Synthesis and Biological Evaluation of Ginsenoside Compound K Derivatives as a Novel Class of LXRα Activator

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
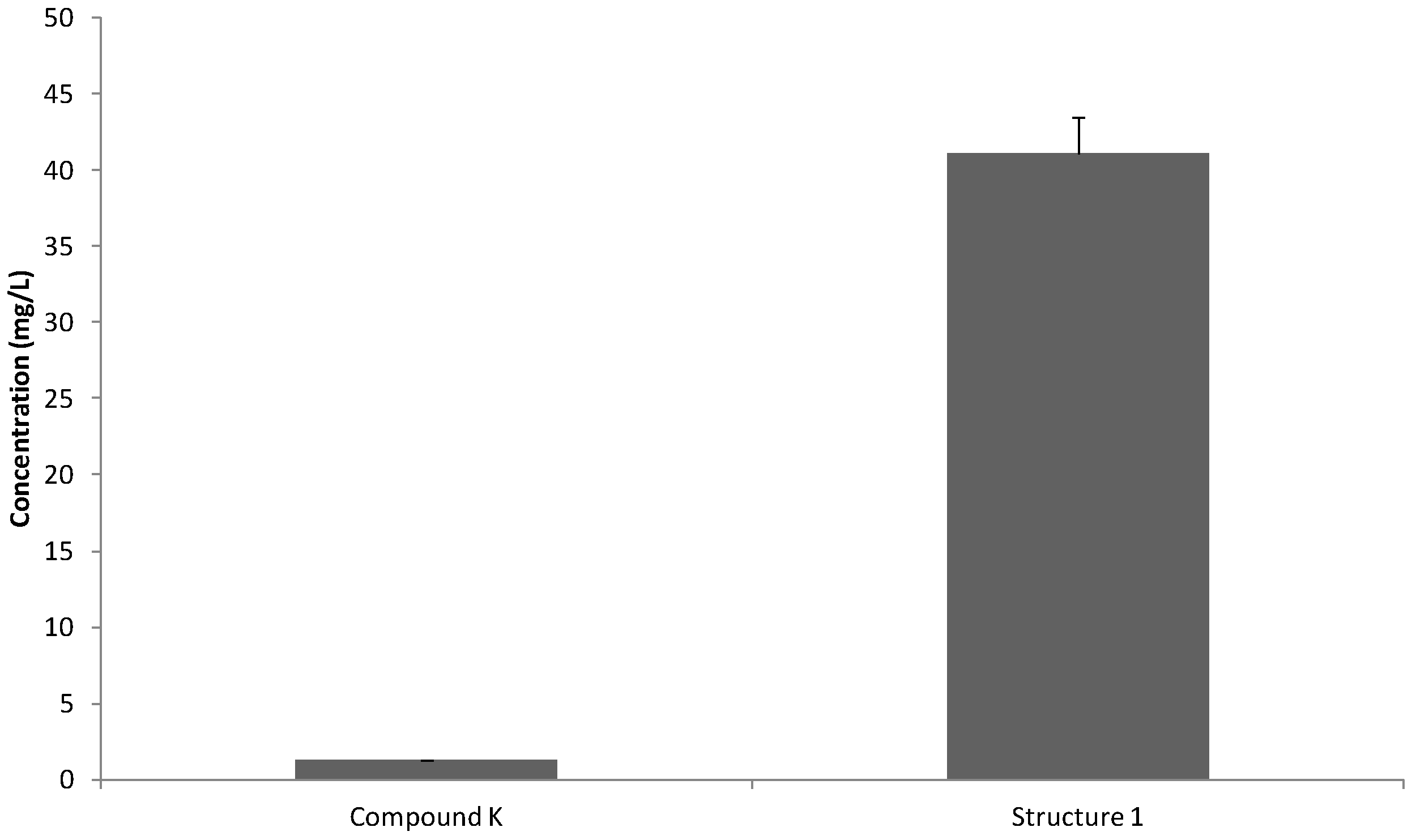
2.1. Water Solubility Measurements
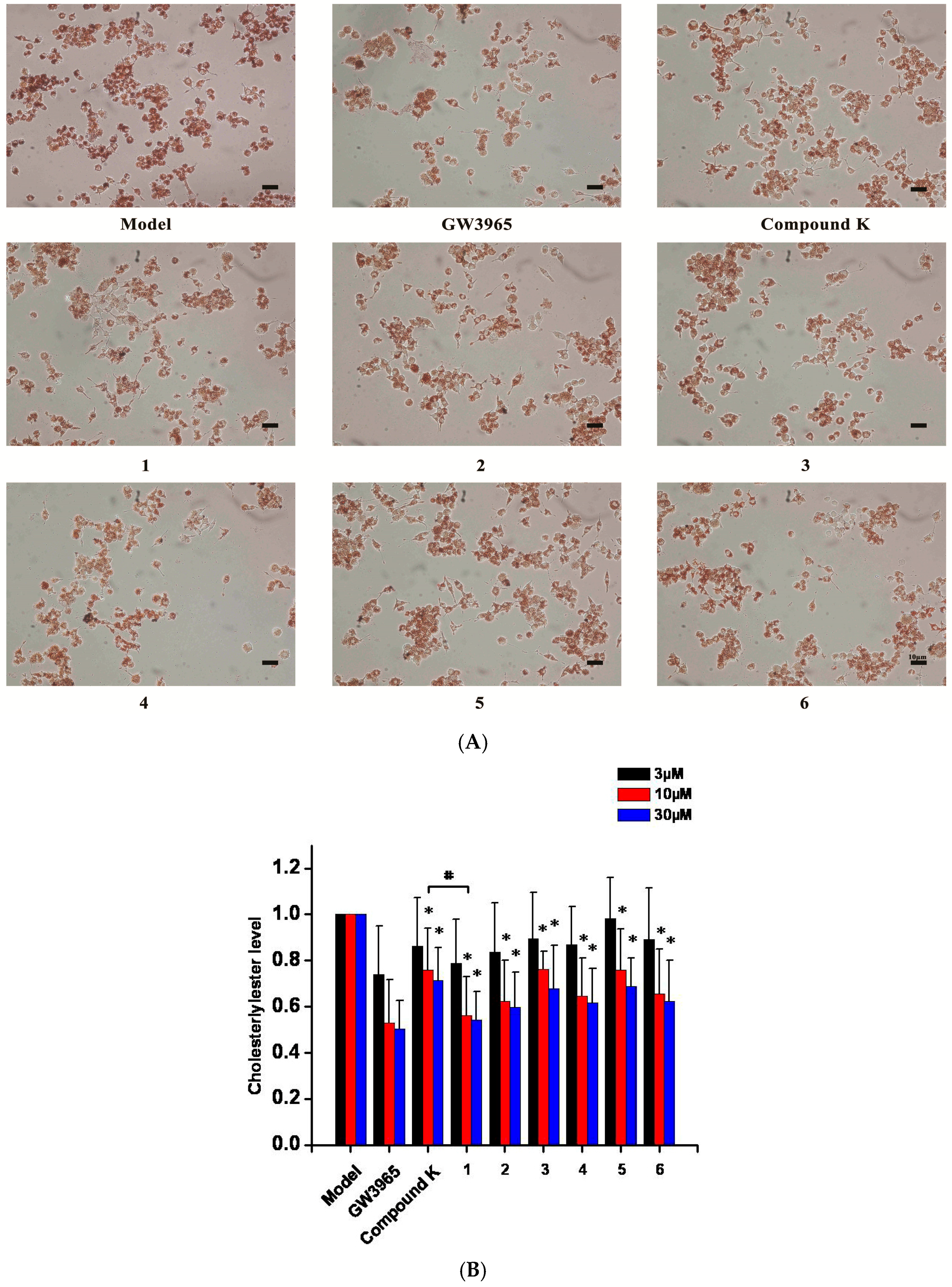
2.2. Inhibition of the Formation of Foam Cells
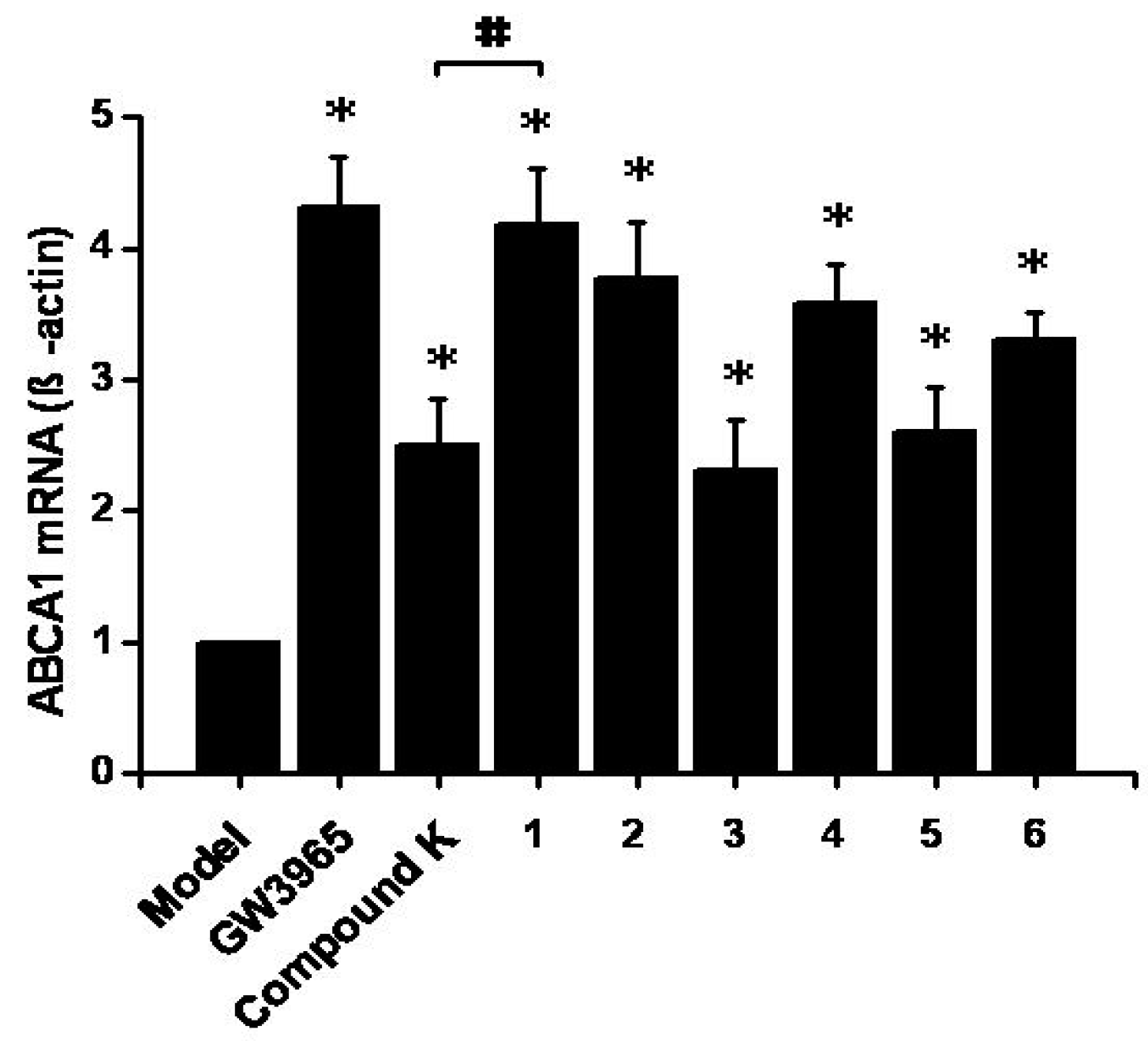
2.3. Effects on ABCA1 mRNA Expression
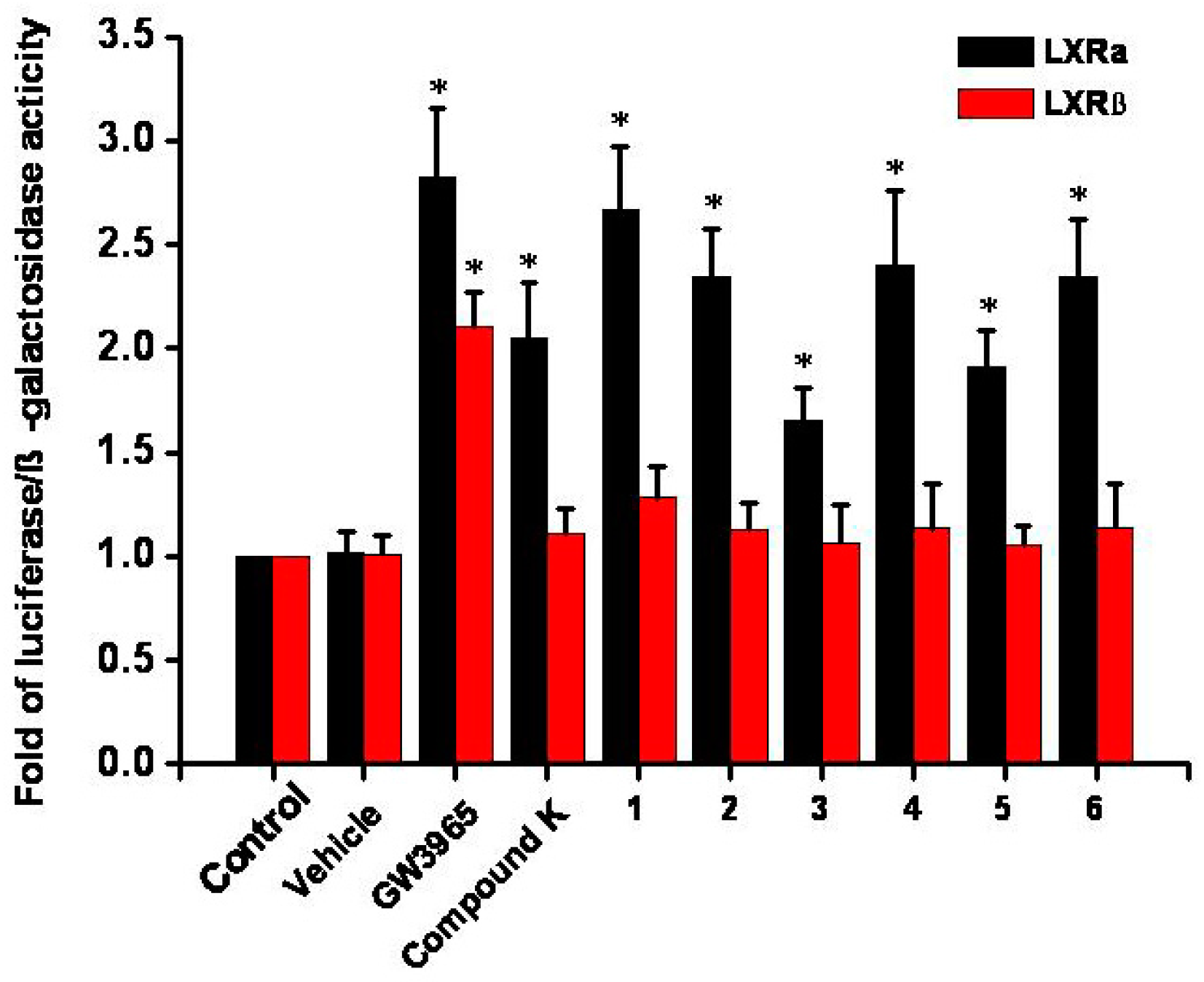
2.4. Luciferase Reporter Assay
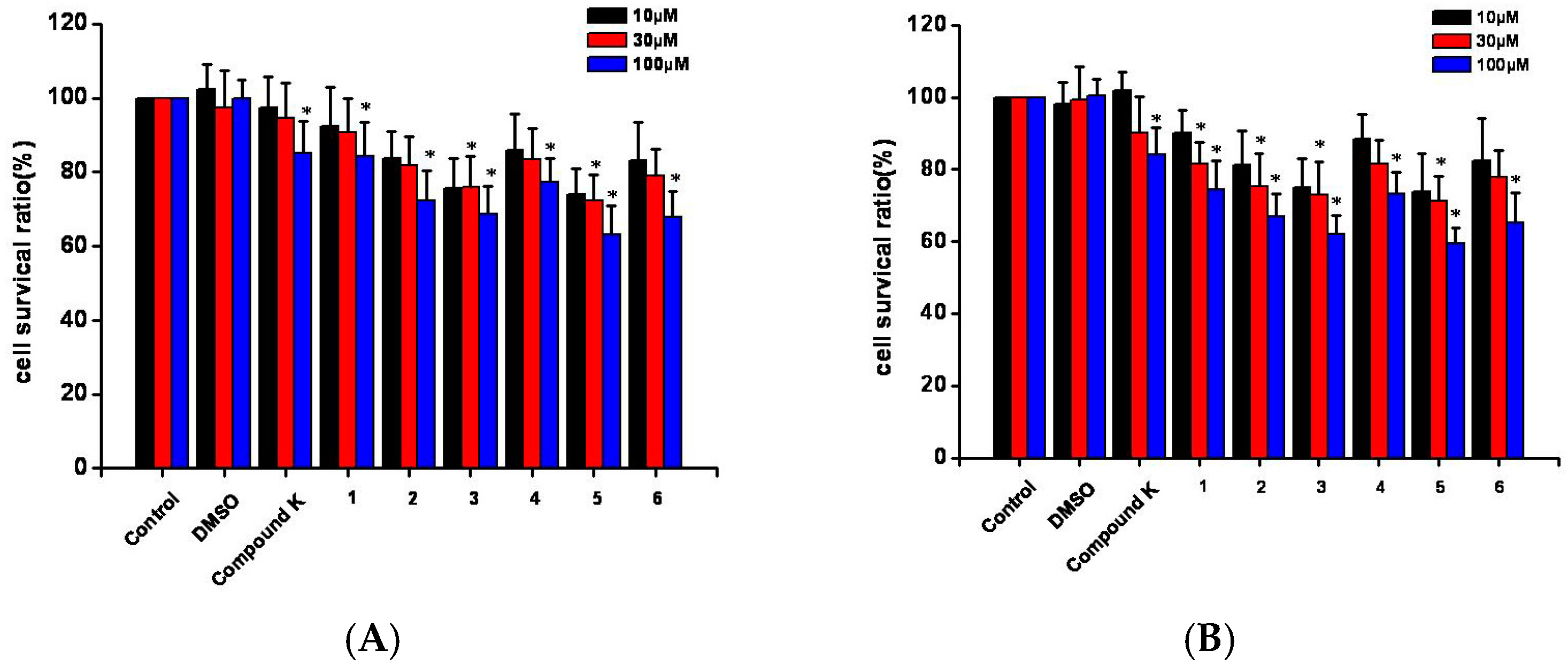
2.5. Cell Toxicity
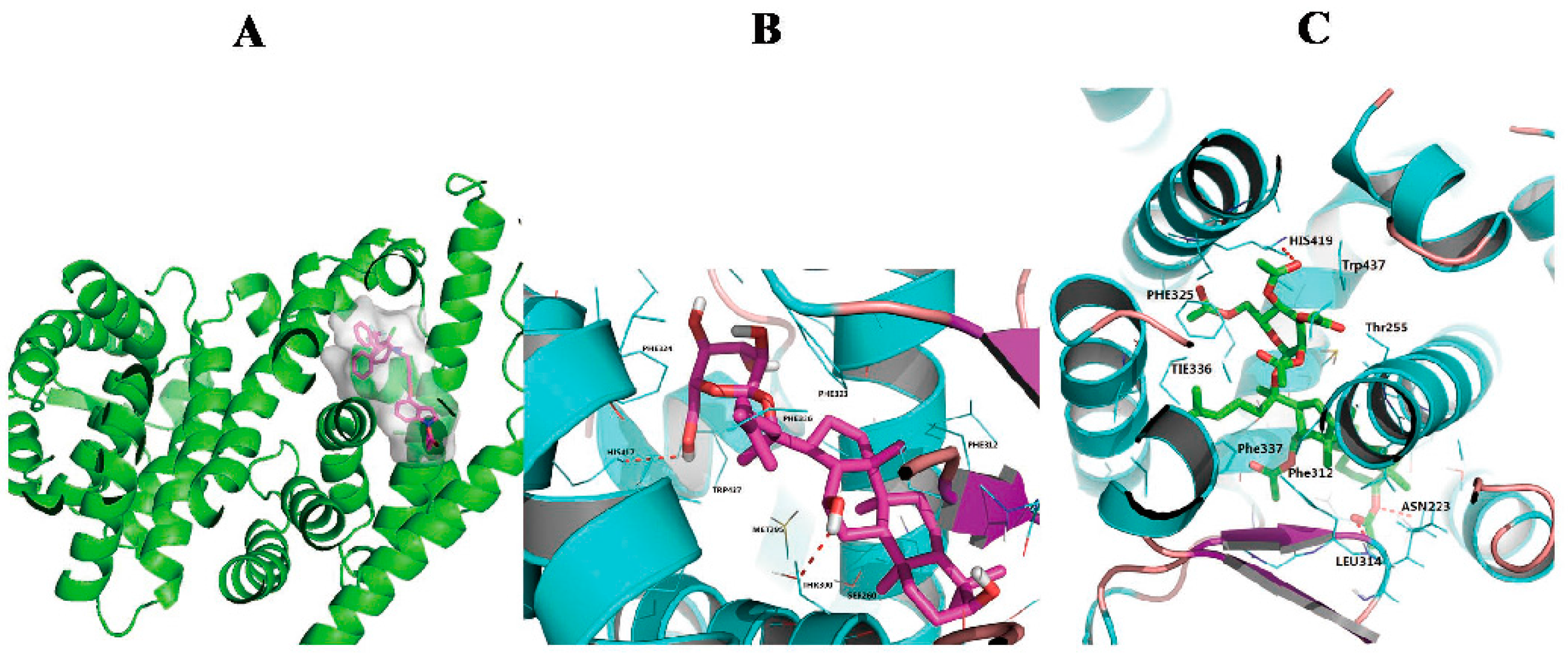
2.6. Compound K and Structure 1 Dock into the LXRα
2.7. Discussion
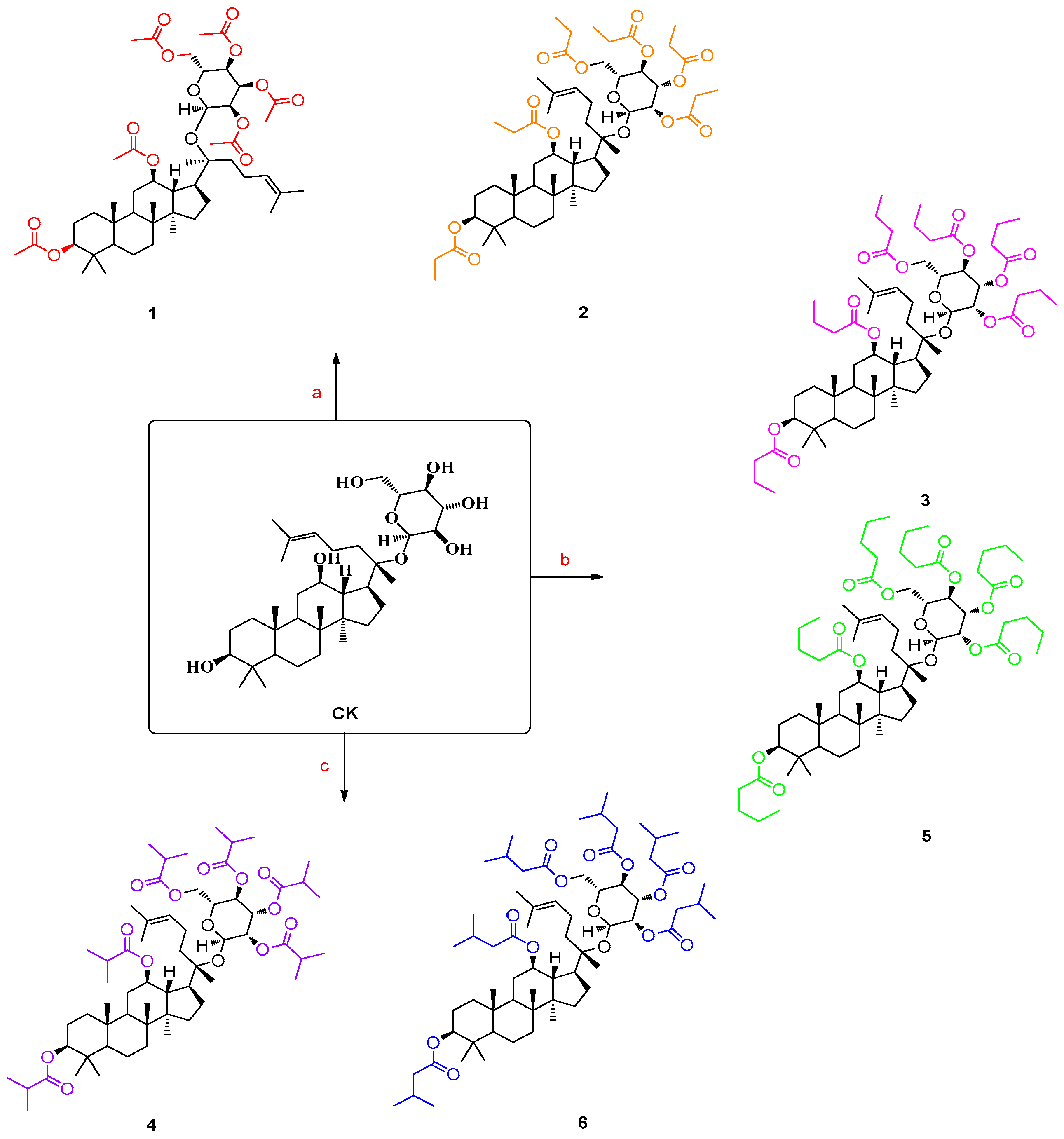
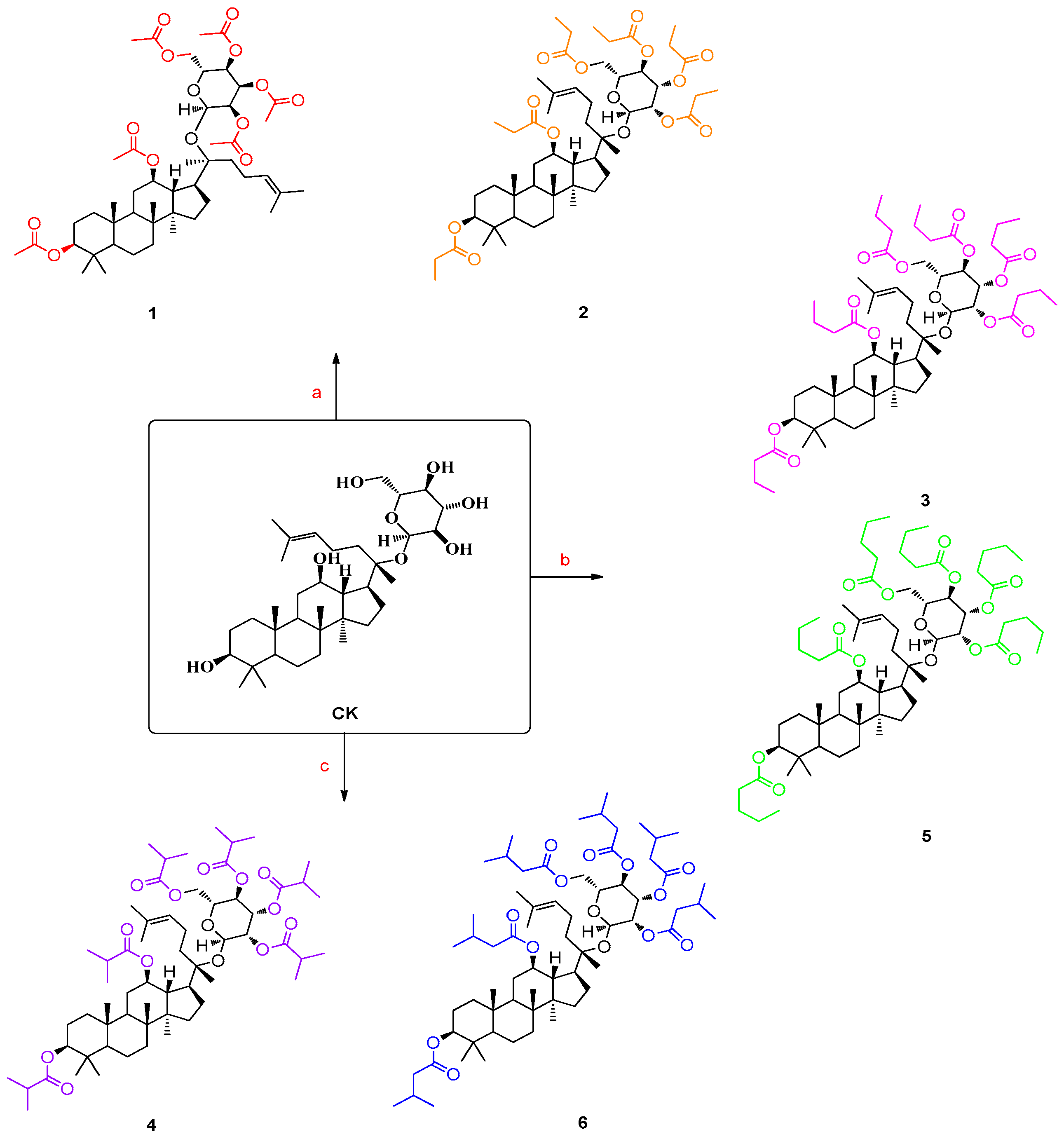
3. Design and Syntheses
4. Experimental
4.1. General
4.2. General Synthetic Procedure for Structures 1–6 [25,26,27,28]
4.2.1. Ginsenoside Compound K Derivative 1 (Structure 1)
4.2.2. Ginsenoside Compound K Derivative 2 (Structure 2)
4.2.3. Ginsenoside Compound K Derivative 3 (Structure 3)
4.2.4. Ginsenoside Compound K Derivative 4 (Structure 4)
4.2.5. Ginsenoside Compound K Derivative 5 (Structure 5)
4.2.6. Ginsenoside Compound K Derivative 6 (Structure 6)
4.3. Water Solubility Measurements
4.4. Formation of Foam Cells (Oil Red O Staining) [29]
4.5. ABCA1 mRNA Expression in RAW264.7 [4]
4.6. Cellular Toxicity
4.7. Luciferase Reporter Assay
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Logue, J.; Murray, H.M. Obesity is associated with fatal coronary heart disease independently of traditional risk factors and deprivation. Heart 2011, 97, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C.; Sacks, F.M. The Residual risk reduction initiative: A call to action to reduce residual vascular risk in dyslipidaemic patient. Diabetes Vasc. Dis. Res. 2008, 5, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Zhang, H.G. Panaxnotoginsengsaponins attenuate atherosclerosis in rats by regulating the blood lipid profile and an anti-inflammatory action. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, Z.Y. Panaxnotoginsengsaponins decrease cholesterol ester via up-regulating ATP-binding cassette transporter A1 in foam cells. J. Ethnopharmacol. 2010, 132, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Maqdasy, S.; Trousson, A. Once and for all, LXRα and LXRβ are gatekeepers of the endocrine system. Mol. Asp. Med. 2016, 49, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K. Pharmacokinetics of ginsenoside Rb1 and its metabolite compound K after oral administration of Korean Red Ginseng extract. J. Ginseng Res. 2013, 37, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Song, G. Intestinal metabolite compound K of panaxoside inhibits the growth of gastric carcinoma by augmenting apoptosis via bid-mediated mitochondrial pathway. J. Cell. Mol. Med. 2012, 16, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.Z.; Ming, Y.L. Compound K-induced apoptosis of human hepatocellular carcinoma MHCC97-H cells in vitro. Oncol. Rep. 2014, 32, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Lee, K.P. Compound K, an intestinal metabolite of ginsenosides, inhibits PDGF-BB-induced VSMC proliferation and migration through G1 arrest and attenuates neointimal hyperplasia after arterial injury. Atherosclerosis 2013, 228, 53–60. [Google Scholar] [CrossRef]
- Yang, C.S.; Ko, S.R. The ginsenoside metabolite compound K, a novel agonist of glucocorticoid receptor, induces tolerance to endotoxin-induced lethal shock. J. Cell. Mol. Med. 2008, 12, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Q. The ginsenoside metabolite compound K exerts its anti-inflammatory activity by downregulating memory B cell in adjuvant-induced arthritis. Pharm. Biol. 2016, 54, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zheng, Y. Compound K attenuates the development of atherosclerosis in ApoE−/− mice via LXRα activation. Int. J. Mol. Sci. 2016, 17, 1054. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Laffitte, B.A. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef] [PubMed]
- Bełtowski, J. Liver X receptors (LXR) as therapeutic targets in dyslipidemia. Cardiovasc. Ther. 2008, 26, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tong, D. Ascorbylpalmitate/d-α-tocopheryl polyethylene glycol 1000 succinate monoester mixed micelles for prolonged circulation and targeted delivery of compound K for antilung cancer therapy in vitro and in vivo. Int. J. Nanomed. 2017, 12, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.; Ostberg, T. Crystal structure of the heterodimeric complex of LXRalpha and RXRbeta ligand-binding domains in a fully agonistic conformation. EMBO J. 2003, 22, 4625–4633. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, C.P.; Baffic, J. A potent synthetic LXR agonist is more effective than cholesterol loading at inducing ABCA1 mRNA and stimulating cholesterol efflux. J. Biol. Chem. 2002, 277, 10021–10027. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; McKilligin, E. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7604–7609. [Google Scholar] [CrossRef] [PubMed]
- Calpe-Berdiel, L.; Rotllan, N. Liver X receptor-mediated activation of reverse cholesterol transport from macrophages to feces in vivo requires ABCG5/G8. J. Lipid Res. 2008, 49, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R. Natural Products as a Robust Source of New Drugs and Drug Leads: Past Successes and Present Day Issues. Am. J. Cardiol. 2008, 101, 43D–49D. [Google Scholar]
- Ryu, S.K.; King, T.J. Effect of an oral astaxanthinprodrug (CDX-085) on lipoprotein levels and progression of atherosclerosis in LDLR−/− and ApoE−/− mice. Atherosclerosis 2012, 222, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.S.; Partridge, E.A. Complex N-Glycan Number and Degree of Branching Cooperate to Regulate Cell Proliferation and Differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T.; Aich, U. Targeting Pro-Invasive Oncogenes with Short Chain Fatty Acid-Hexosamine Analogues Inhibits the Mobility of Metastatic MDA-MB-231 Breast Cancer Cells. J. Med. Chem. 2008, 51, 8135–8147. [Google Scholar] [CrossRef] [PubMed]
- Elmouelhi, N.; Aich, U. The Hexosamine Template—A Platform for Modulating Gene Expression and for Sugar-based Drug Discovery. J. Med. Chem. 2009, 52, 2515–2530. [Google Scholar] [CrossRef] [PubMed]
- Naoshima, Y.; Daijro, H. Synthesis of Both Enantiomers of 6-Methyl-3-octanone, An Alarm Pheromone of Ants of the Genus Crematogaster. Agric. Biol. Chem. 1988, 52, 1605–1606. [Google Scholar] [CrossRef]
- Liu, J.H.; Chen, D. Discovery, synthesis, and structure activity relationships of 20(S)-protopanaxadiol (PPD) derivatives as a novel class of AMPKα2β1γ1 activators. Eur. J. Med. Chem. 2014, 79, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Lou, Q.H.; Meng, X. Design, Synthesis and Antifibrotic Activities of Carbohydrate-Modified 1-(Substituted aryl)-5-trifluoromethyl-2(1H) Pyridones. Molecules 2012, 17, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.; Thompson, K.E. Synthesis and biological evaluation of quinic acid derivatives as anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2009, 19, 5458–5460. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.S.; Liu, D.N. Panax notoginseng saponins attenuate atherosclerosis via reciprocal regulation of lipid metabolism and inflammation by inducing liver X receptor alpha expression. J. Ethnopharmacol. 2012, 142, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Xu, M.D. BCL6 is a negative prognostic factor and exhibits pro-oncogenic activity in ovarian cancer. Am. J. Cancer Res. 2014, 5, 255–266. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Liu, H.; Zhang, Y.; Li, J.; Wang, C.; Zhou, L.; Jia, Y.; Li, X. Synthesis and Biological Evaluation of Ginsenoside Compound K Derivatives as a Novel Class of LXRα Activator. Molecules 2017, 22, 1232. https://doi.org/10.3390/molecules22071232
Huang Y, Liu H, Zhang Y, Li J, Wang C, Zhou L, Jia Y, Li X. Synthesis and Biological Evaluation of Ginsenoside Compound K Derivatives as a Novel Class of LXRα Activator. Molecules. 2017; 22(7):1232. https://doi.org/10.3390/molecules22071232
Chicago/Turabian StyleHuang, Yan, Hongmei Liu, Yingxian Zhang, Jin Li, Chenping Wang, Li Zhou, Yi Jia, and Xiaohui Li. 2017. "Synthesis and Biological Evaluation of Ginsenoside Compound K Derivatives as a Novel Class of LXRα Activator" Molecules 22, no. 7: 1232. https://doi.org/10.3390/molecules22071232