
6H, Pyr), 8.32 (bs, 1H, C
3H, Pyr), 10.34 (bs, 1H, CON
H), 12.72 (bs, 1H, N
H) ppm;
13C-NMR (DMSO-
d6): δ = 15.1 (S
CH
3), 25.3 (COCH
2CH
2), 36.0 (CO
CH
2CH
2), 55.2 (C
5O
CH
3), 55.8 (C
2O
CH
3), 110.6 (
C3H, Pyr), 111.2 (
C4H, (OCH
3)
2-Phe), 111.5 (
C3H, (OCH
3)
2-Phe), 115.6 (
C3/5H, F-Phe), 115.9 (
C6H, (OCH
3)
2-Phe), 116.5 (
C5H, Pyr), 126.7 (
C1, F-Phe), 130.0 (
C1, (OCH
3)
2-Phe), 130.6 (
C5, Imdz and
C2/6H, F-Phe), 134.5 (
C4, Imdz), 142.3 (
C2, Imdz), 147.7 (
C4 and
C6H, Pyr), 151.2 (
C2, (OCH
3)
2-Phe), 152.5 (
C2, Pyr), 153.0 (
C5, (OCH
3)
2-Phe), 161.9 (d,
1JCF = 247.3,
CF), 171.3 (
CO) ppm; MS (ESI, 70 eV)
m/
z 494 [MH]
+; HRMS (EI, 70 eV)
m/
z [M]
+ calcd. for C
26H
25FN
4O
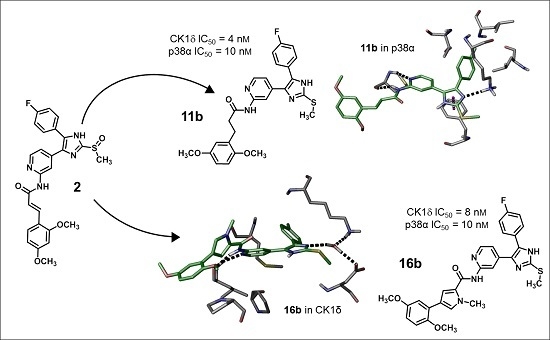
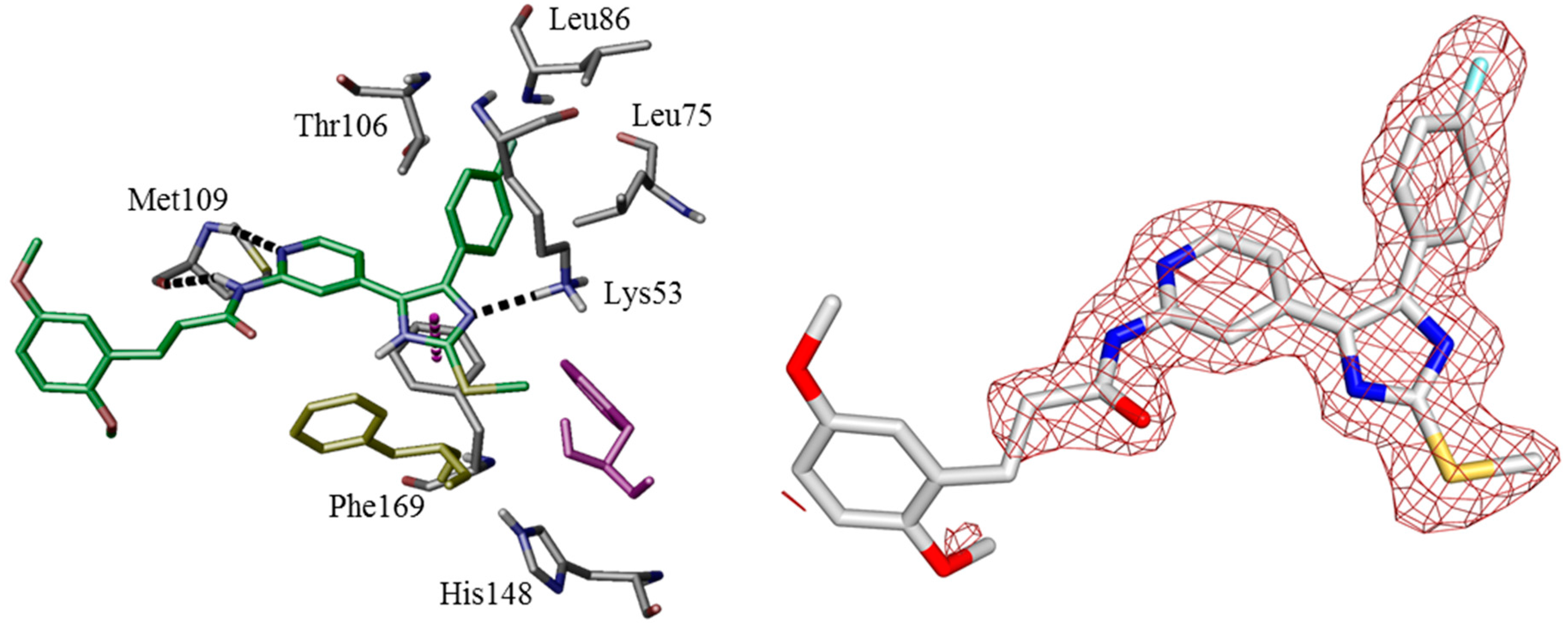
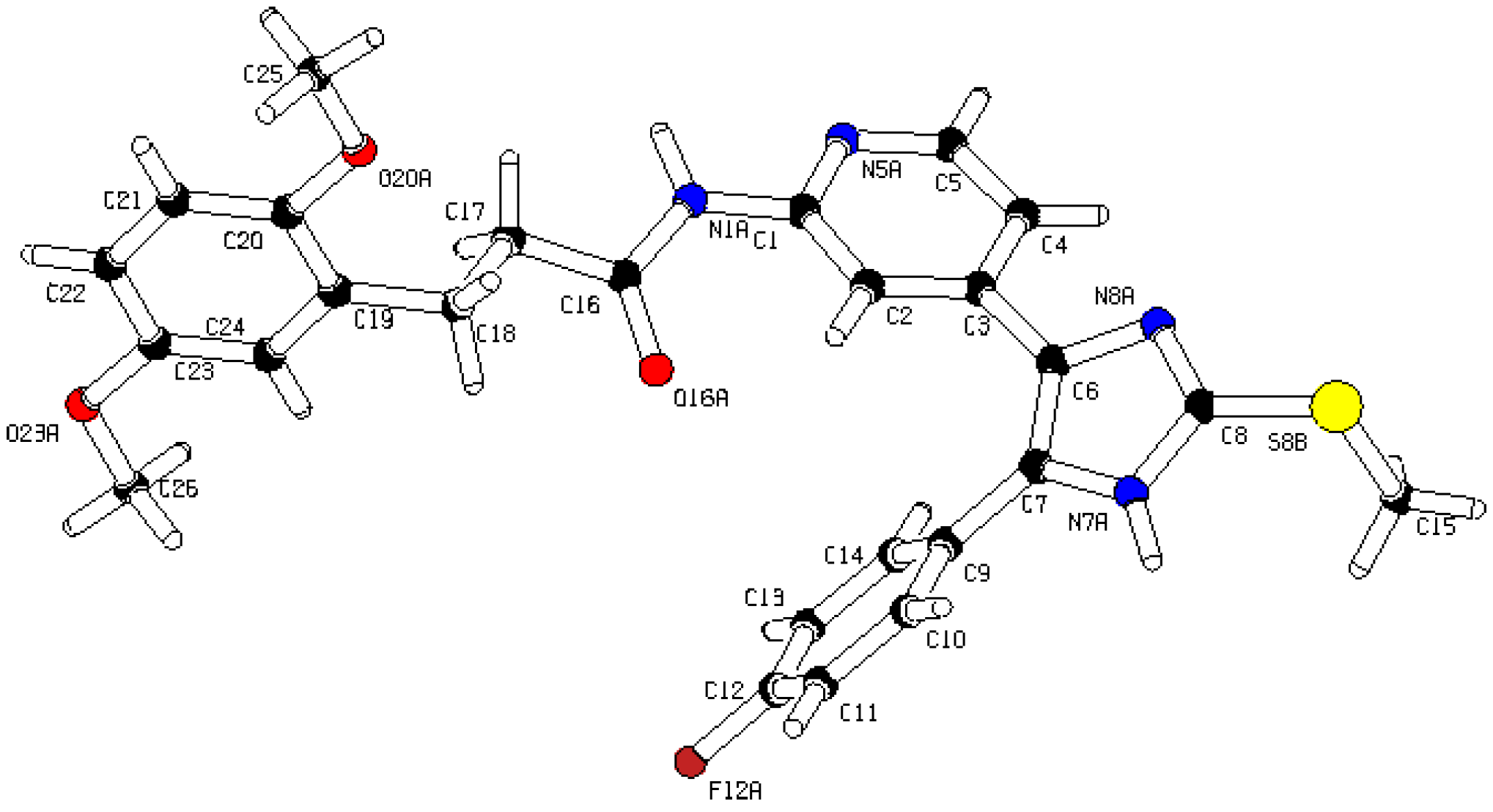
3S, 492.1631; found, 492.1631. The compound was demonstrated to have the desired structure by small molecule X-ray structure determination (
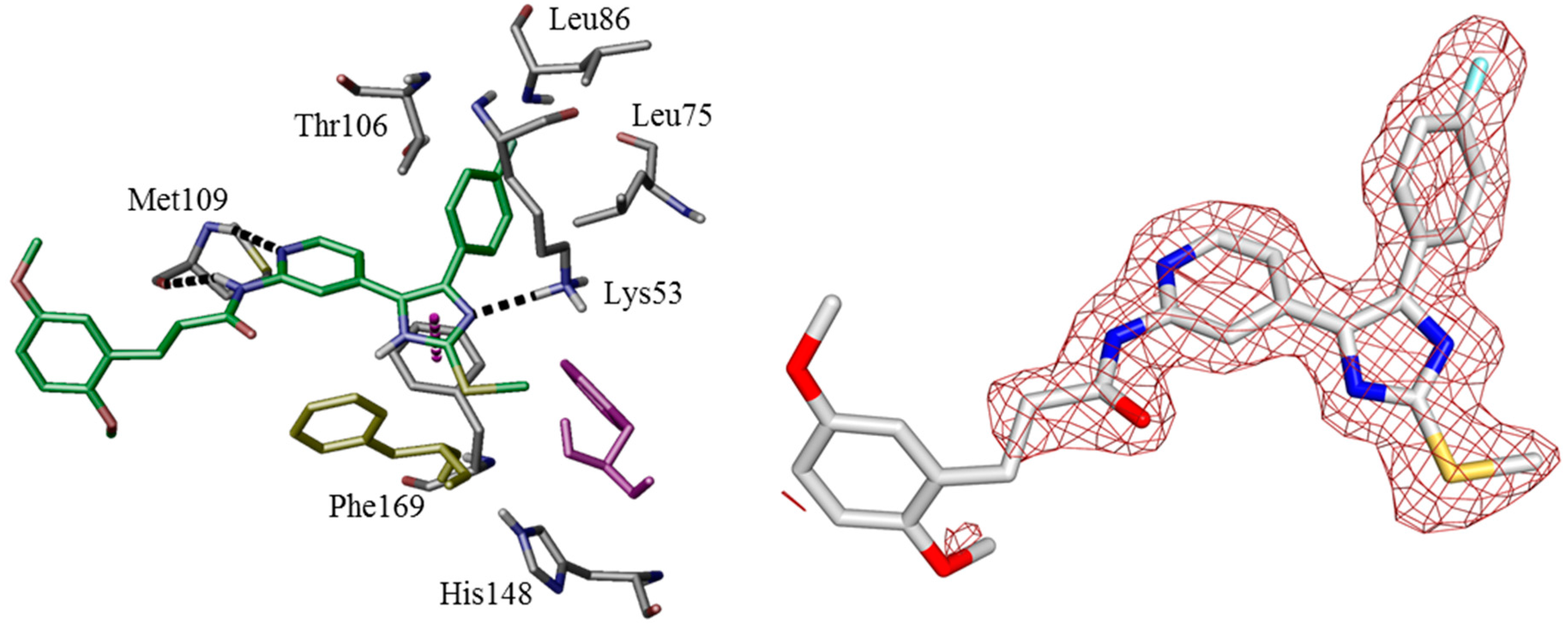
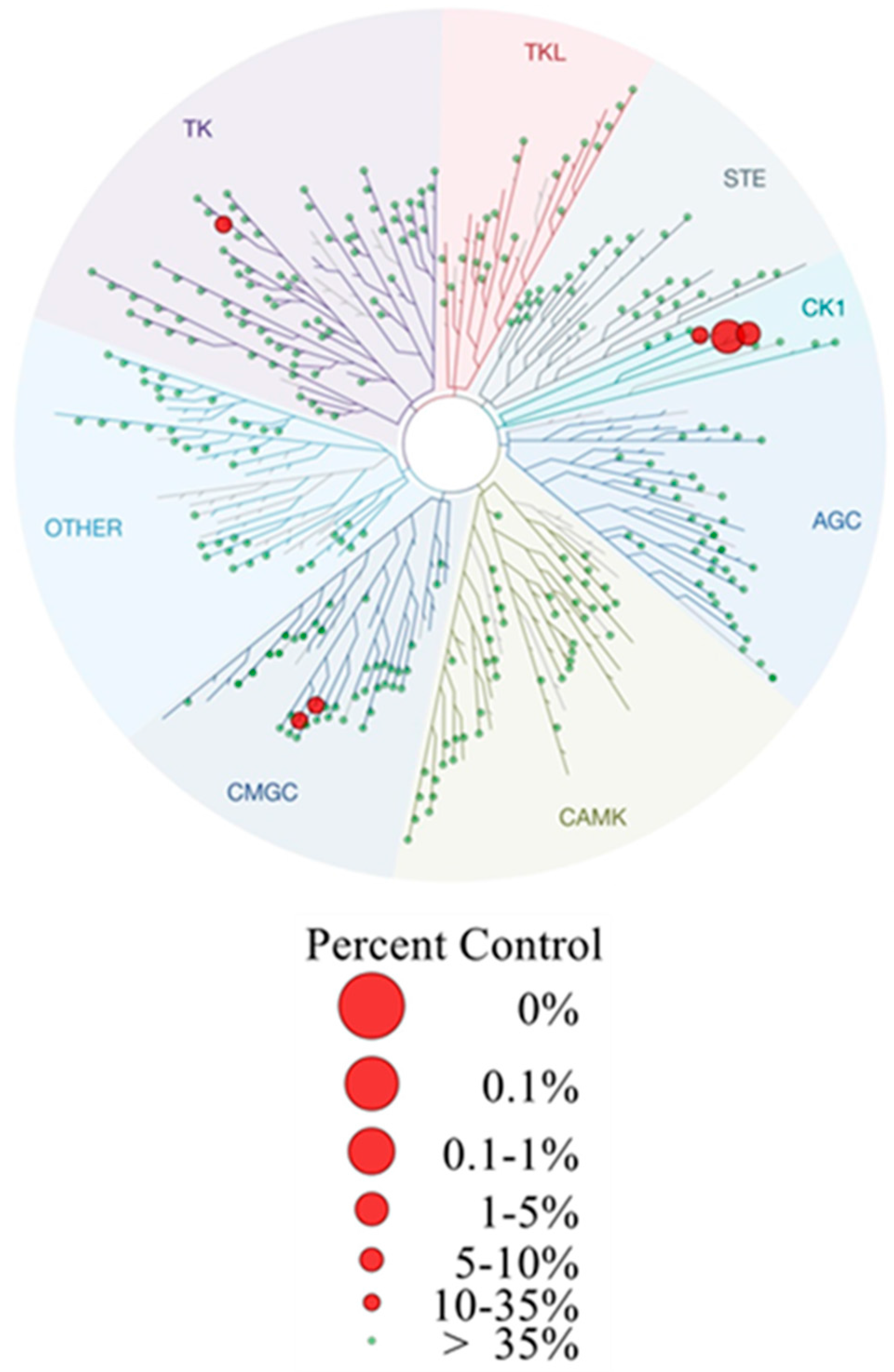
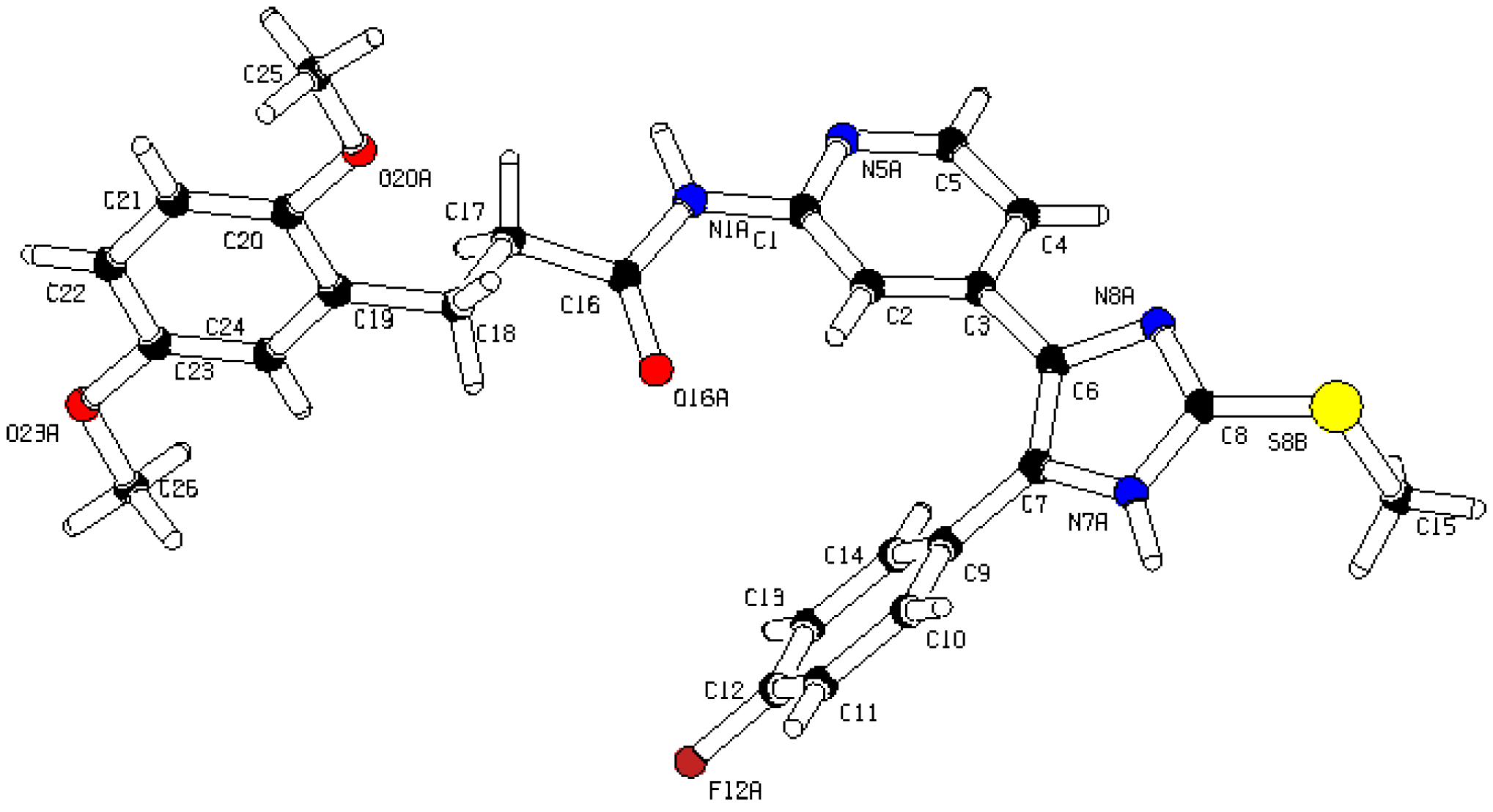
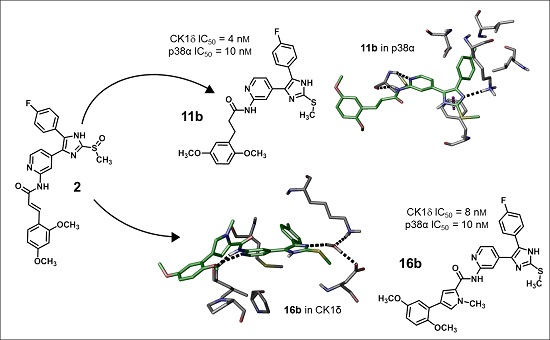
Figure 6). For further details see CCDC and Supporting Information.
3-(2,3-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-propanamide (11c). Synthesis was performed according to the general procedure from 3-(2,3-dimethoxyphenyl)propionic acid (700 mg, 3.33 mmol) and 7 (500 mg, 1.67 mmol). Purification was achieved by flash chromatography (SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 11c as colorless solid. Yield 515 mg (63%); C26H25FN4O3S (Mr 492.57); 1H-NMR (DMSO-d6): δ = 2.62 (s, 3H, SCH3), 2.59–2.66 (m, 2H, COCH2CH2), 2.84 (t, 3J = 7.7 Hz, 2H, COCH2CH2), 3.73 (s, 3H, C2OCH3), 3.78 (s, 3H, C3OCH3), 6.79 (dd, 3J = 7.5 Hz, 4J = 1.6 Hz, 1H, C6H, (OCH3)2-Phe), 6.88 (dd, 3J = 8.1 Hz, 4J = 1.2 Hz, 1H, C4H, (OCH3)2-Phe), 6.97 (t, 3J = 7.8 Hz, 1H, C5H, (OCH3)2-Phe), 7.01 (dd, 3J = 5.3 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.26–7.32 (m, 2H, C3/5H, F-Phe), 7.45–7.51 (m, 2H, C2/6H, F-Phe), 8.11 (d, 3J = 5.3 Hz, 1H, C6H, Pyr), 8.34 (bs, 1H, C3H, Pyr), 10.35 (s, 1H, CONH), 12.70 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 24.8 (COCH2CH2), 36.8 (COCH2CH2), 55.3 (C3OCH3), 60.0 (C2OCH3), 110.6 (C3H, Pyr), 110.9 (C4H, (OCH3)2-Phe), 115.8 (d, 2JCF = 21.8 Hz, C3/5H, F-Phe), 116.4 (C5H, Pyr), 121.4 (C6H, (OCH3)2-Phe), 123.7 (C5H, (OCH3)2-Phe), 126.6 (d, 4JCF = 3.1 Hz, C1, F-Phe), 130.1 (C5, Imdz), 130.7 (d, 3JCF = 8.4 Hz, C2/6H, F-Phe), 134.4 (C1, (OCH3)2-Phe), 134.5 (C4, Imdz), 142.2 (C2, Imdz), 143.8 (C4, Pyr), 146.6 (C2OCH3), 147.6 (C6H, Pyr), 152.4 (C3OCH3), 152.5 (C2, Pyr), 162.0 (d, 1JCF = 244.7 Hz, CF), 171.1 (CO) ppm; MS (ESI, 70 eV) m/z 493 [MH]+.
3-(3,4-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-propanamide (11d). Synthesis was performed according to the general procedure from 3-(3,4-dimethoxyphenyl)propionic acid (700 mg, 3.33 mmol) and 7 (500 mg, 1.67 mmol). Purification was achieved by flash chromatography (SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 11d as colorless solid. Yield 468 mg (57%); 1H-NMR (DMSO-d6): δ = 2.62 (s, 3H, SCH3), 2.62–2.67 (m, 2H, COCH2CH2), 2.81 (t, 3J = 7.4 Hz, 2H, COCH2CH2), 3.70 (s, 3H, C4OCH3), 3.72 (s, 3H, C3OCH3), 6.73 (dd, 3J = 8.2 Hz, 4J = 1.9 Hz, 1H, C6H, (OCH3)2-Phe), 6.84 (d, 3J = 8.3 Hz, 1H, C5H, (OCH3)2-Phe), 6.85 (bs, 1H, C2H, (OCH3)2-Phe), 7.00 (dd, 3J = 5.2 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.26–7.31 (m, 2H, C3/5H, F-Phe), 7.46–7.50 (m, 2H, C2/6H, F-Phe), 8.10–8.34 (m, 2H, C3/6H Pyr), 10.33 (s, 1H, CONH), 12.71 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 30.4 (COCH2CH2), 38.0 (COCH2CH2), 55.4 (C4OCH3), 55.5 (C3OCH3), 110.6 (C3H, Pyr), 111.9 (C5H, (OCH3)2-Phe), 112.3 (C2H, (OCH3)2-Phe), 115.8 (d, 2JCF = 22.5 Hz, C3/5H, F-Phe), 116.4 (C5H, Pyr), 120.0 (C6H, (OCH3)2-Phe), 130.6 (C2/6H, F-Phe), 133.5 (C1, (OCH3)2-Phe), 147.1 (C4OCH3), 147.6 (C6H, Pyr), 148.6 (C3OCH3), 152.5 (C2, Pyr), 165.8 (d, 1JCF = 242.6 Hz, CF), 171.2 (CO) ppm; MS (ESI, 70 eV) m/z 493 [MH]+.
3-(3,4,5-Trimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-propanamide (11e). Synthesis was performed according to the general procedure from 3-(3,4,5-dimethoxyphenyl)propionic acid (800 mg, 3.33 mmol) and 7 (500 mg, 1.67 mmol). Purification was achieved by flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 11e as pale yellowish solid. Yield 422 mg (49%); C27H27FN4O4S (Mr 522.60); 1H-NMR (DMSO-d6): δ = 2.62 (s, 3H, SCH3), 2.62–2.69 (m, 2H, COCH2CH2), 2.80–2.85 (m, 2H, COCH2CH2), 3.61 (s, 3H, C4OCH3), 3.74 (bs, 6H, C3/5OCH3), 6.56 (s, 2H, C2/6H, (OCH3)3-Phe), 7.01 (dd, 3J = 5.3 Hz, 4J = 1.6 Hz, 1H, C5H, Pyr), 7.26–7.32 (m, 2H, C3/5H, F-Phe), 7.46–7.51 (m, 2H, C2/6H, F-Phe), 8.11 (dd, 3J = 5.3 Hz, 5J = 0.5 Hz, 1H, C6H, Pyr), 8.36 (bs, 1H, C3H, Pyr), 10.36 (s, 1H, CONH), 12.70 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 31.3 (COCH2CH2), 37.9 (COCH2CH2), 55.7 (C3/5OCH3), 59.9 (C4OCH3), 105.5 (C2/6H, (OCH3)3-Phe), 110.6 (C3H, Pyr), 115.8 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 116.4 (C5H, Pyr), 126.6 (d, 4JCF = 3.3 Hz, C1, F-Phe), 130.1 (C5, Imdz), 130.7 (d, 3JCF = 8.4 Hz, C2/6H, F-Phe), 134.5 (C4, Imdz), 135.7 (C4OCH3), 136.8 (C1, (OCH3)3-Phe), 142.2 (C2, Imdz), 143.8 (C4, Pyr), 147.6 (C6H, Pyr), 152.5 (C2, Pyr), 152.7 (C3/5OCH3), 162.0 (d, 1JCF = 245.4 Hz, CF), 171.2 (CO) ppm; MS (ESI, 70 eV) m/z 523 [MH]+.
3.2.4. Synthesis of 4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-carbamides 12a–g
A solution of 7 (1.0 equiv), the appropriate isocyanate (1.1 equiv) and DIPEA (1.2 equiv) in 5 mL anhyd. DMF was stirred under a nitrogen atmosphere at rt for 12 h. The solvent was removed under reduced pressure, the residue resuspended in ethyl acetate, and washed with 0.1 M aq. HCl, sat. aq. NaHCO3 solution, and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and concentrated under reduced pressure. The crude product was purified by flash chromatography (SiO2 and RP-18, eluent and mixing ratio given for each compound, respectively) to afford the particular compound.
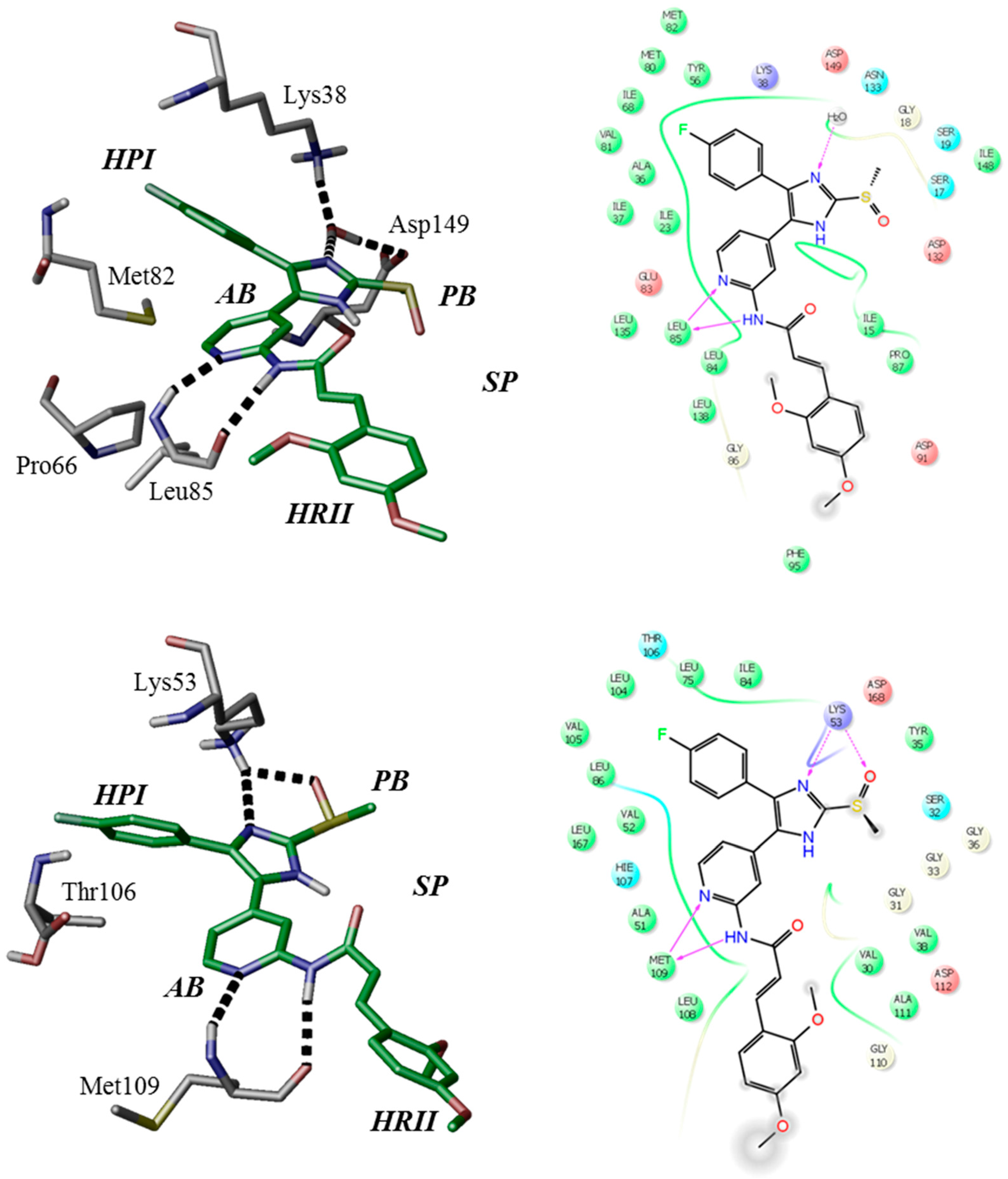
1-(2,4-Dimethoxyphenyl)-3-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)carbamide (12a). Synthesis was performed according to the general procedure from 2,4-dimethoxyphenyl isocyanate (262 mg, 1.47 mmol). Purification was achieved by flash chromatography (SiO2, 0%–10% methanol/DCM and RP-18, 50%–100% methanol/H2O) to afford 12a as colorless solid. Yield 135 mg (21%); C24H22FN5O3S (Mr 479.53); m.p. 214 °C; 1H-NMR (DMSO-d6): δ = 2.63 (s, 3H, SCH3), 3.74 (s, 3H, C4OCH3), 3.88 (s, 3H, C2OCH3), 6.48 (dd, 3J = 8.9 Hz, 4J = 2.7 Hz, 1H, C5H, (OCH3)2-Phe), 6.62 (d, 4J = 2.7 Hz, 1H, C3H, (OCH3)2-Phe), 6.92 (dd, 3J = 5.4 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.30–7.51 (m, 5H, C3/5H, F-Phe and C3H, Pyr and C2/6H, F-Phe), 8.03 (d, 3J = 8.9 Hz, 1H, C6H, (OCH3)2-Phe), 8.12 (bs, 1H, C6H, Pyr), 9.64 (bs, 1H, NH), 10.89–11.26 (m, 1H, NH), 12.74 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 55.3 (C4OCH3), 56.0 (C2OCH3), 98.8 (C3H, (OCH3)2-Phe), 104.1 (C5H, (OCH3)2-Phe), 108.6 (C3H, Pyr), 114.6 (C5H, Pyr), 115.7 (C2/6H, F-Phe), 119.7 (C6H, (OCH3)2-Phe), 121.9 (C1, (OCH3)2-Phe), 130.7 (C3/5H, F-Phe), 134.2 (C4, Pyr), 142.3 (C2, Imdz), 146.1 (C6H, Pyr), 149.4 (C2, (OCH3)2-Phe), 152.2 (CO), 153.6 (C2, Pyr), 155.2 (C4, (OCH3)2-Phe), 161.8 (d, 1JCF = 241.6 Hz, CF) ppm; MS (ESI, 70 eV) m/z 480 [MH]+.
1-(2,5-Dimethoxyphenyl)-3-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)carbamide (12b). Synthesis was performed according to the general procedure from 2,5-dimethoxyphenyl isocyanate (300 mg, 1.67 mmol). Purification was achieved by flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether) to afford 12b as pale yellowish solid. Yield 79.9 mg (15%); C24H22FN5O3S (Mr 479.53); m.p. 184 °C; 1H-NMR (DMSO-d6): δ = 2.63 (s, 3H, SCH3), 3.70 (s, 3H, C5OCH3), 3.84 (s, 3H, C2OCH3), 6.52 (dd, 3J = 8.8 Hz, 4J = 2.9 Hz, 1H, C4H, (OCH3)2-Phe), 6.91–6.96 (m, 1H, C3H, (OCH3)2-Phe), 6.95 (dd, 3J = 5.5 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.28–7.38 (m, 2H, C3/5H, F-Phe), 7.47–7.51 (m, 3H, C2/6H, F-Phe and C3H, Pyr), 7.92 (d, 4J = 2.9 Hz, 1H, C6H, (OCH3)2-Phe), 8.13 (d, 3J = 5.4 Hz, 1H, C6H, Pyr), 9.76 (bs, 1H, NH), 11.52 (vbs, 1H, NH), 12.74 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 55.3 (C5OCH3), 55.6 (C2OCH3), 105.6 (C6H, (OCH3)2-Phe), 105.8 (C4H, (OCH3)2-Phe), 108.5 (C3H, Pyr), 111.7 (C3H, (OCH3)2-Phe), 114.7 (C5H, Pyr), 115.9 (d, 2JCF = 22.0 Hz, C3/5H, F-Phe), 126.5 (d, 4JCF = 3.3 Hz, C1, F-Phe), 129.6 (C1, (OCH3)2-Phe), 130.4 (C5, Imdz), 130.8 (d, 3JCF = 8.4 Hz, C2/6H, F-Phe), 134.1 (C4, Imdz), 142.3 (C2, Imdz), 142.4 (C2OCH3), 144.2 (C4, Pyr), 146.1 (C6H, Pyr), 152.2 (CO), 153.4 (C5OCH3), 153.4 (C2, Pyr), 162.1 (d, 1JCF = 245.9 Hz, CF) ppm; MS (ESI, 70 eV) m/z 480 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(4-methoxy-phenyl)carbamide (12c). Synthesis was performed according to the general procedure from 4-methoxyphenyl isocyanate (165 µL, 1.27 mmol). Purification was achieved by flash chromatography (SiO2, 50%–100% ethyl acetate/petrol ether and RP-18, 60%–100% methanol/H2O) to afford 12c as pale yellow solid. Yield 137 mg (26%); C23H20FN5O2S (Mr 449.50); m.p. 201 °C; 1H-NMR (DMSO-d6): δ = 2.62 (s, 3H, SCH3), 3.72 (s, 3H, OCH3), 6.86–6.90 (m, 2H, C3/5H, H3CO-Phe), 6.92 (dd, 3J = 5.4 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.30 (bs, 2H, C3/5H, F-Phe), 7.39–7.44 (m, 2H, C2/6H, H3CO-Phe), 7.47–7.51 (m, 2H, C2/6H, F-Phe), 7.58 (bs, 1H, C3H, Pyr), 8.11 (bs, 1H, C6H, Pyr), 9.40 (bs, 1H, NH), 10.35–10.71 (m, 1H, NH), 12.74 (s, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 55.2 (OCH3), 105.3 (C4, Pyr), 108.7 (C3H, Pyr), 114.0 (C3/5H, H3CO-Phe), 114.6 (C5H, Pyr), 115.8 (d, 2JCF = 22.0 Hz, C3/5H, F-Phe), 120.6 (C2/6H, H3CO-Phe), 130.6–130.8 (C2/6H, F-Phe), 132.0 (C1, H3CO-Phe), 142.3 (C2, Imdz), 146.4 (C6H, Pyr), 152.3 (CO), 153.5 (C2, Pyr), 154.9 (C4, H3CO-Phe), 162.1 (d, 1JCF = 252.6 Hz, CF) ppm; MS (ESI, 70 eV) m/z 450 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(m-tolyl)-carbamide (12d). Synthesis was performed according to the general procedure from 3-methylphenyl isocyanate (284 µL, 2.20 mmol). Purification was achieved by flash chromatography (SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 60%–80% methanol/H2O) to afford 12d as colorless solid. Yield 241 mg (28%); C23H20FN5OS (Mr 433.51); m.p. 211 °C; 1H-NMR (DMSO-d6): δ = 2.29 (s, 3H, CH3), 2.63 (s, 3H, SCH3), 6.83 (d, 3J = 7.4 Hz, 1H, C4H, Tol), 6.95 (dd, 3J = 5.4 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.15–7.35 (m, 2H, C3/5H, F-Phe), 7.17 (t, 3J = 7.6 Hz, 1H, C5H, Tol), 7.31 (d, 3J = 8.0 Hz, 1H, C6H, Tol), 7.35 (s, 1H, C2H, Tol), 7.47–7.52 (m, 2H, C2/6H, F-Phe), 7.61 (bs, 1H, C3H, Pyr), 8.13 (bs, 1H, C6H, Pyr), 9.44 (s, 1H, NH), 10.42–10.78 (m, 1H, NH), 12.74 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 21.2 (CH3), 108.7 (C3H, Pyr), 114.8 (C5H, Pyr), 115.8 (d, 2JCF = 22.3 Hz, C3/5H, F-Phe), 116.0 (C6H, Tol), 119.3 (C2H, Tol), 123.2 (C4H, Tol), 126.6 (d, 4JCF = 2.6 Hz, C1, F-Phe), 128.7 (C5H, Tol), 129.7 (C5, Imdz), 130.8 (C2/6H, F-Phe), 134.2 (C4, Imdz), 138.1 (C3, Tol), 139.0 (C1, Tol), 142.4 (C2, Imdz), 144.3 (C4, Pyr), 146.4 (C6H, Pyr), 152.1 (CO), 153.4 (C2, Pyr), 162.0 (d, 1JCF = 243.0 Hz, CF) ppm; MS (ESI, 70 eV) m/z 434 [MH]+.
1-(3-Chloro-4-methylphenyl)-3-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-carbamide (12e). Synthesis was performed according to the general procedure from 3-chloro-4-methylyphenyl isocyanate (205 µL, 1.47 mmol). Purification was achieved by flash chromatography (SiO2, 0%–10% methanol/DCM and RP-18, 70%–100% methanol/H2O and SiO2, 20%–100% ethyl acetate/petrol ether) to afford 12e as colorless solid. Yield 210 mg (34%); C23H19ClFN5OS (Mr 467.95); m.p. 210 °C; 1H-NMR (DMSO-d6): δ = 2.27 (s, 3H, CH3), 2.63 (s, 3H, SCH3), 6.96 (dd, 3J = 5.5 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.26 (m, 2H, C5/6H, Cl-Tol), 7.28–7.34 (m, 2H, C3/5H, F-Phe), 7.46–7.50 (m, 2H, C2/6H, F-Phe), 7.59 (bs, 1H, C3H, Pyr), 7.77 (s, 1H, C2H, Cl-Tol), 8.12 (d, 3J = 5.4 Hz, 1H, C6H, Pyr), 9.51 (s, 1H, NH), 10.99 (s, 1H, NH), 12.73 (s, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 18.8 (CH3), 108.7 (C3H, Pyr), 114.8 (C5H, Pyr), 115.9 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 117.6 (C6H, Cl-Tol), 118.7 (C2H, Cl-Tol), 126.5 (d, 4JCF = 3.1 Hz, C1, F-Phe), 128.9 (C4, Cl-Tol), 130.4 (C5, Imdz), 130.8 (d, 3JCF = 8.3 Hz, C2/6H, F-Phe), 131.2 (C5H, Cl-Tol), 133.2 (C3, Cl-Tol), 134.1 (C4, Imdz), 138.2 (C1, Cl-Tol), 142.3 (C2, Imdz), 144.3 (C4, Pyr), 146.4 (C6H, Pyr), 152.2 (CO), 153.2 (C2, Pyr), 162.1 (d, 1JCF = 245.5 Hz, CF) ppm; MS (ESI, 70 eV) m/z 468 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(3-(trifluoromethyl)phenyl)-carbamide (12f). Synthesis was performed according to the general procedure from 3-(trifluoromethyl)phenyl isocyanate (300 µL, 2.20 mmol). Purification was achieved by flash chromatography (SiO2, 40% methanol/DCM) and subsequent filtration to afford 12f as colorless solid. Yield 255 mg (26%); C23H17FN5OS (Mr 487.48); m.p. 234 °C; 1H-NMR (DMSO-d6): δ = 2.63 (s, 3H, SCH3), 6.99 (dd, 3J = 5.6 Hz, 4J = 1.2 Hz, 1H, C5H, Pyr), 7.28–7.37 (m, 3H, C3/5H, F-Phe and C4H, F3C-Phe), 7.46–7.66 (m, 5H, C3H, Pyr and C2/6H, F-Phe and C5/6H, F3C-Phe), 8.08 (bs, 1H, C2H, F3C-Phe), 8.14 (d, 3J = 5.5 Hz, 1H, C6H, Pyr), 9.56 (s, 1H, NH), 11.12 (s, 1H, NH), 12.74 (s, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 108.7 (C3H, Pyr), 114.7 (C2H, F3C-Phe), 115.0 (C5H, Pyr), 115.9 (d, 2JCF = 21.8 Hz, C3/5H, F-Phe), 118.7 (C4H, F3C-Phe), 122.5 (C6H, F3C-Phe), 124.2 (q, 1JCF = 272.1 Hz, CF3, F3C-Phe), 126.5 (d, 4JCF = 2.9 Hz, C1, F-Phe), 129.6 (q, 2JCF = 31.3 Hz, C3, F3C-Phe), 130.0 (C5H, F3C-Phe), 130.5 (C5, Imdz), 130.8 (d, 3JCF = 8.3 Hz, C2/6H, F-Phe), 134.0 (C4, Imdz), 139.9 (C1, F3C-Phe), 142.3 (C2, Imdz), 144.3 (C4, Pyr), 146.6 (C6H, Pyr), 152.2 (CO), 153.0 (C2, Pyr), 162.0 (d, 1JCF = 245.2 Hz, CF) ppm; MS (ESI, 70 eV) m/z 488 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C23H17FN5OS, 487.1090; found 487.1090.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(naphthalen-1-yl)carbamide (12g). Synthesis was performed according to the general procedure from naphthyl isocyanate (211 µL, 1.47 mmol). Purification was achieved by flash chromatography (SiO2, 0%–10% methanol/DCM and SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 12g as pale yellow solid. Yield 257 mg (41%); C26H20FN5OS (Mr 469.54); m.p. 249 °C; 1H-NMR (DMSO-d6): δ = 2.64 (s, 3H, SCH3), 7.01 (dd, 3J = 5.5 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.30–7.36 (m, 2H, C3/5H, F-Phe), 7.42–7.59 (m, 5H, C3H, Pyr and C2/6H, F-Phe and C3/6H, Naph), 7.63–7.68 (m, 2H, C4/7H, Naph), 7.95 (d, 3J = 8.1 Hz, 1H, C5H, Naph), 8.16–8.22 (m, 2H, C2/8H, Naph), 8.27 (d, 3J = 5.5 Hz, 1H, C6H, Pyr), 9.87 (s, 1H, NH), 11.86 (bs, 1H, NH), 12.77 (s, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 108.7 (C3H, Pyr), 114.7 (C5H, Pyr), 115.9 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 116.7 (C2H, Naph), 120.9 (C8H, Naph), 123.0 (C4H, Naph), 125.4 (C1, Naph), 126.0 (C3/6H, Naph), 126.3 (C7H, Naph), 126.5 (d, 4JCF = 3.2 Hz, C1, F-Phe), 128.6 (C5H, Naph), 130.6 (C5, Imdz), 130.8 (d, 3JCF = 8.3 Hz, C2/6H, F-Phe), 133.7 (C4a, Naph), 134.0 (C8a, Naph), 134.2 (C4, Imdz), 142.4 (C2, Imdz), 144.5 (C4, Pyr), 146.1 (C6H, Pyr), 152.6 (CO), 153.5 (C2, Pyr), 162.1 (d, 1JCF = 246.2 Hz, CF) ppm; MS (ESI, 70 eV) m/z 471 [MH]+.
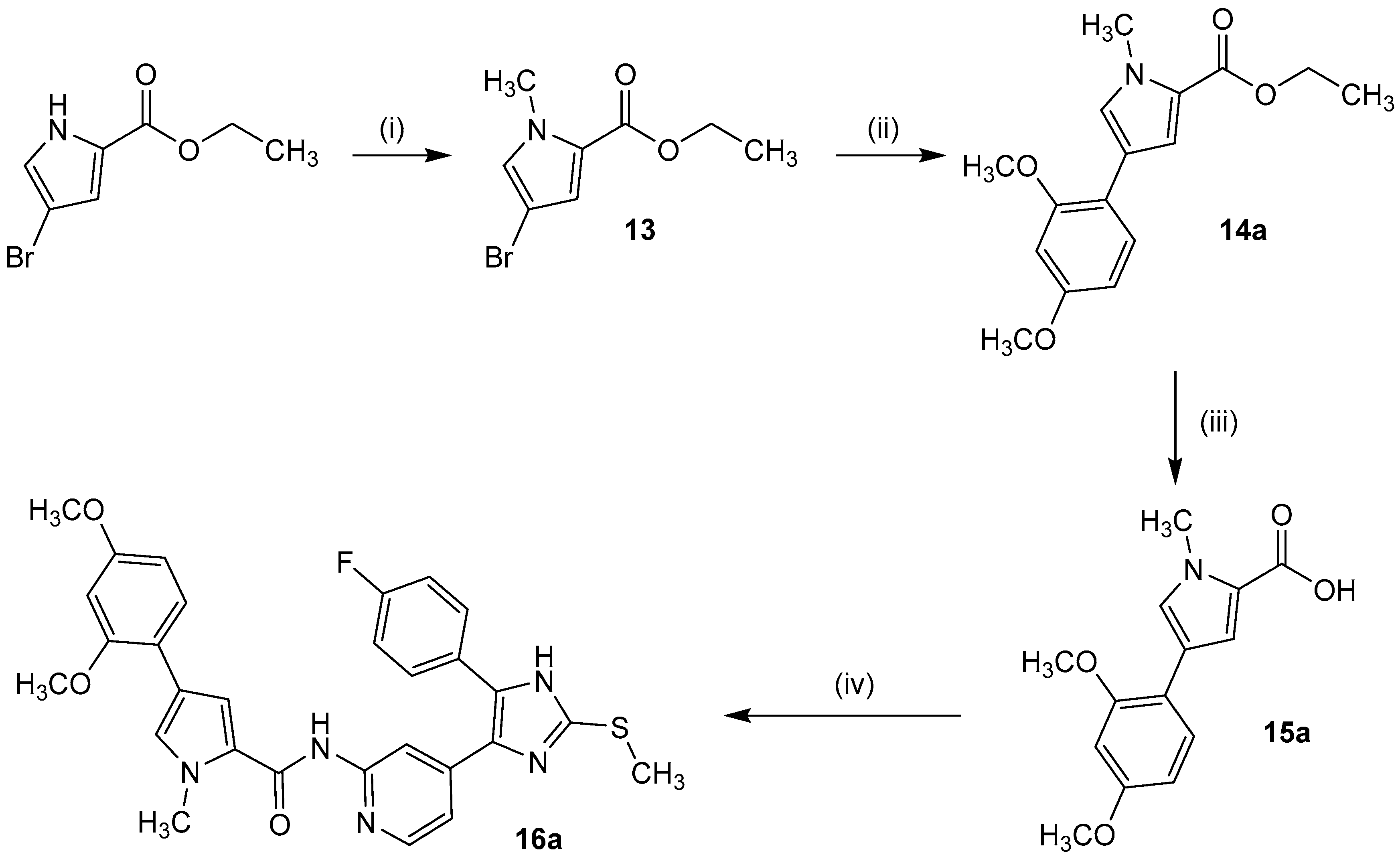
3.2.5. Synthesis of N-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-1-methyl-4-phenyl-1H-pyrrole-2-carboxamides 13, 14a–b, 15a–b, 16a–b
Ethyl 4-bromo-1-methyl-1H-pyrrole-2-carboxylate (13). NaH (240 mg 60% dispersion in mineral oil, 6.05 mmol) was added in one portion to a solution of 4-bromo-1H-pyrrole-2-carboxylate (1.17 g, 5.34 mmol) in 15 mL anhyd. DMF at 0 °C under a nitrogen atmosphere. The suspension was stirred for 20 min at the same temp. before methyl iodide (400 µL, 6.43 mmol) was carefully added and stirring continued at 0 °C for 15 min and another 2.5 h at r.t. The reaction was quenched with H2O and extracted with ethyl acetate. The combined organic phases were washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. Purification was achieved by flash chromatography (SiO2, 2%–10% ethyl acetate/petrol ether) to afford 13 as clear colorless oil that crystallized on standing as colorless needles. Yield 1.22 g (98%); C8H10BrNO2 (Mr 232.08); m.p. 43 °C; 1H-NMR (DMSO-d6): δ = 1.26 (t, 3J = 7.1 Hz, 3H, COOCH2CH3), 3.83 (d, J = 0.2 Hz, 3H, CH3), 4.21 (q, 3J = 7.1 Hz, 2H, COOCH2CH3), 6.84 (d, 4J = 2.0 Hz, 1H, C3H, Pyrrole), 7.28 (d, 4J = 1.7 Hz, 1H, C5H, Pyrrole) ppm; 13C-NMR (DMSO-d6): δ = 14.2 (COOCH2CH3), 36.6 (CH3), 59.8 (COOCH2CH3), 93.8 (CBr), 118.1 (C3H, Pyrrole), 122.7 (C2, Pyrrole), 129.5 (C5H, Pyrrole), 159.5 (COOCH2CH3) ppm.
Ethyl 4-(2,4-dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylate (14a). 27 mL 2 M aq. Na2CO3 solution were added to a stirred solution of 13 (1.22 g, 5.26 mmol), 2,4-dimethoxyphenyl boronic acid (2.89 g, 15.9 mmol), and Pd(PPh3)4 (306 mg, 265 µmol) in 80 mL DMF. Stirring continued for 4 h under reflux and 12 h at r.t. H2O was added and the mixture was extracted with ethyl acetate. The combined organic phases were washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (SiO2, 5%–10% ethyl acetate/petrol ether and RP-18, 50%–70% methanol/H2O) to afford 14a as colorless solid. Yield 1.02 g (67%); C16H19NO4 (Mr 289.33); 1H-NMR (DMSO-d6): δ = 1.29 (t, 3J = 7.1 Hz, 3H, COOCH2CH3), 3.77 (s, 3H, C4OCH3), 3.84 (s, 3H, C2OCH3), 3.87 (s, 3H, CH3), 4.23 (q, 3J = 7.1 Hz, 2H, COOCH2CH3), 6.53 (dd, 3J = 8.5 Hz, 4J = 2.5 Hz, 1H, C5H, (OCH3)2-Phe), 6.60 (d, 4J = 2.5 Hz, 1H, C3H, (OCH3)2-Phe), 7.17 (d, 4J = 2.0 Hz, 1H, C3H, Pyrrole), 7.44 (d, 3J = 8.5 Hz, 1H, C6H, (OCH3)2-Phe), 7.46 (dd, 4J = 2.0 Hz, 5J = 0.4 Hz, 1H, C5H, Pyrrole) ppm; 13C-NMR (DMSO-d6): δ = 14.4 (COOCH2CH3), 36.4 (CH3), 55.2 (C4OCH3), 55.4 (C2OCH3), 59.4 (CH2), 98.8 (C3H, (OCH3)2-Phe), 105.2 (C5H, (OCH3)2-Phe), 115.4 (C3H, Pyrrole), 115.5 (C1, (OCH3)2-Phe), 119.0 (C4, Pyrrole), 121.3 (C2, Pyrrole), 127.7 (C6H, (OCH3)2-Phe), 129.0 (C5H, Pyrrole), 156.7 (C4OCH3), 158.74 (C2OCH3), 160.5 (COOCH2CH3) ppm; MS (ESI, 70 eV) m/z 290 [MH]+.
Ethyl 4-(2,5-dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylate (14b). Synthesis was performed according to the procedure described for 14a starting from 13 (1.11 g, 4.78 mmol), 2,5-dimethoxyphenyl boronic acid (2.65 g, 14.6 mmol), Pd(PPh3)4 (289 mg, 250 µmol) in 80 mL DMF, and 25 mL 2 M aq. Na2CO3 solution. Purification was achieved by flash chromatography (SiO2, 2%–10% ethyl acetate/petrol ether and RP-18, 65% methanol/H2O) to afford 14b as colorless solid. Yield 984 mg (71%); C16H19NO4 (Mr 289.33); 1H-NMR (DMSO-d6): δ = 1.30 (t, 3J = 7.1 Hz, 3H, COOCH2CH3), 3.74 (s, 3H, C5OCH3), 3.79 (s, 3H, C2OCH3), 3.89 (s, 3H, CH3), 4.24 (q, 3J = 7.1 Hz, 2H, COOCH2CH3), 6.74 (dd, 3J = 8.9 Hz, 4J = 3.1 Hz, 1H, C4H, (OCH3)2-Phe), 6.96 (d, 3J = 8.9 Hz, 1H, C3H, (OCH3)2-Phe), 7.09 (d, 4J = 3.1 Hz, 1H, C6H, (OCH3)2-Phe), 7.28 (d, 4J = 2.0 Hz, 1H, C3H, Pyrrole), 7.60 (dd, 4J = 2.0 Hz, 5J = 0.4 Hz, 1H, C5H, Pyrrole) ppm; 13C-NMR (DMSO-d6): δ = 14.3 (COOCH2CH3), 36.5 (CH3), 55.4 (C5OCH3), 55.9 (C2OCH3), 59.4 (COOCH2CH3), 111.7 (C4H, (OCH3)2-Phe), 112.5 (C6H, (OCH3)2-Phe), 112.9 (C3H, (OCH3)2-Phe), 116.1 (C3H, Pyrrole), 118.9 (C4, Pyrrole), 121.6 (C1, Pyrrole), 123.4 (C1, (OCH3)2-Phe), 130.0 (C5H, Pyrrole), 150.0 (C2OCH3), 153.4 (C5OCH3), 160.5 (COOCH2CH3) ppm; MS (ESI, 70 eV) m/z 290 [MH]+.
4-(2,4-Dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylic acid (15a). 7 mL 4 M aq. NaOH were added to a solution of 14a (990 mg, 3.42 mmol) in 18 mL THF and 9 mL methanol and the mixture was stirred at 50 °C for 5 h and then 12 h at r.t. H2O was added to the reaction, the pH was adjusted to 3 using 1 M aq. HCl, and the mixture was extracted with ethyl acetate. The combined organic phases were washed with H2O and sat. aq. NaCl solution, the organic phase was dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure to afford 15a as brown solid. Yield 894 mg (quant.); C14H15NO4 (Mr 261.28); m.p. 164 °C; 1H-NMR (DMSO-d6): δ = 3.76 (s, 3H, C4OCH3), 3.83 (s, 3H, C2OCH3), 3.86 (s, 3H, CH3), 6.53 (dd, 3J = 8.5 Hz, 4J = 2.4 Hz, 1H, C5H, (OCH3)2-Phe), 6.59 (d, 4J = 2.4 Hz, 1H, C3H, (OCH3)2-Phe), 7.14 (d, 4J = 2.0 Hz, 1H, C3H, Pyrrole), 7.41 (d, 4J = 2.0 Hz, 1H, C5H, Pyrrole), 7.42 (d, 3J = 8.4 Hz, 1H, C6H, (OCH3)2-Phe), 12.17 (bs, 1H, COOH) ppm; 13C-NMR (DMSO-d6): δ = 36.3 (CH3), 55.2 (C4OCH3), 55.4 (C2OCH3), 98.8 (C3H, (OCH3)2-Phe), 105.2 (C5H, (OCH3)2-Phe), 115.7 (C3H, Pyrrole), 115.7 (C1, (OCH3)2-Phe), 118.8 (C4, Pyrrole), 122.0 (C2, Pyrrole), 127.6 (C6H, (OCH3)2-Phe), 128.6 (C5H, Pyrrole), 156.7 (C2OCH3), 158.7 (C4OCH3), 162.1 (COOH) ppm; MS (ESI, 70 eV) m/z 262 [MH]+.
4-(2,5-Dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylic acid (15b). Synthesis was performed according to the procedure described for 15a starting from 14b (980 mg, 3.39 mmol) in 19 mL THF and 10 mL methanol, and 7 mL 4 M aq. NaOH to afford 15b as brown solid. Yield 885 mg (quant.); C14H15NO4 (Mr 261.28); m.p. 149 °C; 1H-NMR (DMSO-d6): δ = 3.74 (s, 3H, C2OCH3), 3.79 (s, 3H, C5OCH3), 3.88 (CH3), 6.72 (dd, 3J = 8.9 Hz, 4J = 3.1 Hz, 1H, C4H, (OCH3)2-Phe), 6.95 (d, 3J = 8.9 Hz, 1H, C3H, (OCH3)2-Phe), 7.08 (d, 4J = 3.1 Hz, 1H, C6H, (OCH3)2-Phe), 7.25 (d, 4J = 2.0 Hz, 1H, C3H, Pyrrole), 7.55 (d, 4J = 2.0 Hz, 1H, C5H, Pyrrole), 12.15 (bs, 1H, COOH) ppm; 13C-NMR (DMSO-d6): δ = 36.4 (CH3), 55.4 (C2OCH3), 55.9 (C5OCH3), 111.5 (C4H, (OCH3)2-Phe), 112.5 (C6H, (OCH3)2-Phe), 112.9 (C3H, (OCH3)2-Phe), 116.2 (C3H, Pyrrole), 118.7 (C4, Pyrrole), 122.4 (C2, Pyrrole), 123.6 (C1, (OCH3)2-Phe), 129.6 (C5H, Pyrrole), 150.1 (C5OCH3), 153.4 (C2OCH3), 162.1 (COOH) ppm; MS (ESI, 70 eV) m/z 262 [MH]+.
4-(2,4-Dimethoxyphenyl)-N-(4-(5-(4-fluorphenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-1-methyl-1H-pyrrole-2-carboxamide (16a). A solution of 15a (1.01 g, 4.07 mmol), PyBOP (2.54 g, 4.88 mmol), and DIPEA (2.15 mL, 12.3 mmol) was stirred in 14 mL anhyd. DMF under a nitrogen atmosphere for 30 min at r.t. 7 (1.60 g, 5.31 mmol) was added in one portion and the mixture was stirred for 12 h at 110 °C. The reaction was quenched with H2O and extracted with ethyl acetate. The combined organic phases were washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 16a as beige solid. Yield 303 mg (24%); C29H26FN5O3S (Mr 543.62); m.p. 236 °C; 1H-NMR (DMSO-d6): δ = 2.64 (s, 3H, SCH3), 3.78 (s, 3H, C2OCH3), 3.86 (s, 3H, C4OCH3), 3.90 (s, 3H, CH3), 6.57 (dd, 3J = 8.5 Hz, 4J = 2.3 Hz, 1H, C5H, (OCH3)2-Phe), 6.61 (d, 4J = 2.3 Hz, 1H, C3H, (OCH3)2-Phe), 7.06 (dd, 3J = 5.3 Hz, 4J = 1.2 Hz, 1H, C5H, Pyr), 7.28–7.34 (m, 2H, C3/5H, F-Phe), 7.41 (d, 4J = 1.3 Hz, 1H, C3H, Pyrrole), 7.46 (d, 3J = 8.4 Hz, 1H, C6H, (OCH3)2-Phe), 7.49–7.54 (m, 2H, C2/6H, F-Phe), 7.66 (d, 4J = 1.4 Hz, 1H, C5H, Pyrrole), 8.18 (d, 3J = 5.2 Hz, 1H, C6H, Pyr), 8.38 (bs, 1H, C3H, Pyr), 10.10 (bs, 1H, CONH), 12.73 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.3 (SCH3), 36.5 (CH3), 55.2 (C2OCH3), 55.5 (C4OCH3), 98.8 (C3H, (OCH3)2-Phe), 105.2 (C5H, (OCH3)2-Phe), 111.3 (C3H, Pyr), 113.2 (C5H, Pyrrole), 115.9 (d, 2JCF = 21.9 Hz, C3/5H, F-Phe), 115.9 (C1, (OCH3)2-Phe), 116.3 (C5H, Pyr), 118.5 (C4, Pyrrole), 124.1 (C2, Pyrrole), 126.7 (d, 4JCF = 3.5 Hz, C1, F-Phe), 127.5 (C6H, (OCH3)2-Phe), 128.5 (C3H, Pyrrole), 130.8 (d, 3JCF = 8.4 Hz, C2/6H, F-Phe), 134.5 (C5, Imdz), 142.1 (C2, Imdz), 143.6 (C4, Imdz), 147.6 (C6H, Pyr), 152.7 (C2, Pyr), 156.7 (C4OCH3), 158.6 (C2OCH3), 159.8 (CONH), 162.0 (d, 1JCF = 245.6 Hz, CF) ppm; MS (ESI, 70 eV) m/z 544 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C29H26FN5O3S, 543.1740; found, 543.1740.
4-(2,5-Dimethoxyphenyl)-N-(4-(5-(4-fluorphenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-1-methyl-1H-pyrrole-2-carboxamide (16b). Synthesis was performed according to the procedure described for 16a starting from 15b (604 mg, 2.31 mmol), PyBOP (1.45 g, 2.79 mmol), DIPEA (1.20 mL, 6.87 mmol), and 7 (904 mg, 3.01 mmol) in 12 mL anhyd. DMF. The crude product was purified by flash chromatography (SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 16b as pale yellowish solid. Yield 255 mg (20%); m.p. 127 °C; 1H-NMR (DMSO-d6): δ = 2.64 (s, 3H, SCH3), 3.76 (s, 3H, C5OCH3), 3.81 (s, 3H, C2OCH3), 3.92 (s, 3H, CH3), 6.73 (dd, 3J = 8.9 Hz, 4J = 3.1 Hz, 1H, C4H, (OCH3)2-Phe), 6.96 (d, 3J = 9.0 Hz, 1H, C3H, (OCH3)2-Phe), 7.06 (dd, 3J = 5.3 Hz, 4J = 1.6 Hz, 1H, C5H, Pyr), 7.17 (d, 4J = 3.1 Hz, 1H, C6H, (OCH3)2-Phe), 7.24–7.30 (m, 2H, C3/5H, F-Phe), 7.50–7.55 (m, 2H, C2/6H, F-Phe), 7.56 (d, 4J = 1.7 Hz, 1H, C5H, Pyrrole), 7.80 (d, 4J = 1.8 Hz, 1H, C3H, Pyrrole), 8.22 (d, 3J = 5.2 Hz, 1H, C6H, Pyr), 8.35 (bs, 1H, C3H, Pyr), 10.15 (bs, 1H, CONH), 12.77 (vbs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.2 (SCH3), 36.7 (CH3), 55.4 (C5OCH3), 55.9 (C2OCH3), 111.3 (C4H, (OCH3)2-Phe and C3H, Pyr), 112.5 (C6H, (OCH3)2-Phe), 112.8 (C3H, (OCH3)2-Phe), 113.6 (C3H, Pyrrole), 115.7 (d, 2JCF = 21.5 Hz, C3/5H, F-Phe), 116.5 (C5H, Pyr), 118.3 (C4, Pyrrole), 123.8 (C1, (OCH3)2-Phe), 124.3 (C2, Pyrrole), 129.7 (C5H, Pyrrole), 130.42 (C2/6H, F-Phe), 142.5 (C2, Imdz and C4, Pyr), 147.7 (C6H, Pyr), 150.1 (C2OCH3), 152.7 (C1, Pyr), 153.4 (C5OCH3), 159.8 (CONH), 161.9 (d, 1JCF = 247.7 Hz, CF) ppm; MS (ESI, 70 eV) m/z 544 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C29H26FN5O3S, 543.1740; found, 543.1740.
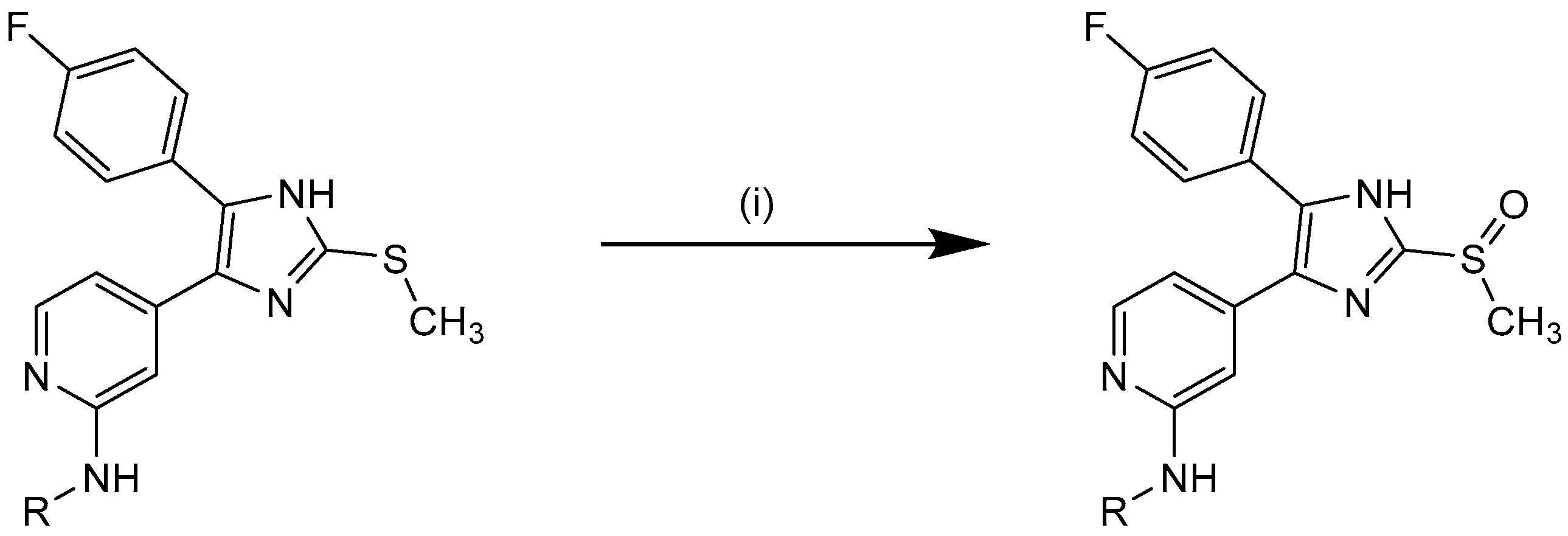
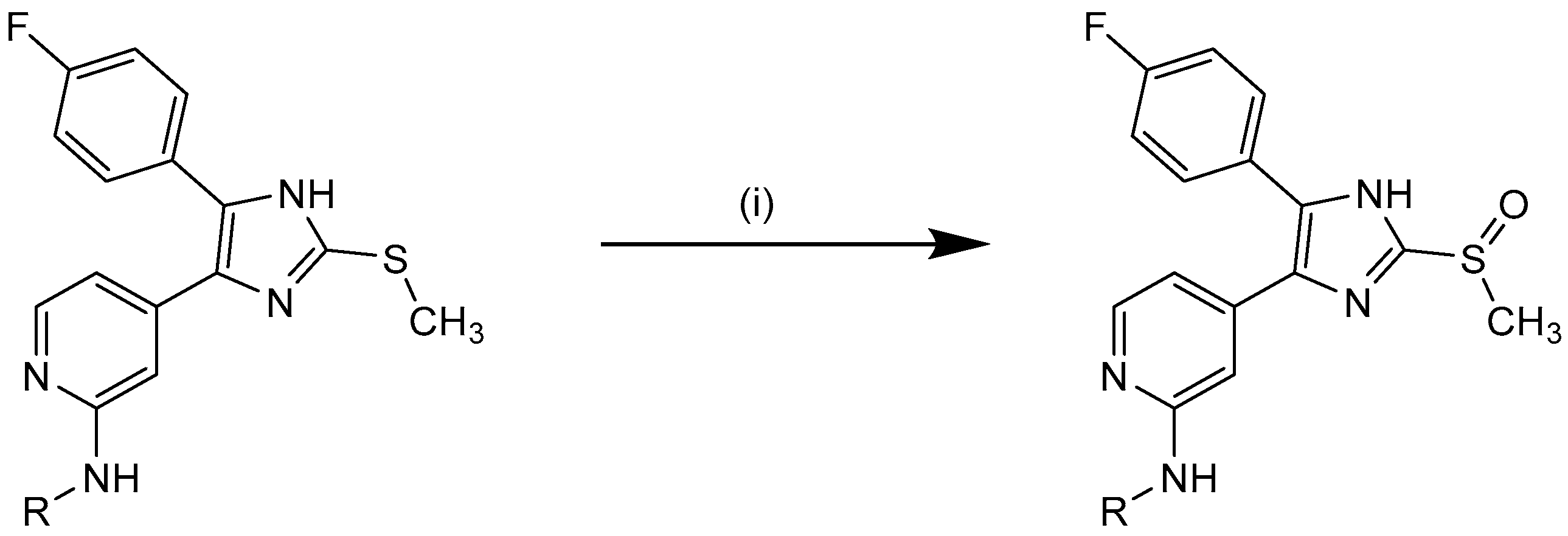
3.2.6. Synthesis of Sulfoxides 10j–k, 11f–g, 12h–m, 16c
The sulfide (1.0 equiv) was dissolved in THF and H2O was added (approx. 3:1). The mixture was stirred at 0 °C for 10 min before an ice-cold aq. solution of potassium peroxomonosulfate (Oxone®, 0.6 equiv) was added and stirring continued for 0.5–2 h at the same temp. After completion of the reaction sat. aq. NaHCO3 solution, H2O, and ethyl acetate were added and the phases were separated. The organic layer was washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. Purification of the crude products was achieved by crystallization from ethyl acetate or flash chromatography (SiO2 and RP-18, eluent and mixing ratio given for each compound) to afford the appropriate compound.
N-(2-Ethoxyphenyl)-4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)pyridin-2-amine (10j). Synthesis was performed according to the general procedure for sulfoxidation starting from 10c (300 mg, 713 µmol) in 5 mL THF and 2 mL H2O. Flash chromatography (RP-18, 20%–90% methanol/H2O) afforded 10j as voluminous yellow solid. Yield 255 mg (82%); C23H21FN4O2S (Mr 436.51); m.p. 152 °C; 1H-NMR (CDCl3): δ = 1.42 (t, 3J = 7.0 Hz, 3H, CH3), 3.03 (s, 3H, SCH3), 4.07 (q, 3J = 7.0 Hz, 2H, CH2), 6.94–6.78 (m, 4H, C4–6H, EtO-Phe and C5H, Pyr), 7.03 (dd, 4J = 1.4 Hz, 5J = 0.7 Hz, 1H, C3H, Pyr), 7.07–7.13 (m, 3H, C3/5H, F-Phe and NH), 7.46–7.50 (m, 2H, C2/6H, F-Phe), 7.62 (dd, 3J = 7.8 Hz, 4J = 1.5 Hz, 1H, C3H, EtO-Phe), 8.14 (dd, 3J = 5.3 Hz, 5J = 0.7 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 14.9 (CH3), 40.8 (SCH3), 64.1 (CH2), 106.9 (C3H, Pyr), 111.5 (C3H, EtO-Phe), 113.3 (C5H, Pyr), 116.0 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 118.2 (C6H, EtO-Phe), 120.6 (C5H, EtO-Phe), 121.9 (C4H, EtO-Phe), 129.9 (C1, EtO-Phe), 130.6 (d, 3JCF = 8.2 Hz, C2/6H, F-Phe), 146.7 (C2, Imdz), 148.2 (C2, EtO-Phe), 148.5 (C6H, Pyr), 156.0 (C2, Pyr), 162.9 (d, 1JCF = 248.8 Hz, CF) ppm; MS (ESI, 70 eV) m/z 437 [MH]+.
N-(3,4-Dimethoxyphenethyl)-4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)-pyridin-2-amine (10k). Synthesis was performed according to the general procedure for sulfoxidation starting from 10d (500 mg, 1.08 mmol) in 5 mL H2O in 9 mL THF and 3 mL H2O. Flash chromatography (RP-18, 20%–90% methanol/H2O) afforded 10d as pale yellow solid. Yield 208 mg (39%); C25H25FN4O3S (Mr 480.56); m.p. 183 °C; 1H-NMR (CDCl3): δ = 2.74 (t, 3J = 6.8 Hz, 1H, CH2CH2NH), 2.97 (s, 3H, SCH3), 3.36 (m, 2H, CH2CH2NH), 3.76 (s, 6H, 2 OCH3), 5.00 (bs, 1H, CH2CH2NH), 6.49 (bs, 1H, C3H, Pyr), 6.52 (dd, 3J = 5.4 Hz, 4J = 1.3 Hz, 1H, C5H, Pyr), 6.60–6.63 (m, 2H, C2/6H, (OCH3)2-Phe), 6.71 (d, 3J = 8.7 Hz, 1H, C5H, (OCH3)2-Phe), 6.96–7.02 (m, 2H, C3/5H, F-Phe), 7.36–7.41 (m, 2H, C2/6H, F-Phe), 7.80 (d, 3J = 5.5 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 35.1 (CH2CH2NH), 40.7 (SCH3), 43.3 (CH2CH2NH), 55.9 (C3OCH3), 55.9 (C4OCH3), 104.6 (C3H, Pyr), 111.2 (C5H, Pyr), 111.4 (C5H, (OCH3)2-Phe), 112.1 (C2H, (OCH3)2-Phe), 115.9 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 120.7 (C6H, (OCH3)2-Phe), 127.2 (C1, F-Phe), 130.5 (d, 3JCF = 8.2 Hz, C2/6H, F-Phe), 131.5 (C1, (OCH3)2-Phe), 135.3 (C4/5, Imdz), 141.6 (C4, Pyr), 146.8 (C2, Imdz), 147.4 (C6H, Pyr), 147.7 (C4OCH3), 149.0 (C3OCH3), 158.6 (C2, Pyr), 162.9 (d, 1JCF = 249.2 Hz, CF) ppm; MS (ESI, 70 eV) m/z 481 [MH]+.
3-(2,4-Dimethoxyphenyl)-N-(4-(5-(4-fluorphenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)-pyridin-2-yl)-propanamide (11f). Synthesis was performed according to the general procedure for sulfoxidation starting from 11a (100 mg, 203 µmol) in 2 mL THF and 0.6 mL H2O. Flash chromatography (SiO2, 40%–100% ethyl acetate/petrol ether) afforded 11f as colorless solid. Yield 96.1 mg (93%); C26H25FN4O4S (Mr 508.57); m.p. 209 °C; 1H-NMR (DMSO-d6): δ = 2.58 (t, 3J = 7.5 Hz, 2H, CH2CH2CO), 2.75 (t, 3J = 7.4 Hz, 2H, CH2CH2CO), 3.08 (s, 3H, SCH3), 3.72 (s, 3H, C2OCH3), 3.77 (s, 3H, C4OCH3), 6.42 (dd, 3J = 8.3 Hz, 4J = 2.4 Hz, 1H, C5H, (OCH3)2-Phe), 6.51 (d, 4J = 2.4 Hz, 1H, C3H, (OCH3)2-Phe), 7.01–7.05 (m, 2H, C5H, Pyr and C6H, (OCH3)2-Phe), 7.26–7.32 (m, 2H, C3/5H, F-Phe), 7.51–7.55 (m, 2H, C2/6H, F-Phe), 8.19 (d, 3J = 5.2 Hz, 1H, C6H, Pyr), 8.34 (bs, 1H, C3H, Pyr), 10.40 (s, 1H, CONH), 13.89 (bs, NH) ppm; 13C-NMR (DMSO-d6): δ = 24.7 (CH2CH2CO), 36.3 (CH2CH2CO), 39.1 (SCH3), 55.1 (C2OCH3), 55.3 (C4OCH3), 98.3 (C3H, (OCH3)2-Phe), 104.3 (C5H, (OCH3)2-Phe), 111.1 (C3H, Pyr), 115.8 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 117.0 (C5H, Pyr), 120.9 (C1, (OCH3)2-Phe), 127.3 (C1, F-Phe), 129.8 (C6H, (OCH3)2-Phe), 130.8 (d, 3JCF = 8.2 Hz, C2/6H, F-Phe), 133.4 (C4, Imdz), 133.9 (C5, Imdz), 142.2 (C4, Pyr), 147.9 (C6H, Pyr), 148.9 (C2, Imdz), 152.6 (C2, Pyr), 157.9 (C4OCH3), 159.0 (C2OCH3), 162.1 (d, 1JCF = 245.5 Hz, CF), 171.54 (CO) ppm; MS (ESI, 70 eV) m/z 509 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C26H25FN4O4S, 508.1581; found, 508.1581.
3-(2,5-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)-pyridin-2-yl)propanamide (11g). Synthesis was performed according to the general procedure for sulfoxidation starting from 11b (100 mg, 203 µmol) in 2 mL THF and 0.6 mL H2O. Crystallization from ethyl acetate afforded 11g as colorless solid. Yield 26.6 mg (26%); m.p. 204 °C; 1H-NMR (DMSO-d6): δ = 2.62 (t, 3J = 7.7 Hz, 2H, CH2CH2CO), 2.80 (t, 3J = 7.5 Hz, 2H, CH2CH2CO), 3.09 (s, 3H, SCH3), 3.66 (s, 3H, C5OCH3), 3.72 (s, 3H, C2OCH3), 6.72 (dd, 3J = 8.8 Hz, 4J = 3.1 Hz, 1H, C4H, (OCH3)2-Phe), 6.77 (d, 4J = 3.0 Hz, 1H, C6H, (OCH3)2-Phe), 6.86 (d, 3J = 8.8 Hz, 1H, C3H, (OCH3)2-Phe), 7.02 (dd, 3J = 5.2 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.25–7.31 (m, 2H, C3/5H, F-Phe), 7.51–7.55 (m, 2H, C2/6H, F-Phe), 8.19 (d, 3J = 5.0 Hz, 1H, C6H, Pyr), 8.33 (s, 1H, C3H, Pyr), 10.45 (s, 1H, CONH), 14.00 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 25.3 (CH2CH2CO), 36.0 (CH2CH2CO), 39.0 (SCH3), 55.2 (C5OCH3), 55.8 (C2OCH3), 111.2 (C4H, (OCH3)2-Phe), 111.3 (C3H, Pyr), 111.5 (C3H, (OCH3)2-Phe), 115.8 (d, 2JCF = 21.3 Hz, C3/5H, F-Phe), 115.9 (C6H, (OCH3)2-Phe), 126.5 (C1, F-Phe), 130.8 (d, 3JCF = 8.3 Hz, C2/6H, F-Phe), 142.1 (C4, Pyr), 148.0 (C6H, Pyr), 148.8 (C2, Imdz), 151.2 (C2OCH3), 152.58 (C2, Pyr), 153.0 (C5OCH3), 162.1 (d, 1JCF = 245.6 Hz, CF), 171.42 (CO) ppm; MS (ESI, 70 eV) m/z 509 [MH]+.
1-(2,4-Dimethoxyphenyl)-3-(4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)-pyridin-2-yl)-carbamide (12h). Synthesis was performed according to the general procedure for sulfoxidation starting from 12a (85.0 mg, 177 µmol) in 3.4 mL THF and 1 mL H2O. Flash chromatography (SiO2, 35%–100% ethyl acetate/petrol ether and RP-18, 55%–100% methanol/H2O) afforded 12h as colorless solid. Yield 18.9 mg (22%); C24H22FN5O4S (Mr 495.53); m.p. 227 °C; 1H-NMR (DMSO-d6): δ = 3.08 (s, 3H, SCH3), 3.74 (s, 3H, C4OCH3), 3.88 (s, 3H, C2OCH3), 6.48 (dd, 3J = 8.9 Hz, 4J = 2.7 Hz, 1H, C5H, (OCH3)2-Phe), 6.62 (d, 4J = 2.7 Hz, 1H, C3H, (OCH3)2-Phe), 6.95 (dd, 3J = 5.4 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.27–7.33 (m, 2H, C3/5H, F-Phe), 7.46 (bs, 1H, C3H, Pyr), 7.51–7.56 (m, 2H, C2/6H, F-Phe), 8.02 (d, 3J = 8.9 Hz, 1H, C6H, (OCH3)2-Phe), 8.19 (d, 3J = 5.3 Hz, 1H, C6H, Pyr), 9.70 (s, 1H, NH), 11.07 (bs, 1H, NH), 13.87 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 39.1 (SCH3), 55.3 (C2OCH3), 56.0 (C4OCH3), 98.8 (C3H, (OCH3)2-Phe), 104.1 (C5H, (OCH3)2-Phe), 109.2 (C3H, Pyr), 115.1 (C5H, Pyr), 115.8 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 119.8 (C6H, (OCH3)2-Phe), 121.8 (C1, (OCH3)2-Phe), 127.2 (C1, F-Phe), 130.8 (d, 3JCF = 8.4 Hz, C2/6H, F-Phe), 132.7 (C4, Imdz), 134.1 (C5, Imdz), 142.8 (C4, Pyr), 146.5 (C6H, Pyr), 149.1 (C2, Imdz), 149.4 (C2OCH3), 152.2 (CO), 153.6 (C2, Pyr), 155.2 (C4OCH3), 162.14 (d, 1JCF = 244.6 Hz, CF) ppm; MS (ESI, 70 eV) m/z 496 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)pyridin-2-yl)-3-(4-meth-oxyphenyl)carbamide (12i). Synthesis was performed according to the general procedure for sulfoxidation starting from 12c (100 mg, 225 µmol) in 2 mL THF and 0.6 mL H2O. Flash chromatography (RP-18, 50%–70% methanol/H2O) afforded 12i as colorless solid. Yield 62.6 mg (61%); C23H20FN5O3S (Mr 465.50); m.p. 233 °C; 1H-NMR (DMSO-d6): δ = 3.09 (s, 3H, SCH3), 3.73 (s, 3H, C4OCH3), 6.89 (d, 3J = 9.1 Hz, 2H, C3/5H, H3CO-Phe), 6.95 (dd, 3J = 5.4 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.28–7.34 (m, 2H, C3/5H, F-Phe), 7.41 (d, 3J = 9.1 Hz, 2H, C2/6H, H3CO-Phe), 7.51–7.56 (m, 2H, C2/6H, F-Phe), 7.65 (bs, 1H, C3H, Pyr), 8.18 (d, 3J = 5.4 Hz, 1H, C6H, Pyr), 9.43 (s, 1H, NH), 10.47 (bs, 1H, NH), 13.92 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 39.1 (SCH3), 55.2 (C4OCH3), 109.4 (C3H, Pyr), 114.0 (C3/5H, H3CO-Phe), 115.2 (C5H, Pyr), 115.9 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 120.6 (C2/6H, H3CO-Phe), 127.5 (C1, F-Phe), 130.9 (d, 3JCF = 8.7 Hz, C2/6H, F-Phe), 132.0 (C1, H3CO-Phe), 146.9 (C6H, Pyr), 148.8 (C2, Imdz), 152.2 (CO), 153.5 (C2, Pyr), 154.9 (C4OCH3), 162.2 (d, 1JCF = 246.7 Hz, CF) ppm; MS (ESI, 70 eV) m/z 466 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)pyridin-2-yl)-3-(m-tolyl)-carbamide (12j). Synthesis was performed according to the general procedure for sulfoxidation starting from 12d (100 mg, 231 µmol) in 2 mL THF and 0.6 mL H2O. Flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether) afforded 12j as colorless solid. Yield 74.8 mg (72%); C23H20FN5O2S (Mr 449.50); m.p. 235 °C; 1H-NMR (DMSO-d6): δ = 2.29 (s, 3H, CH3), 3.09 (s, 3H, SCH3), 6.83 (d, 3J = 7.4 Hz, 1H, C4H, Tol), 6.97 (dd, 3J = 5.4 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.18 (t, 3J = 7.7 Hz, 1H, C5H, Tol), 7.29–7.35 (m, 4H, C3/5H, F-Phe and C2/6H, Tol), 7.52–7.57 (m, 2H, C2/6H, F-Phe), 7.69 (s, 1H, C3H, Pyr), 8.19 (d, 3J = 5.1 Hz, 1H, C6H, Pyr), 9.48 (s, 1H, NH), 10.54 (bs, 1H, NH), 13.92 (bs, 1H, NH, Imdz) ppm; 13C-NMR (75 MHz, DMSO-d6): δ = 21.2 (CH3), 39.1 (SCH3), 109.3 (C3H, Pyr), 115.3 (C5H, Pyr), 115.9 (d, 2JCF = 21.5 Hz, C3/5H, F-Phe), 116.0 (C6H, Tol), 119.3 (C2H, Tol), 123.2 (C4H, Tol), 128.7 (C5H, Tol), 130.9 (C2/6H, F-Phe), 138.1 (C3, Tol), 138.9 (C1, Tol), 143.1 (C4, Pyr), 147.0 (C6H, Pyr), 148.7 (C2, Imdz), 152.1 (CO), 153.4 (C2, Pyr), 162.2 (d, 1JCF = 244.0 Hz, CF) ppm; MS (ESI, 70 eV) m/z 450 [MH]+.
1-(3-Chloro-4-methylphenyl)-3-(4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)pyridin-2-yl)-carbamide (12k). Synthesis was performed according to the general procedure for sulfoxidation starting from 12e (100 mg, 214 µmol) in 4 mL THF and 1 mL H2O. Flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) afforded 12k as beige solid. Yield 78.0 mg (75%); C23H19ClFN5O2S (Mr 483.95); 1H-NMR (DMSO-d6): δ = 2.27 (s, 3H, CH3), 3.09 (s, 3H, SCH3), 6.99 (dd, 3J = 5.4 Hz, 4J = 1.3 Hz, 1H, C5H, Pyr), 7.26 (s, 1H, C5H, Cl-Tol), 7.26 (s, 1H, C6H, Cl-Tol), 7.29–7.35 (m, 2H, C3/5H, F-Phe), 7.52–7.56 (m, 2H, C2/6H, F-Phe), 7.65 (bs, 1H, C3H, Pyr), 7.76 (bs, 1H, C2H, Cl-Tol), 9.55 (s, 1H, NH), 10.77 (bs, 1H, NH), 13.93 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 18.8 (CH3), 39.1 (SCH3), 109.2 (C3H, Pyr), 115.4 (C5H, Pyr), 115.9 (d, 2JCF = 21.5 Hz, C3/5H, F-Phe), 117.5 (C6H, Cl-Tol), 118.7 (C2H, Cl-Tol), 129.0 (C4, Cl-Tol), 131.1 (C2/6H, F-Phe), 131.2 (C5, Imdz and C5H, Cl-Tol), 133.2 (C3, Cl-Tol), 134.6 (C4, Imdz), 138.2 (C1, Cl-Tol), 147.0 (C6H, Pyr), 148.7 (C2, Imdz), 152.1 (CO), 153.2 (C2, Pyr), 162.3 (d, 1JCF = 249.9 Hz, CF) ppm; MS (ESI, 70 eV) m/z 484 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)pyridin-2-yl)-3-(3-(trifluormethyl)phenyl)carbamide (12l). Synthesis was performed according to the general procedure for sulfoxidation starting from 12f (20.0 mg, 41.0 µmol) in 0.4 mL THF and 0.1 mL water. Flash chromatography (SiO2, 40%–100% ethyl acetate/petrol ether) afforded 12l as a colorless solid. Yield 15.1 mg (73%); m.p. 235 °C; 1H-NMR (DMSO-d6): δ = 3.10 (s, 3H, SCH3), 7.02 (dd, 3J = 5.4 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.30–7.37 (m, 2H, C3/5H, F-Phe), 7.36 (d, 3J = 7.7 Hz, 1H, C4H, F3C-Phe), 7.51–7.57 (m, 3H, C2/6H, F-Phe and C5H, F3C-Phe), 7.65 (d, 3J = 8.3 Hz, 1H, C6H, F3C-Phe), 7.71 (s, 1H, C2H, F3C-Phe), 8.08 (bs, 1H, C3H, Pyr), 8.22 (bs, 1H, C6H, Pyr), 9.59 (s, 1H, NH), 10.90 (bs, 1H, NH), 13.93 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 39.1 (SCH3), 109.3 (C2H, F3C-Phe), 114.7 (q, 3JCF = 2.5 Hz, C3H, Pyr), 115.5 (C5H, Pyr), 115.9 (d, 3JCF = 21.9 Hz, C3/5H, F-Phe), 118.7 (q, 3JCF = 2.1 Hz, C4H, F3C-Phe), 122.5 (C6H, F3C-Phe), 124.2 (q, 1JCF = 272.2 Hz, CF3), 125.9 (d, 4JCF = 3.2 Hz, C1, F-Phe), 129.6 (q, 2JCF = 31.4 Hz, C3, F3C-Phe), 130.0 (C5H, F3C-Phe), 130.8 (C5, Imdz), 131.1 (d, 4JCF = 11.4 Hz, C2/6H, F-Phe), 134.5 (C4, Imdz), 139.9 (C1, F3C-Phe), 143.4 (C4, Pyr), 147.1 (C6H, Pyr), 148.7 (C2, Imdz), 152.2 (CO), 162.4 (d, 1JCF = 242.12 Hz, CF) ppm; MS (ESI, 70 eV) m/z 504 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(naphthalen-1-yl)carbamide (12m). Synthesis was performed according to the general procedure for sulfoxidation starting from 12g (51.0 mg, 109 µmol) in 5 mL THF and 1.5 mL H2O. Flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether) afforded 12m as colorless solid. Yield 37.0 mg (70%); C26H20FN5O2S (Mr 485.54); m.p. 244 °C; 1H-NMR (DMSO-d6): δ = 3.10 (s, 3H, SCH3), 7.04 (dd, 3J = 5.4 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.31–7.36 (m, 2H, C3/5H, F-Phe), 7.49 (t, 3J = 7.9 Hz, 1H, C3H, Naph), 7.54–7.59 (m, 4H, C3H, Pyr and C2/6H, F-Phe and C6H, Naph), 7.63–7.68 (m, 2H, C4/7H, Naph), 7.96 (dd, 3J = 8.1 Hz, 4J = 0.8 Hz, 1H, C5H, Naph), 8.16–8.20 (m, 2H, C2/8H, Naph), 8.34 (d, 3J = 5.4 Hz, 1H, C6H, Pyr), 8.93 (s, 1H, NH), 11.58 (bs, 1H, NH), 13.95 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 39.1 (SCH3), 109.4 (C3H, Pyr), 115.3 (C5H, Pyr), 115.9 (d, 2JCF = 21.8 Hz, C3/5H, F-Phe), 116.9 (C2H, Naph), 120.9 (C8H, Naph), 123.1 (C4H, Naph), 125.5 (C1, Naph), 126.0 (C6H, Naph), 126.0 (C3H, Naph), 126.3 (C7H, Naph), 128.6 (C5H, Naph), 130.7 (C5, Imdz), 130.9 (C2/6H, F-Phe), 133.7 (C4a, Naph), 134.1 (C8a, Naph), 143.5 (C4, Pyr), 146.7 (C6H, Pyr), 148.9 (C2, Imdz), 152.6 (CO), 153.6 (C2, Pyr), 162.3 (d, 1JCF = 246.9 Hz, CF) ppm; MS (ESI, 70 eV) m/z 486 [MH]+.
4-(2,4-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)-pyridin-2-yl)-1-methyl-1H-pyrrol-2-carboxamide (16c). Synthesis was performed according to the general procedure for sulfoxidation starting from 16a (196 mg, 368 µmol) in 4 mL THF and 1 mL H2O. Flash chromatography (SiO2, 40%–100% ethyl acetate/petrol ether) afforded 16c as yellow solid. Yield 182 mg (88%); C29H26FN5O4S (Mr 559.62); m.p. 136 °C; 1H-NMR (DMSO-d6): δ = 3.11 (s, 3H, SCH3), 3.78 (s, 3H, C2OCH3), 3.86 (s, 3H, C4OCH3), 3.90 (s, 3H, CH3), 6.57 (dd, 3J = 8.5 Hz, 4J = 2.4 Hz, 1H, C5H, (OCH3)2-Phe), 6.61 (d, 4J = 2.3 Hz, 1H, C3H, (OCH3)2-Phe), 7.07 (d, 3J = 4.8 Hz, 1H, C5H, Pyr), 7.23–7.38 (m, 2H, C3/5H, F-Phe), 7.42 (bs, 1H, C3H, Pyrrole), 7.46 (d, 3J = 8.4 Hz, 1H, C6H, (OCH3)2-Phe), 7.54–7.59 (m, 2H, C2/6H, F-Phe), 7.68 (bs, 1H, C5H, Pyrrole), 8.22 (d, 3J = 4.5 Hz, 1H, C6H, Pyr), 8.42 (s, 1H, C3H, Pyr), 10.17 (s, 1H, CONH), 13.89 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 36.6 (CH3), 39.2 (SCH3), 55.2 (C2OCH3), 55.4 (C4OCH3), 98.8 (C3H, (OCH3)2-Phe), 105.2 (C5H, (OCH3)2-Phe), 111.8 (C3H, Pyr), 113.3 (C5H, Pyrrole), 115.9 (C1, (OCH3)2-Phe), 116.0 (d, 2JCF = 22.4 Hz, C3/5H, F-Phe), 116.1 (C5H, Pyr), 118.5 (C4, Pyrrole), 124.0 (C2, Pyrrole), 125.8 (d, 4JCF = 3.3 Hz, C1, F-Phe), 127.5 (C6H, (OCH3)2-Phe), 128.6 (C3H, Pyrrole), 131.3 (d, 3JCF = 8.3 Hz, C2/6H. F-Phe), 131.9 (C4, Imdz), 135.1 (C5, Imdz), 143.0 (C4, Pyr), 147.8 (C6H, Pyr), 148.5 (C2, Imdz), 152.9 (C2, Pyr), 156.7 (C4OCH3), 158.6 (C2OCH3), 159.9 (CO), 162.4 (d, 1JCF = 244.3 Hz, CF) ppm; MS (ESI, 70 eV) m/z 560 [MH]+.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





















