
One–Pot Phosphate-Mediated Synthesis of Novel 1,3,5-Trisubstituted Pyridinium Salts: A New Family of S. aureus Inhibitors

Abstract
:
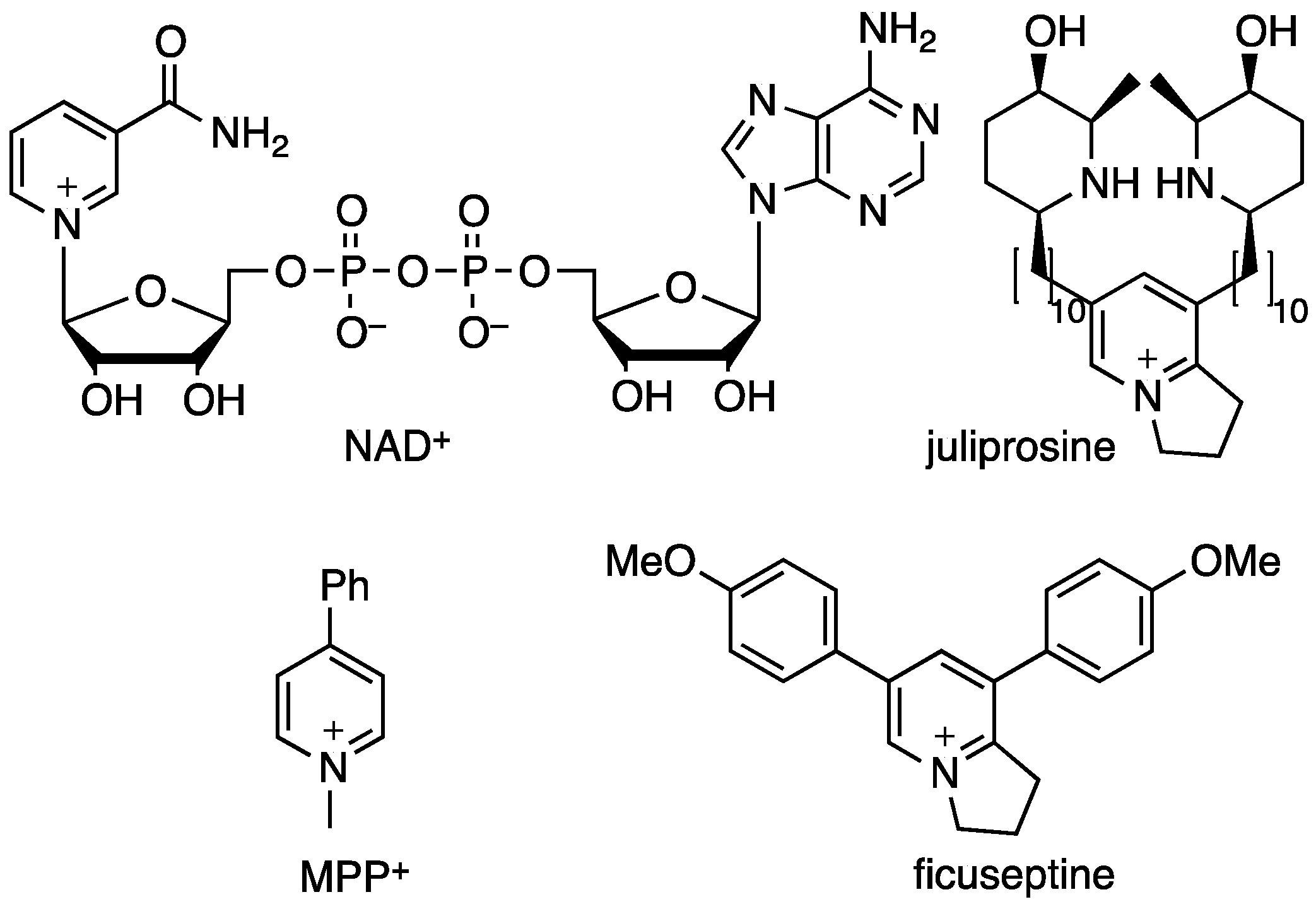
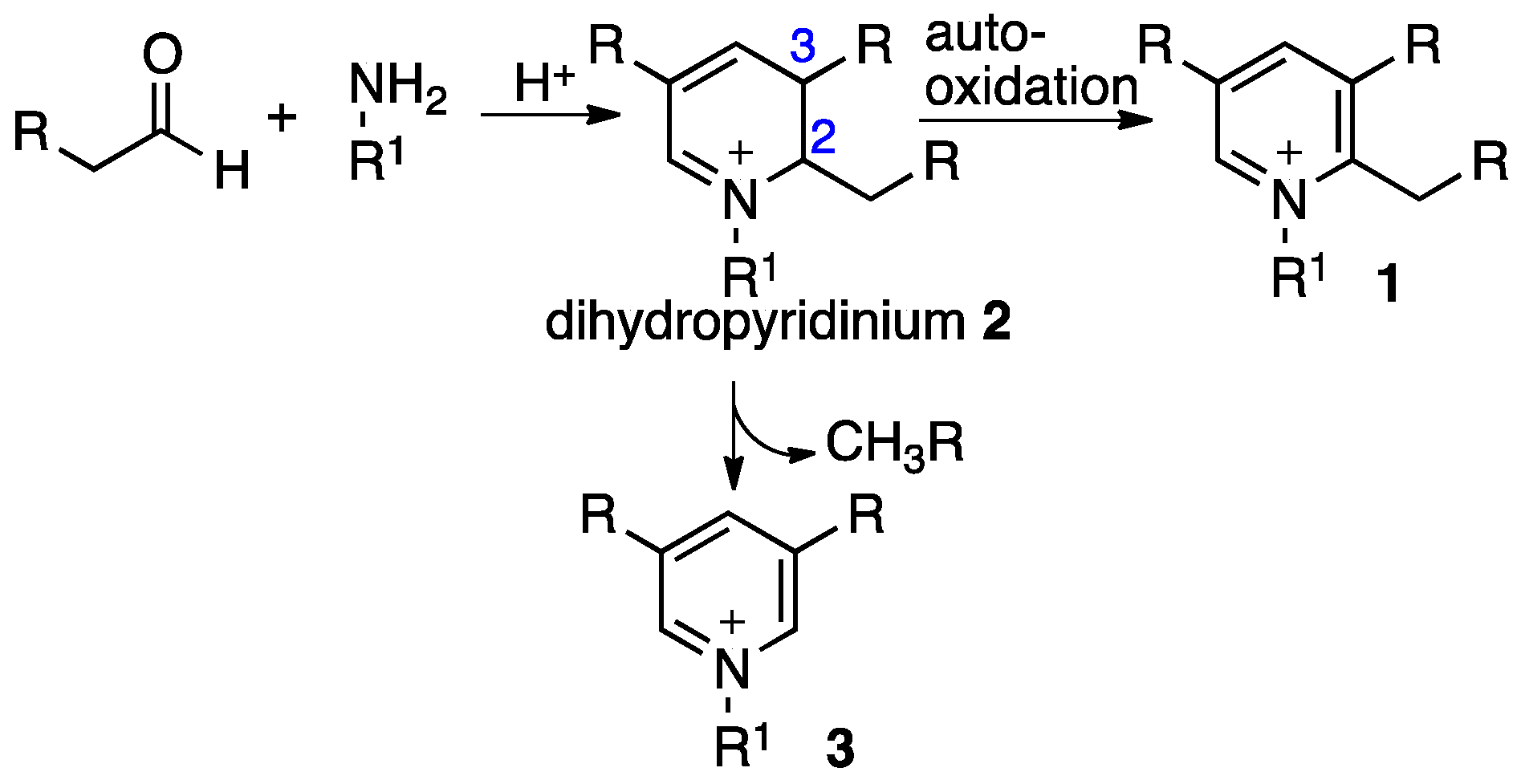
1. Introduction
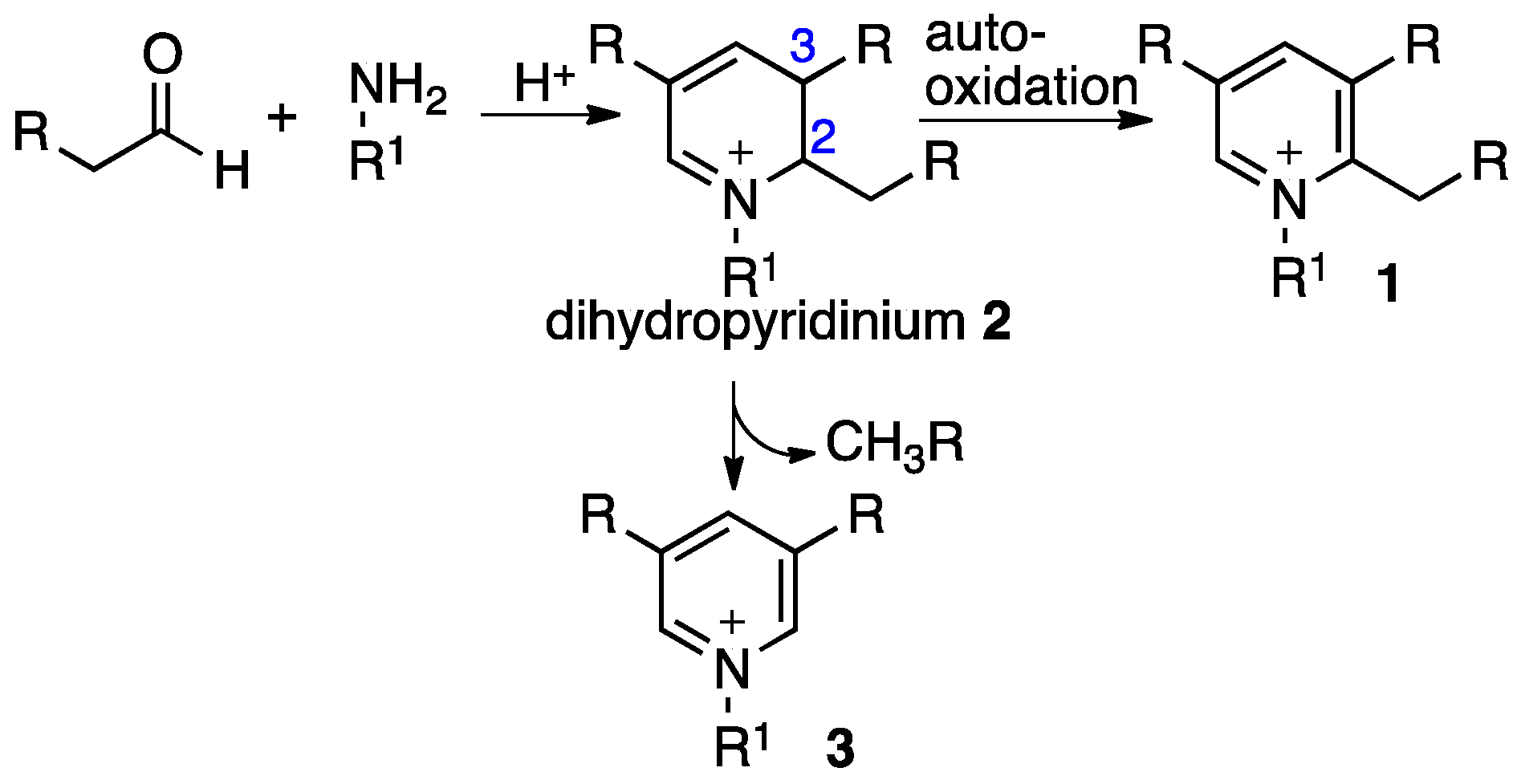
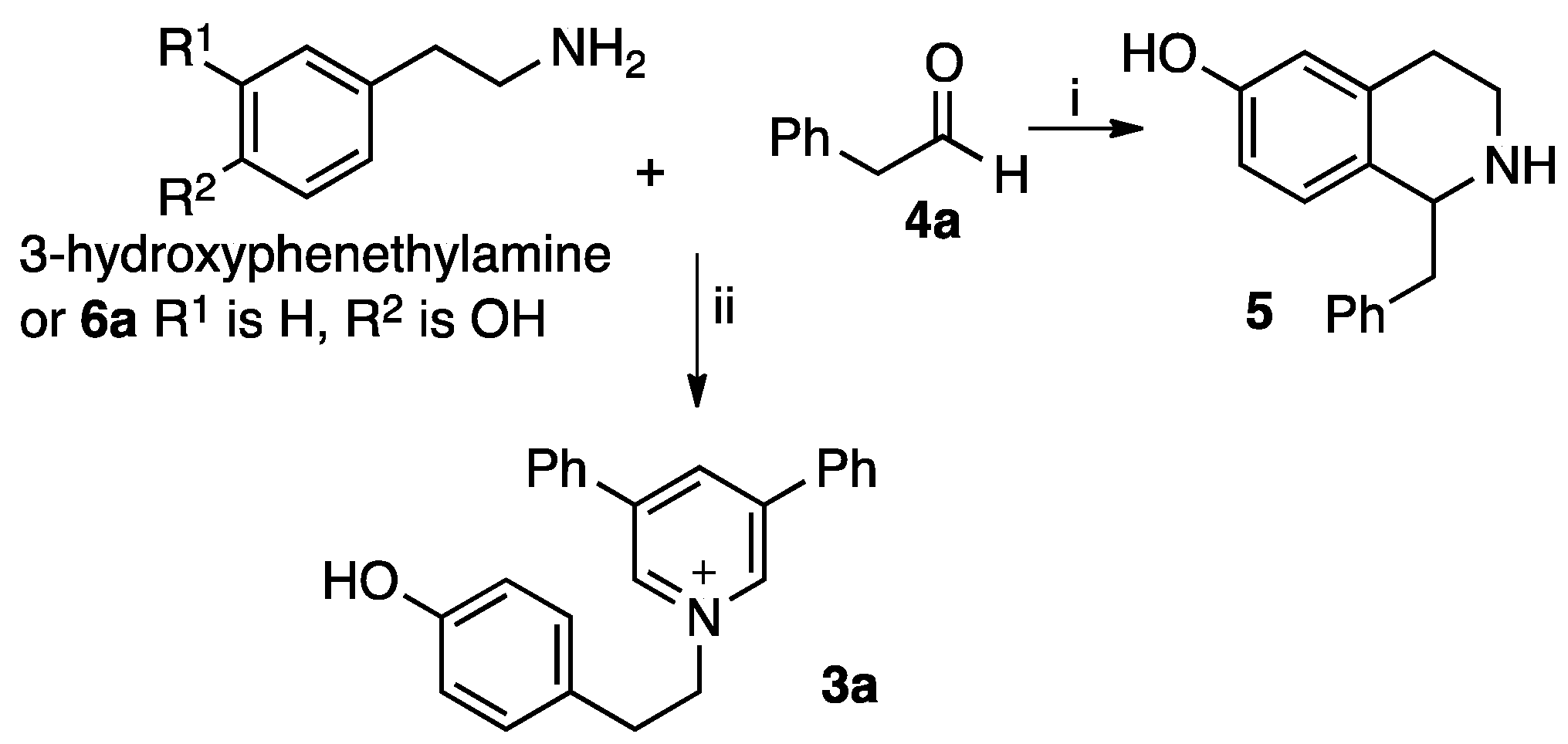
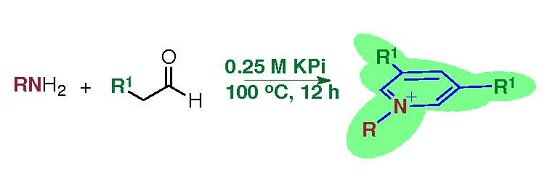
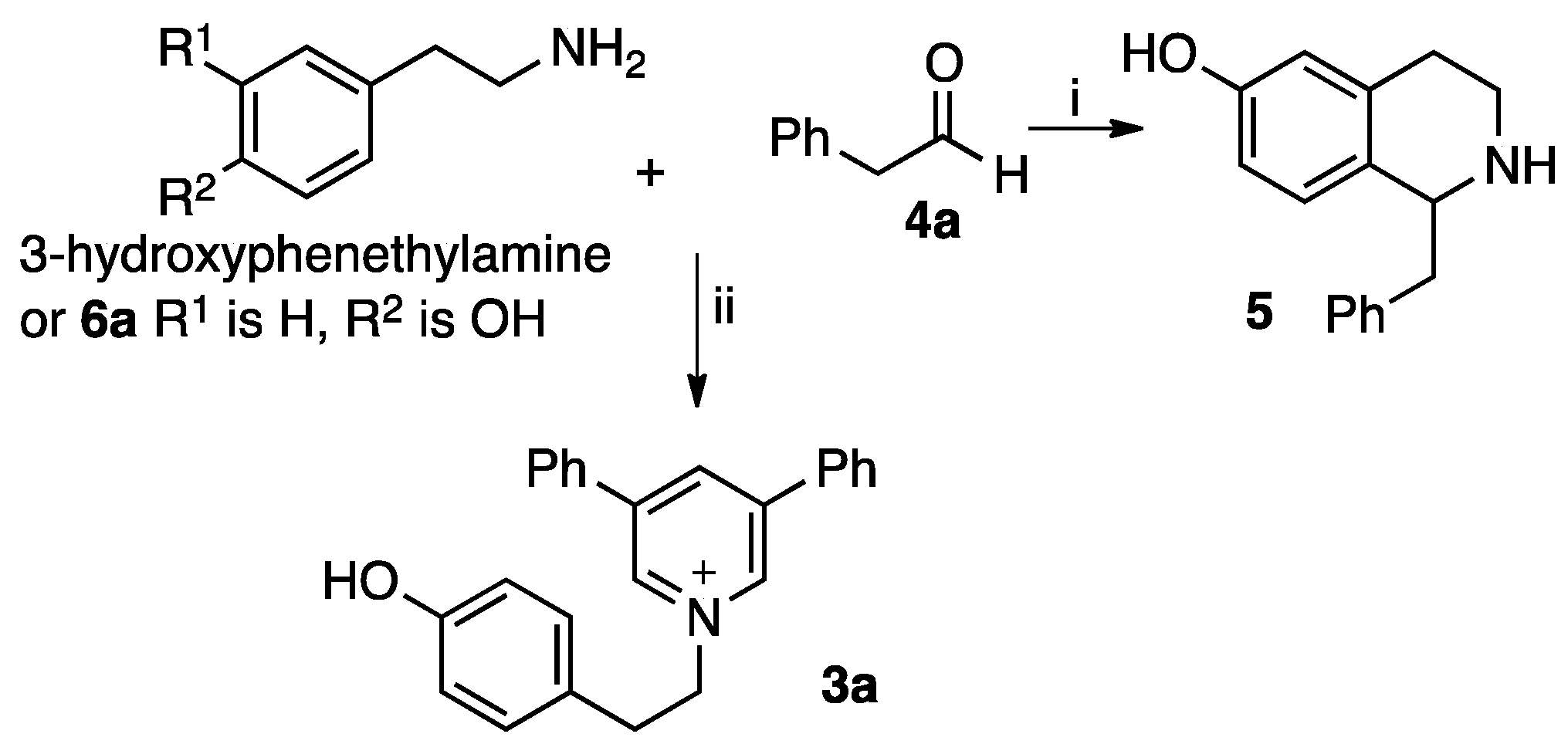
2. Results and Discussion
3. Experimental Section
3.1. General Information and Methods
3.1.1. Chemistry
3.1.2. Biological Assays
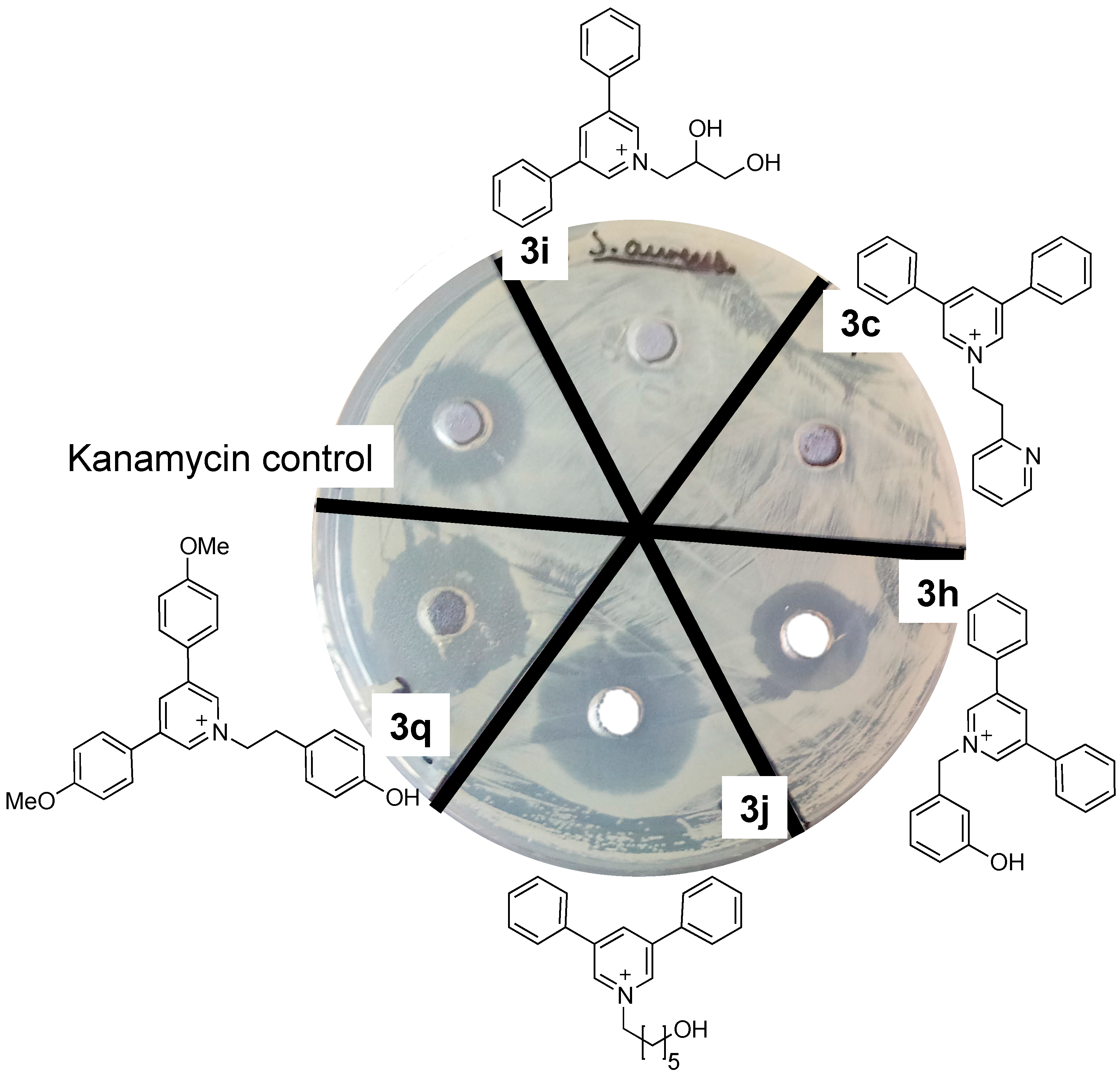
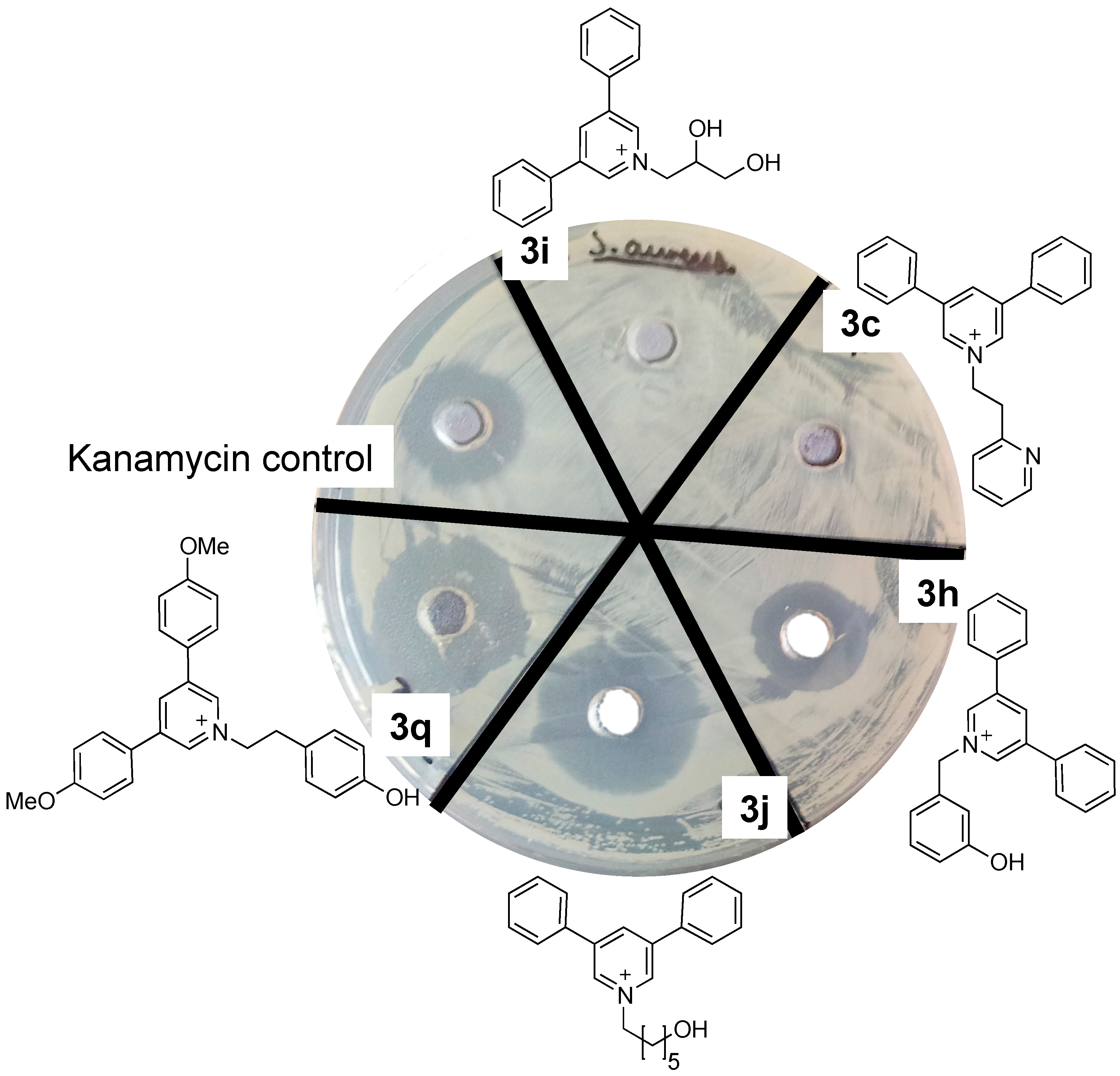
Plate Zone Assays
Minimimal Inhibitory Concentration (MIC)
3.2. Procedure A for Synthesis of the 1,3,5-Substituted Pyridinium Salts
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pollack, N.; Dölle, C.; Ziegler, M. The power to reduce: Pyridine nucleotides—Small molecules with a multitude of functions. Biochem. J. 2007, 402, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Von Dätwyler, P.; Ott-Longoni, R.; Schöpp, E.; Hesse, M. Juliprosine, a further alkaloid isolated from Prosopis juliflora. Helv. Chim. Acta 1981, 64, 1959–1963. [Google Scholar]
- Baumgartner, B.; Erdelmeier, C.A.J.; Wright, A.D.; Rali, T.; Sticher, O. An antimicrobial alkaloid from Ficus septica. Phytochemistry 1990, 29, 3327–3330. [Google Scholar] [CrossRef]
- Bracher, F.; Daab, J. Total synthesis of the indolizidinium alkaloid ficuseptine. Eur. J. Org. Chem. 2002, 2002, 2288–2291. [Google Scholar] [CrossRef]
- Snider, B.B.; Neubert, B.J. Synthesis of ficuseptine, juliprosine, and juliprosopine by biomimetic intramolecular chichibabin pyridine syntheses. Org. Lett. 2005, 7, 2715–2718. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.K.; Riachi, N.J.; Fiedler, G.C.; Singh, M.P.; Abdallah, F.; Harik, S.I.; Sayre, L.M. Structure-neurotoxicity trends of analogues of 1-methyl-4-phenylpyridinium (MPP+) the cytotoxic metabolite of the dopaminergic neurotoxin MPTP. Life Sci. 1990, 46, 379–390. [Google Scholar] [CrossRef]
- Wimalasena, D.S.; Perera, R.P.; Heyen, B.J.; Balasooriya, I.S.; Wimalasena, K. Vesicular Monoamine Transporter Substrate/Inhibitor Activity of MPTP/MPP+ Derivatives: A structure-activity study. J. Med. Chem. 2008, 51, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Pijper, D.; Bulten, E.; Šmisterová, J.; Wagenaar, A.; Hoekstra, D.; Engberts, J.B.F.N.; Hulst, R. Novel biodegragable pyridinium amphiphiles for gene delivery. Eur. J. Org. Chem. 2003, 4406–4412. [Google Scholar] [CrossRef]
- Trova, M.P.; Wissner, A.; Carroll, M.L.; Kerwar, S.S.; Pickett, W.C.; Schaub, R.E.; Torley, L.W.; Kohler, C.A. Analogues of platelet activating factor. 8. Antagonists of PAF containing an aromatic ring linked to a pyridinium ring. J. Med. Chem. 1993, 36, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Alptüzün, V.; Parlar, S.; Tash, H.; Erciyas, E. Synthesis and antimicrobial activity of some pyridinium salts. Molecules 2009, 14, 5203–5215. [Google Scholar] [CrossRef] [PubMed]
- Chichibabin, A.E. Uber condensationen der aldehyde mit ammonok zu pyridinbasen. J. Prakt. Chem. 1924, 107, 122–128. [Google Scholar]
- Frank, R.L.; Seven, R.P. Pyridines. IV. A study of the Chichibabin synthesis. J. Am. Chem. Soc. 1949, 71, 2629–2635. [Google Scholar] [CrossRef]
- Eliel, E.L.; McBride, R.T.; Kaufmann, S. Abnormal Chichibabin reactions. The condensation of phenylacetaldehyde and homoveratric aldehyde with ammonia. J. Am. Chem. Soc. 1953, 75, 4291–4296. [Google Scholar] [CrossRef]
- Farley, C.P.; Eliel, E.L. Chichibabin reactions with phenylacetaldehyde. J. Am. Chem. Soc. 1956, 78, 3477–3484. [Google Scholar] [CrossRef]
- Charman, H.B.; Rowe, J.M. Condensation of aldehydes with ammonium salts to give substituted pyridines. Chem. Commun. 1971, 476–477. [Google Scholar] [CrossRef]
- Suyama, K.; Adachi, S. Reaction of alkanals and amino acids or primary amines. Synthesis of 1,2,3,5- and 1,3,4,5-substituted quaternary pyridinium salts. J. Org. Chem. 1979, 44, 1417–1420. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, S.; Li, Y.; Yang, F.; Gong, C.; Zhao, J. Synthesis and characterisation of soluble polyimides based on a new fluorinated diamine: 4-Phenyl-2,6-bis[3-(4′amino-2′trifluoromethyl-phenoxy)phenyl] pyridine. J. Fluor. Chem. 2010, 131, 724–730. [Google Scholar] [CrossRef]
- Yu, L.-B.; Chen, D.; Li, J.; Ramirez, J.; Wang, P.G. Lanthanide-promoted reactions of aldehydes and amine hydrochlorides in aqueous solution. Synthesis of 2,3-dihydropyridinium and pyridinium derivatives. J. Org. Chem. 1997, 62, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Burns, N.Z.; Baran, P.S. On the origin of the haouamine alkaloids. Angew. Chem. Int. Ed. 2008, 47, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Burns, N.Z.; Jessing, M.; Baran, P.S. Total synthesis of haouamine A: The indeno-tetrahydropyridine core. Tetrahedron 2009, 65, 6600–6610. [Google Scholar] [CrossRef] [PubMed]
- Dagorn, F.; Yan, L.-H.; Gravel, E.; Leblanc, K.; Maciuk, A.; Poupon, E. Particular behavior of ‘C6C2 units’ in the Chichibabin pyridine synthesis and biosynthetic implications. Tetrahedron Lett. 2011, 52, 3523–3526. [Google Scholar] [CrossRef]
- Pesnot, T.; Gershater, M.C.; Ward, J.M.; Hailes, H.C. Phosphate mediated biomimetic synthesis of tetrahydroisoquinolines. Chem. Commun. 2011, 47, 3242–3244. [Google Scholar] [CrossRef] [PubMed]
- Lichman, B.R.; Lamming, E.D.; Pesnot, T.; Smith, J.M.; Hailes, H.C.; Ward, J.M. One-pot triangular chemoenzymatic cascades for the syntheses of chiral alkaloids from dopamine. Green Chem. 2015, 17, 852–855. [Google Scholar] [CrossRef]
- Pesnot, T.; Gershater, M.C.; Ward, J.M.; Hailes, H.C. The Catalytic Potential of Coptis japonica NCS2 Revealed—Development and Utilisation of a Fluorescamine-Based Assay. Adv. Syn. Catal. 2012, 354, 2997–3008. [Google Scholar] [CrossRef]
- Davril, M.; Han, K.-K. Isolation and characterization of a highly cross-linked peptide from elastin of porcine aorta. FEBS Lett. 1974, 43, 331–336. [Google Scholar] [CrossRef]
- Umeda, H.; Takeuchi, M.; Suyama, K. Two new elastin cross-links having pyridine skeleton. J. Biol. Chem. 2001, 276, 12579–12587. [Google Scholar] [CrossRef] [PubMed]
- Powner, M.W.; Gerland, B.; Sutherland, J.D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 2009, 459, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.M.; Abreu, A.C.; Simões, M. Action of kanamycin against single and dual species biofilms of Escherichia coli and Staphylococcus aureus. J. Microbiol. Res. 2012, 2, 84–88. [Google Scholar] [CrossRef]
- Liang, J.T.; Liu, J.; Shireman, B.T.; Tran, V.; Deng, X.; Mani, N.S. A practical synthesis of regioisomeric 6- and 7-methoxytetrahydro-3-benzazepines. Org. Process Res. Dev. 2010, 14, 380–385. [Google Scholar] [CrossRef]
- Chao, H.J.; Tuerdi, R.T.; Herpin, T.; Roberge, J.Y.; Liu, Y.; Lawrence, R.M.; Rehfuss, R.P.; Clark, C.G.; Qiao, J.X.; Gungor, T.; et al. Urea antagonists of p2y1 receptor useful in the treatment of thrombotic conditions. WO/2005/113511, 5 Novermber 2005. [Google Scholar]
Sample Availability: Samples of compounds 3 are available from the authors upon reasonable requests (where materials are available). |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Buffer | Yield of 3a |
|---|---|---|
| 1 | KH2PO4 (KPi) | 48% |
| 2 | Glc-1-P | 40% |
| 3 | PPi | 23% |
| 4 | UMP | 53% |
| 5 | B(OH)3 | <1% |
| 6 | HEPES | 3% |
| 7 | Tris | <1% |
| 8 | water only | <1% |

| Amine | R | Product | Isolated Yield | Amine | R | Product | Isolated Yield |
|---|---|---|---|---|---|---|---|
| 6a |  | 3a | 70% | 6h | 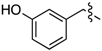 | 3h | 65% |
| 6b | PhCH2CH2 | 3b | 54% | 6i | 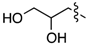 | 3i | 72% |
| 6c | 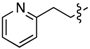 | 3c | 52% | 6j | (CH2)6OH | 3j | 69% |
| 6d | 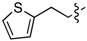 | 3d | 38% | 6k | (CH2)3CO2H | 3k | 63% |
| 6e | 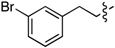 | 3e | 50% | 6l | 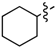 | 3l | 53% |
| 6f | 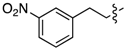 | 3f | 45% | 6m | Ph | 3m | 0% |
| 6g | 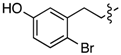 | 3g | 13% 2 | 6n |  | 3n | 0% |
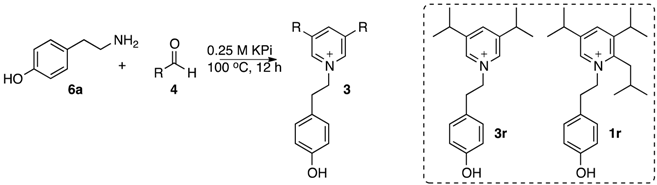
| Aldehyde | R | Product | Yield |
|---|---|---|---|
| 4a | PhCH2 | 3a | 70% |
| 4o | 4-HOC6H4CH2 | 3o | 66% |
| 4p |  | 3p | 42% |
| 4q | 4-MeOC6H4CH2 | 3q | 58% |
| 4r | CH(CH3)2 | 3r (1r) 2 | 5(10)% |
| 4s | C2H5 | 3s | 0% |
| Compound | 1 MIC (μg/mL) | Compound | 1 MIC (μg/mL) |
|---|---|---|---|
| 3a | n.d. | 3h | 64 |
| 3b | 32 | 3i | n.d. |
| 3c | n.d. | 3j | 64 |
| 3d | 64 | 3k | n.d. |
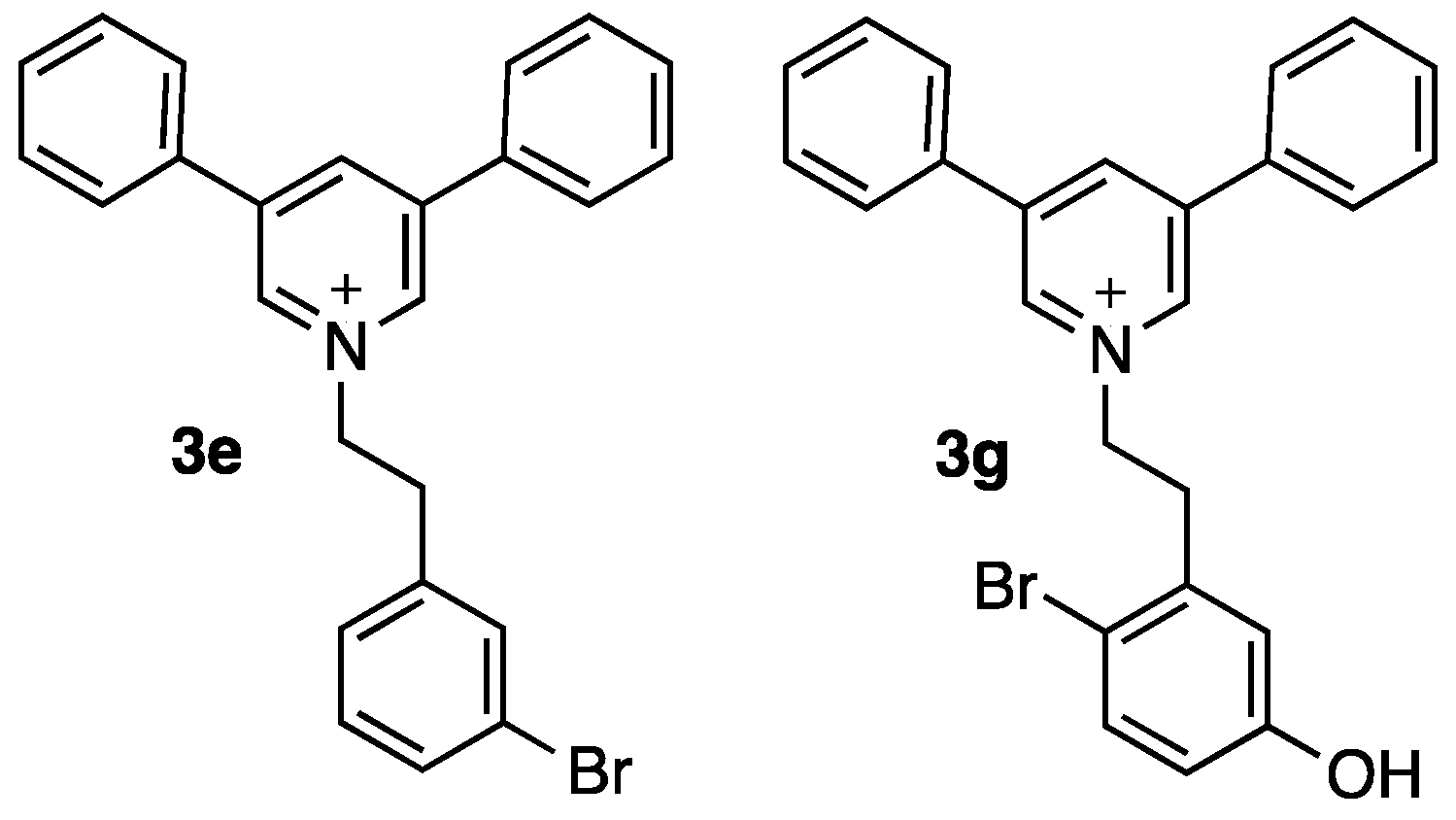
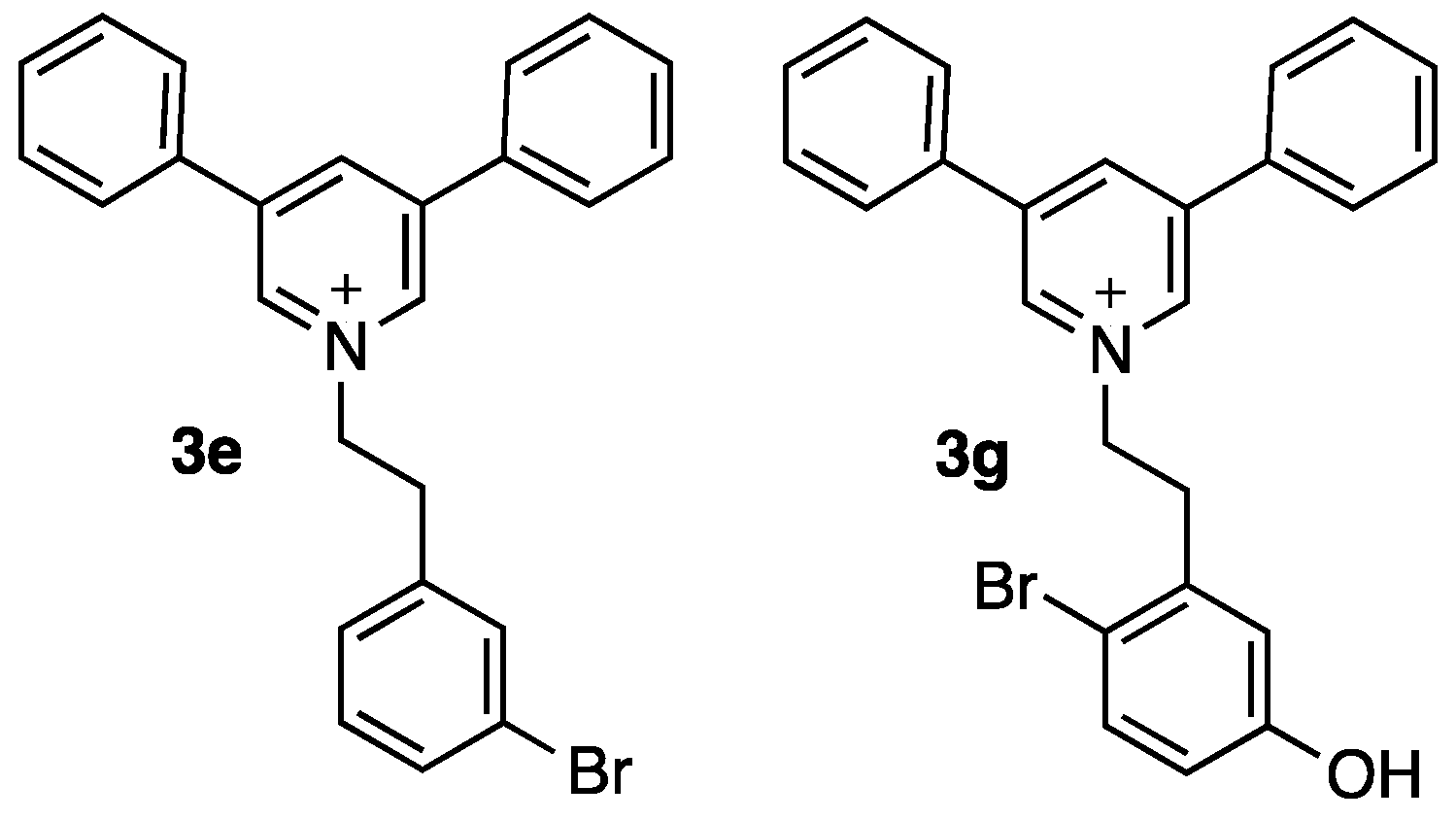
| 3e | 16 | 3o | 32 |
| 3f | 64 | 3q | 32 |
| 3g | 16 | 3r | n.d. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pesnot, T.; Gershater, M.C.; Edwards, M.; Ward, J.M.; Hailes, H.C. One–Pot Phosphate-Mediated Synthesis of Novel 1,3,5-Trisubstituted Pyridinium Salts: A New Family of S. aureus Inhibitors. Molecules 2017, 22, 626. https://doi.org/10.3390/molecules22040626
Pesnot T, Gershater MC, Edwards M, Ward JM, Hailes HC. One–Pot Phosphate-Mediated Synthesis of Novel 1,3,5-Trisubstituted Pyridinium Salts: A New Family of S. aureus Inhibitors. Molecules. 2017; 22(4):626. https://doi.org/10.3390/molecules22040626
Chicago/Turabian StylePesnot, Thomas, Markus C. Gershater, Martin Edwards, John M. Ward, and Helen C. Hailes. 2017. "One–Pot Phosphate-Mediated Synthesis of Novel 1,3,5-Trisubstituted Pyridinium Salts: A New Family of S. aureus Inhibitors" Molecules 22, no. 4: 626. https://doi.org/10.3390/molecules22040626