Enantioselective Michael Addition of Cyclic β-Diones to α,β-Unsaturated Enones Catalyzed by Quinine-Based Organocatalysts


Abstract
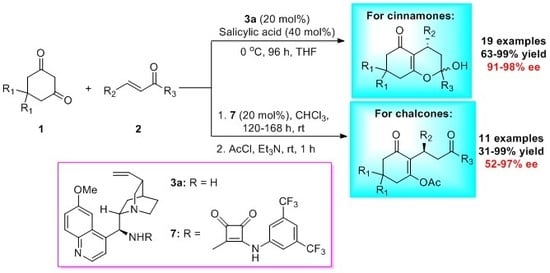
:
1. Introduction
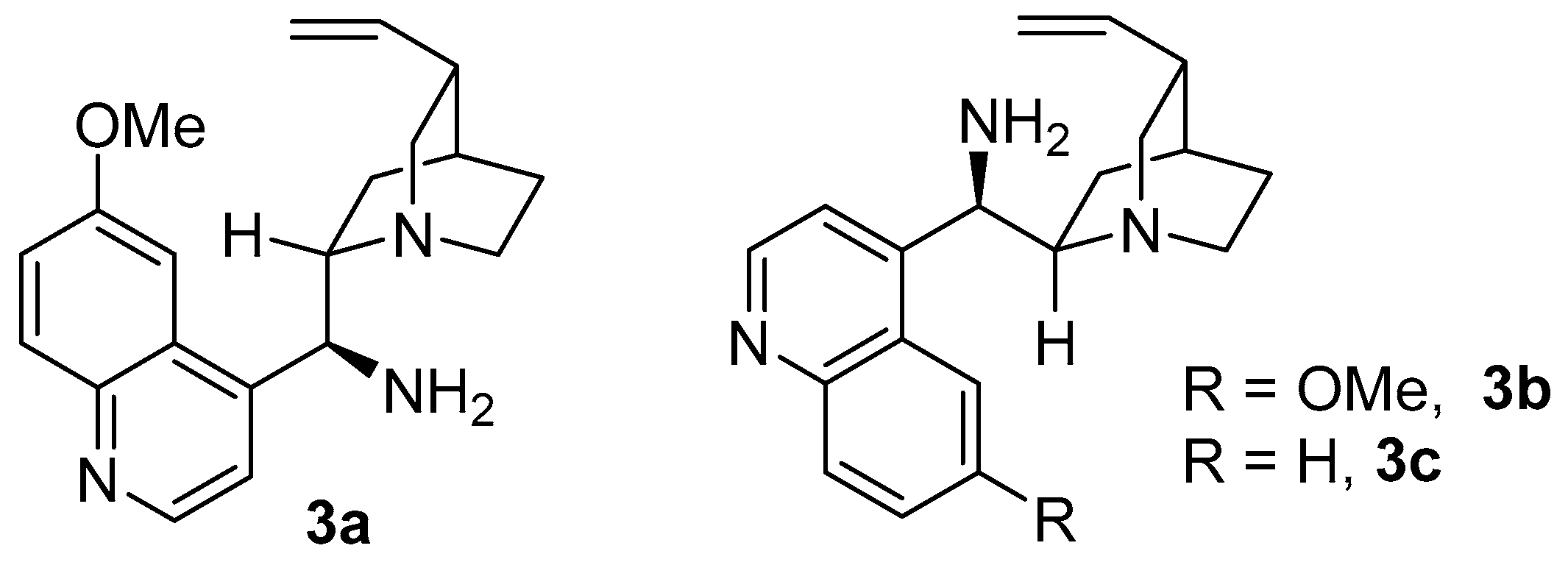
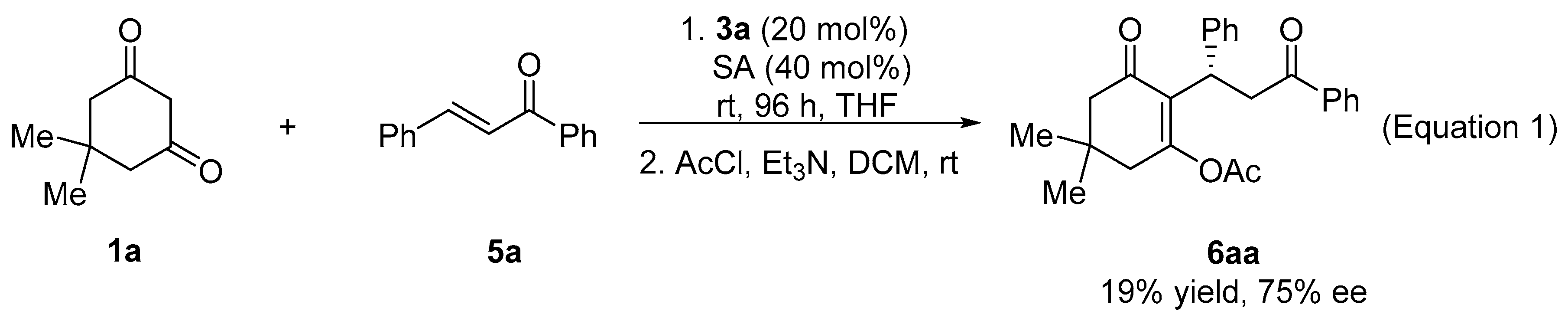
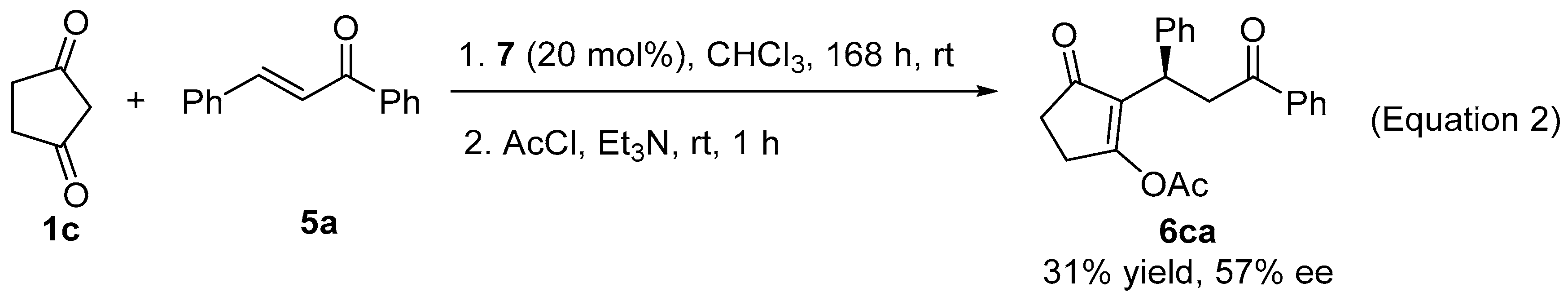
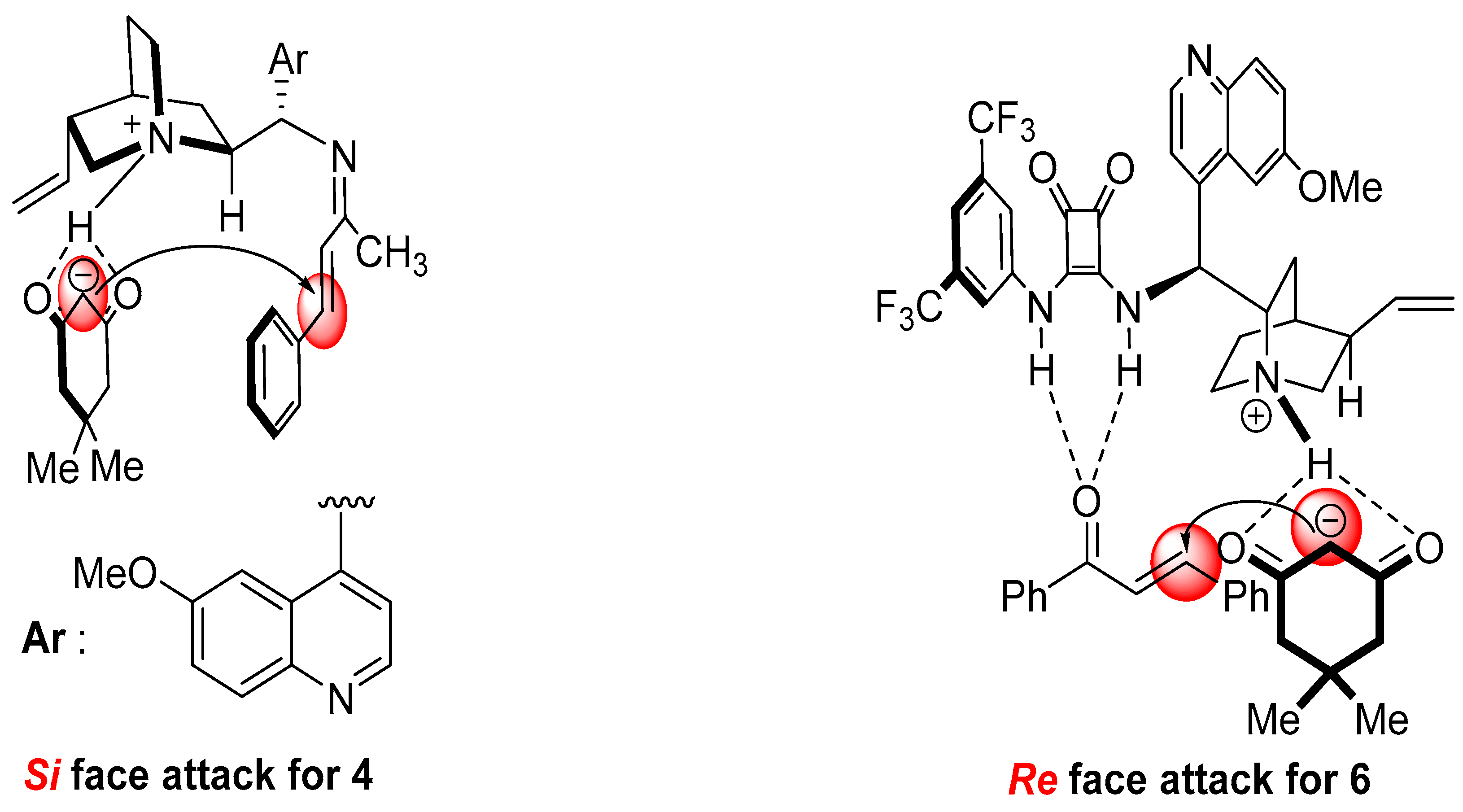
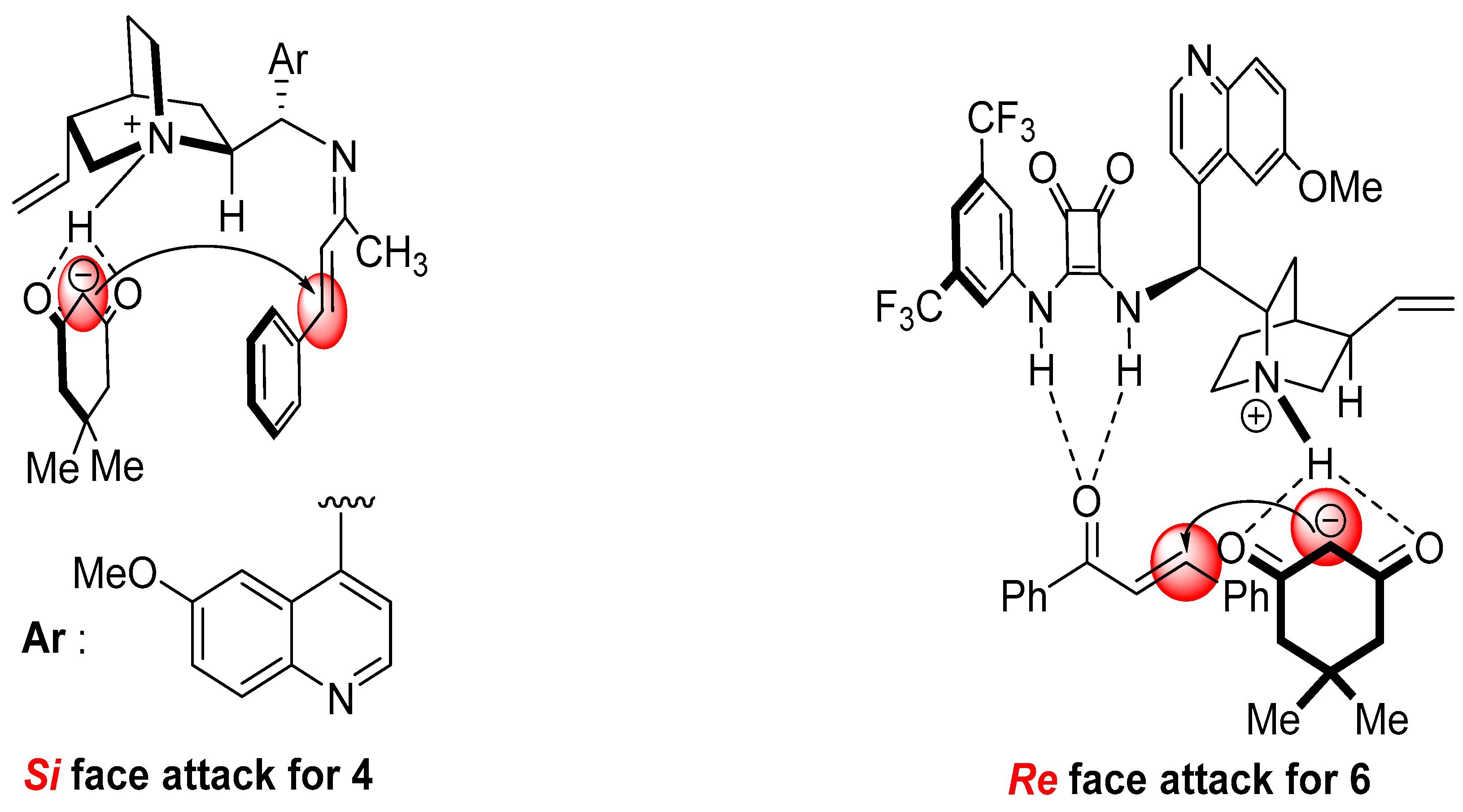
2. Results and Discussion
3. Materials and Methods
3.1. General Remarks
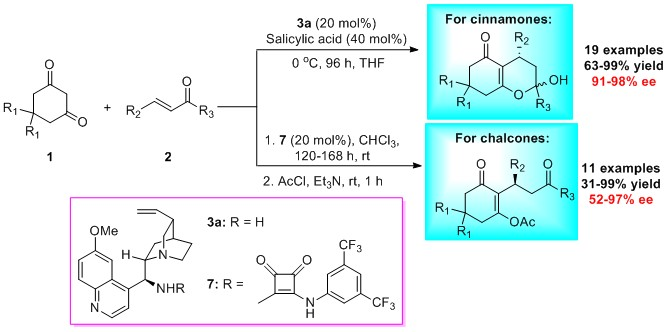
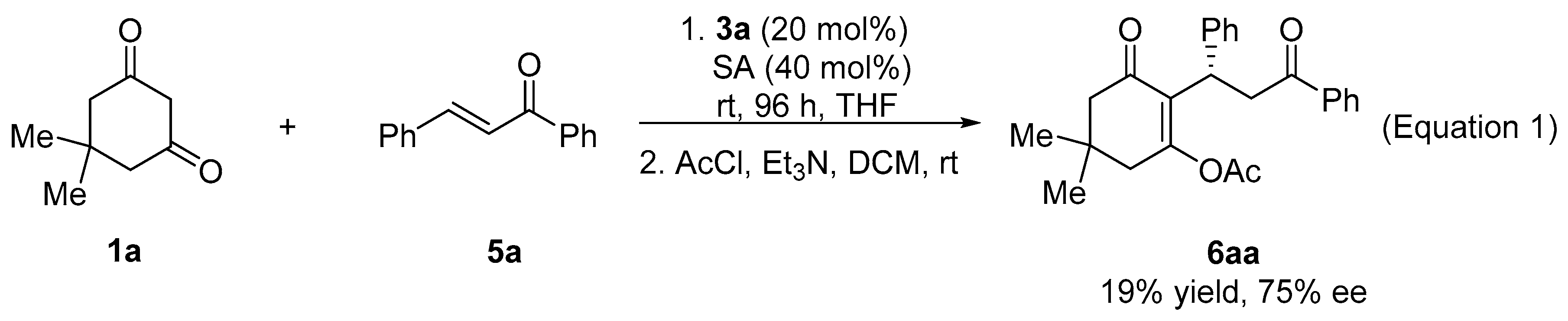
3.2. General Procedure for the Asymmetric Michael Reaction of Cinnamones
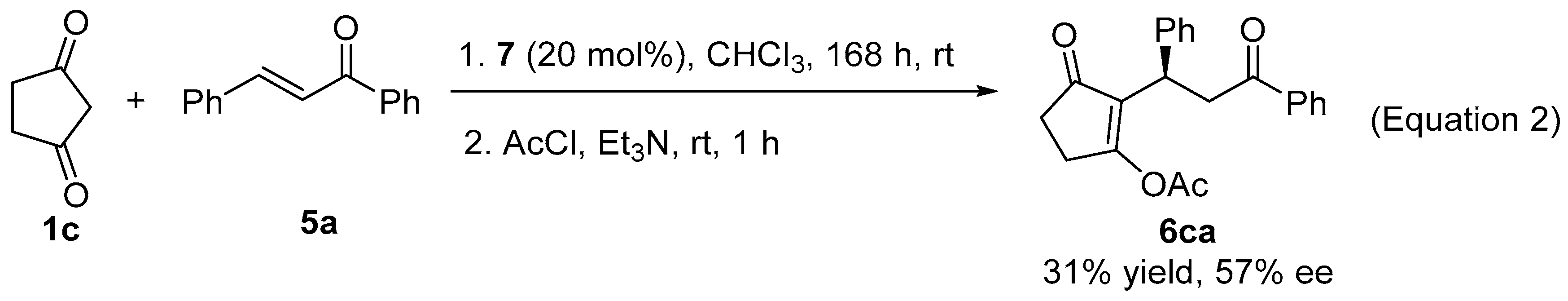
3.3. Procedure for the Asymmetric Michael Reaction of Chalcones
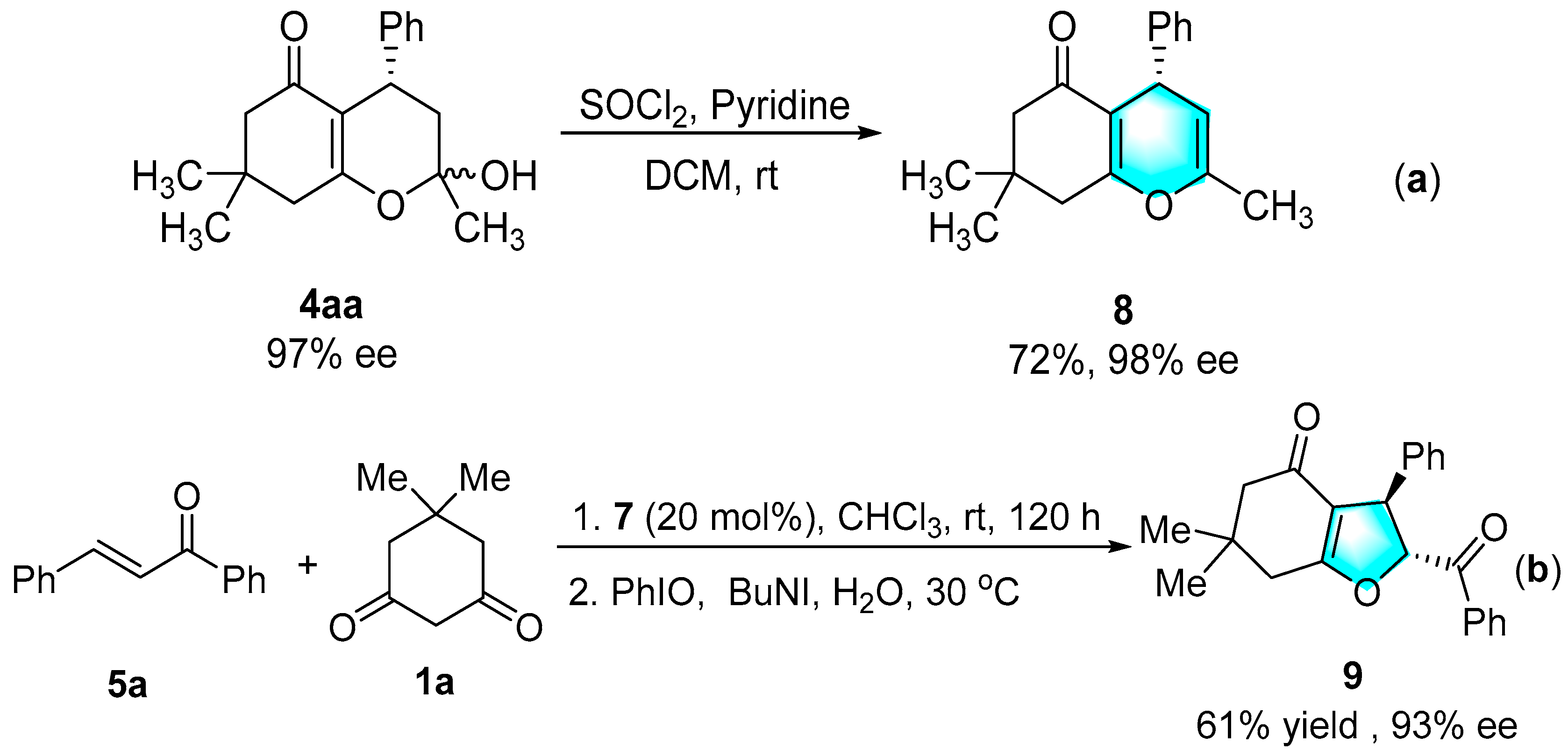
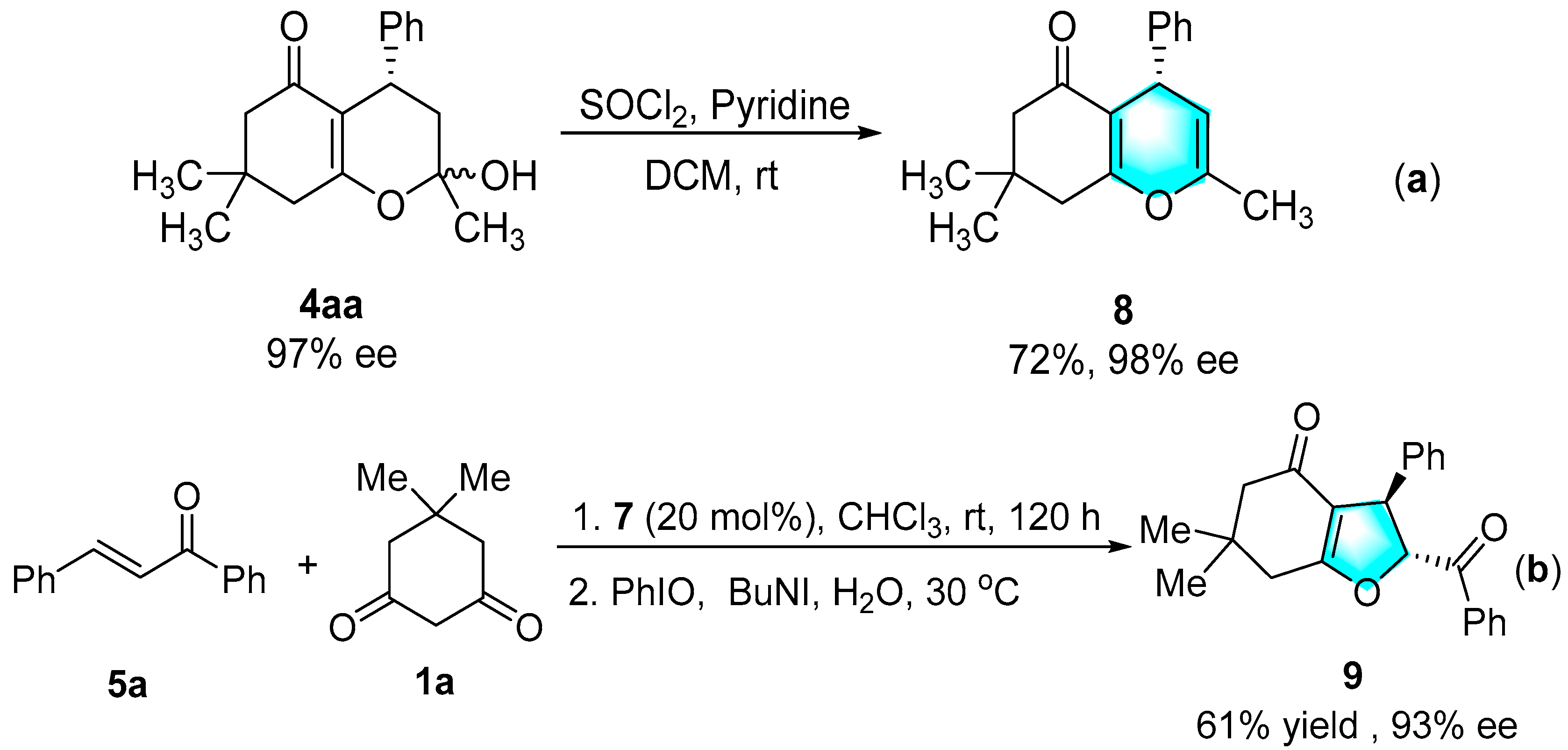
3.4. Preparation of 4H-Pyran via Dehydrating
3.5. Preparation of Fused Dihydrofuran via Stereoselective Oxidative Cyclization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sibi, M.P.; Manyem, S. Enantioselective conjugate additions. Tetrahedron 2000, 56, 8033–8061. [Google Scholar] [CrossRef]
- Christoffers, J.; Koripelly, G.; Rosiak, A.; Rössle, M. Recent advances in metal-catalyzed asymmetric conjugate additions. Synthesis 2007, 9, 1279–1300. [Google Scholar] [CrossRef]
- Krause, N.; Hoffmann-Röder, A. Recent advances in catalytic enantioselective Michael additions. Synthesis 2001, 2, 171–196. [Google Scholar] [CrossRef]
- Almaşi, D.; Alonso, D.A.; Najera, C. Organocatalytic asymmetric conjugate additions. Tetrahedron Asymmetry 2007, 18, 299–365. [Google Scholar] [CrossRef]
- Halland, N.; Hazell, R.G.; Jørgensen, K.A. Organocatalytic asymmetric conjugate addition of nitroalkanes to α,β-unsaturated enones using novel imidazoline catalysts. J. Org. Chem. 2002, 67, 8331–8338. [Google Scholar] [CrossRef] [PubMed]
- Halland, N.; Aburel, P.S.; Jørgensen, K.A. Highly Enantioselective Organocatalytic Conjugate Addition of Malonates toAcyclic α,β-Unsaturated Enones. Angew. Chem. Int. Ed. 2003, 42, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Halland, N.; Hansen, T.; Jørgensen, K.A. Organocatalytic Asymmetric Michael Reaction of Cyclic 1,3-Dicarbonyl Compounds and α,β-Unsaturated Ketones—A Highly Atom-Economic Catalytic One-Step Formation of Optically Active Warfarin Anticoagulant. Angew. Chem. Int. Ed. 2003, 42, 4955–4957. [Google Scholar] [CrossRef] [PubMed]
- Halland, N.; Aburel, P.S.; Jørgensen, K.A. Highly Enantio- and Diastereoselective Organocatalytic Asymmetric Domino Michael-Aldol Reaction of β-Ketoesters and α,β-Unsaturated Ketones. Angew. Chem. Int. Ed. 2004, 43, 1272–1277. [Google Scholar]
- Pulkkinen, J.; Aburel, P.S.; Halland, N.; Jørgensen, K.A. Highly Enantio-and Diastereoselective Organocatalytic Domino Michael-Aldol Reactions of β-Diketone and β-Ketosulfone Nucleophiles with α,β-Unsaturated Ketones. Adv. Synth. Catal. 2004, 346, 1077–1080. [Google Scholar] [CrossRef]
- Prieto, A.; Halland, N.; Jørgensen, K.A. Novel imidazolidine-tetrazole organocatalyst for asymmetric conjugate addition of nitroalkanes. Org. Lett. 2005, 7, 3897–3900. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, P.; Dudzinski, K.; Łyżwa, D. Effect of High Pressure on the Organocatalytic Asymmetric Michael Reaction: Highly Enantioselective Synthesis of γ-Nitroketones with Quaternary Stereogenic Centers. Org. Lett. 2011, 13, 3624–3627. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, P. Cinchona-based Primary Amine Catalysis in the Asymmetric Functionalization of Carbonyl Compounds. Angew. Chem. Int. Ed. 2012, 51, 9748–9770. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-W.; Luo, J.; Lu, Y. Asymmetric catalysis with chiral primary amine-based organocatalysts. Chem. Commun. 2009, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C. The Development of AsymmetricPrimary Amine Catalysts Based on Cinchona Alkaloids. Synlett 2008, 2008, 1919–1930. [Google Scholar] [CrossRef]
- Xie, J.W.; Chen, W.; Li, R.; Zeng, M.; Du, W.; Yue, L.; Chen, Y.C.; Wu, Y.; Zhu, J.; Deng, J.G. Highly Asymmetric Michael Addition to α,β-Unsaturated Ketones Catalyzed by 9-Amino-9-deoxyepiquinine. Angew. Chem. Int. Ed. 2007, 46, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, G.; Bosco, M.; Carlone, A.; Pesciaioli, F.; Sambri, L.; Melchiorre, P. Organocatalytic asymmetric Friedel-Crafts alkylation of indoles with simple α,β-unsaturated ketones. Org. Lett. 2007, 9, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Pesciaioli, F.; De Vincentiis, F.; Galzerano, P.; Bencivenni, G.; Bartoli, G.; Mazzanti, A.; Melchiorre, P. Organocatalytic Asymmetric Aziridination of Enones. Angew. Chem. Int. Ed. 2008, 47, 8703–8706. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Liu, Y.; Sun, B.; Cindric, B.; Deng, L. Catalytic Enantioselective Peroxidation of α,β-Unsaturated Ketones. J. Am. Chem. Soc. 2008, 130, 8134–8135. [Google Scholar] [CrossRef] [PubMed]
- McCooey, S.H.; Connon, S.J. Readily accessible 9-epi-amino cinchona alkaloid derivatives promote efficient, highly enantioselective additions of aldehydes and ketones to nitroolefins. Org. Lett. 2007, 9, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Calter, M.A.; Wang, J. Catalytic, asymmetric Michael reactions of cyclic diketones with β, γ-unsaturated α-ketoesters. Org. Lett. 2009, 11, 2205–2208. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zheng, F.; Ye, H.; Zhong, W. Enantioselective synthesis of functionalized 3,4-dihydropyran derivatives organocatalyzed by a novel fluorinated-diphenylprolinolether. Tetrahedron 2009, 65, 10016–10021. [Google Scholar] [CrossRef]
- Dong, Z.; Feng, J.; Fu, X.; Liu, X.; Lin, L.; Feng, X. Highly Enantioselective Conjugate Addition of Cyclic Diketones to β,γ-Unsaturated α-Ketoesters Catalyzed by an N,N′-Dioxide-Cu(OTf)2 Complex. Chem. Eur. J. 2011, 17, 1118–1121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, Y.; Zhang, W.; Xu, X.; Zhong, A.; Xu, D. Organocatalytic Michael Addition of Naphthoquinone with α,β-Unsaturated Ketones: Primary Amine Catalyzed Asymmetric Synthesis of Lapachol Analogues. Eur. J. Org. Chem. 2011, 2011, 2142–2147. [Google Scholar] [CrossRef]
- Tan, B.; Lu, Y.; Zeng, X.; Chua, P.J.; Zhong, G. Facile Domino Access to Chiral Bicyclo[3.2.1]octanes and Discovery of a New Catalytic Activation Mode. Org. Lett. 2010, 12, 2682–2685. [Google Scholar] [CrossRef] [PubMed]
- Tsakos, M.; Elsegood, M.R.J.; Kokotos, C.G. Organocatalytic asymmetric domino Michael-Henry reaction for the synthesis of substituted bicyclo[3.2.1]octan-2-ones. Chem. Commun. 2013, 49, 2219–2221. [Google Scholar] [CrossRef] [PubMed]
- Franke, P.T.; Richter, B.; Jørgensen, K.A. Organocatalytic Asymmetric Synthesis of Functionalized 3,4-Dihydropyran Derivatives. Chem. Eur. J. 2008, 14, 6317–6321. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Sugiono, E.; Merino, E. Asymmetric Organocatalysis: An Efficient Enantioselective Access to Benzopyranes and Chromenes. Chem. Eur. J. 2008, 14, 6329–6332. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yen, C.; Preston, P.; Chin, J. Substrate-directed stereoselectivity in vicinal diamine-catalyzed synthesis of warfarin. Org. Lett. 2006, 8, 5239–5242. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.-W.; Yue, L.; Chen, W.; Du, W.; Zhu, J.; Deng, J.-G.; Chen, Y.-C. Highly enantioselective Michael addition of cyclic 1, 3-dicarbonyl compounds to α,β-unsaturated ketones. Org. Lett. 2007, 9, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wang, L.; Chen, X.; Liu, X.; Lin, L.; Feng, X. Organocatalytic Enantioselective Michael Addition of 4-Hydroxycoumarin to α,β-Unsaturated Ketones: A Simple Synthesis of Warfarin. Eur. J. Org. Chem. 2009, 2009, 5192–5197. [Google Scholar] [CrossRef]
- Kristensen, T.E.; Vestli, K.; Hansen, F.K.; Hansen, T. New Phenylglycine-Derived Primary Amine Organocatalysts for the Preparation of Optically Active Warfarin. Eur. J. Org. Chem. 2009, 2009, 5185–5191. [Google Scholar] [CrossRef]
- Zhu, X.; Lin, A.; Shi, Y.; Guo, J.; Zhu, C.; Cheng, Y. Enantioselective Synthesis of Polycyclic Coumarin Derivatives Catalyzed by an in Situ Formed Primary Amine-Imine Catalyst. Org. Lett. 2011, 13, 4382–4385. [Google Scholar] [CrossRef] [PubMed]
- Mei, R.-Q.; Xu, X.-Y.; Li, Y.-C.; Fu, J.-Y.; Huang, Q.-C.; Wang, L.-X. Highly effective and enantioselective Michael addition of 4-hydroxycoumarin to α,β-unsaturated ketones promoted by simple chiral primary amine thiourea bifunctional catalysts. Tetrahedron Lett. 2011, 52, 1566–1568. [Google Scholar] [CrossRef]
- Ray, S.K.; Singh, P.K.; Molleti, N.; Singh, V.K. Enantioselective Synthesis of Coumarin Derivatives by PYBOX-DIPH-Zn(II) Complex Catalyzed Michael Reaction. J. Org. Chem. 2012, 77, 8802–8808. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Du, D.-M. Highly enantioselective synthesis of Warfarin and its analogs catalysed by primary amine-phosphinamide bifunctional catalysts. Org. Biomol. Chem. 2012, 10, 8125–8131. [Google Scholar] [CrossRef] [PubMed]
- Işık, M.; Akkoca, H.U.; Akhmedov, İ.M.; Tanyeli, C. A bis-Lewis basic 2-aminoDMAP/prolinamide organocatalyst for application to the enantioselective synthesis of Warfarin and derivatives. Tetrahedron Asymmetry 2016, 27, 384–388. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, X.; Wang, M.; He, P.; Lin, L.; Feng, X. Enantioselective Synthesis of 3,4-Dihydropyran Derivatives via Organocatalytic Michael Reaction of α,β-Unsaturated Enones. J. Org. Chem. 2012, 77, 4136–4142. [Google Scholar] [CrossRef] [PubMed]
- Molleti, N.; Allu, S.; Ray, S.K.; Singh, V.K. Bifunctional chiral urea catalyzed highly enantioselective Michael addition of cyclic 1,3-dicarbonyl compounds to 2-enoylpyridines. Tetrahedron Lett. 2013, 54, 3241–3244. [Google Scholar] [CrossRef]
- Ray, S.K.; Rout, S.; Singh, V.K. Enantioselective synthesis of 3, 4-dihydropyran derivatives via a Michael addition reaction catalysed by chiral pybox–diph–Zn (II) complex. Org. Biomol. Chem. 2013, 11, 2412–2416. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, S.; Tripathi, V.D.; Maurya, R.A.; Srivastava, S.P.; Bhatia, G.; Tamrakar, A.K.; Srivastava, A.K. Design and synthesis of 2,4-disubstituted polyhydroquinolines as prospective antihyperglycemic and lipid modulating agents. Biorg. Med. Chem. 2010, 18, 4138–4148. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, G.; Standen, N. ATP-sensitive potassium channels. Curr. Pharm. Des. 2005, 11, 1915–1940. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cun, L.; Lian, C.; Zhong, L.; Chen, Y.; Liao, J.; Zhu, J.; Deng, J. Highly enantioselective Michael addition of malononitrile to alpha,beta-unsaturated ketones. Org. Biomol. Chem. 2008, 6, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, Y.; Xing, Z.; Tang, N.; Zhu, J.; Deng, J. The asymmetric addition of malononitrile to α,β-unsaturated ketones catalyzed by RuCl2[(R,R)-DPEN](PPh3)2 as the precatalyst. Tetrahedron Lett. 2014, 55, 3868–3872. [Google Scholar] [CrossRef]
- Zheng, Y.; Yao, Y.; Ye, L.; Shi, Z.; Li, X.; Zhao, Z.; Li, X. Highly enantioselective Michael addition of malononitrile to α,β-unsaturated pyrazolamides catalyzed by a bifunctional thiourea. Tetrahedron 2016, 72, 973–978. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Q.; Ye, L.; Shi, Z.; Zhao, Z.; Yang, X.; Ding, K.; Li, X. A general, highly enantioselective Michael addition of nitroalkanes to α,β-unsaturated enones catalyzed by 9-amino (9-deoxy)-epi-quinine: A remarkable additive effect. Tetrahedron 2016, 72, 5115–5120. [Google Scholar] [CrossRef]
- Chen, X.K.; Zheng, C.W.; Zhao, S.L.; Chai, Z.; Yang, Y.Q.; Zhao, G.; Cao, W.G. Highly Enantioselective Michael Addition of Cyclic 1, 3-Dicarbonyl Compounds to β, γ-Unsaturated α-Keto Esters. Adv. Synth. Catal. 2010, 352, 1648–1652. [Google Scholar] [CrossRef]
- Wang, Y.F.; Wang, K.; Zhang, W.; Zhang, B.B.; Zhang, C.X.; Xu, D.Q. Enantioselective Asymmetric Michael Addition of Cyclic Diketones to β, γ-Unsaturated α-Keto Esters. Eur. J. Org. Chem. 2012, 2012, 3691–3696. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.; Yan, J.; Chen, R.; Wang, S.; Ma, Y.; Wang, R. One-Pot Enantioselective Synthesis of Functionalized Pyranocoumarins and 2-Amino-4H-chromenes: Discovery of a Type of Potent Antibacterial Agent. J. Org. Chem. 2012, 77, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Du, D.-M. Enantioselective synthesis of 2-amino-5,6,7,8-tetrahydro-5-oxo-4H-chromene-3-carbonitriles using squaramide as the catalyst. Tetrahedron Asymmetry 2012, 23, 1343–1349. [Google Scholar] [CrossRef]
- Feng, X.; Zhou, Z.; Zhou, R.; Zhou, Q.-Q.; Dong, L.; Chen, Y.-C. Stereodivergence in Amine-Catalyzed Regioselective [4 + 2] Cycloadditions of β-Substituted Cyclic Enones and Polyconjugated Malononitriles. J. Am. Chem. Soc. 2012, 134, 19942–19947. [Google Scholar] [CrossRef] [PubMed]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Parra, A.; Uria, U.; Besselievre, F.; Merino, E. Catalytic asymmetric domino Michael addition– alkylation reaction: Enantioselective synthesis of dihydrofurans. Org. Lett. 2010, 12, 5680–5683. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-W.; Lu, Q.-Q.; Xia, J.-J. Three Types of Products Obtained Unexpectedly from the Reaction of Dimedone with Chalcones. Eur. J. Org. Chem. 2011, 2011, 4429–4438. [Google Scholar] [CrossRef]
- Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K.A. Squaramides: Bridging from Molecular Recognition to Bifunctional Organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Tsakos, M.; Kokotos, C.G. Primary and secondary amine-(thio)ureas and squaramides and their applications in asymmetric organocatalysis. Tetrahedron 2013, 69, 10199–10222. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Zu, L.; Jiang, W.; Xie, H.; Duan, W.; Wang, W. Organocatalytic enantioselective conjugate additions to enones. J. Am. Chem. Soc. 2006, 128, 12652–12653. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Wang, L.; Fan, R. Aqueous iodine (III)-mediated stereoselective oxidative cyclization for the synthesis of functionalized fused dihydrofuran derivatives. J. Org. Chem. 2010, 75, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Jia, Y.; Du, D.-M. Squaramide-catalyzed enantioselective Michael addition of malononitrile to chalcones. Org. Biomol. Chem. 2012, 10, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 7th ed.; Butterworth-Heinemann: Oxford, UK, 2012. [Google Scholar]
- Jiang, L.; Wang, B.; Li, R.-R.; Shen, S.; Yu, H.-W.; Ye, L.-D. Catalytic promiscuity of Escherichia coli BioH esterase: Application in the synthesis of 3,4-dihydropyran derivatives. Process Biochem. 2014, 49, 1135–1138. [Google Scholar] [CrossRef]
- Ye, Y.; Wang, L.; Fan, R. Aqueous Iodine(III)-Mediated Stereoselective Oxidative Cyclization for the Synthesis of Functionalized Fused Dihydrofuran Derivatives. J. Org. Chem. 2010, 75, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 4, 6, 8 and 9 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Cat. | Acid | Solvent | Time (h) | Yield (%) b | ee (%) c |
|---|---|---|---|---|---|---|
| 1 | 3a | TsOH | toluene | 48 | 60 | 66 |
| 2 | 3a | TFA | toluene | 48 | 65 | 75 |
| 3 | 3a | AcOH | toluene | 48 | 82 | 85 |
| 4 | 3a | BA | toluene | 24 | 89 | 90 |
| 5 | 3a | ONBA | toluene | 36 | 91 | 90 |
| 6 | 3a | PNBA | toluene | 36 | 96 | 89 |
| 7 | 3a | OFBA | toluene | 24 | 96 | 88 |
| 8 | 3a | p-MeOC6H4CO2H | toluene | 36 | 91 | 87 |
| 9 | 3a | SA | toluene | 24 | 99 | 90 |
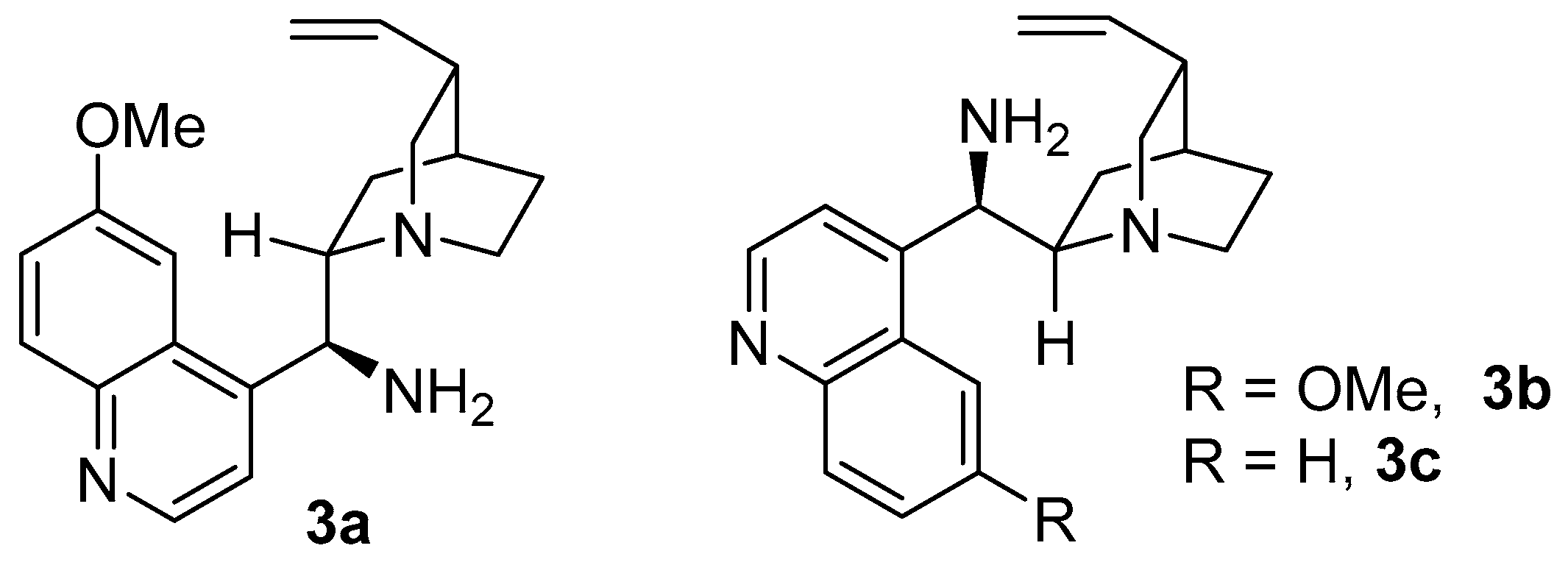
| 10 | 3b | SA | toluene | 96 | 89 | −82 |
| 11 | 3c | SA | toluene | 36 | 89 | −82 |
| 12 | 3a | SA | PhCF3 | 24 | 96 | 85 |
| 13 | 3a | SA | DCM | 24 | 55 | 83 |
| 14 | 3a | SA | EtOH | 24 | 99 | 69 |
| 15 | 3a | SA | THF | 24 | 99 | 94 |
| 16 d | 3a | SA | THF | 24 | 99 | 94 |
| 17 d,e | 3a | SA | THF | 96 | 99 | 97 |

| Entry | 1 | R2 | R3 | 2 | 4 | Yield (%) b | ee (%) c |
|---|---|---|---|---|---|---|---|
| 1 | 1a | Ph | Me | 2a | 4aa | 99 (95) d | 97 (94) d |
| 2 | 1a | o-ClC6H4 | Me | 2b | 4ab | 87 | 91 |
| 3 | 1a | m-ClC6H4 | Me | 2c | 4ac | 99 | 96 |
| 4 | 1a | p-ClC6H4 | Me | 2d | 4ad | 99 | 97 (S) e |
| 5 | 1a | p-FC6H4 | Me | 2e | 4ae | 96 | 96 |
| 6 | 1a | p-BrC6H4 | Me | 2f | 4af | 99 | 97 |
| 7 | 1a | p-MeC6H4 | Me | 2g | 4ag | 95 | 97 |
| 8 | 1a | p-MeOC6H4 | Me | 2h | 4ah | 89 | 97 |
| 9 | 1a | 1-naphthyl | Me | 2i | 4ai | 89 | 96 |
| 10 | 1a | 2-naphthyl | Me | 2j | 4aj | 98 | 98 |
| 11 | 1a | 2-furanyl | Me | 2k | 4ak | 78 | 95 |
| 12 | 1a | 2-thiophenyl | Me | 2l | 4al | 97 | 91 |
| 13 | 1a | Me | Me | 2m | 4am | 69 | 91 |
| 14 | 1a | n-Bu | Me | 2n | 4an | 75 | 94 |
| 15 | 1a | -C3H6- | 2o | 4ao | 91 | 98 | |
| 16 | 1a | Ph | Et | 2p | 4ap | 63 | 98 |
| 17 | 1b | Ph | Me | 2a | 4ba | 71 | 94 |
| 18 | 1b | p-ClC6H4 | Me | 2d | 4bd | 78 | 95 |
| 19 | 1b | p-BrC6H4 | Me | 2f | 4bf | 67 | 96 |

| Entry | 1 | R2 | Ar1 | 5 | 6 | Yield (%) b | ee (%) c |
|---|---|---|---|---|---|---|---|
| 1 | 1a | Ph | Ph | 5a | 6aa | 95 (99) d | 93 (91) d (R) e |
| 2 | 1a | p-ClC6H4 | Ph | 5b | 6ab | 99 | 94 |
| 3 | 1a | p-MeC6H4 | Ph | 5c | 6ac | 96 | 93 |
| 4 f | 1a | 2-thiophenyl | Ph | 5d | 6ad | 95 | 91 |
| 5 f | 1a | 2-naphthyl | Ph | 5e | 6ae | 68 | 91 |
| 6 | 1a | Ph | p-ClC6H4 | 5f | 6af | 98 | 87 |
| 7 | 1a | Ph | p-MeC6H4 | 5g | 6ag | 99 | 95 |
| 8 | 1a | Ph | 2-thiophenyl | 5h | 6ah | 99 | 97 |
| 9 f | 1b | Ph | Ph | 5a | 6ba | 93 | 91 |
| 10 g | 1a | n-Pr | Ph | 5i | 6ai | 93 | 52 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Wang, W.; Ye, L.; Yang, X.; Li, X.; Zhao, Z.; Li, X. Enantioselective Michael Addition of Cyclic β-Diones to α,β-Unsaturated Enones Catalyzed by Quinine-Based Organocatalysts. Molecules 2017, 22, 1096. https://doi.org/10.3390/molecules22071096
Wang Q, Wang W, Ye L, Yang X, Li X, Zhao Z, Li X. Enantioselective Michael Addition of Cyclic β-Diones to α,β-Unsaturated Enones Catalyzed by Quinine-Based Organocatalysts. Molecules. 2017; 22(7):1096. https://doi.org/10.3390/molecules22071096
Chicago/Turabian StyleWang, Qingqing, Wei Wang, Ling Ye, Xuejun Yang, Xinying Li, Zhigang Zhao, and Xuefeng Li. 2017. "Enantioselective Michael Addition of Cyclic β-Diones to α,β-Unsaturated Enones Catalyzed by Quinine-Based Organocatalysts" Molecules 22, no. 7: 1096. https://doi.org/10.3390/molecules22071096