
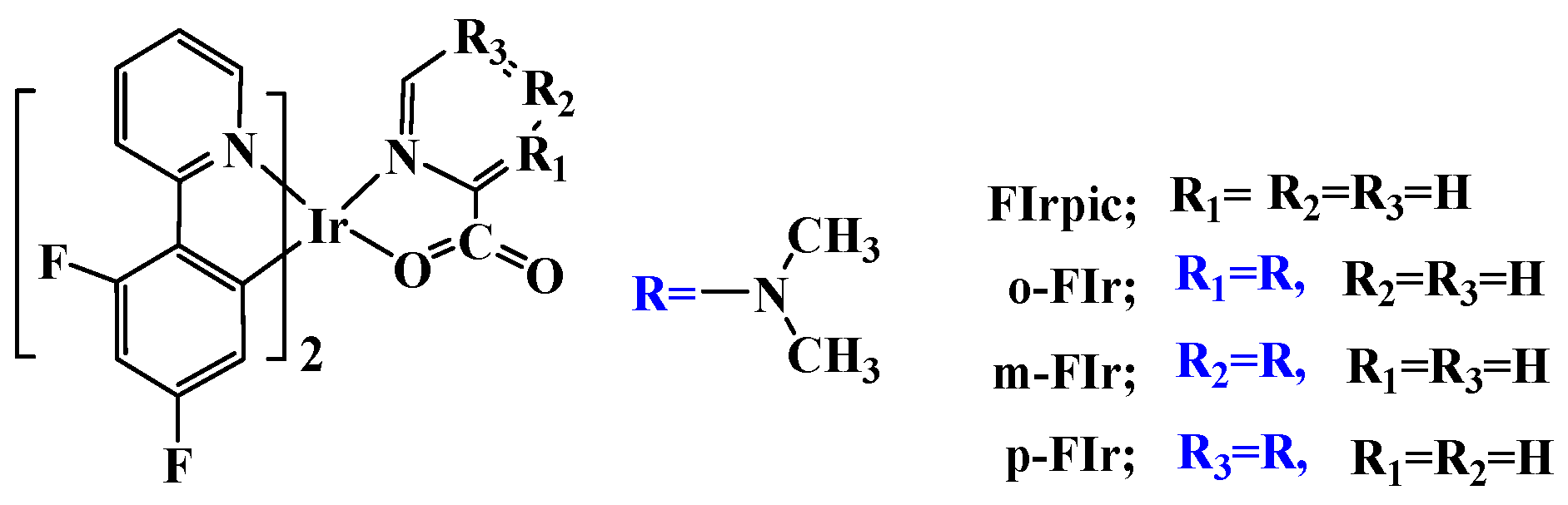
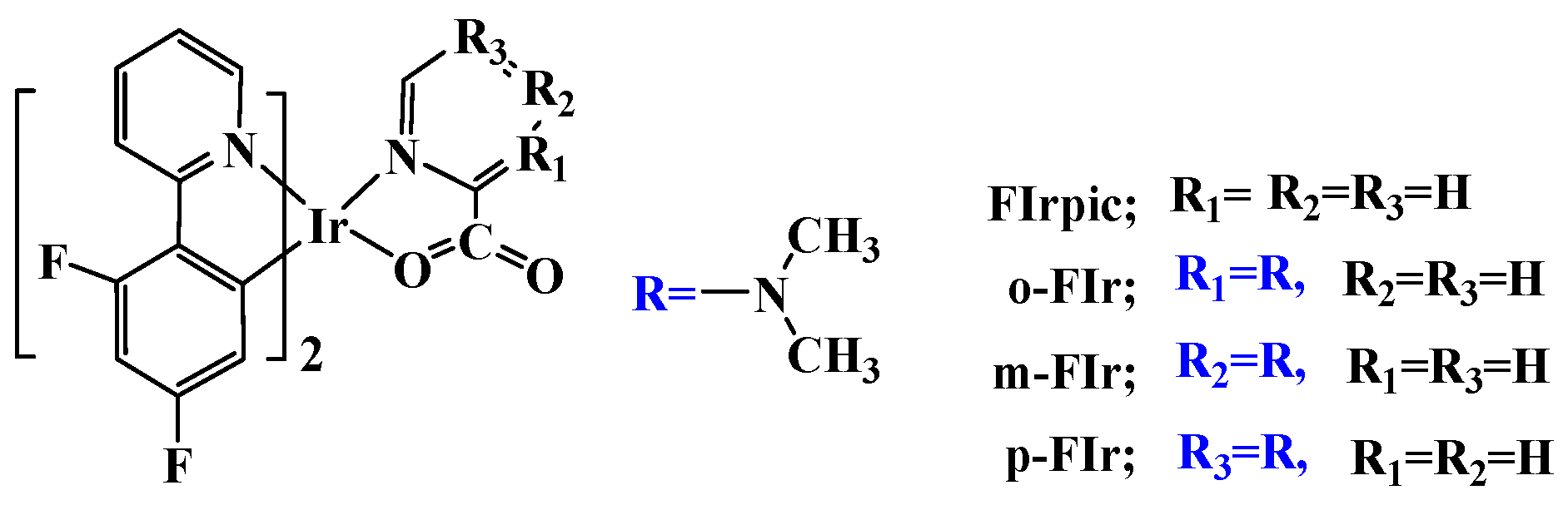
Tuning the Geometrical Structures and Optical Properties of Blue-Emitting Iridium(III) Complexes through Dimethylamine Substitutions: A Theoretical Study

Abstract
:1. Introduction
2. Results and Discussion
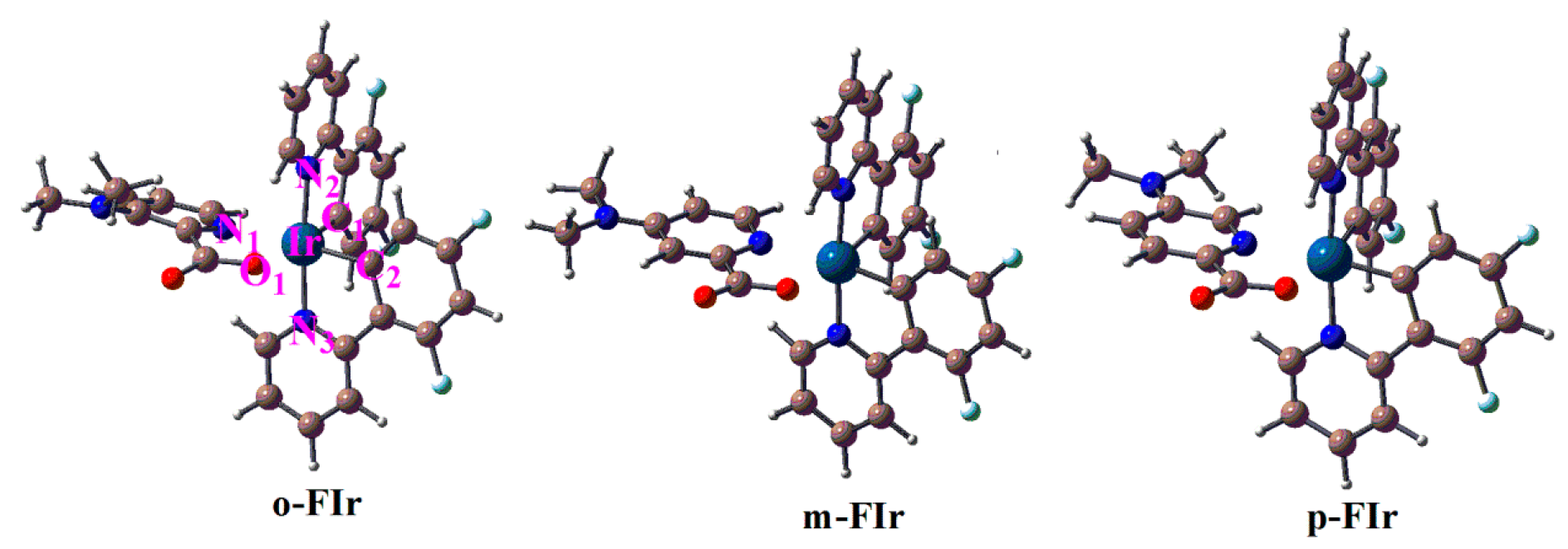
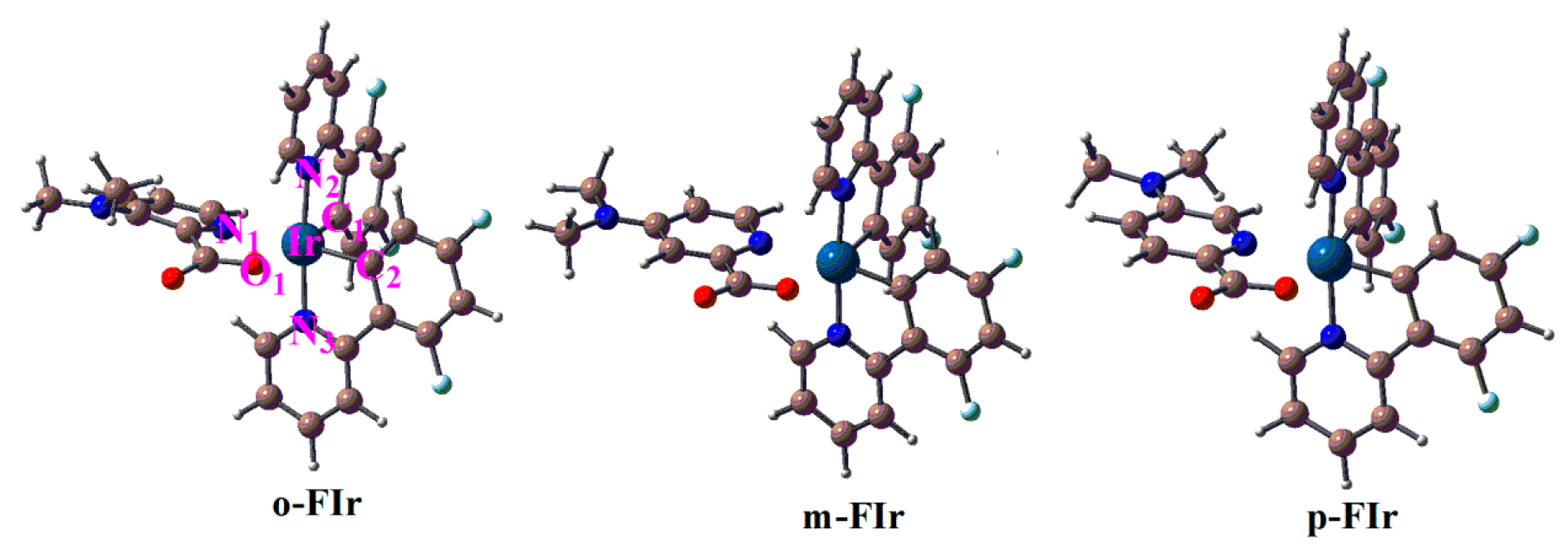
2.1. The Optimized Geometries in the Ground and Lowest Lying Triplet Excited States
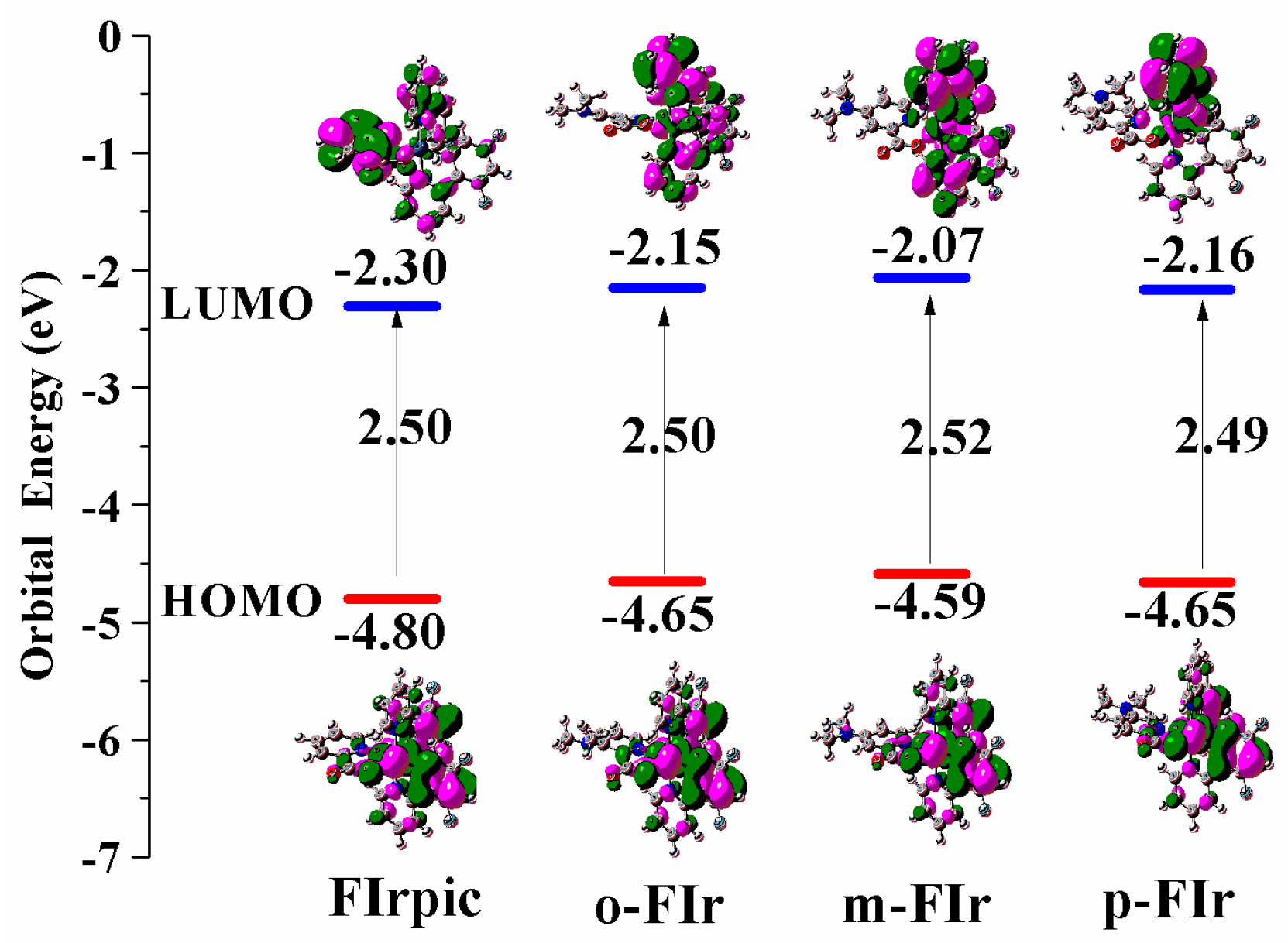
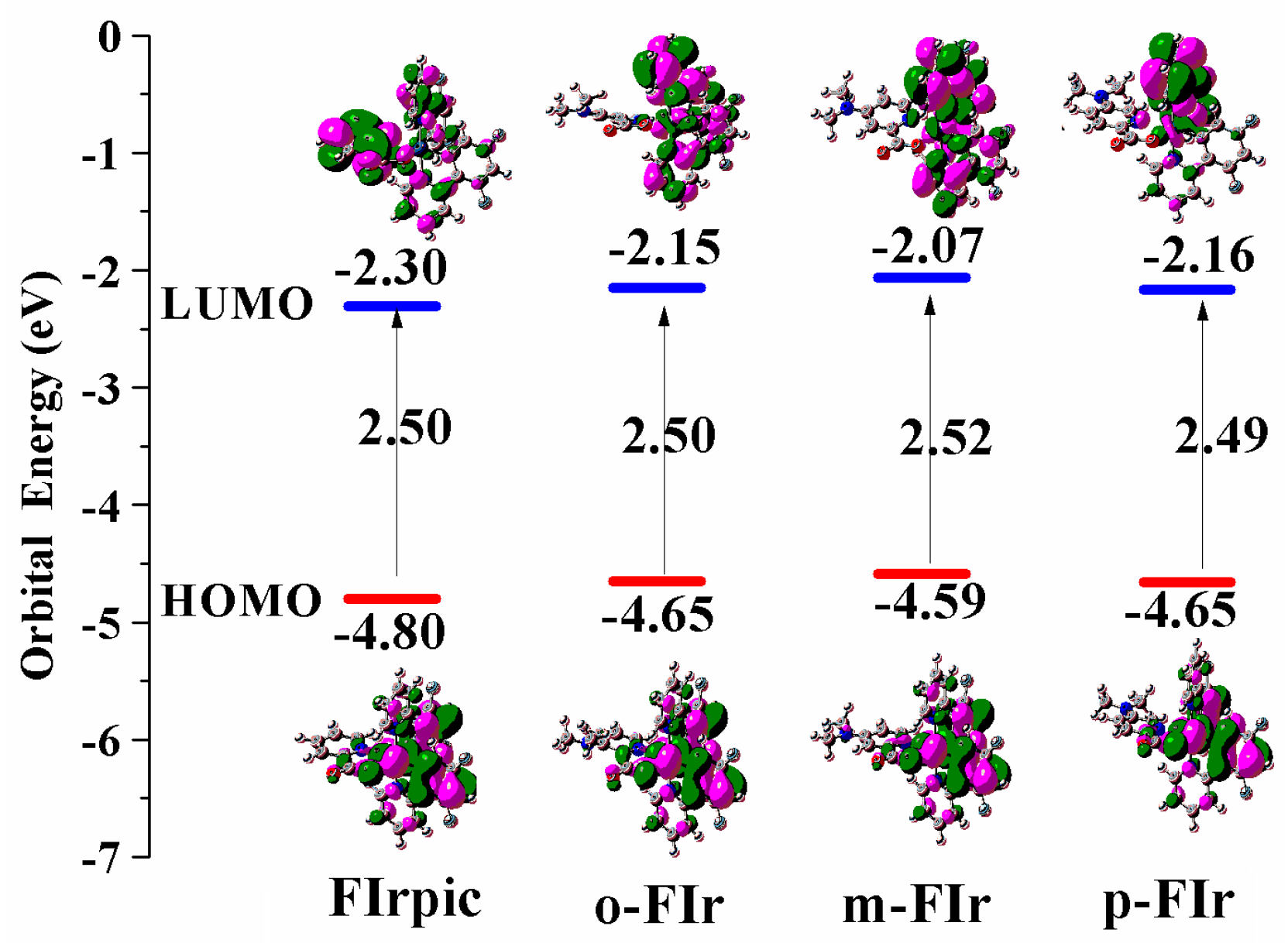
2.2. Froniter Molecular Orbtials
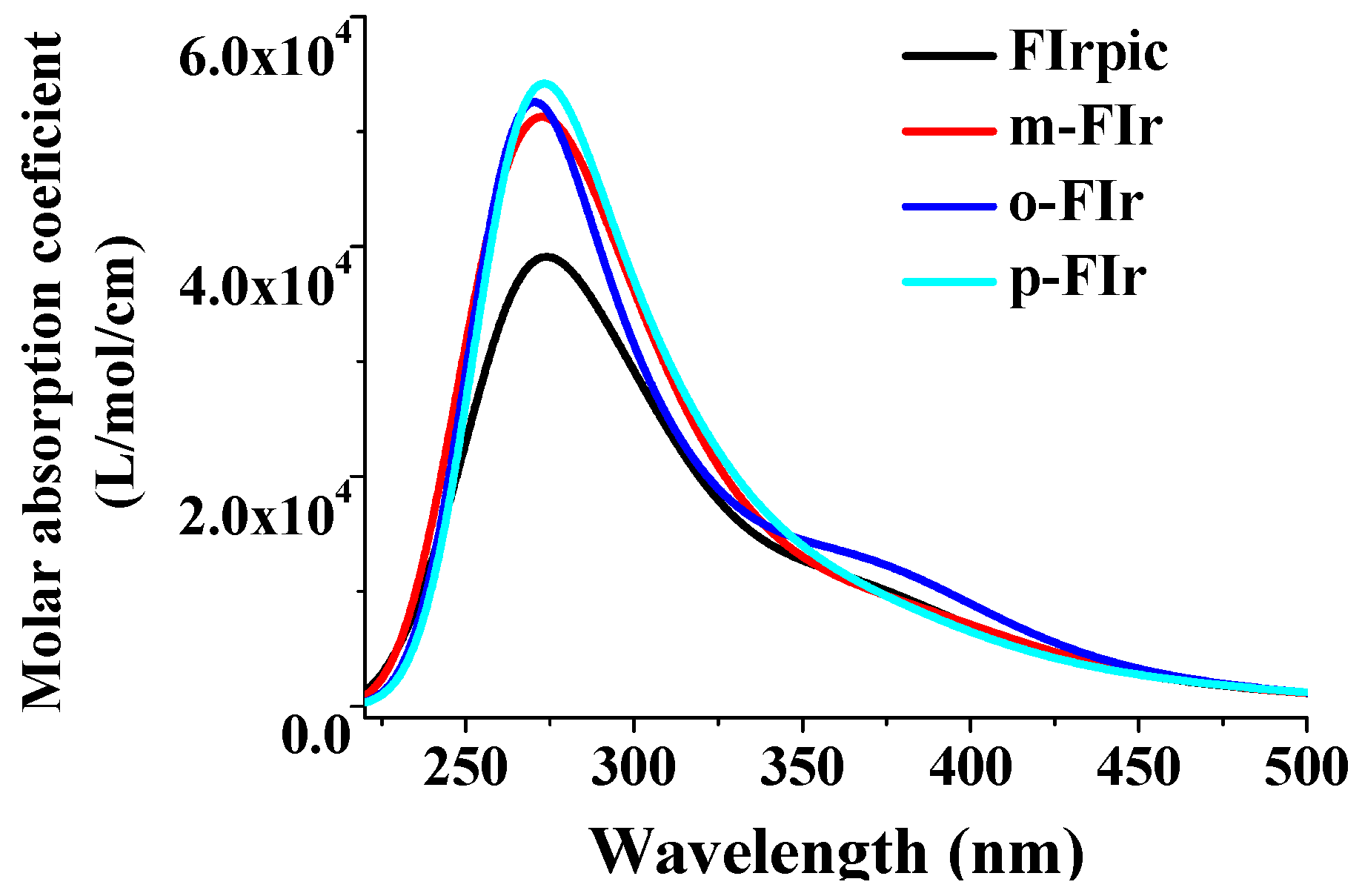
2.3. Absorption Spectra
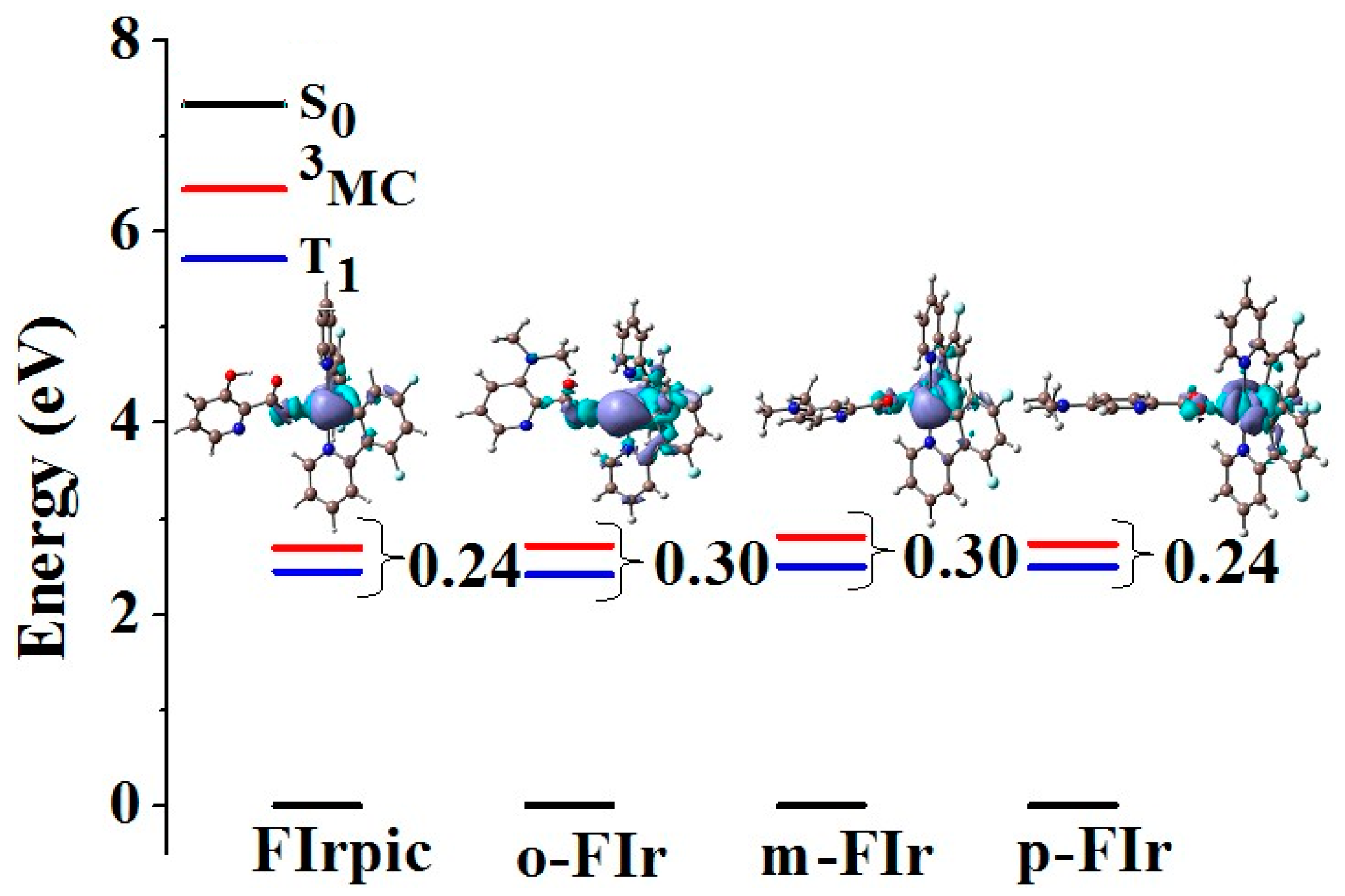
2.4. Phosphorescence Emission Spectra
2.5. Performance in OLEDs
3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lin, C.-H.; Chiu, Y.-C.; Chi, Y.; Tao, Y.-T.; Liao, L.-S.; Tseng, M.-R.; Lee, G.-H. Mechanistic Investigation of Improved Syntheses of Iridium(III)-Based OLED Phosphors. Organometallics 2012, 31, 4349–4355. [Google Scholar] [CrossRef]
- Xia, H.; Zhu, Y.; Lu, D.; Li, M.; Zhang, C.; Yang, B.; Ma, Y. Ruthenium(II) Complexes with the Mixed Ligands 2,2′-Bipyridine and 4,4′-Dialkyl Ester-2,2′-bipyridine as Pure Red Dopants for a Single-Layer Electrophosphorescent Device. J. Phys. Chem. 2006, 110, 18718–18723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-T.; Jia, J.-F.; Ren, Y.; Wu, H.-S. Ligand Effects on Structures and Spectroscopic Properties of Pyridine-2-aldoxime Complexes of Re(CO)3+: DFT/TDDFT Theoretical Studies. J. Phys. Chem. 2011, 115, 3174–3181. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-L.; Wong, W.-Y. Small-molecular blue phosphorescent dyes for organic light-emitting devices. New J. Chem. 2013, 37, 1665–1683. [Google Scholar] [CrossRef]
- Won, D.-I.; Lee, J.-S.; Ji, J.-M.; Jung, W.-J.; Son, H.-J.; Pac, C.; Kang, S.O. Highly Robust Hybrid Photocatalyst for Carbon Dioxide Reduction: Tuning and Optimization of Catalytic Activities of Dye/TiO2/Re(I) Organic–Inorganic Ternary Systems. J. Am. Chem. Soc. 2015, 137, 13679–13690. [Google Scholar] [CrossRef] [PubMed]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Lee, H.-E.; Adachi, C.; Burrows, P.E.; Forrest, S.R.; Thompson, M.E. Highly Phosphorescent Bis-Cyclometalated Iridium Complexes: Synthesis, Photophysical Characterization, and Use in Organic Light Emitting Diodes. J. Am. Chem. Soc. 2001, 123, 4304–4312. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, G.; Ramamoorthi, B.K.; Venuvanalingam, P. Are Re(I) phenanthroline complexes suitable candidates for OLEDs? Answers from DFT and TD-DFT investigations. Phys. Chem. Chem. Phys. 2014, 16, 21157–21171. [Google Scholar] [CrossRef] [PubMed]
- Rausch, A.F.; Thompson, M.E.; Yersin, H. Matrix Effects on the Triplet State of the OLED Emitter Ir(4,6-dFppy)2(pic) (FIrpic): Investigations by High-Resolution Optical Spectroscopy. Inorg. Chem. 2009, 48, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Kozhevnikov, V.N.; Zheng, Y.H.; Clough, M.; Al-Attar, H.A.; Griffiths, G.C.; Abdullah, K.; Raisys, S.; Jankus, V.; Bryce, M.R.; Monkman, A.P. Cyclometalated Ir(III) Complexes for High-Efficiency Solution-Processable Blue PhOLEDs. Chem. Mater. 2013, 25, 2352–2358. [Google Scholar] [CrossRef]
- Rausch, A.F.; Thompson, M.E.; Yersin, H. Blue Light Emitting Ir(III) Compounds for OLEDs - New Insights into Ancillary Ligand Effects on the Emitting Triplet State. J. Phys. Chem. 2009, 113, 5927–5932. [Google Scholar] [CrossRef] [PubMed]
- Sajoto, T.; Djurovich, P.I.; Tamayo, A.B.; Oxgaard, J.; Goddard, W.A.; Thompson, M.E. Temperature Dependence of Blue Phosphorescent Cyclometalated Ir(III) Complexes. J. Am. Chem. Soc. 2009, 131, 9813–9822. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Kim, J.-H.; Cho, Y.-J.; Lee, J.; Choi, T.-S.; Cho, D.W.; Pac, C.; Han, W.-S.; Son, H.-J.; Kang, S.O. Stable Blue Phosphorescence Iridium(III) Cyclometalated Complexes Prompted by Intramolecular Hydrogen Bond in Ancillary Ligand. Inorg. Chem 2016, 55, 3324–3331. [Google Scholar] [CrossRef] [PubMed]
- Stringer, B.D.; Quan, L.M.; Barnard, P.J.; Wilson, D.J.D.; Hogan, C.F. Iridium Complexes of N‑Heterocyclic Carbene Ligands: Investigation into the Energetic Requirements for Efficient Electrogenerated Chemiluminescence. Organometallics 2014, 33, 4860–4872. [Google Scholar] [CrossRef]
- Chu, W.-K.; Yiu, S.-M.; Ko, C.-C. Neutral Luminescent Bis(bipyridyl) Osmium(II) Complexes with Improved Phosphorescent Properties. Organometallics 2014, 33, 6771–6777. [Google Scholar] [CrossRef]
- You, Y.; Park, S.Y. Phosphorescent iridium(III) complexes: toward high phosphorescence quantum efficiency through ligand control. Dalton Trans. 2009, 1269, 1267–1282. [Google Scholar] [CrossRef]
- Kang, G.J.; Ren, X.F.; He, Q.Q. Theoretical study on effect of thiophene substitution on the structure and phosphorescence quantum yields of red-emitting iridium(III) emitters in OLEDs. J. Photochem. Photobiol. 2016, 319, 25–33. [Google Scholar] [CrossRef]
- Shang, X.H.; Han, D.M.; Zhou, D.F.; Zhang, G. Shedding light on the photophysical properties of iridium(III) complexes with a dicyclometalated phosphate ligand via N-substitution from a theoretical viewpoint. New J. Chem. 2017, 41, 1645–1652. [Google Scholar] [CrossRef]
- Baranoff, E.; Curchod, B.F.E.; Monti, F.; Steimer, F.; Accorsi, G.; Tavernelli, I.; Rothlisberger, U.; Scopelliti, R.; Grätzel, M.; Nazeeruddin, M.K. Influence of Halogen Atoms on a Homologous Series of Bis-Cyclometalated Iridium(III) Complexes. Inorg. Chem. 2012, 51, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.-O.; Shin, H.; Yun, H.-J.; Yang, K.; Kwon, S.-K.; Kim, J.-J.; Kim, Y.-H. Deep-Blue Phosphorescence from Perfluoro Carbonyl-Substituted Iridium Complexes. J. Am. Chem. Soc. 2013, 135, 14321–14328. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Q.; He, H.; Wang, L.; Zhang, J. Tuning the electronic and phosphorescence properties of blue-emitting iridium(III) complexes through different cyclometalated ligand substituents: A theoretical investigation. Dalton Trans. 2015, 44, 8577–8589. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-L.; Che, G.-B.; Li, X.-Y.; Xiao, Q. Bis[3,5-difluoro-2-(2-pyridyl)phenyl]-(picolinato)iridium(III). Acta Cryst. 2009, E65, m28. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Liu, Y. You have full text access to this content, Recent Progress in n-Channel Organic Thin-Film Transistors. Adv. Mater. 2010, 22, 1331–1345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-F.; Chen, X.-K.; Fan, J.-X.; Ren, A.-M. Charge transport properties in a series of five-ring-fused thienoacenes: A quantum chemistry and molecular mechanic study. Org. Electron. 2013, 14, 607–620. [Google Scholar] [CrossRef]
- Baranoff, E.; Curchod, B.F.E. FIrpic: Archetypal blue phosphorescent emitter for Electroluminescence. Dalton Trans. 2015, 44, 8318–8329. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Cheng, C.P.; Lao, Z.P.M. Reorganization Energies in the Transports of Holes and Electrons in Organic Amines in Organic Electroluminescence Studied by Density Functional Theory. J. Phys. Chem. 2003, 107, 5241–5251. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101–194118. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision B.04; Gaussian: Wallingford, CT, USA, 2009. [Google Scholar]
Sample Availability: Not available. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
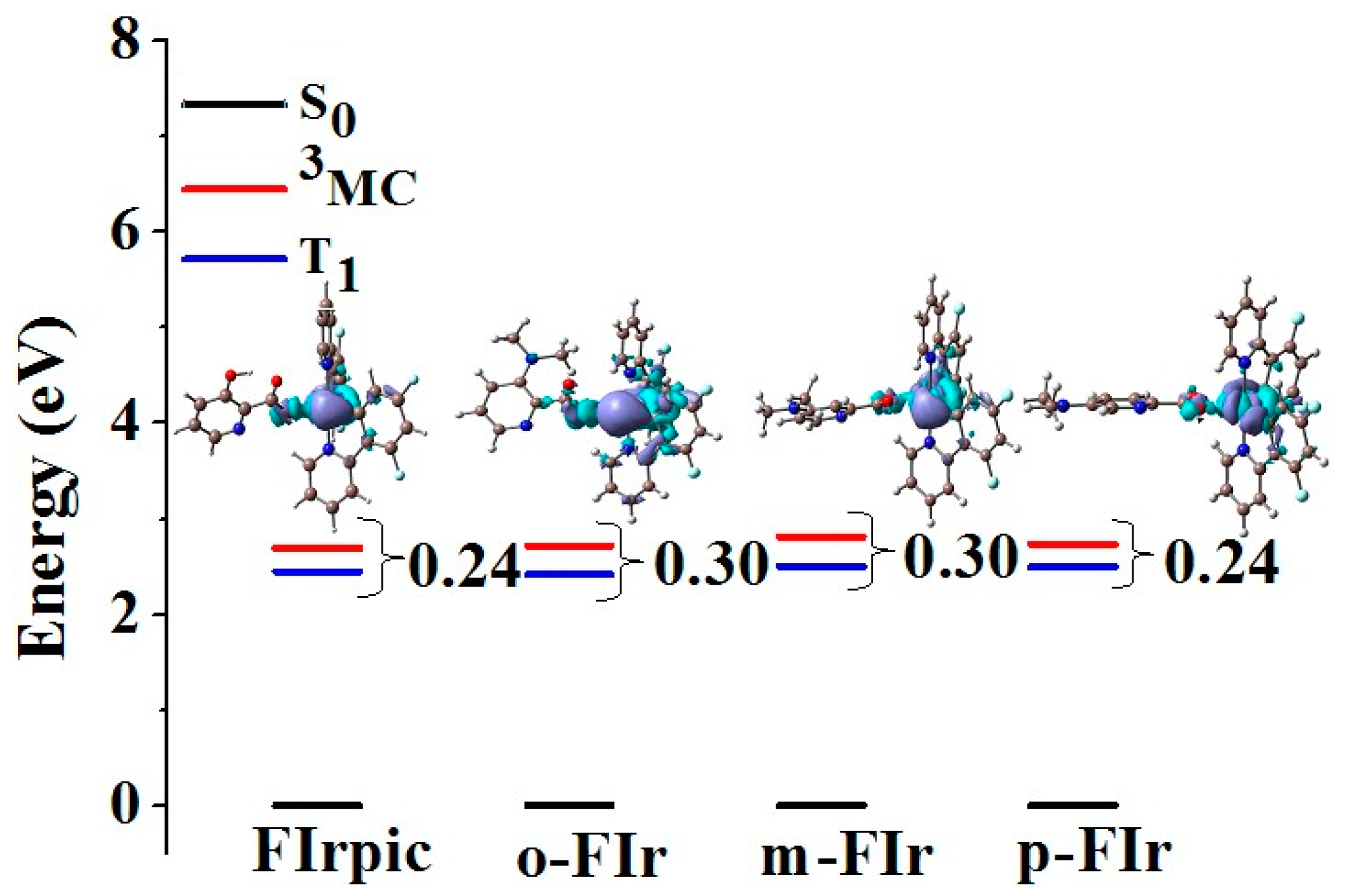{kind=link}
| Molecule | State | Ir-N1 | Ir-O1 | Ir-N2 | Ir-C1 | Ir-N3 | Ir-C2 | N1-Ir-N2 | O1-Ir-N2 | N1-O1-C2-C1 |
|---|---|---|---|---|---|---|---|---|---|---|
| FIrpic | S0 | 2.215 | 2.190 | 2.066 | 2.001 | 2.058 | 1.997 | 88.4 | 93.5 | 6.8 |
| T1 | 2.313 | 2.172 | 2.069 | 1.979 | 2.060 | 1.975 | 97.1 | 88.7 | −3.4 | |
| o-FIr | S0 | 2.210 | 2.180 | 2.066 | 2.002 | 2.054 | 1.997 | 87.3 | 94.0 | 5.0 |
| T1 | 2.316 | 2.154 | 2.057 | 1.981 | 2.067 | 1.977 | 84.4 | 93.7 | 3.1 | |
| m-FIr | S0 | 2.206 | 2.193 | 2.064 | 2.002 | 2.056 | 1.995 | 88.6 | 93.0 | 7.2 |
| T1 | 2.285 | 2.171 | 2.069 | 1.979 | 2.057 | 1.978 | 95.9 | 89.0 | −4.6 | |
| p-FIr | S0 | 2.165 | 2.177 | 2.055 | 2.010 | 2.045 | 2.001 | 87.6 | 92.9 | 6.3 |
| T1 | 2.305 | 2.160 | 2.068 | 1.982 | 2.058 | 1.977 | 97.4 | 88.7 | −3.0 |
| Molecule | Transition | λ(nm) | f | Composition | CI | Exp. [24] (nm) |
|---|---|---|---|---|---|---|
| FIrpic | S0→S2 | 453.00 | 0.0250 | HOMO→LUMO + 1 | 0.68971 | 455 |
| S0→S7 | 380.95 | 0.0614 | HOMO − 1→LUMO + 2 | 0.55936 | 379 | |
| S0→S50 | 265.86 | 0.2104 | HOMO − 8→LUMO + 2 | 0.38300 | 256 | |
| HOMO→LUMO + 7 | 0.24359 | |||||
| o-FIr | S0→S1 | 463.87 | 0.0258 | HOMO→LUMO | 0.70168 | |
| S0→S7 | 395.15 | 0.0266 | HOMO − 2→LUMO + 2 | 0.56244 | ||
| HOMO − 1→LUMO + 2 | −0.40890 | |||||
| S0→S52 | 269.04 | 0.1135 | HOMO − 9→LUMO + 1 | 0.25480 | ||
| HOMO − 5→LUMO + 5 | 0.42176 | |||||
| HOMO − 1→LUMO + 7 | 0.23321 | |||||
| S0→S53 | 268.66 | 0.1642 | HOMO − 7→LUMO + 4 | −0.35667 | ||
| HOMO − 1→LUMO + 7 | 0.36355 | |||||
| S0→S54 | 266.76 | 0.1318 | HOMO − 9→LUMO + 2 | 0.21920 | ||
| HOMO − 2→LUMO + 7 | 0.26962 | |||||
| HOMO − 1→LUMO + 7 | −0.21838 | |||||
| HOMO→LUMO + 7 | 0.41903 | |||||
| S0→S55 | 266.32 | 0.0836 | HOMO − 5→LUMO + 5 | 0.42068 | ||
| S0→S56 | 264.54 | 0.1094 | HOMO − 9→LUMO + 2 | 0.48856 | ||
| HOMO − 8→LUMO + 2 | −0.19242 | |||||
| HOMO − 1→LUMO + 5 | 0.27865 | |||||
| S0→S57 | 262.33 | 0.1716 | HOMO − 9→LUMO + 2 | −0.24371 | ||
| HOMO − 5→LUMO + 5 | −0.24091 | |||||
| HOMO − 4→LUMO + 5 | −0.25407 | |||||
| HOMO − 2→LUMO + 5 | 0.20626 | |||||
| HOMO − 2→LUMO + 7 | 0.30782 | |||||
| HOMO − 1→LUMO + 5 | 0.32002 | |||||
| m-FIr | S0→S1 | 462.95 | 0.0263 | HOMO→LUMO | 0.70166 | |
| S0→S6 | 393.55 | 0.0423 | HOMO − 2→LUMO | −0.41398 | ||
| HOMO − 2→LUMO + 1 | 0.42182 | |||||
| HOMO − 1→LUMO + 1 | 0.31148 | |||||
| S0→S53 | 267.06 | 0.3060 | HOMO − 8→LUMO + 2 | −0.24031 | ||
| HOMO→LUMO+7 | 0.25154 | |||||
| p-FIr | S0→S1 | 463.29 | 0.0286 | HOMO→LUMO | 0.70148 | |
| S0→S8 | 388.02 | 0.0581 | HOMO − 1→LUMO + 1 | −0.43685 | ||
| HOMO→LUMO + 3 | 0.47613 | |||||
| S0→S52 | 271.08 | 0.2512 | HOMO − 7→LUMO + 4 | −0.27219 | ||
| HOMO − 3→LUMO + 5 | 0.21898 | |||||
| HOMO − 2→LUMO + 5 | 0.29024 | |||||
| HOMO→LUMO + 7 | −0.21723 |
| Molecule | Evert | E0-0 | Exp [24] | ||||
|---|---|---|---|---|---|---|---|
| λ (nm) | E (eV) | Composition | 3MLCT (%) | λ (nm) | E (eV) | λ (nm) | |
| FIrpic | 490.49 | 2.53 | 0.49 (HOMO→LUMO) | 20.31 | 480.86 | 2.58 | 468 |
| o-FIr | 492.78 | 2.52 | 0.45 (HOMO − 4→LUMO) 0.43 (HOMO→LUMO) | 18.78 | 482.58 | 2.60 | |
| m-FIr | 484.32 | 2.56 | −0.36 (HOMO − 4→LUMO) 0.40 (HOMO→LUMO) | 9.11 | 481.78 | 2.57 | |
| p-FIr | 485.83 | 2.55 | −0.36 (HOMO − 4→LUMO) 0.49 (HOMO→LUMO) | 11.56 | 481.76 | 2.57 | |
| Molecule | IP(v) | HEP | EA(v) | EEP | λhole | λelectron |
|---|---|---|---|---|---|---|
| FIrpic | 6.56 | 6.29 | 0.72 | 0.98 | 0.27 | 0.26 |
| o-FIr | 6.32 | 6.06 | 0.48 | 0.69 | 0.26 | 0.21 |
| m-FIr | 6.39 | 6.09 | 0.56 | 0.75 | 0.30 | 0.19 |
| p-FIr | 6.34 | 6.11 | 0.53 | 0.73 | 0.23 | 0.20 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, X.-F.; Tang, H.-Q.; Kang, G.-J. Tuning the Geometrical Structures and Optical Properties of Blue-Emitting Iridium(III) Complexes through Dimethylamine Substitutions: A Theoretical Study. Molecules 2017, 22, 758. https://doi.org/10.3390/molecules22050758
Ren X-F, Tang H-Q, Kang G-J. Tuning the Geometrical Structures and Optical Properties of Blue-Emitting Iridium(III) Complexes through Dimethylamine Substitutions: A Theoretical Study. Molecules. 2017; 22(5):758. https://doi.org/10.3390/molecules22050758
Chicago/Turabian StyleRen, Xue-Feng, Hong-Qu Tang, and Guo-Jun Kang. 2017. "Tuning the Geometrical Structures and Optical Properties of Blue-Emitting Iridium(III) Complexes through Dimethylamine Substitutions: A Theoretical Study" Molecules 22, no. 5: 758. https://doi.org/10.3390/molecules22050758