2. Results and Discussion
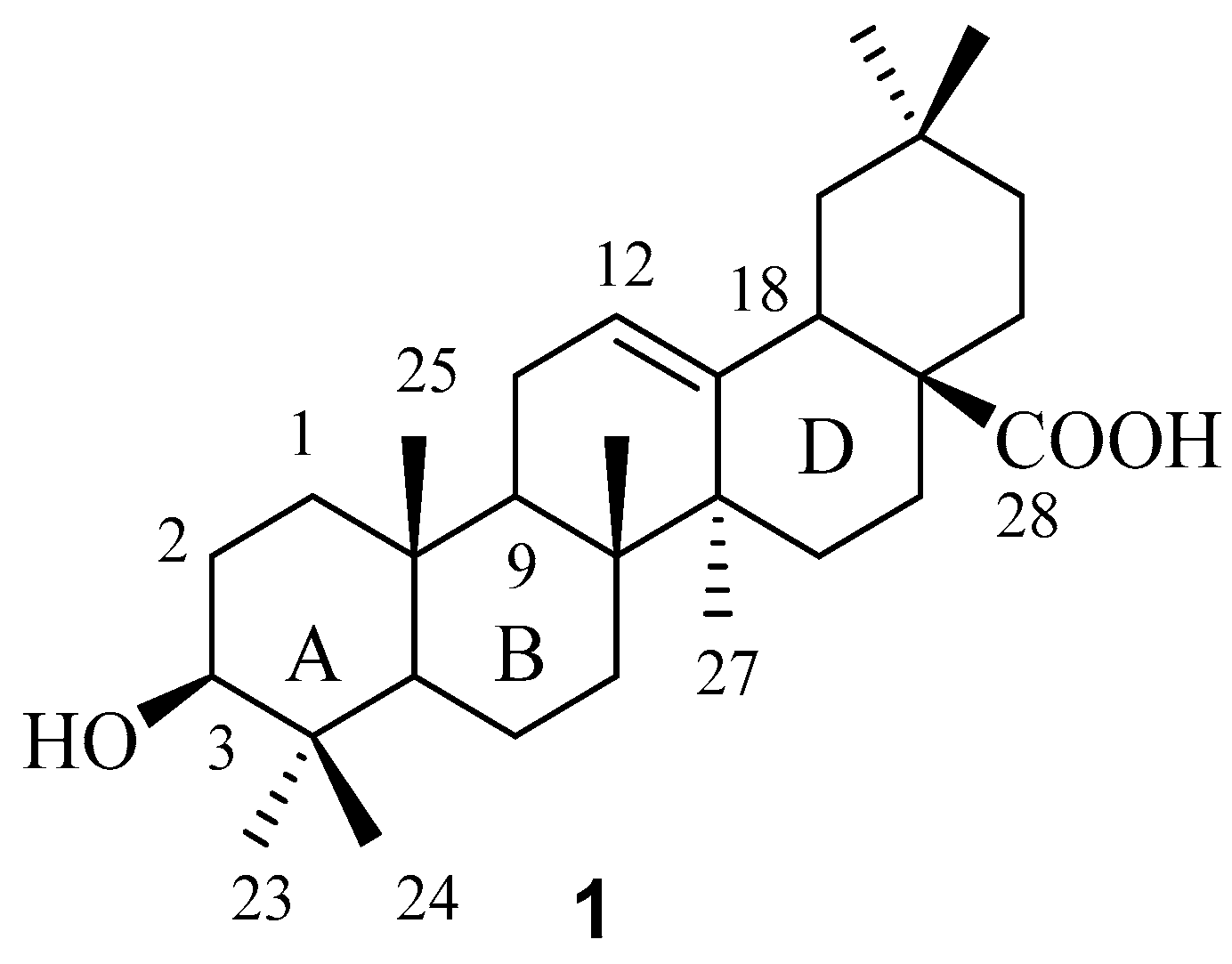
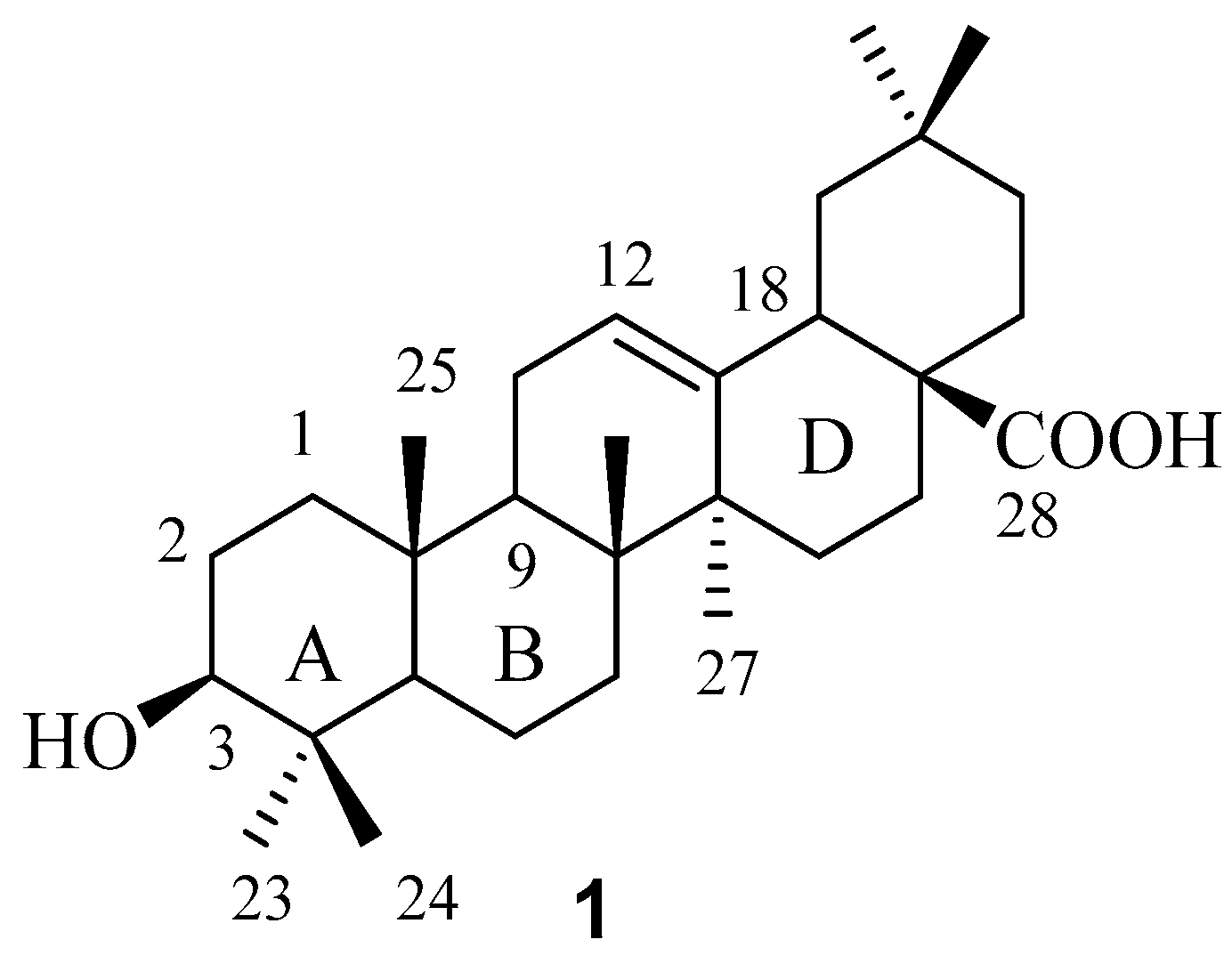
In order to maximize our chance of finding better FXR-interacting compounds during our exploration in the chemical space around the C-3 position in OA, we decided to introduce diverse substituents, among which a limited number of representative structures were selected with the aid of different filters, for this position.
To build up a small diverse OA 3-
O-ester library consisting of various carboxyl acids from a commercial compounds database, MDL
® Available Chemicals Directory (ACD) containing a total of 34,895 carboxylic acids was utilized. Non-compliant compounds according to Linpinski’s “Rule of 5” [
17] were first filtered out of the ~35 K acids in the library to minimize the total number of the building blocks by using the Discovery Studio 2.0 software (Accelrys Inc., San Diego, CA, USA). Considering the frequently observed violation of this empirical rule by natural products, and also with the intention of including the physiochemical properties of the parent compound
1 itself in the parameter settings, we modified this rule with our own customized criteria:
- (1)
For a balance between more choice of building blocks and drug-like properties, i.e., not too large a molecular weight (MW), we set the upper MW limit of the carboxylic acids to 200. In Lipinkski’s rule, the MW is set to below 500 for orally active compounds. Considering the MW of 1 itself is 456, using MW 500 for 3-O-esters of OA would significantly limit the choice of carboxylic acids.
- (2)
For other parameter settings, e.g., hydrogen bond donors (HBD) and hydrogen bond acceptors (HBA), the functional groups presenting already in OA (at C-3 and C-28) are also considered, so these parameters were set as follows: HBD equal or less than 4, HBA equal or less than 8, the number of rotatable bonds equal or less than 7, the number of rings equal or less than 2, the number of aromatic rings equal or less than 2.
After filtering the ~35 K acids using the above criteria, only 6106 compounds were left in the library. Next, the compounds with highly reactive or unstable groups, such as aminonitrile, thionitrile, epoxide, aziridine, disulfide, nitrogen oxides, azo compounds, β-lactams, etc., were also filtered out. In addition, the molecules which seemed not to be very “drug-like”, such as long-chain fatty acids, gem-dicarboxylic acids, compounds with boron, selenium or silicon atoms, ion exchange resins, quaternary ammonium salt, etc., as well as inorganic or organic salts of some carboxylic acids were also removed. After all the above filtering steps, 4454 compounds remained and were subjected to the diversity analysis.
A 2D fingerprint [
18] is a set of binary strings encoding the presence or absence of sub-structural fragments in a certain molecule, which can be used for the measurement of inter-molecular structural similarity and for the evaluation of structural and biological diversity of a compound library. We chose molecular fingerprint FCFP_4 as the descriptor and clustered all of the 4454 carboxylic acids to 285 clusters using Accelrys Discovery Studio 2.0. One acid was randomly selected from each cluster to assemble a sub-library of 285 acids. Those acids were used as the building blocks to create a 3-
O-ester library of OA, for the next step virtual screening by docking.
A total of 285 OA 3-
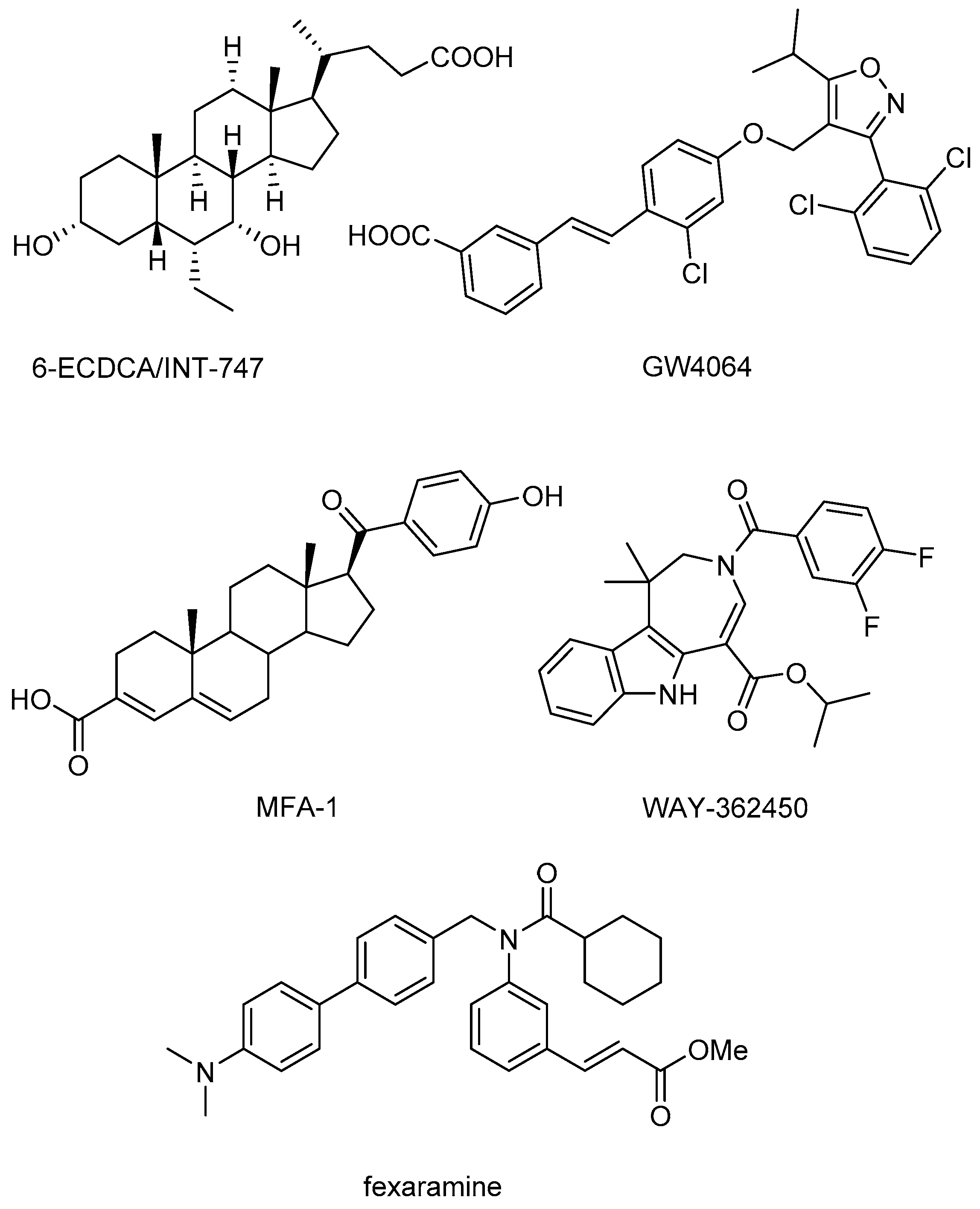
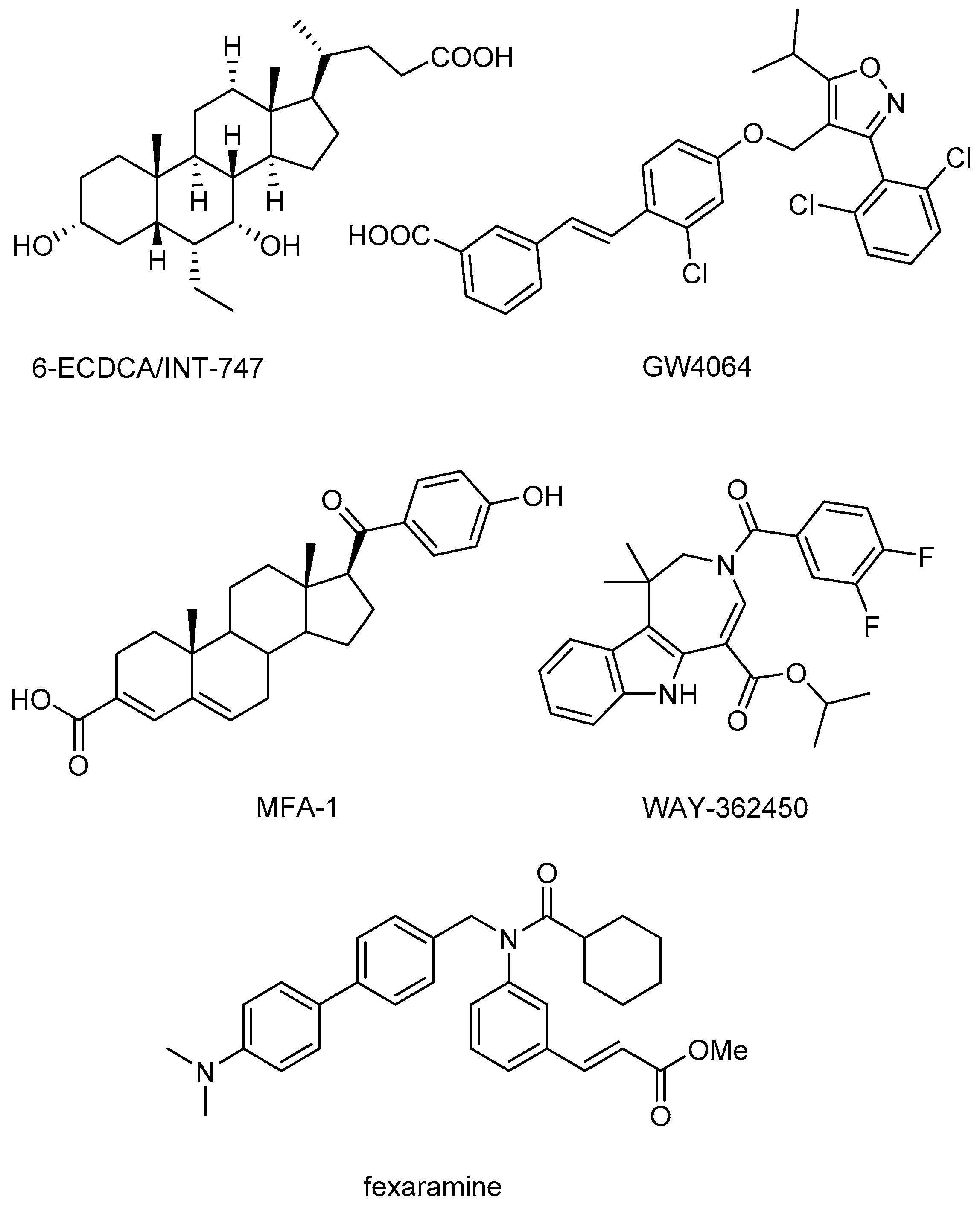
O-esters and one positive control molecule (fexaramine,
Figure 1) were screened against FXR (PDB code 1OSH) using Autodock 4.2 (The Scripps Research Institute, La Jolla, CA, USA). The positive control fexaramine scored the highest among those compounds with a p
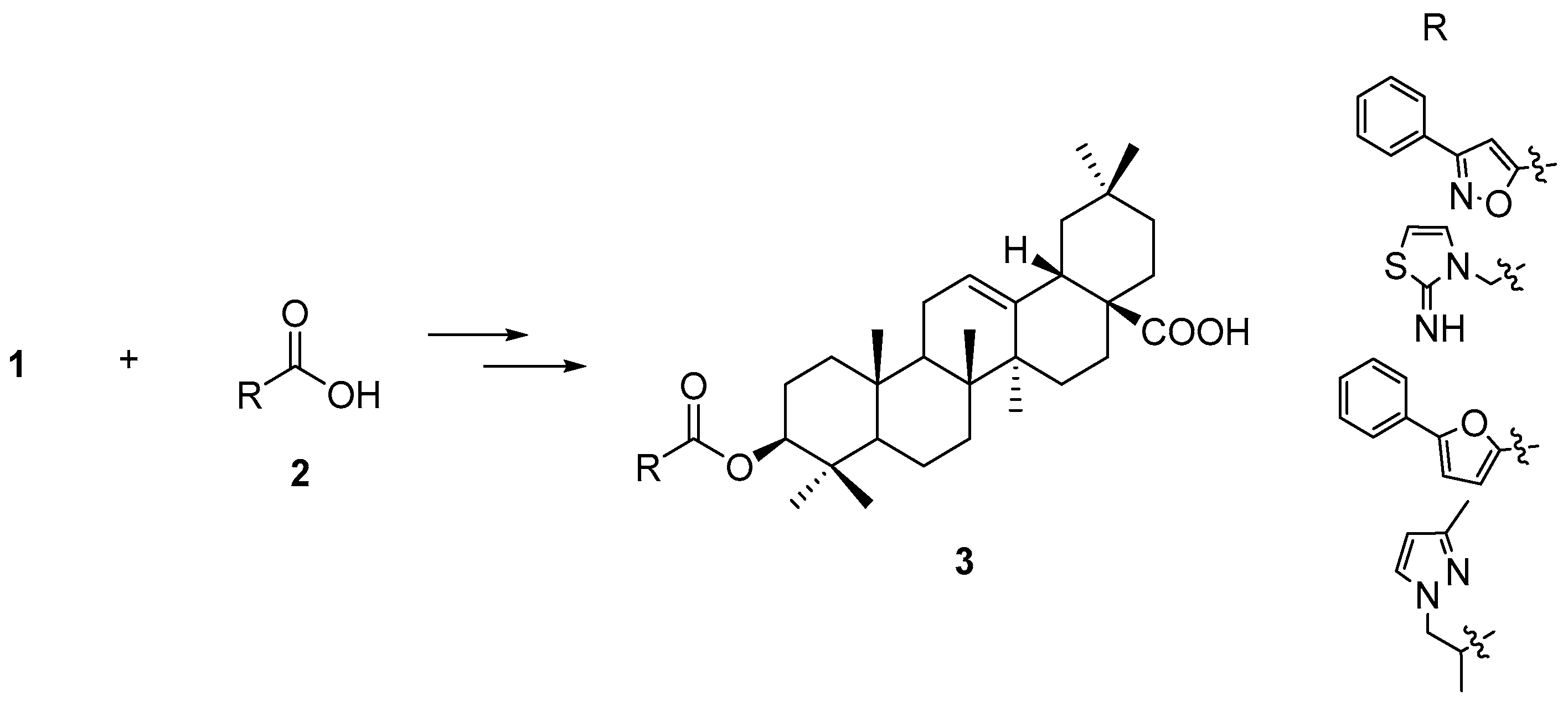
Ki of 10.39, and its binding mode is consistent with the experimental determination. Further examination of the positive control and other four top-scored compounds
3a–
d (
Figure 3) using the Autodock Vina software showed a similar scoring list and binding position in FXR compared with the docking results from Autodock 4.2. The best compound in the triterpenoid library has a p
Ki of 9.68 while the p
Ki values of the other three compounds ranged from 8.00 to 9.35 (
Table 1).
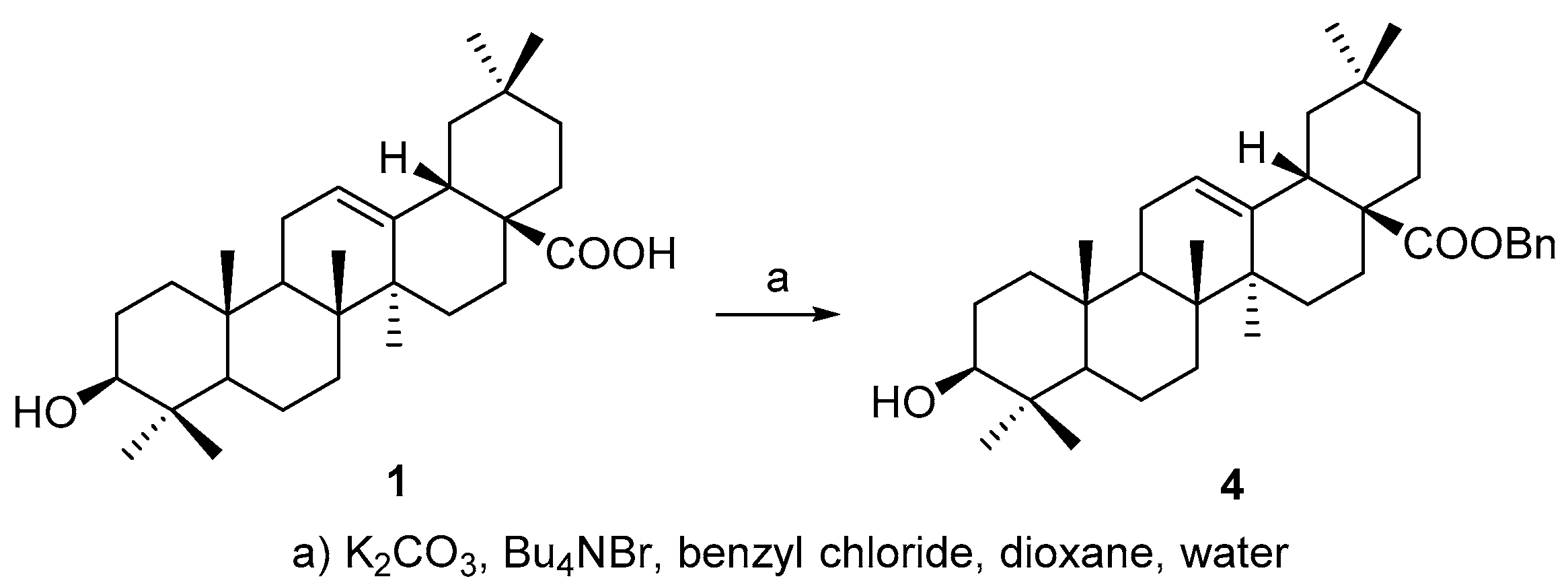
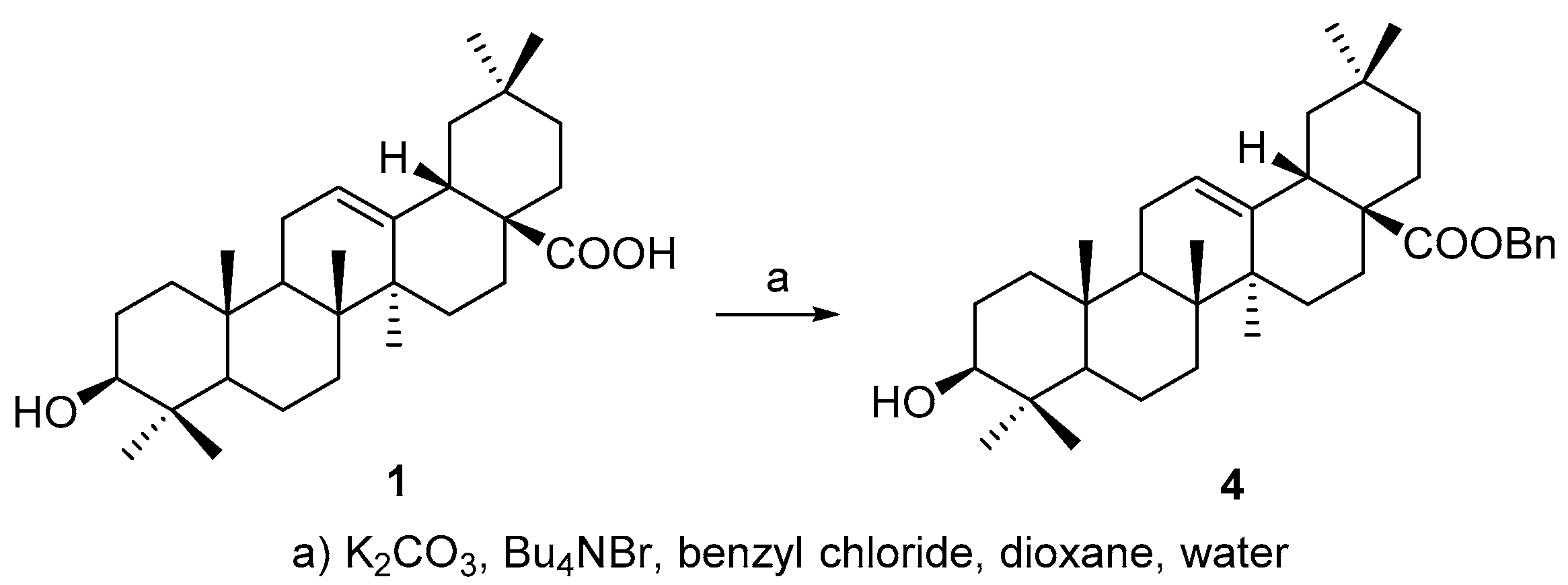
All above four triterpenoids were synthesized as below. The C-28 carboxyl group was protected before C-3 derivatization, according to a modified procedure of literature [
19], to afford benzyl 3β-hydroxyolean-28-oate (
4) (
Scheme 1) so as to improve the overall yield of the OA 3-
O-esters, based on our experience on triterpene chemistry.
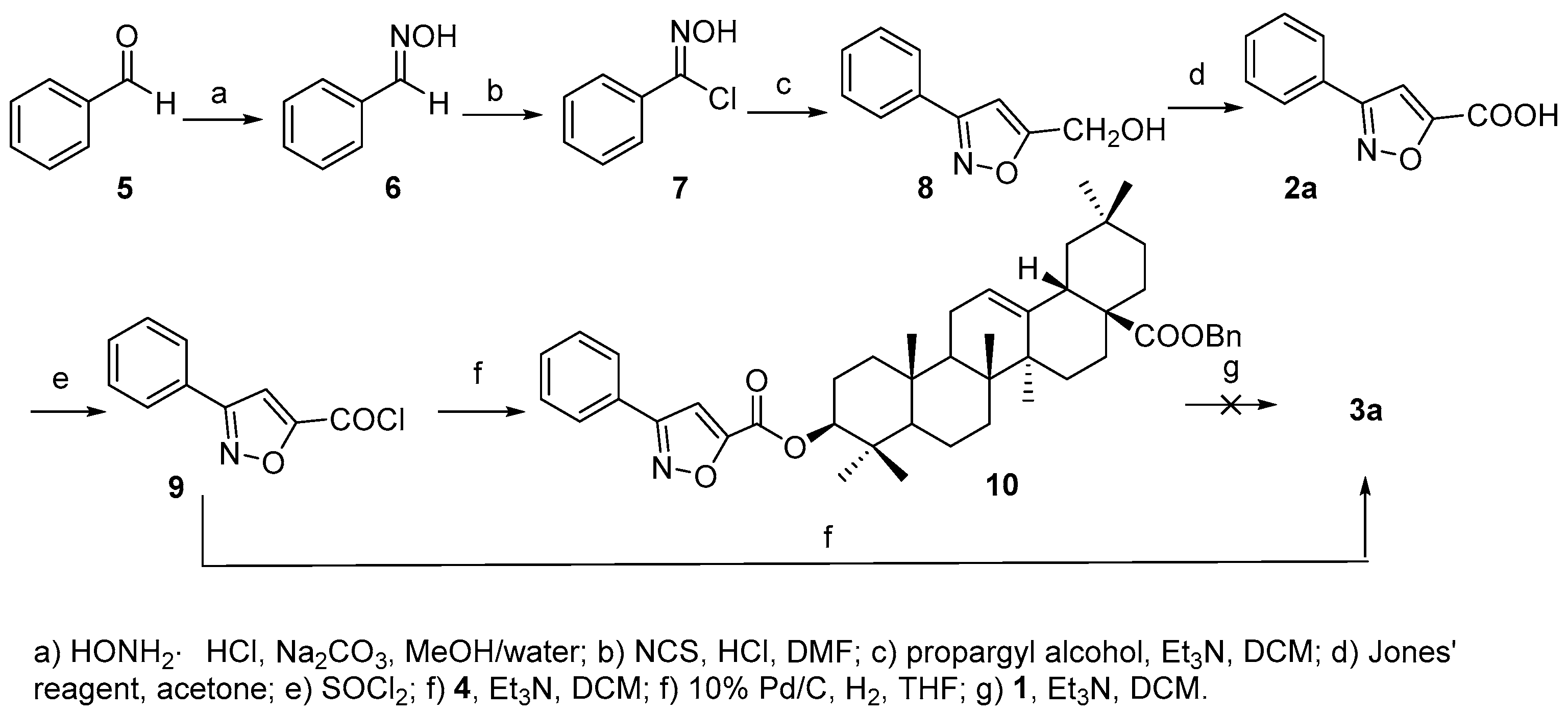
Compound
3a was prepared as shown in
Scheme 2. Acid
2a was first synthesized according to [
20]. Starting from benzaldehyde (
5), consequential transformations including a [3 + 2] cycloaddition of chlorobenzaldoxime (
7) and propargyl alcohol to afford phenyl substituted isoxazole acid
2a, which was further converted to acyl chloride
9 that reacted with
4 to furnish the 3-
O-acylated analogue
10. However, the debenzylation of
10 to afford the desired product
3a proved troublesome, maybe due to the ring-opening of the isoxazole group under hydrogenolysis conditions as reported [
21]. Thus,
3a was finally prepared in reasonable yield (63.0%) by direct acylation of
1 with
9.
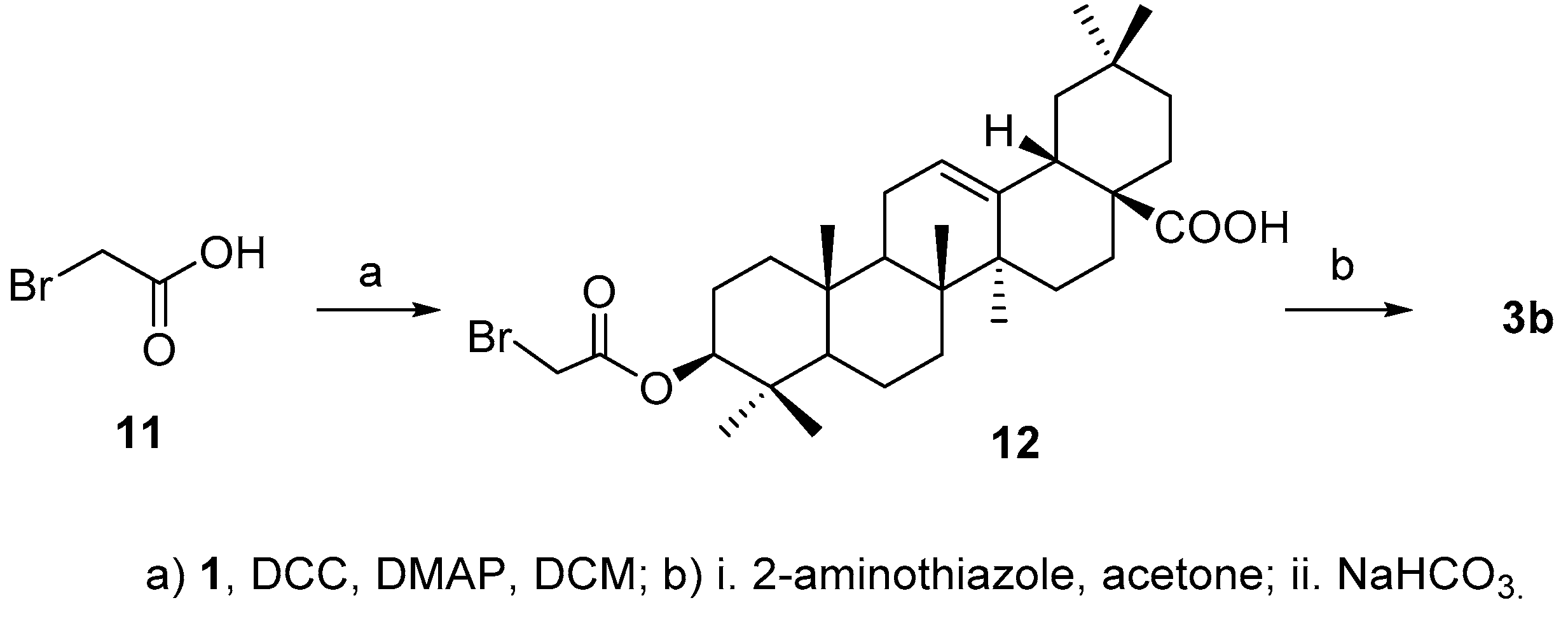
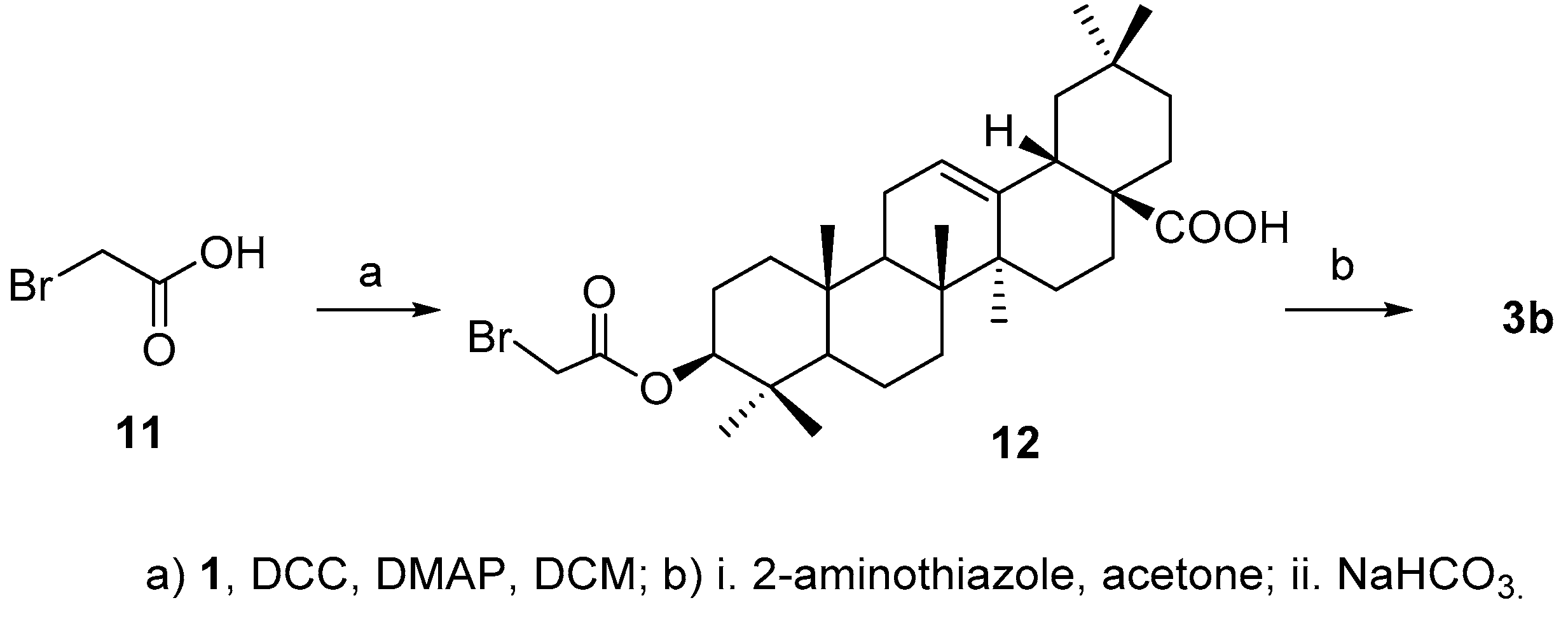
For the synthesis of
3b, acid
2b was initially prepared successfully by reacting 2-aminothiazole with bromoacetic acid (
11) [
22], but the aminothiazole acetic acid failed to attach to 3-OH in
4 either by coupling reaction in the presence of
N,
N′-dicyclohexylcarbodiimide (DCC) or
N-(3-dimethylaminopropyl)-
N′-ethylcarbodiimide (EDCI) in the presence of 4-dimethylaminopyridine (DMAP) or using the corresponding acyl chloride. Alternatively, bromoacetic acid
11 was coupled with
1 to afford
12 [
23], which was then treated with 2-aminothiazole to afford the desired product
3b (
Scheme 3) in 50.6% yield over two steps.
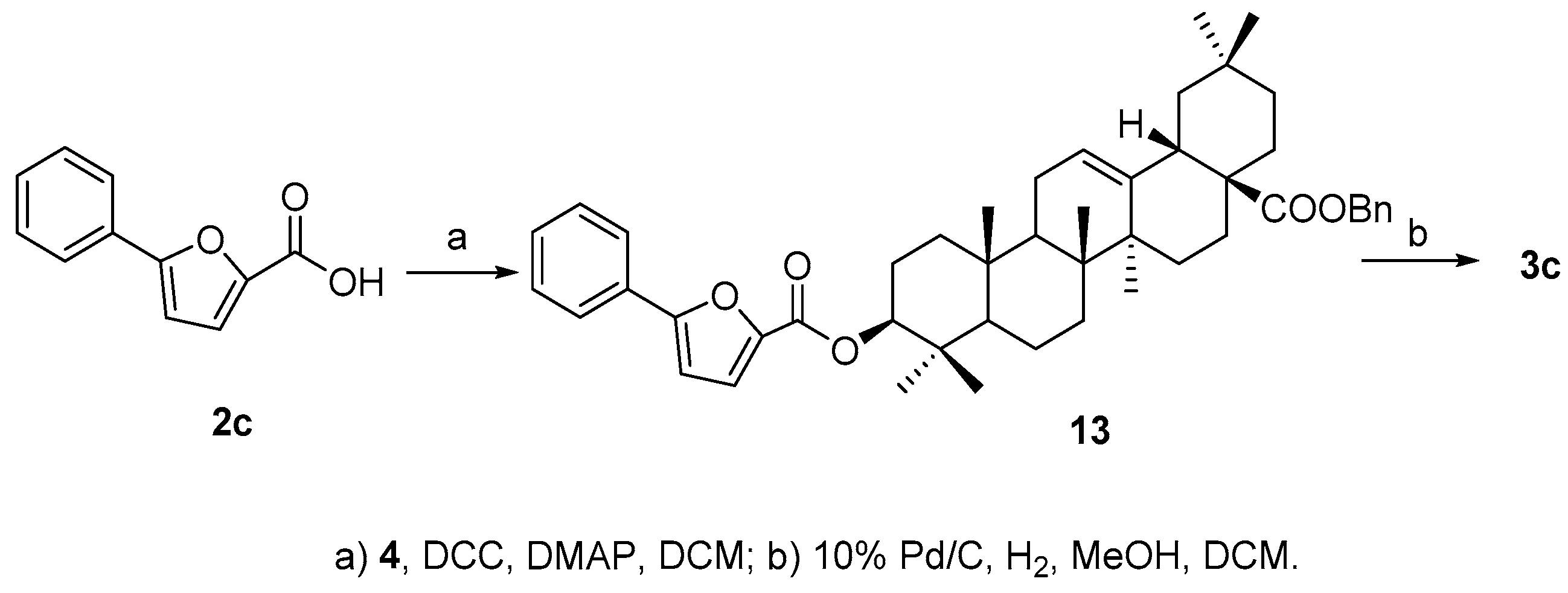
Compound
3c was much easier to synthesize since acid
2c is commercially available. Ester
13 was obtained by coupling of
2c and
4 in the presence of DCC and DMAP, and then the benzyl group was removed readily by catalytic hydrogenolysis to afford the final product
3c in good overall yield (89.6%) (
Scheme 4).
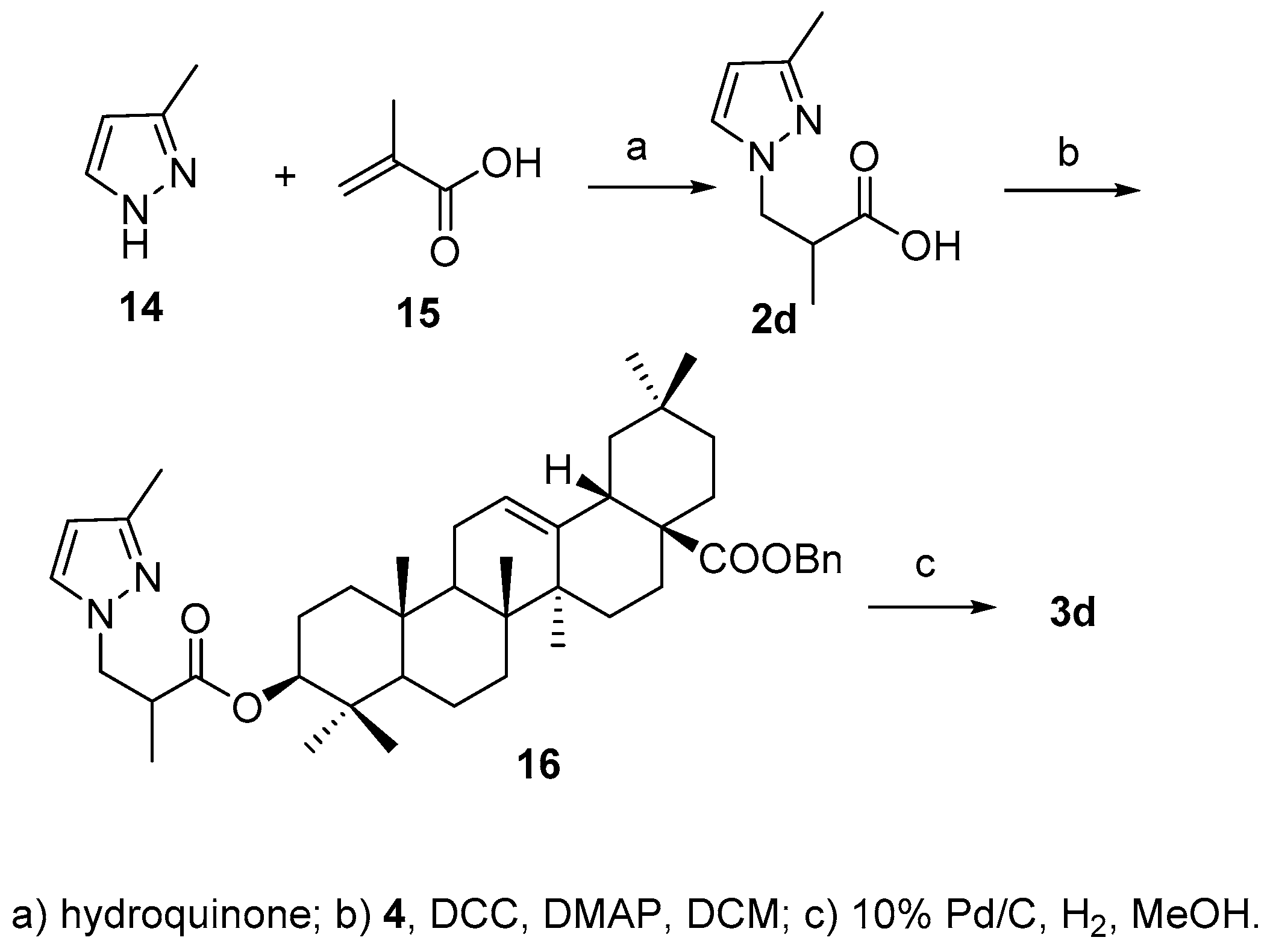
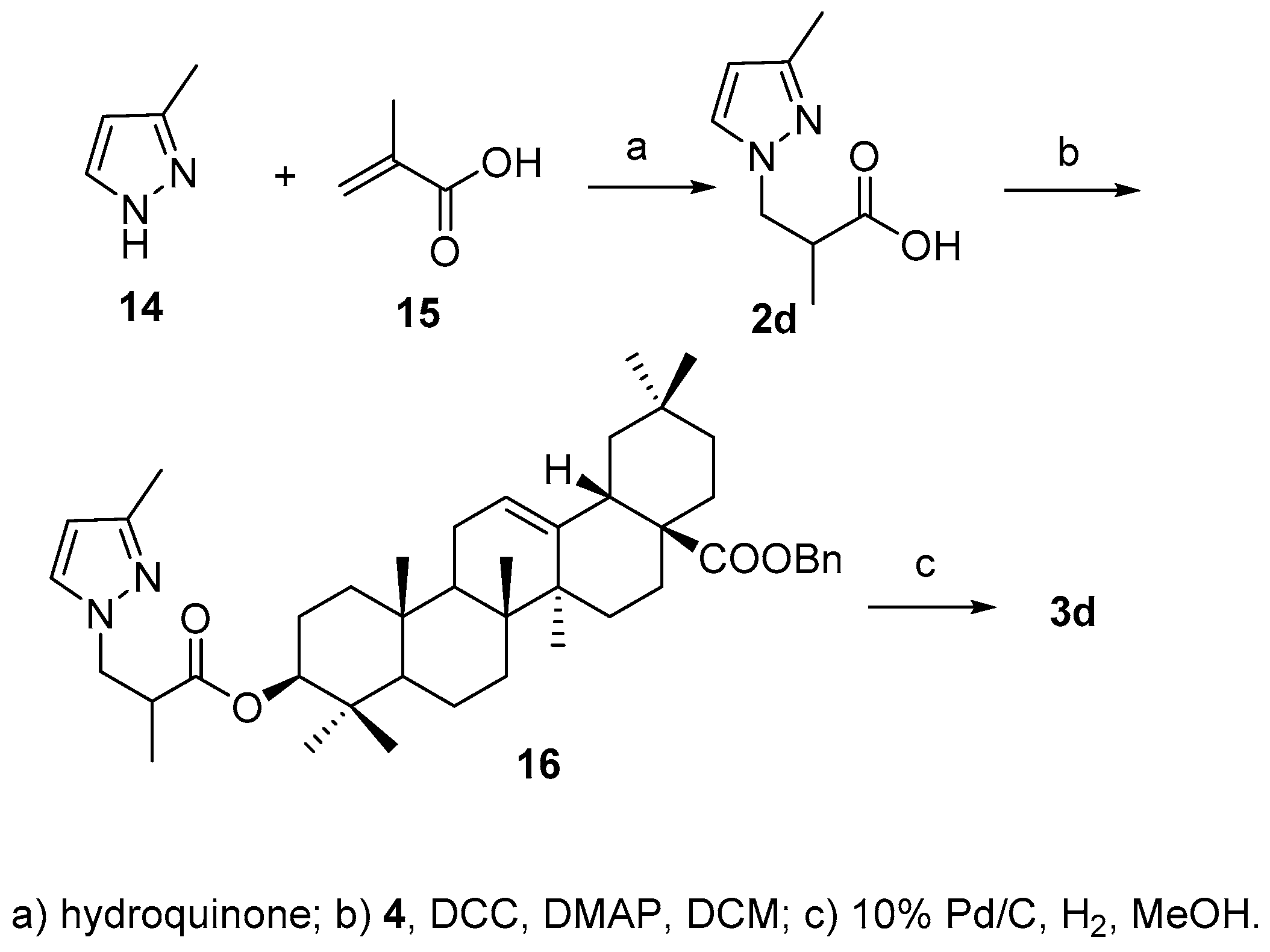
According to the literature method [
24], acid
2d was synthesized by Michael addition reaction between 3-methylpyridine (
14) and α-methylacrylic acid (
15) in the presence of hydroquinone in 81.0% yield., After attachment of acid to 3-OH and debenzylation of 28-COOH, the final product
3d was obtained in 41.6% yield over two steps (
Scheme 5).
The biological activities of these OA analogues against FXR were evaluated using a co-transfection assay in human embryonic kidney 293T cells. None of the compounds had any effect when incubated with the cells alone, but they could inhibit CDCA-induced FXR transactivation in a concentration-dependent manner. The IC
50 values of compounds
3a–
d were measured as 19.41, 7.03, 13.74, and 9.03 μM, respectively (
Table 1). It is worth noting that the trend of measured IC
50 values was consistent with the predicted p
Ki from the virtual screening, indicating the feasibility of this ligand-FXR model.
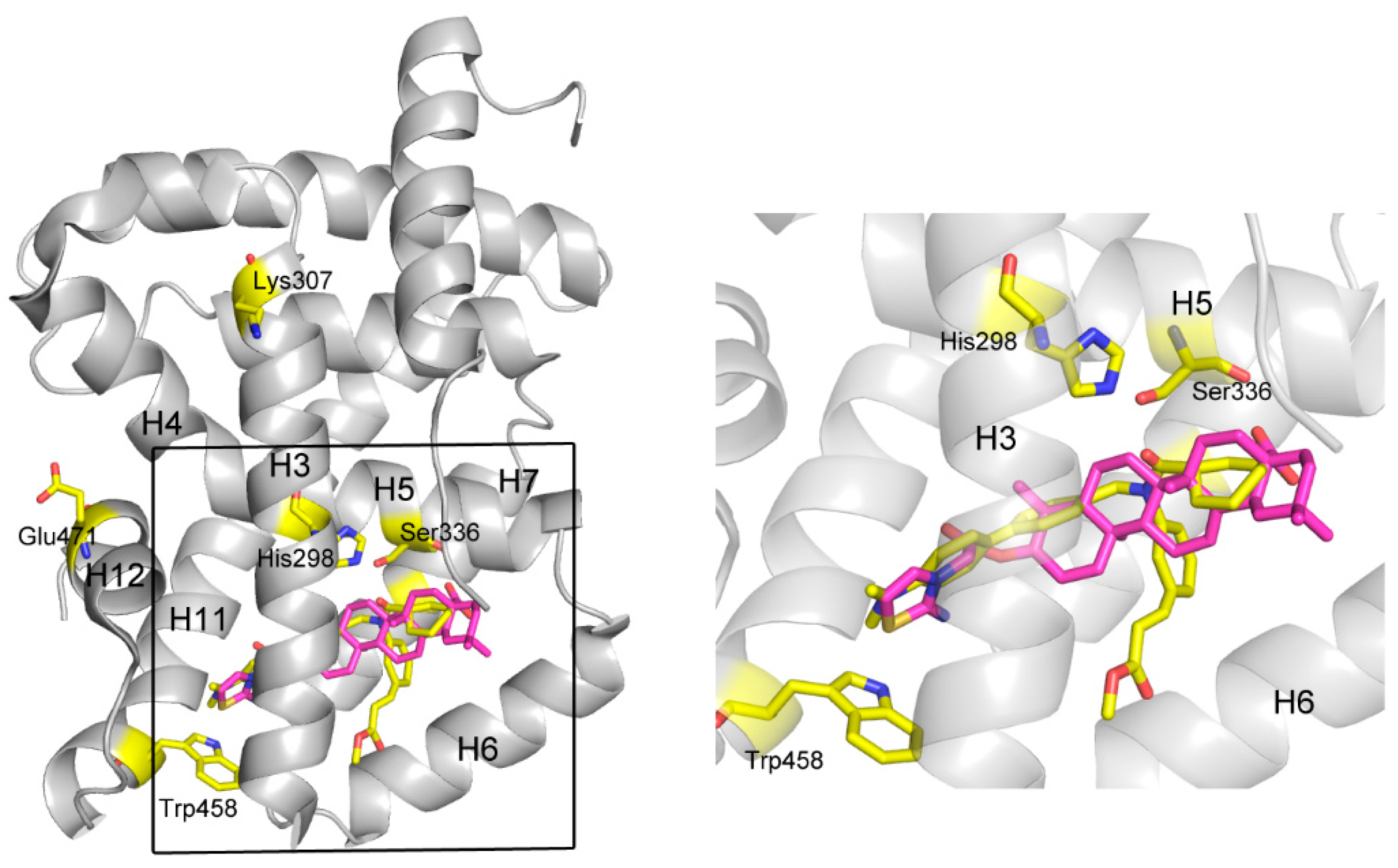
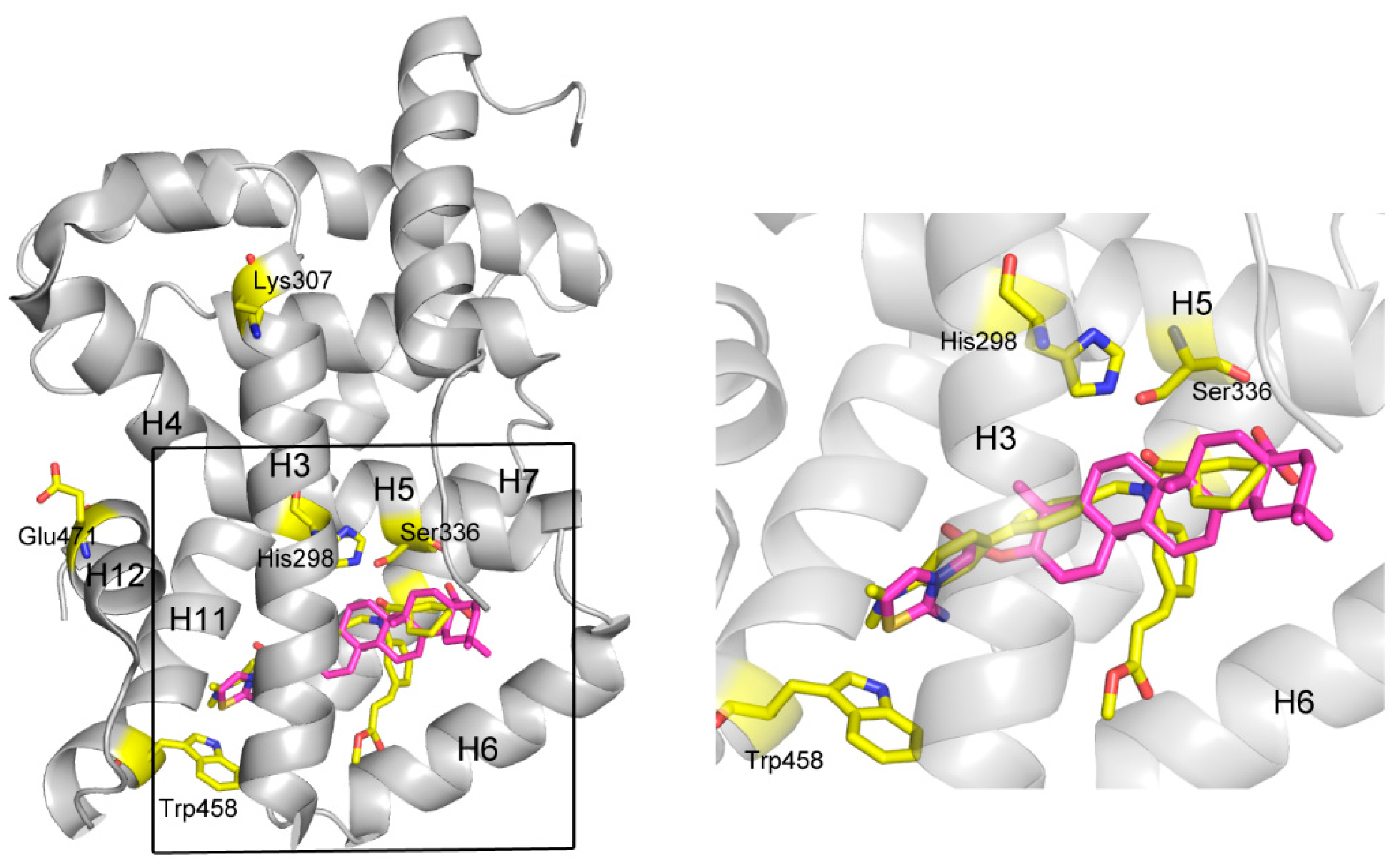
A further, more exhaustive docking was carried out to study the mechanism of action for these ligands by docking them into FXR (PDB code 1OSH) using Autodock 4.2. For
3b, it was shown that two pairs of T-shaped π-π interactions exist between the thiazoline ring and one aromatic residue (Trp458, H11,
Figure 4). This interaction seems important for anchoring the compound in the binding site, since other atoms of
3b contribute to interactions only by shape matching and hydrophobic interaction with H3 (H=helix), H5, H6, and H7. It is believed that H12 plays a crucial role in the activation of FXR by both natural or unnatural agonists. Agonists such as CDCA could activate FXR by directly stabilize H12 through forming two important H-bonds with the protein [
25] However, in some cases, not only H12 but also the other helices, especially H3, are necessary to enable coactivator recruitment [
26]. A so-called “charge clamp”, formed by a glutamic acid residue from H12 (Glu471 in FXR) and a lysine residue from H3 (Lys307 in FXR) in the coactivator-interacting surface, exists in many NRs and is able to stabilize their active conformations [
27]. In addition, there are a significant number of contacts between the methyl ester group in fexaramine and H3/H6 of FXR, which significantly contribute for its potency [
28]. In contrast, these interactions are absent in the model of
3b binding to FXR, although the binding positions of these two compounds are similar. Moreover, the amide CO of fexaramine formed two H-bonds with His298 in the middle section of H3 and with Ser336 in H5, respectively, strengthening the stabilizing effect of the ligand. Although there is a hydrophobic interaction between compound
3b and His298, it might not be strong enough to stabilize the overall conformation of H3, especially hold the remote key residue Lys307 (one of the component of the “charge clamp”) in the proper position, thus compromising the whole stabilizing effect of the compound. The binding mode of compound
3d was very similar with
3b, so that they have similar activities. The lack of some important interactions, such as the H-bonds with His298 and Ser336, and the hydrophobic interaction with H3/H6, may account for their inability to exert agonist activity, so that they exhibited only antagonistic effect.
In contrast, the binding poses for the other analogs are different. For OA, since the molecule is smaller than other derivatives, there is lack of some interactions with H5 and H6 in FXR (
Figure S1). However, an existing H-bond between C-28 COOH of OA and Tyr373 at H7 might compensate some lost interactions. For
3a (
Figure S1,
3c is similar), although there is an H-bond between this compound and Tyr373 in a subpocket, which is encompassed by H3, H11, and H12, the binding pose and position are quite different from those of the above mentioned trierpenoids and fexaramine. Thus, the binding affinities of compounds OA,
3a, and
3c, which are lower than the more active compounds
3b and
3d, made them less potent for the FXR modulation.
3. Materials and Methods
3.1. General Information
Oleanolic acid (purity > 98%) was purchased from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). All chemical reagents were of standard quality and used without further purification. Column chromatography was carried out with silica gel as the stationary phase. 1H- and 13C-NMR spectra were measured on Mercury-500 or Mercury-300 spectrometers (Varian, Palo Alto, CA, USA). The mass spectra (MS) were measured on an 1100 LC/MSD high performance ion trap mass spectrometer and an ESI mass spectrometer (LCQ) (Agilent, Santa Clara, CA, USA).
3.2. Synthesis of Compound 10
To a stirred solution of 4 (20 mg, 0.036 mmol) in dry dichloromethane (DCM) (0.5 mL) was added Et3N (15.6 μL, 0.108 mmol) and a solution of 9 (0.072 mmol) in dry DCM (0.5 mL) under Ar. The mixture was stirred for 3 h at room temperature, then DCM (10 mL) was added, and the organic layer was washed with 2 M HCl (aq.) (5 mL × 3), sat. NaHCO3 (aq.) (5 mL × 3) and sat. NaCl (aq.) (5 mL × 3) sequentially, dried over Na2SO4 and concentrated. The residue was purified through a silica chromatography column (petroleum ether−EtOAc 20:1) to afford the product 10 as a white solid (16 mg, 61.5%). 1H-NMR (300 MHz, CDCl3): δ 7.84 (2H, m, H-2, 6 of Ph), 7.48 (3H, m, H-3, 4, 5 of Ph), 7.35 (5H, m, Bn), 7.20 (1H, s, H of isoxazole), 5.30 (1H, br s, H-12), 5.08 (2H, ABq, J = 12.6 Hz, CH2 of Bn), 4.79 (1H, t, J = 8.1 Hz, H-3α), 2.92 (1H, d, J = 9.6 Hz, H-18), 1.14 (3H, s, CH3), 1.00 (3H, s, CH3), 0.96 (6H, s, 2CH3), 0.93 (3H, s, CH3), 0.91 (3H, s, CH3). ESI-MS: m/z [M + H]+ 718.7.
3.3. Synthesis of Compound 3a
To a stirred solution of 1a (25 mg, 0.055 mmol) in dry DCM (0.5 mL) was added Et3N (23.8 μL, 0.165 mmol) and a solution of 9 (0.11 mmol) in dry DCM (0.5 mL) under Ar. The mixture was stirred for 10 h at room temperature, then DCM (10 mL) was added, and the organic layer was washed with 2 M HCl (aq.) (5 mL × 3), sat. NaHCO3 (aq.) (5 mL × 3) and sat. NaCl (aq.) (5 mL × 3) sequentially, dried over Na2SO4 and concentrated. The residue was purified through a silica chromatography column (petroleum ether−EtOAc−HOAc 15:1:0.1) to afford the product (3a) as a white solid (19 mg, 63.0%). 1H-NMR (300 MHz, CDCl3): δ 7.84 (2H, m, H-2, 6 of Ph), 7.48 (3H, m, H-3, 4, 5 of Ph), 7.21 (1H, s, H of isoxazole), 5.30 (1H, brs, H-12), 4.80 (1H, t, H-3α), 2.83 (1H, d, J = 12 Hz, H-18), 1.16 (3H, s, CH3), 1.01 (3H, s, CH3), 1.00 (3H, s, CH3), 0.97 (3H, s, CH3), 0.94 (3H, s, CH3), 0.92 (3H, s, CH3), 0.79 (3H, s, CH3). 13C-NMR (125 MHz, CDCl3): δ 183.8, 163.0, 161.3, 156.7, 143.6, 130.5, 129.1 (2C), 128.1, 126.9 (2C), 122.5, 107.1, 83.6, 55.3, 47.6, 46.5, 45.8, 41.6, 40.9, 39.3, 38.1, 37.0, 33.8, 33.1, 32.5, 32.4, 30.7, 28.2, 27.7, 25.9, 23.6, 23.5, 23.4, 22.9, 18.2, 17.2, 16.8, 15.4. ESI-MS: m/z [M − H]− 626.4.
3.4. Synthesis of Compound 12
To a stirred solution of 1a (30 mg, 0.066 mmol) in DCM (1 mL) was added bromoacetic acid (18 mg, 0.13 mmol), DCC (54 mg, 0.26 mmol) and DMAP (0.8 mg, 0.0066 mmol). Then, the mixture was stirred at room temperature for 1 h. Then the precipitate was filtered and the filtrate was concentrated. The residue was purified through a silica chromatography column (petroleum ether−EtOAc 20:1) to afford the product (12) as a white solid (35 mg, 92.0%). 1H-NMR (300 MHz, CDCl3): δ 5.27(1H, brs, H-12), 4.55 (1H, t, J = 8.1 Hz, H-3α), 3.82 (2H, ABq, J = 12.3 Hz, CH2), 2.81 (1H, dd, H-18), 1.18 (3H, s, CH3), 0.94 (3H, s, CH3), 0.92(3H, s, CH3), 0.90 (6H, br s, 2CH3), 0.88 (3H, s, CH3), 0.79 (3H, s, CH3). ESI-MS: m/z [M + H]− 577.4.
3.5. Synthesis of Compound 3b
To a stirred solution of 12 (35 mg, 0.06 mmol) in acetone (1.5 mL) was added 2-aminothiazole (18 mg, 0.18 mmol). The mixture was stirred at 50 °C for 20 h, then DCM (20 mL) and NaHCO3 (aq.) (10 mL) were added. And the mixture was stirred for another 30 min at room temperature. Then the aqueous layer was extracted with DCM (5 mL × 3). The organic layer was collected, dried over Na2SO4 and concentrated. The residue was purified through a silica chromatography column (CHCl3−MeOH−Et3N 25:1:0.2) to afford the product (3b) as a white solid (20 mg, 55.0%). 1H-NMR (500 MHz, C5D5N): δ 6.25 (1H, d, J = 5.0 Hz, H of thiazolimine), 5.96 (1H, d, J = 5.0 Hz, H of thiazolimine), 5.44 (1H, br s, H-12), 4.83 (2H, q, J = 10.2 Hz, CH2), 4.75 (1H, dd, J = 5.0, 12.0 Hz, H-3α), 3.33 (1H, d, J = 10.5 Hz, H-18), 1.24 (3H, s, CH3), 0.99 (3H, s, CH3), 0.94 (9H, br s, 3CH3), 0.85 (3H, s, CH3), 0.77 (3H, s, CH3). 13C-NMR (125 MHz, C5D5N): δ 180.3, 168.9, 163.0, 144.9, 128.3, 122.3, 97.4, 82.1, 55.4, 47.8, 47.3, 46.7, 46.5, 42.1, 42.0, 39.7, 38.1, 38.0, 37.1, 34.2, 33.3, 33.2, 33.0, 31.0, 28.3, 28.2, 26.2, 23.8, 23.7, 23.7, 18.4, 17.4, 16.9, 15.3. ESI-MS: m/z [M − H]− 595.5.
3.6. Synthesis of Compound 13
To a stirred solution of 4 (40 mg, 0.07 mmol) in DCM (1 mL) was added 2c (55 mg, 0.29 mmol), EDCI (70 mg, 0.37 mmol) and DMAP (5 mg, 0.04 mmol). The mixture was stirred at 50 °C for 4 h. Then the reaction solution was concentrated. The residue was purified through a silica chromatography column (petroleum ether−EtOAc 20:1) to afford the product (13) as white solid (41 mg, 78.0%). 1H-NMR (300 MHz, CDCl3): δ 7.78 (2H, d, J = 7.5 Hz, H-2, 6 of Ph) 7.42 (3H, m, H-3, 4, 5 of Ph), 7.34 (5H, s, Bn), 7.10 (1H, d, J = 1.8 Hz, H of furan), 6.73 (1H, d, J = 1.8 Hz, H of furan), 5.30 (1H, br s, H-12), 5.08 (2H, ABq, J = 12.0 Hz, CH2 of Bn), 4.72 (1H, t, J = 8.7 Hz, H-3α), 2.91 (1H, d, J = 12.9 Hz, H-18), 1.14 (3H, s, CH3), 0.99 (3H, s, CH3), 0.95 (6H, br s, 2CH3), 0.92 (3H, s, CH3), 0.90 (3H, s, CH3), 0.63 (3H, s, CH3). ESI-MS: m/z [M + Na]+ 739.6.
3.7. Synthesis of Compound 3c
To a stirred solution of 13 (41 mg, 0.057 mmol) in a mixed solvent of MeOH (0.4 mL) and DCM (0.1 mL) was added 10% Pd/C (4 mg) and H2. The mixture was stirred for 20 h at room temperature, then filtered and the filtrate was concentrated. The residue was purified through a silica chromatography column (petroleum ether−acetone 15:1) to afford the product (3c) as a white solid (29 mg, 89.6% based on 4 mg recovery of substrate). 1H-NMR (300 MHz, CDCl3): δ 7.78 (2H, d, J = 7.5 Hz, H-2, 6 of Ph), 7.41 (2H, t, H-3, 5 of Ph), 7.33 (1H, t, H-4 of Ph),7.20 (1H, d, J = 3.3 Hz, H of furan), 6.73 (1H, d, J = 3.6 Hz, H of furan), 5.29 (1H, br s, H-12), 4.74 (1H, t, H-3α), 2.83 (1H, d, J = 10.5 Hz, H-18), 1.16 (3H, s, CH3), 1.00 (6H, br s, 2CH3), 0.96 (3H, s, CH3), 0.94 (3H, s, CH3), 0.92 (3H, s, CH3), 0.78 (3H, s, CH3). 13C-NMR (75 MHz, CDCl3): δ 184.1, 158.7, 157.4, 144.1, 143.6, 129.6, 128.8 (3C), 124.8 (2C), 122.6, 119.4, 106.7, 81.5, 55.3, 47.5, 46.5, 45.8, 41.6, 40.9, 39.3, 38.1, 37.0, 33.8, 33.1, 32.5, 32.4, 30.7, 28.2, 27.7, 25.9, 23.6, 23.4, 22.9, 18.2, 17.2, 16.8, 15.4. ESI-MS: m/z [M − H]− 625.5.
3.8. Synthesis of Compound 16
To a stirred solution of 4 (10 mg, 0.018 mmol) in DCM (0.4 mL) was added 2d (6 mg, 0.036 mmol), DCC (14.8 mg, 0.072 mmol) and DMAP (0.2 mg, 0.0018 mmol). The mixture was stirred for 2 h at room temperature. Then the precipitate was filtered and the filtrate was concentrated. The residue was purified through a silica chromatography column (petroleum ether−EtOAc 20:1) to afford the product (16) as a white solid (8 mg, 63.0%). 1H-NMR (300 MHz, CDCl3): δ 7.29 (5H, s, Bn), 7.24 (1H, s, H-4 of pyrazole), 5.96 (1H, s, H-5 of pyrazole), 5.28 (1H, br s, H-12), 5.12–5.02 (2H, ABq, J = 12.6 Hz, CH2 of Bn), 4.48 (1H, t, J = 8.1 Hz, H-3α), 4.36 (1H, dd, -N-CH2-CH-, J = 6.9, 13.5 Hz), 4.03 (1H, m, -N-CH2-CH-), 3.08 (1H, m, -CH2-CH-COOH), 2.90 (1H, d, J = 9.6 Hz, H-18), 2.25 (3H, s, CH3 of pyrazole), 1.16 (3H, d, CH3-CH-COOH), 1.12 (3H, s, CH3), 0.91 (3H, s, CH3), 0.89 (6H, br s, 2CH3), 0.82 (6H, br s, 2CH3), 0.60 (3H, s, CH3). ESI-MS: m/z [M + H]+ 697.8.
3.9. Synthesis of Compound 3d
To a stirred solution of 16 (8 mg, 0.011 mmol) in MeOH (0.5 mL) was added 10% Pd/C (1 mg) and H2. The mixture was stirred for 2 h at room temperature. After another portion of 10% Pd/C (4 mg) was added, the reaction was maintained for another 12 h, then filtered and the filtrate was concentrated. The residue was purified through a silica chromatography column (petroleum ether−acetone 15:1) to afford the product (3d) as a white solid (4 mg, 66.0%). 1H-NMR (300 MHz, CDCl3): δ 7.24 (1H, s, H-4 of pyrazole), 5.95 (1H, s, H-5 of pyrazole), 5.27 (1H, br s, H-12), 4.48 (1H, m, H-3α), 4.36 (1H, m, -N-CH2-C-), 4.04 (1H, m, -N-CH2-C-), 3.06 (1H, m, -CH2-CH-COOH), 2.82 (1H, d, J = 10.2 Hz, H-18), 2.25 (3H, s, CH3 of pyrazole), 1.16 (3H, d, CH3-CH-COOH), 1.13 (3H, s, CH3), 0.93 (6H, s, 2CH3), 0.90 (3H, s, CH3), 0.84 (3H, s, CH3), 0.82 (3H, s, CH3), 0.74 (3H, s, CH3). 13C-NMR (125 MHz, CDCl3): δ 182.9, 174.2, 148.8, 128.9, 126.8, 122.5, 104.8, 81.2, 55.3, 47.5, 46.5, 45.8, 41.6, 41.0, 39.3, 38.0, 37.0, 33.8, 33.0, 32.5, 32.4, 30.7, 28.0, 27.8, 27.7, 25.9, 23.6, 23.4, 22.9, 18.1, 17.1, 15.3, 15.1. ESI-MS: m/z [M − H]− 605.8.
3.10. Assembling the Database of Carboxylic Acids
A database of all the commercial available carboxylic acids was extracted from the MDL® ACD package as an .sdf file. The file was input into Accelrys Discovery Studio 2.0 (Accelrys Inc., San Diego, CA, USA) to calculate the molecular properties, such as Molecular Weight, H-bond Acceptor, H-bond Donor, etc. The output file was opened in Discovery Studio Visualizer, and the undesired molecules were filtered out manually from the file according to the criterions, which were demonstrated in the context.
3.11. Evaluating the Diversity of the Carboxylic Acids Database
The database file generated in the last step was input into Accelrys Discovery Studio 2.0 (Accelrys Inc., San Diego, CA, USA) to be clustered into 285 classes using a 2D fingerprint descriptor FCFP_4. One molecule was randomly selected from each cluster and put into an .sdf file to consist of a sub-library consisting of 285 members.
3.12. Docking
Autodock 4.2 was employed to screen a library of OA-containing structurally diverse 3-
O-esters [
29]. The crystal structure of FXR in complex with its agonist fexaramine (PDB code 1OSH) was used as the receptor. As a positive control, fexaramine was first docked against the FXR structure to validate the docking method. Docking was performed with a 60 × 60 × 60 grid covering the entire ligand binding site and 10 runs per ligand were done using the Lamarckian Genetic Algorithm (LGA) method. Other parameters were set as defaults. Top five compounds (including positive control) from the above screening were selected and further examined using Autodock Vina with default parameters [
30]. To obtain more precise docking poses, a second run of Autodock was performed on these selected compounds and the parameter of LGA runs was set to 100. Docking figures were prepared using the PyMOL program (DeLano Scientific LLC: Palo Alto, CA, USA).
3.13. Cell Culture
293T cells were maintained in MEM media with 10% fetal bovine serum (FBS), 100 U/mL penicillin and streptomycin and 1 mM non-essential amino acid under humidified air containing 5% CO2 at 37 °C.
3.14. Transient Transfection and Luciferase Reporter Assay
293T cells were plated in 96-well plates at 2.5 × 104 per well. Five ng/well pCMV-GAL4-DBD-hFXR-LBD expression vector, 25 ng/well pFRLuciferase and 5 ng/well Renilla-Luciferase reporter plasmids were transiently transfected into cells using 0.25 μL/well lipofectamine (Invitrogen, Carlsbad, CA, USA). Eighteen hours after transfection, cells were treated with CDCA at 25 μM in fresh MEM containing 0.5% charcoal-stripped FBS. Chemical derivatives dissolved in dimethyl sulfoxide (DMSO) were additionally added in concentrations indicated. Luciferase activities were measured after an additional 24 h using Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA). Relative activity was defined as pFRLuciferase activity/Renilla-Luciferase activity. The relative activity of FXR in the presence of 25 μM of CDCA was set as 100%.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}