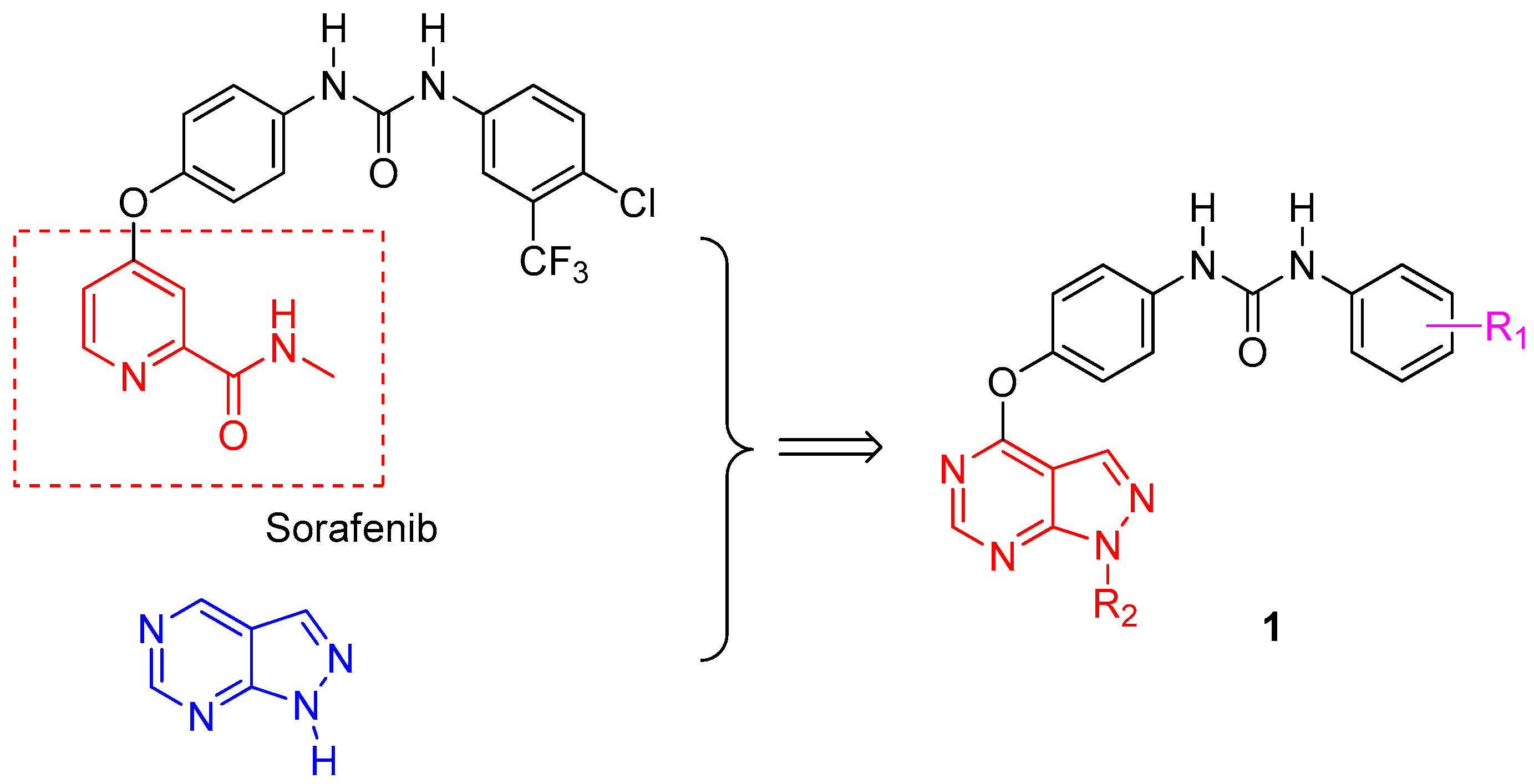
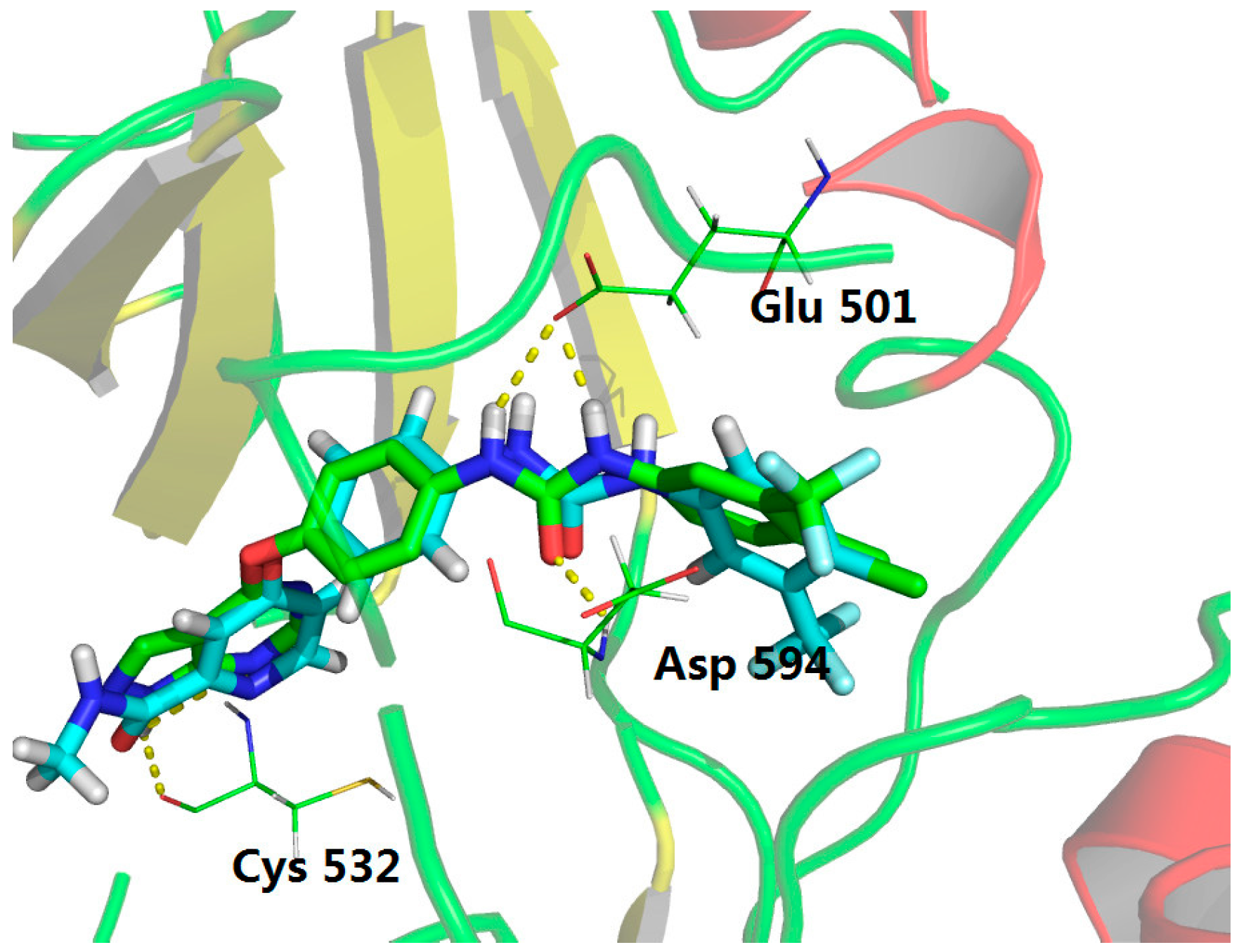
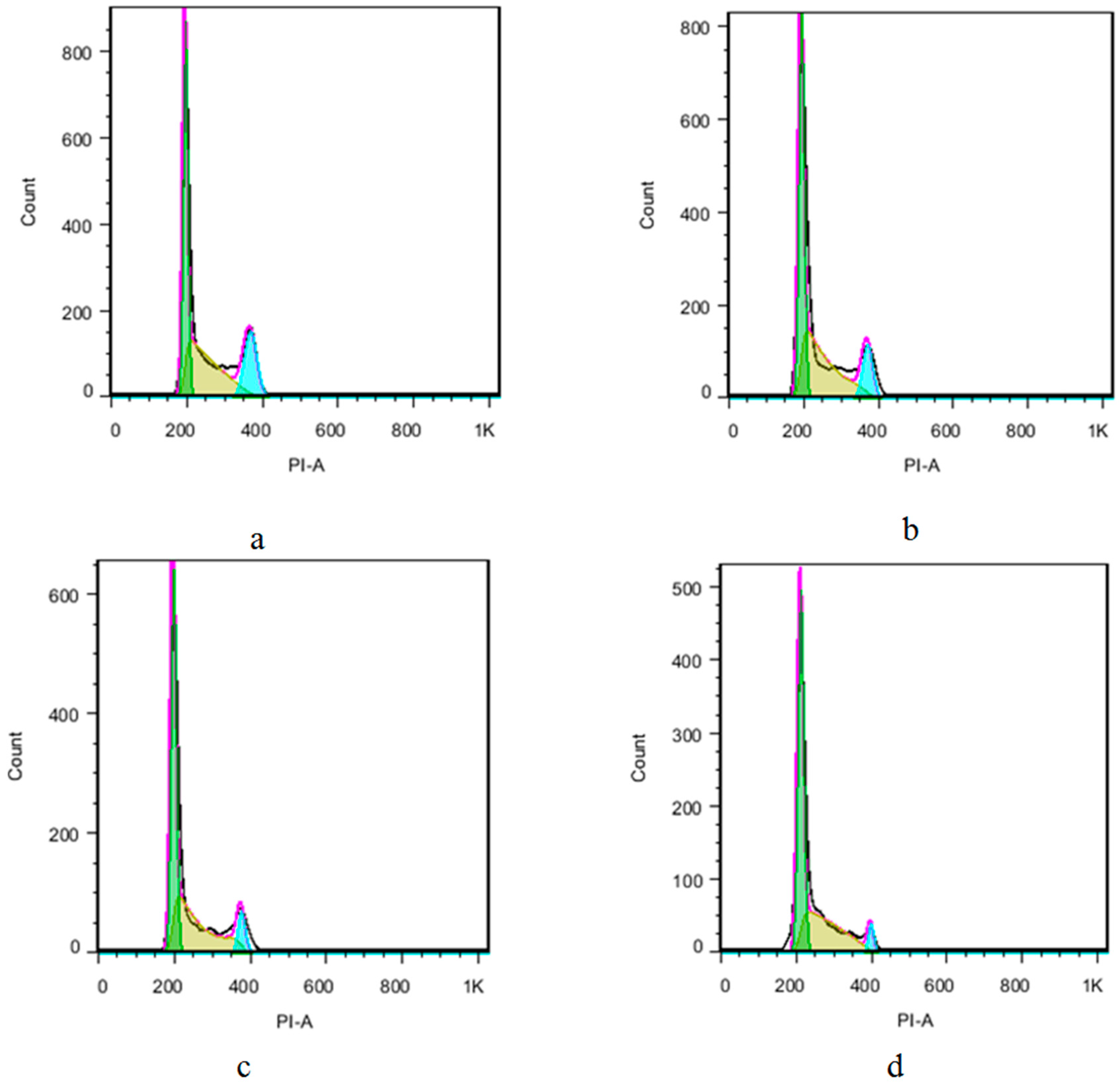
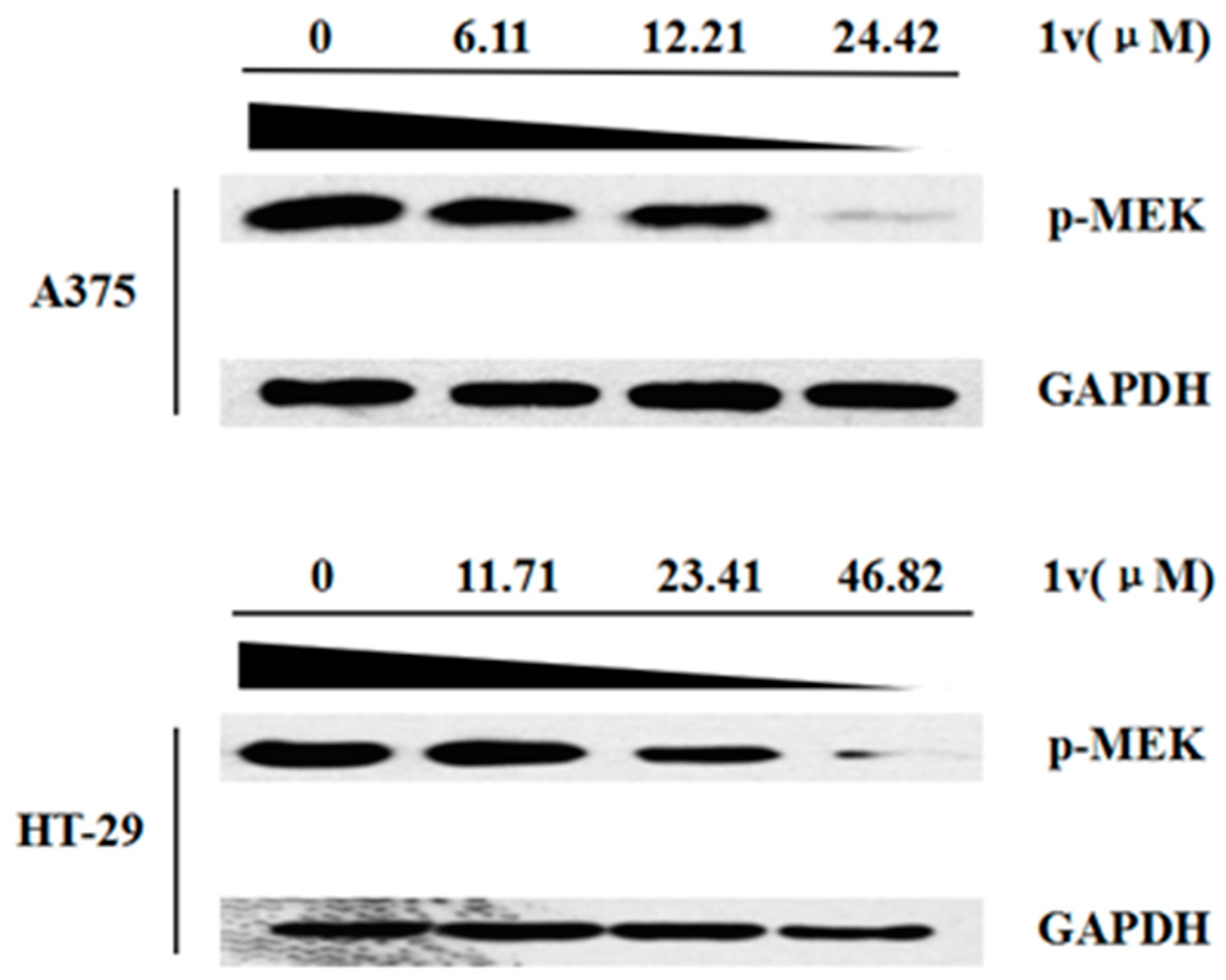
3.1. Chemistry
Melting points were determined using an X-4 Melting-point Apparatus with Microscope (Yuhua Instrument Co., Ltd., Henan, China) and were uncorrected. 1H- and 13C-NMR spectra were recorded on a Bruker AVIII 400 MHz or Bruker AVIII 600 MHz and 100 MHz spectrometer (Bruker Co., Fällanden, Switzerland) with tetramethylsilane (TMS) as the internal standard, the values of the chemical shifts (δ) are given in ppm, and coupling constants (J) are given in Hz. Mass spectra (ESI-MS) were performed on Waters ZQ4000 (Waters Co., Milford, MA, USA). All reactions were monitored using thin-layer chromatography (TLC) on silica gel plates at 254 nm under an ultraviolet (UV) light. Flash column chromatography separations were performed on normal phase silica gel (200–300 mesh, Merck Co., Elkton, VA, USA) or reverse phase silica gel by using Yamazen AI-580 flash chromatography (Yamazen Co., Osaka, Japan) with UV detection at 254 nm. All reagents were purchased from commercial vendors and were used without further purification. All oxygen-sensitive or moisture-sensitive reactions were run under nitrogen atmosphere. Yields were not optimized.
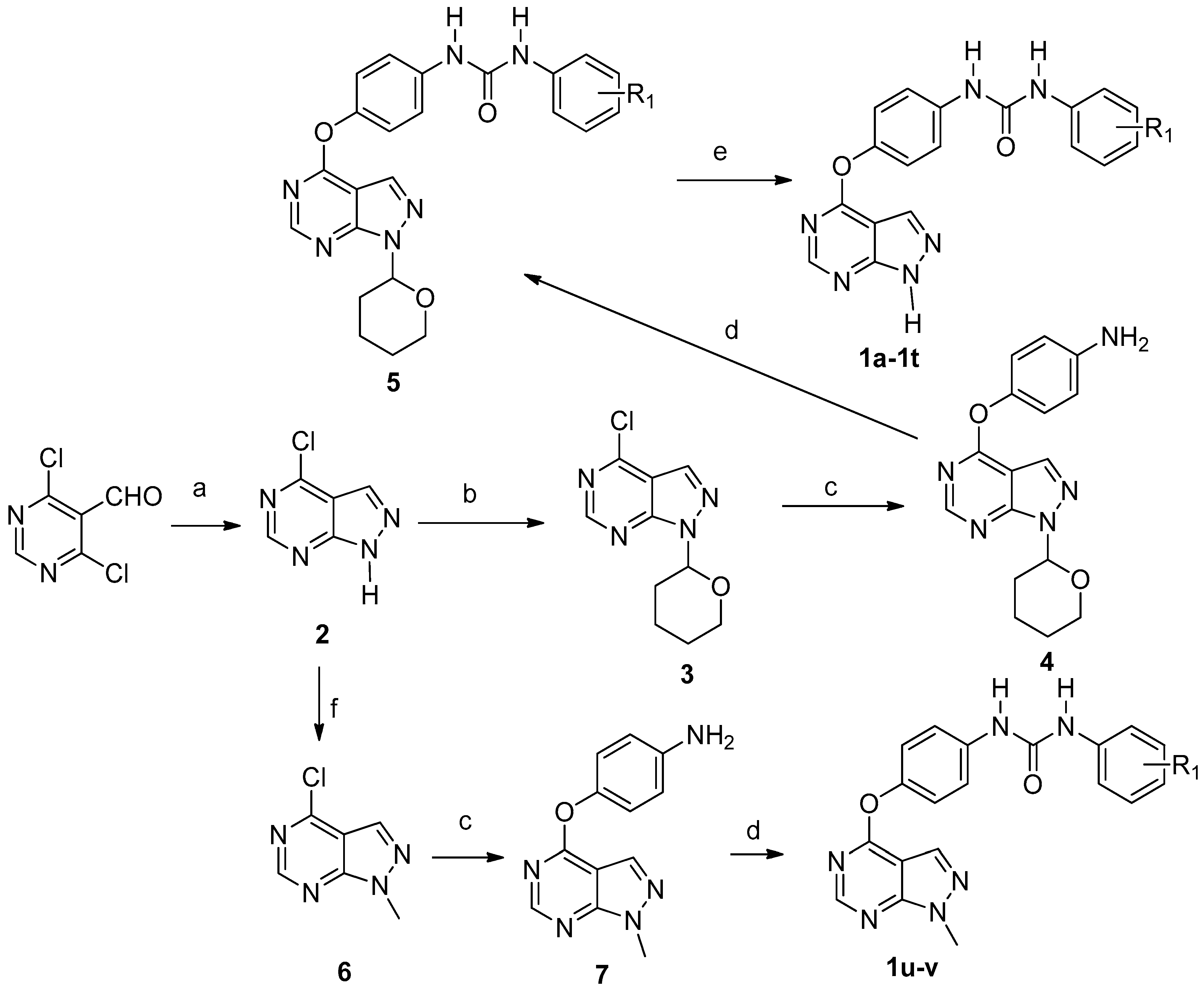
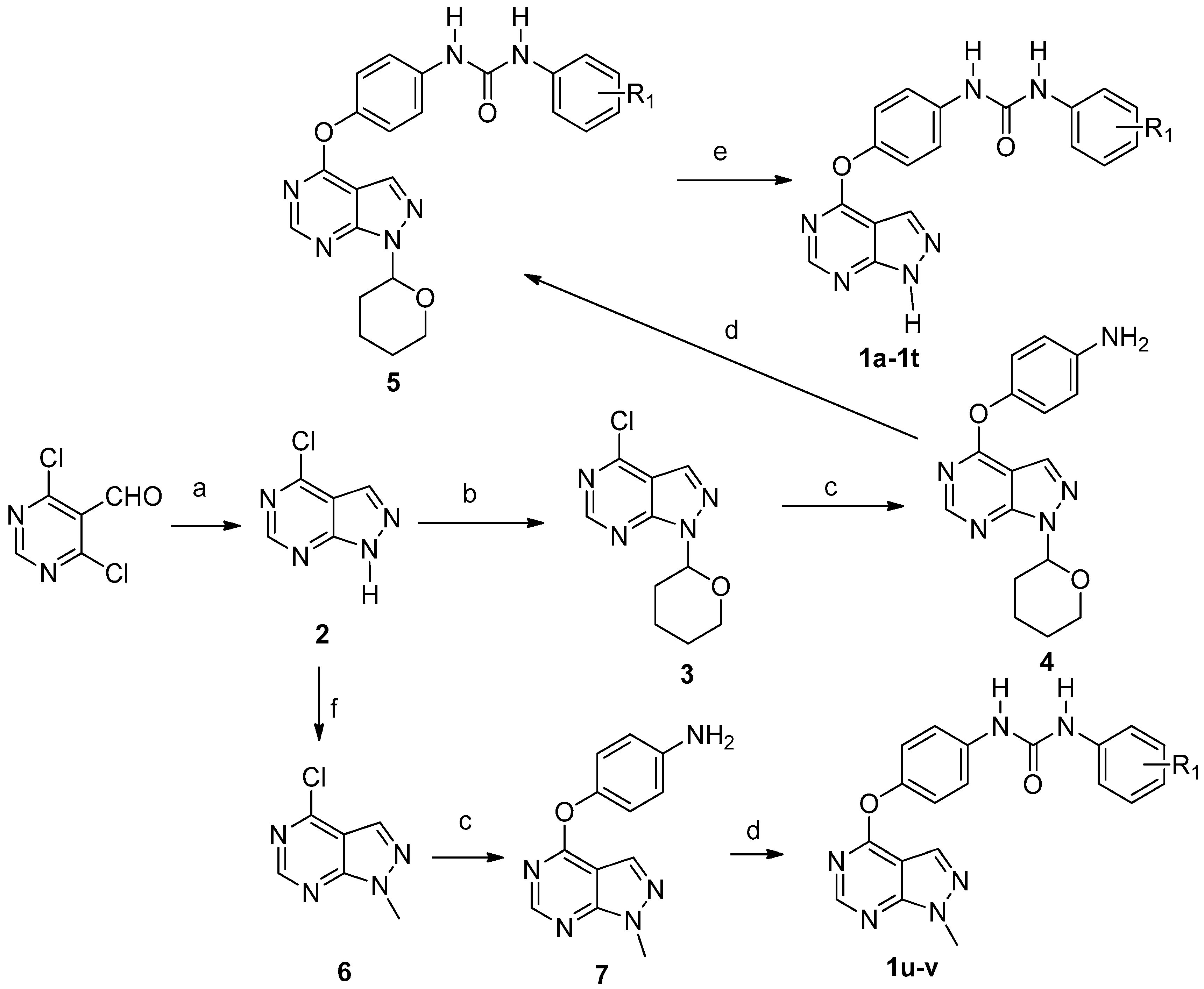
4-Chloro-1H-pyrazolo[3,4-d]pyrimidine (2). To the solution of 4,6-dichloropyrimidine-5-carbaldehyde (1.0 g, 0.006 mol) in methanol (20 mL) at −65 °C, triethylamine (0.97 mL,1.2 equivalents (eq.)) was added. A solution of hydrazine monohydrate (0.274 mL 1.0 eq.) in methanol (10 mL) was slowly dripped into above stirred solution by using a constant-pressure dropping funnel. The mixture was allowed to warm to room temperature and stirred for 2–3 h. The reaction mixture was concentrated in vacuo and crude product was diluted with water (20 mL), and extracted with EtOAc (60 mL × 3). The combined organic layer was washed with saturated solution of NaCl (60 mL × 3), dried over MgSO4 and concentrated to give compound 2. Yield: 68.9%. 1H-NMR (400 MHz, deuteriated dimethyl sulfoxide (DMSO-d6)) δ 14.51 (s, 1H), 8.84 (s, 1H), 8.45 (s, 1H). ESI-MS m/z: 153.00 [M − H]−.
4-Chloro-1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidine (3). Compound 2 (1.88 g, 0.012 mol) was dissolved in EtOAc (50 mL) and heated to 50 °C. After 10 min pyridinium 4-toluenesulfonate (PPTs) (50 mg) were added, followed by the addition of 3,4-dihydro-2H-pyran. The resulting reaction mixture was at stirred 50 °C. After the reaction was complete according to the TLC detection, the mixture was cooled to room temperature, washed with water (60 mL × 1), and a saturated solution of NaCl (50 mL × 2), and dried over MgSO4. The ethyl acetate was removed and petroleum ether (60 mL × 2) was added. The mixture was heated and filtered through cotton. Removal of petroleum ether in vacuo gives compound 3 as light yellow colored solid. Yield: 76.5%. 1H-NMR (400 MHz, DMSO-d6) δ 8.92 (s, 1H), 8.55 (s, 1H), 6.02 (dd, J = 10.4, 2.4 Hz, 1H), 3.97 (d, J = 12.0 Hz, 1H), 3.76–3.70 (m, 1H), 2.49–2.42 (m, 1H), 2.07–2.08 (m, 1H), 1.98–1.94 (m, 1H), 1.85–1.73 (m, 1H), 1.64–1.58 (m, 2H). ESI-MS m/z: 239.06 [M + H]+.
4-((1-(Tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)aniline (4). To the mixture of para-aminophenol (0.28 g) in dimethylformamide (DMF, 2 mL), Cs2CO3 (1.2 eq.) was added under the protection of nitrogen. The reaction mixture was stirred at room temperature for 1.5–2 h, and then compound 3 was added slowly. After stirring overnight, the reaction mixture was diluted with EtOAc (50 mL) and washed with 1 M NaOH (60 mL × 1), water (80 mL × 1), 5% LiCl (50 mL × 2) and brine. The organic layer was dried over MgSO4 and concentrated to give compound 4 as a brown oily solid. Yield: 79.5%. 1H-NMR (400 MHz, DMSO-d6) δ 8.57 (s, 1H), 7.75 (s, 1H), 6.99–6.95 (m, 2H), 6.67–6.31 (m, 2H), 5.96 (dd, J = 10.0, 2.4 Hz, 1H), 5.19 (s, 2H), 3.96 (d, J = 12.4 Hz, 1H), 3.73–3.67 (m, 1H), 2.48–2.40 (m, 1H), 2.06–2.00 (m, 1H), 1.92–1.88 (m, 1H), 1.79–1.71 (m, 1H), 1.61–1.56 (m, 2H). ESI-MS m/z: 310.14 [M − H]−.
1-(4-Chloro-3-(trifluoromethyl)phenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5a). To the solution of compound 4 in CH2Cl2 at 0 °C 4-chloro-3-(trifluoromethyl)phenyl isocyanate (1.0 eq.) was added. The mixture was stirred overnight at room temperature. To the resulting suspension, petroleum ether (60 mL) was added. The solid material was collected by filtration to provide the title compound as a white solid. Yield: 66.6%. 1H-NMR (400 MHz, DMSO-d6) δ 9.22 (s, 1H), 8.99 (s, 1H), 8.58 (s, 1H), 8.14 (s, 2H), 7.69–7.57 (m, 4H), 7.27 (d, J = 8.8 Hz, 2H), 5.98 (d, J = 10.0 Hz, 1H), 3.97 (d, J = 11.6 Hz, 1H), 3.74–3.68 (m, 1H), 2.05 (d, J = 12.4 Hz, 1H), 1.93 (d, J = 12.4 Hz, 1H), 1.77 (d, J = 8.0 Hz, 1H), 1.59 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 163.7, 155.7, 152.9, 147.1, 139.8, 137.6, 132.4, 132.3, 127.3, 123.5, 122.7, 121.9, 120.3, 117.3, 102.7, 82.7, 67.5, 29.1, 25.0, 22.6. ESI-MS m/z: 533.12 [M + H]+.
1-(4-((1-(Tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(m-tolyl)urea (5b). Compound 5b was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 3-methyl phenyl isocyanate. Yield: 80.0%. 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 8.82 (s, 1H), 8.66 (s, 1H), 8.51 (s, 1H), 8.01 (s, 1H), 7.56 (d, J = 8.9 Hz, 2H), 7.32 (s, 1H), 7.25 (d, J = 8.9 Hz, 3H), 7.17 (t, J = 7.7 Hz, 1H), 6.80 (d, J = 7.7 Hz, 1H), 2.29 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 163.68, 157.13, 155.39, 153.00, 146.77, 139.99, 138.36, 138.11, 132.26, 129.04, 123.02, 122.66, 120.47, 119.63, 117.67, 117.24, 102.01. ESI-MS m/z: 445.19 [M − H]−.
1-(3,4-Dichlorophenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl) urea (5c). Compound 5c was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 3,4-dichlorophenyl isocyanate. Yield: 67.0%. 1H-NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 8.97 (s, 1H), 8.58 (s, 1H), 8.14 (s, 1H), 7.90 (s, 1H), 7.58–7.52 (m, 3H), 7.37 (d, J = 8.4 Hz, 1H), 7.27 (d, J = 8.8 Hz, 2H), 5.99 (d, J = 9.6 Hz, 1H), 3.97 (d, J = 10.8 Hz, 1H), 3.71 (s, 1H), 2.05 (d, J = 12.8 Hz, 1H), 1.93 (d, J = 12.4 Hz, 1H), 1.79 (s, 1H), 1.59 (s, 2H), 1.24 (s, 1H). ESI-MS m/z: 499.10 [M + H]+.
1-(4-Chlorophenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5d). Compound 5d was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 4-chlorophenyl isocyanate. Yield: 68.3%. 1H-NMR (400 MHz, DMSO-d6) δ 8.87 (s, 1H), 8.85 (s, 1H), 8.58 (s, 1H), 8.13 (s, 1H), 7.56 (d, J = 8.9 Hz, 2H), 7.51 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 8.9 Hz, 2H), 5.98 (dd, J = 10.1, 1.9 Hz, 1H), 3.97 (d, J = 11.2 Hz, 1H), 3.77–3.65 (m, 1H), 2.49–2.41 (m, 1H), 2.05 (d, J = 12.5 Hz, 1H), 1.93 (dd, J = 12.9, 2.3 Hz, 1H), 1.83–1.68 (m, 1H), 1.67–1.53 (m, 2H). ESI-MS m/z: 465.14 [M + H]+.
1-Phenyl-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5e). Compound 5e was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with phenyl isocyanate. Yield: 60.2%. 1H-NMR (400 MHz, DMSO-d6) δ 8.81 (s, 1H), 8.71 (s, 1H), 8.58 (s, 1H), 8.12 (s, 1H), 7.57 (d, J = 7.7 Hz, 2H), 7.48 (d, J = 7.7 Hz, 2H), 7.33–7.28 (t, 2H), 7.26 (d, J = 8.9 Hz, 2H), 6.99 (t, J = 7.3 Hz, 1H), 5.99 (d, J = 12.5 Hz, 1H), 3.97 (d, J = 11.2 Hz, 1H), 3.76–3.66 (m, 1H), 2.45 (m, 1H), 2.03 (m, 1H), 1.93 (m, 1H), 1.86–1.69 (m, 1H), 1.66–1.53 (m, 2H). ESI-MS m/z: 431.18 [M + H]+.
1-(2-Chloro-5-methylphenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy) phenyl)urea (5f). Compound 5f was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 2-chloro-5-methylphenyl isocyanate. Yield: 69.4%. 1H-NMR (400 MHz, DMSO-d6) δ 9.54 (s, 1H), 8.59 (s, 1H), 8.28 (s, 1H), 8.13 (s, 1H), 8.03 (s, 1H), 7.58 (d, J = 8.8 Hz, 2H), 7.34 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 8.0 Hz, 1H), 5.99 (d, J = 10.0 Hz, 1H), 3.97 (d, J = 11.2 Hz, 1H), 3.75–3.68 (m, 1H), 2.30 (s, 3H), 2.08–2.00 (m, 1H), 1.93 (d, J = 11.6 Hz, 1H), 1.77 (s, 1H), 1.60 (s, 2H), 1.24 (s, 1H). ESI-MS m/z: 477.15 [M − H]−.
1-(3-Chlorophenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5g). Compound 5g was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 3-chlorophenyl isocyanate. Yield: 72.3%. 1H-NMR (400 MHz, DMSO-d6) δ 8.94 (s, 1H), 8.90 (s, 1H), 8.60 (s, 1H), 8.14 (s, 1H), 7.73 (s, 1H), 7.57 (d, J = 9.0 Hz, 2H), 7.30 (m, 5H), 7.03 (m, 1H), 5.99 (dd, J = 10.2, 2.3 Hz, 1H), 3.97 (d, J = 12.0 Hz, 1H), 3.76–3.66 (m, 1H), 2.45 (m, 1H), 2.05 (m, 1H), 1.93 (m, 1H), 1.83–1.72 (m, 1H), 1.61 (m, 2H). ESI-MS m/z: 463.14 [M − H]−.
1-(2,3-Dimethylphenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl) urea (5h). Compound 5h was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 2,3-dimethylphenyl isocyanate. Yield: 65.5%. 1H-NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 8.58 (s, 1H), 8.10 (s, 1H), 8.00 (s, 1H), 7.56 (t, J = 8.6 Hz, 3H), 7.25 (d, J = 8.4 Hz, 1H), 7.05 (t, J = 7.8 Hz, 1H), 6.92 (d, J = 7.2 Hz, 1H), 5.99 (d, J = 9.2 Hz, 1H), 3.97 (d, J = 11.2 Hz, 1H), 3.75–3.68 (m, 1H), 2.45 (d, J = 10.4 Hz, 1H), 2.27 (s, 3H), 2.16 (s, 3H), 2.05 (d, J = 10.8 Hz, 1H), 1.93 (d, J = 12.0 Hz, 1H), 1.79 (s, 1H), 1.60 (s, 2H), 1.46 (s, 1H). ESI-MS m/z: 481.21 [M + Na]+.
1-(2-Chloro-5-(trifluoromethyl)phenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5i). Compound 5i was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 2-chloro-5-(trifluoromethyl)phenyl isocyanate. Yield: 74.5%. 1H-NMR (400 MHz, DMSO-d6) δ 9.22 (s, 1H), 8.99 (s, 1H), 8.58 (s, 1H), 8.14 (s, 2H), 7.69–7.57 (m, 4H), 7.27 (d, J = 8.8 Hz, 2H), 5.98 (d, J = 10.0 Hz, 1H), 3.97 (d, J = 11.6 Hz, 1H), 3.74–3.68 (m, 1H), 2.05 (d, J = 12.4 Hz, 1H), 1.93 (d, J = 12.4 Hz, 1H), 1.77 (d, J = 8.0 Hz, 1H), 1.59 (s, 3H). ESI-MS m/z: 533.12 [M + H]+.
1-(3-Fluoro-5-(trifluoromethyl)phenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5j). Compound 5j was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 3-fluoro-5-(trifluoromethyl)phenyl isocyanate. Yield: 60.2%. 1H-NMR (400 MHz, DMSO-d6) δ 9.31 (s, 1H), 9.06 (s, 1H), 8.58 (s, 1H), 8.15 (s, 1H), 7.73 (s, 1H), 7.64 (d, J = 11.2 Hz, 1H), 7.58 (d, J = 8.8 Hz, 2H), 7.28 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.5 Hz, 1H), 5.99 (dd, J = 10.1, 2.1 Hz, 1H), 3.97 (d, J = 11.6 Hz, 1H), 3.76–3.66 (m, 1H), 2.45 (dd, J = 12.8, 4.0 Hz, 1H), 2.05 (d, J = 12.5 Hz, 1H), 1.93 (dd, J = 13.0, 2.6 Hz, 1H), 1.84–1.70 (m, 1H), 1.61 (d, J = 10.0 Hz, 2H). ESI-MS m/z: 517.15 [M + H]+.
1-(4-Fluoro-3-methylphenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5k). Compound 5k was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 4-fluoro-3-methylphenyl isocyanate. Yield: 79.4%. 1H-NMR (400 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.69 (s, 1H), 8.58 (s, 1H), 8.11 (s, 1H), 7.56 (d, J = 8.8 Hz, 2H), 7.38 (dd, J = 6.8, 2.4 Hz,1H), 7.30–7.24 (m, 3H), 7.06 (t, J = 9.2 Hz, 1H), 5.98 (dd, J = 10.4, 2.4 Hz, 1H), 3.97 (d, J = 10.8 Hz, 1H), 3.74–3.66 (m, 1H), 2.22 (s, 3H), 2.05 (d, J = 11.6 Hz,1H), 1.95–1.90 (m, 1H), 1.80–1.76 (m, 1H), 1.59 (s, 2H), 1.24 (s, 1H). ESI-MS m/z: 485.15 [M + Na]+.
1-(3-Ethylphenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5l). Compound 5l was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 3-ethylpheny isocyanate. Yield: 69.4%. 1H-NMR (400 MHz, DMSO-d6) δ 8.79 (s, 1H), 8.65 (s, 1H), 8.58 (s, 1H), 8.11 (s, 1H), 7.56 (d, J = 8.9 Hz, 2H), 7.37 (d, J = 9.2 Hz, 1H), 7.29 (s, 1H), 7.25 (d, J = 8.8 Hz, 2H), 7.06 (t, J = 9.2 Hz, 1H), 5.98 (dd, J = 9.9, 1.4 Hz, 1H), 3.97 (d, J = 11.4 Hz, 1H), 3.72 (m, 1H), 2.50–2.41 (m, 1H), 2.23 (s, 3H), 2.05 (m, 1H), 1.92 (m, 1H), 1.85–1.70 (m, 1H), 1.59 (m, 2H). ESI-MS m/z: 459.21 [M + H]+.
1-(3-Nitrophenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5m). Compound 5m was s prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 3-nitrophenyl isocyanate. Yield: 62.3%. 1H-NMR (400 MHz, DMSO-d6) δ 9.41 (s, 1H), 9.11 (s, 1H), 8.59 (s, 2H), 8.16 (s, 1H), 7.85 (s, 1H), 7.74 (s, 1H), 7.59 (s, 2H), 7.29–7.00 (m, 2H), 5.99 (d, J = 12.4 Hz, 1H), 5.31 (s, 1H), 3.96 (s, 2H), 3.72 (s, 2H), 2.02 (s, 2H), 1.77 (s, 1H), 1.59 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 167.36, 163.69, 155.70, 152.91, 148.58, 147.07, 141.45, 137.66, 132.23, 131.95, 130.46, 129.04, 124.77, 122.64, 120.27, 116.72, 112.61, 102.74, 82.72, 67.82, 28.77, 22.79. ESI-MS m/z: 474.16 [M − H]−.
1-(2-Fluoro-5-(trifluoromethyl)phenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5n). Compound 5n was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 2-fluoro-5-(trifluoromethyl)phenyl isocyanate. Yield: 70.2%. 1H-NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.96 (s, 1H), 8.61 (d, J = 22.8 Hz, 2H), 8.16 (s, 1H), 7.55 (d, J = 26.4 Hz, 3H), 7.41 (s, 1H), 7.29 (d, J = 5.6 Hz, 2H), 5.98 (d, J = 8.8 Hz, 1H), 3.95 (s, 1H), 3.71 (s, 1H), 2.03 (s, 1H), 1.92 (d, J = 12.4 Hz, 2H), 1.78 (s, 1H), 1.59 (s, 2H). ESI-MS m/z: 517.15 [M + H]+.
1-(3-(Methylthio)phenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5o). Compound 5o was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 3-(methylthio) phenyl isocyanate. Yield: 67.8%. 1H-NMR (400 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.77 (s, 1H), 8.58 (s, 1H), 8.12 (s, 1H), 7.57 (d, J = 9.0 Hz, 2H), 7.49 (t, J = 1.7 Hz, 1H), 7.26 (d, J = 8.9 Hz, 2H), 7.22 (d, J = 7.8 Hz, 1H), 7.17 (d, J = 8.8 Hz, 1H), 6.88 (d, J = 7.8 Hz, 1H), 5.98 (dd, J = 10.2, 2.2 Hz, 2H), 2.51 (d, J = 1.8 Hz, 2H), 2.47 (s, 2H), 1.99 (dd, J = 42.9, 17.3 Hz, 2H), 1.59 (s, 3H). ESI-MS m/z: 477.16 [M + H]+.
1-(3-Cyanophenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5p). Compound 5p was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 3-cyanophenyl isocyanate. Yield: 69.8%. 1H-NMR (400 MHz, DMSO-d6) δ 9.10 (s, 1H), 9.00 (s, 1H), 8.58 (s, 1H), 8.13 (s, 1H), 8.00 (t, J = 1.8 Hz, 1H), 7.71 (ddd, J = 3.2, 2.0, 1.2 Hz, 1H), 7.59 (d, J = 2.4 Hz, 1H), 7.57 (d, J = 2.0 Hz, 1H), 7.51 (t, J = 8.0 Hz, 1H), 7.43 (dt, J = 7.6, 1.2 Hz, 1H), 7.29–7.27 (m, 2H), 5.99 (dd, J = 10.0, 2.4 Hz, 1H), 3.18 (d, J = 5.2 Hz, 2H), 1.99 (s, 6H). ESI-MS m/z: 456.17 [M + H]+.
Methyl 3-(3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)ureido)benzoate (5q). Compound 5q was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with methyl 3-isocyanatobenzoate. Yield: 74.5%. 1H-NMR (400 MHz, DMSO-d6) δ 9.03 (s, 1H), 8.89 (s, 1H), 8.58 (s, 1H), 8.23 (s, 1H), 8.13 (s, 1H), 7.65 (d, J = 7.9 Hz, 1H), 7.58 (d, J = 8.9 Hz, 3H), 7.44 (t, J = 7.9 Hz, 1H), 7.27 (d, J = 8.9 Hz, 2H), 5.98 (dd, J = 10.2, 2.1 Hz, 1H), 4.03–3.93 (m, 1H), 3.87 (s, 3H), 3.68–3.71 (m, 1H), 2.44–2.51 (m, 1H), 1.99–2.03 (m, 1H), 1.91–1.95 (m, 1H), 1.78 (m, 1H), 1.59 (m, 3H). ESI-MS m/z: 489.18 [M + H]+.
1-(5-Methyl-2-nitrophenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy) phenyl)urea (5r). Compound 5r was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 5-methyl-2-nitrophenylisocyanate. Yield: 66.8%. 1H-NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 9.71 (s, 1H), 8.57 (s, 1H), 8.23 (s, 1H), 8.13 (s, 1H), 8.03 (d, J = 7.7 Hz, 1H), 7.61 (d, J = 6.9 Hz, 2H), 7.28 (d, J = 6.7 Hz, 2H), 7.02 (d, J = 7.3 Hz, 1H), 5.98 (d, J = 9.1 Hz, 1H), 3.96 (d, J = 10.6 Hz, 1H), 3.69 (d, J = 9.5 Hz, 1H), 2.39 (s, 3H), 1.98 (dd, J = 50.4, 13.4 Hz, 2H), 1.42 (d, J = 31.1 Hz, 2H), 0.84 (s, 2H). ESI-MS m/z: 490.18 [M + H]+.
1-(3-Bromophenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (5s). Compound 5s was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 3-bromophenylisocyanate. Yield: 79.0%. 1H-NMR (400 MHz, DMSO-d6) δ 8.92 (s, 1H), 8.89 (s, 1H), 8.58 (s, 1H), 8.13 (s, 1H), 7.87 (s, 1H), 7.57 (d, J = 8.8 Hz, 2H), 7.34 (d, J = 8.1 Hz, 1H), 7.28 (s, 1H), 7.24 (d, J = 8.7 Hz, 2H), 7.16 (d, J = 7.7 Hz, 1H), 5.98 (d, J = 8.8 Hz, 1H), 3.97 (d, J = 11.2 Hz, 1H), 3.71 (t, J = 12.6 Hz, 1H), 1.99 (dd, J = 47.7, 13.1 Hz, 2H), 1.82–1.71 (m, 1H), 1.59 (d, J = 3.0 Hz, 2H), 1.45–1.35 (m, 1H). ESI-MS m/z: 509.09 [M + H]+.
1-(3,4-Dimethylphenyl)-3-(4-((1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazolo[3,4-d]pyri-midin-4-yl)oxy) phenyl)urea (5t). Compound 5t was prepared using the same procedure as described for the synthesis of 5a by replacing 4-chloro-3-(trifluoromethyl)phenyl isocyanate with 3,4-dimethylphenylisocyanate. Yield: 61.8%. 1H-NMR (400 MHz, DMSO-d6) δ 8.74 (s, 1H), 8.58 (s, 1H), 8.52 (s, 1H), 8.10 (s, 1H), 7.55 (d, J = 8.8 Hz, 2H), 7.25 (s, 2H), 7.23 (s, 1H), 7.19 (d, J = 8.1 Hz, 1H), 7.04 (d, J = 8.2 Hz, 1H), 5.98 (d, J = 9.7 Hz, 1H), 3.97 (dd, J = 10.9, 1.0 Hz, 1H), 3.75–3.66 (m, 1H), 2.44 (dd, J = 12.8, 3.9 Hz, 1H), 2.20 (s, 3H), 2.16 (s, 3H), 2.08–2.01 (m, 1H), 1.92 (d, J = 14.7 Hz, 1H), 1.83–1.73 (m, 1H), 1.59 (t, J = 8.3 Hz, 2H). ESI-MS m/z: 459.21 [M + H]+.
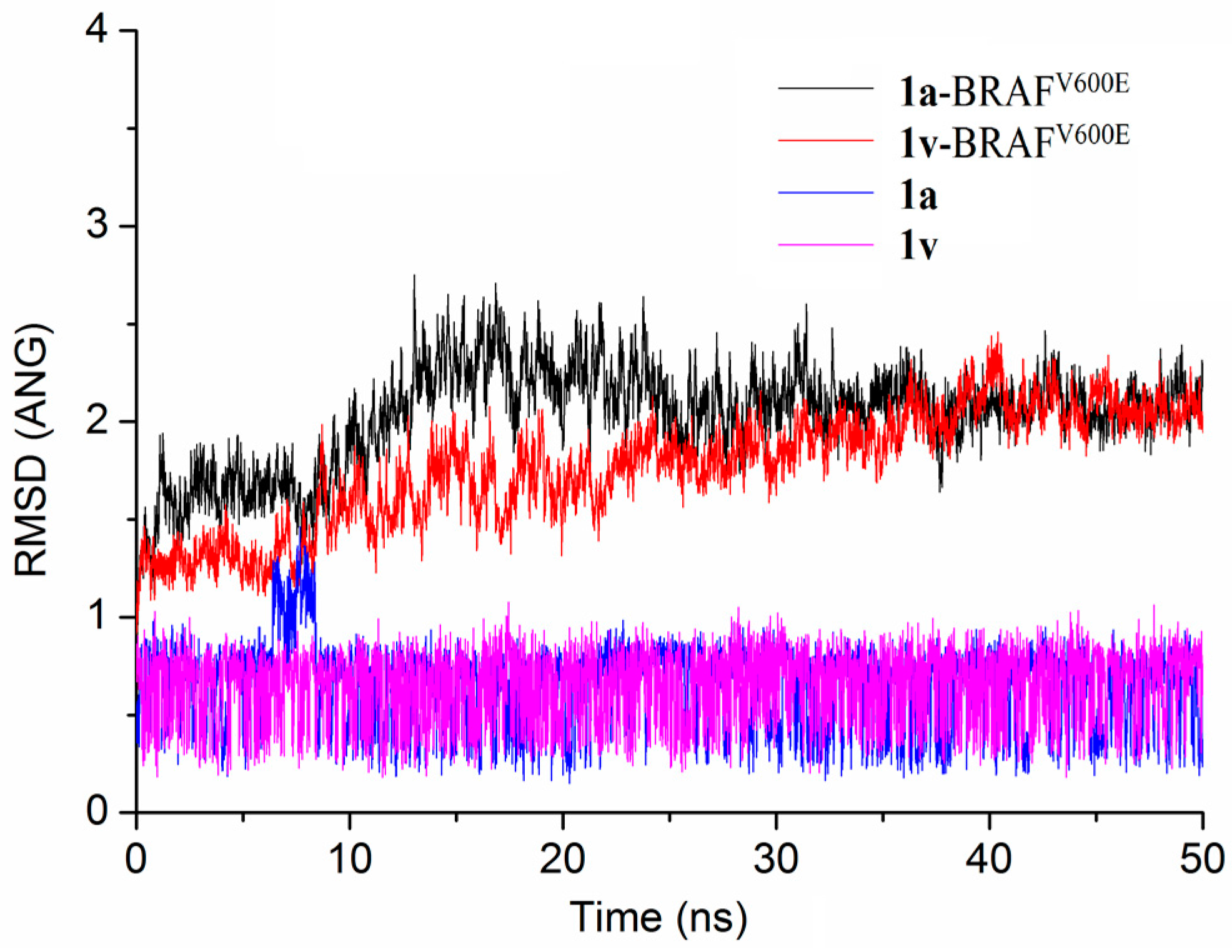
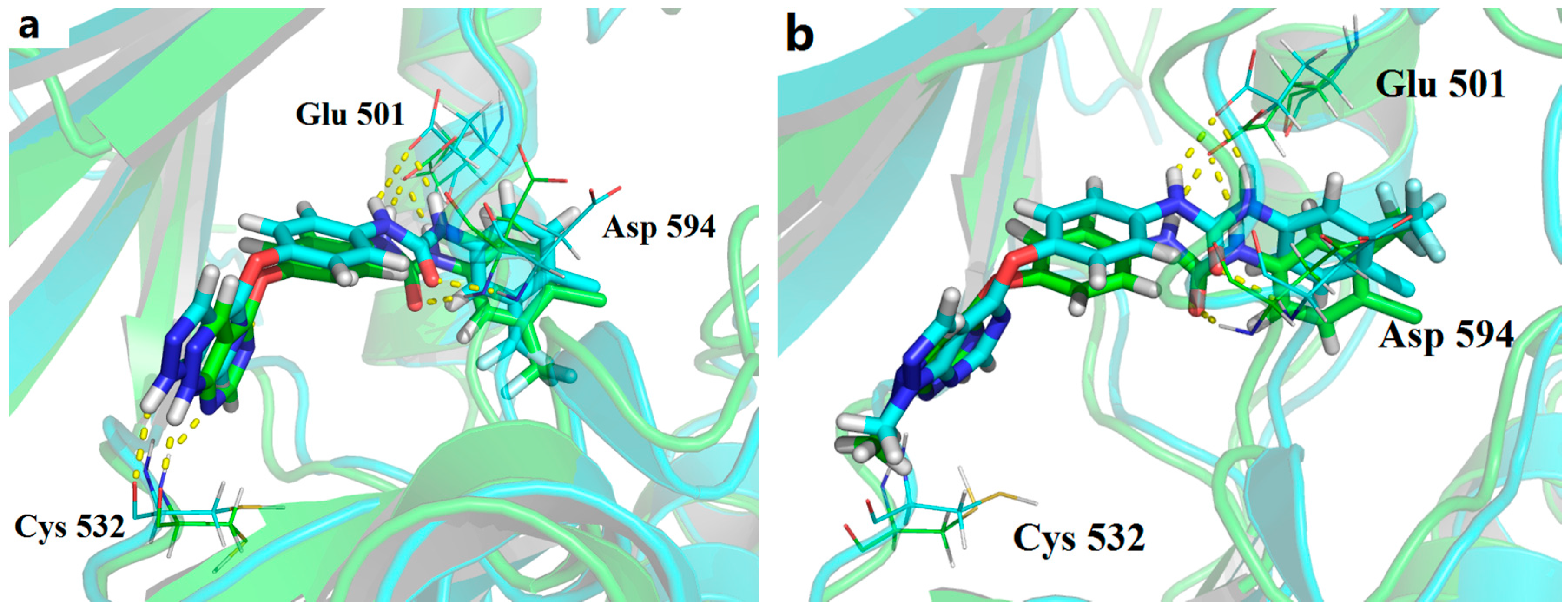
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(4-chloro-3-(trifluoromet-hyl)phenyl)urea (1a). A mixture of 1a (0.1 mmol) in 4 M HCl/1,4-dioxane (5 mL) was stirred at room temperature for 30–70 min. To the resulting suspension, ether (60 mL) was added. The solid material was filtered, washed twice with ether and further purified by flash column chromatography to provide the title compound. Yield: 70.2%; m.p. 222.6–225.8 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 9.25 (s, 1H), 9.01 (s, 1H), 8.51 (s, 1H), 8.13 (s, 1H), 8.04 (s, 1H), 7.71–7.60 (m, 2H), 7.58 (d, J = 8.8 Hz, 2H), 7.27 (d, J = 8.8 Hz, 2H). 13C-NMR (100 MHz, DMSO-d6) δ 163.61, 157.13, 155.36, 152.91 147.18, 139.77, 137.54, 132.41, 132.25, 127.28, 126.98, 124.76, 123.48, 122.71, 120.31, 117.23, 101.76. ESI-MS m/z: 449.40 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(m-tolyl)urea (1b). Compound 1b was prepared using the same procedure as described for the synthesis of 1a. Yield: 71.8%; m.p. 227.4–230.0 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 8.82 (s, 1H), 8.66 (s, 1H), 8.51 (s, 1H), 8.01 (s, 1H), 7.56 (d, J = 8.9 Hz, 2H), 7.32 (s, 1H), 7.25 (d, J = 8.9 Hz, 3H), 7.17 (t, J = 7.7 Hz, 1H), 6.80 (d, J = 7.7 Hz, 1H), 2.29 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 163.76, 157.15, 155.33, 153.15, 148.13, 146.61, 140.15, 138.32, 138.29, 132.05, 128.99, 122.83, 122.62, 119.42, 118.90, 115.59, 101.75, 66.74, 21.63. ESI-MS m/z: 361.60 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(3,4-dichlorophenyl)urea (1c). Compound 1c was prepared using the same procedure as described for the synthesis of 1a. Yield: 92.6%; m.p. 245.0–247.2 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.12 (s, 1H), 9.08 (s, 1H), 8.97 (s, 1H), 8.51 (s, 1H), 8.03 (s, 1H), 7.90 (d, J = 2.4 Hz, 1H), 7.54 (dd, J = 27.2, 8.8 Hz, 3H), 7.36 (dd, J = 8.8, 2.4 Hz, 1H), 7.26 (d, J = 8.8 Hz, 2H). 13C-NMR (100 MHz, DMSO-d6) δ 163.59, 157.10, 155.34, 152.79, 147.08, 140.35, 137.60, 132.23, 131.43, 130.95, 123.53, 122.69, 120.14, 119.71, 118.76, 101.75. ESI-MS m/z: 415.04 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(4-chlorophenyl)urea (1d). Compound 1d was prepared using the same procedure as described for the synthesis of 1a. Yield: 80.5%; m.p. 260.8–262.0 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 8.86 (d, J = 10.2 Hz, 2H), 8.51 (s, 1H), 8.03 (s, 1H), 7.56 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 8.8 Hz, 2H), 7.34 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H). 13C-NMR (100 MHz, DMSO-d6) δ 157.13, 155.38, 152.92, 146.92, 139.10, 137.88, 132.26, 129.04, 125.79, 122.69, 120.18, 119.94, 101.76. ESI-MS m/z: 391.50 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-phenylurea (1e). Compound 1e was prepared using the same procedure as described for the synthesis of 1a. Yield: 81.2%; m.p. 247.3–249.2 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 8.80 (s, 1H), 8.71 (s, 1H), 8.52 (s, 1H), 8.02 (s, 1H), 7.56 (d, J = 8.9 Hz, 2H), 7.48 (d, J = 7.7 Hz, 2H), 7.30 (t, J = 7.7 Hz, 2H), 7.25 (d, J = 8.9 Hz, 2H), 6.98 (t, J = 7.3 Hz, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 163.68, 157.13, 155.39, 153.01, 146.80, 140.07, 138.07, 132.27, 129.20, 122.68, 122.28, 119.79, 118.66, 101.76, 0.51. ESI-MS m/z: 347.40 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(2-chloro-5-methylphenyl)urea (1f). Compound 1f was prepared using the same procedure as described for the synthesis of 1a. Yield: 79.5%; m.p. 230.2–232.4 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 9.53 (s, 1H), 8.52 (s, 1H), 8.28 (s, 1H), 8.03 (s, 1H), 7.58 (d, J = 9.2 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 9.2 Hz, 1H), 2.90 (s, 1H), 2.74 (s, 1H), 2.30 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 163.64, 157.14, 155.39, 152.60, 147.00, 137.81, 137.48, 135.95, 132.27, 129.22, 124.46, 122.79, 122.20, 119.75, 119.44, 101.76, 21.33, 0.51. ESI-MS m/z: 395.30 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(3-chlorophenyl)urea (1g). Compound 1g was prepared using the same procedure as described for the synthesis of 1a. Yield: 75.4%; m.p. 234.8–236.0 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 8.94 (s, 1H), 8.89 (s, 1H), 8.51 (s, 1H), 8.03 (s, 1H), 7.73 (s, 1H), 7.57 (d, J = 8.8 Hz, 2H), 7.34–7.26 (m, 4H), 7.06–7.00 (m, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 163.64, 157.13, 155.37, 152.87, 147.01, 141.66, 137.75, 133.61, 132.26, 130.80, 122.70, 121.89, 120.04, 118.02, 117.10, 101.76, 99.93. ESI-MS m/z: 381.08 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(2,3-dimethylphenyl)urea (1h). Compound 1h was prepared using the same procedure as described for the synthesis of 1a. Yield: 81.4%; m.p. 250.4–252.3 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 9.06 (s, 1H), 8.51 (s, 1H), 8.00 (s, 2H), 7.57 (d, J = 6.8 Hz, 3H), 7.25 (d, J = 7.4 Hz, 2H), 7.06–7.02 (m, 1H), 6.91 (d, J = 6.6 Hz, 1H), 2.27 (s, 3H), 2.16 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 163.82, 157.13, 155.39, 153.40, 146.63, 138.40, 137.34, 137.00, 132.28, 128.05, 125.65, 125.41, 122.67, 120.92, 119.51, 101.75, 20.75, 14.05. ESI-MS m/z: 375.15 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(2-chloro-5-(trifluoromethyl)phenyl)urea (1i). Compound 1i was prepared using the same procedure as described for the synthesis of 1a. Yield: 82.3%; m.p. 239.2–240.3 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 9.70 (s, 1H), 8.66 (d, J = 3.2 Hz, 2H), 8.52 (s, 1H), 8.05 (s, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.8 Hz, 2H), 7.39 (d, J = 8.4 Hz, 1H), 7.29 (d, J = 8.8 Hz, 2H). 13C-NMR (100 MHz, DMSO-d6) δ 163.29, 157.14, 155.68, 152.91, 147.56, 137.32, 137.25, 132.22, 130.54, 128.80, 128.48, 125.49, 123.25, 120.47, 119.63, 117.67, 117.24, 102.01. ESI-MS m/z: 449.07 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(3-fluoro-5-(trifluoromethyl)phenyl)urea (1j). Compound 1j was prepared using the same procedure as described for the synthesis of 1a. Yield: 88.0%; m.p. 249.2–250.6 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 9.30 (s, 1H), 9.04 (s, 1H), 8.51 (s, 1H), 8.04 (s, 1H), 7.73 (s, 1H), 7.64 (d, J = 11.3 Hz, 1H), 7.58 (d, J = 8.6 Hz, 2H), 7.27 (d, J = 8.6 Hz, 2H), 7.22 (d, J = 8.2 Hz, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 163.87, 163.58, 161.39, 157.11, 155.33, 152.81, 147.27, 143.05, 137.41, 132.20, 122.69, 120.40, 110.95, 108.89, 105.75, 105.54, 101.76. ESI-MS m/z: 433.10 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(3-fluoro-5-methylphenyl)urea (1k). Compound 1k was prepared using the same procedure as described for the synthesis of 1a. Yield: 81.6%; m.p. 242.8–245.2 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H), 8.45 (s, 1H), 8.28 (s, 1H), 7.33 (dd, J = 6.8, 2.4 Hz, 1H), 7.25–18 (m, 3H), 7.02 (t, J = 9.2 Hz, 1H), 6.69 (d, J = 8.8 Hz, 2H). 13C-NMR (100 MHz, DMSO-d6) δ 163.71, 157.48, 155.37, 155.13, 153.21, 146.68, 138.21, 136.15, 132.24, 124.65, 124.48, 122.62, 121.44, 119.57, 117.63, 115.40, 115.17, 101.73, 14.77. ESI-MS m/z: 379.12 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(3-ethylphenyl)urea (1l). Compound 1l was prepared using the same procedure as described for the synthesis of 1a. Yield: 61.2%; m.p. 219.5–221.5 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 8.78 (s, 1H), 8.65 (s, 1H), 8.52 (s, 1H), 8.01 (s, 1H), 7.57 (d, J = 8.7 Hz, 2H), 7.34 (s, 1H), 7.24–7.20 (m, 4H), 6.83 (d, J = 7.3 Hz, 1H), 2.58 (d, J = 7.2 Hz, 2H), 1.24–1.17 (m, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 157.18, 155.39, 153.05, 146.75, 144.75, 140.08, 138.15, 129.09, 129.02, 122.66, 121.80, 120.74, 119.71, 117.99, 117.74, 116.09, 115.58, 101.76, 28.71, 15.96. ESI-MS m/z: 375.15 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(3-nitrophenyl)urea (1m). Compound 1m was prepared using the same procedure as described for the synthesis of 1a. Yield: 70.3%; m.p. 229.2–231.4 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 9.27 (s, 1H), 8.97 (s, 1H), 8.58 (s, 1H), 8.51 (s, 1H), 8.04 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.60 (d, J = 8.9 Hz, 2H), 7.57 (t, J = 8.3 Hz, 1H), 7.28 (d, J = 8.9 Hz, 2H). 13C-NMR (100 MHz, DMSO-d6) δ 155.35, 152.91, 148.52, 147.11, 141.43, 137.55, 130.46, 124.74, 122.73, 120.23, 116.71, 112.55, 101.74, 40.50, 40.29, 40.08, 39.87, 39.67, 39.46, 39.25. ESI-MS m/z: 392.10 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(2-fluoro-5-(trifluoromethyl)phenyl)urea (1n). Compound 1n was prepared using the same procedure as described for the synthesis of 1a. Yield: 78.6%; m.p. 178.4–179.0 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 9.70 (s, 1H), 8.66 (d, J = 3.2 Hz, 2H), 8.52 (s, 1H), 8.05 (s, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.4 Hz, 1H), 7.29 (d, J = 8.8 Hz, 2H). 13C-NMR (100 MHz, DMSO-d6) δ 155.34, 153.38, 152.64, 152.56, 152.44, 147.24, 137.30, 130.76, 129.47, 125.66, 122.82, 120.88, 119.96, 119.19, 116.72, 116.42, 116.21, 115.69, 101.76. ESI-MS m/z: 406.12 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(3-(methylthio)phenyl)urea (1o). Compound 1o was prepared using the same procedure as described for the synthesis of 1a. Yield: 99.6%; m.p. 227.3–228.4 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 8.80 (d, J = 23.2 Hz, 2H), 8.51 (s, 1H), 8.02 (s, 1H), 7.56 (d, J = 8.8 Hz, 2H), 7.49 (s, 1H), 7.26–7.21 (m, 3H), 7.17 (d, J = 8.0 Hz, 1H), 6.88 (d, J = 7.6 Hz, 1H), 2.51 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 163.64, 157.12, 155.36, 152.95, 146.89, 140.61, 139.00, 137.92, 132.24, 129.66, 122.65, 119.92, 119.70, 115.68, 115.26, 101.75, 15.04. ESI-MS m/z: 393.11 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(3-cyanophenyl)urea (1p). Compound 1p was prepared using the same procedure as described for the synthesis of 1a. Yield: 86.7%; m.p. 235.2–237.4 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.15 (s, 1H), 9.05 (d, J = 39.2 Hz, 2H), 8.51 (s, 1H), 8.03 (d, J = 20.4 Hz, 2H), 7.70 (s, 1H), 7.57–7.44 (m, 4H), 7.27 (s, 2H). 13C-NMR (100 MHz, DMSO-d6) δ 163.60, 157.08, 155.36, 152.89, 147.05, 141.01, 137.61, 132.25, 130.59, 125.74, 123.32, 122.74, 121.19, 120.11, 111.99, 101.74. ESI-MS m/z: 372.11 [M + H]+.
Methyl-3-(3-(4-((1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)ureido)benzoate (1q). Compound 1q was prepared using the same procedure as described for the synthesis of 1a. Yield: 66.0%; m.p. 217.3–220.1 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.15 (s, 1H), 9.30 (s, 1H), 9.01 (s, 1H), 8.59 (s, 1H), 8.51 (s, 1H), 8.06 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 8.4 Hz, 3H), 7.28 (d, J = 7.6 Hz, 2H), 3.39 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 166.61, 163.63, 157.09, 155.37, 152.98, 146.92, 140.53, 137.83, 132.26, 130.56, 129.61, 123.19, 122.91, 122.69, 120.01, 119.02, 101.74, 52.58. ESI-MS m/z: 405.12 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(5-methyl-2-nitrophenyl)urea (1r). Compound 1r was prepared using the same procedure as described for the synthesis of 1a. Yield: 54.2%; m.p. 359.7–360.2 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.12 (s, 1H), 9.53 (s, 1H), 8.50 (d, J = 6.8 Hz, 1H), 8.26 (d, J = 6.0 Hz, 1H), 8.03 (s, 2H), 7.57 (s, 2H), 7.33–7.26 (m, 3H), 6.85 (s, 1H), 2.28 (d, J = 6.0 Hz, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 163.61, 157.11, 155.36, 152.58, 146.98, 137.80, 137.45, 135.93, 132.24, 129.18, 124.42, 122.76, 122.18, 119.74, 119.42, 101.75, 21.32. ESI-MS m/z: 406.12 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(3-bromophenyl)urea (1s). Compound 1s was prepared using the same procedure as described for the synthesis of 1a. Yield: 85.5%; m.p. 234.5–237.0 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.16 (s, 1H), 8.95 (s, 1H), 8.91 (s, 1H), 8.51 (s, 1H), 8.06 (s, 1H), 7.88 (s, 1H), 7.56 (d, J = 7.2 Hz, 2H), 7.34 (d, J = 8.0 Hz, 1H), 7.26 (d, J = 6.8 Hz, 3H), 7.16 (d, J = 7.6 Hz, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 155.38, 152.84, 146.95, 141.79, 137.73, 131.13, 124.78, 122.72, 122.13, 120.82, 120.02, 117.47, 101.74. ESI-MS m/z: 425.03 [M + H]+.
1-(4-((1H-Pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)-3-(3,4-dimethylphenyl)urea (1t). Compound 1t was prepared using the same procedure as described for the synthesis of 1a. Yield: 87.7%; m.p. 249.9–250.6 °C; 1H-NMR (400 MHz, DMSO-d6) δ 14.12 (s, 1H), 8.75 (s, 1H), 8.52 (d, J = 5.6 Hz, 2H), 8.00 (s, 1H), 7.55 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.8 Hz, 3H), 7.19 (d, J = 8.0 Hz, 1H), 7.04 (d, J = 8.0 Hz, 1H), 2.20 (s, 3H), 2.16 (s, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 163.67, 157.11, 155.37, 153.02, 146.69, 138.19, 137.69, 136.73, 132.25, 130.04, 129.93, 122.62, 120.02, 119.67, 116.25, 101.74, 20.05, 19.07. ESI-MS m/z: 375.15 [M + H]+.
4-Chloro-1-methyl-1H-pyrazolo[3,4-d]pyrimidine (6). A suspension of compound 2 (1.88 g, 0.012 mol) and NaH (0.864 g, 0.036 mol) in dry DMF (10 mL) was stirred at 0 °C for 30 min. Iodomethane (2.56 g, 0.018 mol) was added, and the resulting mixture was stirred overnight. The reaction mixture was treated with water (100 mL) and extracted with EtOAc (60 mL × 3). The combined organic layers were removed in vacuo to give compound 6. Yield: 70.8%; 1H-NMR (400 MHz, DMSO-d6) δ 8.85 (s, 1H), 8.44 (s, 1H), 4.08 (s, 3H).
4-(1-Methyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)aniline (7). To the mixture of para-aminophenol (0.28 g) in DMF (2 mL), Cs2CO3 (1.2 eq.) was added under the protection of nitrogen. The reaction mixture was stirred at room temperature for 1.5–2 h, and then compound 6 was added slowly. After stirring overnight, the reaction mixture was diluted with EtOAc (50 mL) and was washed with 1 M NaOH (60 mL × 1), water (80 mL × 1), 5% LiCl (50 mL × 2), and brine. The organic layer was dried over MgSO4 and concentrated to give compound 7. Yield: 69.5%; 1H-NMR (400 MHz, DMSO-d6) δ 8.5 (s, 1H), 7.66 (s, 1H), 6.97 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 5.37 (s, 2H), 4.01 (s, 3H).
1-(3-Chlorophenyl)-3-(4-((1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (1u). Compound 1u was prepared from compound 7 and 3-chlorophenyl isocyanate using the same procedure as described for the synthesis of 1a. Yield: 83.4%; m.p. 309.8–312.0 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.94 (s, 1H), 8.89 (s, 1H), 8.55 (s, 1H), 8.04 (s, 1H), 7.73 (s, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.30–7.25 (m, 4H), 7.03 (s, 1H), 4.05 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 163.70, 155.39, 155.27, 152.86, 146.96, 141.65, 137.81, 133.61, 131.32, 130.80, 122.65, 121.90, 120.04, 118.02, 117.10, 102.19. ESI-MS m/z: 395.09 [M + H]+.
1-(4-Chloro-3-(trifluoromethyl)phenyl)-3-(4-((1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)oxy)phenyl)urea (1v). Compound 1v was prepared using the same procedure as described for the synthesis of 1u by replacing 3-chlorophenyl isocyanate with 4-chloro-3-(trifluoromethyl)phenyl isocyanate. Yield: 89.3%; m.p. 212.1–213.0 °C; 1H-NMR (600 MHz, DMSO-d6) δ 9.21 (s, 1H), 8.98 (s, 1H), 8.54 (s, 1H), 8.12 (s, 1H), 8.04 (s, 1H), 7.62-7.68 (m, 2H), 758 (d,J = 9.0 Hz, 2H), 7.26 (d,J = 9.0 Hz, 2H), 4.04 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 163.65, 155.35, 155.24, 152.88, 147.10, 139.73, 137.57, 132.37, 131.29, 127.26, 126.95, 123.47, 122.63, 120.29, 117.17, 102.17, 34.43. ESI-MS m/z: 461.81 [M − H]−.


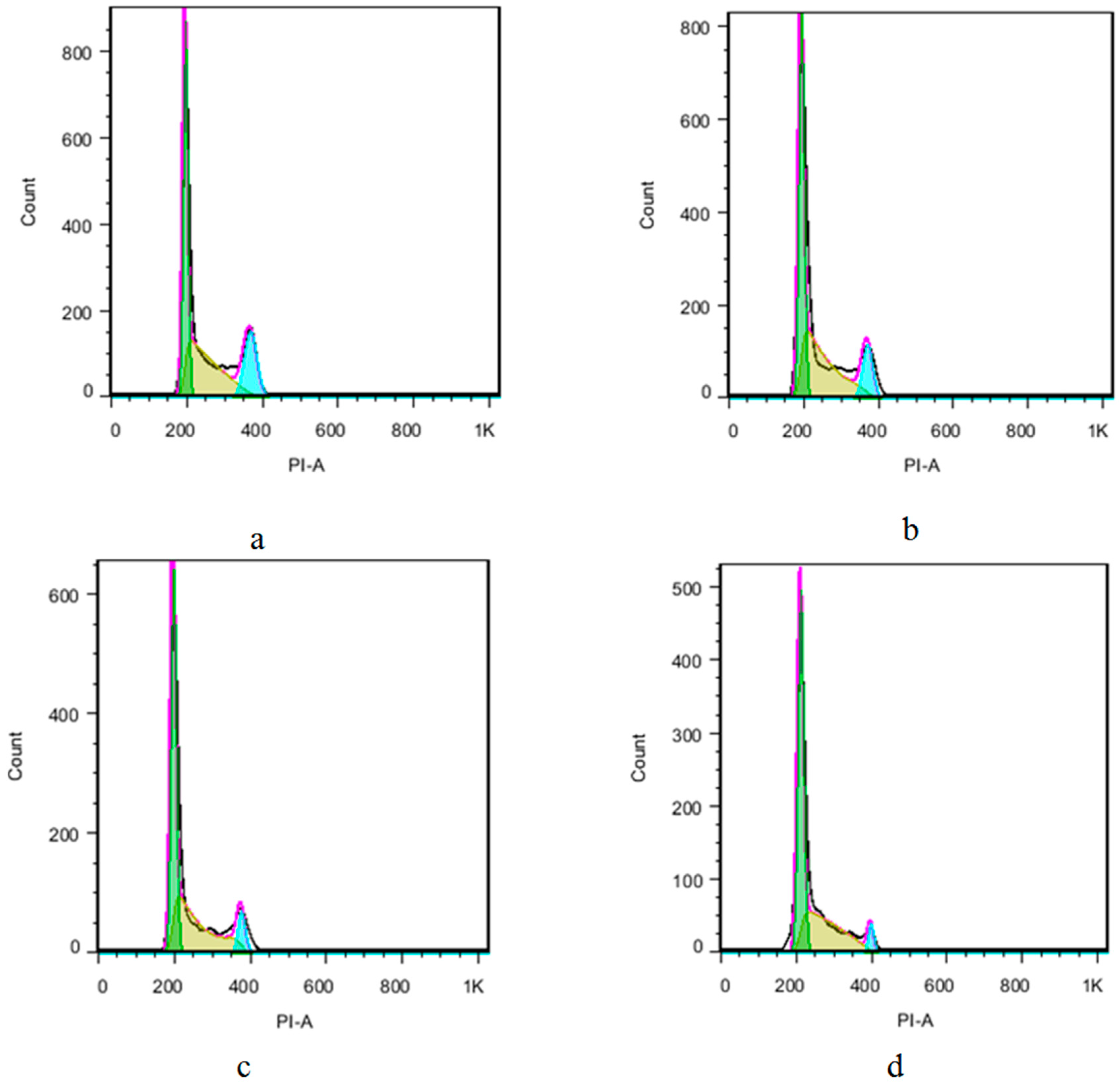

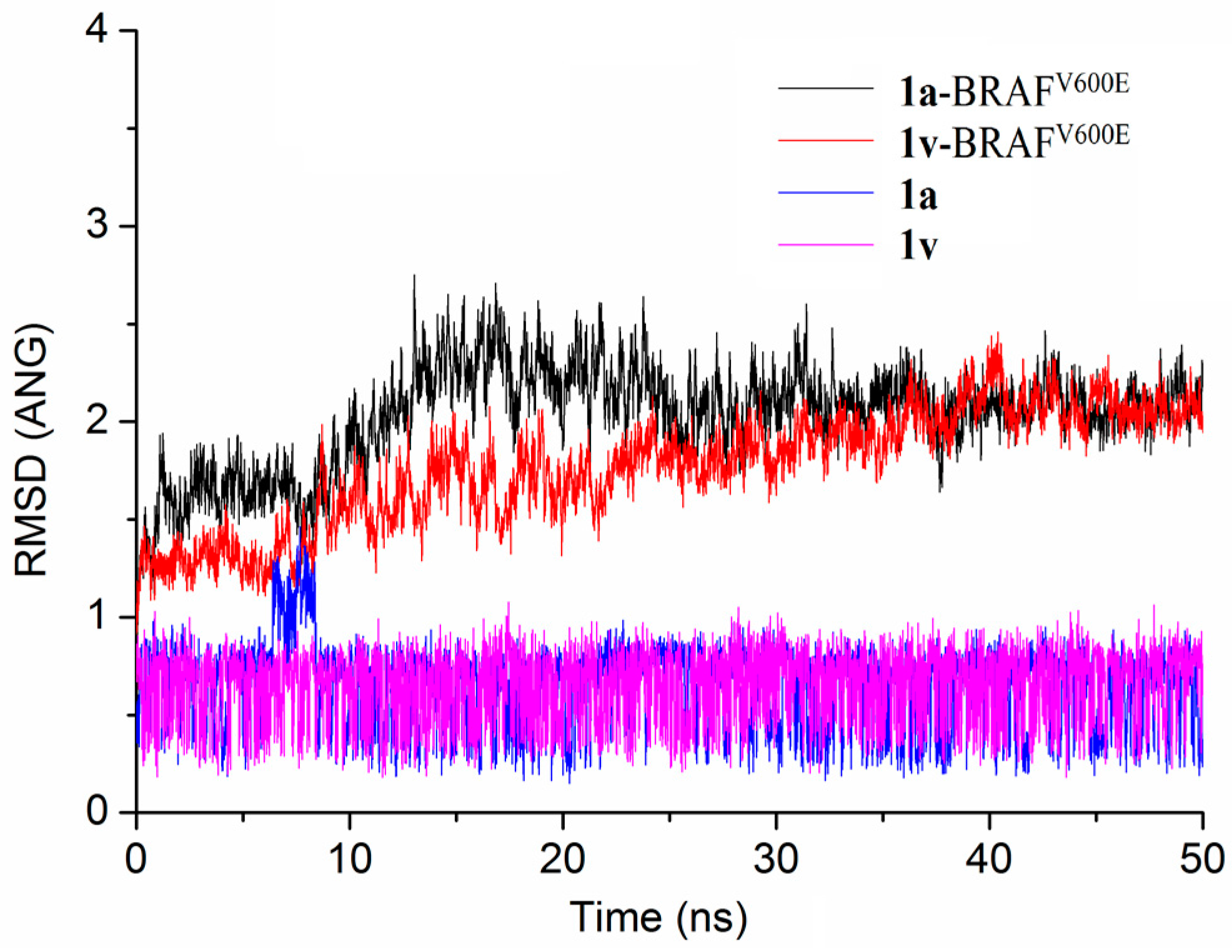
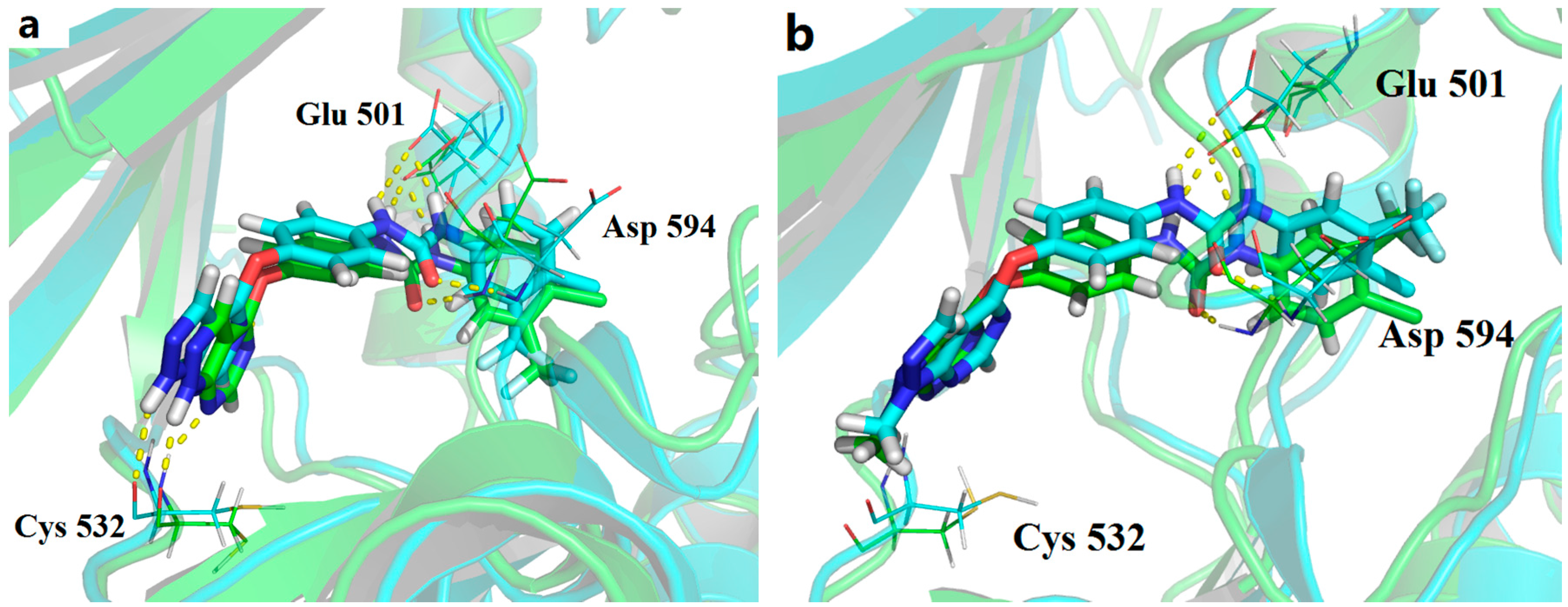

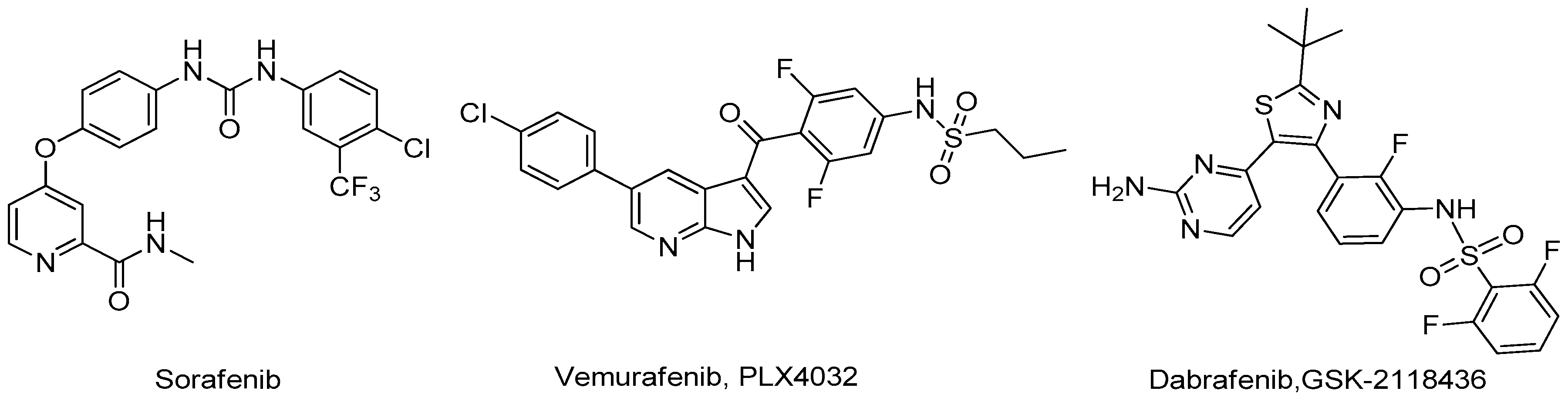{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
