
Identification of Non-Electrophilic Nrf2 Activators from Approved Drugs
by
 ,
,
Qing-Ye Zhang
1,*,†, 
Xin-Yi Chu
1,†,
Ling-Han Jiang
1,
Meng-Yuan Liu
1,
Zhi-Ling Mei
2 and
Hong-Yu Zhang
1,* 1
Hubei Key Laboratory of Agricultural Bioinformatics, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China
2
Shanghai Thinkgene Biotech Co., LTD., Shanghai 200000, China
*
Authors to whom correspondence should be addressed.
†
The first two authors contributed equally.
Molecules 2017, 22(6), 883; https://doi.org/10.3390/molecules22060883
Submission received: 5 May 2017
/
Revised: 22 May 2017
/
Accepted: 24 May 2017
/
Published: 26 May 2017
Abstract
:Oxidative damage can lead to a wide range of diseases. Nrf2 is an important transcription factor that regulates many of the cytoprotective enzymes involved in the oxidative stress response. Therefore, targeting the regulation of Nrf2 activation is one logical and effective strategy to prevent or lower the risk of oxidative stress-related diseases. Until now, most research has focused on electrophilic indirect Nrf2 activators, but the risk of ‘off-target’ effects may be associated with these activators. To find novel small non-electrophilic modulators of Nrf2, we started from chemical agents derived from a connectivity map (cMap) and identified 22 non-electrophilic potential Nrf2-activating drugs through a drug repositioning tactic. By determining the expression changes of antioxidant genes in MCF7 cells that were treated with the potential Nrf2 activators using quantitative real-time polymerase chain reaction RT-PCR (real-time polymerase chain reaction) (qRT-PCR), astemizole was found to have a greater scale of upregulating antioxidant genes NQO1, HO-1, and GCLM than the positive control d,l-sulforaphane, although the testing concentration was lower than that of the control. Astemizole is a good potential redox regulator and deserves more pharmacodynamic experimentation to test and verify its feasibility for use as an Nrf2 activator.
1. Introduction
Developing effective means to protect against metabolic oxidative stress and exposure to environmental toxicants is an essential requirement for the evolution of early eukaryotic life [1]. Oxidative stress is aroused by the imbalance in reactive species and the antioxidative defense systems in living cells, which can damage all cellular macromolecules, including proteins, lipids, and DNA [2]. This oxidative damage could be a common mechanism for a wide range of disorders, which include chronic inflammation, cancers, cardiovascular diseases, neurodegenerative diseases, and aging [3]. To counteract oxidative stress, cells switch on the antioxidative defense system to protect against and recover from the damage to the cell by upregulating a battery of cytoprotective genes, including antioxidant and detoxifying enzymes [4].
Nrf2 (nuclear factor (erythroid-derived 2)-like 2) is an important transcription factor that regulates many of the cytoprotective enzymes that are involved in the adaptive oxidative stress response [5]. Nrf2 is a member of the cap ‘n’ collar (CNC) subfamily of basic region leucine zipper (bZIP) transcription factors. Kelch-like ECH (chicken homologous to Nrf2)-associated protein 1 (Keap1) is used as a stress sensor of Nrf2. Under unstressed conditions, Keap1 binds to Nrf2, which promotes the ubiquitination and subsequent proteolysis of Nrf2. This mechanism maintains a low level of Nrf2 protein in the cell. Upon activation in response to oxidative and electrophilic stress, Nrf2 detaches from Keap1, and the degradation is stopped. Nrf2 rapidly accumulates, migrates to the nucleus, forms heterodimers with small musculoaponeurotic fibrosarcoma (Maf) proteins, and binds to the antioxidant response element (ARE) sequence to activate the transcription of a wide array of multiple antioxidant, detoxifying, and cell survival genes against oxidative and xenobiotic stress [6,7,8]. The Keap1-Nrf2-ARE pathway can activate ARE-dependent genes, encoding a group of approximately 500 proteins that are expressed in various isoforms and distributed in various organelles and subcellular compartments. These antioxidant and detoxifying enzymes such as NAD(P)H quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO1), and glutamate-cysteine ligase (GCL), which consists of a catalytic subunit (GCLC) and a modulatory subunit (GCLM), are upregulated by Nrf2 and have important roles in protecting cells against oxidative stress; thus they serve as common biomarkers for evaluating the Nrf2 dependent transcription [3,9,10].
Based on the comprehensive and critical models of Nrf2-regulated genes, the Keap1-Nrf2-ARE pathway has served as a high-value therapeutic target for diseases and conditions involving oxidative stress [11]. Targeting the regulation of the activation of Nrf2 could be one logical and effective strategy to prevent and lower the risk of a number of diseases [12]. The protective roles of Nrf2 activation in cancers [13,14,15,16], Alzheimer’s disease [17], Parkinson’s disease [18], Huntington’s disease [19,20], and amyotrophic lateral sclerosis [21,22] have been well established. To date, the activation of Nrf2 can take place through either a direct antioxidant or indirect antioxidant mechanism, and most of the research has focused on electrophilic indirect Nrf2 activators [3,23,24]. Up until now, indirect activators have been classified into several classes based on their chemical structures and modes of interaction with cysteine sulfhydryl groups: (1) oxidizable phenols and quinones; (2) dithiolethiones; (3) isothiocyanates and sulfoxythiocarbamates; (4) trivalent arsenicals; (5) vicinal dimercaptans and diallyl sulfides; (6) Michael acceptors; (7) polyenes; (8) heavy metals and metal complexes; (9) hydroperoxides; and (10) selenium-based compounds [3,10,25,26,27,28,29,30,31].
Although several successful electrophilic indirect Nrf2 activators have been approved and used as first-line oral drugs, the risk of ‘off-target’ effects may be associated with them due to the complex molecular mechanisms of action of the traditional electrophilic Nrf2 activators, which covalently bind to the thiol of the cysteine in Keap1. This may lead to unwanted binding between the activators and cysteine-containing proteins [32,33]. Recently, novel small direct or non-electrophilic modulators of the Keap1-Nrf2-ARE pathway have been discovered [4,34,35,36,37,38,39,40,41,42], but it is still urgent to develop more small-molecule antioxidants as therapies for many diseases [43,44].
De novo drug discovery is a very expensive and time-consuming process. Therefore, drug repositioning has played a critical role in the modern pharmaceutical industry. The gene expression data, in particular those contained in the connectivity map (cMap), have been widely used in drug repurposing and lead discovery [45,46]. cMap is a database comprising gene expression profiles for human cell lines treated with 1309 agents. A recent biclustering analysis of cMap data generated a total of 49 modules for gene expression profiles, and module 36 is regulated by Nrf2 [47]. This module was linked with 189 chemical agents, which were considered potential redox regulators through activating Nrf2 [47]. This provides us an with opportunity to quickly and effectively find novel small non-electrophilic modulators of Nrf2. In this study, all the agents coupled with module 36 were analyzed and screened based on their chemical characteristics and approved information for further biological testing.
2. Results and Discussion
2.1. Screening of Potential Redox Regulators
According to our previous study [47], there are 189 agents attached to the Nrf2 transcription factor. Some of them are known antioxidants such as quercetin, butein, nordihydroguaiaretic acid, myricetin, kaempferol, genistein, ascorbic acid, and ebselen. Moreover, four para-quinones (i.e., tanespimycin, 1,4-chrysenequinone, menadione, and tetroquinone) are also included in this list, which abstract electrons and protons from the thiols of Keap1 and result in the activation of Nrf2 [48]. Therefore, the 189 agents coupled with Nrf2 are likely potential redox regulators.
To reduce the time and money consumption of the drug discovery process, drug repositioning is an important method. In this study, 86 approved drugs were screened out from the 189 agents for further research (Table S1). To find novel small non-electrophilic modulators of Nrf2, all the agents with an electrophilic substructure were deleted from the approved drug list. Finally, 22 approved non-electrophilic drug compounds were selected and divided into seven categories according to their structure characteristics (Table 1). The seven categories are: phenothiazine (type-I), tricyclic antidepressants (type-II), trihexyphenidyl (type-III), phenyl pyridine (type-IV), quinolin-8-substitute (type-V), tamoxifen (type-VI), and hexetidine (type-VII).
2.2. Potency of Candidate Compounds to Activate the Expression of Nrf2-Regulated Cytoprotective Genes
To further inspect the redox regulator candidates’ potency to activate Nrf2, one or two representatives from each category were selected to carry on experimental validation (as shown in Table 2). In order to ensure that the selected structures are representative, the compounds with common forms and large changes in substituents were selected. The time-dependent increases in the expression levels of Nrf2-regulated genes after treatment with known redox regulators and candidates at various time points were sampled and analyzed. In this study, the known redox regulator d,l-sulforaphane served as a positive control, and DMSO or chloroform solvent was used as the negative control. We measured the expression changes of antioxidant genes NQO1, HO-1, and GCLM in cells treated with nine different candidate drugs using quantitative RT-PCR (qRT-PCR). These three genes are well-characterized transcriptional targets of Nrf2 and are widely used in the evaluation of the effect of the Nrf2 regulators [3,9,10]. The expression levels of these genes at 6 h, 12 h, and 24 h after treatment with the nine candidates were measured. The concentrations of the candidates were determined based on the reported information (as shown in Table 2).
All the experimental results are shown in histograms displayed in Figure 1 and Figure S1 to Figure S6. The expression levels of NQO1, HO-1, and GCLM genes were determined using β-actin as a loading control. The experimental results showed that, compared to the negative control, almost all of the nine candidates displayed the function of upregulating antioxidant genes NQO1, HO-1, and GCLM; six of the nine candidates displayed the function of upregulating all of the three genes. Most of the results showed that the expression levels of the three genes increased gradually with the time change. These results preliminarily validated the effectiveness of our drug repositioning tactic.
Among these candidates, astemizole showed the strongest ability to upregulate the antioxidant genes NQO1, HO-1, and GCLM. The scale of the upregulation of the three genes in the cells incubated with astemizole was even larger than that in the cells incubated with d,l-sulforaphane at the 24 h time point, although the testing concentration of astemizole was nearly less than one half of that of d,l-sulforaphane (Figure 2). Further analysis detected that there were similar trends in the upregulation of the antioxidant genes NQO1, HO-1, and GCLM between astemizole and d,l-sulforaphane (Figure 1A,B). They both have the greatest impact on the HO-1 gene and then on the GCLM gene, with the smallest impact being found on the NQO1 gene. From the perspective of the intensity of action with time, d,l-sulforaphane reached a peak in upregulating the antioxidant genes GCLM and HO-1 at 12 h, and astemizole has a significant upregulation of these two genes between 12 h and 24 h. Thus, it may be still not reach its peak at 24 h. The ability of astemizole to upregulate the antioxidant genes has also been observed in a rat module. Lee and colleagues found that oxidative stress response related genes are upregulated in the cardiac tissue and blood mononuclear cells of astemizole treated rats [51].
Astemizole is a second-generation antihistamine drug, which acts as a histamine H1-receptor antagonist and suppresses the formation of edema caused by histamine [52]. Previous researches prevent that edema may result from reactive oxygen species (ROS) and could be relieved by some antioxidants [53]. This suggests that astemizole may serve as an antioxidant. The regulation of ion channels by ROS has been suggested to be associated with some pathological conditions, including liver diseases [54]. Astemizole is also a nonspecific inhibitor of Kv10.1 and Kv11.1 potassium channels, and it could significantly decrease cell proliferation, increase apoptosis, and clearly prevent hepatocellular carcinoma (HCC) development in vivo [55]. HCC represents 80% of primary liver cancers, which are mainly caused by chronic inflammation, with severe oxidative stress leading to fibrosis and then cirrhotic livers [56,57]. Our existing results showed that astemizole is a good potential redox regulator, which could regulate oxidative stress by activating Nrf2. The results may lend evidence to prove that it could be able to regulate histamine receptors and ion channels to achieve the regulation of oxidative stress. Astemizole deserves more pharmacodynamic experimentation to test and verify its feasibility for use as an Nrf2 activator.
Although the abilities of the candidates to upregulate the antioxidant gene GCLM are all lower than that of d,l-sulforaphane ,except for astemizole, several drugs show stronger abilities to upregulate the NQO1 gene than d,l-sulforaphane, such as trifluoperazine (Figure 1C), tamoxifen (Figure 1D), and diphenylpyraline (Figure S4). Moreover, the expression of the HO-1 gene has a larger increase after treatment with trifluoperazine (10 μM) at the 24 h time point, while d,l-sulforaphane has begun to decrease at that time (Figure 1A).
Trifluoperazine is a phenothiazine, and its primary application is for schizophrenia [58]. It has also been described as modifying P-type ATPase activity [59], inhibiting HIV-associated apoptosis [60], and preventing ROS damage by Fe/H2O2 [61]. Trifluoperazine was proven to be able to decrease the intracellular ROS accumulation and alleviate oxidative damage to cells [62]. Therefore, the present finding provides evidence to support the opinion that trifluoperazine may be useful in preventing cell damage in some situations where oxidative stress is expected to take place [62,63].
Tamoxifen is an approved anticancer drug to prevent breast cancer by inducing oxidative damage. However, recently, several clinical resistances to tamoxifen were reported, and the resistance mechanism was demonstrated to be that the breast cancer cells could increase the expression of Nrf2 and the subsequently activated ARE genes could respond to the tamoxifen-induced oxidation [64,65]. Although tamoxifen has the smallest concentration of 1 μM, it still showed the potency to upregulate NQO1, HO-1, and GCLM genes in our experiment (Figure 1D). This is consistent with the reported results and also suggests that reducing the tamoxifen-induced activation of Nrf2 may be a key factor to overcome the clinical resistance to the drug.
3. Materials and Methods
3.1. Data and Reagent Retrieval
The 189 potential redox regulators coupled with Nrf2 were downloaded from the reference [47]. All the tested compounds were purchased from Sigma-Aldrich (St. Louis, MO, USA). The MCF7 cells were purchased from the Cell Bank of the Chinese Academy of Science (http://www.cellbank.org.cn/detail_1.asp?id=152&serial=TCHu%2074).
3.2. Cell Culture Conditions
The MCF7 cells were cultured in minimum essential medium (MEM) (GIBCO, 41500034, Thermo Fisher Scientific Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. Then, the MCF7 cells were seeded in 12-well plates (Corning, 3513) and exposed to different concentrations of test compounds separately. d,l-sulforaphane served as a positive control, while DMSO (dimethyl sulfoxide) or chloroform served as the negative control. After incubation for 6 h, 12 h, or 24 h, the medium was removed and the cells were harvested for further analysis.
3.3. RNA Extraction and qRT-PCR Analysis
The total RNA of the MCF7 cells was isolated using an RNeasy Mini kit (Qiagen, 74104, Hilden, North Rhine-Westphalia, Germany). The quantity and purity of RNA samples were assessed by the a NanoDrop One Microvolume UV-Vis Spectrophotometer (Thermo Scientific, Thermo Fisher Scientific Inc., Waltham, MA, USA), and RNA samples with an A260/A280 ratio of 1.9–2.2 were stored at −80 °C for further analysis. The RNA was reverse transcribed by the TOYOBO (FSQ-101, Kita, Osaka, Japan) reagent kit following the manufacturer’s instructions.
Quantitative real-time RT-PCR analyses of NQO1, HO-1, and GCLM were performed on the LightCycler@96 Real Time PCR system (Roche, Aigle, Vaud, Switzerland) using iTaq Universal SYBR Green Suoermix (Bio-Rad, Hercules, CA, USA). The primers used for qRT-PCR are listed in Table 1. β-actin was used for normalization. The PCR conditions were as follows: 95 °C for 1 min; 40 cycles of degeneration at 95 °C for 10 s, annealing at 57 °C for 10 s, and extension at 72 °C for 15 s; then degeneration at 95 °C for 1 min and annealing at 55 °C for 1 min. Finally, the dissociation curves were generated by increasing the temperature from 65 to 95 °C stepwise by 0.5 °C every 10 s. All the qPCR analyses were performed in triplicate.
4. Conclusions
In this study, 86 approved drugs were screened from the 189 potential Nrf2-related agents derived from cMap data. Among these compounds, 22 non-electrophilic drugs were selected and divided into seven categories for further analysis. Nine candidate compounds from the seven categories were selected to validate their potencies to activate Nrf2. The expression changes of antioxidant genes NQO1, HO-1, and GCLM in MCF7 cells treated with the nine candidate drugs were analyzed at various time points using qRT-PCR. The experimental results showed that almost all of the candidates indeed displayed the ability to upregulate the expressions of the antioxidant genes to a certain extent, and most of the expression quantities increase gradually with time. Astemizole was found to have a greater scale upregulation of the antioxidant genes than the known redox regulator d,l-sulforaphane at a lower testing concentration than that of the control. In addition, several drugs upregulate the NQO1 gene to a greater extent than d,l-sulforaphane, such as trifluoperazine, diphenylpyraline, and tamoxifen. Moreover, the expression of the HO-1 gene has a larger increase after treatment with trifluoperazine (10 μM) at the 24 h time point. The above results show the application value of the drug repositioning tactic in novel non-electrophilic modulators of Nrf2.
Supplementary Materials
Supplementary materials are available online.
Acknowledgments
This work was supported by the Fundamental Research Funds for the Central Universities (Grant number 2662015PY004).
Author Contributions
Hong-Yu Zhang and Qing-Ye Zhang conceived and designed the project; Qing-Ye Zhang, Xin-Yi Chu, Ling-Han Jiang and Meng-Yuan Liu performed and analyzed the data; Qing-Ye Zhang and Xin-Yi Chu designed the experiments; Zhi-Ling Mei performed the experiments; Qing-Ye Zhang and Xin-Yi Chu wrote the manuscript; and Hong-Yu Zhang helped in preparing the manuscript and revised the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| cMap | connectivity map |
| qRT-PCR | quantitative real time polymerase chain reaction |
| ARE | antioxidant response element |
| MEM | minimum essential medium |
| FBS | fetal bovine serum |
| DMSO | dimethyl sulfoxide |
| ROS | reactive oxygen species |
References
- Gacesa, R.; Dunlap, W.C.; Long, P.F. Bioinformatics analyses provide insight into distant homology of the Keap1-Nrf2 pathway. Free Radic. Biol. Med. 2015, 88, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Lacher, S.E.; Lee, J.S.; Wang, X.; Campbell, M.R.; Bell, D.A.; Slattery, M. Beyond antioxidant genes in the ancient Nrf2 regulatory network. Free Radic. Biol. Med. 2015, 88, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Magesh, S.; Chen, Y.; Hu, L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Narayanapillai, S.; Zhang, W.; Sham, Y.Y.; Xing, C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B.G.; Jain, A.D.; Speltz, T.E.; Moore, T.W. Non-electrophilic modulators of the canonical Keap1/Nrf2 pathway. Bioorg. Med. Chem. Lett. 2015, 25, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, P.; Zhao, Y.; Yang, C.; Clark, A.; Leung, T.C.; Chen, X.; Sang, S. Synthesis, evaluation, and metabolism of novel [6]-shogaol derivatives as potent Nrf2 activators. Free Radic. Biol. Med. 2016, 95, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Otsuki, A.; Keleku-Lukwete, N.; Yamamoto, M. Overview of redox regulation by Keap1–Nrf2 system in toxicology and cancer. Curr. Opin. Toxicol. 2016, 1, 29–36. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Chen, F.; Fu, J.; Xu, Y.; Hou, Y.; Kou, H.H.; Zhai, C.; Nelson, M.B.; Zhang, Q.; et al. An overview of chemical inhibitors of the Nrf2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic. Biol. Med. 2016, 99, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, H.C.; Schaap, M.; Baird, L.; Georgakopoulos, N.D.; Fowkes, A.; Thiollier, C.; Kachi, H.; Dinkova-Kostova, A.T.; Wells, G. Design, synthesis, and evaluation of triazole derivatives that induce Nrf2 dependent gene products and inhibit the Keap1-Nrf2 protein-protein interaction. J. Med. Chem. 2015, 58, 7186–7194. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kim, I.-S.; More, S.V.; Kim, B.-W.; Choi, D.-K. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat. Prod. Rep. 2014, 31, 109–139. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Johnson, J.A. Nrf2-a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Done, A.J.; Traustadóttir, T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016, 10, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, T.A; Fahey, J.W.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Stephenson, K.K.; Wade, K.L.; Ye, L.; Talalay, P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: A clinical phase I study. Nutr. Cancer 2006, 55, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.T.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.S.A.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 2007, 28, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.; Torti, S. Curcumin: From ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008, 65, 1631–1652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Munday, R. Dithiolethiones for cancer chemoprevention: Where do we stand? Mol. Cancer Ther. 2008, 7, 3470–3479. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Wille, E.; Calingasan, N.Y.; Tampellini, D.; Williams, C.; Gouras, G.K.; Liby, K.; Sporn, M.; Nathan, C.; Flint Beal, M.; et al. Triterpenoid CDDO-methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2009, 109, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Kaidery, N.A.; Banerjee, R.; Yang, L.; Smirnova, N.A.; Hushpulian, D.M.; Liby, K.T.; Williams, C.R.; Yamamoto, M.; Kensler, T.W.; Ratan, R.R.; et al. Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid. Redox Signal. 2013, 18, 139–157. [Google Scholar] [PubMed]
- Yang, L.; Calingasan, N.Y.; Thomas, B.; Charturvedi, R.K.; Kiaei, M.; Wille, E.J.; Liby, K.T.; Williams, C.; Royce, D.; Risingson, R.; et al. Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS ONE 2009, 4, e5757. [Google Scholar] [CrossRef] [PubMed]
- Stack, C.; Ho, D.; Wille, E.; Calingasan, N.Y.; Williams, C.; Liby, K.; Sporn, M.; Dumont, M.; Beal, M.F. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic. Biol. Med. 2010, 49, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Korner, S.; Kiaei, M. Nrf2/ARE signaling pathway: Key mediator in oxidative stress and potential therapeutic target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Kerns, J.K.; Callahan, J.F.; Moody, C.J. Keap calm, and carry on covalently. J. Med. Chem. 2013, 56, 7463–7476. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: Promises and perils of Nrf2. Pharmacol. Rev. 2012, 64, 1055–1081. [Google Scholar] [CrossRef] [PubMed]
- Tkachev, V.O.; Menshchikova, E.B.; Zenkov, N.K. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochem. Mosc. 2011, 76, 407–422. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P.; Sharkey, J.; Zhang, Y.; Holtzclaw, W.D.; Wang, X.J.; David, E.; Schiavoni, K.H.; Finlayson, S.; Mierke, D.F.; et al. An exceptionally potent inducer of cytoprotective enzymes: Elucidation of the structural features that determine inducer potency and reactivity with Keap1. J. Biol. Chem. 2010, 285, 33747–33755. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Dinkova-Kostova, A.T.; Stephenson, K.K.; Talalay, P. The ‘Prochaska’ microtiter plate bioassay for inducers of NQO1. Methods Enzymol. 2004, 382, 243–258. [Google Scholar] [PubMed]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Talalay, P. Chemical structures of inducers of nicotinamide quinone oxidoreductase 1 (NQO1). Methods Enzymol. 2004, 382, 423–448. [Google Scholar] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Prestera, T.; Zhang, Y.; Spencer, S.R.; Wilczak, C.A.; Talalay, P. The electrophile counterattack response: Protection against neoplasia and toxicity. Adv. Enzyme Regul. 1993, 33, 281–296. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Xu, L.L.; Lu, M.C.; Chen, Z.Y.; Yuan, Z.W.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; Sun, H.P.; You, Q.D. Structure-activity and structure-property relationship and exploratory in vivo evaluation of the Nanomolar Keap1-Nrf2 protein-protein interaction inhibitor. J. Med. Chem. 2015, 58, 6410–6421. [Google Scholar] [CrossRef] [PubMed]
- Cleasby, A.; Yon, J.; Day, P.J.; Richardson, C.; Tickle, I.J.; Williams, P.A.; Callahan, J.F.; Carr, R.; Concha, N.; Kerns, J.K.; et al. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS ONE 2014, 9, e98896. [Google Scholar] [CrossRef] [PubMed]
- Steel, R.; Cowan, J.; Payerne, E.; O’Connell, M.A.; Searcey, M. Anti-inflammatory effect of a cell-penetrating peptide targeting the Nrf2/Keap1 interaction. ACS Med. Chem. Lett. 2012, 3, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Magesh, S.; Chen, L.; Wang, L.; Lewis, T.A.; Chen, Y.; Khodier, C.; Inoyama, D.; Beamer, L.J.; Emge, T.J.; et al. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–3043. [Google Scholar] [CrossRef] [PubMed]
- Jnoff, E.; Albrecht, C.; Barker, J.J.; Barker, O.; Beaumont, E.; Bromidge, S.; Brookfield, F.; Brooks, M.; Bubert, C.; Ceska, T.; et al. Binding mode and structure-activity relationships around direct inhibitors of the Nrf2-Keap1 complex. Chem. Med. Chem. 2014, 9, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, D.; Zeng, W.; Hus, J.C.; McKenzie, A.; Hession, C.; Jin, P.; Bergeron, C.; Lugovskoy, A.; Enyedy, I.; Cuervo, H.; et al. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013, 21, 4011–4019. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-P.; Jiang, Z.-Y.; Zhang, M.-Y.; Lu, M.-C.; Yang, T.-T.; Pan, Y.; Huang, H.-Z.; Zhang, X.-J.; You, Q. Novel protein–protein interaction inhibitor of Nrf2–Keap1 discovered by structure-based virtual screening. Med. Chem. Commun. 2014, 5, 93–98. [Google Scholar] [CrossRef]
- Moehlenkamp, J.D.; Johnson, J.A. Activation of antioxidant/electrophile-responsive elements in IMR-32 human neuroblastoma cells. Arch. Biochem. Biophys. 1999, 363, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gidwani, V.; Sun, Z.; Zhang, D.D.; Wong, P.K. Development of a molecular assay for rapid screening of chemopreventive compounds targeting Nrf2. J. Lab. Autom. 2008, 13, 243–248. [Google Scholar] [CrossRef]
- Westerink, W.M.A.; Stevenson, J.C.R.; Horbach, G.J.; Schoonen, W.G.E.J. The development of RAD51C, cystatin A, p53 and Nrf2 luciferase-reporter assays in metabolically competent HepG2 cells for the assessment of mechanism-based genotoxicity and of oxidative stress in the early research phase of drug development. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2010, 696, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Antioxidants as therapies: Can we improve on nature? Free Radic. Biol. Med. 2014, 66, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Radical medicine: Treating ageing to cure disease. Nat. Rev. Mol. Cell Biol. 2005, 6, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Li, B.; Zhu, Q.; Wang, Y.X.; Zhang, H.Y. Identification of transcription factors for drug-associated gene modules and biomedical implications. Bioinformatics 2014, 30, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Abiko, Y.; Miura, T.; Phuc, B.H.; Shinkai, Y.; Kumagai, Y. Participation of covalent modification of Keap1 in the activation of Nrf2 by tert-butylbenzoquinone, an electrophilic metabolite of butylated hydroxyanisole. Toxicol. Appl. Pharmacol. 2011, 255, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Connectivity Map 02. Available online: https://portals.broadinstitute.org/cmap/ (accessed on 15 July 2016).
- Zhang, Y.; Talalay, P.; Chot, C.-G.; Posnert, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Oh, J.H.; Park, H.J.; Kim, D.G.; Lee, J.H.; Kim, C.Y.; Kwon, M.S.; Yoon, S. Simultaneous gene expression signature of heart and peripheral blood mononuclear cells in astemizole-treated rats. Arch. Toxicol. 2010, 84, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.M.; Brogden, R.N.; Heel, R.C. Astemizole. Drugs 1984, 28, 38–61. [Google Scholar] [CrossRef] [PubMed]
- Özben, T. Pathophysiology of Cerebral Ischemia: Mechanisms involved in neuronal damage. In Free Radicals, Oxidative Stress, and Antioxidants: Pathological and Physiological Significance, 1st ed.; Özben, T., Ed.; Plenum Press: New York, NY, USA, 1998; Volume 296, pp. 163–188. [Google Scholar]
- Ramirez, A.; Vazquez-Sanchez, A.Y.; Camacho, J.; Carrion-Robalino, N. Ion channels and oxidative stress as a potential link for the diagnosis or treatment of liver diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3928714. [Google Scholar] [CrossRef] [PubMed]
- De Guadalupe Chávez-López, M.; Pérez-Carreón, J.I.; Zuñiga-García, V.; Díaz-Chávez, J.; Herrera, L.A.; Caro-Sánchez, C.H.; Acuña-Macías, I.; Gariglio, P.; Hernández-Gallegos, E.; Chiliquinga, A.J.; et al. Astemizole-based anticancer therapy for hepatocellular carcinoma (HCC), and Eag1 channels as potential early-stage markers of HCC. Tumor Biol. 2015, 36, 6149–6158. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Reis, H.M.; Feitelson, M.A. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar]
- Sun, B.; Karin, M. Inflammation and liver tumorigenesis. Front. Med. 2013, 7, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Tardy, M.; Dold, M.; Rr, E.; Leucht, S. Trifluoperazine versus low-potency first-generation antipsychotic drugs for schizophrenia. Cochrane Database Syst. Rev. 2014, 7, CD009396. [Google Scholar]
- Khan, S.Z.; Longland, C.L.; Michelangeli, F. The effects of phenothiazines and other calmodulin antagonists on the sarcoplasmic and endoplasmic reticulum Ca2+ pumps. Biochem. Pharmacol. 2000, 60, 1797–1806. [Google Scholar] [CrossRef]
- Pan, G.; Zhou, T.; Radding, W.; Saag, M.S.; Mountz, J.D.; McDonald, J.M. Calmodulin antagonists inhibit apoptosis of CD4+ T-cells from patients with AIDS. Immunopharmacology 1998, 40, 91–103. [Google Scholar] [CrossRef]
- Castilho, R.F.; Carvalho-Alves, P.C.; Vercesi, A.E.; Ferreira, S.T. Oxidative damage to sarcoplasmic reticulum Ca2+-pump induced by Fe2+/H2O2/ascorbate is not mediated by lipid peroxidation or thiol oxidation and leads to protein fragmentation. Mol. Cell. Biochem. 1996, 159, 105–114. [Google Scholar] [CrossRef] [PubMed]
- An, B.; Chen, Y.; Li, B.; Qin, G.; Tian, S. Ca2+-CaM regulating viability of Candida guilliermondii under oxidative stress by acting on detergent resistant membrane proteins. J. Proteom. 2014, 109, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Souza dos Santos, P.; Saraiva, D.F.; Ferraz da Costa, D.C.; Scofano, H.M.; de Carvalho-Alves, P.C. Trifluoperazine protects brain plasma membrane Ca2+-ATPase from oxidative damaging. Exp. Brain Res. 2007, 177, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Zenebe, W.J.; Parihar, A.; Larson, S.K.; Alidema, E.; Choi, J.; Ghafourifar, P. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res. 2007, 67, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Bekele, R.T.; Venkatraman, G.; Liu, R.-Z.; Tang, X.; Mi, S.; Benesch, M.G.K.; MacKey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.W.; et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: Implications for tamoxifen therapy and resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
The expressions of NQO1, HO-1, and GCLM genes after treatment with the positive control and the potential Nrf2-activating drugs at 6 h, 12 h, and 24 h time points. (A) The results of samples treated with the positive control d,l-sulforaphane (15 μM). (B), (C), and (D) The results of samples treated with astemizole (8 μM), trifluoperazine (10 μM), and tamoxifen (1 μM), respectively. The genes’ expression levels at 0 h are normalized to 1. Bars represent the average standard deviations, n = 3. The significance of the expression fold changes between samples treated with drugs and negative control at the same time points are tested using a paired t-test: * p < 0.05; ** p < 0.01; *** p < 0.001.
Figure 1.
The expressions of NQO1, HO-1, and GCLM genes after treatment with the positive control and the potential Nrf2-activating drugs at 6 h, 12 h, and 24 h time points. (A) The results of samples treated with the positive control d,l-sulforaphane (15 μM). (B), (C), and (D) The results of samples treated with astemizole (8 μM), trifluoperazine (10 μM), and tamoxifen (1 μM), respectively. The genes’ expression levels at 0 h are normalized to 1. Bars represent the average standard deviations, n = 3. The significance of the expression fold changes between samples treated with drugs and negative control at the same time points are tested using a paired t-test: * p < 0.05; ** p < 0.01; *** p < 0.001.


Figure 2.
The comparison of NQO1, HO-1, and GCLM expression changes after treatment with d,l-sulforaphane (15 μM) and astemizole (8 μM) at 6 h, 12 h, and 24 h time points. Bars represent the average standard deviations, n = 3. The significance of the expression fold changes between samples treated with d,l-sulforaphane and astemizole at the same time points are tested using a paired t-test: * p < 0.05; *** p < 0.001.
Figure 2.
The comparison of NQO1, HO-1, and GCLM expression changes after treatment with d,l-sulforaphane (15 μM) and astemizole (8 μM) at 6 h, 12 h, and 24 h time points. Bars represent the average standard deviations, n = 3. The significance of the expression fold changes between samples treated with d,l-sulforaphane and astemizole at the same time points are tested using a paired t-test: * p < 0.05; *** p < 0.001.


{kind=link}
{kind=link}
{kind=link}
Table 1.
The seven selected categories of non-electrophilic redox regulators candidates.
| Type-I | 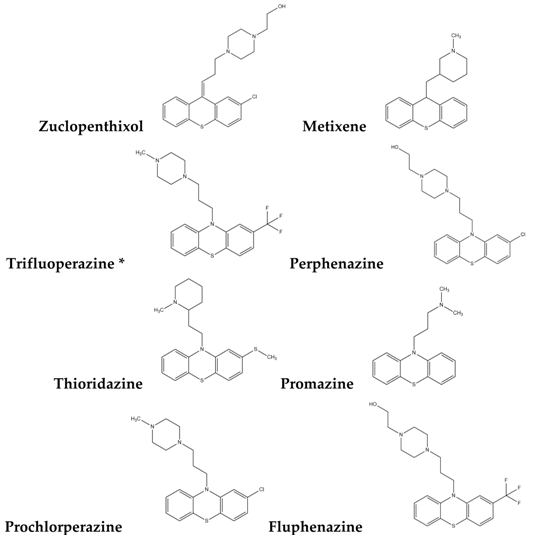 |
| Type-II |  |
| Type-III | 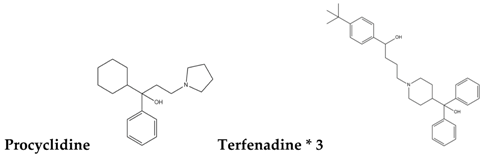 |
| Type-IV |  |
| Type-V |  |
| Type-VI | 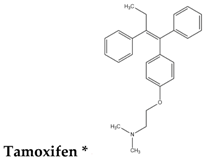 |
| Type-VII |  |
* Selected for further experimental evaluation.
Table 2.
The information about the tested samples.
| Drug | Solvent | Concentration | Sample |
|---|---|---|---|
| Terfenadine | Chloroform | 8 μM [49] | 6 h/12 h/24 h |
| Trimipramine | Chloroform | 10 μM [49] | 6 h/12 h/24 h |
| Quinidine | Chloroform | 10 μM [49] | 6 h/12 h/24 h |
| Hexetidine | Chloroform | 10 μM [49] | 6 h/12 h/24 h |
| Dosulepin | Chloroform | 12 μM [49] | 6 h/12 h/24 h |
| Diphenylpyraline | Chloroform | 12 μM [49] | 6 h/12 h/24 h |
| d,l-Sulforaphane | Chloroform | 15 μM [50] | 6 h/12 h/24 h |
| Tamoxifen | DMSO | 1 μM [49] | 6 h/12 h/24 h |
| Astemizole | DMSO | 8 μM [49] | 6 h/12 h/24 h |
| Trifluoperazine | DMSO | 10 μM [49] | 6 h/12 h/24 h |
| Chloroform (control) | 0 h/6 h/12 h/24 h | ||
| DMSO (control) | 0 h/6 h/12 h/24 h |
All of the data are obtained from MCF7 cell line.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, Q.-Y.; Chu, X.-Y.; Jiang, L.-H.; Liu, M.-Y.; Mei, Z.-L.; Zhang, H.-Y. Identification of Non-Electrophilic Nrf2 Activators from Approved Drugs. Molecules 2017, 22, 883. https://doi.org/10.3390/molecules22060883
AMA Style
Zhang Q-Y, Chu X-Y, Jiang L-H, Liu M-Y, Mei Z-L, Zhang H-Y. Identification of Non-Electrophilic Nrf2 Activators from Approved Drugs. Molecules. 2017; 22(6):883. https://doi.org/10.3390/molecules22060883
Chicago/Turabian StyleZhang, Qing-Ye, Xin-Yi Chu, Ling-Han Jiang, Meng-Yuan Liu, Zhi-Ling Mei, and Hong-Yu Zhang. 2017. "Identification of Non-Electrophilic Nrf2 Activators from Approved Drugs" Molecules 22, no. 6: 883. https://doi.org/10.3390/molecules22060883