Development of 2-Methoxyhuprine as Novel Lead for Alzheimer’s Disease Therapy

 , , ,
, , ,
Abstract
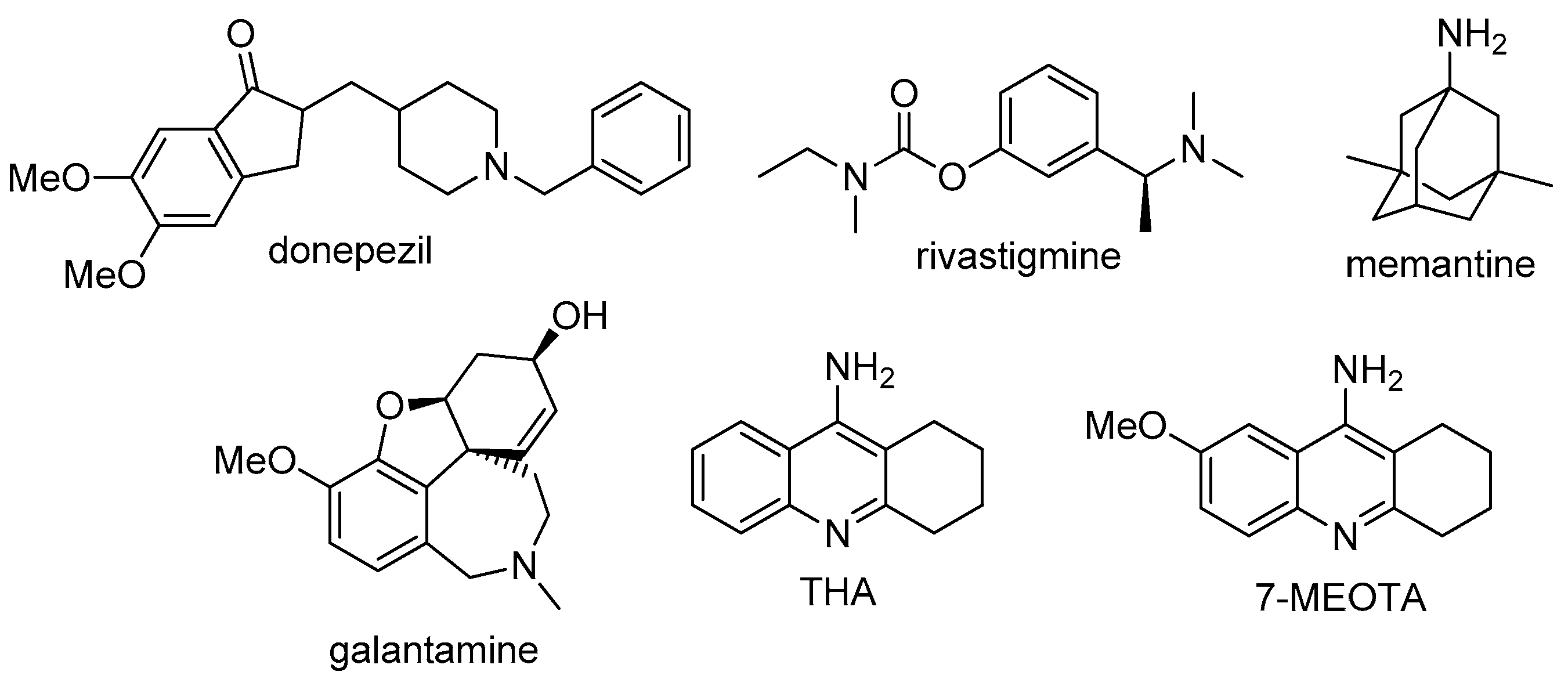
:1. Introduction
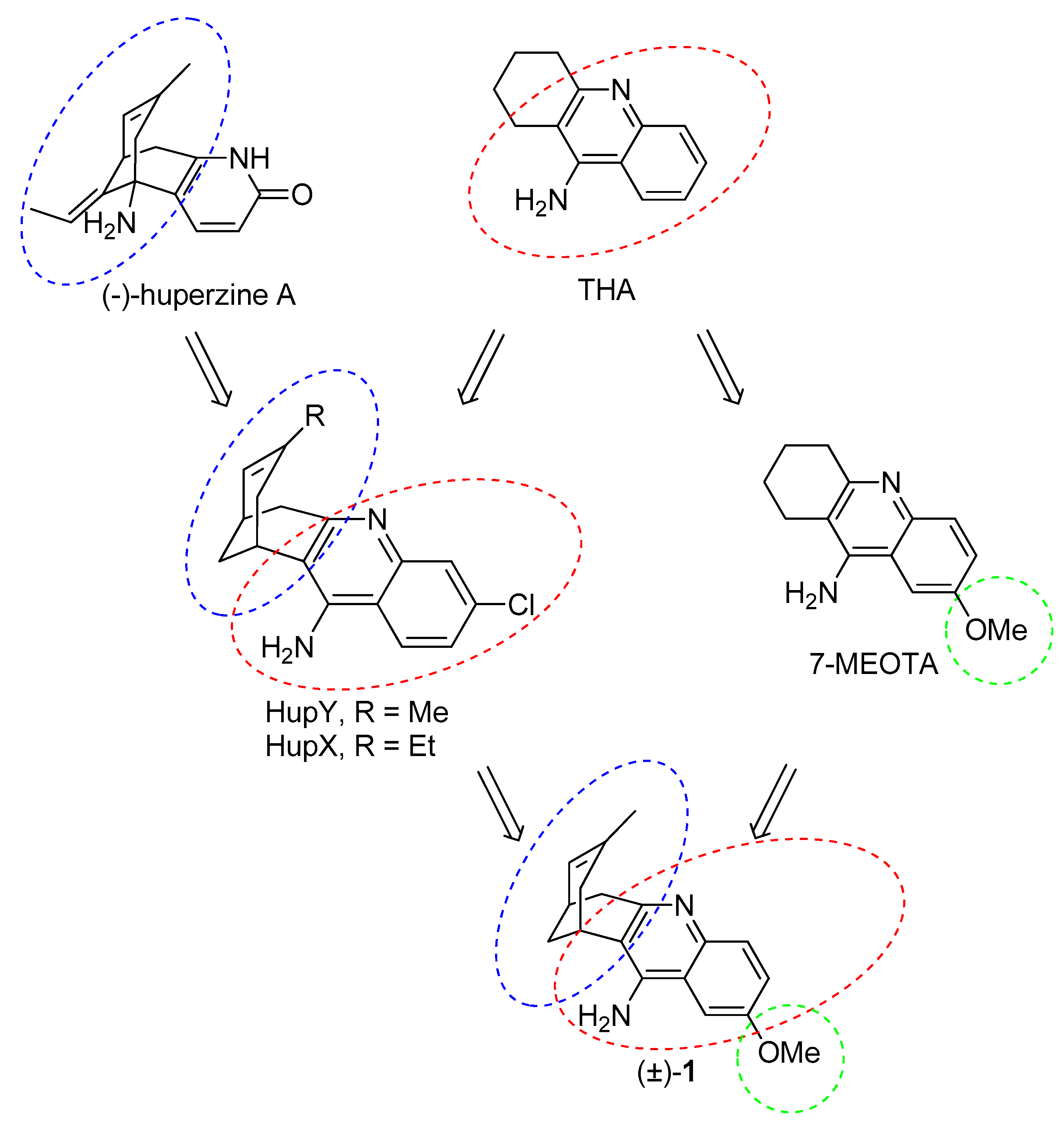
2. Design
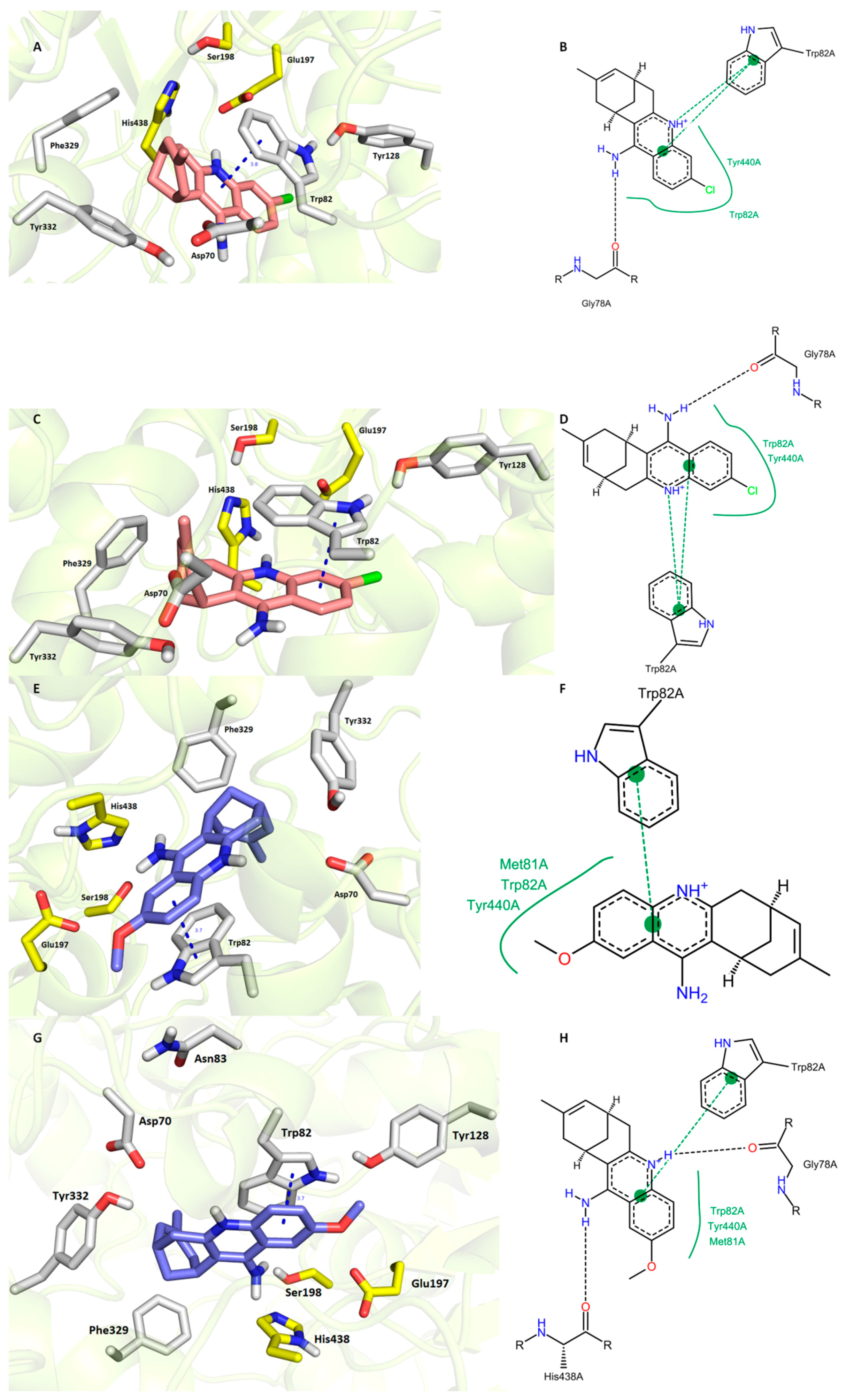
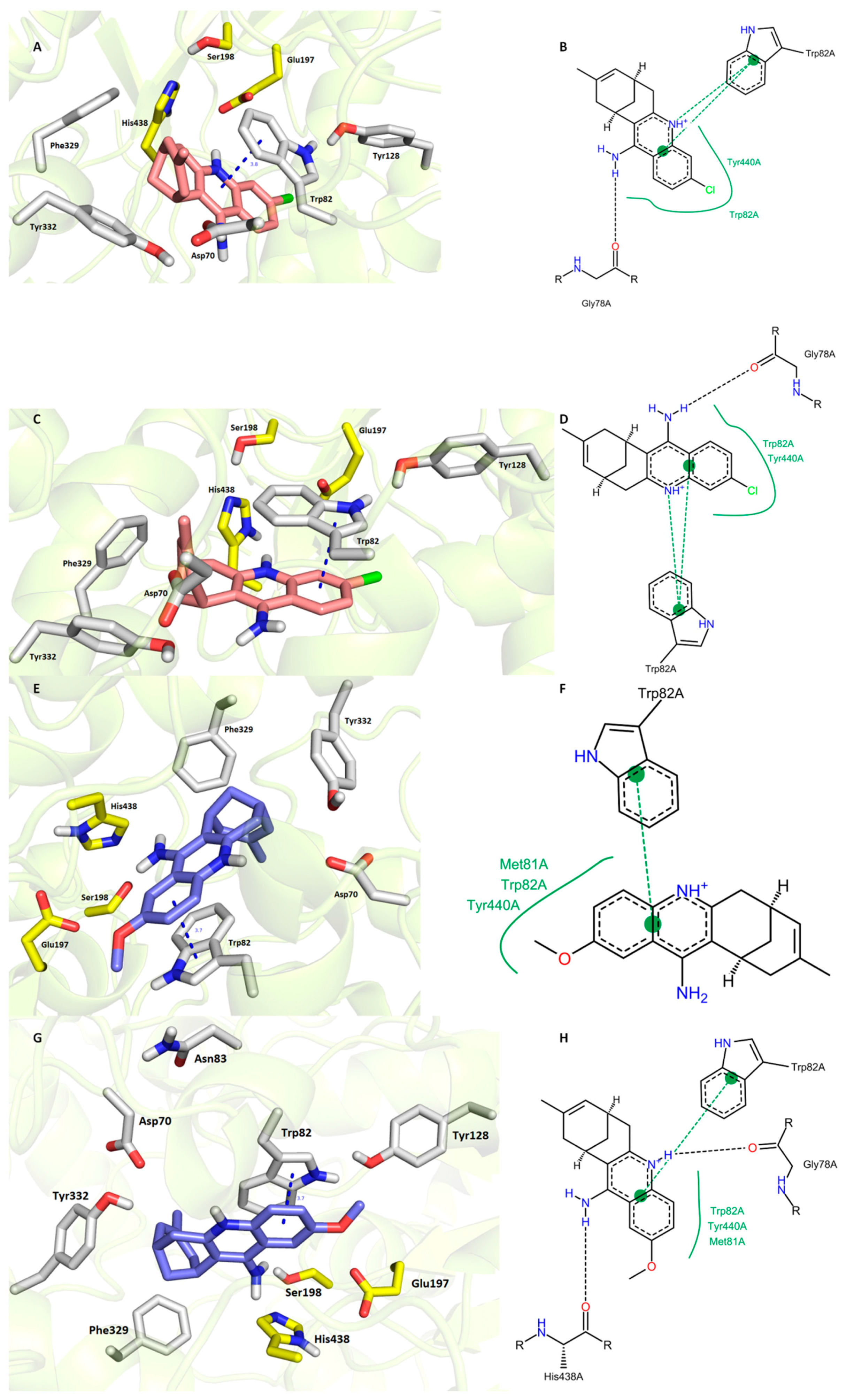
3. Results and Discussion
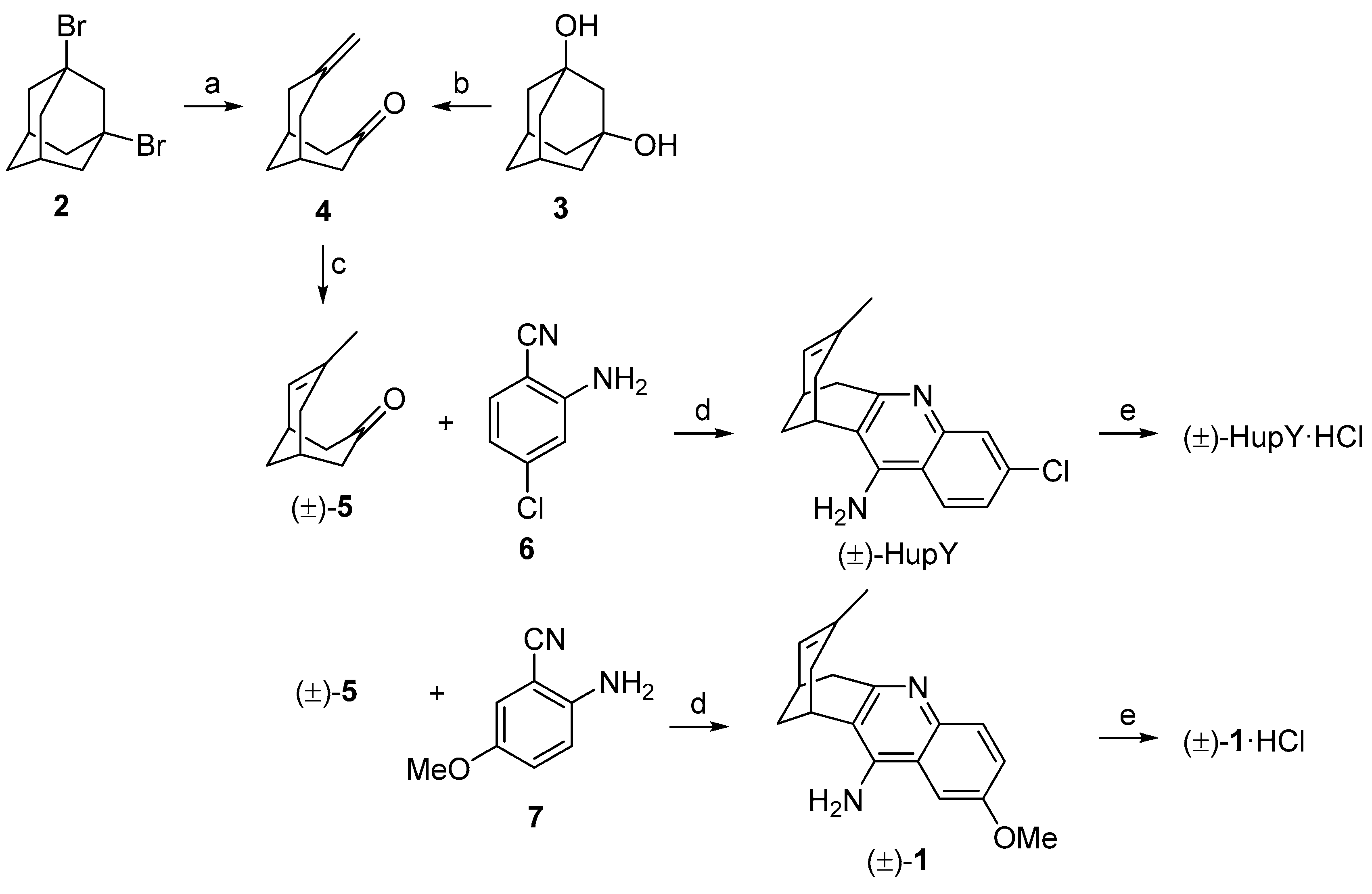
3.1. Synthesis
3.2. Evaluation of AChE and BChE Inhibition Activity
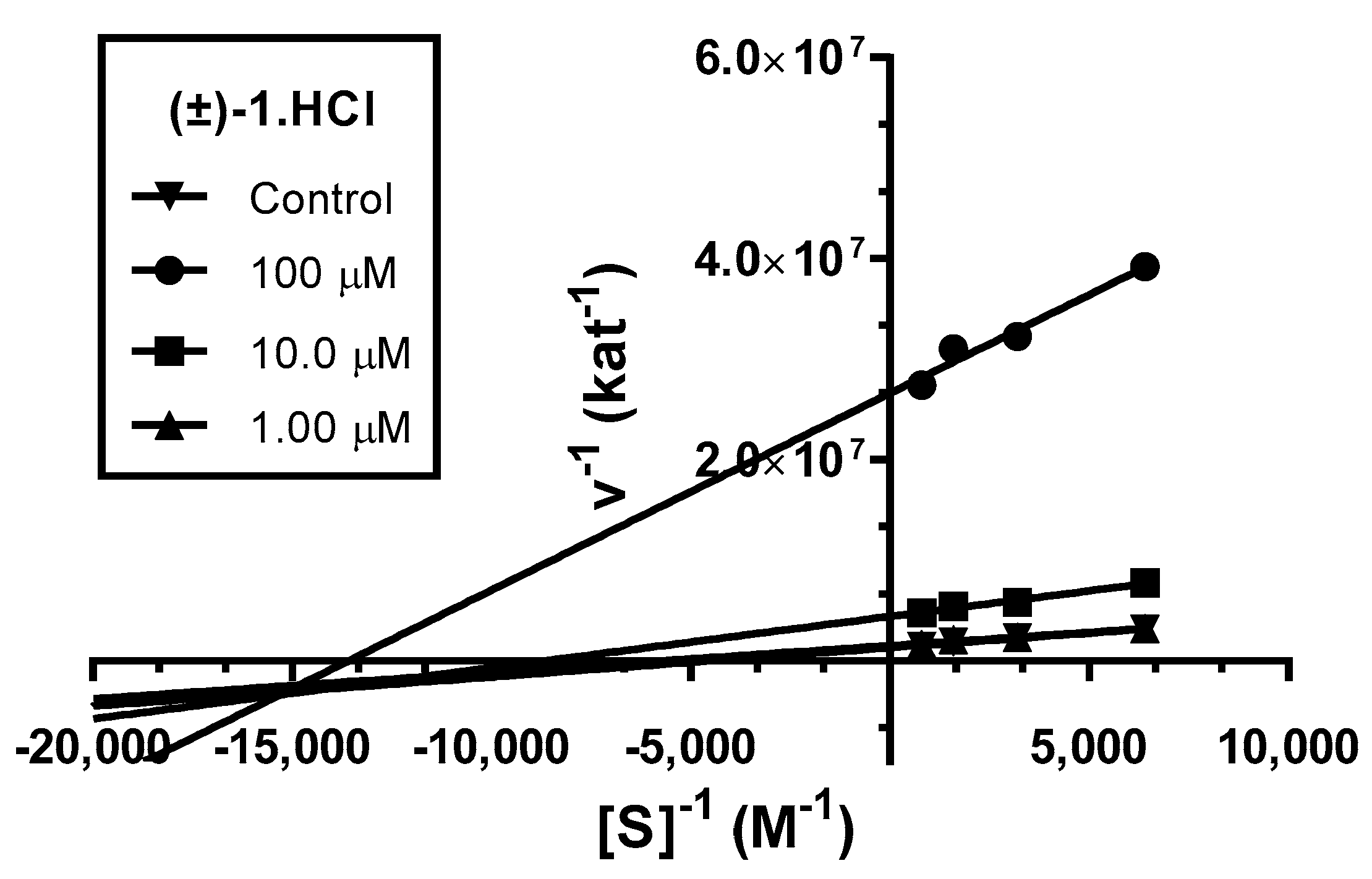
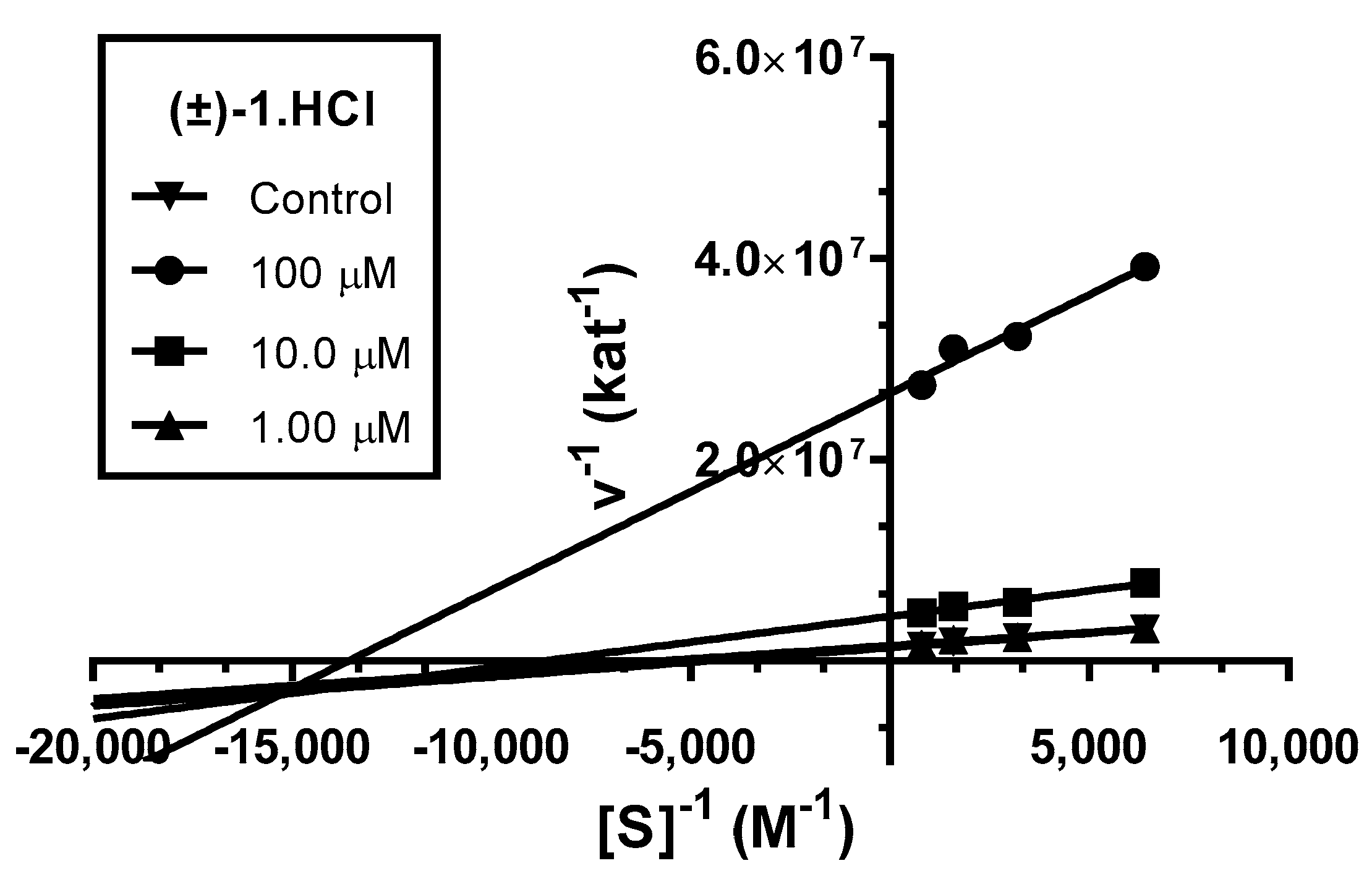
3.3. Kinetic Study of AChE Inhibition
3.4. In Vitro Blood-Brain Barrier Permeation and Druglikeness
3.5. Cytotoxicity
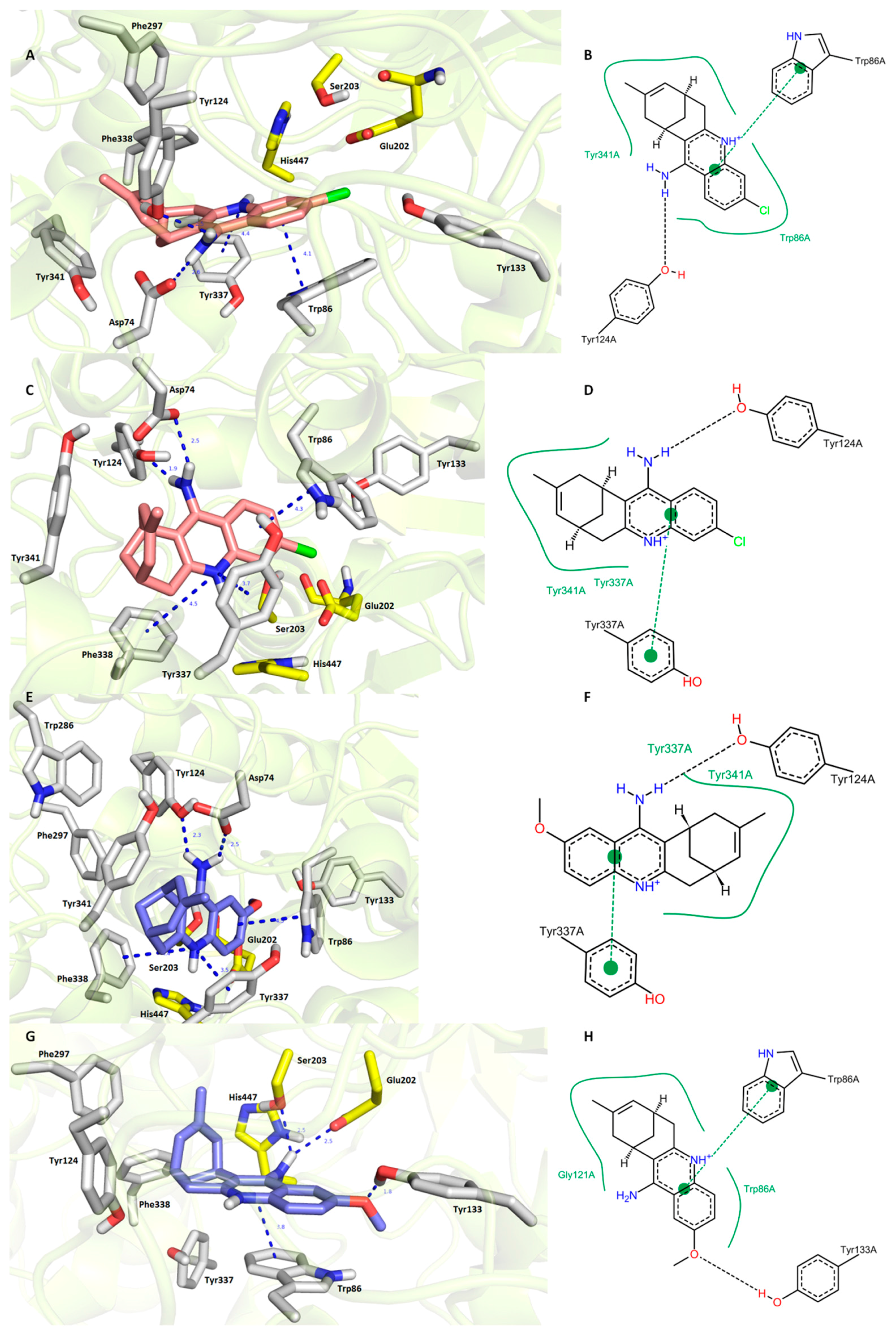
3.6. In Silico Docking Studies
4. Materials and Methods
4.1. General Chemistry Methods
4.1.1. Preparation of 7-Methylenebicyclo[3.3.1]nonan-1-one (4)
4.1.2. Preparation of rac-7-Chloro-15-methyl-10-azatetracyclo[11.3.1.02,11.04,9]heptadeca-2(11),3,5,7,9,14-hexaen-3-amine hydrochloride ((±)-HupY·HCl)
4.1.3. Preparation of rac-6-Methoxy-15-methyl-10-azatetracyclo[11.3.1.02,11.04,9]heptadeca-2(11),3,5,7,9,14-hexaen-3-amine hydrochloride ((±)-1·HCl)
4.2. Biochemical Studies
4.2.1. In Vitro Anti-Cholinesterase Assay
4.2.2. Kinetic Study of AChE Inhibition
4.2.3. Determination of In Vitro Blood-Brain Barrier Permeation
4.2.4. Evaluation of Cytotoxicity by MTT Assay
4.3. Molecular Modeling Studies
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Maresova, P.; Klimova, B.; Novotny, M.; Kuca, K. Alzheimer’s and Parkinson’s Diseases: Expected Economic Impact on Europe-A Call for a Uniform European Strategy. J. Alzheimers Dis. JAD 2016, 54, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Marešová, P.; Mohelská, H.; Dolejš, J.; Kuča, K. Socio-economic Aspects of Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Zemek, F.; Korabecny, J.; Nepovimova, E.; Soukup, O.; Windisch, M.; Kuca, K. Adamantane-A Lead Structure for Drugs in Clinical Practice. Curr. Med. Chem. 2016, 23, 3245–3266. [Google Scholar] [CrossRef] [PubMed]
- Horak, M.; Holubova, K.; Nepovimova, E.; Krusek, J.; Kaniakova, M.; Korabecny, J.; Vyklicky, L.; Kuca, K.; Stuchlik, A.; Ricny, J.; et al. The pharmacology of tacrine at N-methyl-d-aspartate receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 75, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xiao, K.; Ma, L.; Xiong, B.; Fu, Y.; Yu, H.; Wang, W.; Wang, X.; Hu, D.; Peng, H.; et al. Design, synthesis and biological evaluation of novel dual inhibitors of acetylcholinesterase and beta-secretase. Bioorg. Med. Chem. 2009, 17, 1600–1613. [Google Scholar] [CrossRef] [PubMed]
- Rampa, A.; Mancini, F.; De Simone, A.; Falchi, F.; Belluti, F.; Di Martino, R.M.C.; Gobbi, S.; Andrisano, V.; Tarozzi, A.; Bartolini, M.; et al. From AChE to BACE1 inhibitors: The role of the amine on the indanone scaffold. Bioorg. Med. Chem. Lett. 2015, 25, 2804–2808. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, P.; Cariati, L.; Desiderio, D.; Sgammato, R.; Lamberti, A.; Arcone, R.; Salerno, R.; Nardi, M.; Masullo, M.; Oliverio, M. Design, Synthesis, and Evaluation of Donepezil-Like Compounds as AChE and BACE-1 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Viayna, E.; Sabate, R.; Muñoz-Torrero, D. Dual inhibitors of β-amyloid aggregation and acetylcholinesterase as multi-target anti-Alzheimer drug candidates. Curr. Top. Med. Chem. 2013, 13, 1820–1842. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.; Rincón, Á.; Jiménez-Aliaga, K.L.; Bermejo-Bescós, P.; Martín-Aragón, S.; Molina, M.T.; Csákÿ, A.G. Synthesis and evaluation of arylquinones as BACE1 inhibitors, β-amyloid peptide aggregation inhibitors, and destabilizers of preformed β-amyloid fibrils. Bioorg. Med. Chem. Lett. 2011, 21, 2183–2187. [Google Scholar] [CrossRef] [PubMed]
- Panek, D.; Więckowska, A.; Wichur, T.; Bajda, M.; Godyń, J.; Jończyk, J.; Mika, K.; Janockova, J.; Soukup, O.; Knez, D.; et al. Design, synthesis and biological evaluation of new phthalimide and saccharin derivatives with alicyclic amines targeting cholinesterases, beta-secretase and amyloid beta aggregation. Eur. J. Med. Chem. 2016, 125, 676–695. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer’s disease. Indian J. Psychiatry 2009, 51, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Babkova, K.; Korabecny, J.; Soukup, O.; Nepovimova, E.; Jun, D.; Kuca, K. Prolyl oligopeptidase and its role in the organism: Attention to the most promising and clinically relevant inhibitors. Future Med. Chem. 2017, 9, 1015–1038. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Soukup, O.; Maresova, P.; Korabecny, J.; Nepovimova, E.; Klimova, B.; Honegr, J.; Ramalho, T.C.; França, T.C.C. Current Approaches Against Alzheimer’s Disease in Clinical Trials. J. Braz. Chem. Soc. 2016, 27, 641–649. [Google Scholar] [CrossRef]
- Soukup, O.; Jun, D.; Zdarova-Karasova, J.; Patocka, J.; Musilek, K.; Korabecny, J.; Krusek, J.; Kaniakova, M.; Sepsova, V.; Mandikova, J.; et al. A resurrection of 7-MEOTA: A comparison with tacrine. Curr. Alzheimer Res. 2013, 10, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Musilek, K.; Zemek, F.; Horova, A.; Holas, O.; Nepovimova, E.; Opletalova, V.; Hroudova, J.; Fisar, Z.; Jung, Y.-S.; et al. Synthesis and in vitro evaluation of 7-methoxy-N-(pent-4-enyl)-1,2,3,4-tetrahydroacridin-9-amine-new tacrine derivate with cholinergic properties. Bioorg. Med. Chem. Lett. 2011, 21, 6563–6566. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Musilek, K.; Holas, O.; Binder, J.; Zemek, F.; Marek, J.; Pohanka, M.; Opletalova, V.; Dohnal, V.; Kuca, K. Synthesis and in vitro evaluation of N-alkyl-7-methoxytacrine hydrochlorides as potential cholinesterase inhibitors in Alzheimer disease. Bioorg. Med. Chem. Lett. 2010, 20, 6093–6095. [Google Scholar] [CrossRef] [PubMed]
- Patocka, J.; Jun, D.; Kuca, K. Possible role of hydroxylated metabolites of tacrine in drug toxicity and therapy of Alzheimer’s disease. Curr. Drug Metab. 2008, 9, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Torrero, D.; Camps, P. Huprines for Alzheimer’s disease drug development. Expert Opin. Drug Discov. 2008, 3, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [PubMed]
- Raves, M.L.; Harel, M.; Pang, Y.P.; Silman, I.; Kozikowski, A.P.; Sussman, J.L. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (-)-huperzine A. Nat. Struct. Biol. 1997, 4, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Dvir, H.; Wong, D.M.; Harel, M.; Barril, X.; Orozco, M.; Luque, F.J.; Muñoz-Torrero, D.; Camps, P.; Rosenberry, T.L.; Silman, I.; et al. 3D structure of Torpedo californica acetylcholinesterase complexed with huprine X at 2.1 A resolution: Kinetic and molecular dynamic correlates. Biochemistry (Mosc.) 2002, 41, 2970–2981. [Google Scholar] [CrossRef]
- Gabrielson, K.L.; Hogue, B.A.; Bohr, V.A.; Cardounel, A.J.; Nakajima, W.; Kofler, J.; Zweier, J.L.; Rodriguez, E.R.; Martin, L.J.; de Souza-Pinto, N.C.; et al. Mitochondrial toxin 3-nitropropionic acid induces cardiac and neurotoxicity differentially in mice. Am. J. Pathol. 2001, 159, 1507–1520. [Google Scholar] [CrossRef]
- Canudas, A.M.; Pubill, D.; Sureda, F.X.; Verdaguer, E.; Camps, P.; Muñoz-Torrero, D.; Jiménez, A.; Camins, A.; Pallàs, M. Neuroprotective effects of (±)-huprine Y on in vitro and in vivo models of excitoxicity damage. Exp. Neurol. 2003, 180, 123–130. [Google Scholar] [CrossRef]
- Yu, J.; Jin, Y.; Lu, M. 3-Methyl-4-oxa-5-azahomoadamantane as an Organocatalyst for the Aerobic Oxidation of Primary Amines to Oximes in Water. Adv. Synth. Catal. 2015, 357, 1175–1180. [Google Scholar] [CrossRef]
- Denmark, S.E.; Henke, B.R. Investigations on transition-state geometry in the aldol condensation. J. Am. Chem. Soc. 1989, 111, 8032–8034. [Google Scholar] [CrossRef]
- Camps, P.; Contreras, J.; Font-Bardia, M.; Morral, J.; Muñoz-Torrero, D.; Solans, X. Enantioselective synthesis of tacrine–huperzine A hybrids. Preparative chiral MPLC separation of their racemic mixtures and absolute configuration assignments by X-ray diffraction analysis. Tetrahedron Asymmetry 1998, 9, 835–849. [Google Scholar] [CrossRef]
- Lynch, T.J.; Mattes, C.E.; Singh, A.; Bradley, R.M.; Brady, R.O.; Dretchen, K.L. Cocaine detoxification by human plasma butyrylcholinesterase. Toxicol. Appl. Pharmacol. 1997, 145, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Cravatt, B.F. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev. 2011, 111, 6022–6063. [Google Scholar] [CrossRef] [PubMed]
- Komloova, M.; Musilek, K.; Dolezal, M.; Gunn-Moore, F.; Kuca, K. Structure-activity relationship of quaternary acetylcholinesterase inhibitors-outlook for early myasthenia gravis treatment. Curr. Med. Chem. 2010, 17, 1810–1824. [Google Scholar] [CrossRef] [PubMed]
- Čolović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Pohanka, M.; Karasova, J.Z.; Kuca, K.; Pikula, J.; Holas, O.; Korabecny, J.; Cabal, J. Colorimetric dipstick for assay of organophosphate pesticides and nerve agents represented by paraoxon, sarin and VX. Talanta 2010, 81, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Sepsova, V.; Karasova, J.Z.; Korabecny, J.; Dolezal, R.; Zemek, F.; Bennion, B.J.; Kuca, K. Oximes: Inhibitors of human recombinant acetylcholinesterase. A structure-activity relationship (SAR) study. Int. J. Mol. Sci. 2013, 14, 16882–16900. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M.; Jun, D.; Kuca, K. Improvement of acetylcholinesterase-based assay for organophosphates in way of identification by reactivators. Talanta 2008, 77, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Recanatini, M.; Cavalli, A.; Belluti, F.; Piazzi, L.; Rampa, A.; Bisi, A.; Gobbi, S.; Valenti, P.; Andrisano, V.; Bartolini, M.; et al. SAR of 9-amino-1,2,3,4-tetrahydroacridine-based acetylcholinesterase inhibitors: Synthesis, enzyme inhibitory activity, QSAR, and structure-based CoMFA of tacrine analogues. J. Med. Chem. 2000, 43, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Camps, P.; El Achab, R.; Morral, J.; Muñoz-Torrero, D.; Badia, A.; Baños, J.E.; Vivas, N.M.; Barril, X.; Orozco, M.; Luque, F.J. New Tacrine-Huperzine A Hybrids (Huprines): Highly Potent Tight-Binding Acetylcholinesterase Inhibitors of Interest for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2000, 43, 4657–4666. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Selective inhibitors of butyrylcholinesterase: A valid alternative for therapy of Alzheimer’s disease? Drugs Aging 2001, 18, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim. Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Farlow, M.R.; Hintz, N.; Utsuki, T.; Greig, N.H. Cholinesterase inhibitors, beta-amyloid precursor protein and amyloid beta-peptides in Alzheimer’s disease. Acta Neurol. Scand. Suppl. 2000, 176, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: Possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef]
- Pang, Y.P.; Quiram, P.; Jelacic, T.; Hong, F.; Brimijoin, S. Highly potent, selective, and low cost bis-tetrahydroaminacrine inhibitors of acetylcholinesterase. Steps toward novel drugs for treating Alzheimer’s disease. J. Biol. Chem. 1996, 271, 23646–23649. [Google Scholar] [CrossRef] [PubMed]
- Nepovimova, E.; Korabecny, J.; Dolezal, R.; Babkova, K.; Ondrejicek, A.; Jun, D.; Sepsova, V.; Horova, A.; Hrabinova, M.; Soukup, O.; et al. Tacrine-Trolox Hybrids: A Novel Class of Centrally Active, Nonhepatotoxic Multi-Target-Directed Ligands Exerting Anticholinesterase and Antioxidant Activities with Low In Vivo Toxicity. J. Med. Chem. 2015, 58, 8985–9003. [Google Scholar] [CrossRef] [PubMed]
- Nepovimova, E.; Uliassi, E.; Korabecny, J.; Peña-Altamira, L.E.; Samez, S.; Pesaresi, A.; Garcia, G.E.; Bartolini, M.; Andrisano, V.; Bergamini, C.; et al. Multitarget drug design strategy: Quinone-tacrine hybrids designed to block amyloid-β aggregation and to exert anticholinesterase and antioxidant effects. J. Med. Chem. 2014, 57, 8576–8589. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Korabecny, J.; Horova, A.; Musilek, K.; Nepovimova, E.; Drtinova, L.; Gazova, Z.; Siposova, K.; Dolezal, R.; Jun, D.; et al. Design, synthesis and in vitro testing of 7-methoxytacrine-amantadine analogues: A novel cholinesterase inhibitors for the treatment of Alzheimer’s disease. Med. Chem. Res. 2015, 1–11. [Google Scholar] [CrossRef]
- Spilovska, K.; Korabecny, J.; Kral, J.; Horova, A.; Musilek, K.; Soukup, O.; Drtinova, L.; Gazova, Z.; Siposova, K.; Kuca, K. 7-Methoxytacrine-adamantylamine heterodimers as cholinesterase inhibitors in Alzheimer’s disease treatment—Synthesis, biological evaluation and molecular modeling studies. Molecules 2013, 18, 2397–2418. [Google Scholar] [CrossRef] [PubMed]
- Alcalá, M. del M.; Vivas, N.M.; Hospital, S.; Camps, P.; Muñoz-Torrero, D.; Badia, A. Characterisation of the anticholinesterase activity of two new tacrine-huperzine A hybrids. Neuropharmacology 2003, 44, 749–755. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Lemes, L.F.N.; de Andrade Ramos, G.; de Oliveira, A.S.; da Silva, F.M.R.; de Castro Couto, G.; da Silva Boni, M.; Guimarães, M.J.R.; Souza, I.N.O.; Bartolini, M.; Andrisano, V.; et al. Cardanol-derived AChE inhibitors: Towards the development of dual binding derivatives for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 108, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Korabecny, J.; Sepsova, V.; Jun, D.; Hrabinova, M.; Jost, P.; Muckova, L.; Soukup, O.; Janockova, J.; Kucera, T.; et al. Novel Tacrine-Scutellarin Hybrids as Multipotent Anti-Alzheimer’s Agents: Design, Synthesis and Biological Evaluation. Molecules 2017, 22, 1006. [Google Scholar] [CrossRef] [PubMed]
- Gazova, Z.; Soukup, O.; Sepsova, V.; Siposova, K.; Drtinova, L.; Jost, P.; Spilovska, K.; Korabecny, J.; Nepovimova, E.; Fedunova, D.; et al. Multi-target-directed therapeutic potential of 7-methoxytacrine-adamantylamine heterodimers in the Alzheimer’s disease treatment. Biochim. Biophys. Acta 2017, 1863, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Spaldin, V.; Madden, S.; Pool, W.F.; Woolf, T.F.; Park, B.K. The effect of enzyme inhibition on the metabolism and activation of tacrine by human liver microsomes. Br. J. Clin. Pharmacol. 1994, 38, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Monteith, D.K.; Theiss, J.C. Comparison of tacrine-induced cytotoxicity in primary cultures of rat, mouse, monkey, dog, rabbit, and human hepatocytes. Drug Chem. Toxicol. 1996, 19, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Galisteo, M.; Rissel, M.; Sergent, O.; Chevanne, M.; Cillard, J.; Guillouzo, A.; Lagadic-Gossmann, D. Hepatotoxicity of tacrine: Occurrence of membrane fluidity alterations without involvement of lipid peroxidation. J. Pharmacol. Exp. Ther. 2000, 294, 160–167. [Google Scholar] [PubMed]
- Terrell, P.S.; Bredenkamp, V.L.; Lammers, J.E. Null Late-onset alanine aminotransferase increase with tacrine. Ann. Pharmacother. 1996, 30, 301. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J.; Ford, J.M.; Truman, C.A.; Scott, M.; Mäkelä, P.M.; Wilcock, G.K. Assessment of the value of therapeutic monitoring of tacrine in Alzheimer’s disease. Eur. J. Clin. Pharmacol. 1998, 54, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Jeřábek, J.; Uliassi, E.; Guidotti, L.; Korábečný, J.; Soukup, O.; Sepsova, V.; Hrabinova, M.; Kuča, K.; Bartolini, M.; Peña-Altamira, L.E.; et al. Tacrine-resveratrol fused hybrids as multi-target-directed ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2017, 127, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K.; Hamada, H.; Machida, M.; Ushio, H. High throughput prediction of oral absorption: Improvement of the composition of the lipid solution used in parallel artificial membrane permeation assay. J. Biomol. Screen. 2001, 6, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Wohnsland, F.; Faller, B. High-throughput permeability pH profile and high-throughput alkane/water log P with artificial membranes. J. Med. Chem. 2001, 44, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Lee Kwang, S.; Park Kyun, M.; Yi, J.; Won Cho, D.; Sup Ra, C.; Musilek, K.; Horova, A.; Korabecny, J.; Dolezal, R.; Jun, D.; Kuca, K. Preparation of 7-methoxy tacrine dimer analogs and their in vitro/in silico evaluation as potential choliensterase inhibitors. Bull. Korean Chem. Soc. 2015. [Google Scholar] [CrossRef]
- Dolezal, R.; Korabecny, J.; Malinak, D.; Honegr, J.; Musilek, K.; Kuca, K. Ligand-based 3D QSAR analysis of reactivation potency of mono- and bis-pyridinium aldoximes toward VX-inhibited rat acetylcholinesterase. J. Mol. Graph. Model. 2015, 56, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Dolezal, R.; Cabelova, P.; Horova, A.; Hruba, E.; Ricny, J.; Sedlacek, L.; Nepovimova, E.; Spilovska, K.; Andrs, M.; et al. 7-MEOTA-donepezil like compounds as cholinesterase inhibitors: Synthesis, pharmacological evaluation, molecular modeling and QSAR studies. Eur. J. Med. Chem. 2014, 82, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Musilek, K.; Holas, O.; Nepovimova, E.; Jun, D.; Zemek, F.; Opletalova, V.; Patocka, J.; Dohnal, V.; Nachon, F.; et al. Synthesis and in vitro evaluation of N-(Bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine as a cholinesterase inhibitor with regard to Alzheimer’s disease treatment. Molecules 2010, 15, 8804–8812. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Andrs, M.; Nepovimova, E.; Dolezal, R.; Babkova, K.; Horova, A.; Malinak, D.; Mezeiova, E.; Gorecki, L.; Sepsova, V.; et al. 7-Methoxytacrine-p-Anisidine Hybrids as Novel Dual Binding Site Acetylcholinesterase Inhibitors for Alzheimer’s Disease Treatment. Molecules 2015, 20, 22084–22101. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Holas, O.; Musilek, K.; Pohanka, M.; Opletalova, V.; Dohnal, V.; Kuca, K. Synthesis and In Vitro Evaluation of New Tacrine Derivates-Bis-Alkylene Linked 7-MEOTA. Lett. Org. Chem. 2010, 7, 327–331. [Google Scholar] [CrossRef]
- Kristofikova, Z.; Ricny, J.; Soukup, O.; Korabecny, J.; Nepovimova, E.; Kuca, K.; Ripova, D. Inhibitors of Acetylcholinesterase Derived from 7-Methoxytacrine and Their Effects on the Choline Transporter CHT1. Dement. Geriatr. Cogn. Disord. 2017, 43, 45–58. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 ± SEM (µM) a | Selectiviy for hAChE b | |
|---|---|---|---|
| hAChE | hBChE | ||
| (±)-1·HCl | 2.63 ± 0.36 | 3.76 ± 0.12 | 1.43 |
| (±)-HupY·HCl | 0.00164 ± 0.00014 | 0.358 ± 0.025 | 218.29 |
| THA | 0.32 ± 0.013 c | 0.08 ± 0.001 c | 0.68 |
| 7-MEOTA | 10.00 ± 0.974 c | 17.56 ± 0.795 c | 1.76 |
| 6-chlorotacrine | 0.02 ± 0.001 c | 1.78 ± 0.097 c | 100.68 |
| Compound | BBB Penetration Estimation | |
|---|---|---|
| Pe ± SEM (10−6 cm·s−1) a | CNS (+/−) b | |
| (±)-1·HCl | 7.64 ± 0.22 | CNS + |
| (±)-HupY·HCl | 5.89 ± 0.36 | CNS + |
| THA | 5.30 ± 0.20 c | CNS + |
| Donepezil | 7.3 ± 0.9 | CNS + |
| 7-MEOTA | 6.50 ± 1.85 | CNS + |
| 6-chlorotacrine | 5.00 ± 0.45 c | CNS + |
| Cefuroxim | 2.70 ± 0.1 | CNS − |
| Piroxicam | 2.20 ± 0.15 | CNS − |
| Compound | Physicochemical Properties | CNS MPO Score b | |||||
|---|---|---|---|---|---|---|---|
| ClogP a | ClogD7.4 a | TPSA a | MW a | HBD a | CpKa a | ||
| (±)-1·HCl | 2.91 (3.58 ± 0.02) c | 1.56 (1.734) c | 49.39 | 280.36 | 2 | 8.91 (9.54 ± 0.02) c | 5.0 |
| (±)-HupY·HCl | 3.67 | 2.95 | 40.16 | 285.79 | 2 | 8.07 | 5.0 |
| THA | 2.63 | 1.25 | 40.16 | 199.27 | 2 | 8.95 | 4.7 |
| 7-MEOTA | 2.47 | 1.08 | 49.39 | 229.30 | 2 | 8.98 | 5.0 |
| 6-chlorotacrine | 3.23 | 2.41 | 40.16 | 233.72 | 2 | 8.20 | 5.1 |
| Compound | IC50 ± SEM (µM) a | ||
|---|---|---|---|
| HepG2 | ACHN | SH-SY5Y | |
| (±)-1·HCl | 14.91 ± 0.11 | 32.22 ± 0.72 | 17.84 ± 0.09 |
| (±)-HupY·HCl | 15.87 ± 1.17 | 24.62 ± 0.32 | 17.49 ± 0.44 |
| THA | 168.47 ± 3.63 | 155.20 ± 0.67 | 120.63 ± 1.97 |
| 7-MEOTA | 44.37 ± 3.35 | 49.27 ± 2.69 | 26.83 ± 1.04 |
| 6-chlorotacrine | 43.20 ± 1.17 | 55.11 ± 1.76 | 50.40 ± 1.28 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezeiova, E.; Korabecny, J.; Sepsova, V.; Hrabinova, M.; Jost, P.; Muckova, L.; Kucera, T.; Dolezal, R.; Misik, J.; Spilovska, K.; et al. Development of 2-Methoxyhuprine as Novel Lead for Alzheimer’s Disease Therapy. Molecules 2017, 22, 1265. https://doi.org/10.3390/molecules22081265
Mezeiova E, Korabecny J, Sepsova V, Hrabinova M, Jost P, Muckova L, Kucera T, Dolezal R, Misik J, Spilovska K, et al. Development of 2-Methoxyhuprine as Novel Lead for Alzheimer’s Disease Therapy. Molecules. 2017; 22(8):1265. https://doi.org/10.3390/molecules22081265
Chicago/Turabian StyleMezeiova, Eva, Jan Korabecny, Vendula Sepsova, Martina Hrabinova, Petr Jost, Lubica Muckova, Tomas Kucera, Rafael Dolezal, Jan Misik, Katarina Spilovska, and et al. 2017. "Development of 2-Methoxyhuprine as Novel Lead for Alzheimer’s Disease Therapy" Molecules 22, no. 8: 1265. https://doi.org/10.3390/molecules22081265