
Synthesis of Pyrrolo[1,2-a]pyrimidine Enantiomers via Domino Ring-Closure followed by Retro Diels-Alder Protocol



1
Institute of Pharmaceutical Chemistry, University of Szeged, Eötvös utca 6, Szeged H-6720, Hungary
2
Department of Chemistry, University of Jyväskylä, FIN-40014 Turku, Finland
3
MTA-SZTE Stereochemistry Research Group, Hungarian Academy of Sciences, Eötvös utca 6, Szeged H-6720, Hungary
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(4), 613; https://doi.org/10.3390/molecules22040613
Submission received: 10 March 2017
/
Revised: 4 April 2017
/
Accepted: 6 April 2017
/
Published: 13 April 2017
(This article belongs to the Special Issue Asymmetric Synthesis 2017)
Abstract
:From 2-aminonorbornene hydroxamic acids, a simple and efficient method for the preparation of pyrrolo[1,2-a]pyrimidine enantiomers is reported. The synthesis is based on domino ring-closure followed by microwave-induced retro Diels-Alder (RDA) protocols, where the chirality of the desired products is transferred from norbornene derivatives. The stereochemistry of the synthesized compounds was proven by X-ray crystallography. The absolute configuration of the product is determined by the configuration of the starting amino hydroxamic acid.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The first hydroxamic acid (oxalohydroxamic acid [1]) was discovered by Lossen as early as 1869. However, hydroxamic acids attracted further attention at the beginning of the 1980s because of their bioactivity [2]. Their pharmacological properties are related to their ability to scavenge metal ions [3]. In addition, they are able to generate nitric oxide [3,4,5] in living systems. In this way, they can act as antimicrobial [6,7,8,9], antitumour [6,10], antihypertensive [11], anti-inflammatory [6,12], and neuroleptic agents, among others [3,13].
According to the literature data, aromatic hydroxamic acids are useful compounds [14,15,16,17,18,19,20,21,22,23]. However, only limited information is available about their reactions [24,25] and, in particular, about the properties of alicyclic derivatives [26,27].
Though the hydroxamic acid moiety is an important key pharmacophore in most cases, it is usually built up just in the last step of the synthesis [28,29,30]. Previously, we have examined the reactivity and stereoselectivity of the domino reaction of 2-aminonorbornene hydroxamic acids with 2-formylbenzoic acid and 2,3-dimethoxy-6-formylbenzoic acid to form new isoindolo[2,1-a]quinazolines and pyrimido[2,1-a]isoindoles [31].
Focusing on the biological potential of fused quinazolinones, and continuing our work on the synthesis of novel N-heterocycles, herein, we report the synthesis of a new series of pyrrolo[1,2-a]quinazolinones and pyrrolo[1,2-a]pyrimidines starting from hydroxamic acids. Pyrroloquinazolines and pyrrolopyrimidines are important heterocyclic ring systems that occur as a core structure in a variety of naturally occurring alkaloids and synthetic compounds. Pyrrolo[1,2-a]quinazolines are tricyclic compounds with great potential and combine the quinazoline substructure, a privileged structure in medicinal chemistry, with a pyrrole [32]. Their synthetic methods are rather scarce but the past five years have brought efficient new synthetic strategies which could lead to an increased interest in pyrrolo[1,2-a]quinazolines in the near future, mainly for their potential applications in medicinal chemistry [33,34,35,36,37,38].
Pyrrolopyrimidines display a broad applicability in medicinal chemistry exhibiting antimicrobial [39,40,41,42,43], antitumour [44,45,46,47,48,49,50,51,52,53,54,55,56,57,58], antiasthmatic [59], antihypertensive [60], and anti-inflammatory [61] activities. Several method have been developed for synthesizing pyrrolopyrimidines in the last few years [62,63,64,65,66,67,68,69,70].
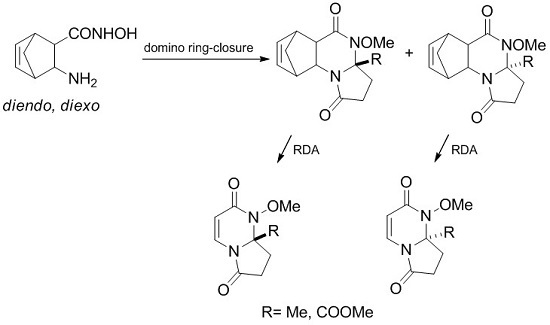
We recently reported an efficient and convenient procedure for the preparation of pyrrolo[1,2-a]pyrimidines starting from 2-aminonorbornene hydroxamic acids [31]. The synthesis was based on the domino ring-closure reaction of hydroxamic acids with oxocarboxylic acids, followed by RDA reaction of the tetracyclic intermediates.
The present target derivatives were planned to be prepared by a two-step strategy: first, the domino ring-closure reaction of diendo- and diexo-2-aminonorbornene hydroxamic acid with levulinic acid and α-ketoglutaric acid was carried out, followed by the second step involving the RDA reaction of the formed tetracycles by the loss of cyclopentadiene.
Domino reactions with hydroxamic acids are not well-documented in the literature. There are examples for the main simple ring-closures with cyclic anhydrides [19], aldehydes [71], sodium nitrite [20], carbon disulfide [72], formic acid, or acetyl chloride [17].
Our present aim was (i) to examine the domino ring-closure reaction of diendo- and diexo-aminonorbornene hydroxamic acids 1 and 2, (ii) to develop the retro Diels-Alder reaction of the tetracyclic heterocycles formed, and to extend this methodology to obtain novel racemic and enantiomeric pyrrolo[1,2-a]pyrimidine derivatives.
2. Results and Discussion
Racemic diendo- and diexo-2-aminonorbornene hydroxamic acids (±)-1 and (±)-2 were prepared from the appropriate ester bases with aqueous hydroxylamine solution according to an earlier procedure [31]. The enantiomers of 2-aminonorbornene hydroxamic acid (+)-1, (–)-1, (+)-2, and (–)-2 were prepared from racemic esters via diastereomeric salt formation with O,O’-di-p-toluoyl-tartaric acid (DPTTA) and O,O’-di-benzoyl-tartaric acid (DBTA) as previously described [31].
In the optimization experiments of the domino reactions of levulinic acid and α-ketoglutaric acid with racemic diendo-2-aminonorbornene hydroxamic acid (±)-1 and diexo-2-aminonorbornene hydroxamic acid (±)-2, conventional heating or microwave irradiation was applied. The reaction time was significantly shorter in the microwave reactor: the best results were achieved in ethanol stirred at 100 °C for 1 h.
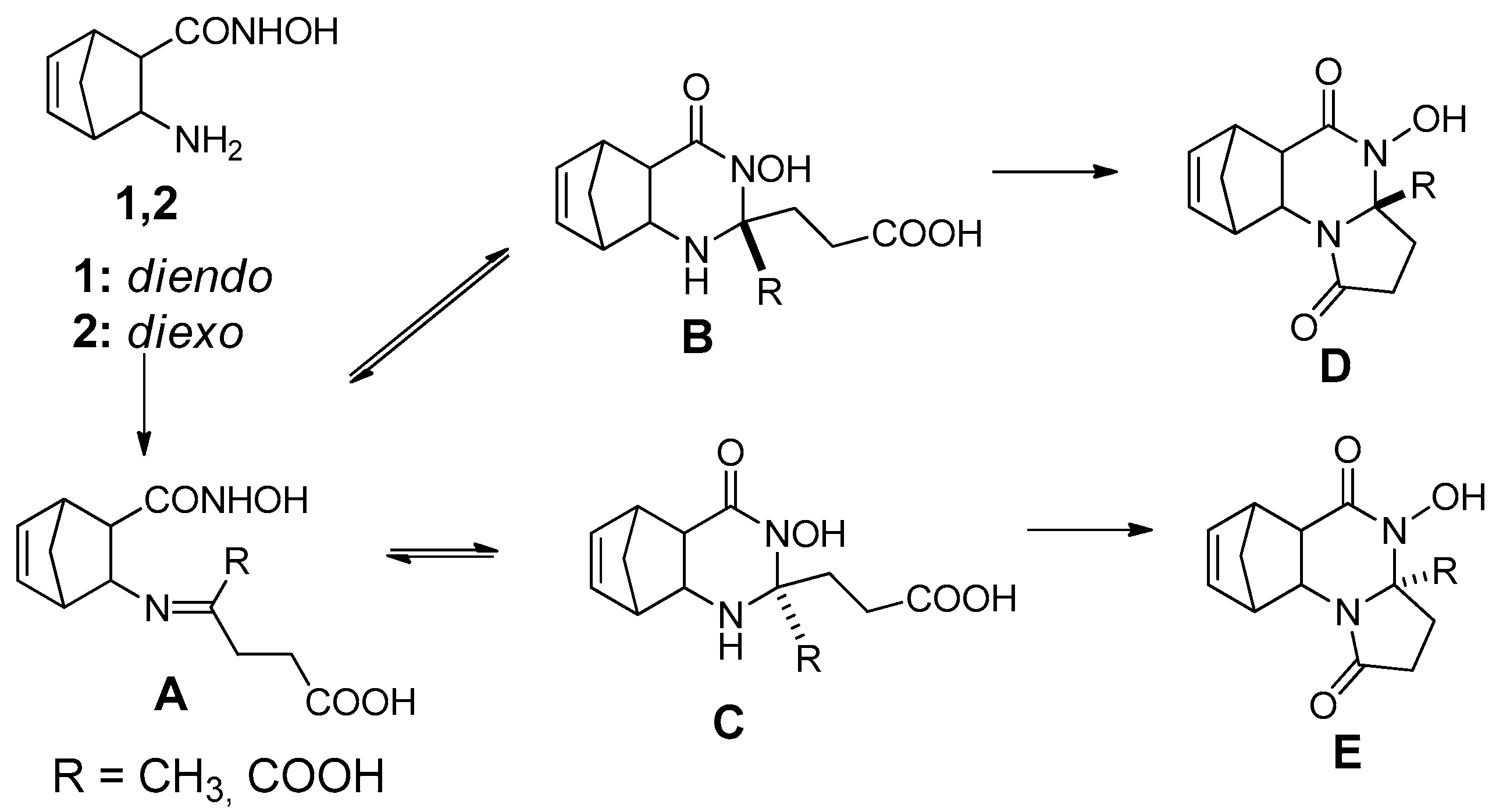
In the domino reaction of 1 and 2 with levulinic acid and α-ketoglutaric acid, the first Schiff base A is produced, which undergoes a ring-closure reaction to produce the quinazoline epimers B and C, which are formed from A through a ring-chain tautomerism. The second ring-closure involves epimers B and C that yields compounds D and E (Scheme 1) [68].
The NMR spectra revealed the formation of two diastereomers of the methanopyrrolo[1,2-a]quinazolines D and E (Scheme 1). Unfortunately, we were not able to separate the diastereomers by column chromatography despite the use of a range of eluent combinations (for example: EtOAc, EtOAc/hexane = 1:1, EtOAc/MeOH = 9:1, CHCl3/MeOH = 9:1, or toluene/MeOH = 4:1). Fortunately, after derivatization with diazomethane, the diastereomers (±)-3a, (±)-3b and (±)-4a, (±)-4b could be easily separated by column chromatography eluted with EtOAc (Scheme 2).
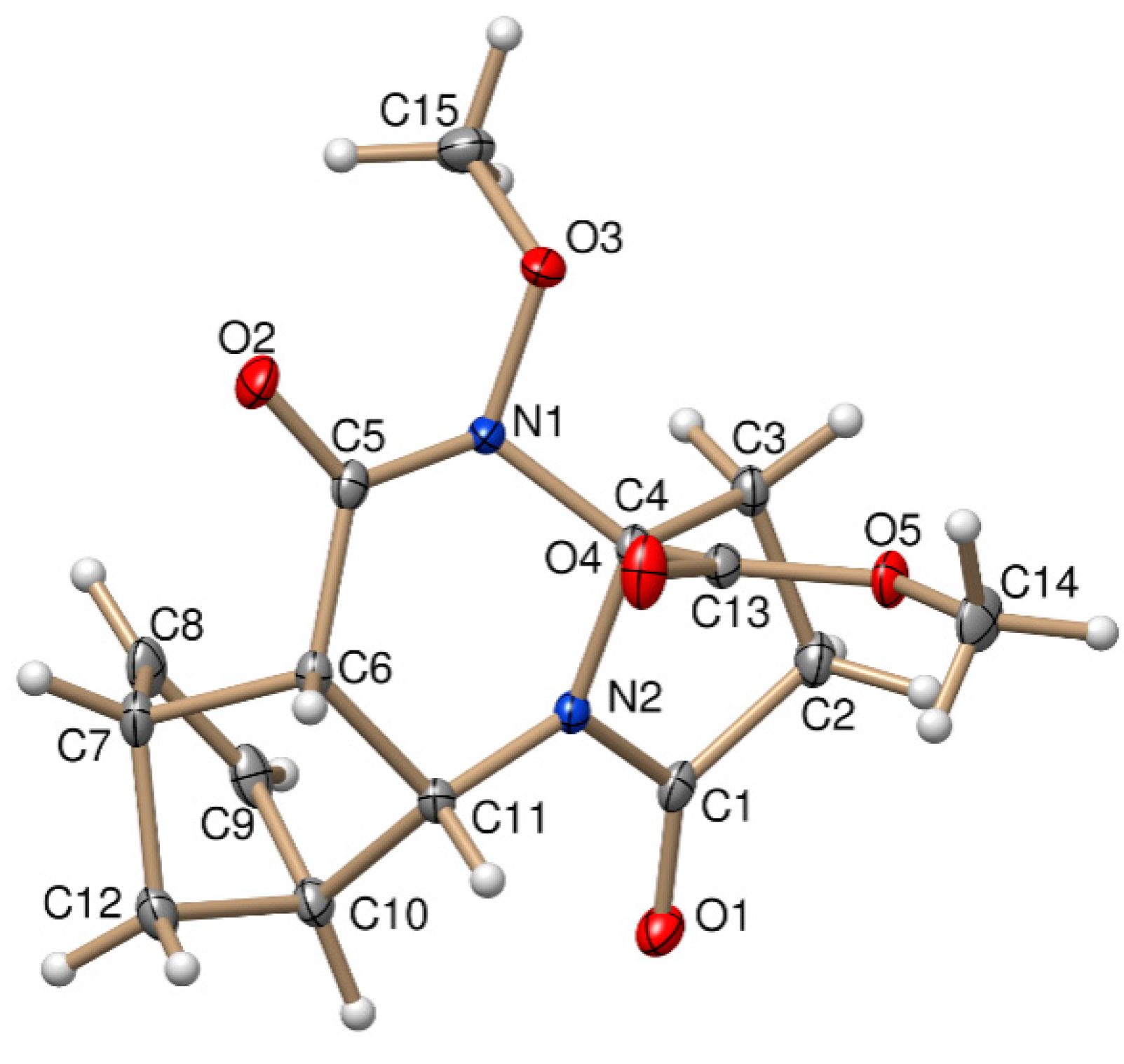
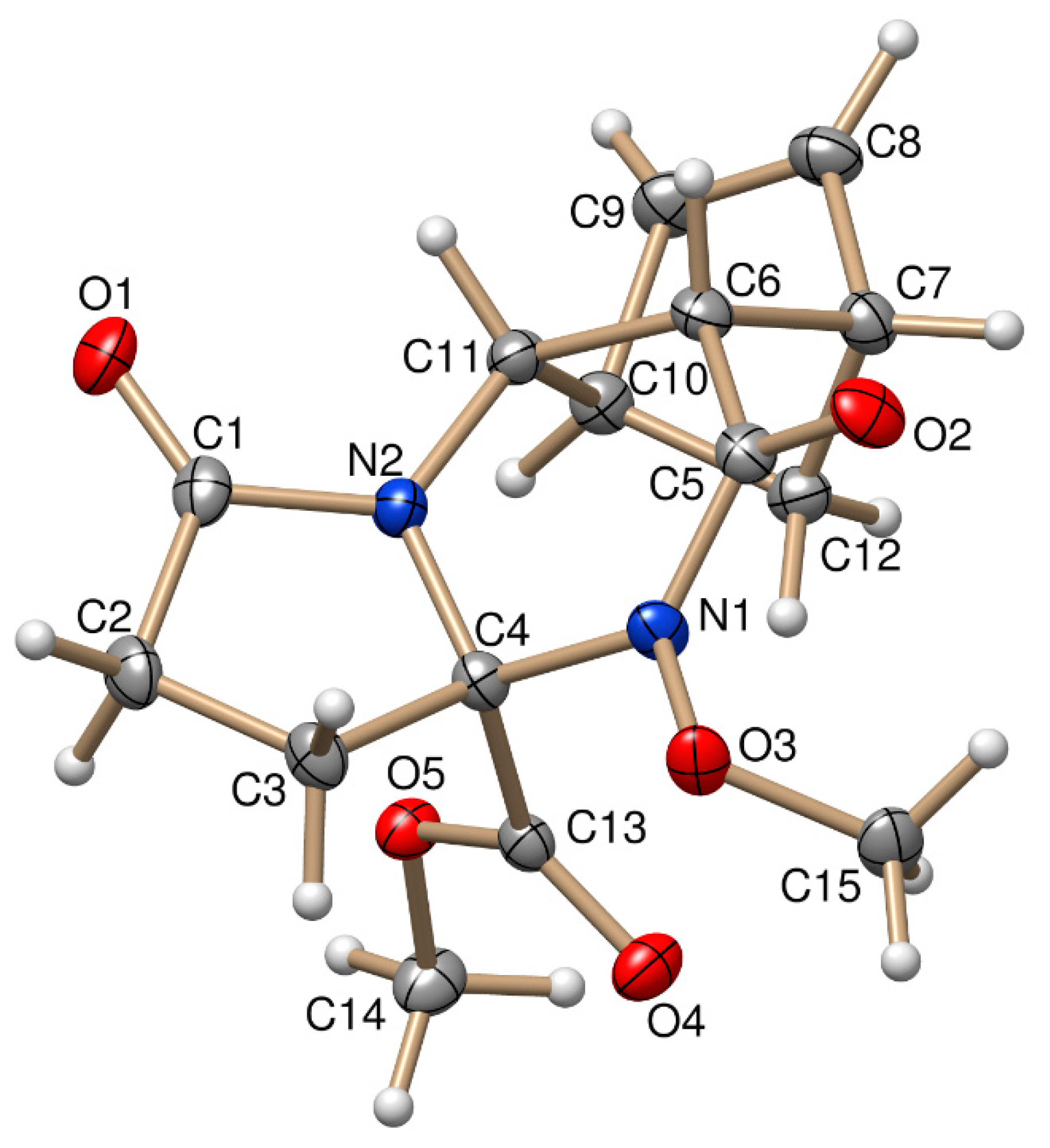
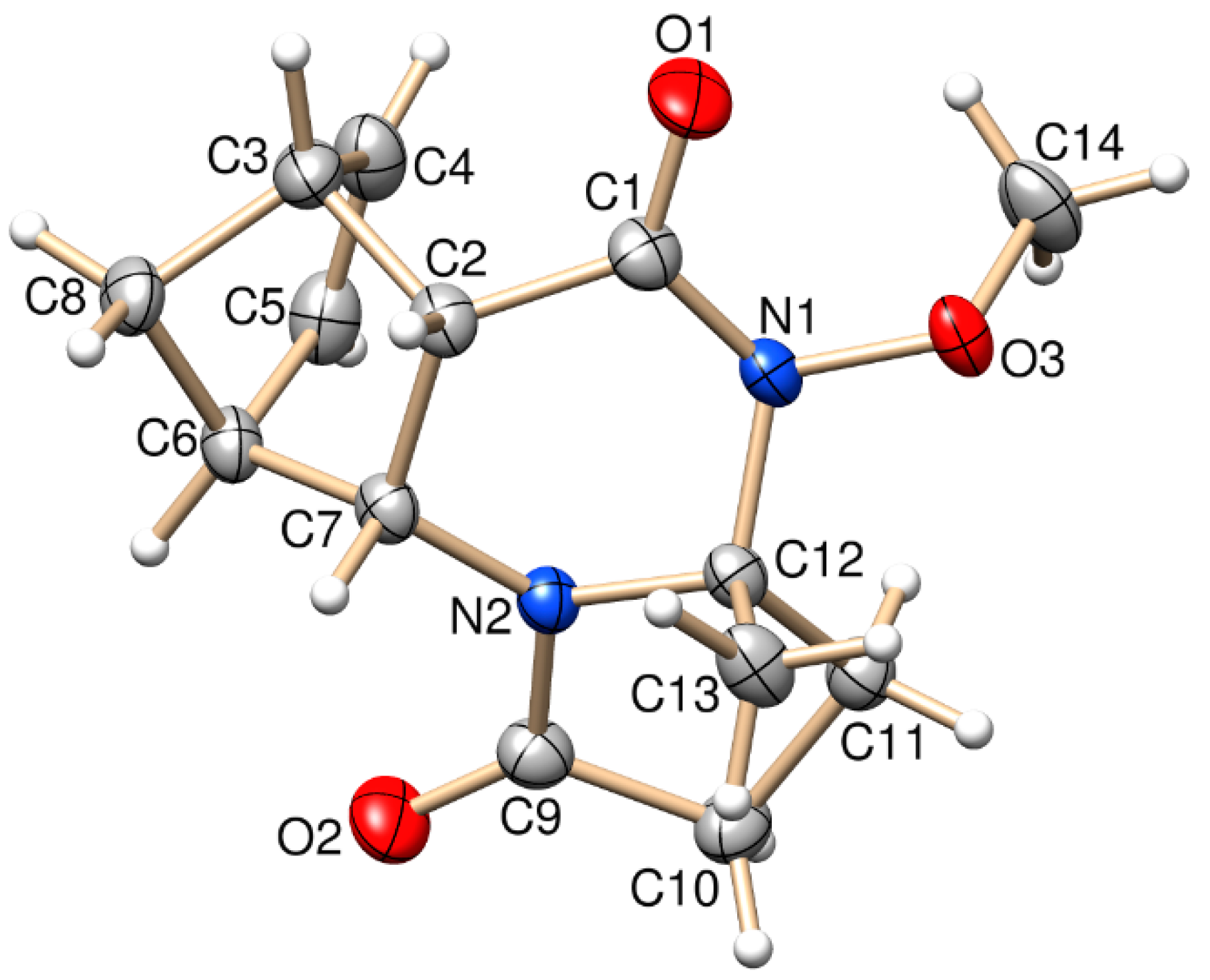
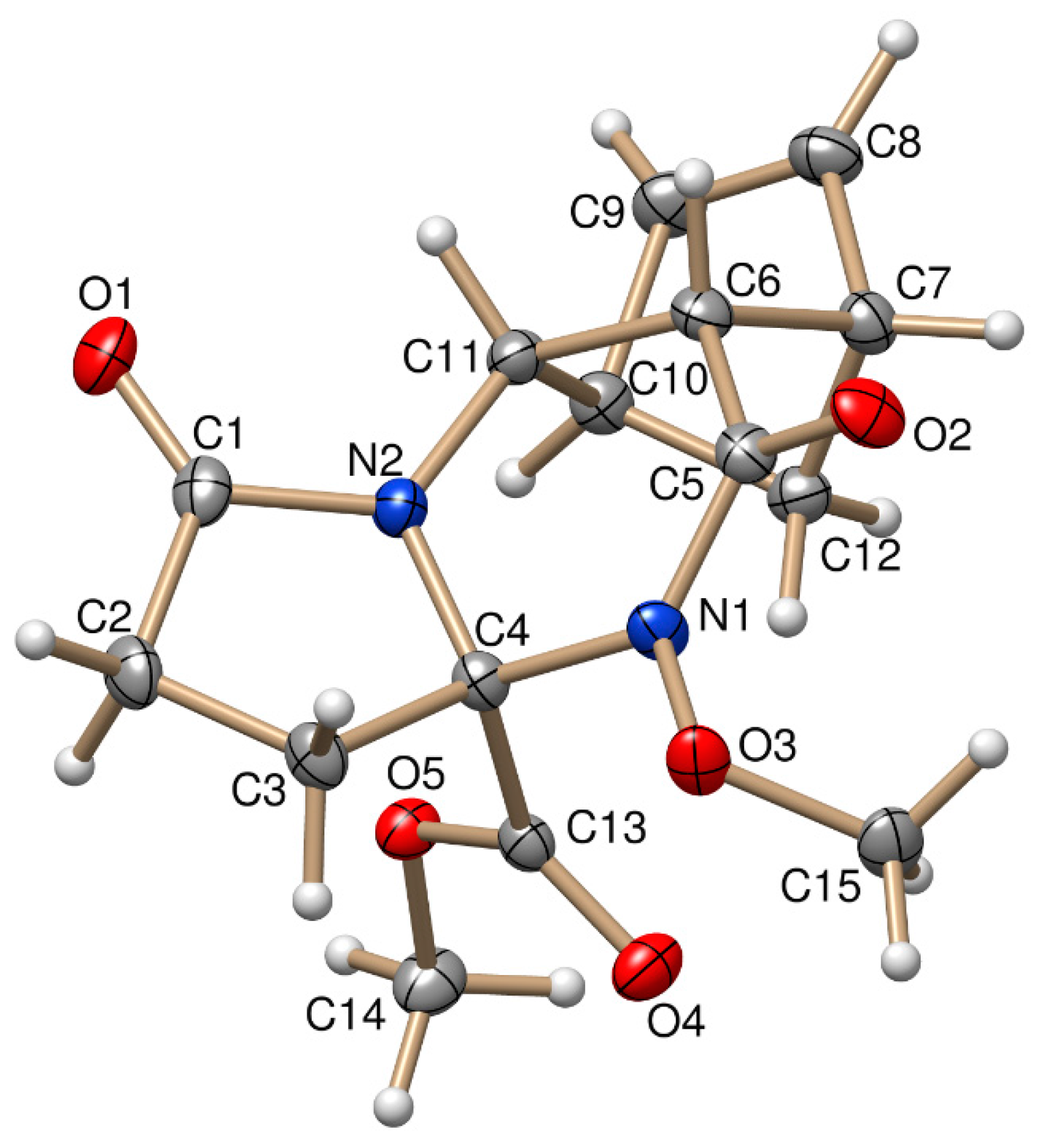
The stereochemistry of (±)-3a and (±)-4b was confirmed by X-ray diffraction analysis. The relative configuration of the COOCH3 group at C-3a and the bridgehead hydrogens of C-5a and C-9a have the same steric orientation in the (±)-3a and (±)-4b tetracyclic derivatives (Figure 1 and Figure 2).
In order to produce racemic pyrrolo[1,2-a]pyrimidine 5, the retro Diels-Alder reaction of the methanopyrrolo[1,2-a]quinazoline derivatives was examined in the microwave reactor under varied conditions. The best results for the cycloreversion were achieved in 1,2-dichlorobenzene (DCB) at 250 °C (20 min).
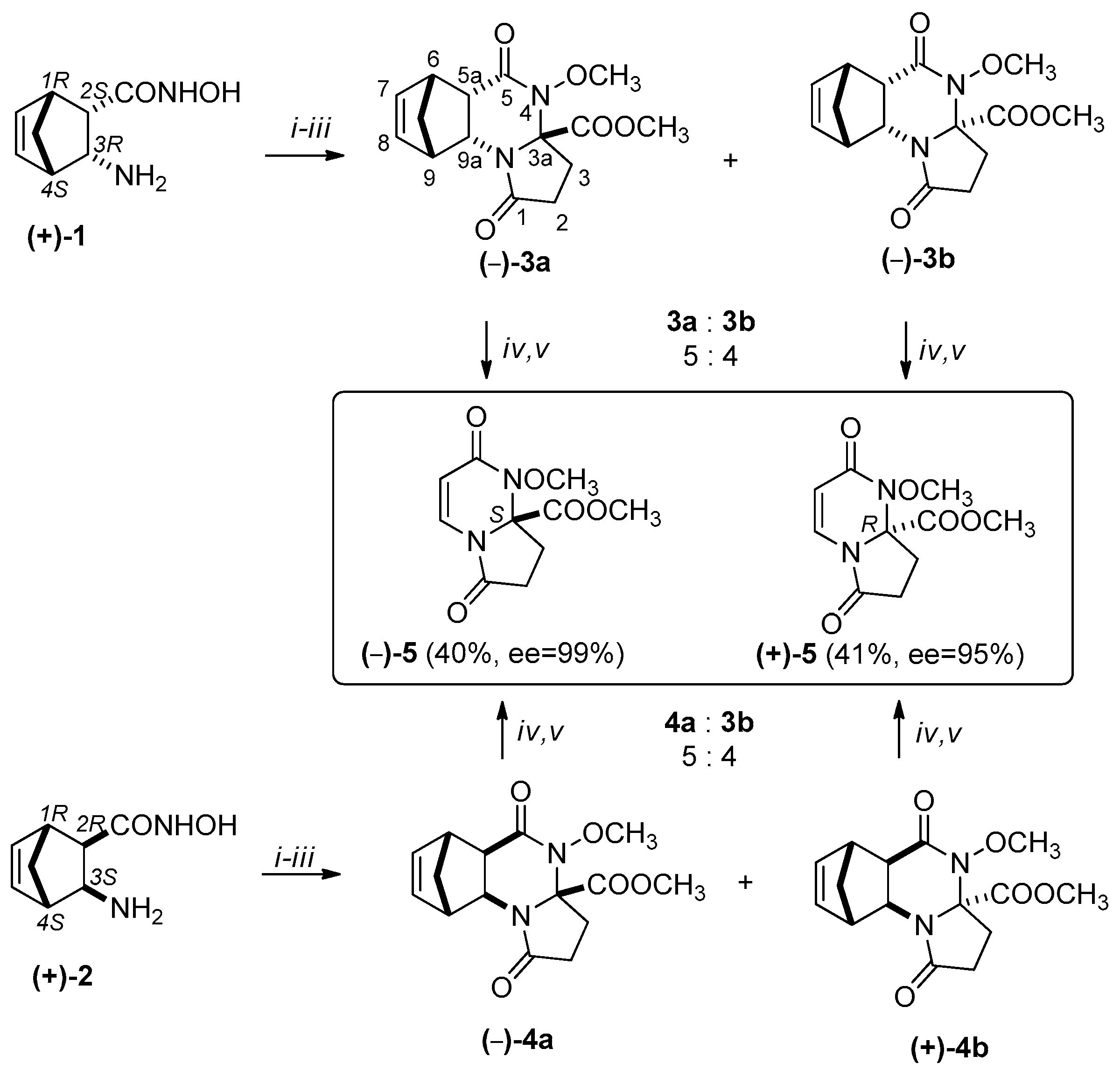
The synthetic method described above was extended for the preparation of the enantiomerically pure substances, via the protocol described for the racemic substances. The domino ring-closure reaction of diendo-2-aminonorbornene hydroxamic acid enantiomer (+)-1 resulted in (–)-3a and (–)-3b, while that with the diexo-2-aminonorbornene hydroxamic acid enantiomer (+)-2 resulted in epimers (–)-4a and (+)-4b. After their separation, the RDA reactions of tetracycles could easily be achieved, resulting in pyrrolo[1,2-a]pyrimidine (+)-5 and (–)-5 (Scheme 2).
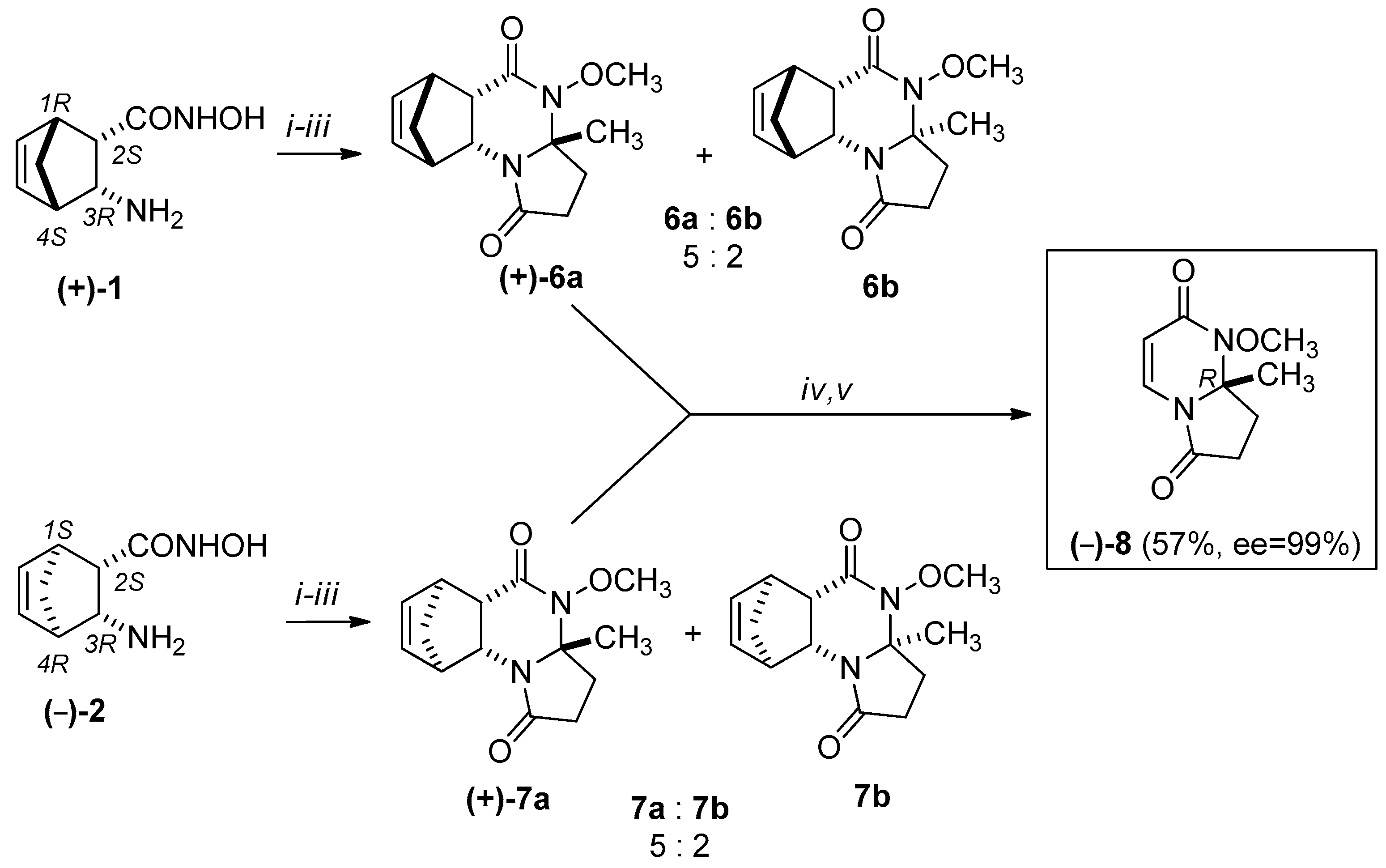
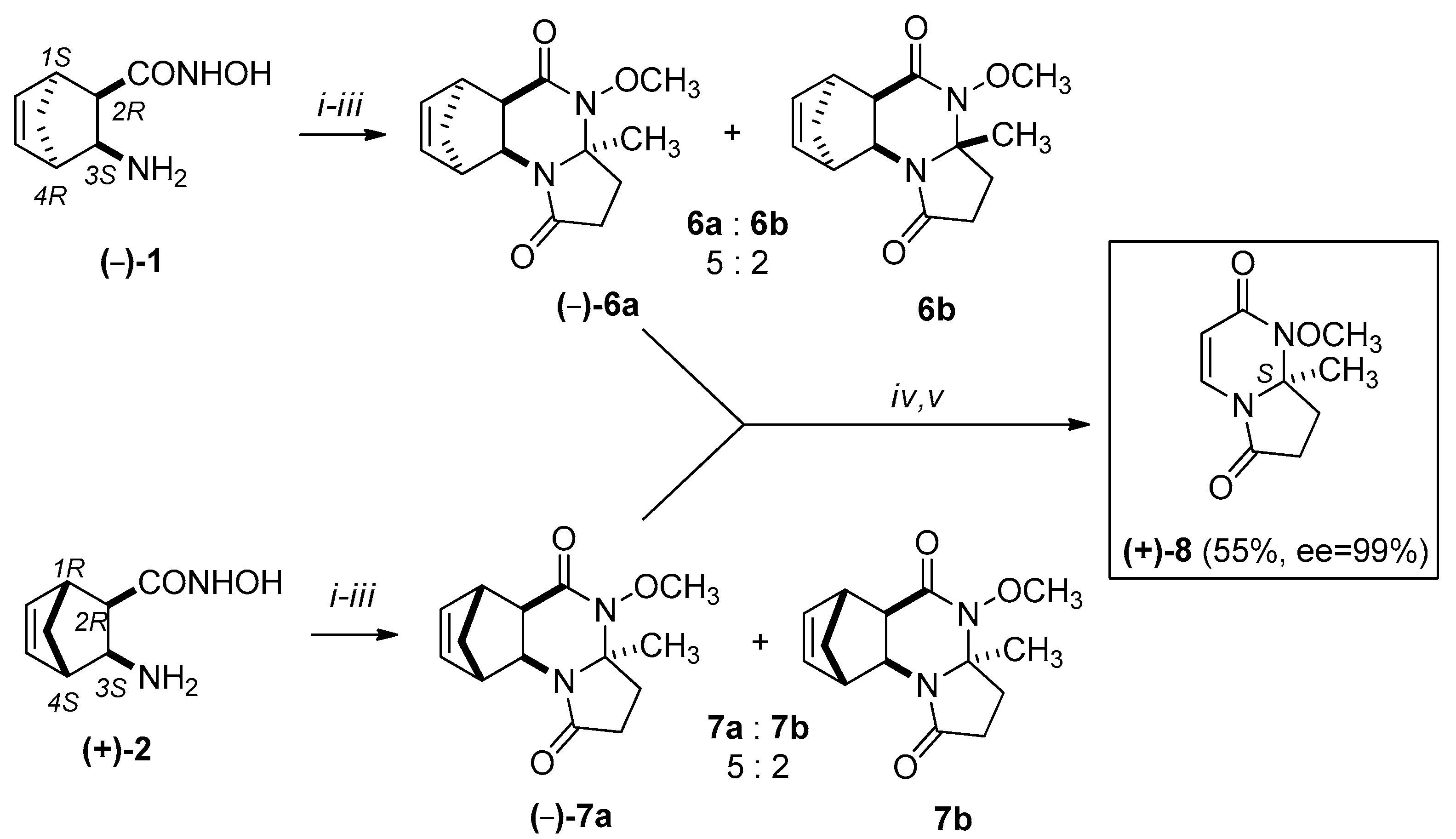
We attempted to use the same methodology to synthesize racemic 8. In this case, however, the ring-closure reaction with levulinic acid resulted in only a small quantity of the minor diastereomers (±)-6b and (±)-7b, which could not be isolated. The stereochemistry of (±)-6a was confirmed by X-ray diffraction analysis. The relative configuration of the CH3 group at the C-3a asymmetric centre and the bridgehead hydrogens of C-5a and C-9a have the same steric orientation in (±)-6a (Figure 3).
We selected an economically better approach for the synthesis of (+)-8 and (–)-8. Namely, we carried out the cycloreversions of the major methanopyrrolo[1,2-a]quinazoline derivatives (+)-6a, (–)-6a, (+)-7a, and (–)-7a and isolated the pyrrolo[1,2-a]pyrimidines (+)-8 and (–)-8 in 55–57% yields (see Scheme 3 and Scheme 4).
The absolute configuration of the newly built asymmetric centre of (+)-7a and (–)-7a was determined by chemical correlation: the absolute configurations of the starting 2-aminonorbornene hydroxamic acids, and the relative configuration of (±)-6a from the X-ray diffraction analysis was known. When (+)-7a was heated under the RDA reaction conditions, (–)-8 was gained. In addition, the RDA product of (+)-6a also afforded (–)-8. In contrast, when (–)-7a and (–)-6a were treated under the RDA reaction conditions, (+)-8 was formed. The NMR and HPLC data (retention times: (–)-8: 41.91 min, opposite enantiomer (+)-8: 34.19 min) and comparison of the optical rotations revealed that the final compound was identical to that of (R)-1-methoxy-8a-methyl-1,7,8,8a-tetrahydropyrrolo[1,2-a]pyrimidine-2,6-dione (–)-8. Since this asymmetric centre was not affected during the RDA reaction, these results allow for the assumption that the absolute configuration of position 3a of (+)-7a is R, and that of (–)-7a is S.
3. Materials and Methods
3.1. General Methods
1H-NMR spectra were recorded at 400.13 MHz or 600.20 MHz and the 13C-NMR spectra were recorded at 100.62 MHz or 150.92 MHz in CDCl3 at ambient temperature, with a Bruker AM 400 or Bruker AV 600 spectrometer (Bruker Biospin, Karlsruhe, Germany). Chemical shifts are given in δ (ppm) relative to TMS as the internal standard. Microwave-promoted reactions were performed in sealed reaction vials (10 mL) in a microwave (CEM, Discover SP) cavity (CEM Corporation, Matthwes, NC, USA). Optical rotations were measured with a Perkin-Elmer 341 polarimeter (Perkin Elmer, Shelton, CT, USA). Mass spectra were recorded with a Micromass Q-TOF Premier mass spectrometer (Waters Corporation, Milford, MA, USA). Melting points were measured with a Hinotek-X4 micro melting point apparatus (Hinotek, Ningbo, China) and are uncorrected. Racemic 2-aminonorbornene hydroxamic acids (±)-1 and (±)-2 and enantiomeric 2-aminonorbornene hydroxamic acids (+)-1, (–)-1, (+)-2, and (–)-2 were prepared by a literature method [31]. The ee values of (+)-1, (–)-1, (+)-2, and (–)-2 were determined by HPLC by a literature method [73]. CCDC-1508562-1508564 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
The ee values of (–)-3a, (–)-3b, (–)-4a, and (+)-4b were determined by HPLC using a Chiralcel-OD-H column (Daicel corporation, Tokyo, Japan). The analytical conditions were as follows: eluent: a mixture of hexane and EtOH (75:25) with 0.1% diethylamine, flow rate: 0.4 mL∙min−1, detection at 220 nm retention times: (−)-3a: 18.82 min (antipode: 22.16 min), (–)-3b: 19.84 min (antipode: 24.23 min), (+)-4a: 20.74 min (antipode: 17.48 min), (–)-4b: 24.22 min (antipode: 20.97 min). (+)-6a, (–)-6a, (+)-7a, and (–)-7a were determined using a Phenomenex-IA column (Phenomenex, Torrance, CA, USA) eluted by a mixture of hexane and IPA (70:30), flow rate: 0.4 mL∙min−1, detection at 220 nm, retention times: (+)-6a: 32.94 min, (–)-6a: 39.46 min, (+)-7a: 31.39 min, (–)-7a: 35.39 min). The ee values of the final products of (+)-5, (–)-5, (+)-8, and (–)-8 were determined by HPLC using a Phenomenex-IA column. The analytical conditions were as follows: eluent: a mixture of hexane and IPA (70:30), flow rate: 0.4 mL∙min−1, detection at 220 nm, retention times: (+)-5: 47.97 min, (–)-5: 57.79 min, (+)-8: 34.19 min, (–)-8: 41.91 min.
1H-NMR and 13C-NMR spectra of compounds (–)-3a, (–)-3b, (–)-4a, (+)-4b, (+)-5 (+)-6a, (+)-7a and (+)-8, HPLC chromatogram of compounds (±)-5, (–)-5, (+)-5 (±)-8 (–)-8, (+)-8, and table for X-ray crystallography data for (±)-3a, (±)-4a and (±)-6a are available in the Supplementary materials.
3.2. Synthesis of New Compounds
3.2.1. Synthesis of Methanopyrrolo[1,2-a]quinazoline Derivatives (–)-3a, (–)-3b, (–)-4a, and (+)-4b
A mixture of 2-aminonorbornene hydroxamic acid (+)-1 or (+)-2 (336 mg, 2 mmol), and α-ketoglutaric acid (292 mg, 2 mmol) was dissolved in 6 mL EtOH in a 10 mL pressurized reaction vial and the solution was stirred at 100 °C for 1 h at max 300 W microwave irradiation. Then the reaction mixture was evaporated to dryness and the crude product was crystallized from Et2O. The crystals isolated were dissolved in MeOH and a diazomethane/Et2O mixture was added dropwise to the solution at ambient temperature and was stirred for 30 min. The reaction was followed by TLC. (Diazomethane is a very harmful and hazardous reagent and must be handled with caution! This reaction should be performed in a well-ventilated hood!) The solvent was then removed by evaporation and the residue was dissolved in 2 mL EtOAc and transferred to a silica gel column (Merck, Darmstadt, Germany) and eluted with EtOAc. The product was crystallized from iPr2O to produce white crystals.
Methyl (3aS,5aS,6R,9S,9aR)-4-methoxy-1,5-dioxo-1,2,3,3a,4,5,5a,6,9a-decahydro-6,9-methanopyrrolo[1,2-a]quinazoline-3a-carboxylate [(–)-3a)]: White crystals (39% yield), m.p. 125–128 °C, = –92.5 (c = 0.33, EtOH), ee 87%, 1H-NMR (400 MHz, CDCl3, 30 °C): 1.36–1.38 (m, 1H, 11-H), 1.58–1.60 (m, 1H, 11-H), 2.13–2.21 (m, 1H, CH2), 2.45–2.51 (m, 1H, CH2), 2.58–2.74 (m, 2H, CH2), 3.09–3.12 (m, 1H, 5a-H), 3.43 (m, 1H, 9-H), 3.82 (s, 3H, COOCH3), 3.85 (s, 3H, OCH3), 3.93–3.95 (m, 1H, 9a-H), 4.06 (m, 1H, 6-H), 6.07–6.09 (m, 1H, 8-H), 6.20–6.22 (m, 1H, 7-H), 13C-NMR (100 MHz, CDCl3, 30 °C): δ = 28.1, 30.1, 41.1, 46.6, 46.9, 46.9, 47.3, 54.0, 54.6, 65.3, 82.3, 135.7, 136.1, 170.0, 172.3, 173.9, HRMS calcd. for [M + H+] m/z = 307.1294, measured: m/z = 307.1288.
Methyl (3aR,5aS,6R,9S,9aR)-4-methoxy-1,5-dioxo-1,2,3,3a,4,5a,6,9a-decahydro-6,9-methanopyrrolo[1,2-a]quinazoline-3a-carboxylate [(–)-3b)]: White crystals (44% yield), m.p. 170–172 °C, = −3.8 (c = 0.30, EtOH), ee 97%, 1H-NMR (600 MHz, CDCl3, 30 °C): 1.44–1.45 (m, 1H, 11-H), 1.56–1.63 (m, 1H, 11-H), 2.24–2.30 (m, 1H, CH2), 2.46–2.50 (m, 2H, CH2), 2.79–2.85 (m, 1H, CH2), 3.25–3.28 (m, 2H, 5a-H, 9-H), 3.42 (m, 1H, 6-H), 3.78 (s, 3H, COOCH3), 3.89 (s, 3H, OCH3), 4.68–4.69 (m, 1H, 9a-H), 5.73–5.75 (m, 1H, 8-H), 6.19–6.20 (m, 1H, 7-H), 13C-NMR (100 MHz, CDCl3, 30 °C): δ = 29.2, 32.1, 45.1, 46.3, 47.2, 48.9, 52.9, 53.1, 64.3, 81.6, 134.2, 138.6, 168.4, 170.3, 174.9, HRMS calcd. for [M + H+] m/z = 307.1294, found m/z = 307.1288.
Methyl (3aS,5aR,6R,9S,9aS)-4-methoxy-1,5-dioxo-1,2,3,3a,4,5a,6,9a-decahydro-6,9-methanopyrrolo[1,2-a]quinazoline-3a-carboxylate [(–)-4a)]: White crystals, (44% yield), m.p. 125–128 °C, = −64 (c = 0.30, EtOH), ee 97% 1H-NMR (600 MHz, CDCl3, 30 °C): 1.39–1.40 (m, 1H, 11-H), 1.49–1.50 (m, 1H, 11-H), 2.40–2.56 (m, 3H, CH2), 2.65–2.81 (m, 3H, CH2, 5a-H, 9-H), 3.34 (m, 1H, 6-H), 3.84 (s, 3H, COOCH3), 4.02 (s, 3H, OCH3), 4.15 (m, 1H, 9a-H), 6.23–6.25 (m, 1H, 7-H), 6.31–6.33 (m, 1H, 8-H), 13C-NMR (150 MHz, CDCl3, 30 °C): δ = 29.1, 30.9, 44.4, 44.8, 46.9, 47.5, 51.9, 53.4, 64.0, 81.6, 137.6, 138.4, 167.7, 170.9, 173.9, HRMS calcd. for [M + H+] m/z = 307.1294, found m/z = 307.1288.
Methyl (3aR,5aR,6R,9S,9aS)-4-methoxy-1,5-dioxo-1,2,3,3a,4,5a,6,9a-decahydro-6,9-methanopyrrolo[1,2-a]quinazoline-3a-carboxylate [(+)-4b]: White crystals, (39% yield), m.p. 170–172 °C, = + 6.5 (c = 0.33, EtOH), ee 81%, 1H-NMR (600 MHz, CDCl3, 30 °C): 1.51–1.52 (m, 2H, 11-H), 2.25–2.30 (m, 1H, CH2), 2.58–2.82 (m, 4H, CH2, 5a-H), 3.35 (m, 1H, 9-H), 3.39–3.40 (m, 1H, 6-H), 3.81 (s, 3H, COOCH3), 3.94 (s, 3H, OCH3), 3.99 (m, 1H, 9a-H), 6.17–6.19 (m, 1H, 7-H), 6.33–6.34 (m, 1H, 8-H), 13C-NMR (150 MHz, CDCl3, 30 °C): δ = 27.9, 30.1, 43.7, 44.6, 46.6, 47.7, 53.7, 54.3, 65.0, 82.0, 135.7, 138.6, 169.4, 173.2, 174.1, HRMS calcd. for [M + H+] m/z = 307.1294, found m/z = 307.1288.
3.2.2. Synthesis of Pyrrolo[1,2-a]pyrimidines (+)-5 and (–)-5
Tetracyclic compound (–)-3a, (–)-4a, or (–)-3b and (+)-4b (50 mg, 0.16 mmol,) was dissolved in 2 mL DCB in a 10 mL sealed reaction vial. The solution was stirred at 250 °C for 20 min at max 300 W microwave irradiation. Then the solvent was evaporated, the residue was dissolved in 2 mL EtOAc/MeOH = 9:1, and was purified by column chromatography on silica gel eluted by EtOAc/MeOH = 9:1. The product was crystallized from iPr2O to produce white crystals.
Methyl (R)-1-methoxy-2,6-dioxo-1,2,6,7,8,8a-hexahydropyrrolo[1,2-a]pyrimidine-8a-carboxylate [(+)-5]: White crystals, (41% yield), m.p. 156–158 °C, = +145 (c = 0.16, EtOH), ee 95%, 1H-NMR (600 MHz, CDCl3, 30 °C): 2.46–2.52 (m, 1H, CH2), 2.70–2.74 (m, 1H, CH2), 2.83–2.98 (m, 2H, CH2), 3.82 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 5.41 (d, J = 7.39 Hz, 1H, 3-H), 7.33 (d, J = 7.61 Hz, 1H, 4-H) 13C-NMR (150 MHz, CDCl3, 30 °C): δ = 28.8, 30.0, 53.7, 66.0, 81.1, 104.7, 131.1, 165.5, 168.8, 170.6, HRMS calcd. for [M + H+] m/z = 241.0825, found m/z = 241.0819.
Methyl (S)-1-methoxy-2,6-dioxo-1,2,6,7,8,8a-hexahydropyrrolo[1,2-a]pyrimidine-8a-carboxylate [(–)-5]: White crystals, (40% yield), m.p. 155–156 °C, = −139 (c = 0.16, EtOH), ee 99% 1H- and 13C-NMR is similar to (+)-5.
3.2.3. Synthesis of Methanopyrrolo[1,2-a]quinazoline Derivatives (+)-6a, (–)-6a, (+)-7a, and (–)-7a
A mixture of 2-aminonorbornene hydroxamic acid (+)-1, (–)-1, (+)-2, or (–)-2 (336 mg, 2 mmol) and levulinic acid (232 mg, 2 mmol) was dissolved in 6 mL EtOH and heated in a 10 mL pressurized reaction vial and the solution was stirred at 100 °C for 1 h at max 300 W microwave irradiation. After that the reaction mixture was evaporated to dryness and the resulting oil was dissolved in dichloromethane and was extracted with 15 mL 20% NaOH solution. The water phase was acidified with 20% HCl solution and extracted with dichloromethane. The organic phase was dried (Na2SO4) and evaporated. The gained yellow oil was dissolved in MeOH and a solution of diazomethane in Et2O was added in a flask protected from light, at room temperature for about 30 min, until no starting material could be observed by TLC. (Diazomethane is a very harmful and hazardous reagent and must be handled with caution! This reaction should be performed in a well-ventilated hood!) The solvent was then removed by evaporation and the residue was dissolved in EtOAc:MeOH = 9:1 (3 mL). The resulting solution was transferred to a silica gel column and eluted with EtOAc:MeOH = 9:1 and the diastereomers were separated. The products were crystallized from iPr2O to afford white crystals.
(3aR,5aS,6R,9S,9aR)-4-methoxy-3a-methyl-2,3,3a,4,5a,6,9a-octahydro-6,9-methanopyrrolo[1,2-a]quinazoline-1,5-dione [(+)-6a]: White crystals (29% yield), m.p. 158–161 °C, = +23.1 (c = 0.44, EtOH), ee 99%, 1H-NMR (400 MHz, CDCl3, 30 °C): 1.39 (m, 1H, 11-H), 1.53 (s, 3H, CH3) 1.57–1.60 (m, 1H, 11-H), 2.12–2.16 (m, 2H, CH2), 2.38–2.44 (m, 2H, CH2), 3.00–3.04 (m, 1H, 5a-H), 3.45 (m, 1H, 9-H), 3.84 (s, 3H, OCH3), 4.03–4.06 (m, 1H, 9a-H), 4.11 (m, 1H, 6-H), 6.09–6.11 (m, 1H, 8-H), 6.18–6.20 (m, 1H, 7-H), 13C-NMR (100 MHz, CDCl3, 30 °C): δ = 23.9, 29.5, 30.8, 45.8, 46.4, 47.0, 48.6, 52.8, 64.8, 80.3, 135.3, 135.6, 170.8, 172.3, HRMS calcd. for [M + H+] m/z = 263.1396, found m/z = 263.1390.
(3aS,5aR,6S,9R,9aS)-4-methoxy-3a-methyl-2,3,3a,4,5a,6,9a-octahydro-6,9-methanopyrrolo[1,2-a]quinazoline-1,5-dione [(–)-6a]: White crystals (32% yield), m.p. 157–159 °C, = −22.9 (c = 0.44, EtOH), ee 98%, 1H- and 13C-NMR is similar to (+)-6a.
(3aR,5aS,6S,9R,9aR)-4-methoxy-3a-methyl-2,3,3a,4,5a,6,9a-octahydro-6,9-methanopyrrolo[1,2-a]quinazoline-1,5-dione [(+)-7a]: White crystals, (28% yield), m.p. 141–142 °C, = +62.3 (c = 1.12, EtOH), ee =87%, 1H-NMR (400 MHz, CDCl3, 30 °C): 1.61–1.64 (m, 1H, 11-H), 1.68 (m, 3H, CH3), 1.80–1.83 (m, 1H, 11-H), 2.17–2.33 (m, 2H, CH2), 2.42–2.55 (m, 2H, CH2), 2.60–2.63 (m, 1H, 5a-H), 2.81 (m, 1H, 9-H), 3.36 (m, 1H, 6-H), 3.91 (s, 3H, OCH3), 4.17–4.19 (m, 1H, 9a-H), 6.19–6.20 (m, 1H, 7-H), 6.39–6.41 (m, 1H, 8-H), 13C-NMR (100 MHz, CDCl3, 30 °C): δ = 22.9, 30.2, 31.3, 44.0, 45.0, 46.3, 48.5, 53.5, 65.3, 81.1, 136.5, 138.7, 166.4, 173.2, HRMS calcd. for [M + H+] m/z = 263.1396, found m/z = 263.1390.
(3aS,5aR,6R,9S,9aS)-4-methoxy-3a-methyl-2,3,3a,4,5a,6,9a-octahydro-6,9-methanopyrrolo[1,2-a]quinazoline-1,5-dione [(–)-7a]: White crystals, (31% yield), m.p. 138–141 °C = −72.3 (c = 1.18, EtOH), ee 91% 1H- and 13C-NMR is similar to (+)-7a.
3.2.4. Synthesis of Pyrrolo[1,2-a]pyrimidines (+)-8 and (–)-8
Tetracyclic compound (+)-6a, (+)-7a, (–)-6a, or (–)-7a (50 mg, 0.19 mmol) was dissolved in 2 mL DCB in a 10 mL sealed reaction vial. The solution was stirred at 240 °C for 20 min at max 300 W microwave irradiation. Then the solvent was evaporated, and the residue was dissolved in 2 mL EtOAc and purified by column chromatography on silica gel eluted by EtOAc. The product was crystallized from iPr2O to produce white crystals.
(S)-1-methoxy-8a-methyl-1,7,8,8a-tetrahydropyrrolo[1,2-a]pyrimidine-2,6-dione [(+)-8]: White crystals, (55% yield), m.p. 137–140 °C, = +158 (c = 0.23, EtOH), ee 99 %, 1H-NMR (400 MHz, CDCl3, 30 °C): 1.49 (m, 3H, CH3), 2.37–2.46 (m, 2H, CH2), 2.51–2.69 (m, 2H, CH2), 3.89 (s, 3H, OCH3), 5.35 (d, J = 7.71 Hz, 1H, 3-H), 7.31 (d, J = 7.62 Hz, 1H, 4-H), 13C-NMR (100 MHz, CDCl3, 30 °C): δ = 18.4, 30.0, 32.4, 65.5, 80.8, 104.3, 131.4, 166.3, 171.6, HRMS calcd. for [M + H+] m/z = 197.0926, measured: m/z = 197.0916.
(R)-1-methoxy-8a-methyl-1,7,8,8a-tetrahydropyrrolo[1,2-a]pyrimidine-2,6-dione[(–)-8]: White crystals, (57% yield), m.p. 139–142 °C; = –152 (c = 0.23, EtOH), ee 99%, 1H- and 13C-NMR is similar to (+)-8.
4. Conclusions
In conclusion, new racemic and enantiomeric pyrrolo[1,2-a]pyrimidines were synthesized starting from diendo- and diexo-2-aminonorbornene hydroxamic acids. Their domino ring closure reactions with α-ketoglutaric acid and levulinic acid, and the formation of two diastereomers were observed in each case. After separation by column chromatography, single diastereomers were subjected to a microwave-mediated RDA reaction and gained bicyclic pyrrolo[1,2-a]pyrimidines through the loss of cyclopentadiene. When enantiomeric diexo- or diendo-3-amino-N-hydroxybicyclo[2.2.1]hept-5-ene-2-carboxamides were used, the products were enantiomeric heterocycles with ee = 95–99%, demonstrating that the starting compounds are excellent chiral sources, and the stereochemical information can be effectively transferable to the newly formed stereogenic centre.
Supplementary Materials
Supplementary materials are available online.
Acknowledgments
The authors are grateful for financial support from the Hungarian Research Foundation (OTKA K-115731) and the Ministry of National Economy, National Research Development and Innovation Office (GINOP-2.3.2-15-2016-00038.
Author Contributions
F.F. and M.P. designed and planned the research. B.F. and M.P. performed the synthetic work and characterized the compounds. M.P. prepared the single crystals of the title compounds, M.H. carried out the X-ray analysis of the title molecules. B.F. prepared the manuscript for publication, all authors discussed the results and commented on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lossen, H. Über die Oxalohydroxamsäure. Liebigs Ann. 1869, 150, 314–322. [Google Scholar] [CrossRef]
- Marmion, C.J.; Parker, J.P.; Nolan, K.B. Hydroxamic acids: An important class of metalloenzyme inhibitors. In Comprehensive Inorganic Chemistry II; Reedijk, J., Poeppelmeier, K., Eds.; Royal College of Surgeons: Dublin, Ireland, 2013; Volume 3, pp. 683–708. [Google Scholar]
- Bertrand, S.; Hélesbeux, J.-J.; Larcher, G.; Duval, O. Hydroxamate, a key pharmacophore exhibiting a wide range of biological activities. Mini-Rev. Med. Chem. 2013, 13, 1311–1326. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Samuni, U.; Goldstein, S. The mechanism underlying nitroxyl and nitric oxide formation from hydroxamic acids. Biochim. Biophys. Acta 2012, 1820, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.M.; Almaraza, A.E.; Barib, S.E.; Olabeb, J.A.; Amorebietaa, V.T. The HNO donor ability of hydroxamic acids upon oxidation with cyanoferrates (III). J. Coord. Chem. 2015, 68, 3236–3246. [Google Scholar] [CrossRef]
- Pinto, D.C.G.A.; Silva, A.M.S. Hydroxamic acids, recent breakthroughs in stereoselective synthesis and biological evaluations. Curr. Org. Synth. 2016, 13, 659–668. [Google Scholar] [CrossRef]
- Mishra, R.C.; Tripathi, R.; Katiyar, D.; Tewari, N.; Singh, D.; Tripathia, R.P. Synthesis of glycosylated β-Amino hydroxamates as new class of antimalarials. Bioorg. Med. Chem. 2003, 11, 5363–5374. [Google Scholar] [CrossRef] [PubMed]
- Zarember, K.A.; Cruz, A.R.; Huang, C.; Gallin, J.I. Antifungal activities of natural and synthetic iron chelators alone and in combination with azole and polyene antibiotics against Aspergillus fumigates. Antimicrob. Agents Chemother. 2009, 53, 2654–2656. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K. Ciclopirox: An overview. Int. J. Dermatol. 2001, 40, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Grassadonia, A.; Cioffi, P.; Simiele, F.; Iezzi, L.; Zilli, M.; Natoli, C. Role of hydroxamate-based histone deacetylase inhibitors (Hb-HDACIs) in the treatment of solid malignancies. Cancers 2013, 5, 919–942. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, C.-N.; Hajji, N.; Oliver, E.; Cotroneo, E.; Wharton, J.; Wang, D.; Li, M.; McKinsey, T.A.; Stenmark, K.R. Histone deacetylation inhibition in pulmonary hypertension: Therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012, 126, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Matsuo, K.; Nakanishi, A.; Hatano, T.; Izeki, H.; Ishida, Y.; Mori, W. Syntheses and anti-inflammatory and analgesic activities of hydroxamic acids and acid hydrazides. Chem. Pharm. Bull. 1983, 31, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Hyman, S.E. Target practice: HDAC inhibitors for schizophrenia. Nat. Neurosci. 2012, 15, 1180–1181. [Google Scholar] [CrossRef] [PubMed]
- Rani, R.; Granchi, C. Bioactive heterocycles containing endocyclic N-hydroxy groups. Eur. J. Med. Chem. 2015, 97, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Hölzl, W.; Rotzinger, B. Isoindolo[2,1-a]quinazoline Derivatives for Stabilization of Organic Materials. US 9321902 B2, 26 April 2016. [Google Scholar]
- Alagha, A.; Parthasarathi, L.; Gaynor, D.; Müller-Bunz, H.; Starikova, Z.A.; Farkas, E.; O’Brien, E.C.; Gil, M.-J.; Nolan, K.B. Metal complexes of cyclic hydroxamates. Synthesis and crystal structures of 3-hydroxy-2-methyl-3H-quinazolin-4-one (ChaH) and of its Fe(III), Co(II), Ni(II), Cu(II) and Zn(II) complexes. Inorg. Chim. Acta 2011, 368, 58–66. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Synthesis and application of N-hydroxylamine derivatives as potential replacements for HOBt. Eur. J. Org. Chem. 2009, 10, 1499–1501. [Google Scholar] [CrossRef]
- Tardibono, L.P.; Miller, M.J. Synthesis and anticancer activity of new hydroxamic acid containing 1,4-benzodiazepines. Org. Lett. 2009, 11, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Shemchuk, L.A.; Chernykh, V.P.; Krys’kiv, O.S. Reaction of anthranilic acid amides with cyclic anhydrides. Russ. J. Org. Chem. 2006, 42, 382–387. [Google Scholar] [CrossRef]
- Shpernat, Y.; Mizhiritskii, M. 3-Hydroxy-4-oxo-1,2,3-triazines and Derivatives thereof for Amide and Ester Bond Formation. WO 2005007634 A1, 27 January 2005. [Google Scholar]
- Tanaka, K.; Matsuo, K.; Nakanishi, A.; Kataoka, Y.; Takase, K.; Otsuki, S. Syntheses of cyclic hydroxamic acid derivatives, and their chelating abilities and biological activities. Chem. Pharm. Bull. 1988, 36, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Seitz, M.; Raymond, K.N. Efficient route to highly water-soluble aromatic cyclic hydroxamic acid ligands. Eur. J. Org. Chem. 2008, 16, 2697–2700. [Google Scholar] [CrossRef]
- Coutts, R.T; Pound, N.J. Preparation of an aromatic hydroxylamine and some cyclic hydroxamic acids, and their reaction with hydrochloric acid. Can. J. Chem. 1970, 48, 1859–1864. [Google Scholar] [CrossRef]
- Bauer, L.; Nambury, C.N.V. The Synthesis of trans-2,4-Dioxo-3-hydroxydecahydroquinazoline. J. Org. Chem. 1961, 26, 1106–1109. [Google Scholar] [CrossRef]
- Giglio, B.C.; Alexanian, E.J. Alkene hydrofunctionalization using hydroxamic acids: A radical-mediated approach to alkene hydration. Org. Lett. 2014, 16, 4304–4307. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.; Defoin, A.; Salomon, E.; Tarnus, C.; Wetterholm, A.; Haeggström, J.Z. Synthesis and structure activity relationships of novel non-peptidic metallo-aminopeptidase inhibitors. Bioorg. Med. Chem. 2006, 14, 7241–7257. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, J.; Kunisch, F.; Matzke, M.; Militzer, H.-C.; Schmidt, A.; Schönfeld, W. Novel antifungal β-amino acids: Synthesis and activity against Candida albicans. Bioorg. Med. Chem. Lett. 2003, 13, 433–436. [Google Scholar] [CrossRef]
- Wang, Y.; Papamichelakis, M.; Chew, W.; Sellstedt, J.; Noureldin, R.; Tadayon, S.; Daigneault, S. Development of a suitable process for the preparation of a TNF-α converting enzyme inhibitor, WAY-281418. Org. Proc. Res. Dev. 2008, 12, 1253–1260. [Google Scholar] [CrossRef]
- Sengupta, P.; Puri, C.S.; Chokshi, H.A.; Sheth, C.K.; Midha, A.S.; Chitturi, T.R.; Thennati, R.; Murumkar, P.R.; Yadav, M.R. Synthesis, preliminary biological evaluation and molecular modeling of some new heterocyclic inhibitors of TACE. Eur. J. Org. Med. Chem. 2011, 46, 5549–5555. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.J.-W.; Chen, L.; Lu, Z.; Xue, C.-B.; Liu, R.-Q.; Covington, M.B.; Qian, M.; Wasserman, Z.R.; Vaddi, K.; Christ, D.D.; et al. Discovery of β-benzamido hydroxamic acids as potent, selective, and orally bioavailable TACE inhibitors. Biorg. Med. Chem. Lett. 2008, 18, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Fekete, B.; Palkó, M.; Mándity, I.; Haukka, M.; Fülöp, F. A domino ring-closure followed by retro-Diels-Alder reaction for the preparation of pyrimido[2,1-a]isoindole enantiomers. Eur. J. Org. Chem. 2016, 21, 3519–3527. [Google Scholar] [CrossRef]
- Dumitrascu, F.; Popa, M.M. Pyrrolo[1,2-a]quinazolines. Synthesis and biological properties. ARKIVOC 2014, 2014, 428–452. [Google Scholar]
- Reddy, S.B.V.; Reddy, B.P.; Reddy, V.G.P.; Siriwardena, A. An efficient lactamisation/N-acyliminium Pictet—Spengler domino strategy for the diasteroselective synthesis of polyhydroxylated quinoxalinone, β-carboline and quinazolinone derivatives. Org. Biomol. Chem. 2016, 14, 4276–4282. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, S.S.; Keivanloo, A.; Nasr-Isfahani, H.; Bamoniri, A. Synthesis of novel 1,5-disubstituted pyrrolo[1,2-a]quinazolines and their evaluation for anti-bacterial and anti-oxidant activities. RSC Adv. 2016, 6, 92663–92669. [Google Scholar] [CrossRef]
- Acosta, P.; Ortiz, A.; Insuasty, B.; Abonia, R.; Quiroga, J. Synthesis and study of the electronic properties of pyrazolo[1,5-c]pyrrolo[1,2-a]quinazoline and pyrazolo[1,5-c]pyrido[1,2-a]quinazoline derivatives. Monatsh Chem. 2017, 148, 237–244. [Google Scholar] [CrossRef]
- Jin, R.-Z.; Zhang, W.-T.; Zhou, Y.-J.; Wang, X.-S. Iodine-catalyzed synthesis of 5H-phthalazino[1,2-b]quinazoline and isoindolo[2,1-a]quinazoline derivatives via a chemoselective reaction of 2-aminobenzohydrazide and 2-formylbenzoic acid in ionic liquids. Tetrahedron Lett. 2016, 57, 2515–2519. [Google Scholar] [CrossRef]
- Zhang, W.-T.; Qiang, W.-W.; Yao, C.-S.; Wang, X.-S. Iodine-catalyzed synthesis of fused tetracyclic pyridazino[6,1-b]pyrrolo [1,2-a]quinazolin-9(1H)-one derivatives via a tandem reaction. Tetrahedron 2016, 72, 2178–2185. [Google Scholar] [CrossRef]
- Zhang, M.-M.; Lu, L.; Zhou, Y.-J.; Wang, X.-S. Iodine-catalyzed synthesis of pyrrolo[1,2-a]quinazoline-3a-carboxylic acid derivatives in ionic liquids. Res. Chem. Intermed. 2013, 39, 3327–3335. [Google Scholar] [CrossRef]
- Bradar, B.; Reich, E. Biochemical and biological properties of 5-bromotubercidin: Differential effects on cellular DNA-directed and viral RNA-directed RNA synthesis. Bioorg. Med. Chem. 2008, 16, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Glunz, P.W.; Douty, B.P.; Decicco, C.P. Design and synthesis of bicyclic pyrimidinone-based HCV NS3 protease inhibitors. Bioorg. Med. Chem. 2003, 13, 785–788. [Google Scholar] [CrossRef]
- Ivanov, M.A.; Aleksandrova, L.A. Bicyclic furano-, pyrrolo-, and thiopheno[2,3-d] derivatives of pyrimidine nucleosides: Synthesis and antiviral properties. Russ. J. Bioorg. Chem. 2013, 39, 22–39. [Google Scholar] [CrossRef]
- Kumar, V.P.; Cisneros, J.A.; Frey, K.M.; Castellanos-Gonzalez, A.; Wang, Y.; Gangjee, A.; White, A.C., Jr.; Jorgensen, W.L.; Anderson, K.S. Structural studies provide clues for analog design of specific inhibitors of Cryptosporidium hominis thymidylate synthase—Dihydrofolate reductase. Bioorg. Med. Chem. Lett. 2014, 24, 4158–4161. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.S.; El-Domany, R.A.; El-Hameed, R.H.A. Synthesis of certain pyrrole derivatives as antimicro-bial agents. Acta Pharm. 2009, 59, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Awazu, Y.; Mizutani, A.; Nagase, Y.; Iwata, H.; Oguro, Y.; Miki, H.; Imamura, S.; Hori, A. A novel pyrrolo[3, 2-d]pyrimidine derivative, as a vascular endothelial growth factor receptor and platelet-derived growth factor receptor tyrosine kinase inhibitor, shows potent antitumor activity by suppression of tumor angiogenesis. Cancer Sci. 2012, 103, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.D.; Wilson, J.W.; Deanda, F.; Patnaik, S.; Redman, A.M.; Yang, B.; Shewchuk, L.; Sabbatini, P.; Leesnitzer, M.A.; Groy, A.; et al. Discovery of 4,6-bis-anilino-1H-pyrrolo[2,3-d]pyrimidines: Potent inhibitors of the IGF-1R receptor tyrosine kinase. Bioorg. Med. Chem. Lett. 2009, 19, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Cherian, C.S.; Desmoulin, K.; Wang, L.; Polin, L.; White, K.; Kushner, J.; Stout, M.; Hou, Z.; Gangjee, A.; Matherly, L.H. Therapeutic targeting malignant mesothelioma with a novel 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate via its selective uptake by the proton-coupled folate transporter. Cancer Chemother. Pharmacol. 2013, 71, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Coumar, M.S.; Wu, J.-S.; Leou, J.-S.; Tan, U.-K.; Chang, C.-Y.; Chang, T.-Y.; Lin, W.-H.; Hsu, J.T.-A.; Chao, Y.-S.; Wua, S.-Y.; et al. Aurora kinase A inhibitors: Identification, SAR exploration and molecular modeling of 6,7-dihydro-4H-pyrazolo-[1,5-a]pyrrolo[3,4-d]pyrimidine-5,8-dione scaffold. Bioorg. Med. Chem. Lett. 2008, 18, 1623–1627. [Google Scholar] [CrossRef] [PubMed]
- Cuny, G.D.; Yu, P.B.; Laha, J.K.; Xing, X.; Liu, J.-F.; Lai, C.S.; Deng, D.Y.; Sachidanandan, C.; Bloch, K.D.; Peterson, R.T. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4388–4392. [Google Scholar] [CrossRef] [PubMed]
- Ella, D.A.A E.; Ghorab, M.M.; Noaman, E.; Heibab, H.I.; Khalil, A.I. Molecular modeling study and synthesis of novel pyrrolo[2,3-d]pyrimidines and pyrrolotriazolopyrimidines of expected antitumor and radioprotective activities. Bioorg. Med. Chem. 2008, 16, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Gerspacher, M.; Pissot-Soldermann, C. Design of two new chemotypes for inhibiting the Janus kinase 2 by scaffold morphing. Bioorg. Med. Chem. Lett. 2010, 20, 1858–1860. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A.; Kurup, S.; Ihnat, M.A.; Thorpe, J.E.; Shenoy, S.S. Synthesis and biological activity of N4-phenylsubstituted-6-(2,4-dichloro phenylmethyl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines as vascular endothelial growth factor receptor-2 inhibitors and antiangiogenic and antitumor agents. Bioorg. Med. Chem. 2010, 18, 3575–3587. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A; Pavana, R.K.; Li, W.; Hamel, E.; Westbrook, C.; Mooberry, S.L. Novel water-soluble substituted pyrrolo[3,2-d]pyrimidines: Design, synthesis, and biological evaluation as antitubulin antitumor agents. Pharm. Res. 2012, 29, 3033–3039. [Google Scholar] [CrossRef] [PubMed]
- Kaspersen, S.J.; Han, J.; Nřrsett, K.G.; Rydså, L.; Kjřbli, E.; Bugge, S.; Bjřrkřy, G.; Sundby, E.; Hoff, B.H. Identification of new 4-N-substituted 6-aryl-7H-pyrrolo[2,3-d]pyrimidine-4-amines as highly potent EGFR-TK inhibitors with Src-family activity. Eur. J. Pharm. Sci. 2014, 59, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, Y.; Miwa, K.; Seto, M.; Banno, H.; Ohta, Y.; Tamura, T.; Yusa, T.; Miki, H.; Kamiguchi, H.; Ikeda, Y.; et al. Design and synthesis of pyrrolo[3,2-d]pyrimidine HER2/EGFR dual inhibitors: Improvement of the physicochemical and pharmacokinetic profiles for potent in vivo anti-tumor efficacy. Bioorg. Med. Chem. 2012, 20, 6171–6180. [Google Scholar] [CrossRef] [PubMed]
- Kumari, K.M.; Yamini, L.; Vijjulathae, M. 3D QSAR of pyrrolo pyrimidine and thieno pyrimidines as human thymidylate synthase inhibitors. E-J. Chem. 2012, 9, 1699–1710. [Google Scholar] [CrossRef]
- Lauria, A.; Patella, C.; Abbate, I.; Martorana, A.; Almerico, A.M. Lead optimization through VLAK protocol: New annelated pyrrolo-pyrimidine derivatives as antitumor agents. Eur. J. Med. Chem. 2012, 55, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Temburnikar, K.W.; Ross, C.R.; Wilson, G.M.; Balzarini, J.; Cawrse, B.M.; Seley-Radtke, K.L. Antiproliferative activities of halogenated pyrrolo[3,2-d]pyrimidines. Bioorg. Med. Chem. 2015, 23, 4354–4363. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-Y.; Chen, W.-H.; Wu, S.-G.; Tian, Y.-X.; Zhang, J.-J. Pyrrolo[3,2-d]pyrimidine derivatives as type II kinase insert domain receptor (KDR) inhibitors: CoMFA and CoMSIA studies. Int. J. Mol. Sci. 2012, 13, 2387–2404. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Hondo, T.; Nagata, H.; Ogiyama, T.; Maeda, J.; Hoshii, H.; Kontani, T.; Kuromitsu, S.; Ohga, K.; Orita, M.; et al. Novel 7H-pyrrolo[2,3-d]pyrimidine derivatives as potent and orally active STAT6 inhibitors. Bioorg. Med. Chem. 2009, 17, 6926–6936. [Google Scholar] [CrossRef] [PubMed]
- Pittala, V.; Siracusa, M.A.; Modica, M.N.; Salerno, L.; Pedretti, A.; Vistoli, G.; Cagnotto, A.; Mennini, T.; Romeo, G. Synthesis and molecular modeling of 1H-pyrrolopyrimidine-2,4-dione derivatives as ligands for the α1-adrenoceptors. Bioorg. Med. Chem. 2011, 19, 5260–5276. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Romagnoli, R.; Saponaro, G.; Tabrizi, M.A.; Baraldi, S.; Pedretti, P.; Fusi, C.; Nassini, R.; Materazzi, S.; Geppetti, P.; et al. 7-Substituted-pyrrolo[3,2-d]pyrimidine-2,4-dione derivatives as antagonists of the transient receptor potential ankyrin 1 (TRPA1) channel: A promising approach for treating pain and inflammation. Bioorg. Med. Chem. 2012, 20, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Abe, N. Syntheses and some reactions of 4H-cyclohepta[4,5]pyrrolo[1,2-α]pyrimidin-4-ones. Bull. Chem. Soc. Jpn. 1987, 60, 1053–1056. [Google Scholar] [CrossRef]
- Acosta, P.; Insuasty, B.; Abonia, R.; Quiroga, J. Annelation of pyrrolo[1,2-a]pyrimidine and pyrido[1,2-a]pyrimidine systems to a pyrazolopyridine framework by a cascade of two cyclization reactions. Tetrahedron Lett. 2015, 56, 2917–2921. [Google Scholar] [CrossRef]
- Amarnath, V.; Madhav, R. A survey of methods for the preparation of pyrrolopyrimidines. Synthesis 1974, 12, 837–859. [Google Scholar] [CrossRef]
- De Coen, L.M.; Heugebaert, T.S.A.; García, D.; Stevens, C.V. Synthetic entries to and biological activity of pyrrolopyrimidines. Chem. Rev. 2016, 116, 80–139. [Google Scholar] [CrossRef] [PubMed]
- Dasari, R.; Kornienko, A. Multicomponent synthesis of the medicinally important pyrrolo[2,3-d]pyrimidine scaffold. Chem. Heterocycl. Compd. 2014, 50, 139–144. [Google Scholar] [CrossRef]
- Gao, J.; Henry, R.F.; Pagano, T.G.; Duerst, R.W.; Souers, A.J. A cascade reaction sequence en route to 7-substituted 2-aminopyrrolo[1,2-a]pyrimidine-4,6-diones and the corresponding acrylic acid derivatives. Tetrahedron Lett. 2007, 48, 7395–7398. [Google Scholar] [CrossRef]
- Miklós, F.; Tóth, Z.; Hänninen, M.M.; Sillanpää, R.; Forró, E.; Fülöp, F. Retro-Diels-Alder protocol for the synthesis of pyrrolo[1,2-a]pyrimidine and pyrimido[2,1-a]isoindole enantiomers. Eur. J. Org. Chem. 2013, 22, 4887–4894. [Google Scholar] [CrossRef]
- Prieur, V.; Heindler, N.; Rubio-Martínez, J.; Guillaumet, G.; Pujol, M.D. One-pot synthesis of 4-aminated pyrrolo[2,3-d]pyrimidines from alkynylpyrimidines under metal-catalyst-free conditions. Tetrahedron 2015, 71, 1207–1214. [Google Scholar] [CrossRef]
- Vincze, Z.; Pilipecz, M.V.; Scheiber, P.; Varga, T.R.; Tóth, G.; Nemes, P. Simple route to multisubstituted tetrahydropyrimidines. Tetrahedron 2015, 71, 6135–6142. [Google Scholar] [CrossRef]
- Cheng, R.; Guo, T.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. One-pot synthesis of quinazolinones from anthranilamides and aldehydes via p-toluenesulfonic acid catalyzed cyclocondensation and phenyliodine diacetate mediated oxidative dehydrogenation. Synthesis 2013, 45, 2998–3006. [Google Scholar] [CrossRef]
- Gao, Y.; Rubin, P.; Xiaoyi, N.; Zepp, C. Therapeutic Heterocyclic Compounds for the Treatment of Asthma and Allergy and Use Thereof. WO 2001070737 A3, 31 January 2002. [Google Scholar]
- Lajkó, G.; Orosz, T.; Grecsó, N.; Fekete, B.; Palkó, M.; Fülöp, F.; Lindner, W.; Péter, A.; Ilisz, I. High-performance liquid chromatographic enantioseparation of cyclic β-aminohydroxamic acids on zwitterionic chiral stationary phases based on Cinchona alkaloids. Anal. Chim. Acta 2016, 921, 84–94. [Google Scholar]
Sample Availability: Samples of the compounds 1–8 are available from the authors. |
Scheme 1.
The domino reaction pathway.

Figure 1.
Oak Ridge Thermal Ellipsoid Plot (ORTEP) plot of the X-ray structure of (±)-3a.
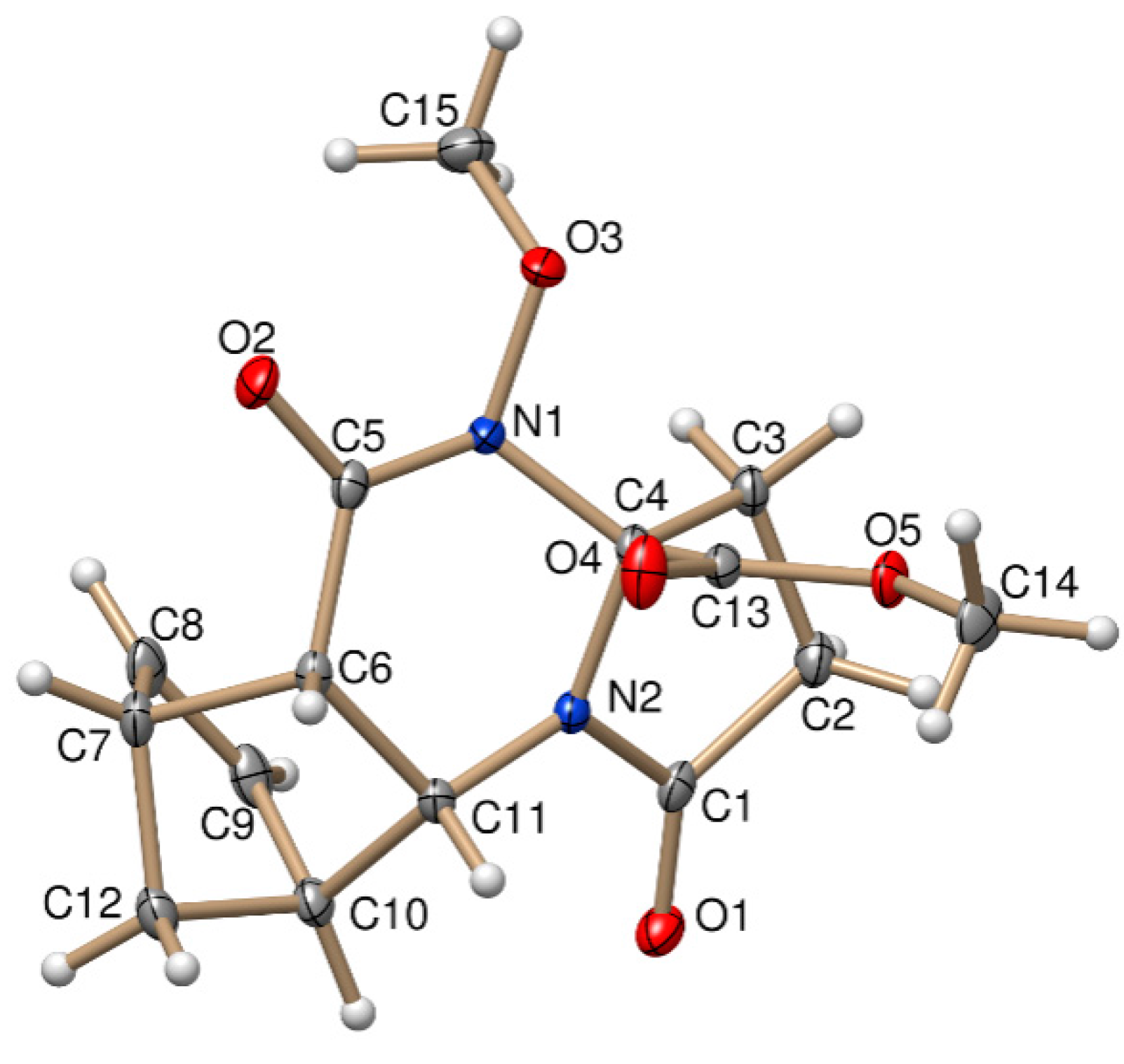
Figure 2.
ORTEP plot of the X-ray structure of (±)-4b.
Scheme 2.
Synthesis of (+)-5 and (–)-5: (i) α-ketoglutaric acid, 1 h, 100 °C, EtOH, Microwave irradiation (MW); (ii) MeOH, diazomethane/Et2O, Room temperature (r.t.) (iii) separation of major (–)-3a, (–)-4a and minor (–)-3b, (+)-4b epimers by column chromatography (epimeric ratio 3a:3b =5:4, 4a:4b = 5:4, eluent: EtOAc) (iv) MW, 20 min, 250 °C, 300 W (v) column chromatography (eluent: EtOAc).
Scheme 2.
Synthesis of (+)-5 and (–)-5: (i) α-ketoglutaric acid, 1 h, 100 °C, EtOH, Microwave irradiation (MW); (ii) MeOH, diazomethane/Et2O, Room temperature (r.t.) (iii) separation of major (–)-3a, (–)-4a and minor (–)-3b, (+)-4b epimers by column chromatography (epimeric ratio 3a:3b =5:4, 4a:4b = 5:4, eluent: EtOAc) (iv) MW, 20 min, 250 °C, 300 W (v) column chromatography (eluent: EtOAc).
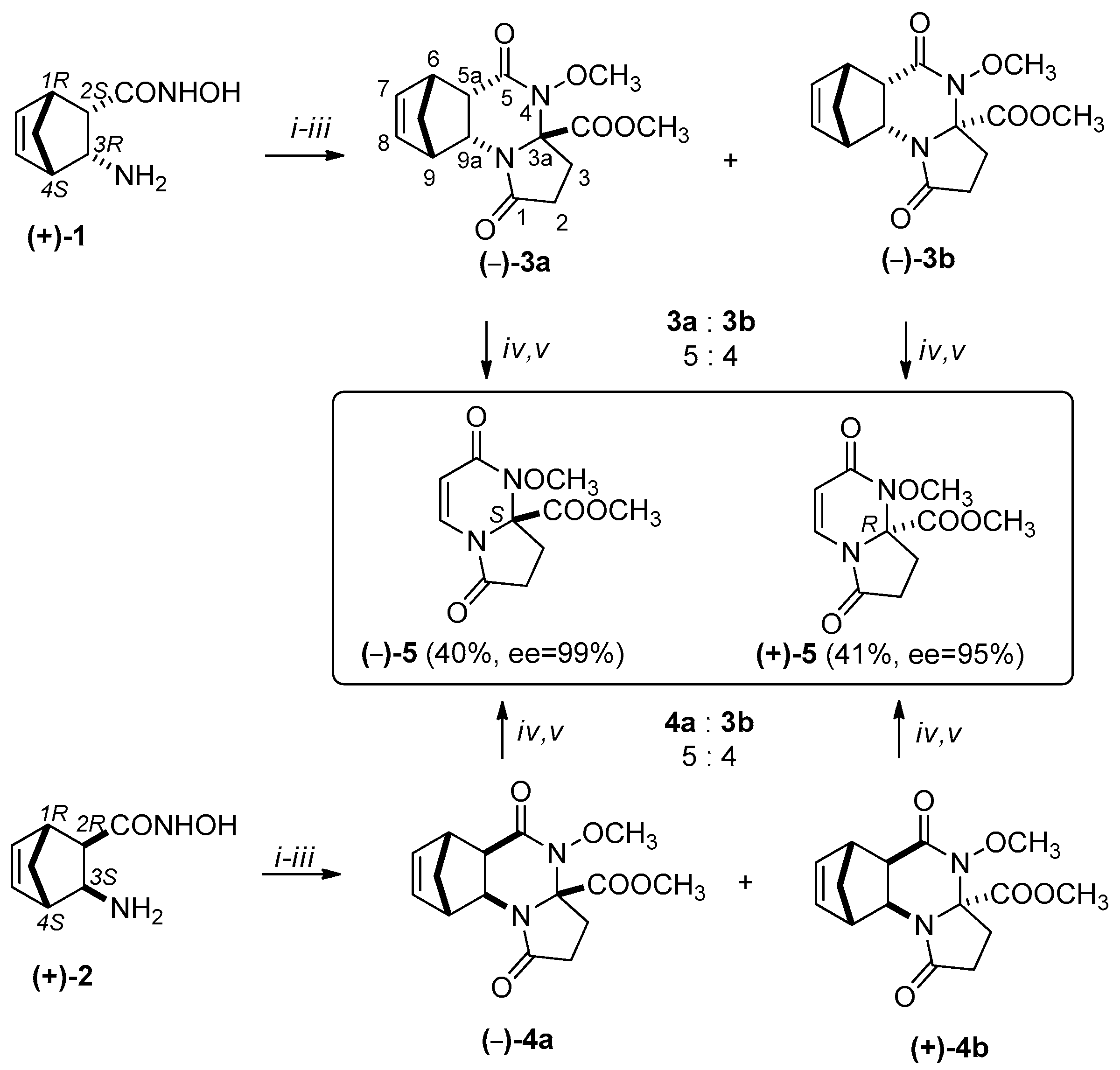
Figure 3.
ORTEP plot of the X-ray structure of (±)-6a.

Scheme 3.
Synthesis of (–)-8: (i) levulinic acid, 1 h, 100 °C, EtOH, MW; (ii) MeOH, diazomethane/Et2O, r.t.; (iii) isolation of major ((+)-6a, (+)-7a) epimers by column chromatography (eluent: EtOAc:MeOH = 9:1) (iv) Retro Diels-Alder (RDA) reaction of major epimers: MW, 220 °C, 20 min, 300 W, (v) column chromatography (eluent: EtOAc:MeOH = 9:1).
Scheme 3.
Synthesis of (–)-8: (i) levulinic acid, 1 h, 100 °C, EtOH, MW; (ii) MeOH, diazomethane/Et2O, r.t.; (iii) isolation of major ((+)-6a, (+)-7a) epimers by column chromatography (eluent: EtOAc:MeOH = 9:1) (iv) Retro Diels-Alder (RDA) reaction of major epimers: MW, 220 °C, 20 min, 300 W, (v) column chromatography (eluent: EtOAc:MeOH = 9:1).
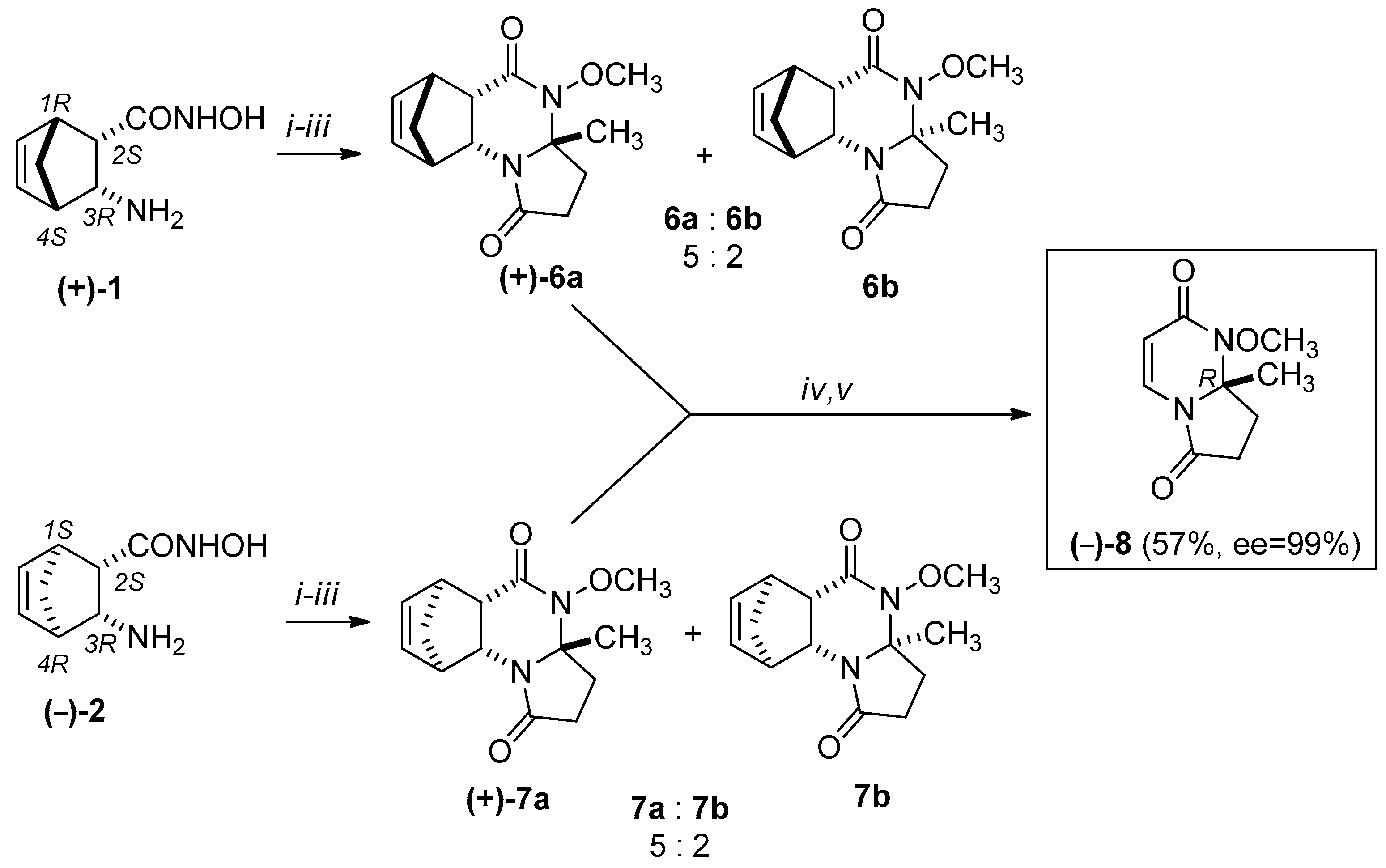
Scheme 4.
Synthesis of (+)-8: (i) levulinic acid, 1 h, 100 °C, EtOH, MW; (ii) MeOH, diazomethane/Et2O, r.t.; (iii) isolation of major (–)-6a, (–)-7a) epimers by column chromatography (eluent: EtOAc:MeOH = 9:1); (iv) RDA reaction of major epimers: MW, 220 °C, 20 min, 300 W, (v) column chromatography (eluent: EtOAc:MeOH =9:1).
Scheme 4.
Synthesis of (+)-8: (i) levulinic acid, 1 h, 100 °C, EtOH, MW; (ii) MeOH, diazomethane/Et2O, r.t.; (iii) isolation of major (–)-6a, (–)-7a) epimers by column chromatography (eluent: EtOAc:MeOH = 9:1); (iv) RDA reaction of major epimers: MW, 220 °C, 20 min, 300 W, (v) column chromatography (eluent: EtOAc:MeOH =9:1).
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fekete, B.; Palkó, M.; Haukka, M.; Fülöp, F. Synthesis of Pyrrolo[1,2-a]pyrimidine Enantiomers via Domino Ring-Closure followed by Retro Diels-Alder Protocol. Molecules 2017, 22, 613. https://doi.org/10.3390/molecules22040613
AMA Style
Fekete B, Palkó M, Haukka M, Fülöp F. Synthesis of Pyrrolo[1,2-a]pyrimidine Enantiomers via Domino Ring-Closure followed by Retro Diels-Alder Protocol. Molecules. 2017; 22(4):613. https://doi.org/10.3390/molecules22040613
Chicago/Turabian StyleFekete, Beáta, Márta Palkó, Matti Haukka, and Ferenc Fülöp. 2017. "Synthesis of Pyrrolo[1,2-a]pyrimidine Enantiomers via Domino Ring-Closure followed by Retro Diels-Alder Protocol" Molecules 22, no. 4: 613. https://doi.org/10.3390/molecules22040613