Cytotoxic and Antiviral Triterpenoids from the Mangrove Plant Sonneratia paracaseolaris

 ,
,
Abstract
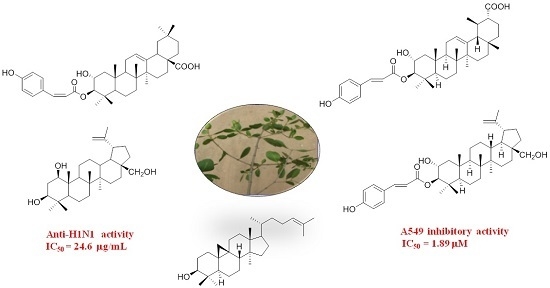
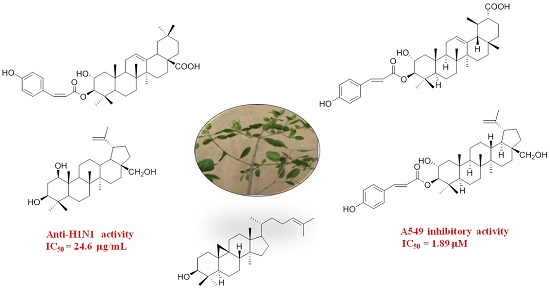
:
1. Introduction
2. Results and Discussion
2.1. Structure Elucidation
2.2. Biological Evaluations
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Cytotoxicity Assay
3.5. Anti-H1N1 Virus Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Qin, H.N.; Graham, S.; Gilbert, M.G. Sonneratia linnaeus f., Suppl. Pl. 38, 252. 1782, nom. cons. In Flora of China; Wu, Z.Y., Ed.; Science Publishers: Beijing, China, 2007; Volume 13, pp. 286–288. [Google Scholar]
- Bandaranayake, W.M. Traditional and medicinal uses of mangroves. Mangroves Salt Marshes 1998, 2, 133–148. [Google Scholar] [CrossRef]
- Wu, S.B.; Wen, Y.; Li, X.W.; Zhao, Y.; Zhao, Z.; Hu, J.F. Chemical constituents from the fruits of Sonneratia caseolaris and Sonneratia ovate (Sonneratiaceae). Biochem. Syst. Ecol. 2009, 37, 1–5. [Google Scholar] [CrossRef]
- Tian, M.Q.; Dai, H.F.; Li, X.M.; Wang, B.G. Chemical constituents of marine medicinal mangrove plant Sonneratia caseolaris. Chin. J. Oceanol. Limnol. 2009, 27, 288–296. [Google Scholar] [CrossRef]
- Liu, H.L.; Huang, X.Y.; Li, J.; Xin, G.R.; Guo, Y.W. Absolute configurations of Integracins A, B and 15′-Dehydroxy-Integracin B. Chirality 2012, 24, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, S.K.; Ahmed, F.; Ohtsuki, T.; Ishibashi, M. Flavonoids from Sonneratia caseolaris. J. Nat. Med. 2006, 60, 264–265. [Google Scholar] [CrossRef]
- Wang, R.J.; Chen, Z.Y. Systematics and biogeography study on the family Sonneratiaceae. Guihaia 2002, 22, 214–219. [Google Scholar]
- Li, H.S.; Chen, G.Z. The distribution status and conservation of mangrove species of Sonneratia paracaseolaris. J. Guangdong Educ. Inst. 2006, 26, 81–83. [Google Scholar]
- Wang, R.J.; Chen, Z.Y.; Chen, E.Y.; Zheng, X.R. Two hybrids of the genus Sonneratia (Sonneratiaceae) from China. Guihaia 1999, 19, 199–204. [Google Scholar]
- Chen, X.L.; Liu, H.L.; Li, J.; Xin, G.R.; Guo, Y.W. Paracaseolide A, first α-alkylbutenolide dimer with an unusual tetraquinane oxacage bislactone skeleton from Chinese mangrove Sonneratia paracaseolaris. Org. Lett. 2011, 13, 5032–5035. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Hafeez, F.; Begum, S.; Siddiqui, B.S. Oleanderol, a new pentacyclic triterpene from the leaves of Nerium oleander. J. Nat. Prod. 1988, 51, 229–233. [Google Scholar] [CrossRef]
- Schmidt, J.; Himmelreich, U.; Adam, G. Brassinosteroids, sterols and Lup-20(29)-en-2α, 3β, 28-triol from Rheum rhabarbarum. Phytochemistry 1995, 40, 527–531. [Google Scholar] [CrossRef]
- Gong, K.K.; Tang, X.L.; Zhang, G.; Cheng, C.L.; Zhang, X.W.; Li, P.L.; Li, G.Q. Polyhydroxylated steroids from the south China sea soft coral Sarcophyton sp. and their cytotoxic and antiviral activities. Mar. Drugs. 2013, 11, 4788–4798. [Google Scholar] [PubMed]
- Giang, P.M.; Son, P.T. A pentacyclic triterpenoid acid from Lonicera Japonica Thunb Caprifoliaceae of Vietnam. J. Chem. 2003, 41, 108–109. [Google Scholar]
- Sholichin, M.; Yamasaki, K.; Kasai, R.; Tanaka, O. 13C nuclear magnetic resonance of lupine-type triterpenes, lupeol, betulin and betulinic acid. Chem. Pharm. Bull. 1980, 28, 1006–1008. [Google Scholar] [CrossRef]
- Fotie, J.; Bohle, D.S.; Leimanis, M.L.; Georges, E.; Rukunga, G.; Nkengfack, A.E. Lupeol long-chain fatty acid esters with antimalarial activity from Holarrhena floribunda. J. Nat. Prod. 2005, 69, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Patra, A.; Chaudhuri, S.K.; Panda, S.K. Betulin-3-caffeate from Quercus suber, 13C-nmr spectra of some lupenes. J. Nat. Prod. 1988, 51, 217–220. [Google Scholar] [CrossRef]
- Reher, G.; Budesinsky, M. Triterpenoids from plants of the sanguisorbeae. Phytochemistry 1992, 31, 3909–3914. [Google Scholar] [CrossRef]
- Aguirre, M.C.; Delporte, C.; Backhouse, N.; Erazo, S.; Letelier, M.E.; Cassels, B.K.; Silva, X.; Alegria, S.; Negrete, R. Topical anti-inflammatory activity of 2α- hydroxyl pentacyclic triterpene acids from the leaves of Ugni molinae. Bioorg. Med. Chem. 2006, 14, 5673–5677. [Google Scholar] [CrossRef] [PubMed]
- Yagi, A.; Okamura, N.; Haraguchi, Y.; Noda, K.; Nishioka, I. Studies on the constituents of Zizyphi fructus. I. structure of three new p-coumaroylates of alphitolic acid. Chem. Pharm. Bull. 1978, 26, 1798–1802. [Google Scholar] [CrossRef]
- Lee, S.M.; Min, B.S.; Lee, C.G.; Kim, K.S.; Kho, Y.H. Cytotoxic triterpenoids from the fruit of Zizyphus jujuba. Planta Med. 2003, 69, 1051–1054. [Google Scholar] [PubMed]
- David, J.P.; Meira, M.; David, J.M.; Guedes, M.L.S. Triterpenos and alkyl ferulates from Maprounea guianensis. Quimca Nova 2004, 27, 62–65. [Google Scholar] [CrossRef]
- Mahato, S.B.; Kundu, A.P. 13C-NMR spectra of pentacyclic triterpenoids—A compilation and some salient features. Phytochemistry 1994, 37, 1517–1575. [Google Scholar] [CrossRef]
- Yagi, A.; Okamura, N.; Haraguchi, Y.; Noda, K.; Nishioka, I. Studies on the constituents of zizyphi fructus. II. Structure of new p-coumaroylates of maslinic acid. Chem. Pharm. Bull. 1978, 26, 3075–3079. [Google Scholar] [CrossRef]
- Lee, S.M.; Park, J.G.; Lee, Y.H.; Lee, C.G.; Byung, S.M.; Kim, J.H.; Lee, H.K. Anti-complementary activity of triterpenoides from fruits of Zizyphus jujuba. Biol. Pharm. Bull. 2004, 27, 1883–1886. [Google Scholar] [CrossRef] [PubMed]
- Teresa, J.P.; Urones, J.G.; Marcos, I.S.; Basabe, P.; Cuadrado, M.J.S.; Moro, R.F. Triterpenes from Euphorbia Broteri. Phytochemistry 1987, 26, 1767–1776. [Google Scholar]
- Wang, W.; Zhang, P.; Yu, G.L.; Li, C.X.; Hao, C.; Qi, X.; Zhang, L.J.; Guan, H.S. Preparation and anti-influenza A virus activity of κ-carrageenan oligosaccharide and its sulphated derivatives. Food. Chem. 2012, 133, 880–888. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1998, 48, 589–601. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
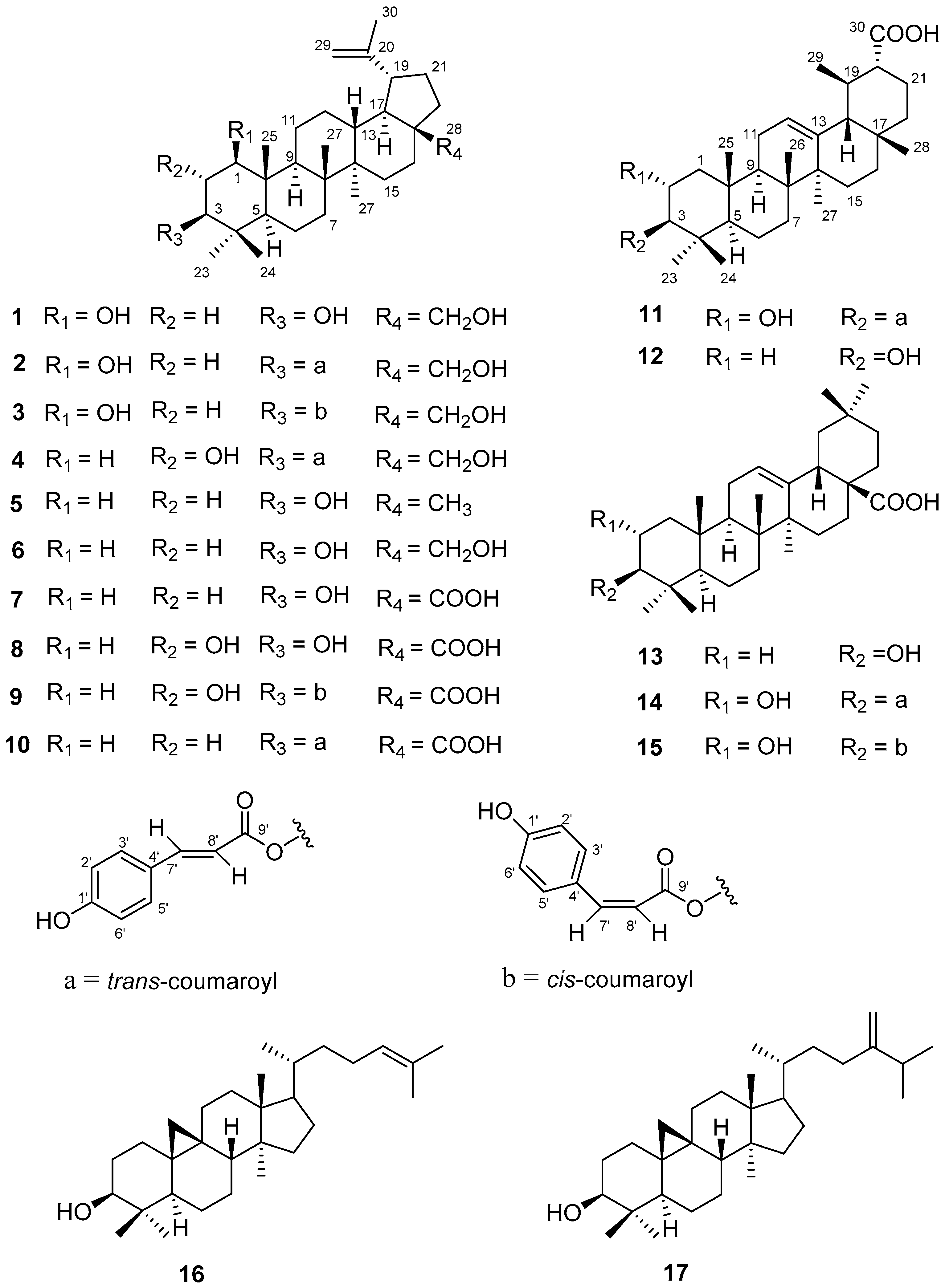
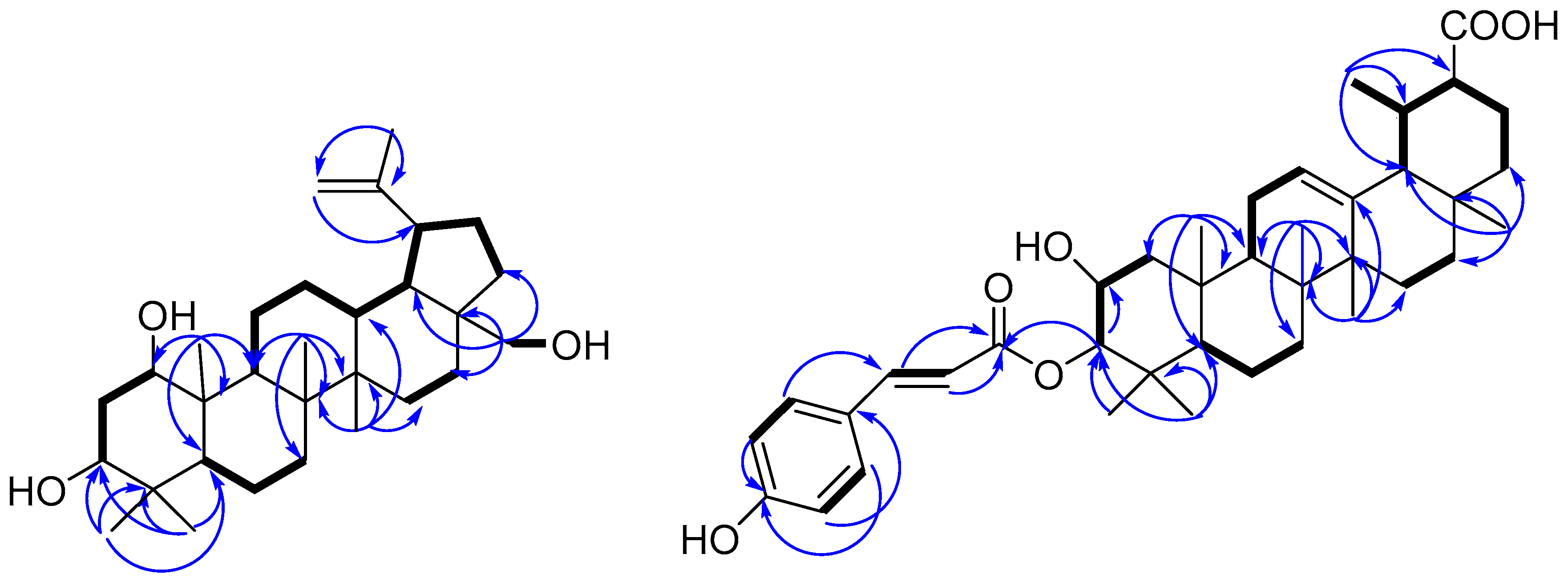
 ), and selected HMBC (
), and selected HMBC (  ) correlations of compounds 1 and 11.
) correlations of compounds 1 and 11.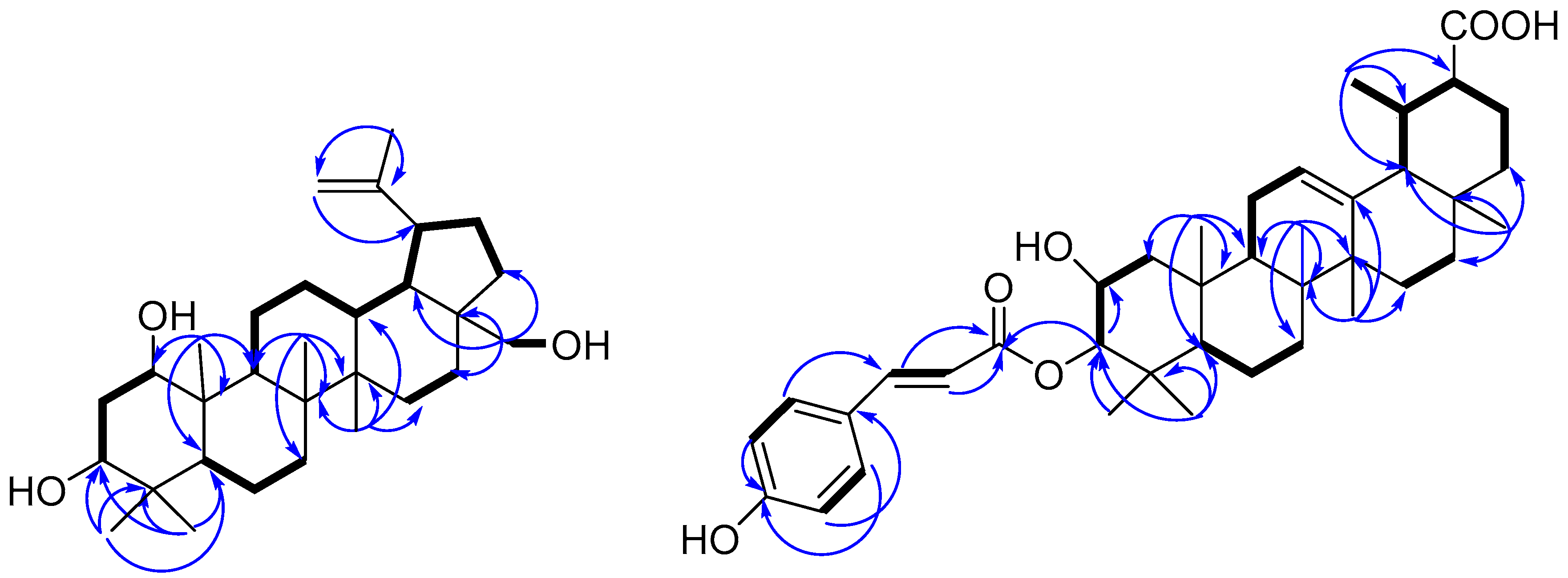

{kind=link}
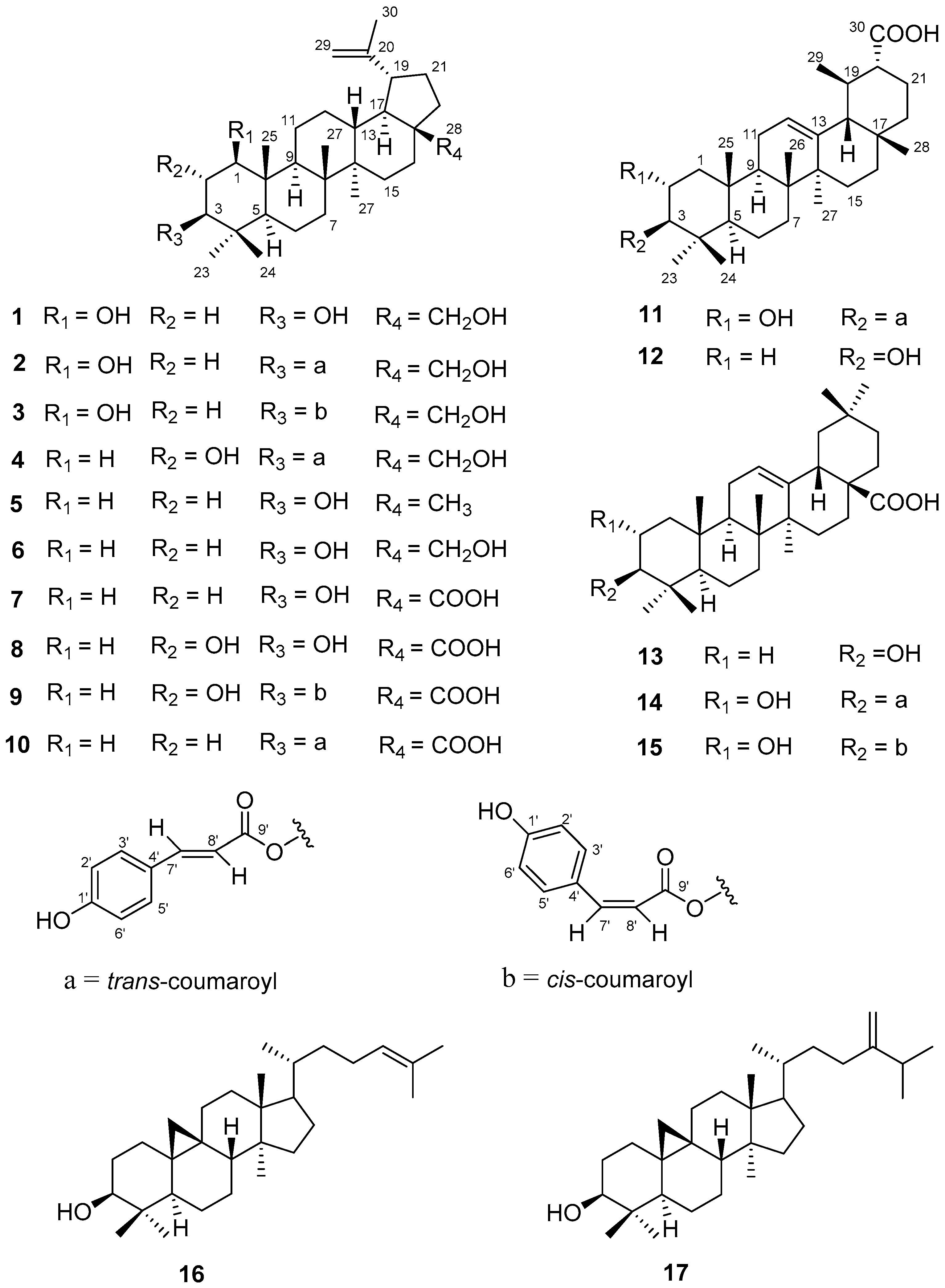{kind=link}
{kind=link}
| Compounds | 1 b,c | 2 b | 3 b | 4 b | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | δH | δC | δH | δC | δH | δC | δH | δC | ||||
| 1 | 3.14 m (W1/2 22.1) | 77.9 | d | 3.30 dd (10.8, 4.7) | 77.2 | d | 3.27 d | 77.2 | d | 1.87 m 0.84m | 47.7 | d |
| 2 | 1.47 m | 38.5 | t | 1.68 m | 34.0 | t | 1.68 m | 33.8 | t | 3.67 m (W1/2 26.6 ) | 64.9 | t |
| 3 | 2.96 m (W1/2 22.8) | 74.0 | d | 4.47 dd (11.9, 4.7) | 76.7 | d | 4.40 dd (12.5, 4.5) | 76.9 | d | 4.48 d (10.0 ) | 83.4 | d |
| 4 | 38.0 | s | 37.6 | s | 37.4 | s | 39.1 | s | ||||
| 5 | 0.46 d (11.0) | 52.6 | d | 0.68 m | 52.2 | d | 0.65 m | 52.2 | d | 0.86 m | 54.5 | d |
| 6 | 1.38 m | 17.6 | t | 1.43 m | 17.4 | t | 1.43 m | 17.4 | t | 1.28 m | 17.8 | t |
| 7 | 1.28 m | 33.9 | t | 1.28 m | 33.8 | t | 1.28 m | 33.8 | t | 1.40 m 1.29 m | 33.6 | t |
| 8 | 41.0 | t | 41.0 | t | 41.0 | t | 40.5 | t | ||||
| 9 | 1.36 m | 51.0 | d | 1.44 m | 50.7 | d | 1.43 m | 50.7 | d | 1.32 m | 49.6 | d |
| 10 | 42.9 | s | 42.8 | s | 42.8 | s | 37.7 | s | ||||
| 11 | 1.13 m 2.35 m | 23.1 | t | 1.15 m 2.35 m | 23.0 | t | 1.11 m 2.32 m | 23.0 | t | 1.29 m 2.34 m | 20.5 | t |
| 12 | 0.87 m 1.48 m | 25.1 | d | 0.90 m 1.49 m | 25.0 | d | 0.97 m 1.49 m | 25.1 | d | 0.93 m 1.60 m | 24.7 | d |
| 13 | 1.59 m | 36.5 | d | 1.59 m | 36.5 | d | 1.60 m | 36.5 | d | 1.60 m | 36.7 | d |
| 14 | 42.3 | s | 42.3 | s | 42.3 | s | 42.3 | s | ||||
| 15 | 1.57 m | 26.7 | t | 1.61 m | 26.8 | t | 1.61 m | 26.7 | t | 1.62 m | 26.6 | t |
| 16 | 1.01 m 1.88 m | 29.1 | t | 1.04 m 1.88 m | 29.1 | t | 1.02 m 1.88 m | 29.1 | t | 1.01 m 1.88 m | 29.0 | t |
| 17 | 47.3 | s | 47.3 | s | 47.3 | s | 47.3 | s | ||||
| 18 | 1.45 m | 48.2 | d | 1.44 m | 48.2 | d | 1.46 m | 48.2 | d | 1.46 m | 48.1 | d |
| 19 | 2.36 m | 47.3 | d | 2.36 m | 47.3 | d | 2.36 m | 47.3 | d | 2.35 m | 47.4 | d |
| 20 | 150.4 | s | 150.4 | s | 150.4 | s | 150.3 | s | ||||
| 21 | 1.22 m 1.83 m | 29.3 | t | 1.22 m 1.83 m | 29.3 | t | 1.24 m 1.84 m | 29.2 | t | 1.24 m 1.84 m | 29.3 | t |
| 22 | 0.87 m 1.85 m | 33.8 | t | 0.86 m 1.85 m | 33.7 | t | 0.83 m1.85 m | 33.7 | t | 0.84 m 1.85 m | 33.8 | t |
| 23 | 0.83 s | 28.0 | q | 0.77 s | 27.6 | q | 0.76 s | 27.5 | q | 0.78 s | 28.3 | q |
| 24 | 0.61 s | 15.4 | q | 0.84 s | 16.0 | q | 0.71 s | 16.0 | q | 0.83 s | 17.6 | q |
| 25 | 0.76 s | 12.2 | q | 0.84 s | 12.2 | q | 0.81 s | 12.1 | q | 0.87 s | 17.0 | q |
| 26 | 0.98 s | 16.0 | q | 1.00 s | 16.2 | q | 0.99 s | 16.1 | q | 0.99 s | 15.7 | q |
| 27 | 0.93 s | 14.5 | q | 0.96 s | 14.5 | q | 0.95 s | 14.5 | q | 0.96 s | 14.4 | q |
| 28 | 3.07 d (10.8) | 58.0 | t | 3.07 d (10.7) | 58.0 | t | 3.07 d (10.8) | 57.9 | t | 3.07 d (10.8) | 57.9 | t |
| 3.51 d (10.8) | 3.51 d (10.7) | 3.51 d (10.8) | 3.51 d (10.8) | |||||||||
| 29 | 4.53 s 4.66 s | 109.6 | t | 4.54 s 4.67 s | 109.5 | t | 4.54 s 4.67 s | 109.6 | t | 4.55 s 4.68 s | 109.6 | t |
| 30 | 1.63 s | 18.7 | q | 1.64 s | 18.7 | q | 1.64 s | 18.7 | q | 1.65 s | 18.8 | q |
| 1′ | 160.0 | s | 159.6 | s | 159.9 | s | ||||||
| 2′,6′ | 7.53 d (8.6) | 130.3 | d | 7.60 d (8.6) | 132.4 | d | 7.49 d (8.6) | 130.1 | d | |||
| 3′,5′ | 6.77 d (8.6) | 115.8 | d | 6.72 d (8.6) | 115.0 | d | 6.74 d (8.6) | 116.0 | d | |||
| 4′ | 124.9 | s | 125.1 | s | 124.3 | s | ||||||
| 7′ | 7.51 d (16.0) | 144.5 | d | 6.82 d (13.0) | 143.1 | d | 7.51 d (16.2) | 144.2 | d | |||
| 8′ | 6.33 d (16.0) | 114.4 | d | 5.73 d (13.0) | 115.0 | d | 6.32 d (16.2) | 114.4 | d | |||
| 9′ | 166.2 | s | 165.8 | s | 166.7 | s | ||||||
| Compound | 11 b | ||
|---|---|---|---|
| No. | δH | δC | |
| 1 | 1.88 m 0.95 m | 47.6 | t |
| 2 | 3.69 m (W1/2 22.9) | 64.8 | d |
| 3 | 4.51 d (9.9) | 83.6 | d |
| 4 | 39.8 | s | |
| 5 | 0.93 m | 54.4 | d |
| 6 | 1.47 m 1.88 m | 17.9 | t |
| 7 | 1.54 m | 32.5 | t |
| 8 | 41.7 | t | |
| 9 | 1.56 m | 46.8 | d |
| 10 | 37.5 | s | |
| 11 | 1.89 m | 23.9 | t |
| 12 | 5.15 br s | 125.1 | d |
| 13 | 138.4 | s | |
| 14 | 41.7 | s | |
| 15 | 1.80 m | 27.5 | t |
| 16 | 1.53 m | 23.0 | t |
| 17 | 30.3 | s | |
| 18 | 2.12 d (11.0) | 52.4 | d |
| 19 | 1.32 m | 38.5 | d |
| 20 | 1.56 m | 38.5 | d |
| 21 | 1.91 m | 36.4 | t |
| 22 | 1.80 m | 39.6 | t |
| 23 | 0.80 s | 28.5 | q |
| 24 | 0.85 s | 17.1 | q |
| 25 | 0.97 s | 17.1 | q |
| 26 | 0.76 s | 16.4 | q |
| 27 | 1.07 s | 23.2 | q |
| 28 | 0.92 s | 21.1 | q |
| 29 | 0.83 d (6.1) | 23.2 | q |
| 30 | 181.4 | s | |
| 1′ | 125.1 | s | |
| 2′,6′ | 7.55 d (8.8) | 130.2 | d |
| 3′,5′ | 6.79 d (8.8) | 115.8 | d |
| 4′ | 159.8 | s | |
| 7′ | 7.52 d (16.4) | 144.1 | d |
| 8′ | 6.38 d (16.4) | 115.0 | d |
| 9′ | 166.7 | s | |
| Compounds | P388 a | HeLa b | A549 b | K562 a |
|---|---|---|---|---|
| 1 | >50 | >50 | >50 | >50 |
| 2 | 27.25 | >50 | >50 | >50 |
| 3 | 22.39 | 33.20 | 14.43 | >50 |
| 4 | 10.56 | 19.13 | 1.89 | >50 |
| 5 | >50 | >50 | >50 | >50 |
| 6 | 44.40 | 42.46 | >50 | >50 |
| 7 | 41.97 | >50 | >50 | >50 |
| 8 | 34.38 | >50 | 27.49 | >50 |
| 9 | 22.36 | 27.15 | 15.43 | >50 |
| 10 | 39.03 | >50 | 37.32 | >50 |
| 11 | >50 | 30.41 | >50 | >50 |
| 12 | 39.77 | >50 | 18 | >50 |
| 13 | >50 | >50 | 23.90 | >50 |
| 14 | 11.04 | 13.10 | >50 | >50 |
| 15 | 23.04 | 24.90 | >50 | 16.28 |
| 16 | >50 | >50 | >50 | >50 |
| 17 | >50 | >50 | >50 | >50 |
| ADM (Adriamycin) c | 0.3 | 0.6 | 0.2 | 0.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, K.-K.; Li, P.-L.; Qiao, D.; Zhang, X.-W.; Chu, M.-J.; Qin, G.-F.; Tang, X.-L.; Li, G.-Q. Cytotoxic and Antiviral Triterpenoids from the Mangrove Plant Sonneratia paracaseolaris. Molecules 2017, 22, 1319. https://doi.org/10.3390/molecules22081319
Gong K-K, Li P-L, Qiao D, Zhang X-W, Chu M-J, Qin G-F, Tang X-L, Li G-Q. Cytotoxic and Antiviral Triterpenoids from the Mangrove Plant Sonneratia paracaseolaris. Molecules. 2017; 22(8):1319. https://doi.org/10.3390/molecules22081319
Chicago/Turabian StyleGong, Kai-Kai, Ping-Lin Li, Dan Qiao, Xing-Wang Zhang, Mei-Jun Chu, Guo-Fei Qin, Xu-Li Tang, and Guo-Qiang Li. 2017. "Cytotoxic and Antiviral Triterpenoids from the Mangrove Plant Sonneratia paracaseolaris" Molecules 22, no. 8: 1319. https://doi.org/10.3390/molecules22081319