Rational Design of Cyclic Antimicrobial Peptides Based on BPC194 and BPC198
by
 , and
, and
Anna D. Cirac
1,2, 
Maria Torné
1,
Esther Badosa
3, 
Emilio Montesinos
3,
Pedro Salvador
2,*,
Lidia Feliu
1,* and
Marta Planas
1,* 1
LIPPSO, Departament de Química, University of Girona, Maria Aurèlia Capmany 69, 17003 Girona, Spain
2
Institut de Química Computacional i Catàlisi i Departament de Química, University of Girona, Maria Aurèlia Capmany 69, 17003 Girona, Spain
3
Laboratory of Plant Pathology, Institute of Food and Agricultural Technology-CIDSAV-XaRTA, University of Girona, Maria Aurèlia Capmany 61, 17003 Girona, Spain
*
Authors to whom correspondence should be addressed.
Molecules 2017, 22(7), 1054; https://doi.org/10.3390/molecules22071054
Submission received: 29 May 2017
/
Revised: 20 June 2017
/
Accepted: 22 June 2017
/
Published: 24 June 2017
(This article belongs to the Special Issue Peptide-Based Drugs and Drug Delivery Systems)
Abstract
:A strategy for the design of antimicrobial cyclic peptides derived from the lead compounds c(KKLKKFKKLQ) (BPC194) and c(KLKKKFKKLQ) (BPC198) is reported. First, the secondary β-structure of BPC194 and BPC198 was analyzed by carrying out molecular dynamics (MD) simulations. Then, based on the sequence pattern and the β-structure of BPC194 or BPC198, fifteen analogues were designed and synthesized on solid-phase. The best peptides (BPC490, BPC918, and BPC924) showed minimum inhibitory concentration (MIC) values <6.2 μM against Pseudomonas syringae pv. syringae and Xanthomonas axonopodis pv. vesicatoria, and an MIC value of 12.5 to 25 μM against Erwinia amylovora, being as active as BPC194 and BPC198. Interestingly, these three analogues followed the structural pattern defined from the MD simulations of the parent peptides. Thus, BPC490 maintained the parallel alignment of the hydrophilic pairs K1–K8, K2–K7, and K4–K5, whereas BPC918 and BPC924 included the two hydrophilic interactions K3–Q10 and K5–K8. In short, MD simulations have proved to be very useful for ascertaining the structural features of cyclic peptides that are crucial for their biological activity. Such approaches could be further employed for the development of new antibacterial cyclic peptides.
1. Introduction
The treatment of plant diseases is a paramount issue in agriculture due to the economic losses caused by bacteria and fungi [1,2]. The main agents used in crop protection, copper derivatives and antibiotics, are environmental contaminants and plant pathogens may develop resistance to them [3,4]. Therefore, the discovery of new compounds that are effective against plant pathogens is essential to meet the challenges of traditional treatments.
Antimicrobial peptides (AMPs) possess favorable properties that make them promising candidates to fulfil the need of useful agents in plant protection [5,6,7,8]. They are short, cationic sequences that contain up to 50% of hydrophobic residues and are able to adopt an amphipathic structure. These peptides display a broad spectrum of activity, selectivity towards microbial targets, and a low frequency in developing microbial resistance [9,10,11,12,13,14,15,16]. Their unique mechanism of action is the basis for these properties. After an initial electrostatic interaction with the anionic phospholipids of the microbial cell membrane, AMPs have been reported to compromise the membrane bilayer via a carpet, barrel stave, toroidal pore or a disordered toroidal pore model. Furthermore, it has been shown that they can interact with intracellular targets [17,18].
The goal in the development of AMPs is to optimize their structural properties in order to enhance their antimicrobial activity. The rational design of AMPs maintaining the crucial features of native antibacterial peptides has led to the development of compounds with remarkable activity. For instance, synthetic cationic peptides V1 to V7 [19] were designed to be structurally similar to cyclic β-sheet peptides, such as protegrin 1, thanatin, and androctonin [20]. To enhance the antimicrobial activity, the common features of AMPs were modulated strategically while preserving the size, symmetry, and amphypathic structure. A similar avenue was pursued by Lee and coworkers [21]. In their study, the cyclic peptide gramicidin S was taken as a reference structure for the design of new biologically active peptides. After a systematic analysis of the effect of the ring size and the alteration of the amphipathicity and hydrophobicity, the authors were able to design a peptide (GS14K4) with a high degree of specificity for microbial cells.
On the other hand, computational tools and, in particular, molecular dynamics (MD) simulations, can provide valuable information on the membrane interaction of AMPs and their subsequent activity. Since the pioneer works of Egberts [22] and Wendoloski [23], significant efforts have been invested in achieving accurate representations of lipid bilayers and lipid–peptide interactions, that have ultimately permitted the elucidation of the mechanism of action of AMPs with atomic resolution [24,25,26,27,28,29,30].
In previous studies, we aimed to find suitable agents to control plant diseases caused by the bacteria Xanthomonas axonopodis pv. vesicatoria, Pseudomonas syringae pv. syringae, and Erwinia amylovora, which are responsible for important economic losses. In particular, X. axonopodis pv. vesicatoria causes the bacterial spot of tomato and pepper, P. syringae pv. syringae is the causal agent of several blight diseases, and E. amylovora causes the fire blight of rosaceous plants [1,2]. In this context, we designed and synthesized a library of 66 cyclic decapeptides with a general structure of c(X5-Phe-X3-Gln), where X is Lys or Leu [31]. The screening of this library against these bacteria led to the identification of the lead peptides c(KKLKKFKKLQ) (BPC194) and c(KLKKKFKKLQ) (BPC198) with minimum inhibitory concentration (MIC) values between 3.1 and 25 μM.
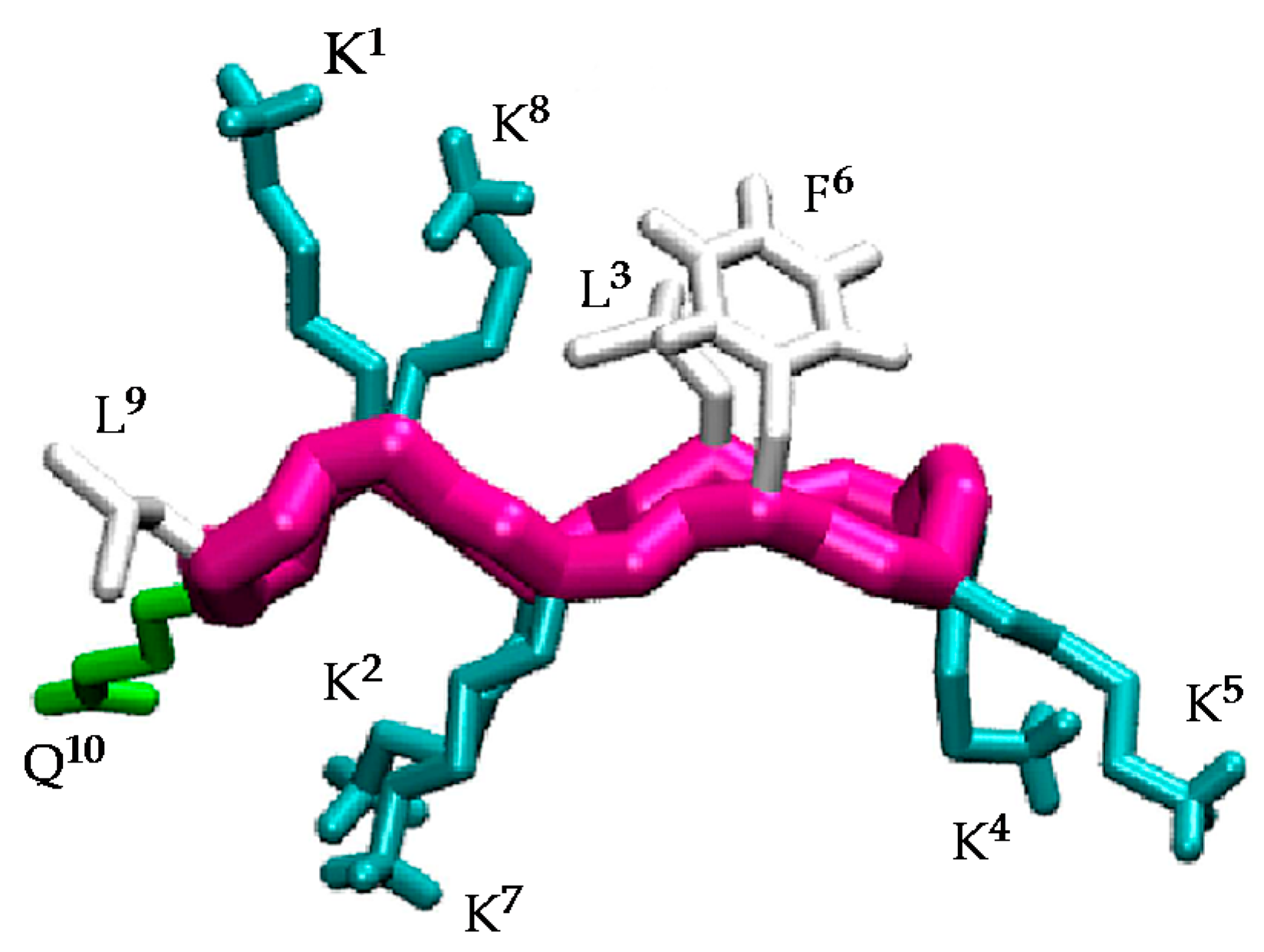
Understanding the structural factors that govern the antimicrobial activity of a library of cyclic peptides is a daunting task. In previous works [30,31,32], a combination of MD simulations and biophysical experiments has highlighted the importance of the specific arrangement of the residues in BPC194 for the formation of stable pores and for inducing the fusion of lipid membranes. In these simulations, a stable β-structure and a symmetric arrangement of the Lys side chains was observed for the peptides inserted in the pore (Figure 1). In particular, the polar side chains of residues K1–K8, K2–K7, and K4–K5 aligned, resulting in a structure reminiscent of an anchor that helped to stabilize the polar head groups of the lipids and the water channel. Based on this, we believe that this structural motif plays a fundamental role in the antimicrobial activity of this peptide and that the precise orientation and alignment of the side chains is essential for its function.
Thus, the focus of this work was to report a strategy for the design of new antimicrobial cyclic peptides. We first analysed the primary structure of a selected number of cyclic peptides of our library to find the structural features that might govern their activity. Then, we performed MD simulations to check the stability and vulnerability of the secondary β-structure of BPC194 and BPC198, with the aim of identifying the factors behind the formation of this structure. Finally, new cyclic peptides were rationally designed following a series of criteria that might help to establish the governing factors for antimicrobial activity.
2. Results and Discussion
2.1. Structural Factors Governing the Activity of Cyclic Peptides BPC194 and BPC198
The β-structure of BPC194 induces the alignment of the polar side-chains of residues K1–K8, K2–K7, and K4–K5, as well as a spatial alignment of the side chains of the hydrophobic residues L3–F6 (Figure 1) [32]. As previously mentioned, this spatial orientation plays an important role in the antimicrobial activity of BPC194. The aligned Lys residues allow the formation of a stable water channel which has been established as the leading mechanism of action of this peptide [32]. Therefore, the rational design of new candidates should be based on a primary structure compatible with the formation of these key motifs.
On the other hand, BPC198 differs from BPC194 in terms of the residues at positions 2 and 3, precluding the alignment of residues K2–K7 and L3–F6, and yet its antimicrobial character is essentially retained [31]. So, it is clear that the primary structure for BPC198 does not follow the same structure–function model induced for BPC194. Thus, it is likely that BPC198 might exhibit an alternative folding that could permit other polar and hydrophobic alignments to take place, whilst maintaining its antimicrobial activity.
In order to validate these assumptions, we undertook MD simulations of both BPC194 and BPC198 peptides in a water environment. In the former case, the aim was to confirm that the active secondary structure in the membrane is also stable in water. It is worth noting that in our previous MD simulations, BPC194 remained in an unfolded state for the first 50 ns, just before the formation of a stable β-structure upon binding to a lipid membrane [30,32].
On the contrary, in the case of BCP198, the aim was to prove that the structure–function model derived from BPC194 is not suitable for this active peptide. It was hoped that the result of these MD simulations would provide an alternative model from which additional new analogues could be rationally designed and tested.
2.2. Stable Secondary Structure of BPC194 and BPC198
All MD simulations were performed for systems containing a single cyclic peptide unit solvated by water. The secondary structure of BPC194 was taken as a reference. The starting peptide structure was characterized by a β-sheet conformation where the polar and hydrophobic side chains were aligned (Figure 2). All systems were simulated in a cubic box of length ca. 4 nm and about 1600–2100 water molecules, with Cl− to neutralize the system.
The spatial arrangement of the side-chain pairs and the stability of the β-structure formation were analyzed along the trajectories. The time evolution of the secondary structure of the peptides was determined with the Dictionary of Secondary Structures of Proteins (DSSP) protocol and through the monitoring of hydrogen bond distances in the backbone. The angle between the normal vector to the (approximate) plane formed by the backbone atoms and the vector representing the side chains was used to represent the spatial arrangement of the side chains, and as a measure of amphipathicity. As shown in Figure 2, small values of this angle (<90°) indicate that the corresponding side chain is arranged on the upper face of the peptide, whereas values larger than 90° correspond to the bottom face.
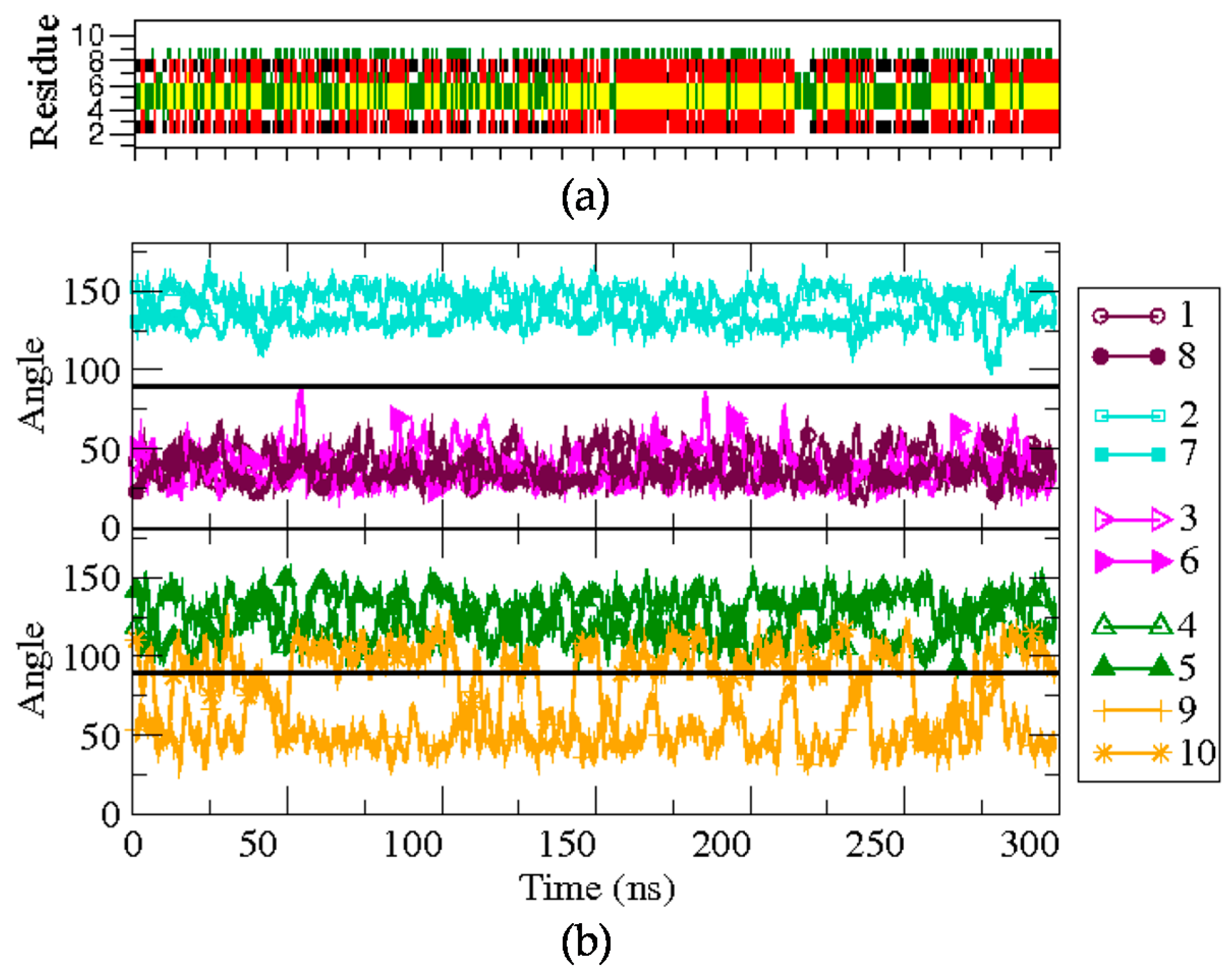
Firstly, BPC194 in its folded state was simulated for over 300 ns in solution. The structural features of the trajectory are depicted in Figure 3 (bottom panel). The side chains of the pair K2–K7 are located at the lower face of the peptide backbone during the whole simulation (>90°), with little angle fluctuations (cyan curves). Similarly, the residues of the pairs K1–K8 and L3–F6 also appear to be aligned, but placed on the upper face of the peptide (<90°). The hydrophilic residues suffer somewhat fewer fluctuations compared to the hydrophobic pair due to the formation of inter-chain hydrogen bonds. The side chains of the remaining pair of residues, K4–K5 and L9–Q10, which are part of the turn regions, show a more flexible behavior. While the values of the angles for the pair K4–K5 often oscillate near 100°, quite close to co-planarity with the backbone, the side chains of residues L9 and Q10 exhibit larger fluctuations and oscillate between the upper and lower face during the whole simulation (orange curves). Despite fluctuations, the overall location of the side-chains gives rise to an amphipathic structure that is stable during the whole simulation.
The DSSP plot (upper panel of Figure 3) shows that the secondary structure in solution is quite stable when starting from the folded state. Indeed, the starting β-structure persists around 39% of the simulation, albeit with minor fluctuations. The hydrogen bond distance between the backbone’s oxygen donor atom of residues K8 or F6 and the hydrogen acceptor of K1 or L3, respectively, was also monitored. The hydrogen bonds between these residues were maintained during most of the simulation time, with average distances of 0.22 ± 0.07 nm (K1–K8) and 0.28 ± 0.06 nm (L3–F6).
As mentioned above, in previously reported MD simulations of BPC194 [30,32], the peptide was essentially in an unfolded state in a water environment for ca 50 ns, prior to forming a β-structure upon binding to lipid membranes. The long simulations in a water environment described here show a seemingly opposite view, where starting from a folded state, the structure remains quite stable for as long as 300 ns. So the lipid membrane and possibly the presence of other peptides induce the formation of a stable specific active structure that is otherwise more difficult to form in solution. Nevertheless, the fact that the active structure is stable in water opens up the possibility of studying other peptides of the library in similar conformations to that of BPC194 in the same conditions.
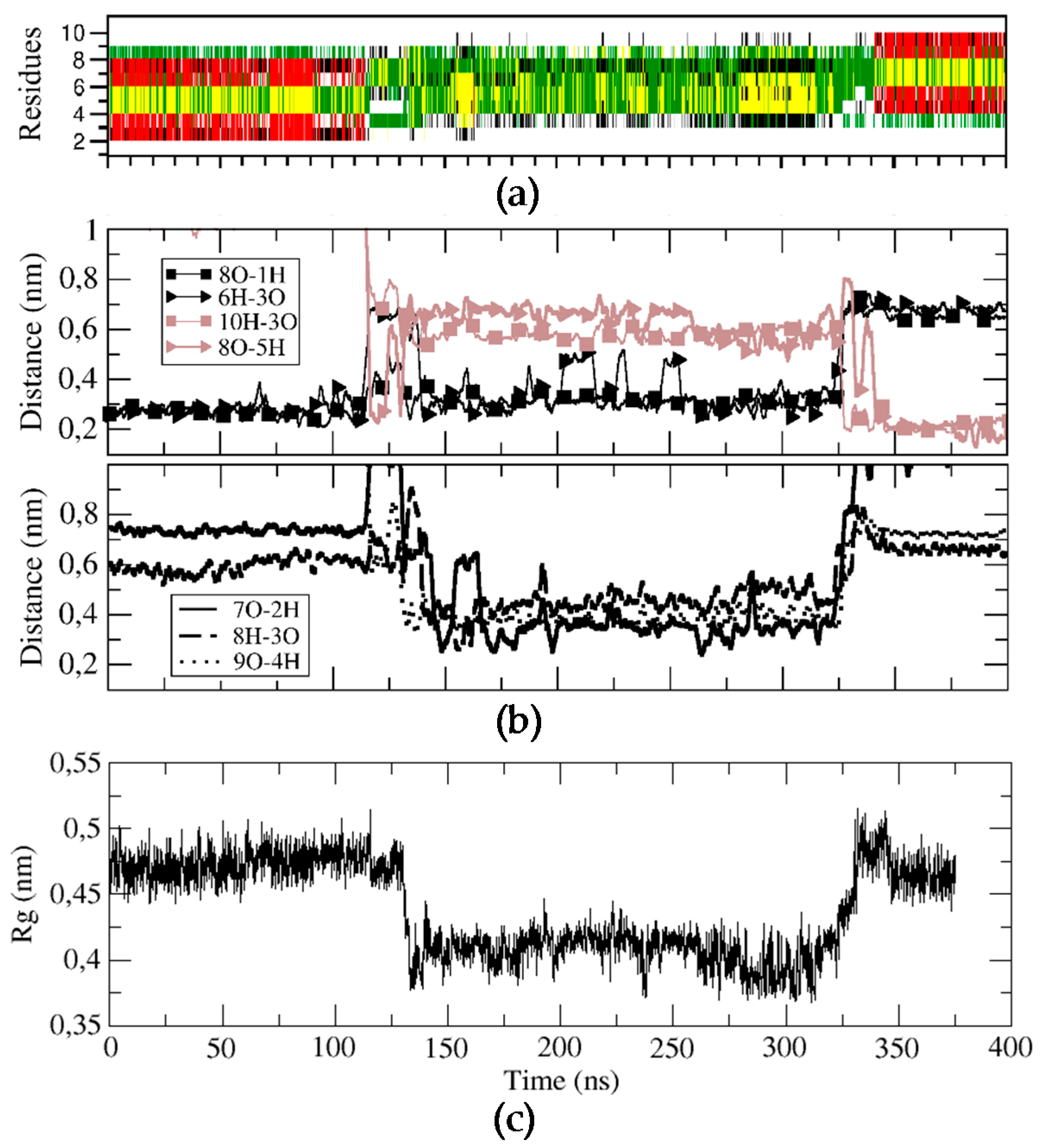
For BPC198, we carried out an analogous simulation starting from the same secondary structure as for BPC194, but replacing the residues at positions 2 and 3 with a Leu and a Lys, respectively. This initial structure was first modeled in silico, followed by energy minimization steps to remove the bad contacts. The qualitative analysis of the possible alignment of the side chains of the amino acids already indicated a mismatch of polar and hydrophobic residues (L2–K7, K3–F6), pointing out that the structure might not be stable (Figure 4c). This fact suggested that BPC198 might exhibit alternative folding with other polar and hydrophobic alignments. This is indeed the case and BPC198 showed a very different behavior in the water phase. As shown in Figure 4a, the peptide unfolded after around 100 ns and the loss of the secondary structure was not immediately followed by an alternative rearrangement of residues to a stable conformation up until 340 ns. During that time lapse, the peptide adopted an unstructured conformation, only presenting turns due to the cyclic nature of the molecule.
To characterize this unfolding/folding process, several key backbone hydrogen bonds were also monitored during the simulation. The initial hydrogen bond interactions between residues K1–K8 and K3–F6 were stable till around 120 ns (Figure 4b). At this point, a sudden fluctuation led to a simultaneous breaking of both hydrogen bonds and a concomitant approach of residues K5–K8 and L3–Q10 to hydrogen bonding distances. Moreover, the structure became more compact, as indicated by the decrease in the radius of gyration of the cyclic peptide (Figure 4, bottom panel) and by the shorter distances observed between residues K7–L2, K8–K3, and L9–K4, which even led to the temporary formation of transient weak hydrogen bonds (Figure 4, third panel). In addition, after around 150 ns, the distances of the initial hydrogen bonds between K1–K8 and K3–F6 slightly increased and oscillated until approximately 340 ns, at which point a large fluctuation occurred leading to a new stable β-structure. This conformation was characterized by strong and stable hydrogen bonds between residues K5–K8 and L3–Q10 (Figure 4, second panel, brown curves).
The role of weak transient interactions on the transition between folded states has already been discussed in the literature. For instance, the folding of a charged 21-amino acid peptide (con-T) showed a multi-step folding pathway, where hydrophobic interactions were not the major driving force of the folding process. Instead, charged–charged interactions and salt-bridge interactions amongst the peptide structure produced different local peptide shapes that, after consecutively broken-formation interactions, led the peptide to its folded state [33].
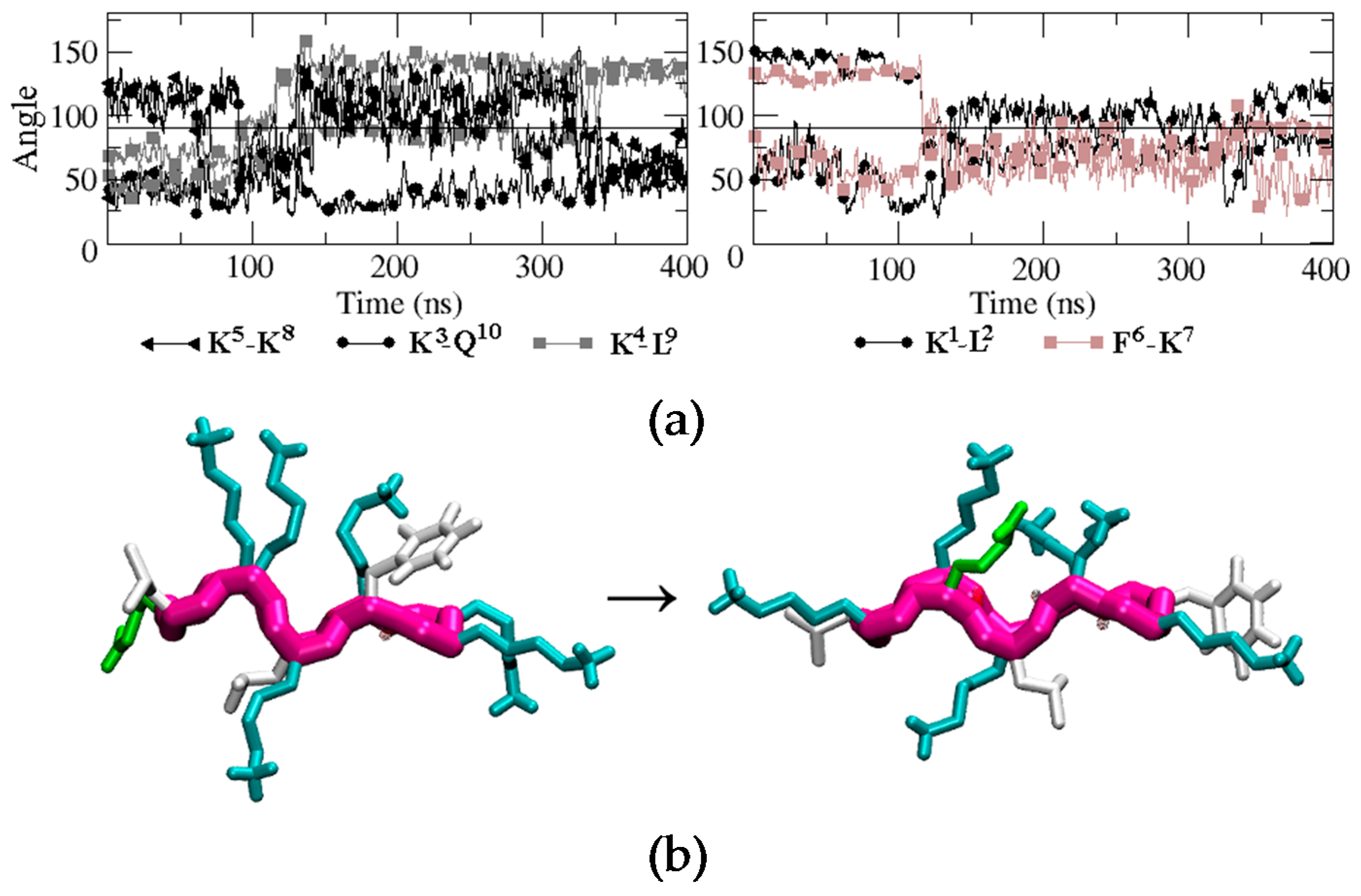
To evaluate how the side chains arrange in space during the unfolding/folding process and whether they align in the upper or in the lower face of the peptide, we analyzed the time evolution of the angles similarly to BPC194. The analysis is displayed in Figure 5. One can observe that the initial structure remained quite constant until around 80 ns. At this point, the side chain of residue Q10 suddenly migrated from the lower face of the peptide to the upper face and temporarily aligned with the K3 side-chain, which remained in the upper face during the whole simulation. A similar process occurred at the same time for the side chain of residue K5. In this case, however, after aligning with the side chain of K8, both started to oscillate around an angle of 90° until the final structure was formed at around 325 ns. These events occurred at a shorter time (80 ns) than the breaking of the intramolecular hydrogen bonds in the backbone (120 ns), suggesting that the latter process was induced by the oscillation of the side chains and not the opposite. In the case of residues K1, L2, F6, and K7, their side chains started to oscillate after 120 ns around a parallel position with respect to the backbone (angle close to 90°). Finally, after 325 ns, just before the formation of the backbone hydrogen bonds that stabilize the final β-structure (340 ns), the side-chain of residue Q10 swung again to the upper face of the peptide and aligned with K3 until the end of the simulation. The residue pair K5–K8 also aligned in the upper face of the conformation, but the angle (around 60°) was larger than that expected for the interaction of two polar residues. The side chains of K1, L2, F6, and K7 ended up in the turn regions of the final β-structure and did not align.
A representative snapshot of the final β-structure of BPC198 is depicted at the bottom of Figure 5. The conformation is reminiscent of the active structure of BPC194 where two pairs of polar side chains are spatially aligned and arranged in one side of the molecule. However, in this case, the side chains of a Lys (polar) and a Leu (hydrophobic) are forced to align in the lower face due to the presence of tight hydrogen bonds in the backbone. So, this structure is not as amphipathic as the one observed for BPC194. To ensure that the new structure was indeed stable in solution, an additional MD simulation was performed starting from the new β-structure, which remained stable along the time course of the simulation (400 ns).
2.3. Design and Synthesis of BPC194 and BPC198 Analogues
With the information gathered from the experiments and simulations described above, we designed new BPC194 and BPC198 analogues to validate our structure–function model and to identify cyclic peptides with antimicrobial activity.
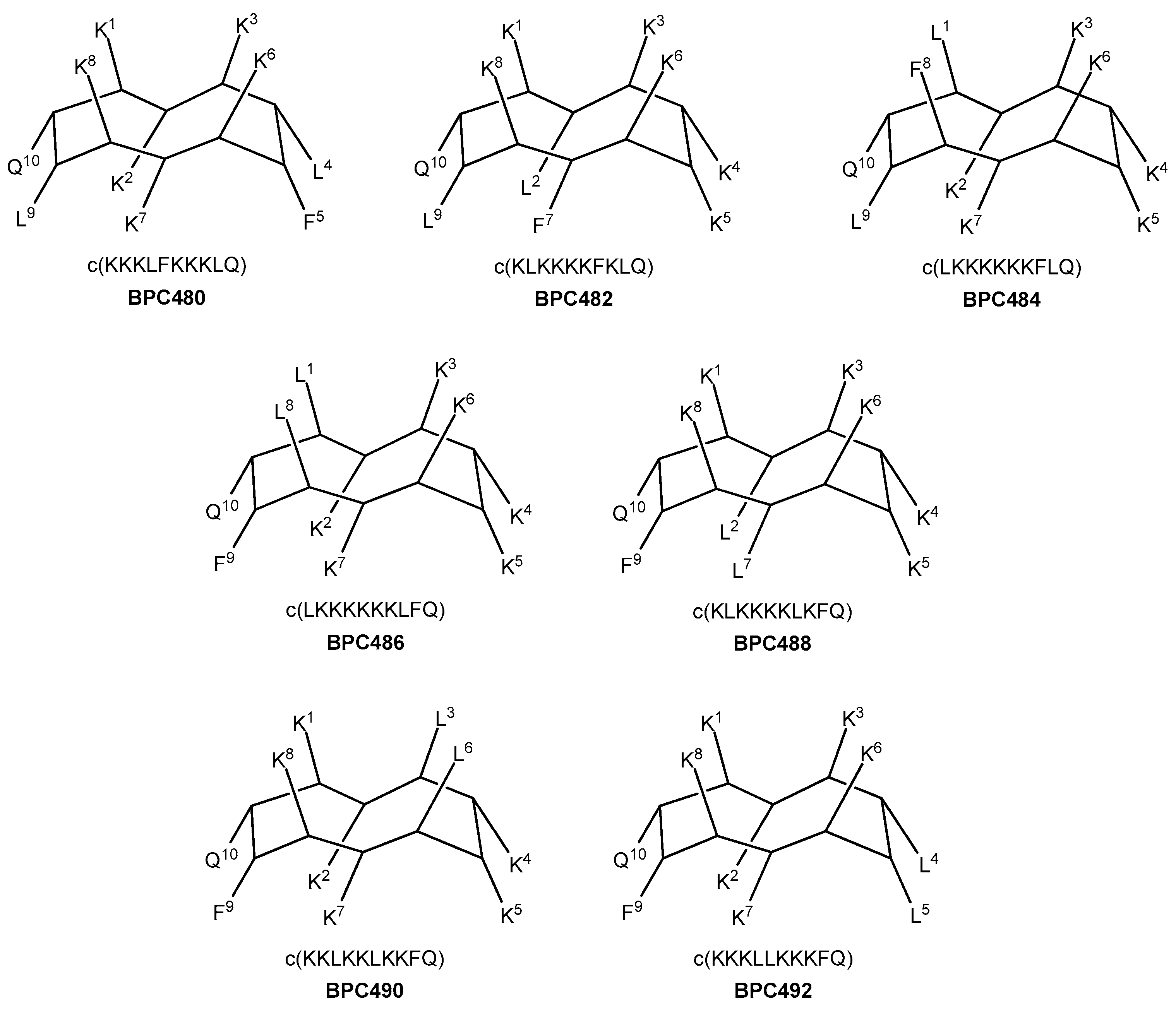
Seven analogues were designed, taking as reference the sequence pattern and active secondary structure of BPC194. The number of hydrophobic and hydrophilic residues, i.e., six Lys, two Leu, one Phe, and one Gln, was maintained, as well as the parallel alignment of the three hydrophilic K–K pairs. Thus, in three analogues (four including BPC194), the hydrophobic residues involved Q–L and L–F pairs (analogues BPC480, BPC482, and BPC484), whereas in the other four analogues, the pairing would take place between the Q–F and L–L residues (analogues BPC486, BPC488, BPC490, and BPC492). The primary structure and the hypothetic conformation of the seven BPC194 analogues are depicted in Figure 6. Peptides differed in terms of the relative distribution of the hydrophilic K–K and hydrophobic pairs on the upper and lower regions of the hypothetic active structure.
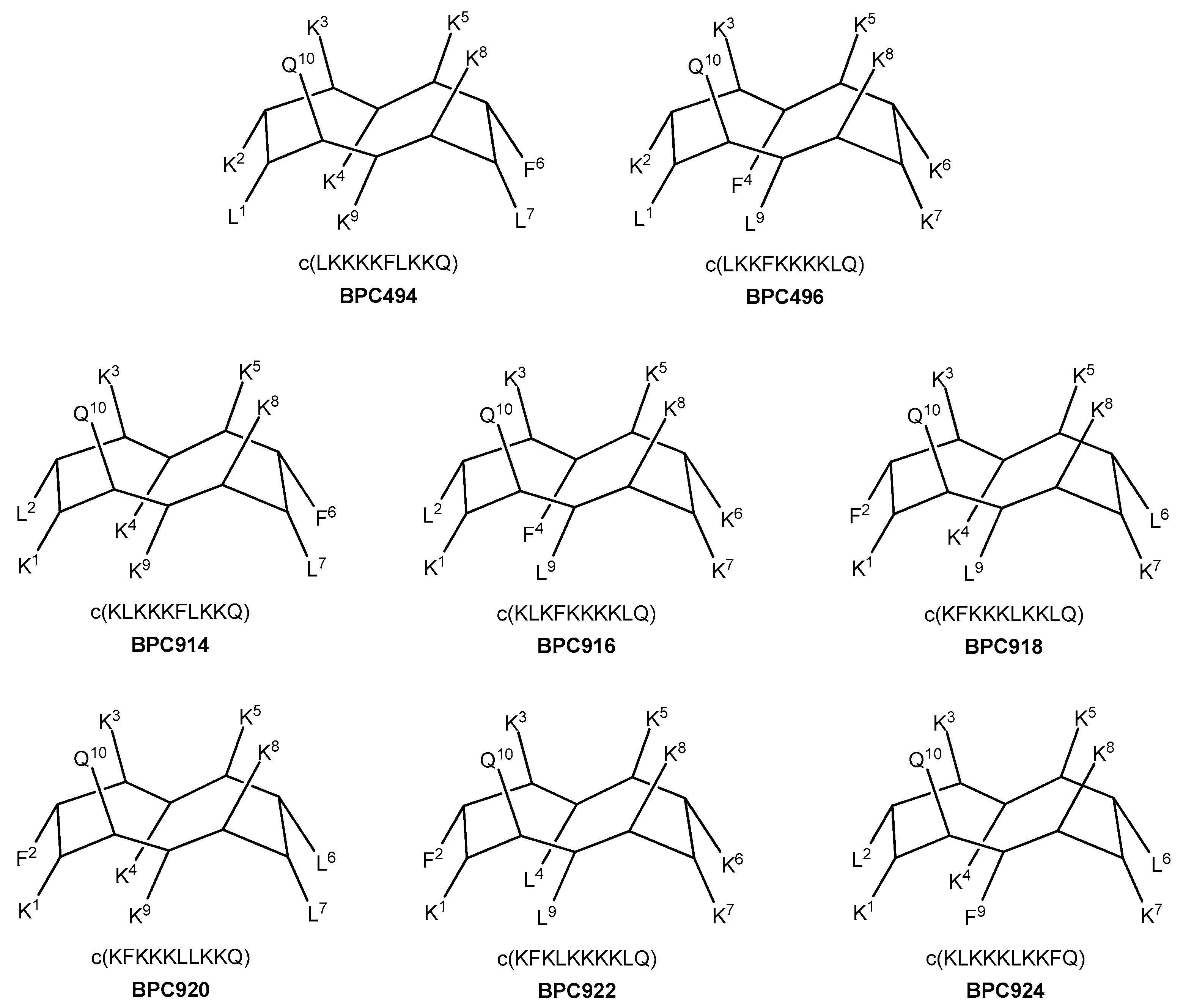
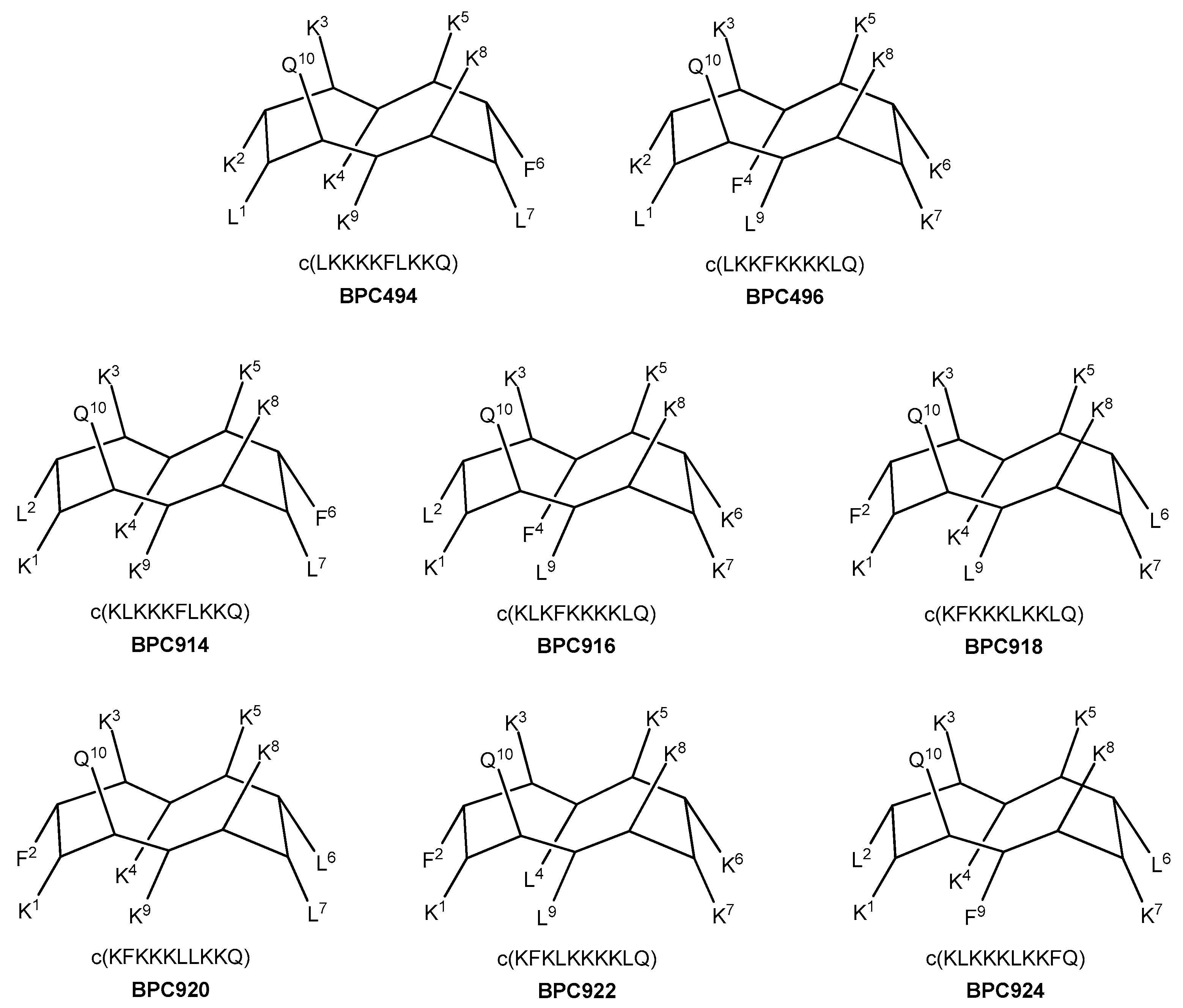
Additional analogues were designed, taking as a reference the new stable β-structure obtained for BPC198 (Figure 7). Six out of the eight analogues designed maintained the K3–Q10 and K5–K8 pairs in the upper face of the molecule (BPC494, BPC496, BPC914, BPC916, BPC920, and BPC922), and differed from BPC198 in the formation of an additional hydrophilic K–K pair, a motif repeatedly observed in this work. The other two analogues, BPC918 and BPC924, included the two hydrophilic interactions present in BPC198, K3–Q10 and K5–K8, and differed only in the position of the hydrophobic residues.
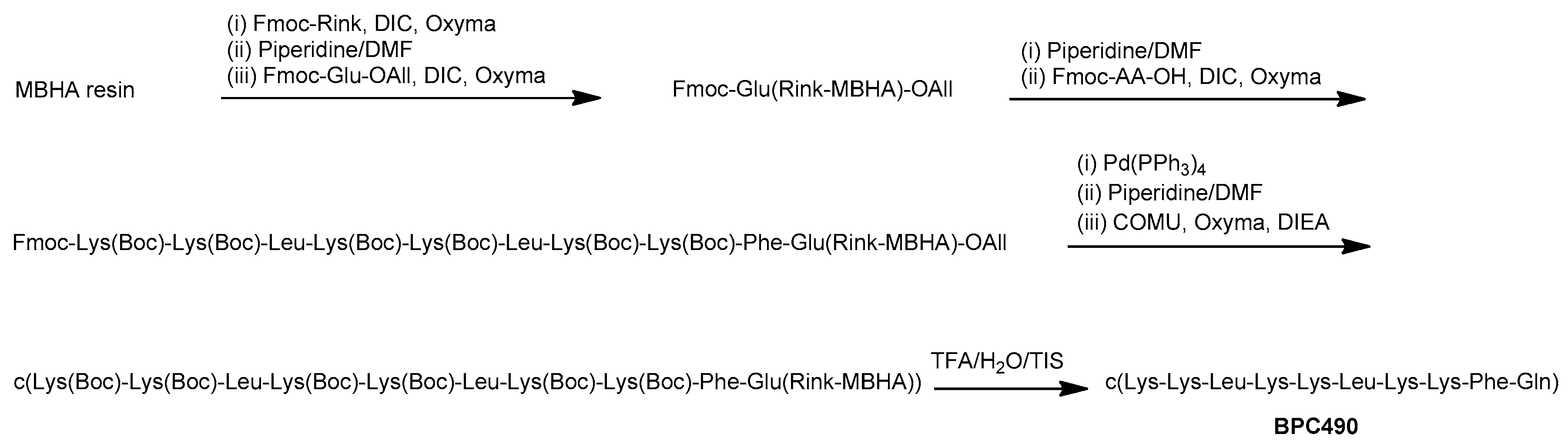
The synthesis of the cyclic decapeptides was carried out on a 4-methylbenzhydrylamine (MBHA) resin (0.4 mmols/g) following a 9-fluorenylmethoxycarbonyl (Fmoc)/tert-butyl (t-Bu)/Allyl strategy by carrying out the solid-phase synthesis of the linear sequence followed by on-resin cyclization (Scheme 1). The tert-butyloxycarbonyl (Boc) group was used as side-chain protection for Lys. Fmoc-Glu-OAll was attached to the solid support through the side chain to serve as a peptide anchoring point to the support, and resulted in a Gln after peptide cleavage from the resin. After the coupling of the Fmoc-Rink linker, the linear peptidyl resin was constructed by sequential Fmoc deprotection and coupling steps. Fmoc group removal was performed with piperidine/DMF (3:7) and amino acid couplings were mediated by N,N-diisopropylcarbodiimide (DIC) and ethyl 2-cyano-2-(hydroxyimino)acetate (Oxyma). Once the synthesis of the linear peptidyl resin was completed, the C-terminal allyl ester cleavage was achieved by treatment with Pd(PPh3)4 in CHCl3-AcOH-N-methylmorpholine (NMM) (37:2:1) and it was followed by Nα-Fmoc group removal. Cyclization was then performed using 1-[(1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylamino-morpholinomethylene)] methanaminium hexafluorophosphate (COMU), Oxyma, and N,N-diisopropylethylamine (DIEA). Finally, the resulting cyclic decapeptides were acidolytically cleaved, analyzed by high-performance liquid chromatography (HPLC), and characterized by mass spectrometry. They were obtained in HPLC purities ranging from 94 to >99%.
2.4. Antibacterial Activity of the Designed Cyclic Decapeptides
The antibacterial activity of the designed cyclic peptides was assayed against P. syringae pv. syringae, X. axonopodis pv. vesicatoria, and E. amylovora at 3.1, 6.2, 12.5, 25, and 50 μM. The results obtained are gathered in Table 1. Peptides BPC194 and BPC198 were also included in these assays for comparison purposes.
All peptides were active against at least one pathogen with MIC < 25 μM. E. amylovora was the least sensitive bacteria to these peptides. This different susceptibility is in agreement with previous reports on this family of cyclic peptides [31,34]. The different membrane composition of the target pathogen would result in a different binding rate of the peptides. Regarding the BPC194 analogues, BPC482, BPC488, and BPC490 displayed significant activity against P. syringae pv. syringae and X. axonopodis pv. vesicatoria with MIC < 12.5 μM. Among them, BPC490 had an MIC value of 3.1 to 6.2 μM against these two bacteria and an MIC value of 12.5 to 25 μM against E. amylovora. This compound was as active as the parent peptide BPC194.
In the case of the eight sequences derived from BPC198, four of them also showed MIC < 12.5 μM against P. syringae pv. syringae and X. axonopodis pv. vesicatoria. Notably, the two sequences BPC918 and BPC924 were as active as BPC198 against P. syringae pv. syringae (MIC of 3.1 to 6.2 μM) and E. amylovora (MIC of 12.5 to 25 μM), and more active against X. axonopodis pv. vesicatoria (MIC of 1.6 to 3.1 μM vs. 3.1 to 6.2 μM).
Noticeably, the analysis of these results reveals that the BPC194 analogue that displays the best biological activity profile, BPC490, is the only one that incorporates the three hydrophilic pairs K1–K8, K2–K7, and K4–K5, differing from BPC194 in only the composition of the hydrophobic pairs. Thus, BPC490 is able to adopt the same spatial disposition of the hydrophilic and hydrophobic pairs as BPC194.
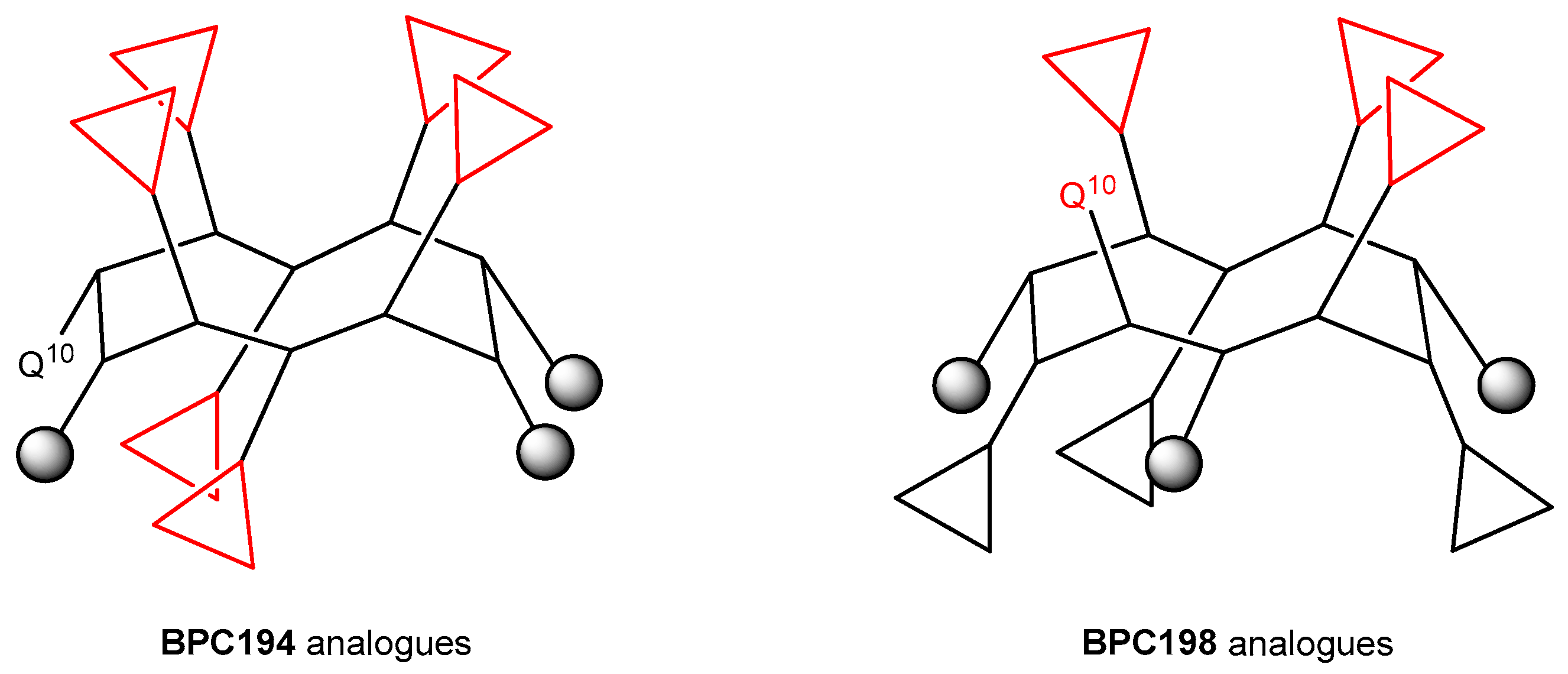
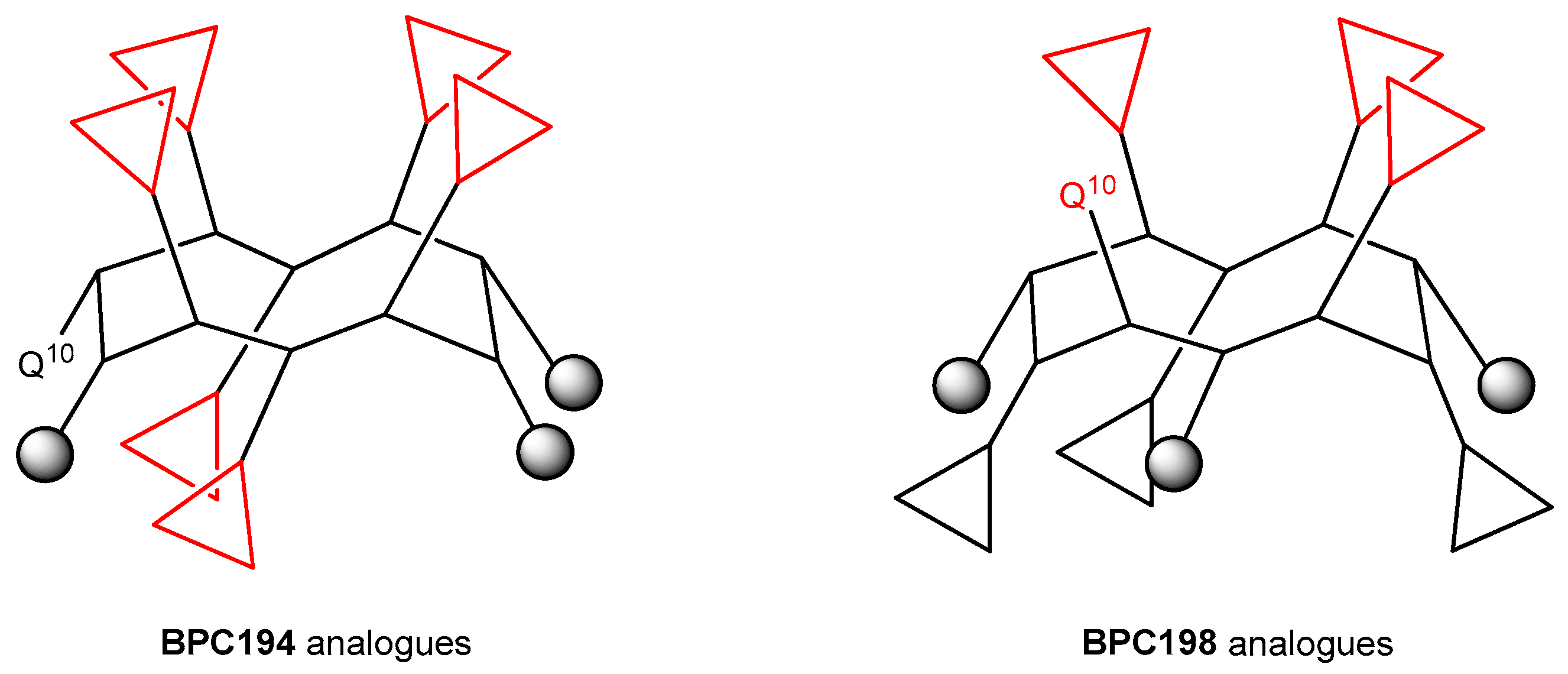
The results from the BPC198 analogues demonstrate that the presence of three hydrophilic K–K pairs leads to a decrease in the antibacterial activity. In fact, the two best analogues, BPC918 and BPC924, contain the two hydrophilic pairs K3–Q10 and K5–K8 present in BPC198 and differ from the parent peptide in the hydrophobic residues at positions 2, 6, and 9 (Leu or Phe). These observations confirm the hypothesis of the structure-function model inferred from our MD simulations for BPC194 and BPC198. The general structure of the active conformation of these two families of cyclic peptides is depicted in Figure 8.
3. Materials and Methods
3.1. Chemicals and Instruments
Commercially available reagents were used throughout, without purification. Solvents were purified and dried by passing them through an activated alumina purification system (MBraun SPS-800, MBraun, Garching, Germany) or by conventional distillation techniques.
All compounds were analyzed under standard analytical HPLC conditions with a Dionex liquid chromatography instrument (Dionex, Germering, Germany). Detection was performed at 220 nm. Analysis was carried out using the Dionex instrument with a Kromasil 100 C18 (40 mm × 4.6 mm, 3.5 μm, Agilent Technologies, Barcelona, Spain) column with a 2%–100% B linear gradient over 17 min at a flow rate of 1 mL/min. Solvent A was 0.1% aqueous trifluoroacetic acid (TFA) and solvent B was 0.1% TFA in CH3CN.
Electrospray ionization (ESI) mass spectrometry (MS) analyses were performed with an Esquire 6000 ESI ion Trap LC/MS (Bruker Daltonics, Madrid, Spain) instrument equipped with an electrospray ion source (Serveis Tècnics de Recerca of the University of Girona). The instrument was operated in the positive ESI(+) ion mode. Samples (5 μL) were introduced into the mass spectrometer ion source directly through an HPLC autosampler (Agilent Technologies). The mobile phase (80:20 CH3CN/H2O at a flow rate of 100 μL/min) was delivered by a 1100 Series HPLC pump (Agilent Technologies). Nitrogen was employed as both the drying and nebulizing gas.
High resolution mass spectrometry (HRMS) spectra were recorded under conditions of ESI with a Bruker MicrOTOF-Q IITM instrument (Bruker, Serveis Tècnics de Recerca of the University of Girona) using a hybrid quadrupole time-of-flight mass spectrometer. Samples were introduced into the mass spectrometer ion source by direct infusion through a syringe pump (Bruker, University of Girona) and were externally calibrated using sodium formate. The instrument was operated in the positive ESI(+) ion mode.
3.2. Synthesis of the Cyclic Peptides
The cyclic peptides were synthesized manually by the solid-phase method using standard Fmoc chemistry on an MBHA resin (0.4 mmol/g). Fmoc-Glu-OAll, Fmoc-Leu-OH, Fmoc-Phe-OH, and Fmoc-Lys(Boc)-OH were used as amino acid derivatives. The resin was swollen with CH2Cl2 (1 × 20 min) and DMF (1 × 20 min), and washed with piperidine/DMF (3:7, 1 × 5 min), DMF (6 × 1 min), and CH2Cl2 (3 × 1 min). Then, the Fmoc-Rink linker (5 eq) was coupled to the resin in the presence of Oxyma (4 eq) and DIC (4 eq) in N,N-dimethylformamide (DMF) for 2 h. After this time, the resin was washed with DMF (6 × 1 min) and CH2Cl2 (3 × 1 min). The linear sequences were prepared by sequential steps of Fmoc group removal, amino acid couplings, and washings. The Fmoc group was removed with piperidine/DMF (3:7, 5 × 10 min). Couplings of the corresponding Fmoc-protected amino acid (4 eq) were mediated by Oxyma (4 eq) and DIC (4 eq) in DMF for 2 h. The completion of the reactions was checked with the Kaiser test [35]. After each coupling and deprotection step, the resin was washed with DMF (6 × 1 min) and CH2Cl2 (3 × 1 min). After the fifth amino acid coupling, N-methyl-2-pyrrolidinone (NMP) was used as the solvent. Once the peptidyl chain was completed, the C-terminal allyl ester was cleaved by treatment with Pd(PPh3)4 (3 eq) in CHCl3/AcOH/NMM (37:2:1) under nitrogen for 3 h. The resin was then washed with tetrahydrofuran (THF) (3 × 2 min), NMP (3 × 2 min), CH2Cl2 (3 × 2 min), DIEA/ CH2Cl2 (1:19, 3 × 2 min), sodium N,N-diethyldithiocarbamate (0.03 M in NMP, 3 × 15 min), NMP (10 × 1 min), CH2Cl2 (3 × 2 min), and NMP (3 × 2 min). Fmoc was removed with piperidine/NMP (3:7, 2 + 10 min), followed by washes with NMP (6 × 1 min) and CH2Cl2 (3 × 1 min). Cyclization was carried out by treating the resin with Oxyma (5 eq), COMU (5 eq), and DIEA (10 eq) in NMP at 25 °C for 24 h. The completion of the reaction was checked with the Kaiser test [35]. Following washes with NMP (6 × 1 min) and CH2Cl2 (3 × 1 min), cyclic decapeptides were cleaved from the resin by treatment with TFA/H2O/triisopropylsilane (TIS) (95:2.5:2.5) for 1 h. The cleavage cocktail was then evaporated and, after diethyl ether extractions, the cyclic peptides were dissolved in H2O, lyophilized, analyzed by reverse-phase (RP)-HPLC, and characterized by mass spectrometry.
BPC480. Retention time, tR = 5.81 min (96% purity). MS (ESI): m/z = 1271.0 [M + H]+, 1293.0 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9424; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8723.
BPC482. tR = 5.96 min (98% purity). MS (ESI): m/z = 1271.0 [M + H]+, 1293.0 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9416; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8700.
BPC484. tR = 5.95 min (97% purity). MS (ESI): m/z = 1271.0 [M + H]+, 1293.0 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9411; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8694.
BPC486. tR = 5.98 min (96% purity). MS (ESI): m/z = 1271.0 [M + H]+, 1293.0 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9427; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8713.
BPC488. tR = 5.92 min (96% purity). MS (ESI): m/z = 1271.0 [M + H]+, 1293.0 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9426; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8721.
BPC490. tR = 6.34 min (95% purity). MS (ESI): m/z = 1271.0 [M + H]+, 1293.0 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9420; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8706.
BPC492. tR = 5.77 min (97% purity). MS (ESI): m/z = 1271.0 [M + H]+, 1293.0 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9426; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8738.
BPC494. tR = 5.80 min (94% purity). MS (ESI): m/z = 1271.0 [M + H]+, 1293.0 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9414; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8723.
BPC496. tR = 5.93 min (97% purity). MS (ESI): m/z = 1271.0 [M + H]+, 1293.0 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9427; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8733.
BPC914. tR = 5.79 min (>99% purity). MS (ESI): m/z = 1270.9 [M + H]+, 1292.9 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9400; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8708.
BPC916. tR = 5.88 min (>99% purity). MS (ESI): m/z = 1270.9 [M + H]+, 1292.9 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9420; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8733.
BPC918. tR = 6.13 min (96% purity). MS (ESI): m/z = 1270.9 [M + H]+, 1292.9 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9401; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8701.
BPC920. tR = 5.91 min (>99% purity). MS (ESI): m/z = 1292.9 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9370; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8683.
BPC922. tR = 6.03 min (99% purity). MS (ESI): m/z = 1270.9 [M + H]+, 1292.9 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9398; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8744.
BPC924. tR = 6.38 min (95% purity). MS (ESI): m/z = 1270.9 [M + H]+, 1292.9 [M + Na]+. HRMS (ESI): calcd. for C62H113N17O11 [M + 2H]2+ 635.9397, found 635.9397; calcd. for C62H112N17O11 [M + H]+ 1270.8722, found 1270.8730.
3.3. Bacterial Strains and Growth Conditions
The following plant pathogenic bacterial strains were used: Erwinia amylovora PMV6076 (Institut National de la Recherche Agronomique, Angers, France); Pseudomonas syringae pv. syringae EPS94 (Institut de Tecnologia Agroalimentària, Universitat de Girona, Spain); and Xanthomonas axonopodis pv. vesicatoria 2133-2 (Instituto Valenciano de Investigaciones Agrarias, Valencia, Spain). All bacteria were stored in Luria Bertani (LB) broth supplemented with glycerol (20%) and maintained at –80 °C. E. amylovora and P. syringae pv. syringae were scrapped from LB agar after growing for 24 h and X. axonopodis pv. vesicatoria after growing for 48 h at 25 °C. The cell material was suspended in sterile water to obtain a suspension of 108 CFU mL−1.
3.4. Antibacterial Activity
Lyophilized peptides were solubilized in sterile Milli-Q water to a final concentration of 1000 μM and sterilized through a 0.22-μm pore filter. For MIC assessment, dilutions of the synthetic peptides were made to obtain a final concentration of 500, 250, 125, 62.5, 31.2, and 15.6 μM. Twenty microlitres of each dilution were mixed in a microtiter plate well with 20 μL of the corresponding suspension of the bacterial indicator and 160 μL of Trypticase Soy Broth (TSB) (BioMèrieux, Craponne, France) to a total volume of 200 μL. Three replicates for each strain, peptide, and concentration were used. Positive controls contained water instead of peptide and negative controls contained peptides without bacterial suspension. Bacterial growth was automatically determined by an optical density measurement at 600 nm (Bioscreen C, Labsystem, Helsinki, Finland). Microplates were incubated at 25 °C with 20 s shaking before hourly absorbance measurements for 48 h. Each experiment was repeated twice. The MIC was taken as the lowest peptide concentration with no growth at the end of the experiment. The inhibition of growth (I) was calculated as a percentage of the positive control using the equation: I = 100 × [(AC − AS)/AC], where AC is the area under the curve of the control, and AS is the area under the curve of a given peptide concentration.
3.5. Molecular Dynamics Simulations
The GROMACS software package [36] was used to perform MD simulations. The GROMOS force-field 43a2 [37] was used to describe the peptide and peptide–solvent interactions. The force-fields were parametrized for use with a group-based twin range cut-off scheme (using cutoffs of 1.0/1.4 nm and a pair-list update frequency of once per 10 steps), including the particle-mesh Ewald (PME) method. The water was modeled using the SPC model [38]. A time step of 2 fs was used. Bond lengths were constrained using the LINCS algorithm [39]. The simulations were performed in the NPT ensemble using periodic boundary conditions. The temperature was weakly coupled (coupling time 0.1 ps) to T = 298 K using the Berendsen thermostat [40]. The pressure was also weakly coupled (coupling time of 1.0 ps and compressibility of 4.5 × 10−5), using an isotropic coupling scheme at 1 bar. The DSSP protocol and the corresponding computer program [41] were employed to analyze the time evolution of the local secondary structure.
4. Conclusions
MD simulations have been used as a tool to recognize the factors that drive the formation of stable β-structures with a partial amphipathic character of cyclic peptides with antimicrobial activity. The stability of the active conformation of peptide BPC194 in the lipid membrane found previously was tested by independent MD simulations in a water solution. Analogous simulations of another active peptide of the library (BPC198) highlighted the key role of the parallel alignment of the side-chain of the residues. At the active conformation of BPC194, the amino acid sequence of BPC198 induced the mismatch of polar and hydrophobic side-chains that eventually led to the unfolding and folding of the peptide to form an alternative compact and stable β-structure. The unfolding pathway of the starting folded structure to the new, amphipathic β-structure is described in atomistic detail. Weak transient interactions assist in the transition between folded states, as already described in the literature for other small peptides.
Based on the stable active structures of peptides BPC194 and BPC198, a set of fifteen cyclic peptides were rationally designed and synthesized. Three of these new analogues exhibited similar antibacterial activity as the parent peptides. These analogues maintain the number and the spatial orientation of the hydrophilic pair interactions of BPC194 and BPC198, differing only in the position of the hydrophobic residues. Therefore, these results confirm the general structure of the active conformation extracted from MD simulations for BPC194 and BPC198. This structure could be the basis of the design of new cyclic decapeptides to be used as antibacterial agents.
Acknowledgments
The authors acknowledge the Serveis Tècnics de Recerca of the University of Girona for the MS analysis. This work was supported by the Ministerio de Economía y Competitividad (MINECO) [grant numbers AGL2009-13255-C02-02/AGR, AGL2012-39880-C02-02 and AGL2015-69876-C2-2-R].
Author Contributions
A.D.C., E.B., E.M., P.S., M.P., and L.F. conceived and designed the experiments; A.D.C., M.T., and E.B. performed the experiments; A.D.C., M.T., E.B., E.M., P.S., M.P., and L.F. analyzed the data; E.B., E.M., P.S., M.P., and L.F. contributed reagents/materials/analysis tools; A.D.C., E.B., E.M., P.S., M.P., and L.F. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Agrios, G.N. Plant Pathology, 5th ed.; Academic Press: San Diego, CA, USA, 2005. [Google Scholar]
- Vidaver, A.K. Uses of antimicrobials in plant agriculture. Clin. Infect. Dis. 2002, 34, 107–110. [Google Scholar] [CrossRef] [PubMed]
- McManus, P.S.; Stockwell, V.O.; Sundin, V.O.; Jones, A.L. Antibiotic use in plant agriculture. Annu. Rev. Phytopathol. 2002, 46, 443–465. [Google Scholar] [CrossRef] [PubMed]
- Sundin, G.W.; Bender, C.L. Ecological and genetic analysis of copper and streptomycin resistance in Pseudomonas syringae pv. syringae. Appl. Environ. Microbiol. 1993, 59, 1018–1024. [Google Scholar] [PubMed]
- Montesinos, E. Antimicrobial peptides and plant disease control. FEMS Microbiol. Lett. 2007, 270, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Marcos, J.F.; Muñoz, A.; Pérez-Payá, E.; Misra, S.; López-García, B. Identification and rational design of novel antimicrobial peptides for plant protection. Annu. Rev. Phytopathol. 2008, 46, 271–301. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, E.; Bardají, E. Synthetic antimicrobial peptides as agricultural pesticides for plant-disease control. Chem. Biodivers. 2008, 5, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Keymanesh, K.; Soltani, S.; Sardari, S. Application of antimicrobial peptides in agriculture and food industry. World J. Microbiol. Biotechnol. 2009, 25, 933–944. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Gorr, S.-U. Antimicrobial peptides: Mechanisms of action and resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [PubMed]
- da Cunha, N.B.; Cobacho, N.B.; Viana, J.F.C.; Lima, L.A.; Sampaio, K.B.O.; Dohms, S.S.M.; Ferreira, A.C.R.; de la Fuente-Núñez, C.; Costa, F.F.; Franco, O.L.; et al. The next generation of antimicrobial peptides (AMPs) as molecular therapeutic tools for the treatment of diseases with social and economic impacts. Drug Discov. Today 2017, 22, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Koh, J.-J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane active antimicrobial peptides: Translating mechanistic insights to design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K. Membrane-active antimicrobial peptides as template structures for novel antibiotic agents. Curr. Top. Med. Chem. 2017, 17, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E. Antimicrobial peptides: An introduction. Methods Mol. Biol. 2017, 1548, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Wang, Y.; Li, G.; Liu, S.; Cui, N.; Liu, S.; Langford, P.R.; Wang, C. The SapA protein is involved in resistance to antimicrobial peptide PR-39 and virulence of Actinobacillus pleuropneumoniae. Front. Microbiol. 2017, 8, 811. [Google Scholar] [CrossRef] [PubMed]
- Raja, Z.; André, S.; Abbassi, F.; Humblot, V.; Lequin, O.; Bouceba, T.; Correia, I.; Casale, S.; Foulon, T.; Sereno, D.; et al. Insight into the mechanism of action of temporin-SHa, a new broad-spectrum antiparasitic and antibacterial agent. PLoS ONE 2017, 12, e0174024. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef] [PubMed]
- Frecer, V.; Ho, B.; Ding, J.L. De novo design of potent antimicrobial peptides. Antimicrob. Agents Chemother. 2004, 48, 3349–3357. [Google Scholar] [CrossRef] [PubMed]
- Dimarcq, J.-L.; Bulet, P.; Hetru, C.; Hoffmann, J. Cysteine-rich antimicrobial peptides in invertebrates. Biopolymers 1998, 47, 465–477. [Google Scholar] [CrossRef]
- Lee, D.L.; Hodges, R.S. Structure activity relationships of de novo designed cyclic antimicrobial peptides based on gramicidin S. Biopolymers 2003, 71, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Egberts, E.; Berendsen, H.J.C. Molecular dynamics simulation of a smectic liquid crystal with atomic detail. J. Chem. Phys. 1988, 89, 3718–3732. [Google Scholar] [CrossRef]
- Wendoloski, J.J.; Kimatian, S.J.; Schutt, C.E.; Salemme, F.R. Molecular dynamics simulation of a phospholipid micelle. Science 1989, 243, 636–638. [Google Scholar] [CrossRef] [PubMed]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial peptides in action. J. Am. Chem. Soc. 2006, 128, 12156–12161. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, S.K.; Larson, R.G. Binding and insertion of alpha-helical antimicrobial peptides in POPC bilayers studied by molecular dynamics simulations. Chem. Phys. Lipids 2004, 132, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Appelt, C.; Eisenmenger, F.; Kühne, R.; Schmieder, P.; Söderhäll, J.A. Interaction of the antimicrobial peptide cyclo(RRWWRF) with membranes by molecular dynamics simulations. Biophys. J. 2005, 89, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Ma, B.; Nussinov, R. Conformational study of the protegrin-1 (PG-1) dimer interaction with lipid bilayers and its effect. BMC Struct. Biol. 2007, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta 2008, 1778, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Orioni, B.; Bocchinfuso, G.; Kim, J.Y.; Palleschi, A.; Grande, G.; Bobone, S.; Park, Y.; Kim, J.I.; Hahm, K.S.; Stella, L. Membrane perturbation by the antimicrobial peptide PMAP-23: A fluorescence and molecular dynamics study. Biochim. Biophys. Acta 2009, 1788, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Cirac, A.D.; Moiset, G.; Mika, J.T.; Koçer, A.; Salvador, P.; Poolman, B.; Marrink, S.J.; Sengupta, D. The molecular basis for antimicrobial activity of pore-forming cyclic peptides. Biophys. J. 2011, 100, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Monroc, S.; Badosa, E.; Besalú, E.; Planas, M.; Bardají, E.; Montesinos, E.; Feliu, L. Improvement of cyclic decapeptides against plant pathogenic bacteria using a combinatorial chemistry approach. Peptides 2006, 27, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Mika, J.T.; Moiset, G.; Cirac, A.D.; Feliu, L.; Bardají, E.; Planas, M.; Sengupta, D.; Marrink, S.J.; Poolman, B. Structural basis for the enhanced activity of cyclic antimicrobial peptides: The case of BPC194. Biochim. Biophys. Acta 2011, 1808, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.C.; Ho, M.H.; Wang, W.H.; Sun, Y.C. Molecular dynamics simulation of folding of a short helical peptide with many charged residues. J. Phys. Chem. B 2005, 109, 19980–19986. [Google Scholar] [CrossRef] [PubMed]
- Güell, I.; Vilà, S.; Badosa, E.; Montesinos, E.; Feliu, L.; Planas, M. Design, synthesis, and biological evaluation of cyclic peptidotriazoles derived from BPC194 as novel agents for plant protection. Pept. Sci. 2017, 108, e23012. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Krüger, P.; Billeter, S.R.; Mark, A.E.; Eising, A.A.; Scott, W.R.P.; Hüneberger, P.H.; Tironi, I.G. Biomolecular Simulation: The GROMOS96 Manual and User Guide; Biomos: Zurich, Switzerland, 1996. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. Intermol. Forces, 1981, 14, 331–342. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1471. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Nola, A.D.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
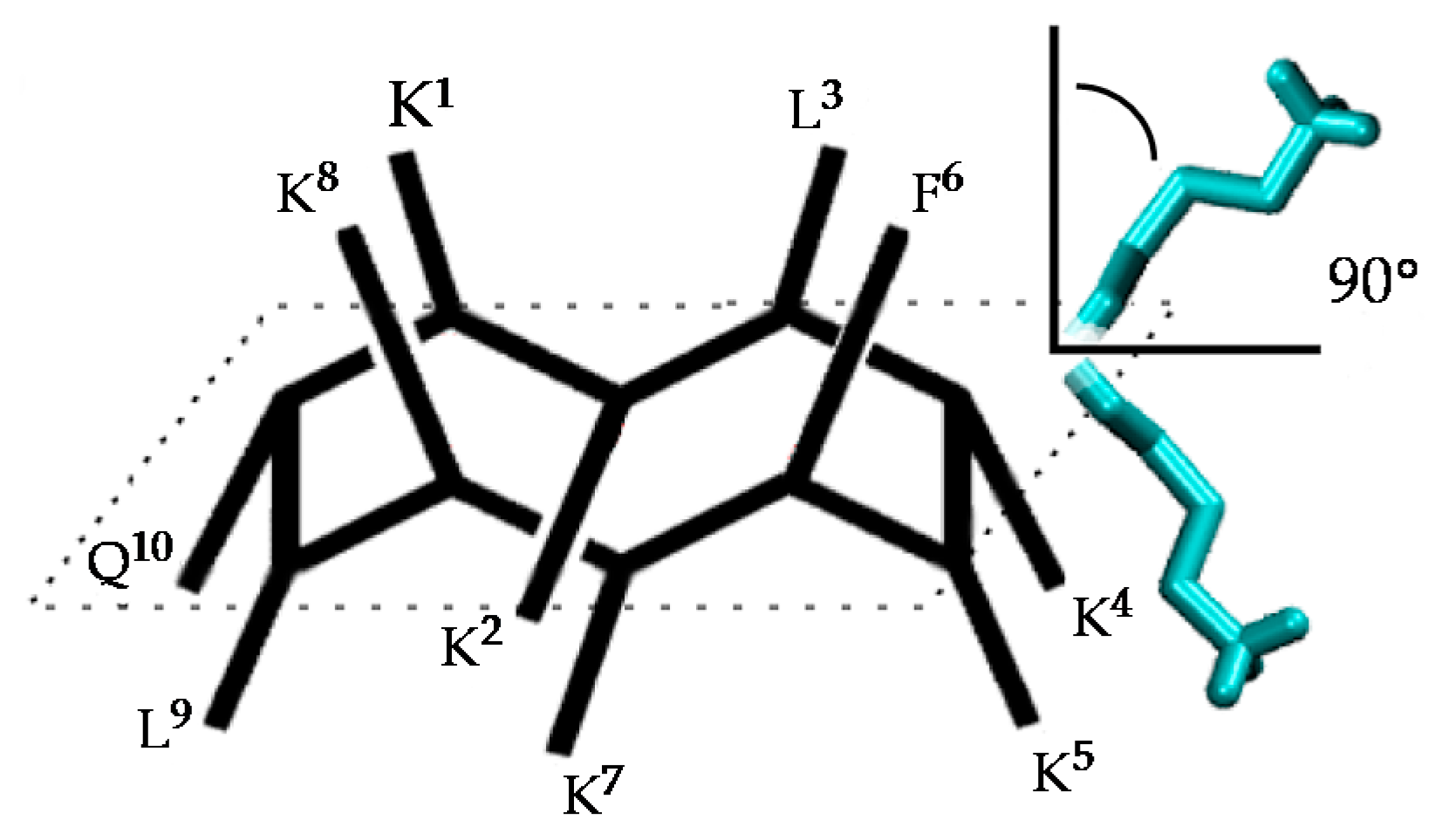
Sample Availability: Not available. |
Figure 1.
Structure of the active conformation of BPC194 showing the side-chain alignments.
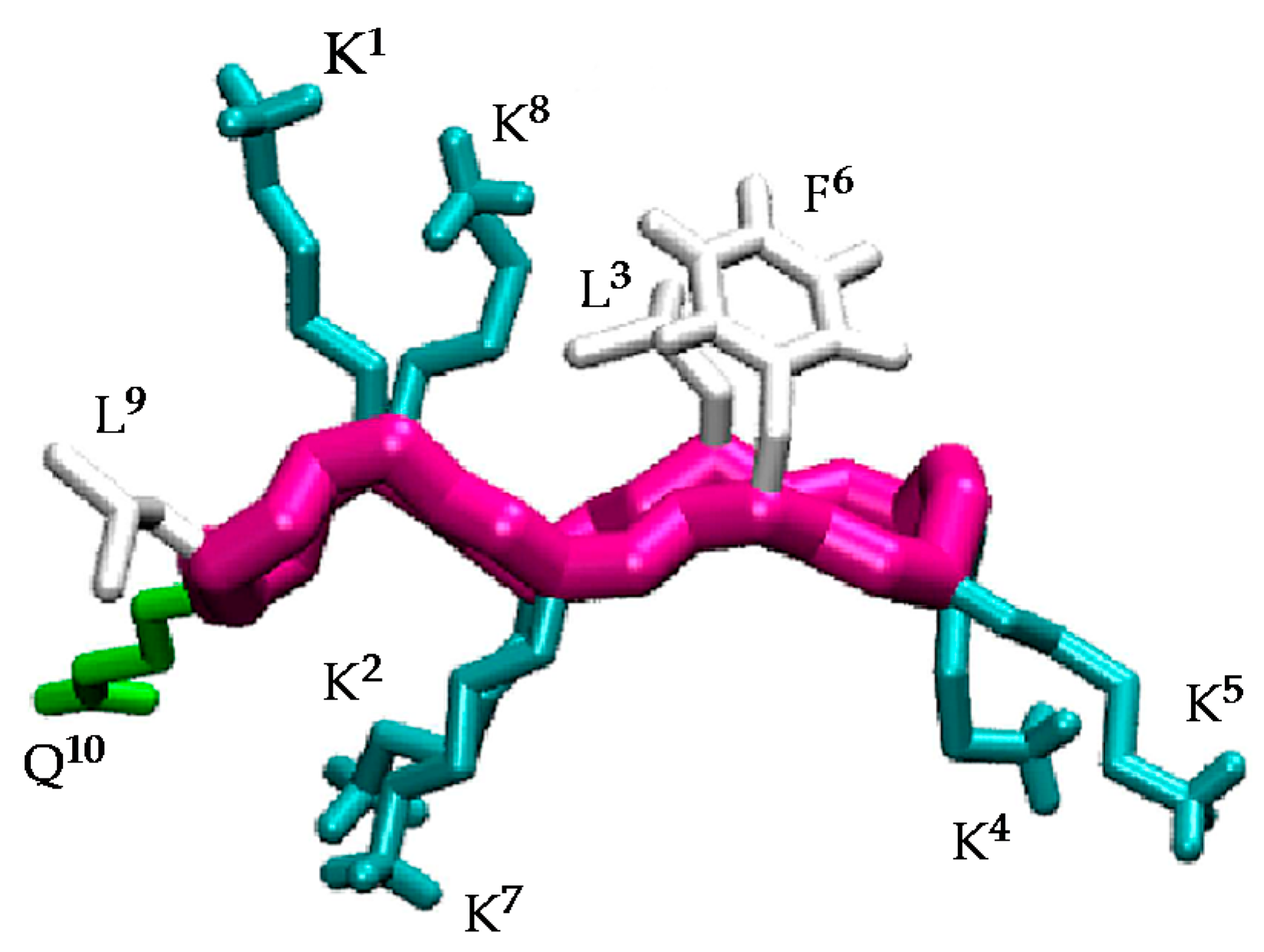
Figure 2.
Structure of the active conformation of BPC194 and representation of the angle formed between the normal vector to the backbone plane and a vector representing a side chain.
Figure 2.
Structure of the active conformation of BPC194 and representation of the angle formed between the normal vector to the backbone plane and a vector representing a side chain.
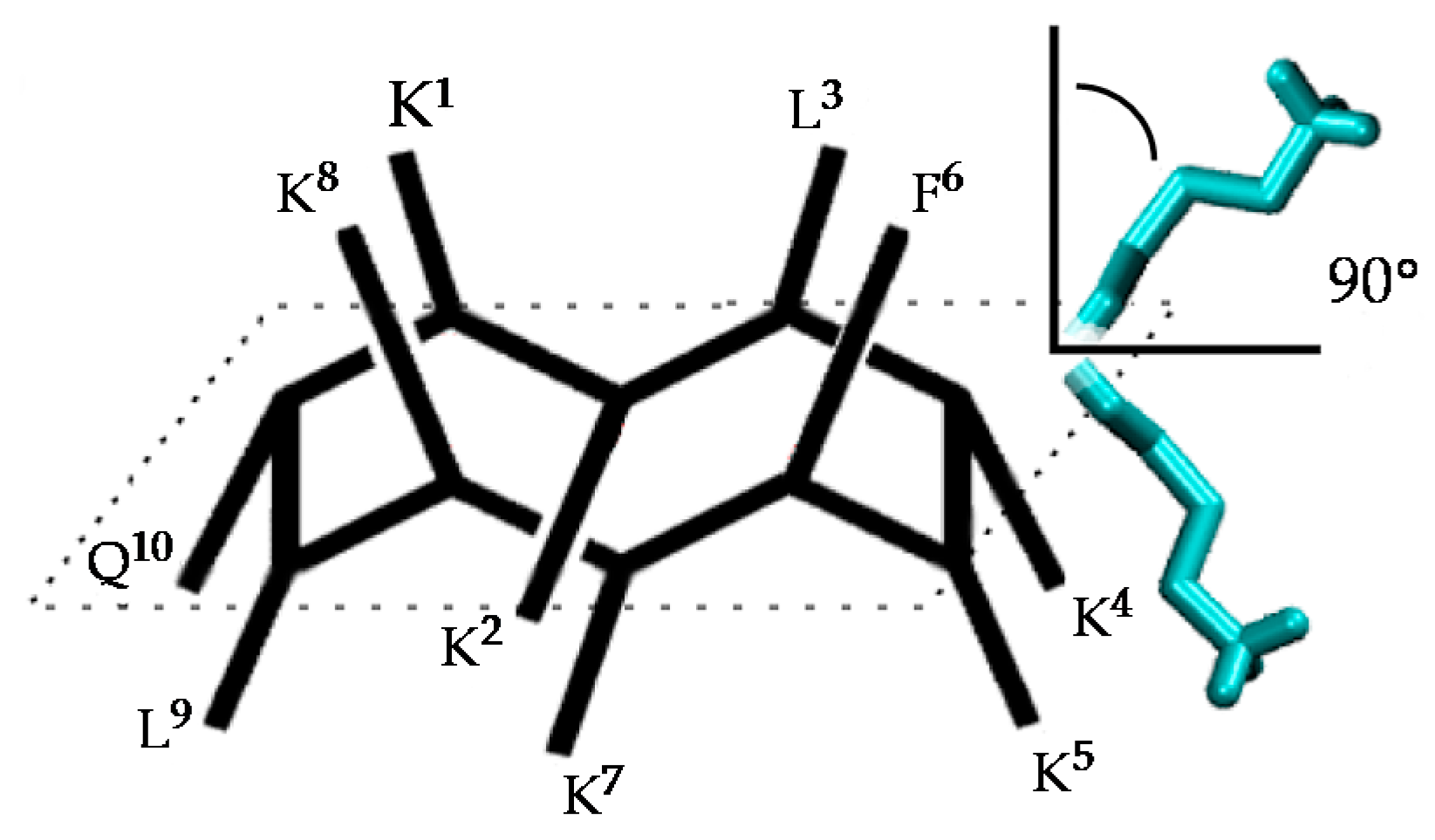
Figure 3.
Structural characterization of the BPC194 simulation. (a) The secondary structure as a function of time is calculated by DSSP. The colors denote the β-sheet (red), the turn (yellow), β-bridge (black), bend (green), and random coil elements (white/empty). (b) Time evolution of the angle formed between the normal vector to the backbone plane and a vector representing each of the side chains.
Figure 3.
Structural characterization of the BPC194 simulation. (a) The secondary structure as a function of time is calculated by DSSP. The colors denote the β-sheet (red), the turn (yellow), β-bridge (black), bend (green), and random coil elements (white/empty). (b) Time evolution of the angle formed between the normal vector to the backbone plane and a vector representing each of the side chains.

Figure 4.
Structural characterization of the BPC198 simulation. (a) DSSP plot, (b) time evolution of the relevant hydrogen bond distances and (c) radius of gyration (nm) of the backbone structure along the trajectory.
Figure 4.
Structural characterization of the BPC198 simulation. (a) DSSP plot, (b) time evolution of the relevant hydrogen bond distances and (c) radius of gyration (nm) of the backbone structure along the trajectory.
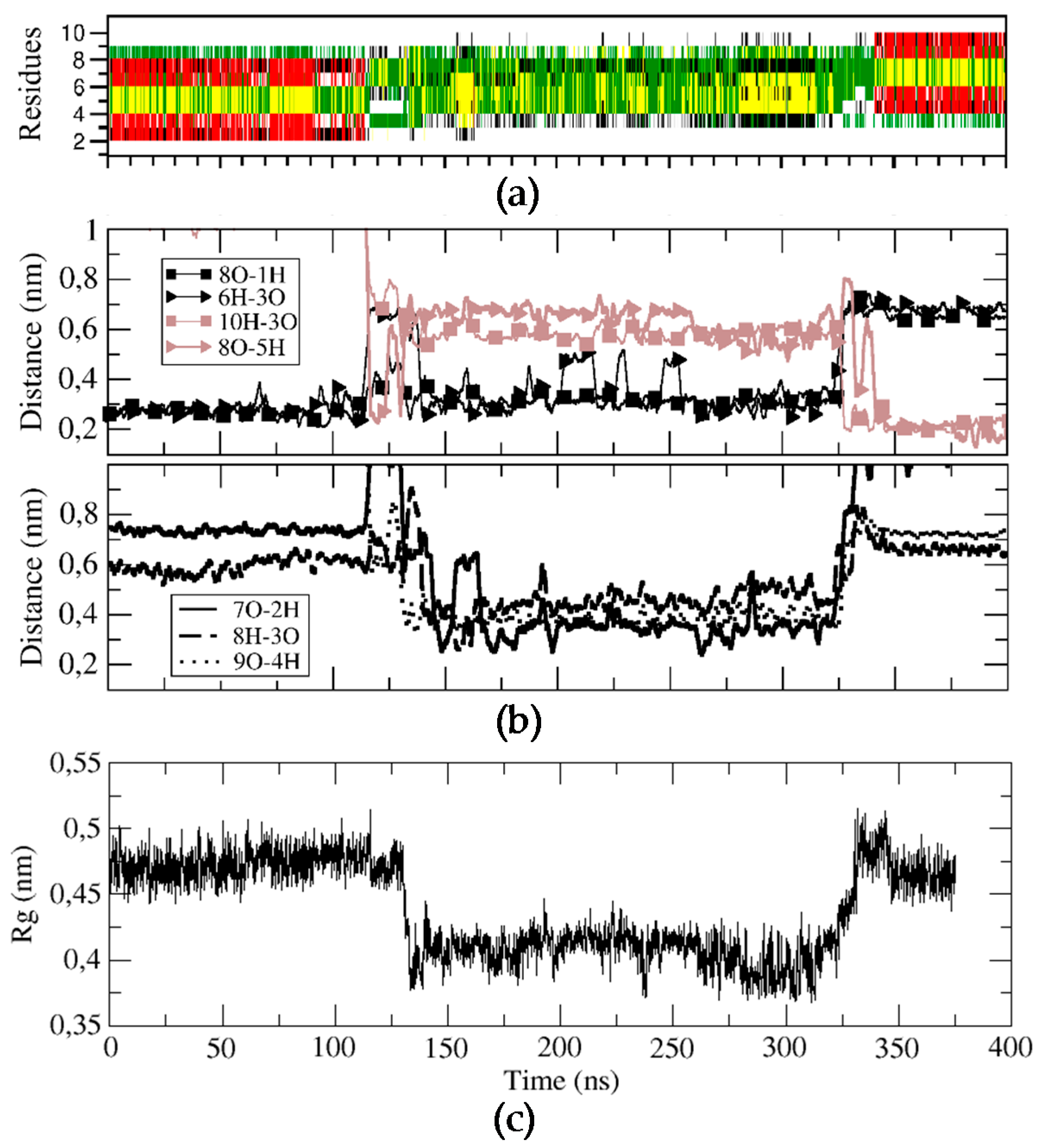
Figure 5.
(a) Time evolution of the angle formed between the normal vector to the backbone plane and the vector representing each of the side chains. Black, brown, and grey curves correspond to hydrophilic, hydrophobic, and hydrophobic-hydrophilic pair residues, respectively. (b) Snapshots of BPC198 at 0 ns (left) and at 350 ns (right).
Figure 5.
(a) Time evolution of the angle formed between the normal vector to the backbone plane and the vector representing each of the side chains. Black, brown, and grey curves correspond to hydrophilic, hydrophobic, and hydrophobic-hydrophilic pair residues, respectively. (b) Snapshots of BPC198 at 0 ns (left) and at 350 ns (right).
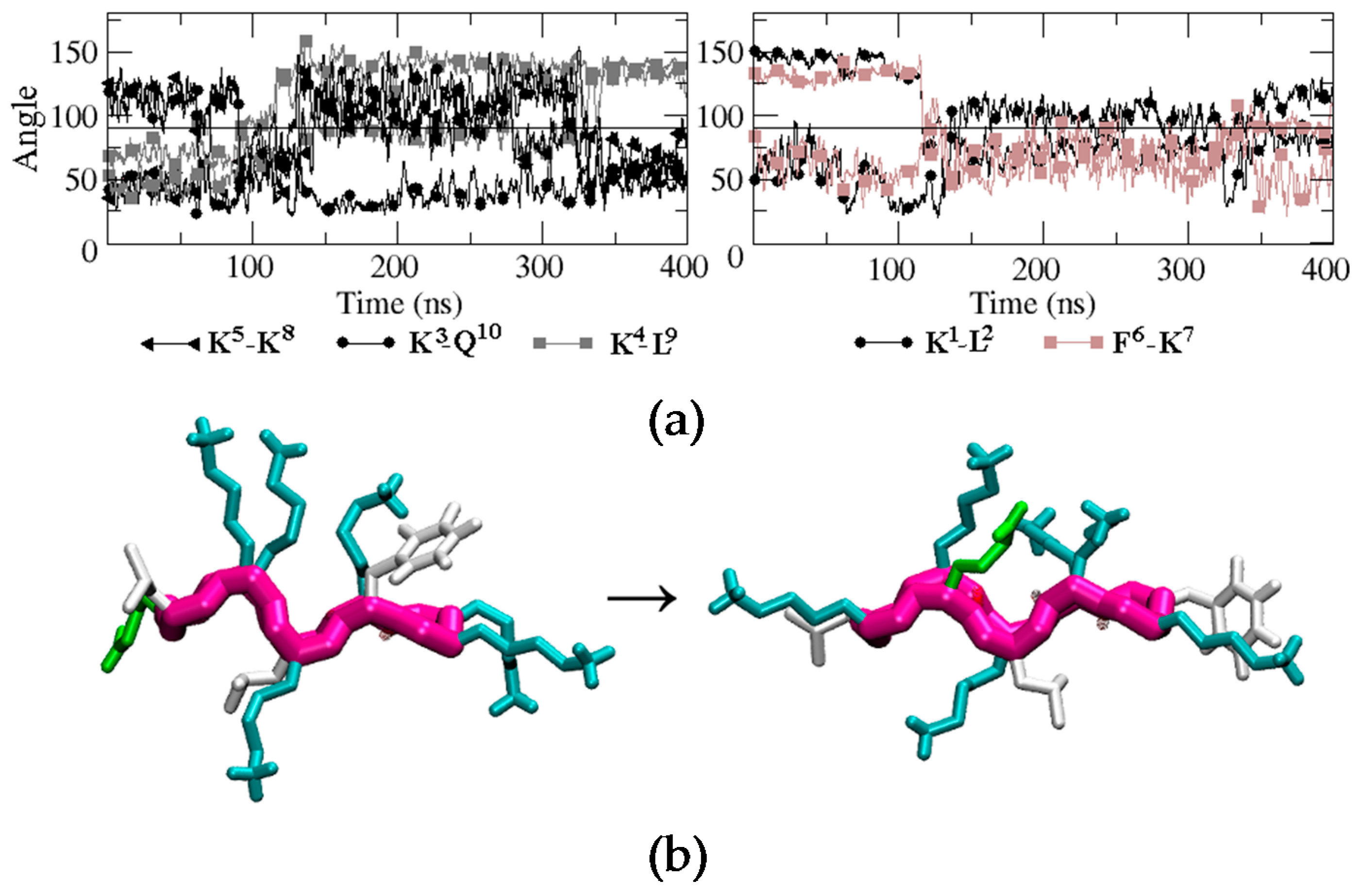
Figure 6.
Sequence and hypothetic structure of the BPC194 analogues.
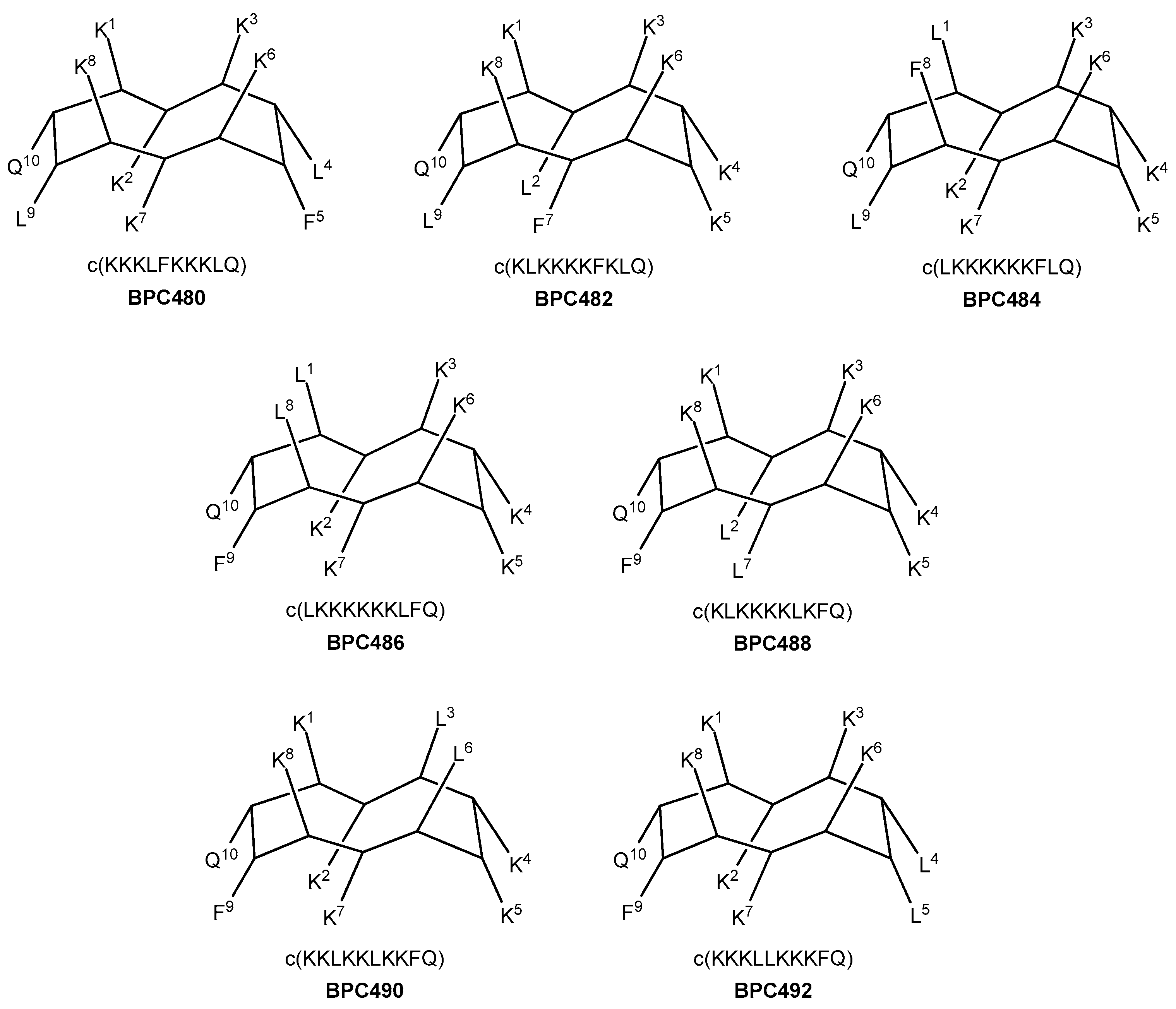
Figure 7.
Sequence and hypothetic structure of the BPC198 analogues.
Scheme 1.
General strategy for the synthesis of the cyclic decapeptides represented for BPC490.

Figure 8.
General structure of the active conformation of BPC194 and BPC198 analogues. Triangles stand for hydrophilic Lys residues and grey circles stand for hydrophobic Leu or Phe residues.
Figure 8.
General structure of the active conformation of BPC194 and BPC198 analogues. Triangles stand for hydrophilic Lys residues and grey circles stand for hydrophobic Leu or Phe residues.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Antibacterial activity (MIC) of the cyclic decapeptides.
| Code | MIC Intervals (μM) | ||
|---|---|---|---|
| Pss a | Xav a | Ea a | |
| BPC194 | 3.1-6.2 | 3.1–6.2 | 6.2–12.5 |
| BPC198 | 3.1–6.2 | 3.1–6.2 | 12.5–25 |
| BPC480 | 12.5–25 | 12.5–25 | >50 |
| BPC482 | 6.2–12.5 | 6.2–12.5 | >50 |
| BPC484 | 12.5–25 | 25–50 | >50 |
| BPC486 | 12.5–25 | >50 | >50 |
| BPC488 | 6.2–12.5 | 6.2–12.5 | >50 |
| BPC490 | 3.1–6.2 | 3.1–6.2 | 12.5–25 |
| BPC492 | 12.5–25 | 12.5–25 | >50 |
| BPC494 | 6.2–12.5 | 6.2–12.5 | >50 |
| BPC496 | 6.2–12.5 | 6.2–12.5 | >50 |
| BPC914 | 12.5–25 | 12.5–25 | >50 |
| BPC916 | 6.2–12.5 | 12.5–25 | >50 |
| BPC918 | 3.1–6.2 | 1.6–3.1 | 12.5–25 |
| BPC920 | 12.5–25 | 25–50 | >50 |
| BPC922 | 12.5–25 | 12.5–25 | >50 |
| BPC924 | 3.1–6.2 | 1.6–3.1 | 12.5–25 |
a Pss stands for Pseudomonas syringae pv. syringae; Xav stands for Xanthomonas axonopodis pv. vesicatoria; Ea stands for Erwinia amylovora.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cirac, A.D.; Torné, M.; Badosa, E.; Montesinos, E.; Salvador, P.; Feliu, L.; Planas, M. Rational Design of Cyclic Antimicrobial Peptides Based on BPC194 and BPC198. Molecules 2017, 22, 1054. https://doi.org/10.3390/molecules22071054
AMA Style
Cirac AD, Torné M, Badosa E, Montesinos E, Salvador P, Feliu L, Planas M. Rational Design of Cyclic Antimicrobial Peptides Based on BPC194 and BPC198. Molecules. 2017; 22(7):1054. https://doi.org/10.3390/molecules22071054
Chicago/Turabian StyleCirac, Anna D., Maria Torné, Esther Badosa, Emilio Montesinos, Pedro Salvador, Lidia Feliu, and Marta Planas. 2017. "Rational Design of Cyclic Antimicrobial Peptides Based on BPC194 and BPC198" Molecules 22, no. 7: 1054. https://doi.org/10.3390/molecules22071054