1H-NMR spectra were recorded at 300 MHz with a Varian VXR-300 spectrometer (Varian Inc, Palo Alto, CA, USA), at 500 MHz with a Bruker AVANCE DRX 500 instrument (Bruker, Billerica, MA, USA), or at 400 MHz with Varian UNITY-Plus 400 spectrometer (Varian Inc, Palo Alto, CA, USA).
13C-NMR‑spectra (proton decoupled) were recorded on a Bruker AVANCE DRX 500 instrument at 125 MHz, or at 100 MHz with Varian UNITY-Plus 400 spectrometer.
19F-NMR spectra were recorded at 188 MHz with a Varian Geminy-200 instrument (Varian Inc., Palo Alto, CA, USA), or at 376 MHz with Varian UNITY-Plus 400 spectrometer. Chemical shifts are given in ppm relative to Me
4Si and CCl
3F, respectively, as internal or external standards.
1H-,
13C-, and
19F-NMR spectra of the compounds can be found in
Supplementary materials. LC-MS spectra were registered on an “Agilent 1100 Series” instrument with diode-matrix and mass-selective detector “Agilent 1100 LS/MSD SL” (ionization method: chemical ionization at atmospheric pressure; ionization chamber operation conditions: simultaneous scanning of positive and negative ions in the range 80–1000
m/
z, Agilent Technologies, Santa Clara, CA, USA). GC-MS spectra were registered on a Hewlett-Packard HP GC/MS 5890/5972 instrument (EI 70 eV) (Philips, Bothell, WA, USA). Melting points were determined in open capillaries using an SMP3 instrument (Stuart Scientific Bibby Sterlin Ltd, Stone, Staffordshire, UK). Elemental analysis was performed in the Analytical Laboratory of the Institute of Organic Chemistry, NAS of Ukraine, Kiev.
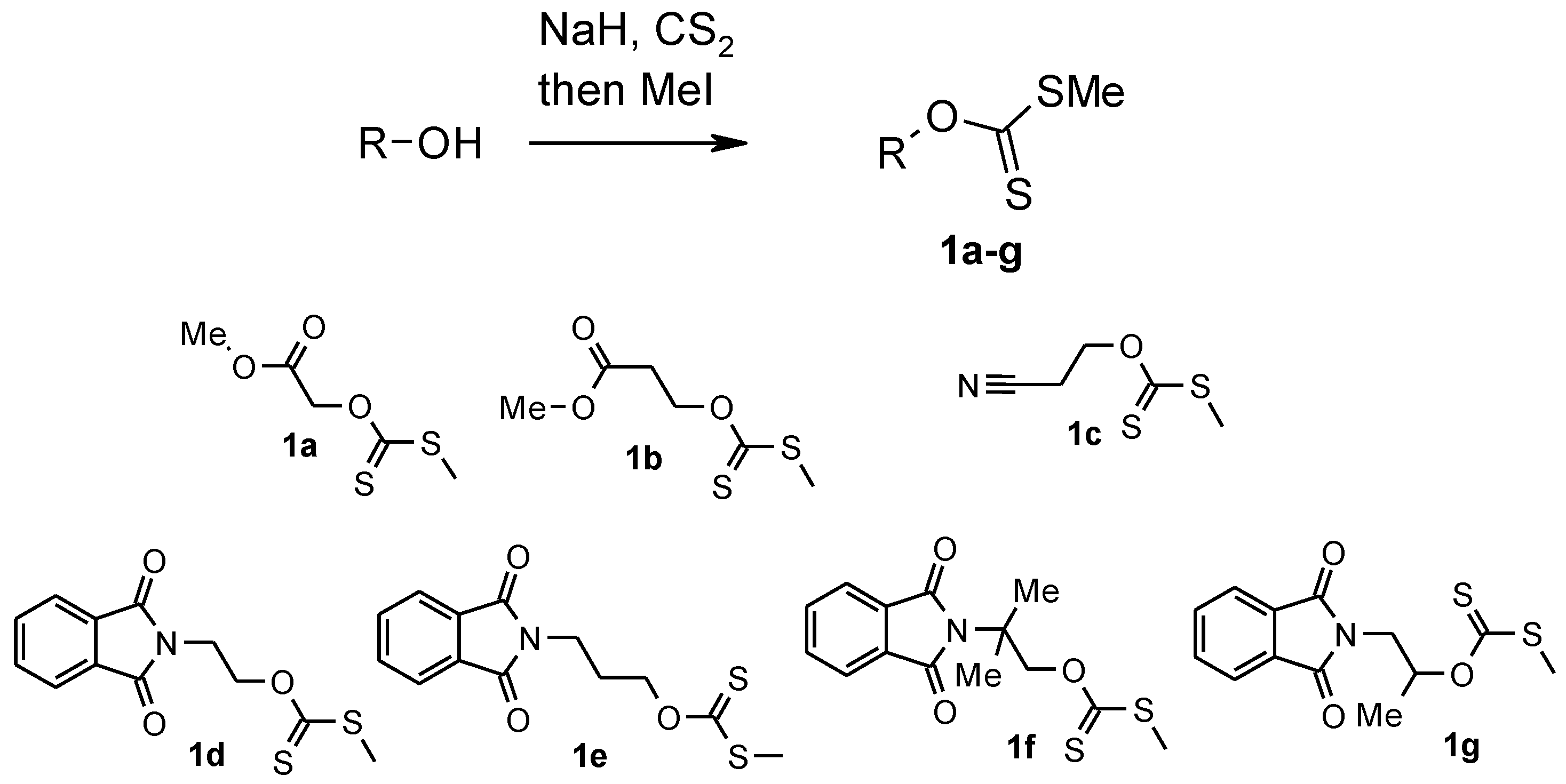
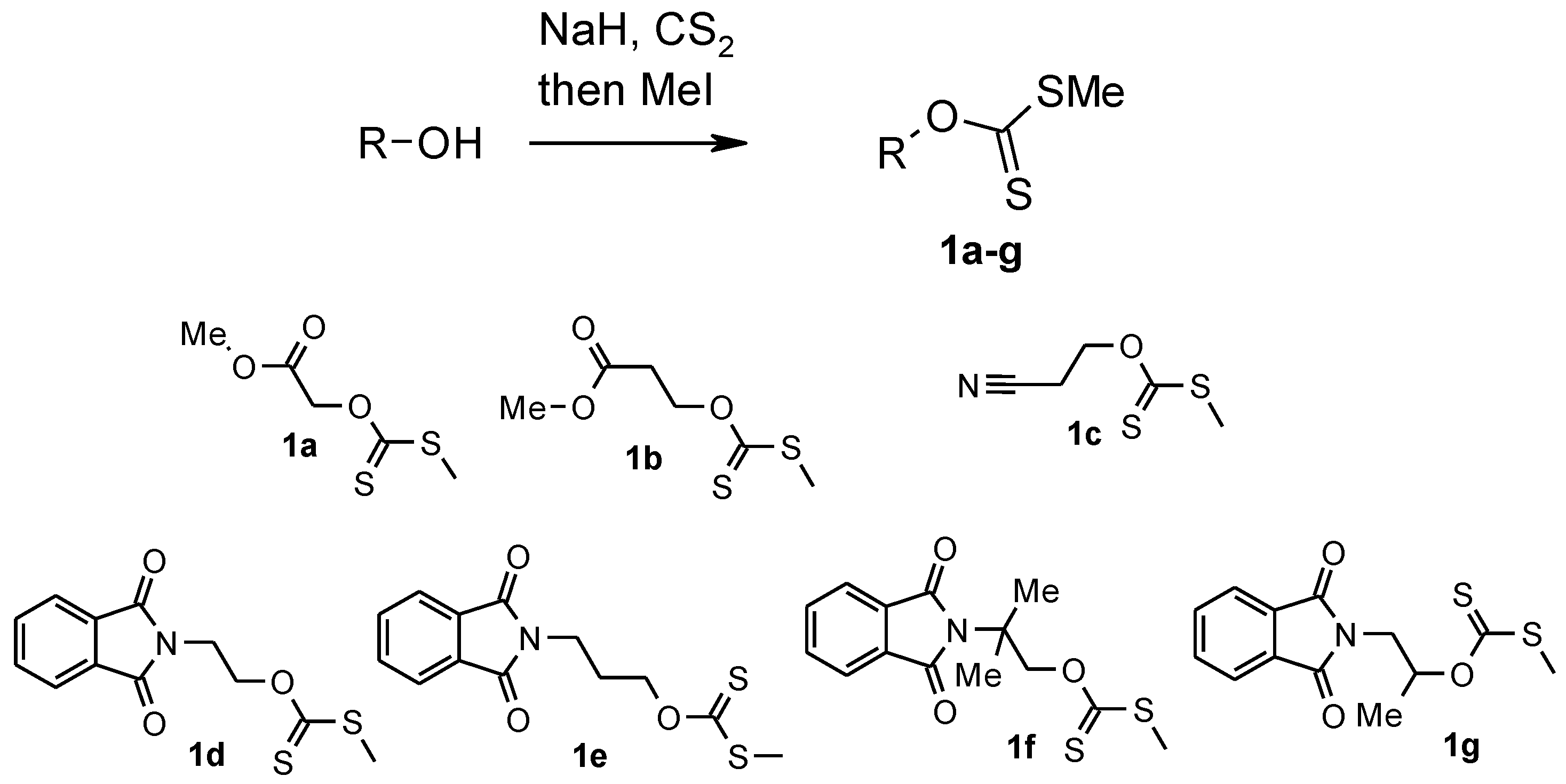
3.1. Synthesis of Xanthates 1a–c: General Procedure
Carbon disulfide (18.1 mL, 0.3 mol) was added dropwise at −30 °C to the suspension of sodium hydride (60% in mineral oil) (4.4 g, 0.11 mol) in anhydrous dimethylformamide (DMF) (200 mL). To this mixture the solution of corresponding hydroxyderivatives (0.1 mol) in anhydrous DMF (15 mL) was added dropwise for 3 h with mechanistic stirring at −30 °C. After addition was completed, the mixture was warmed to room temperature and stirred for 3 h. The color of the mixture gradually changed to dark red. The reaction mixture was cooled to −10 °C and methyl iodide (7.8 mL, 0.12 mol) was added dropwise. The mixture was warmed to room temperature, stirred for 3 h until the color changed from dark red to yellow and poured into ice (600 g), and extracted with tert-buthylmethyl ether (MTBE) (5 × 50 mL). The extract was washed with brine (5 × 50 mL) and dried with MgSO4. The solvent was removed at atmospheric pressure and the residue distilled in vacuum.
3.1.1. Methyl ([(methylsulfanyl)carbonothioyl]oxy)acetate 1a
Yield 15.5 g (86%). Yellow oil: bp 129–130 °C (20 Torr). 1H-NMR (400 MHz, CDCl3): δ 2.60 (s, 3H, SCH3), 3.77 (s, 3H, OCH3), 5.15 (s, 2H, CH2). 13C-NMR (100 MHz, CDCl3): δ 19.5 (s, SCH3), 52.4 (s, OCH3), 67.6 (s, CH2), 167.1 (s, C=O), 215.8 (s, C=S). GC-MS, 70 eV, m/z (rel. int.): 180 (69) [M]+. Anal. calcd for C5H8O3S2: C, 33.32; H, 4.47; S, 35.58; found: C, 33.14; H, 4.51; S, 35.51.
3.1.2. Methyl 3-([(methylsulfanyl)carbonothioyl]oxy)propanoate 1b
Yield 17.8 g (92%). Yellow oil: bp 94–95 °C (0.5 Torr). 1H-NMR (400 MHz, CDCl3): δ 2.52 (s, 3H, SCH3), 2.80 (t, 3J = 6.4 Hz, 2H, CH2), 3.70 (s, 3H, OCH3), 5.15 (t, 3J = 6.4 Hz, 2H, CH2). 13C-NMR (125 MHz, CDCl3): δ 18.7 (s, SCH3), 32.9 (s, CH2), 51.6 (s, OCH3), 68.2 (s, CH2), 170.2 (s, C=O), 215.2 (s, C=S). GC-MS, 70 eV, m/z (rel. int.): 194 (62) [M]+. Anal. calcd for C6H10O3S2: C, 37.09; H, 5.19; S, 33.01; found: C, 37.17; H, 5.29; S, 32.94.
3.1.3. O-(2-Cyanoethyl)-S-methyl-dithiocarbonate 1c
Yield 14.3 g (89%). Yellow oil: bp 94–95 °C (0.3 Torr). 1H-NMR (300 MHz, CDCl3): δ 2.53 (s, 3H, SCH3), 2.82 (t, 3J = 6.3 Hz, 2H, CH2), 4.73 (t, 3J = 6.3 Hz, 2H, CH2). 13C-NMR (125 MHz, CDCl3): δ 17.3 (s, CH2), 18.9 (s, SCH3), 66.2 (s, CH2), 116.1 (s, C≡N), 214.9 (s, C=S). GC-MS, 70 eV, m/z (rel. int.): 161 (67) [M]+. Anal. calcd for C5H7NOS2: C, 37.24; H, 4.38; S, 39.77; found: C, 37.30; H, 5.44; S, 39.70.
3.2. Synthesis of Xanthates 1d–g: General Procedure
To the stirred solution of hydroxyderivatives (0.1 mol) in anhydrous DMF (50 mL) sodium hydride (60% in mineral oil) (4.4 g, 0.11 mol) was added with mechanistic stirring between −5 and 0 °C. After addition, stirring was continued for 2 h at room temperature. Carbon disulfide (9 mL, 0.15 mol) was added dropwise at 0 °C. The reaction mixture was warmed to room temperature and stirred for 5 h. The color of the one was gradually changed to dark red. The reaction mixture was cooled to 0 °C and methyl iodide (7.8 mL, 0.12 mol) was added dropwise. The mixture was warmed to room temperature, stirred for 3 h until the color changed from dark red to yellow and poured into ice (100 g), extracted with CH2Cl2 (3 × 50 mL). The extract was washed with water (3 × 50 mL) and dried with MgSO4. After removal of the solvent the product was washed with hexane and dried in vacuum.
3.2.1. O-[2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)ethyl] S-Methyl Dithiocarbonate 1d
Yield 27.8 g (99%). Yellow powder: mp 105–106 °C. 1H-NMR (400 MHz, CDCl3): δ 2.49 (s, 3H, SCH3), 4.10 (t, 3J = 5.6 Hz, 2H, CH2), 4.82 (t, 3J = 5.6 Hz, 2H, CH2), 7.71–7.73 (m, 2H, arom H), 7.83–7.86 (m, 2H, arom H). 13C-NMR (125 MHz, DMSO-d6): δ 18.4 (s, SCH3), 36.3 (s, CH2), 70.6 (s, CH2), 123.2 (s, arom. C), 131.6 (s, arom. C), 134.6 (s, arom. C), 167.7 (s, C=O), 215.2 (s, C=S). LC-MS, m/z: 282 [M + H]+. Anal. calcd for C12H11NO3S2: C, 51.23; H, 3.94; N, 4.98; S, 22.79; found: C, 51.30; H, 4.00; N, 4.98; S, 22.70.
3.2.2. O-[3-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)propyl] S-Methyl Dithiocarbonate 1e
Yield 29.5 g (100%). Yellow powder: mp 101–102 °C. 1H-NMR (300 MHz, DMSO-d6): δ 2.08–2.12 (m, 2H, CH2), 3.34 (s, 3H, SCH3), 3.71 (t, 3J = 6.3 Hz, 2H, CH2), 4.58 (t, 3J = 6.0 Hz, 2H, CH2), 7.83–7.85 (m, 4H, arom H). 13C-NMR (100 MHz, DMSO-d6): δ 18.8 (s, SCH3), 27.3 (s, CH2), 35.0 (s, CH2), 72.4 (s, CH2), 123.4 (s, arom. C), 132.1 (s, arom. C), 134.7 (s, arom. C), 168.3 (s, C=O), 215.6 (s, C=S). LC-MS, m/z: 296 [M + H]+. Anal. calcd for C13H13NO3S2: C, 52.86; H, 4.44; N, 4.74; S, 21.71; found: C, 52.92; H, 4.52; N, 4.68; S, 21.70.
3.2.3. O-[2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)-2-methylpropyl] S-Methyl Dithiocarbonate 1f
Yield 27.5 g (89%). Yellow powder: mp 85–86 °C. 1H-NMR (300 MHz, CDCl3): δ 1.77 (s, 6H, 2CH2), 2.47 (s, 3H, SCH3), 4.98 (s, 2H, CH2), 7.70–7.73 (m, 2H, arom H), 7.78–7.81 (m, 2H, arom H). 13C-NMR (125 MHz, CDCl3): δ 18.5 (s, SCH3), 26.2 (s, 2CH3), 58.7 (s, C), 77.1 (s, CH2), 122.5 (s, arom. C), 131.4 (s, arom. C), 133.6 (s, arom. C), 168.8 (s, C=O), 214.7 (s, C=S). LC-MS, m/z: 310 [M + H]+. Anal. calcd for C14H15NO3S2: C, 54.35; H, 4.89; N, 4.53; S, 20.73; found: C, 54.42; H, 4.96; N, 4.50; S, 20.75.
3.2.4. O-[2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)-1-methylethyl] S-Methyl Dithiocarbonate 1g
Yield 28.9 g (98%). Yellow powder: mp 85–86 °C. 1H-NMR (300 MHz, CDCl3): δ 1.43 (d, 3J = 6.4 Hz, 3H, CH3), 2.48 (s, 3H, SCH3), 3.89 (dd, 2J = 14.4 Hz, 3J = 3.2 Hz, 1H, CH2), 4.04 (dd, 2J = 14.4 Hz, 3J = 7.6 Hz, 1H, CH2), 6.00–7.10 (m, 1H, CH), 7.70–7.72 (m, 2H, arom H), 7.78–7.81 (m, 2H, arom H). 13C-NMR (125 MHz, CDCl3): δ 17.5 (s, CH3), 18.9 (s, SCH3), 43.7 (s, CH2), 77.6 (s, C), 123.5 (s, arom. C), 132.0 (s, arom. C), 134.2 (s, arom. C), 168.1 (s, C=O), 215.3 (s, C=S). LC-MS, m/z: 296 [M + H]+. Anal. calcd for C13H13NO3S2: C, 52.86; H, 4.44; N, 4.74; S, 21.71; found: C, 52.88; H, 4.86; N, 4.69; S, 21.76.
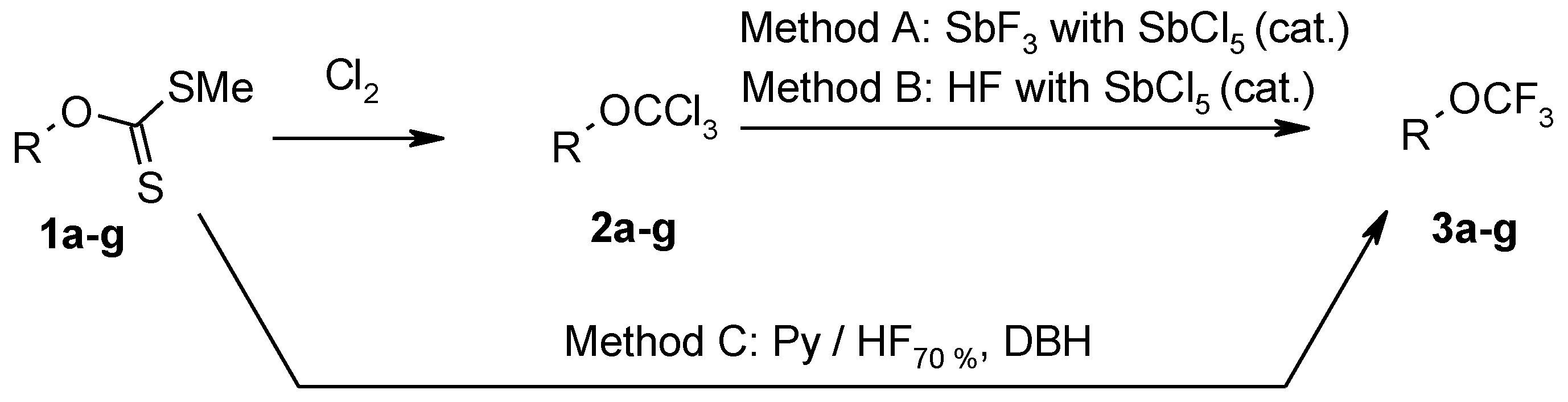
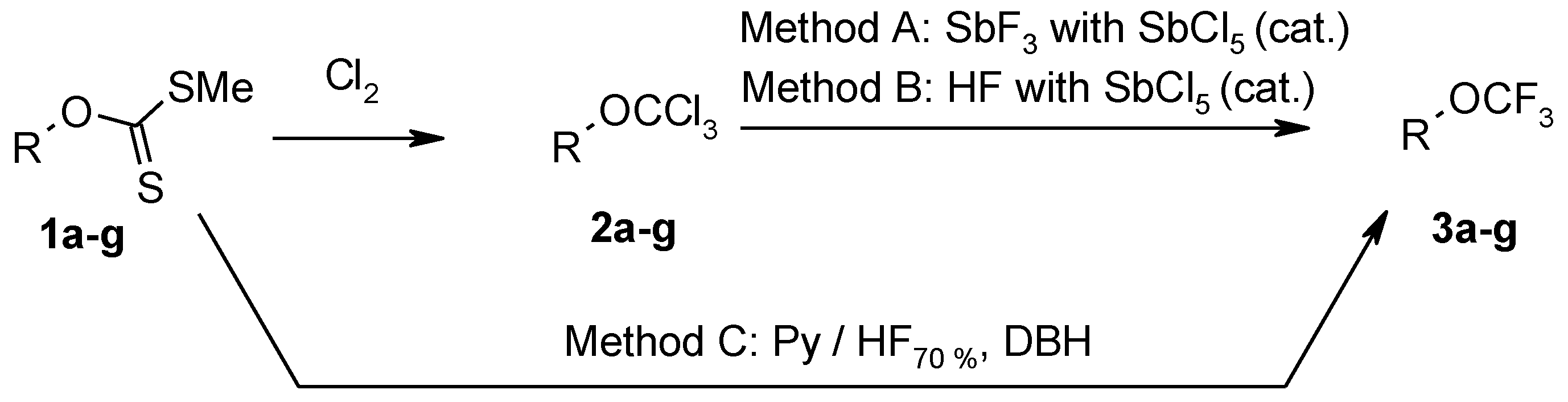
3.3. Synthesis of Trichloroderivatives 2a–g: General Procedure
Chlorine was bubbled through the solution of xanthate 1a–g (50 mmol) in carbon tetrachloride (50 mL) at 0 °C for 1 h. The reaction mixture was warmed to 20 °C and chlorine was bubbled for further 1 h. Methylene chloride (10 mL) was added to the reaction mixture and chlorine was bubbled for further 1 h. Excess of chlorine was removed with stream of nitrogen. After removal of the solvent, product was distilled in vacuum or washed with hexane and dried in vacuum.
3.3.1. Methyl (trichloromethoxy)acetate 2a
Yield 9.0 g (87%). Colorless liquid: bp 87–88 °C (10 Torr). 1H-NMR (400 MHz, CDCl3): δ 3.83 (s, 3H, OCH3), 4.64 (s, 2H, CH2). 13C-NMR (125 MHz, CDCl3): δ 52.7 (s, OCH3), 66.7 (s, CH2), 112.6 (s, CCl3), 166.2 (s, C=O). Anal. calcd for C4H5Cl3O3: C, 23.16; H, 2.43; Cl, 51.27; found: C, 33.12; H, 2.40; Cl, 52.35.
3.3.2. Methyl 3-(trichloromethoxy)propanoate 2b
Yield 9.8 g (89%). Colorless liquid: bp 64–65 °C (0.5 Torr). 1H-NMR (300 MHz, CDCl3): δ 2.76 (t, 3J = 6.4 Hz, 2H, CH2), 3.71 (s, 3H, OCH3), 5.35 (t, 3J = 6.4 Hz, 2H, CH2). 13C-NMR (125 MHz, CDCl3): δ 32.9 (s, CH2), 51.7 (s, OCH3), 66.4 (s, CH2), 111.9 (s, CCl3), 169.7 (s, C=O). Anal. calcd for C5H7Cl3O3: C, 27.12; H, 3.19; Cl, 48.02; found: C, 27.07; H, 3.12; Cl, 48.09.
3.3.3. 3-(Trichloromethoxy)propanenitrile 2c
Yield 8.7 g (92%). Colorless liquid: bp 77–78 °C (0.3 Torr). 1H-NMR (300 MHz, CDCl3): δ 2.83 (t, 3J = 6.6 Hz, 2H, CH2), 4.29 (t, 3J = 6.6 Hz, 2H, CH2). 13C-NMR (125 MHz, CDCl3): δ 17.6 (s, CH2), 64.8 (s, CH2), 111.9 (s, CCl3), 115.5 (s, C≡N). Anal. calcd for C4H4Cl3NO: C, 25.50; H, 2.14; Cl, 56.44; N, 7.43; found: C, 25.55; H, 2.14; Cl, 56.50; N, 7.40.
3.3.4. 2-[2-(Trichloromethoxy)ethyl]-1H-isoindole-1,3(2H)-dione 2d
Yield 14.8 g (96%). Colorless powder: mp 93–94 °C. 1H-NMR (300 MHz, CDCl3): δ 4.06 (t, 3J = 5.3 Hz, 2H, CH2), 4.34 (t, 3J = 5.3 Hz, 2H, CH2), 7.74–7.76 (m, 2H, arom H), 7.83–7.86 (m, 2H, arom H). 13C-NMR (100 MHz, CDCl3): δ 36.4 (s, CH2), 68.1 (s, CH2), 112.5 (s, CCl3), 123.5 (s, arom. C), 131.8 (s, arom. C), 134.2 (s, arom. C), 167.8 (s, C=O). Anal. calcd for C11H8Cl3NO3: C, 42.82; H, 2.61; Cl, 34.47; N, 4.54; found: C, 42.90; H, 2.52; Cl, 34.50; N, 4.44.
3.3.5. 2-[3-(Trichloromethoxy)propyl]-1H-isoindole-1,3(2H)-dione 2e
Yield 15.3 g (95%). Colorless powder: mp 102–103 °C. 1H-NMR (400 MHz, CDCl3): δ 2.12–2.19 (m, 2H, CH2), 3.83 (t, 3J = 6.8 Hz, 2H, CH2), 4.16 (t, 3J = 6.0 Hz, 2H, CH2), 7.60–7.70 (m, 2H, arom H), 7.80–7.90 (m, 2H, arom H). 13C-NMR (125 MHz, CDCl3): δ 27.3 (s, CH2), 35.1 (s, CH2), 69.6 (s, CH2), 112.4 (s, CCl3), 123.3 (s, arom. C), 132.0 (s, arom. C), 134.0 (s, arom. C), 168.2 (s, C=O). Anal. calcd for C12H10Cl3NO3: C, 44.68; H, 3.12; Cl, 32.97; N, 4.34; found: C, 44.75; H, 3.10; Cl, 33.00; N, 4.40.
3.3.6. 2-[1,1-Dimethyl-2-(trichloromethoxy)ethyl]-1H-isoindole-1,3(2H)-dione 2f
Yield 13.8 g (82%). Colorless powder: mp 93–94 °C. 1H-NMR (500 MHz, CDCl3): δ 1.79 (s, 6H, 2CH3), 4.53 (s, 2H, CH2), 7.68–7.71 (m, 2H, arom H), 7.75–7.80 (m, 2H, arom H). 13C-NMR (125 MHz, CDCl3): δ 24.7 (s, 2CH3), 58.8 (s, C), 75.6 (s, CH2), 112.6 (s, CCl3), 123.1 (s, arom. C), 131.9 (s, arom. C), 134.1 (s, arom. C), 169.4 (s, C=O). Anal. calcd for C13H12Cl3NO3: C, 46.39; H, 3.59; Cl, 31.60; N, 4.16; found: C, 46.45; H, 3.50; Cl, 31.68; N, 4.05.
3.3.7. 2-[2-(Trichloromethoxy)propyl]-1H-isoindole-1,3(2H)-dione 2g
Yield 13.7 g (85%). Colorless powder: mp 96–97 °C. 1H-NMR (300 MHz, CDCl3): δ 1.55 (d, 3J = 6.3 Hz, 3H, CH3), 3.79 (dd, 2J = 14.1 Hz, 3J = 3.6 Hz, 1H, CH2), 4.06 (dd, 2J = 14.1 Hz, 3J = 8.1 Hz, 1H, CH2), 4.80–4.95 (m, 1H, CH), 7.70–7.80 (m, 2H, arom H), 7.85–7.95 (m, 2H, arom H). 13C-NMR (125 MHz, CDCl3): δ 18.4 (s, CH3), 42.1 (s, CH2), 78.2 (s, C), 123.5 (s, arom. C), 132.5 (s, arom. C), 134.2 (s, arom. C), 167.9 (s, C=O). Anal. calcd for C12H10Cl3NO3: C, 44.68; H, 3.12; Cl, 32.97; N, 4.34; found: C, 44.75; H, 3.20; Cl, 33.00; N, 4.35.
3.6. Fluorination of Xanthate 1a–g with Pyridine-Hydrogenfluoride and 1,3-Dibromo-5,5-Dimethylhydantoin (DBH) (Method C): General Procedure
Reactions were performed in a polyethylene flask (500 mL) equipped with an additional polyethylene funnel, magnetic stirrer, and BOLA PTFE coated temperature probe.
Complex HF70%-pyridine was added dropwise at −78 °C to a suspension of DBH (42.9 g, 0.15 mol) in CH2Cl2 (130 mL). The mixture was stirred for 10 min and the solution of corresponding xanthate 1a–g (0.05 mol) in CH2Cl2 (70 mL) was added at the same temperature. The reaction mixture was stirred at −78 °C for 1 h, warmed to room temperature, stirred for 5 h and poured into ice (100 g). As saturated aqueous solution of Na2SO3 was added to the mixture until the red color of the mixture changed to light yellow. Then the mixture was neutralized with aqueous K2CO3 solution. The organic layer was separated and the aqueous solution was extracted with CH2Cl2 (3 × 50 mL). Combined organic extracts were washed with water (3 × 500 mL) and dried with MgSO4. After removal of the solvent, the product was distilled or crystallized from hexane and dried in vacuum.
3.6.1. Methyl (trifluoromethoxy)acetate 3a
Yield 2.84 g (36% method C). Colorless liquid: bp 102–104 °C (lit. 110 °C [
11]).
1H-NMR (400 MHz, CDCl
3): δ 3.81 (s, 3H, OCH
3), 4.48 (s, 2H, CH
2).
13C-NMR (125 MHz, CDCl
3): δ 52.5 (s, OCH
3), 62.9 (q,
3JCF = 2.5 Hz, CH
2), 121.4 (q,
JCF = 256.4 Hz, CF
3), 166.5 (s, C=O).
19F-NMR (376.5 MHz, CDCl
3): δ −62.12 (s, CF
3). GC-MS, 70 eV,
m/
z (rel. int.): 158 (46) [M]
+. Anal. calcd for C
4H
5F
3O
3: C, 30.39; H, 3.19; found: C, 30.12; H, 3.04.
3.6.2. Methyl 3-(trifluoromethoxy)propanoate 3b
Yield 4.55 g (53% method A). Yield 5.9 g (69% method B). Yield 5.4 g (63% method B). Colorless liquid: bp 124–125 °C. 1H-NMR (300 MHz, CDCl3): δ 2.64 (t, 3J = 5.4 Hz, 2H, CH2), 3.66 (s, 3H, OCH3), 4.17 (t, 3J = 5.4 Hz, 2H, CH2). 13C-NMR (125 MHz, CDCl3): δ 33.2 (s, CH2), 51.3 (s, OCH3), 62.2 (q, 3JCF = 3.3 Hz, CH2), 121.0 (q, JCF = 253.3 Hz, CF3), 169.8 (s, C=O). 19F-NMR (188 MHz, CDCl3): δ −61.88 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 172 (52) [M]+. Anal. calcd for C5H7F3O3: C, 34.89; H, 4.10; found: C, 34.55; H, 4.02.
3.6.3. 3-(Trifluoromethoxy)propanenitrile 3c
Yield 1.5 g (22% method A). Yield 4.6 g (66% method B). Yield 4.6 g (66% method B). Colorless liquid: bp 100–102 °C (120 Torr). 1H-NMR (300 MHz, CDCl3): δ 2.70 (t, 3J = 6.3 Hz, 2H, CH2), 4.11 (t, 3J = 6.3 Hz, 2H, CH2). 13C-NMR (125 MHz, CDCl3): δ 18.0 (s, CH2), 61.2 (q, 3JCF = 3.4 Hz, CH2), 115.3 (s, C≡N), 120.8 (q, JCF = 255.1 Hz, CF3). 19F-NMR (188 MHz, CDCl3): δ −62.10 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 139 (66) [M]+. Anal. calcd for C4H4F3NO: C, 34.54; H, 2.90; N, 10.07; found: C, 34.40; H, 2.84; N, 9.90.
3.6.4. 2-[2-(Trifluoromethoxy)ethyl]-1H-isoindole-1,3(2H)-dione 3d
Yield 11.65 g (90% method A). Yield 12.0 g (93% method B). Yield 12.4 g (96% method B). Colorless powder: mp 73–74 °C (lit. 77 °C [
16]), bp 100–102 °C (0.2 Torr).
1H-NMR (300 MHz, CDCl
3): δ 3.93 (t,
3J = 5.3 Hz, 2H, CH
2), 4.20 (t,
3J = 5.3 Hz, 2H, CH
2), 7.70–7.77 (m, 2H, arom H), 7.80–7.90 (m, 2H, arom H).
13C-NMR (125 MHz, CDCl
3): δ 36.7 (s, CH
2), 63.9 (s, CH
2), 121.7 (q,
JCF = 255.2 Hz, CF
3), 123.5 (s, arom. C), 131.8 (s, arom. C), 134.2 (s, arom. C), 167.8 (s, C=O).
19F-NMR (188 MHz, CDCl
3): δ −61.55 (s, CF
3). LC-MS,
m/
z: 260 [M + H]
+. Anal. calcd for C
11H
8F
3NO
3: C, 50.98; H, 3.11; N, 5.40; found: C, 50.90; H, 3.23; N, 5.44.
3.6.5. 2-[3-(Trifluoromethoxy)propyl]-1H-isoindole-1,3(2H)-dione 3e
Yield 12.0 g (88% method A). Yield 12.3 g (90% method B). Yield 13.0 g (96% method B). Colorless powder: mp 77–78 °C. 1H-NMR (400 MHz, CDCl3): δ 2.04–2.12 (m, 2H, CH2), 3.70–3.82 (m, 2H, CH2), 4.00–4.12 (m, 2H, CH2), 7.68–7.72 (m, 2H, arom H), 7.80–7.88 (m, 2H, arom H). 13C-NMR (125 MHz, CDCl3): δ 28.0 (s, CH2), 34.8 (s, CH2), 65.1 (s, CH2), 121.6 (q, JCF = 254.0 Hz, CF3), 123.4 (s, arom. C), 132.1 (s, arom. C), 134.2 (s, arom. C), 168.3 (s, C=O). 19F-NMR (376.5 MHz, CDCl3): δ −61.49 (s, CF3). LC-MS, m/z: 274 [M + H]+. Anal. calcd for C12H10F3NO3: C, 52.75; H, 3.69; N, 5.13; found: C, 52.77; H, 3.70; N, 5.22.
3.6.6. 2-[1,1-Dimethyl-2-(trifluoromethoxy)ethyl]-1H-isoindole-1,3(2H)-dione 3f
Yield 8.6 g (60% method A). Yield 8.9 g (62% method B). Yield 12.2 g (85% method B). Colorless powder: mp 73–74 °C. 1H-NMR (300 MHz, CDCl3): δ 1.69 (s, 6H, 2CH3), 4.35 (s, 2H, CH2), 7.60–7.67 (m, 2H, arom H), 7.70–7.77 (m, 2H, arom H). 13C-NMR (125 MHz, CDCl3): δ 24.8 (s, 2CH3), 58.3 (s, C), 70.7 (s, CH2), 120.2 (q, JCF = 253.3 Hz, CF3), 122.5 (s, arom. C), 131.3 (s, arom. C), 133.6 (s, arom. C), 168.9 (s, C=O). 19F-NMR (376.5 MHz, CDCl3): δ −61.11 (s, CF3). LC-MS, m/z: 288 [M + H]+. Anal. calcd for C13H12F3NO3: C, 54.36; H, 4.21; N, 4.88; found: C, 54.47; H, 4.47; N, 4.85.
3.6.7. 2-[2-(Trifluoromethoxy)propyl]-1H-isoindole-1,3(2H)-dione 3g
Yield 3.1 g (23% method A). Yield 4.6 g (34% method B). Yield 13.5 g (99% method B). Colorless powder: mp 71–72 °C. 1H-NMR (500 MHz, CDCl3): δ 1.40–1.43 (m, 3H, CH3), 3.79 (dd, 2J = 14.0 Hz, 3J = 2.0 Hz, 1H, CH2), 3.94–4.00 (m, 1H, CH2), 4.80–4.95 (m, 1H, CH), 7.74 (s, 2H, arom H), 7.87 (s, 2H, arom H). 13C-NMR (125 MHz, CDCl3): δ 18.6 (s, CH3), 42.3 (s, CH2), 72.9 (s, C), 121.5 (q, JCF = 255.1 Hz, CF3), 123.5 (s, arom. C), 131.7 (s, arom. C), 134.2 (s, arom. C), 168.0 (s, C=O). 19F-NMR (376.5 MHz, CDCl3): δ −59.19 (s, CF3). LC-MS, m/z: 274 [M + H]+. Anal. calcd for C12H10F3NO3: C, 52.75; H, 3.69; N, 5.13; found: C, 52.70; H, 3.44; N, 4.12.


 ,
, 

{kind=link}
{kind=link}













