Phosphate-Linked Silibinin Dimers (PLSd): New Promising Modified Metabolites

 ,
,  , ,
, ,
Abstract
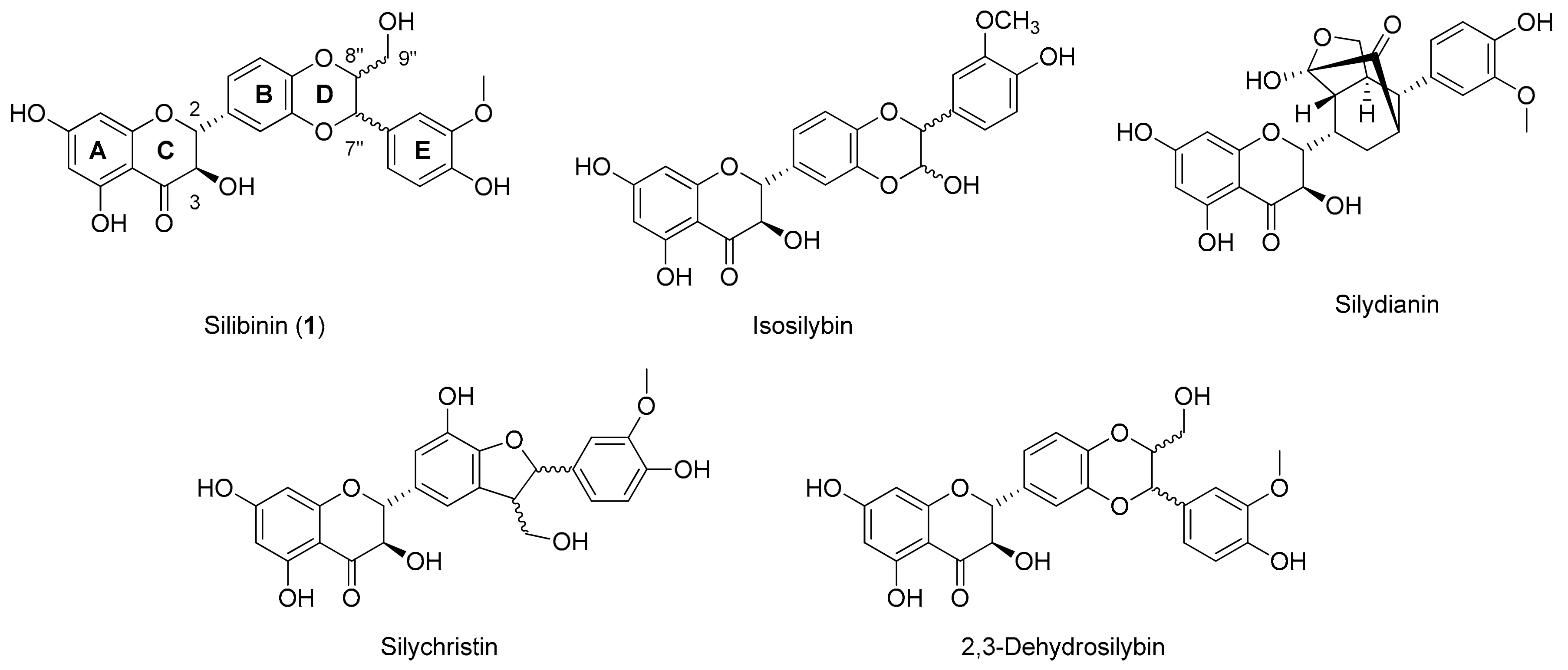
:1. Introduction
2. Results and Discussion
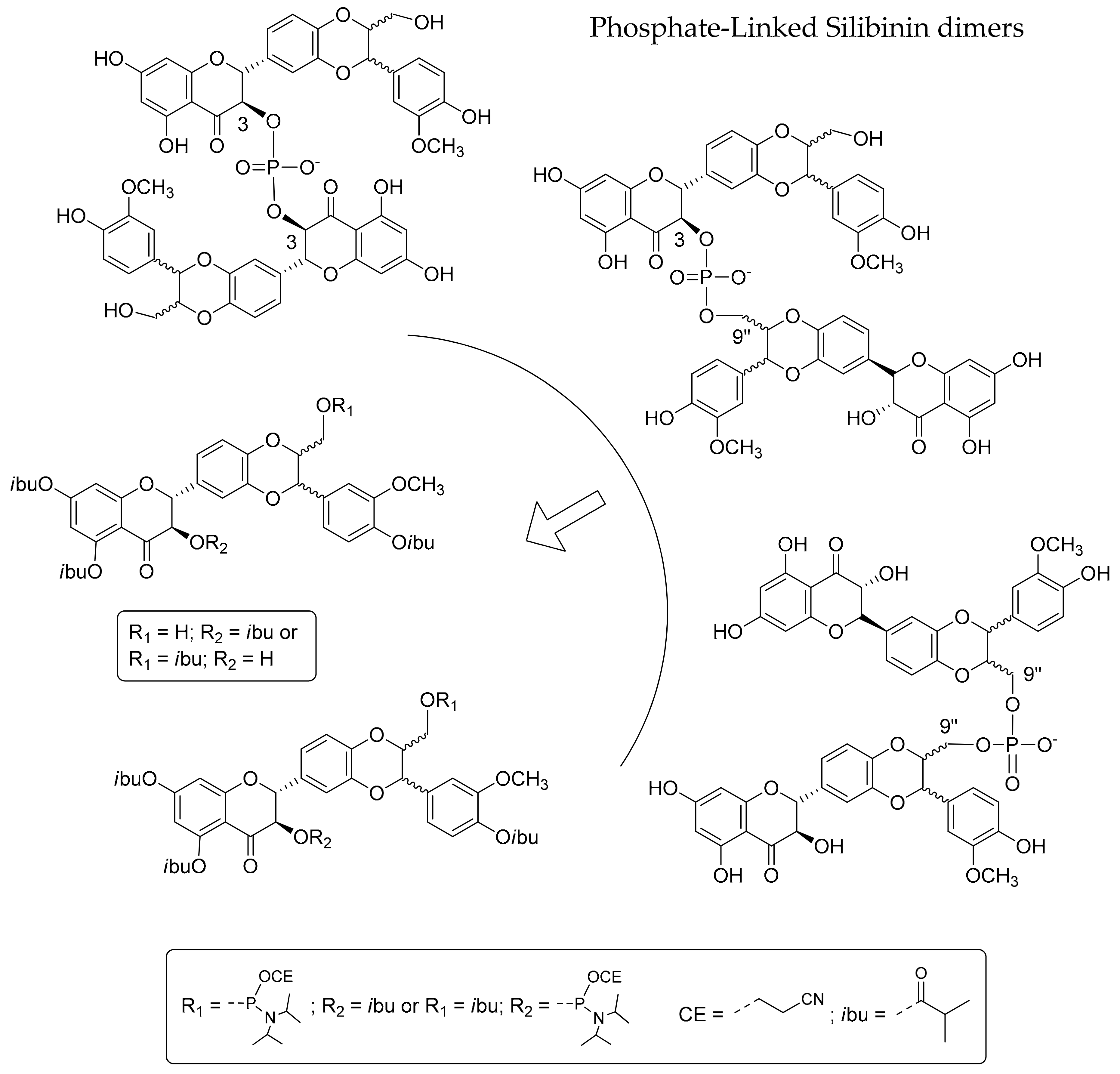
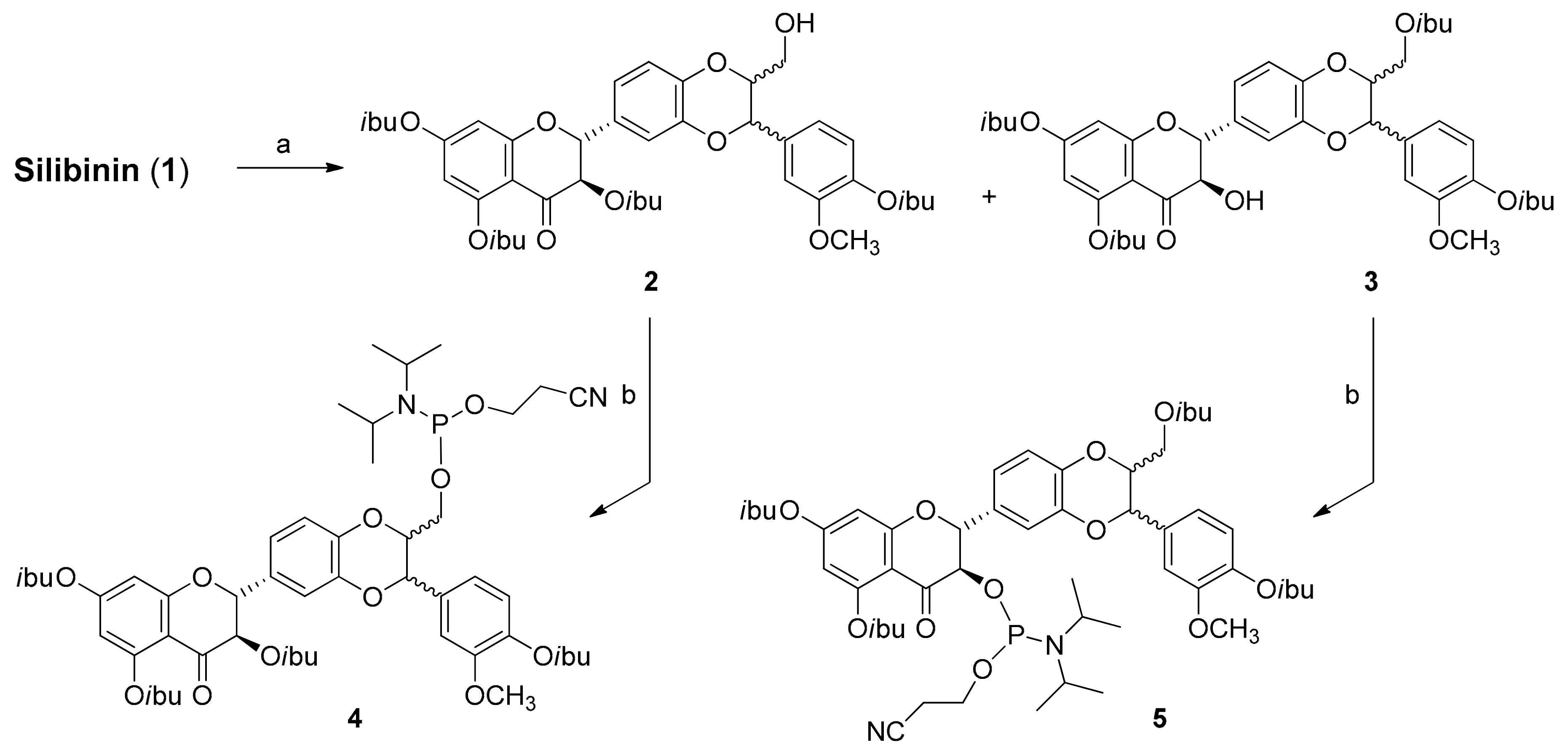
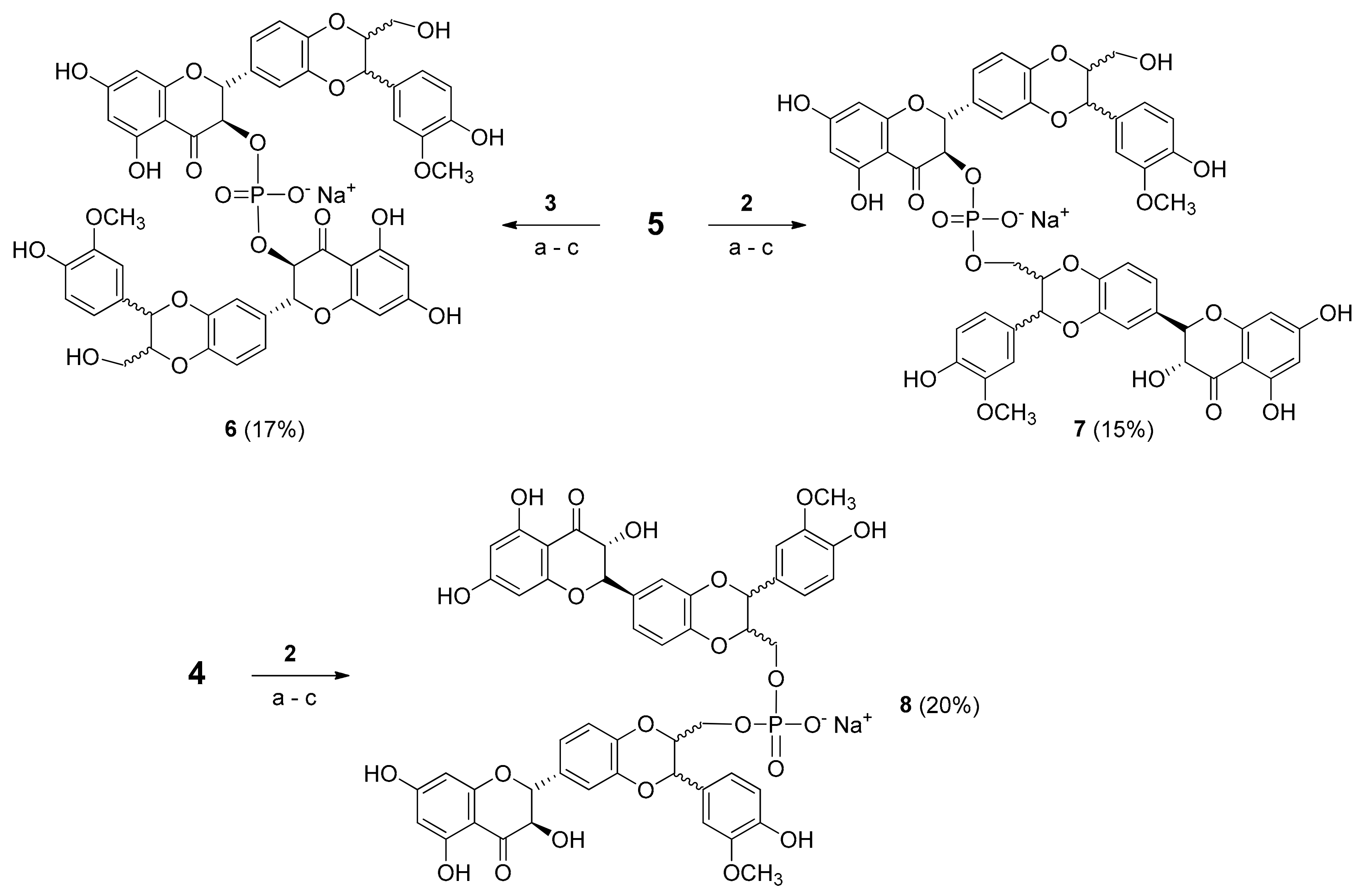
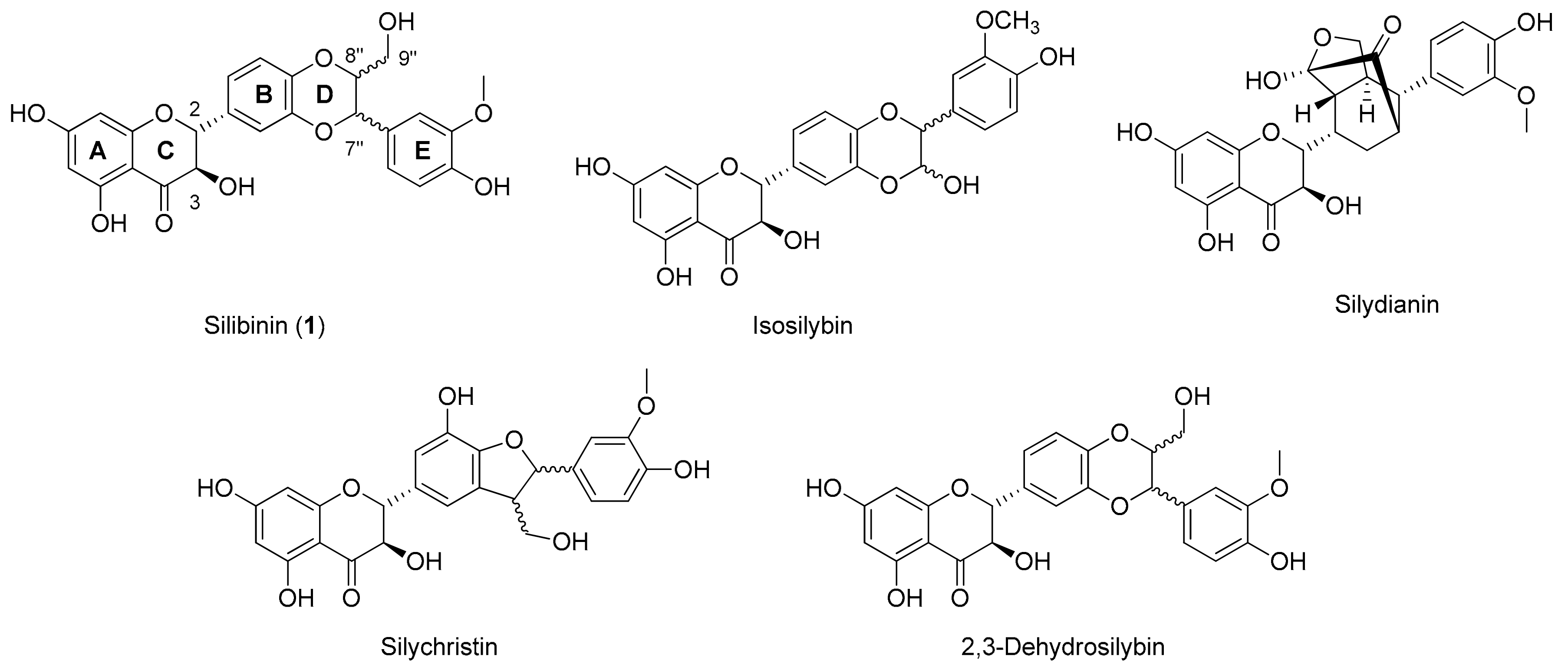
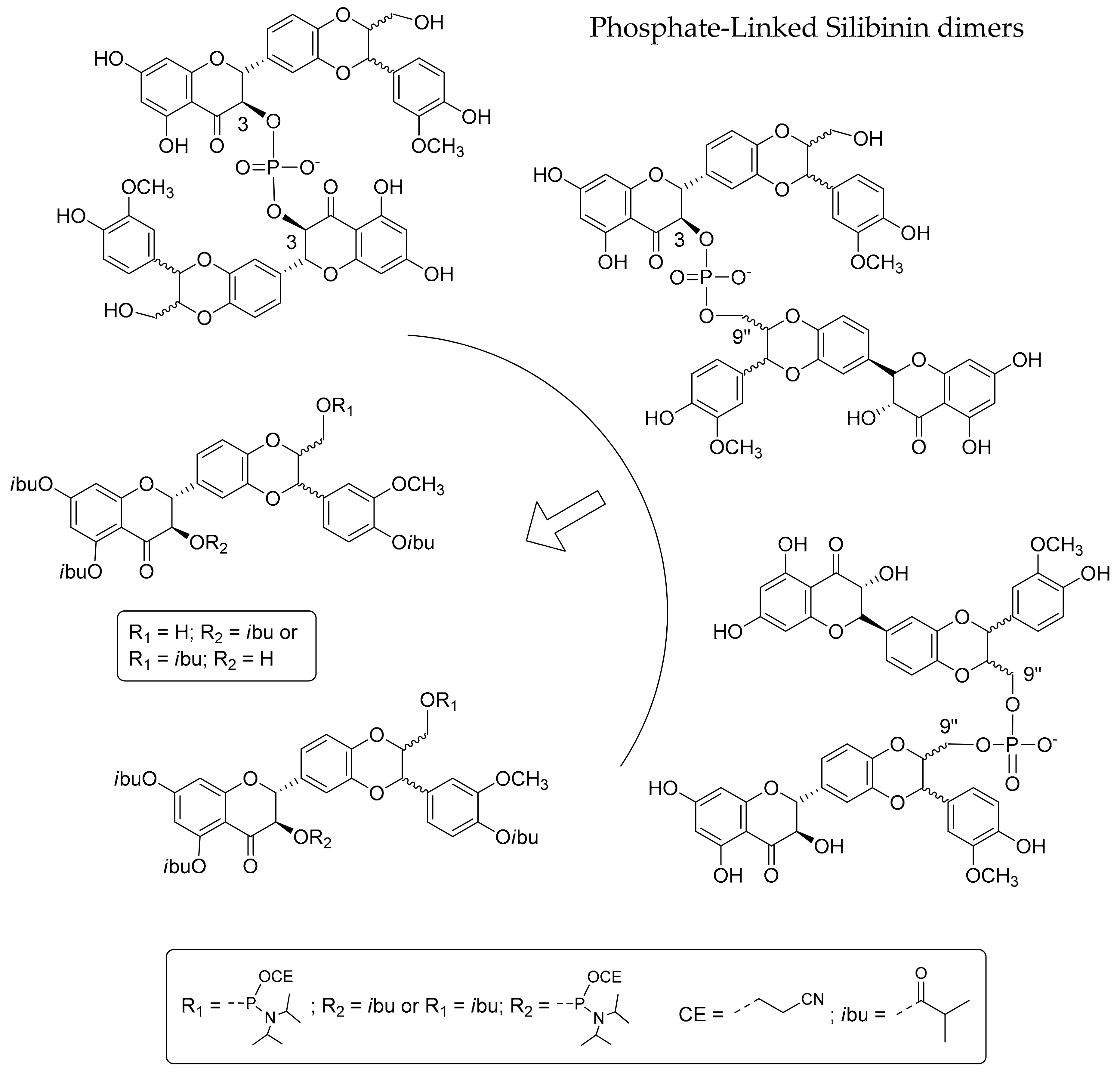
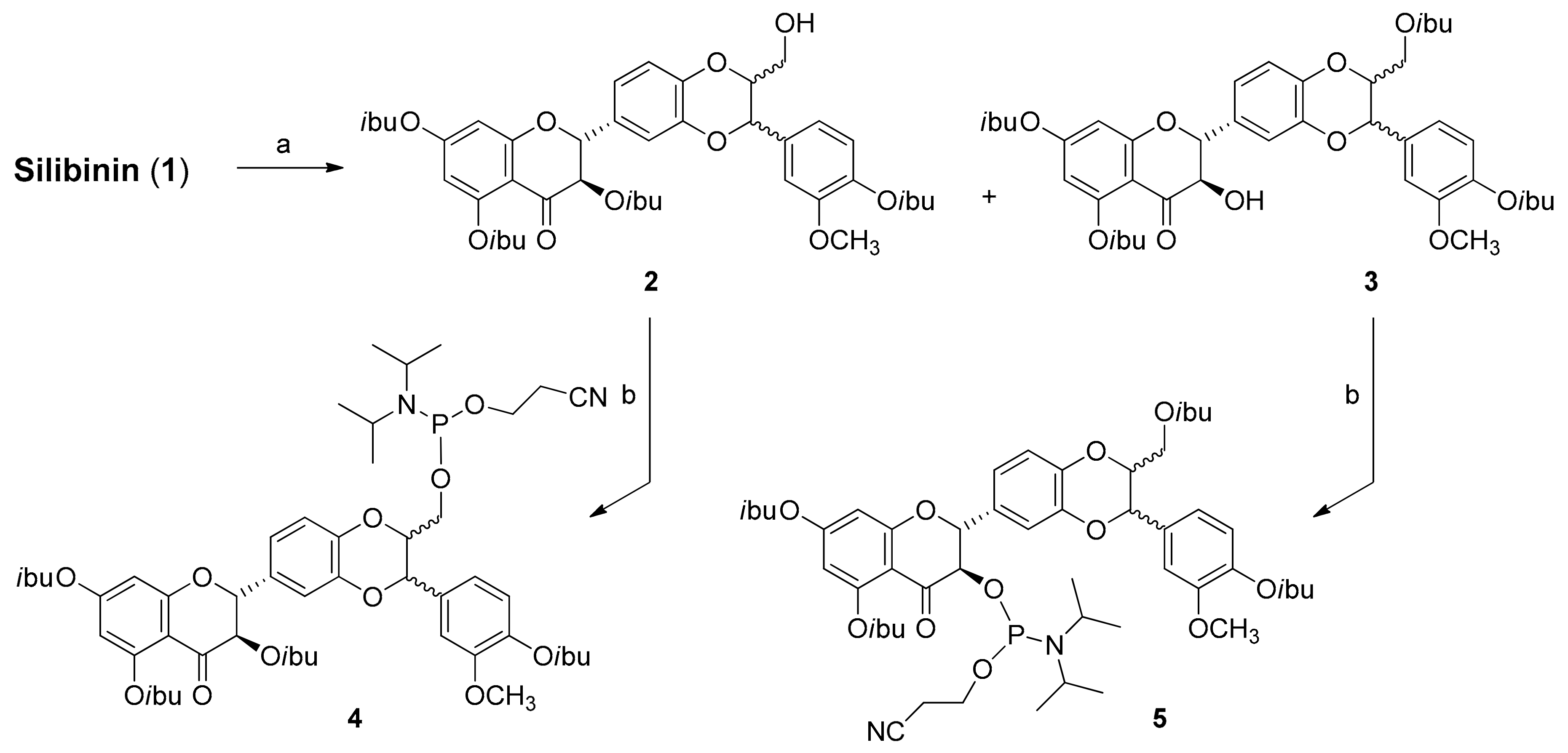
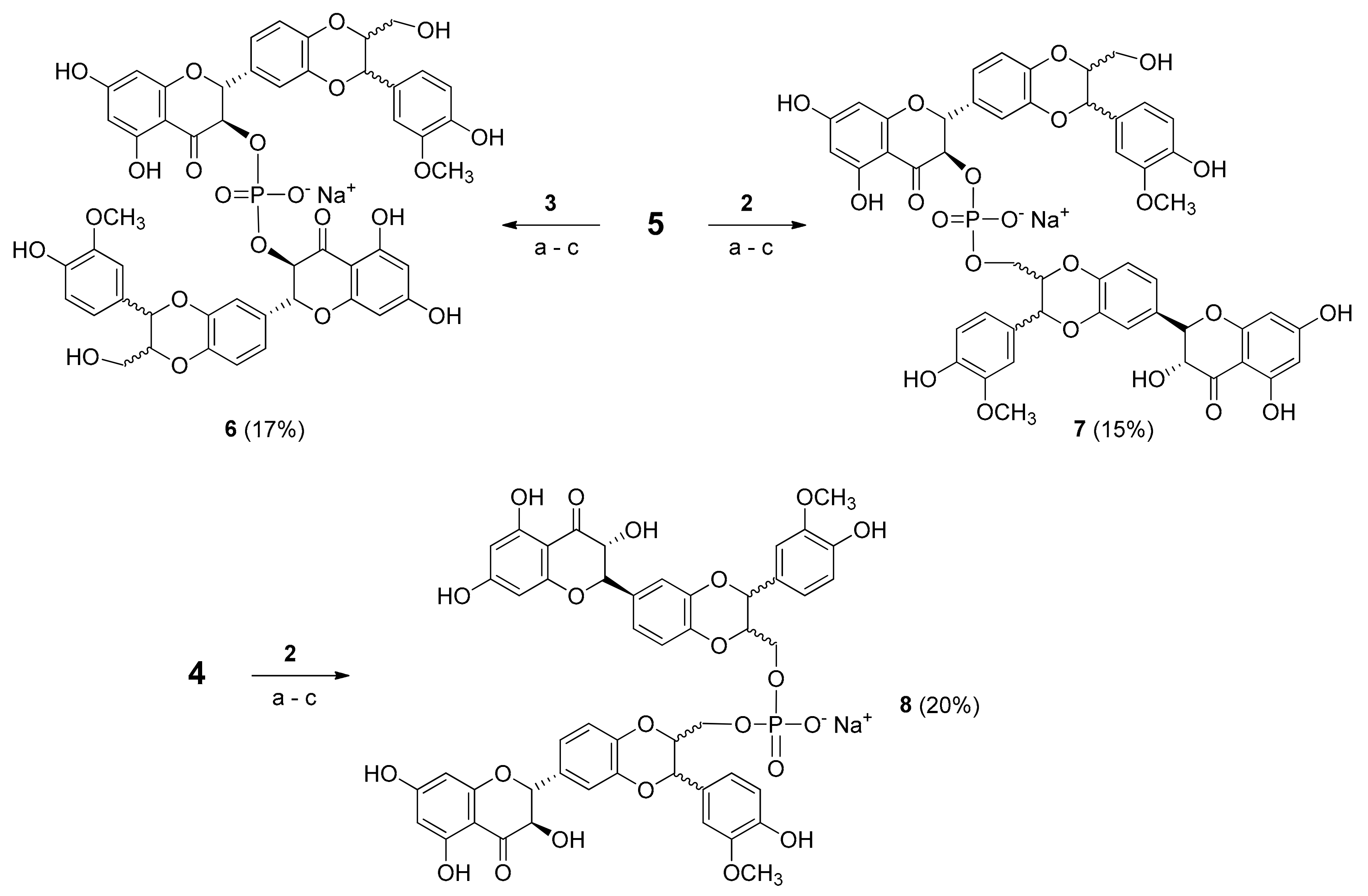
2.1. Synthesis of Phosphate-Linked Silibinin Dimers
2.2. Antioxidant Activity: DPPH Test
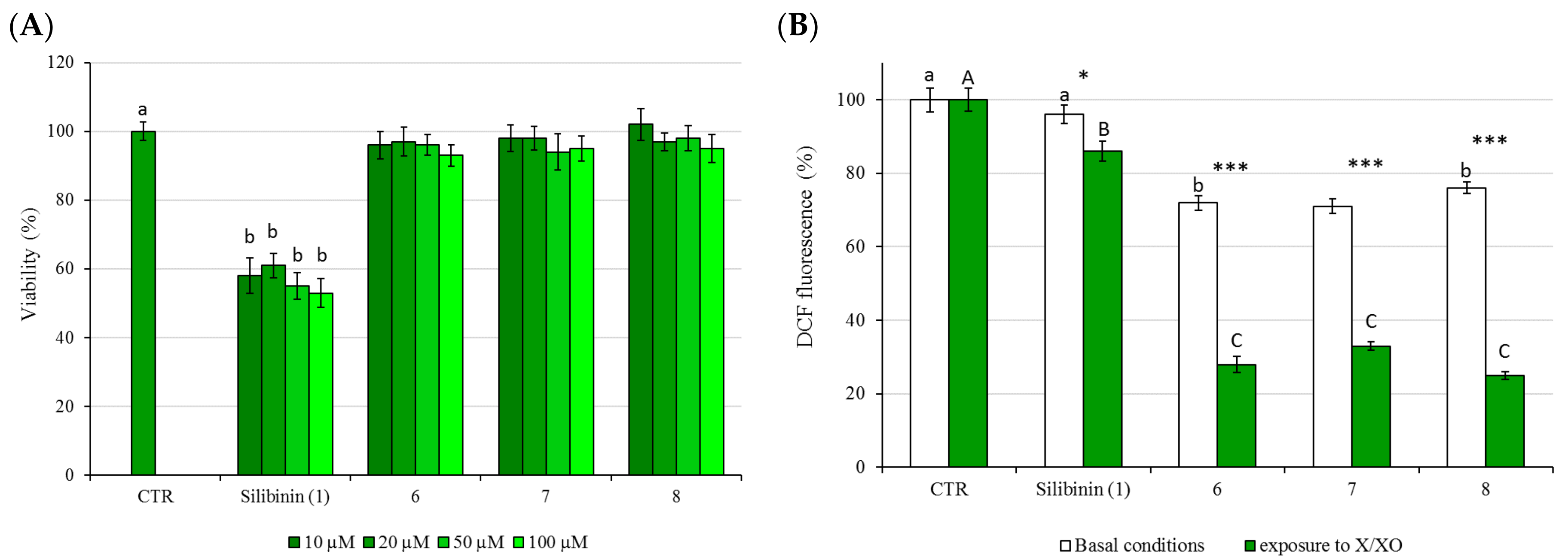
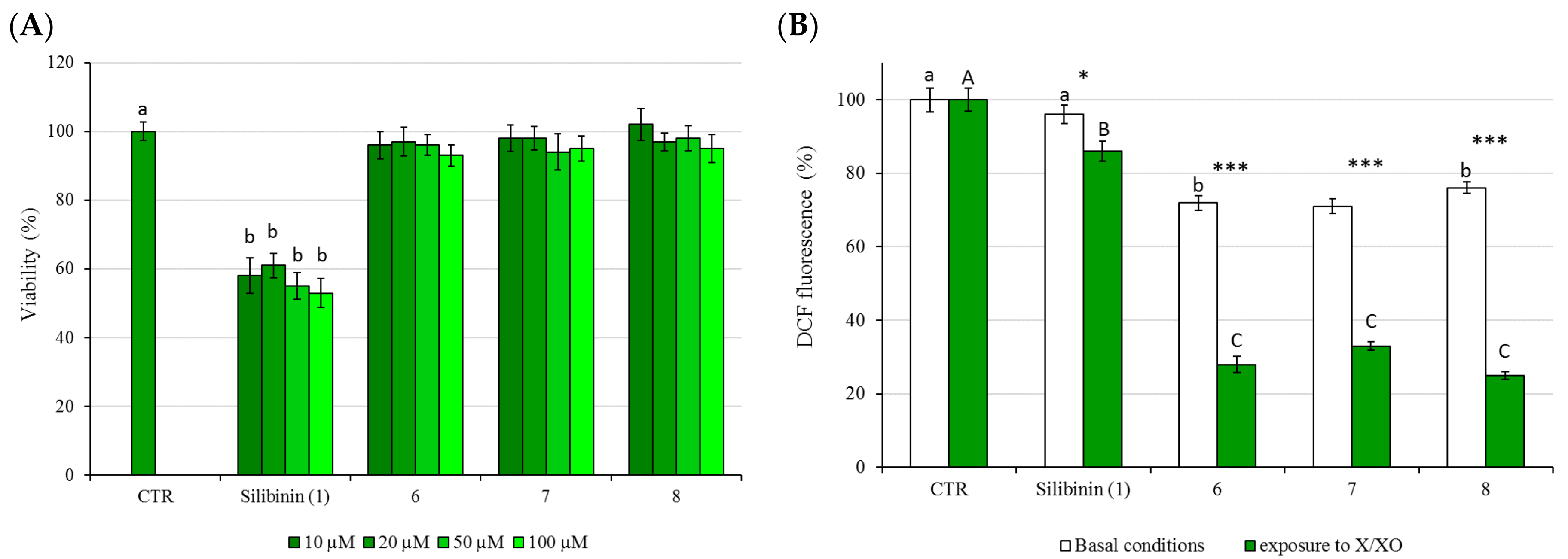
2.3. Cytotoxicity (MMT Test) and Protective Activity (X/XO Assay)
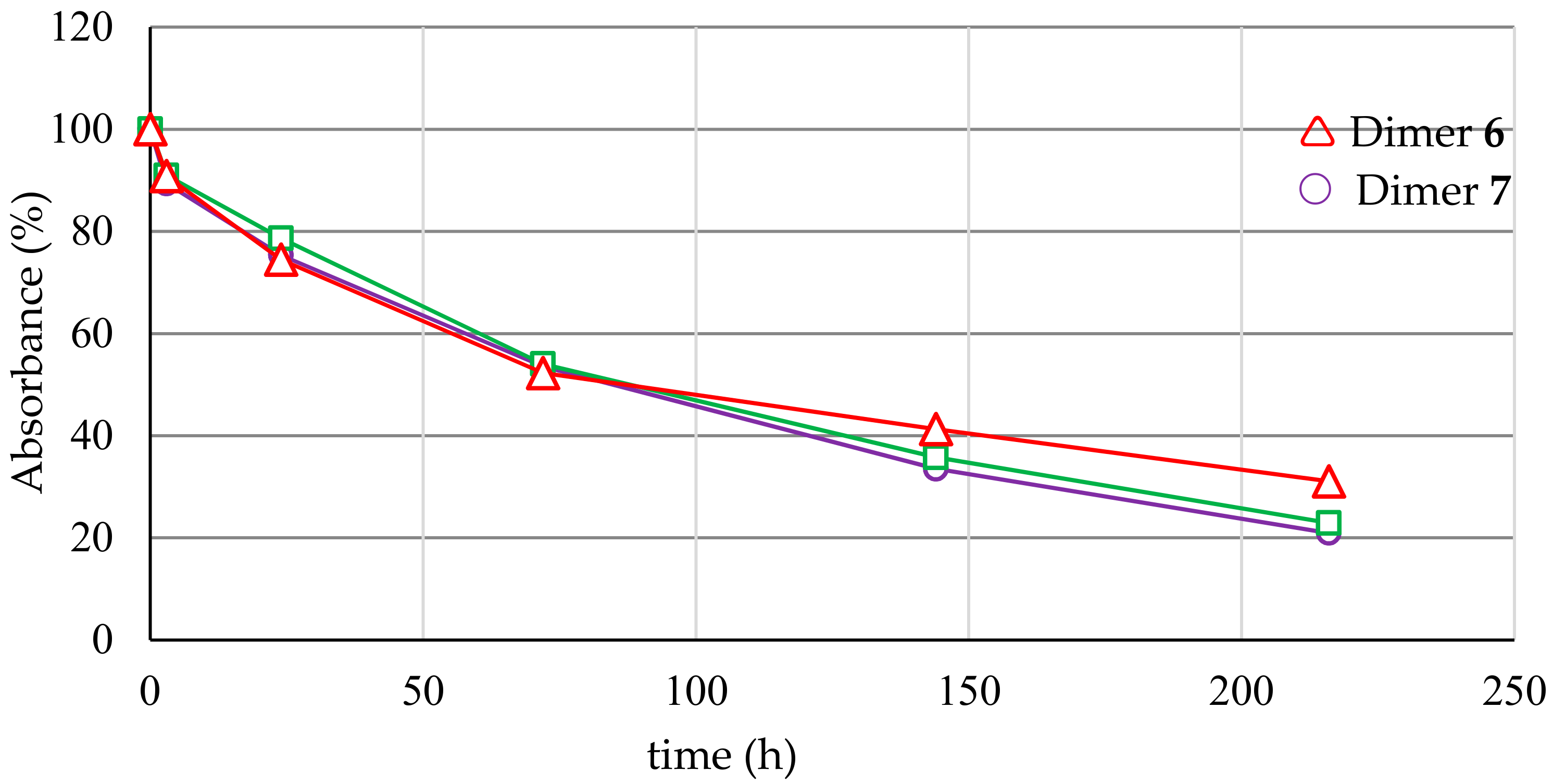
2.4. Serum Stability
2.5. Water Solubility
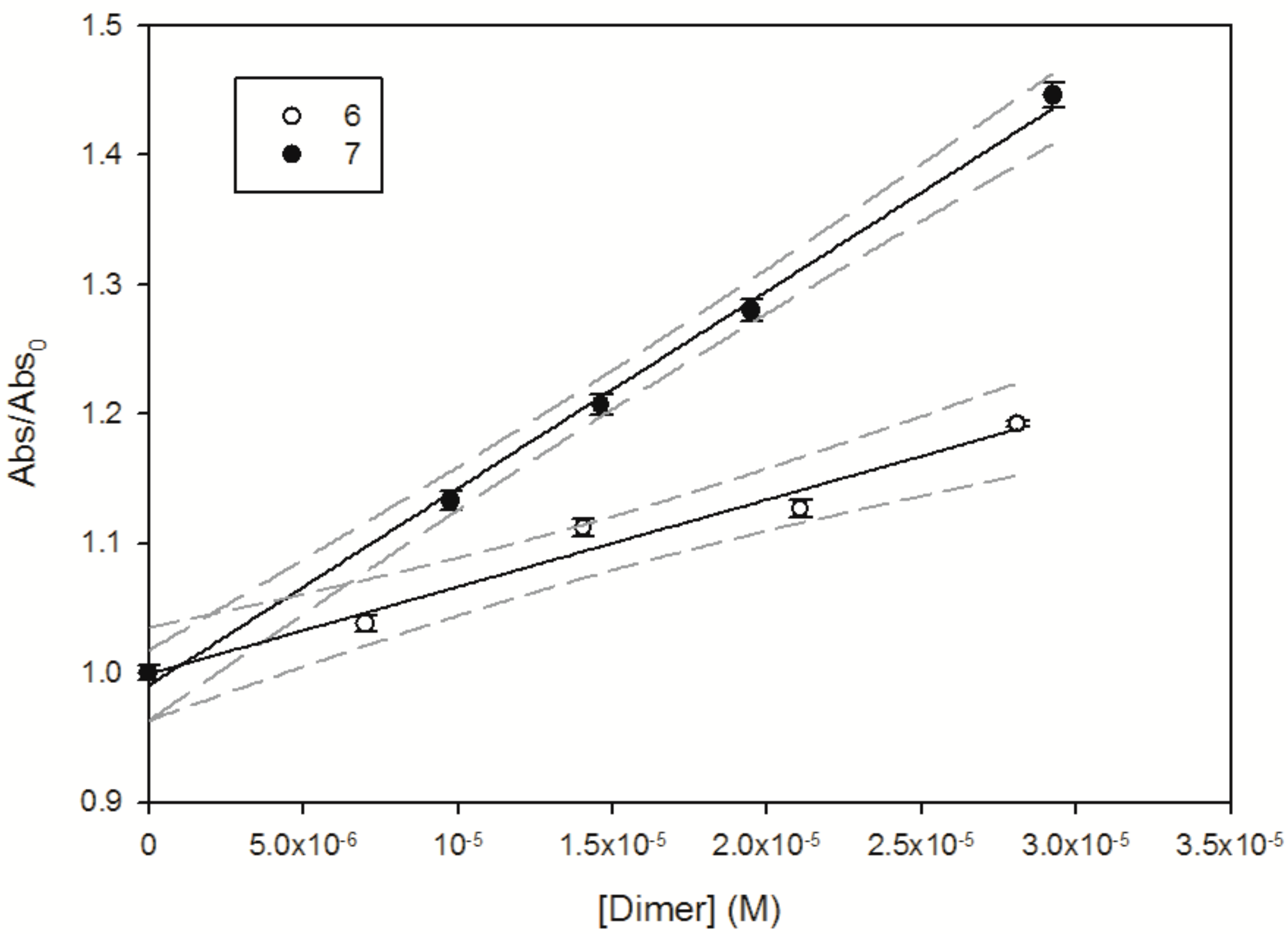
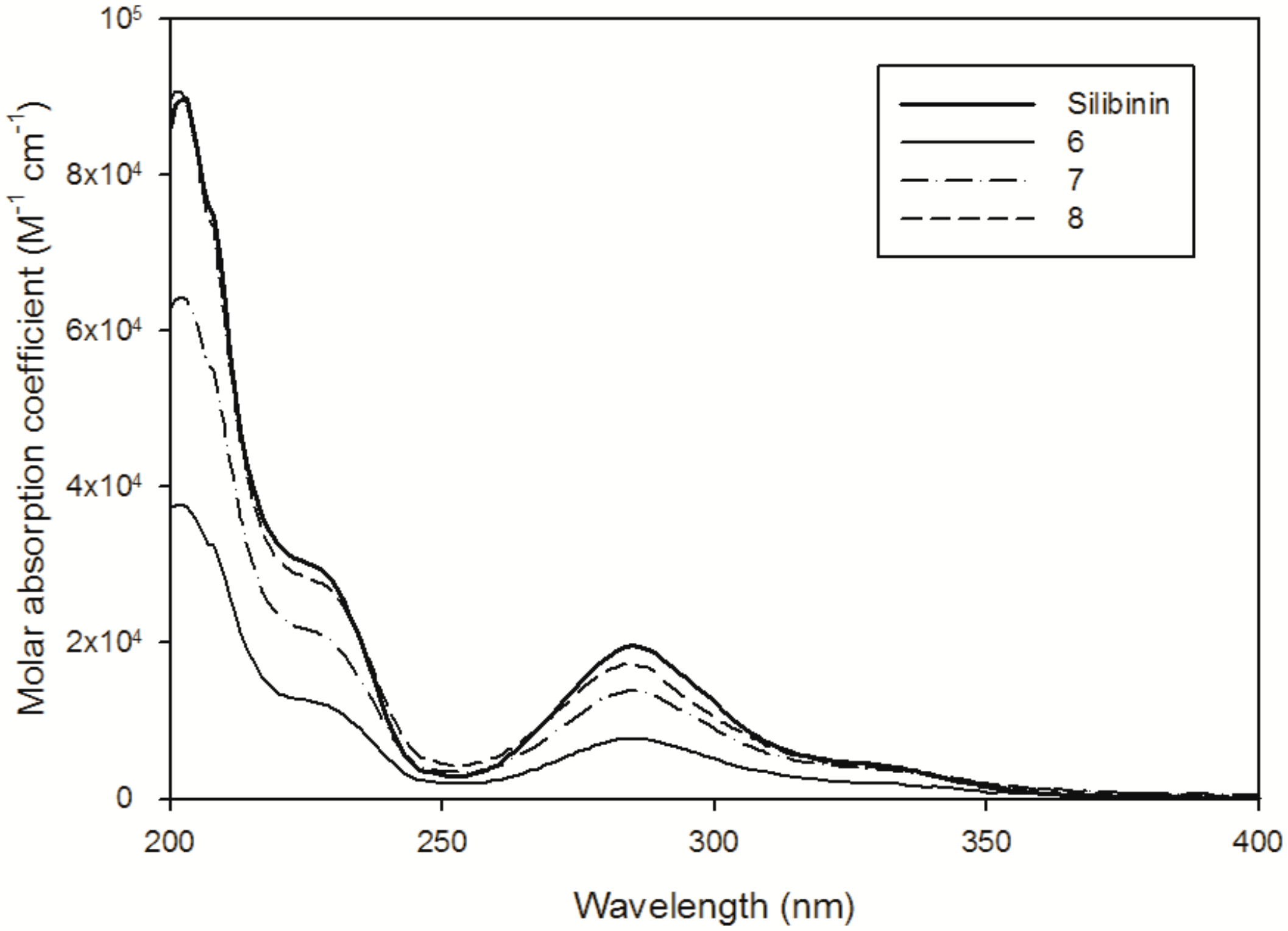
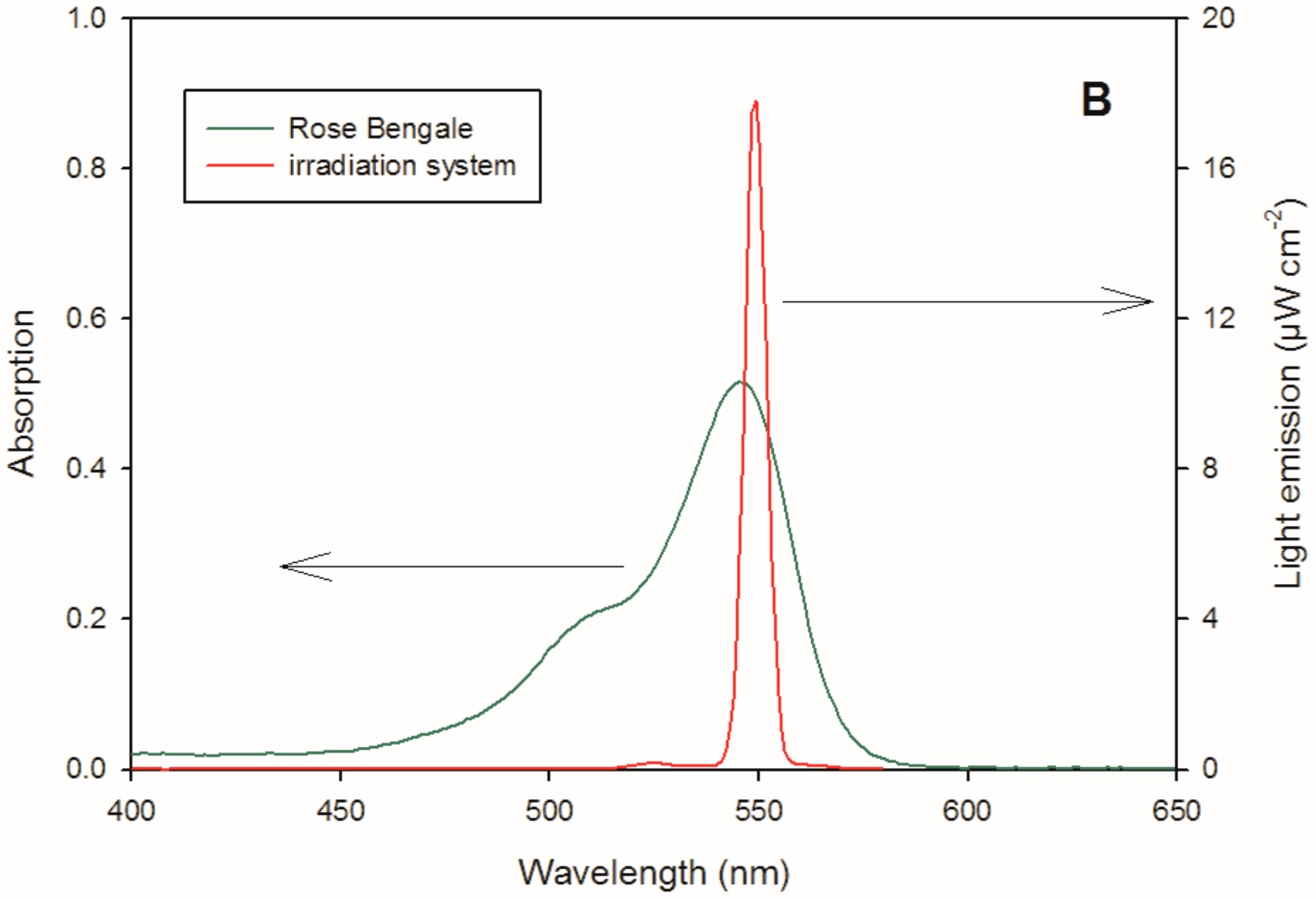
2.6. Reactivity with 1O2 and HO∙
3. Materials and Methods
3.1. General Methods
3.2. Chemicals
3.3. Synthesis of Silibinin Building Block 2
3.4. Synthesis of Silibinin Building Block 3
3.5. Synthesis of Silibinin Building Block 4
3.6. Synthesis of Silibinin Building Block 5
3.7. Synthesis of Phosphate-Linked Silibinin Dimers 6–8
3.8. HPLC Analysis and Purification of 6–8
3.9. DPPH Test
3.10. Evaluation of Cell Viability (MTT Assay)
3.11. Xanthine/Xanthine Oxidase (X–XO) Assay
3.12. Statistical Analysis
3.13. Serum Stability
3.14. Singlet Oxygen Reactivity
3.15. Hydroxyl Radical () Generation and Reactivity Estimation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Plant polyphenols: Chemical properties, biological activities, and synthesis. Angew. Chem. Int. Ed. 2011, 50, 586–621. [Google Scholar] [CrossRef] [PubMed]
- Bernini, R.; Fernanda, C.; Barontini, M.; Tofani, D.; Balducci, V.; Gambacorta, A. Synthesis and structure/antioxidant activity relationship of novel catecholic antioxidant structural analogues to hydroxytyrosol and its lipophilic esters. J. Agric. Food Chem. 2012, 60, 7408–7416. [Google Scholar] [CrossRef] [PubMed]
- Bernini, R.; Merendino, N.; Romani, A.; Velotti, F. Naturally occurring hydroxytyrosol: Synthesis and anticancer potential. Curr. Med. Chem. 2013, 20, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Barontini, M.; Bernini, R.; Carastro, I.; Gentili, P.; Romani, A. Synthesis and DPPH radical scavenging activity of novel compounds obtained from tyrosol and cinnamic acid derivatives. New J. Chem. 2014, 38, 809–816. [Google Scholar] [CrossRef]
- Bernini, R.; Gilardini Montani, M.S.; Merendino, N.; Romani, A.; Velotti, F. Hydroxytyrosol-derived compounds: A basis for the creation of new pharmacological agents for cancer prevention and therapy. J. Med. Chem. 2015, 58, 9089–9107. [Google Scholar] [CrossRef] [PubMed]
- Gazak, R.; Walterova, D.; Křen, V. Silybin and silymarin—New and emerging applications in medicine. Curr. Med. Chem. 2007, 14, 315–338. [Google Scholar] [CrossRef] [PubMed]
- Dixit, N.; Baboota, S.; Kohli, K.; Ahmad, S.; Ali, J. Silymarin: A review of pharmacological aspects and bioavailability enhancement approaches. Indian J. Pharmacol. 2007, 39, 172–179. [Google Scholar] [CrossRef]
- Biedermann, D.; Vavříková, E.; Cvak, L.; Křen, V. Chemistry of silybin. Nat. Prod. Rep. 2014, 31, 1138–1157. [Google Scholar] [CrossRef] [PubMed]
- Skottová, N.; Krecman, V. Silymarin as a potential hypocholesterolaemic drug. Physiol. Res. 1998, 47, 1–7. [Google Scholar] [PubMed]
- Duan, S.; Guan, X.; Lin, R.; Liu, X.; Yan, Y.; Lin, R.; Zhang, T.; Chen, X.; Huang, J.; Sun, X.; et al. Silibinin inhibits acetylcholinesterase activity and amyloid β peptide aggregation: A dual-target drug for the treatment of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 1792–1807. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Liu, J.; Ji, X.; Wang, Y.; Zidichouski, J.; Zhang, J. Silibinin: A novel inhibitor of Aβ aggregation. Neurochem. Int. 2011, 58, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Marrazzo, G.; Bosco, P.; La Delia, F.; Scapagnini, G.; Di Giacomo, C.; Malaguarnera, M.; Galvano, F.; Nicolosi, A.; Li Volti, G. Neuroprotective effect of silibinin in diabetic mice. Neurosci. Lett. 2011, 504, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Di Fabio, G.; Romanucci, V.; De Marco, A.; Zarrelli, A. Triterpenoids from Gymnema sylvestre and their pharmacological activities. Molecules 2014, 19, 10956–10981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fabio, G.; Romanucci, V.; Zarrelli, M.; Giordano, M.; Zarrelli, A. C-4 gem-dimethylated oleanes of Gymnema sylvestre and their pharmacological activities. Molecules 2013, 18, 14892–14919. [Google Scholar] [CrossRef] [PubMed]
- Di Fabio, G.; Romanucci, V.; Di Marino, C.; De Napoli, L.; Zarrelli, A. A Rapid and simple chromatographic separation of diastereomers of silibinin and their oxidation to produce 2,3-dehydrosilybin enantiomers in an optically pure form. Planta Med. 2013, 79, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Di Fabio, G.; Romanucci, V.; De Nisco, M.; Pedatella, S.; Di Marino, C.; Zarrelli, A. Microwave-assisted oxidation of silibinin: A simple and preparative method for the synthesis of improved radical scavengers. Tetrahedron Lett. 2013, 54, 6279–6282. [Google Scholar] [CrossRef]
- Micciarelli, M.; Valadan, M.; Della Ventura, B.; Di Fabio, G.; De Napoli, L.; Bonella, S.; Röthlisberger, U.; Tavernelli, I.; Altucci, C.; Velotta, R. Photophysics and photochemistry of a DNA-protein cross-linking model: A synergistic approach combining experiments and theory. J. Phys. Chem. B 2014, 118, 4983–4992. [Google Scholar] [CrossRef] [PubMed]
- Zarrelli, A.; Romanucci, V.; Della Greca, M.; De Napoli, L.; Previtera, L.; Di Fabio, G. New silybin scaffold for chemical diversification: Synthesis of novel 23-phosphodiester silybin conjugates. Synlett 2012, 24, 45–48. [Google Scholar]
- Zarrelli, A.; Romanucci, V.; Tuccillo, C.; Federico, A.; Loguercio, C.; Gravante, R.; Di Fabio, G. New silibinin glyco-conjugates: Synthesis and evaluation of antioxidant properties. Bioorg. Med. Chem. Lett. 2014, 24, 5147–5149. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Dallio, M.; Di Fabio, G.; Zarrelli, A.; Zappavigna, S.; Stiuso, P.; Tuccillo, C.; Caraglia, M.; Loguercio, C. Silybin-phosphatidylcholine complex protects human gastric and liver cells from oxidative stress. In Vivo 2015, 29, 569–575. [Google Scholar] [PubMed]
- Romanucci, V.; Zarrelli, A.; Guaragna, A.; Di Marino, C.; Di Fabio, G. New phosphorylating reagents for deoxyribonucleosides and oligonucleotides. Tetrahedron Lett. 2017, 58, 1227–1229. [Google Scholar] [CrossRef]
- Di Fabio, G.; Malgieri, G.; Isernia, C.; D’Onofrio, J.; Gaglione, M.; Messere, A.; Zarrelli, A.; De Napoli, L. A novel synthetic strategy for monosubstituted cyclodextrin derivatives. Chem. Commun. 2012, 48, 3875–3877. [Google Scholar] [CrossRef] [PubMed]
- Romanucci, V.; Milardi, D.; Campagna, T.; Gaglione, M.; Messere, A.; D’Urso, A.; Crisafi, E.; La Rosa, C.; Zarrelli, A.; Balzarini, J.; et al. Synthesis, biophysical characterization and anti-HIV activity of d(TG3AG) Quadruplexes bearing hydrophobic tails at the 5’-end. Bioorg. Med. Chem. 2014, 22, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Romanucci, V.; Gaglione, M.; Messere, A.; Potenza, N.; Zarrelli, A.; Noppen, S.; Liekens, S.; Balzarini, J.; Di Fabio, G. Hairpin oligonucleotides forming G-quadruplexes: New aptamers with anti-HIV activity. Eur. J. Med. Chem. 2014, 89, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.P.; Bruner, L.H.; Bassoe, C.F.; Hudson, J.L.; Ward, P.A.; Phan, S.H. Measurement of intracellular fluorescence of human monocytes relative to oxidative metabolism. J. Leukoc. Biol. 1988, 43, 304–310. [Google Scholar] [PubMed]
- Cathcart, R.; Schwiers, E.; Ames, B.N. Detection of picomole levels of hydroperoxides using a fluorescent dichlorofluorescin assay. Anal. Biochem. 1983, 134, 111–116. [Google Scholar] [CrossRef]
- Beetsch, J.W.; Park, T.S.; Dugan, L.L.; Shah, A.R.; Gidday, J.M. Xanthine oxidase-derived superoxide causes reoxygenation injury of ischemic cerebral endothelial cells. Brain Res. 1998, 786, 89–95. [Google Scholar] [CrossRef]
- Abramor, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Fatokuna, A.A.; Stonea, T.W.; Smith, R.A. Hydrogen peroxide mediates damage by xanthine and xanthine oxidase in cerebellar granule neuronal cultures. Neurosci. Lett. 2007, 416, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.X.; Chen, X.; Hughes, R.A.; Williams, S.J.; Woodman, O.L. Understanding the cardioprotective effects of flavonoids: Discovery of relaxant flavonols without antioxidant activity. J. Med. Chem. 2008, 51, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.-C.; Zhu, J.-J.; Zhang, H.-J.; Huang, C.-G. Solubility of silybin in aqueous hydrochloric acid solution. Fluid Ph. Equilib. 2007, 254, 204–210. [Google Scholar] [CrossRef]
- Morales, J.; Günther, G.; Zanocco, A.L.; Lem, E. Singlet oxygen reactions with flavonoids. A theoretical experimental study. PLoS ONE 2012, 7, e40548. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.F.; Luo, J.; Yao, S.D.; Lian, Z.R.; Zhang, J.S.; Lin, N.Y. Interaction of phenolic antioxidants and hydroxyl radicals. Radiat. Phys. Chem. 1993, 42, 985–987. [Google Scholar] [CrossRef]
- Rafat Husain, S.; Cillard, J.; Cillard, P. Hydroxyl radical scavenging activity of flavonoids. Phytochemistry 1987, 26, 2489–2491. [Google Scholar] [CrossRef]
- De Rosa, M.C.; Crutchley, R.J. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233–234, 351–371. [Google Scholar] [CrossRef]
- Wilkinson, F.; Helman, W.P.; Ross, A.B. Rate constants for the decay and reactions of the lowest electronically excited singlet state of molecular oxygen in solution. An expanded and revised compilation. J. Phys. Chem. Ref. Data 1995, 24, 663–677. [Google Scholar] [CrossRef]
- Bertolotti, S.G.; Garcia, N.A.; Arguello, G.A. Effect of the peptide bond on the singlet-molecular-oxygen-mediated sensitized photo-oxidation of tyrosine and tryptophan dipeptides. A kinetic study. J. Photochem. Photobiol. B 1991, 10, 57–70. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Herrmann, H.; Hoffmann, D.; Schaefer, T.; Bräuer, P.; Tilgner, A. Tropospheric aqueous-phase free-radical chemistry: Radical sources, spectra, reaction kinetics and prediction tools. Chem. Phys. Chem. 2010, 11, 3796–3822. [Google Scholar] [CrossRef] [PubMed]
- Dogliotti, L.; Hayon, E. Flash photolysis study of sulfite; thiocyanate, and thiosulfate ions in solution. J. Phys. Chem. 1968, 72, 1800–1807. [Google Scholar] [CrossRef]
- Morgan, M.S.; Van Trieste, P.F.; Garlick, S.M.; Mahon, M.J.; Smith, A.L. Ultraviolet molar absorptivities of aqueous hydrogen peroxide and hydroperoxyl ion. Anal. Chim. Acta 1998, 215, 325–329. [Google Scholar] [CrossRef]
Sample Availability: Not available. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MALDI | tR (min) a | DPPH IC50 (mM) | t1/2 (h) |
|---|---|---|---|---|
| 6 | Calcd. for C50H43O22P = 1026.20 found [M − H]− = 1025.12; [MNa − H]− = 1047.09 | 13.6 | 0.58 ± 0.04 | 86.7 |
| 7 | Calcd. for C50H43O22P = 1026.20 found [M − H]− = 1025.28; [MNa − H]− = 1047.09 | 13.4 | 0.55 ± 0.05 | 85.6 |
| 8 | Calcd. for C50H43O22P = 1026.20 found [M − H]− = 1025.11; [MNa − H]− = 1048.99 | 14.7 | 0.34 ± 0.07 | 81.2 |
| Silibinin (1) | – | – | 1.40 ± 0.06 | – |
| Quercetin | – | – | 0.18 ± 0.01 | – |
| Compound | (M−1∙cm−1) | Suspended in 10 mL | Theoretical Concentration | Abs288 nm after Filtration | Solubility in H2O |
|---|---|---|---|---|---|
| 1 | 22,158 (CH3CN) 19,469 (H2O/CH3CN, 98:2) | 0.44 mg | 91.2 µM * | 0.018 | ≤0.9 µM (0.4 mg·L−1) |
| 6 | 9100 (H2O) | 0.10 mg | 19.5 µM | 0.18 | ≥19.5 µM (20.3 mg·L−1) |
| 7 | 14,435 (H2O) | 0.10 mg | 19.5 µM | 0.30 | ≥19.5 µM (20.3 mg·L−1) |
| 8 | 18,536 (H2O) | 0.10 mg | 19.5 µM | 0.35 | ≥19.5 µM (20.3 mg·L−1) |
| Compound | THF | H2O | ||
|---|---|---|---|---|
| []SS (M) | (M−1s−1) | []SS (M) | (M−1·s−1) | |
| 1 | 3.79 × 10−13 | 6.1 ± 0.4 × 107 | - | - |
| 6 | - | - | 4.07 × 10−13 | 6.8 ± 1.2 × 107 |
| 7 | - | - | 4.07 × 10−13 | 5.4 ± 0.3 × 107 |
| 8 | - | - | 4.07 × 10−13 | 4.0 ± 0.2 × 107 |
| Compound | (M−1·s−1) |
|---|---|
| 1 | 8.26 ± 1.64 × 109 |
| 6 | 6.73 ± 0.32 × 109 |
| 7 | 1.52 ± 0.08 × 1010 |
| 8 | ≥2.0 × 1010 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanucci, V.; Gravante, R.; Cimafonte, M.; Marino, C.D.; Mailhot, G.; Brigante, M.; Zarrelli, A.; Fabio, G.D. Phosphate-Linked Silibinin Dimers (PLSd): New Promising Modified Metabolites. Molecules 2017, 22, 1323. https://doi.org/10.3390/molecules22081323
Romanucci V, Gravante R, Cimafonte M, Marino CD, Mailhot G, Brigante M, Zarrelli A, Fabio GD. Phosphate-Linked Silibinin Dimers (PLSd): New Promising Modified Metabolites. Molecules. 2017; 22(8):1323. https://doi.org/10.3390/molecules22081323
Chicago/Turabian StyleRomanucci, Valeria, Raffaele Gravante, Martina Cimafonte, Cinzia Di Marino, Gilles Mailhot, Marcello Brigante, Armando Zarrelli, and Giovanni Di Fabio. 2017. "Phosphate-Linked Silibinin Dimers (PLSd): New Promising Modified Metabolites" Molecules 22, no. 8: 1323. https://doi.org/10.3390/molecules22081323