One-Pot Green Regioselesctive Synthesis of γ-Lactones from Epoxides and Ketene Silyl Acetals Using 1,3-Dimethylimidazolium Fluoride as a Recoverable Metal-Free Catalyst


Abstract
:1. Introduction
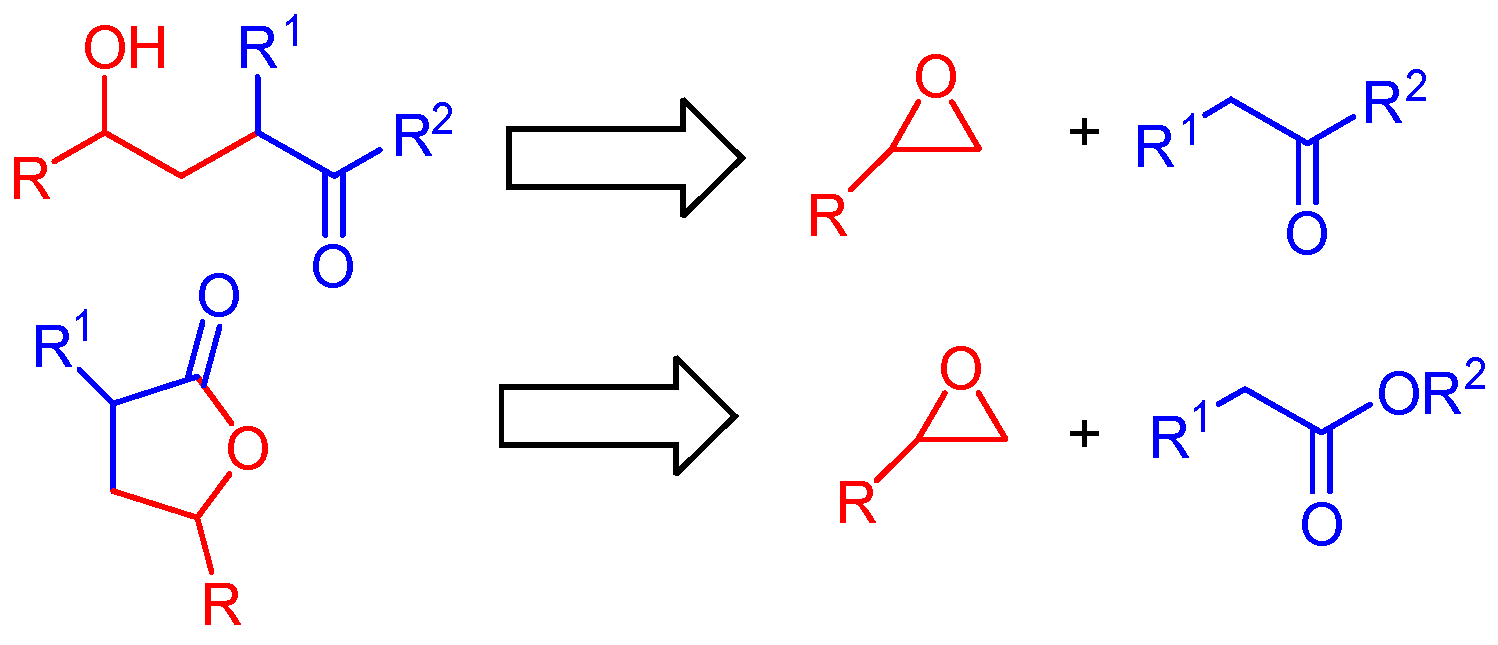
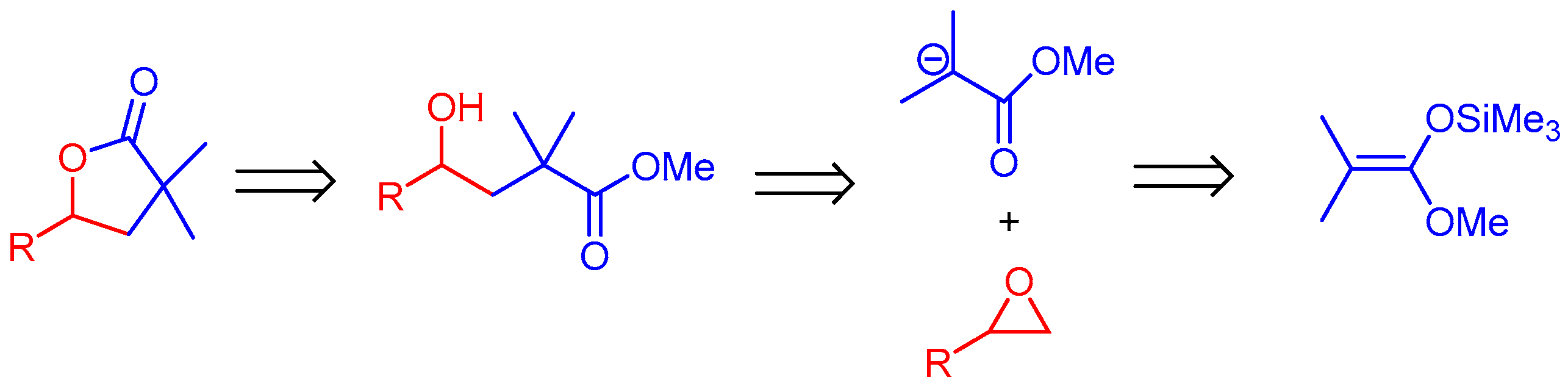
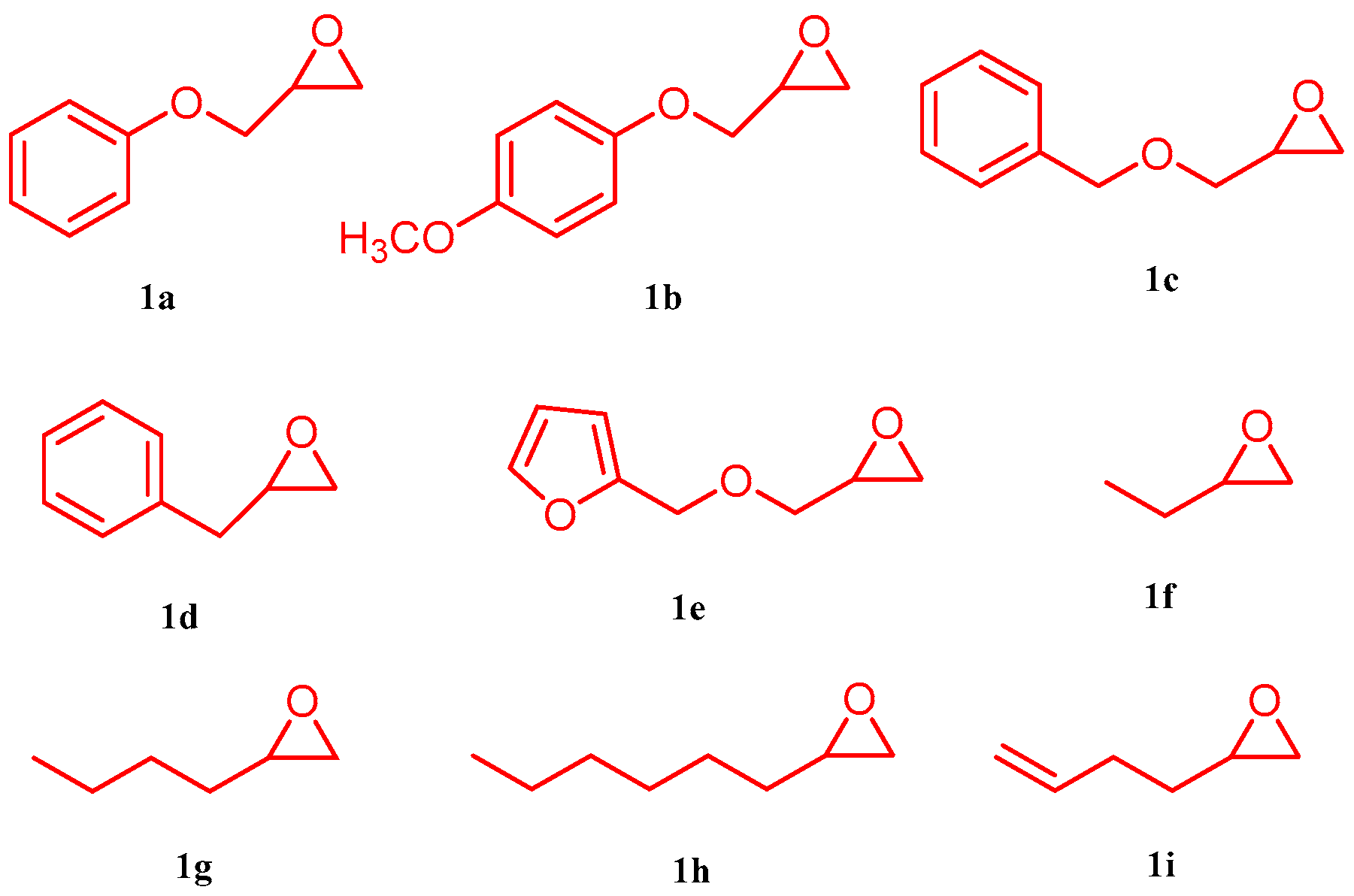
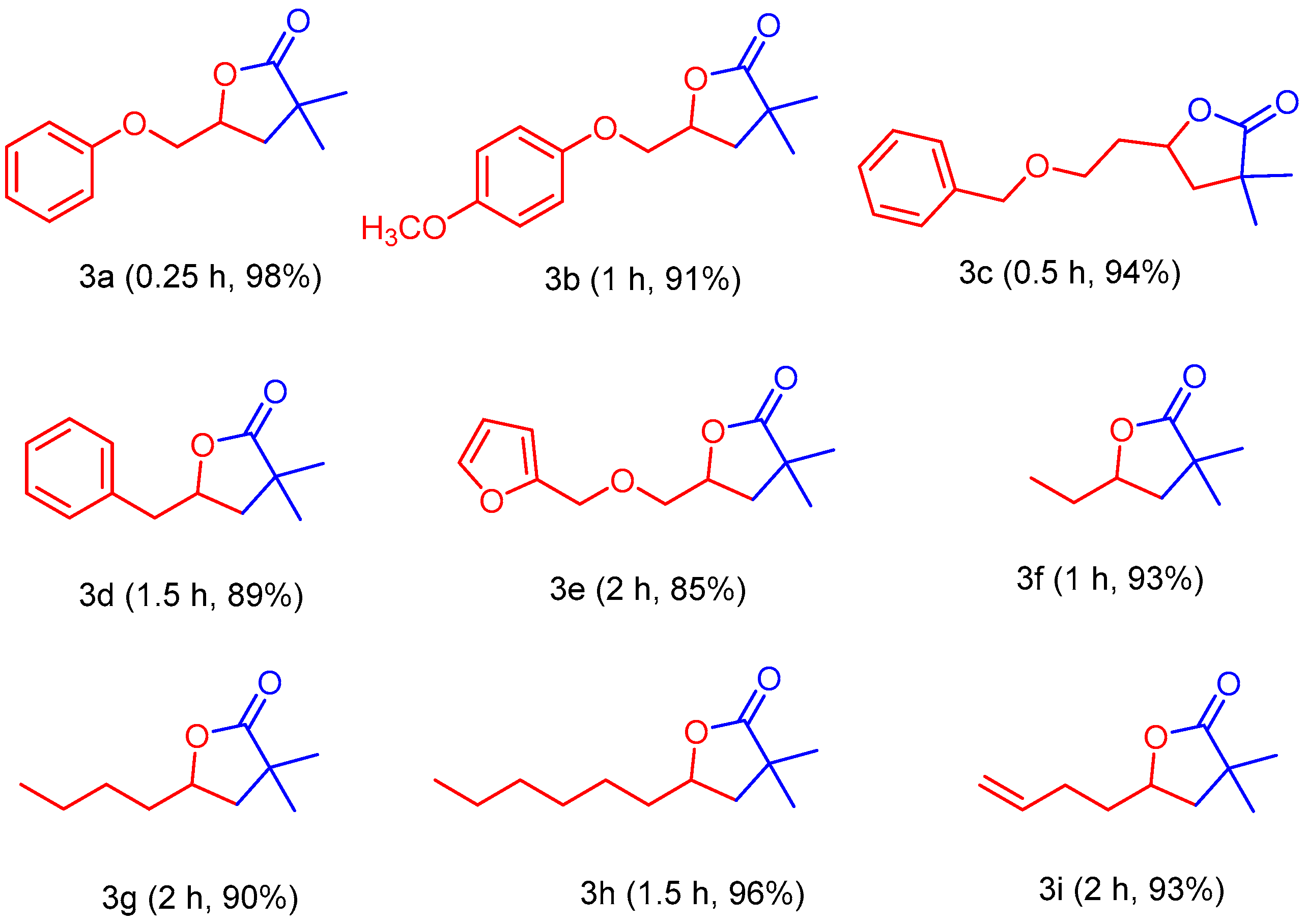
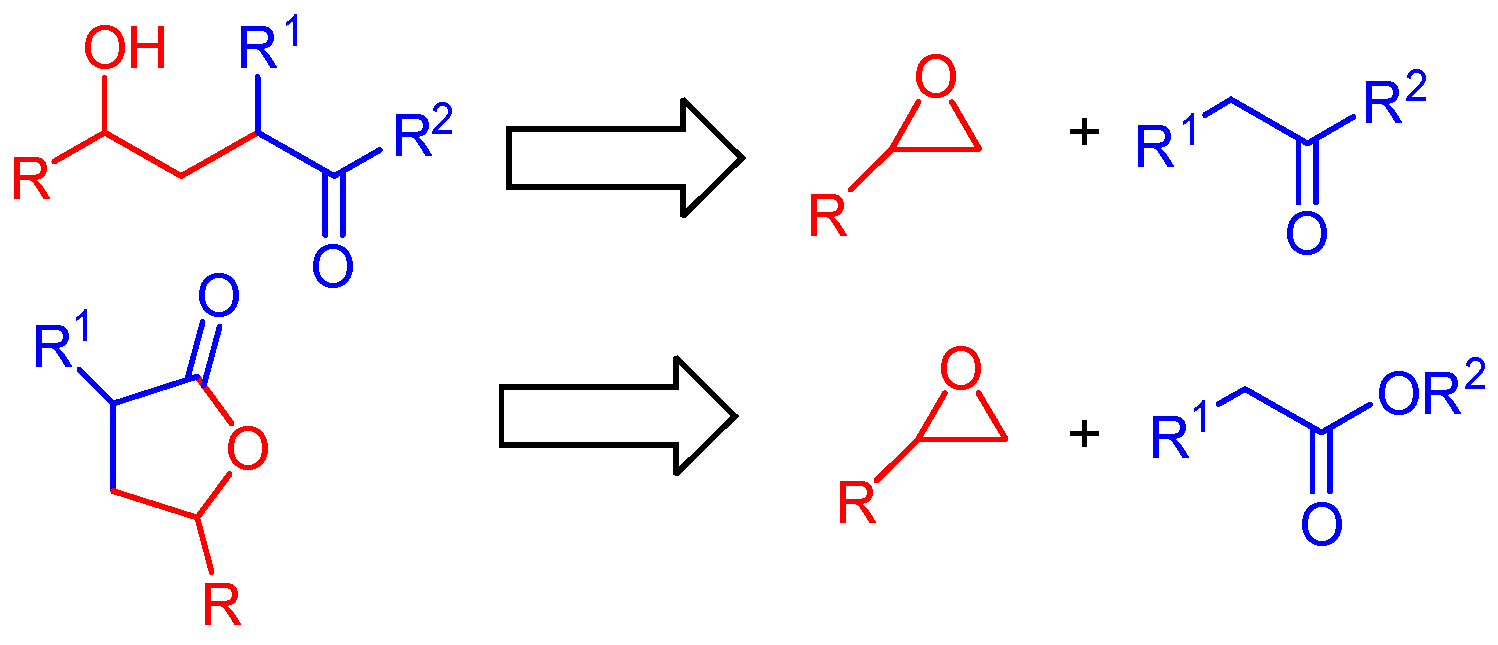
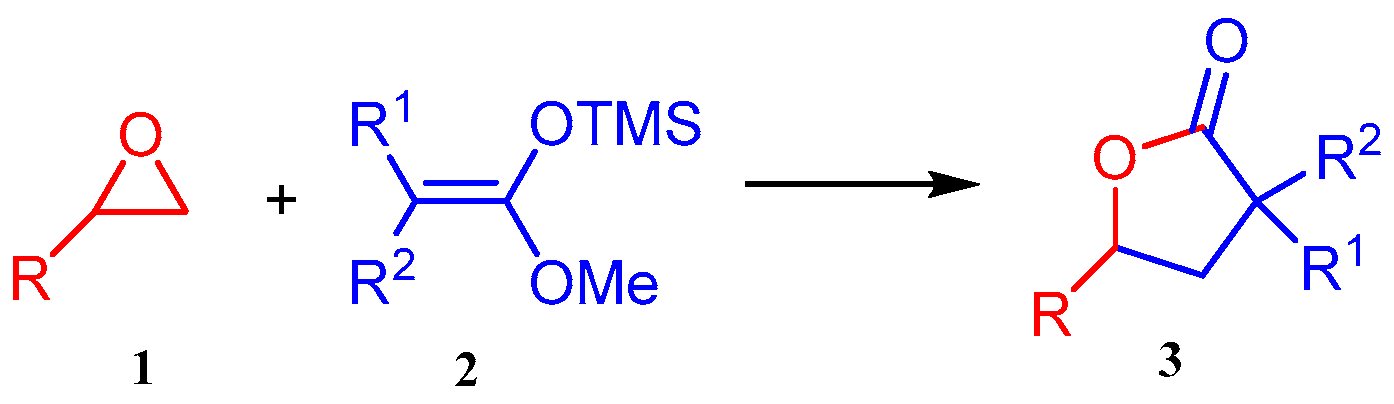
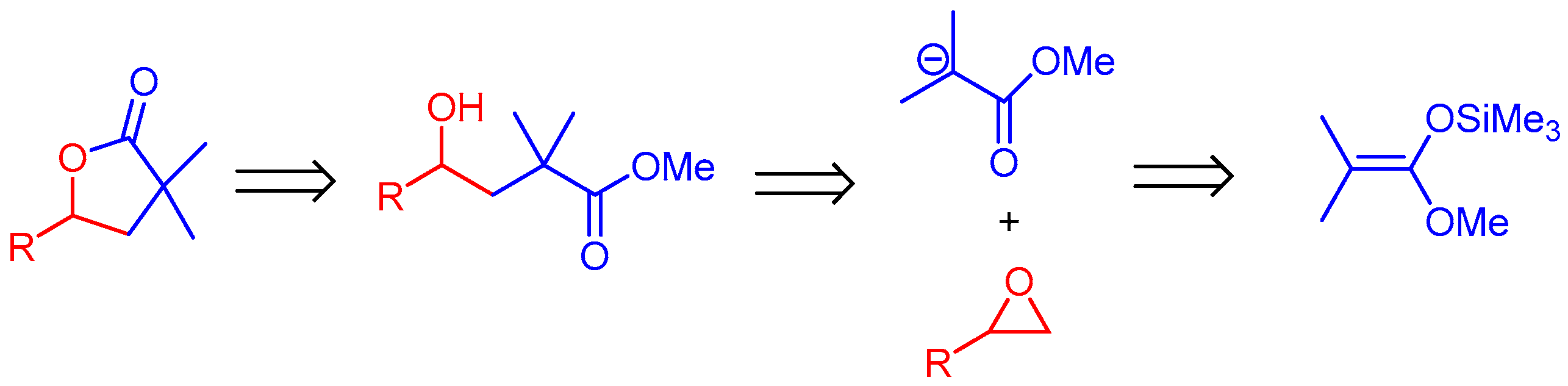
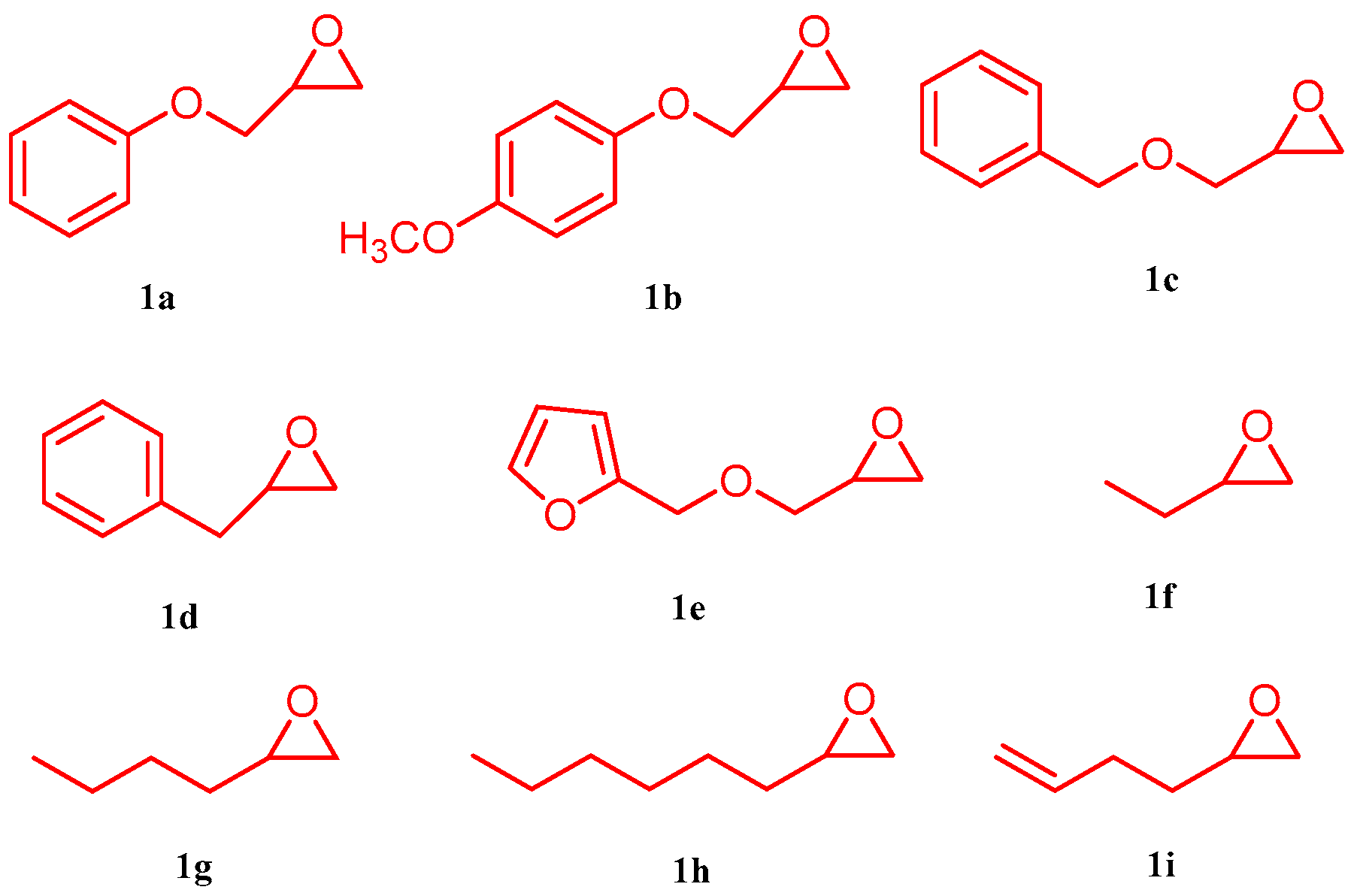
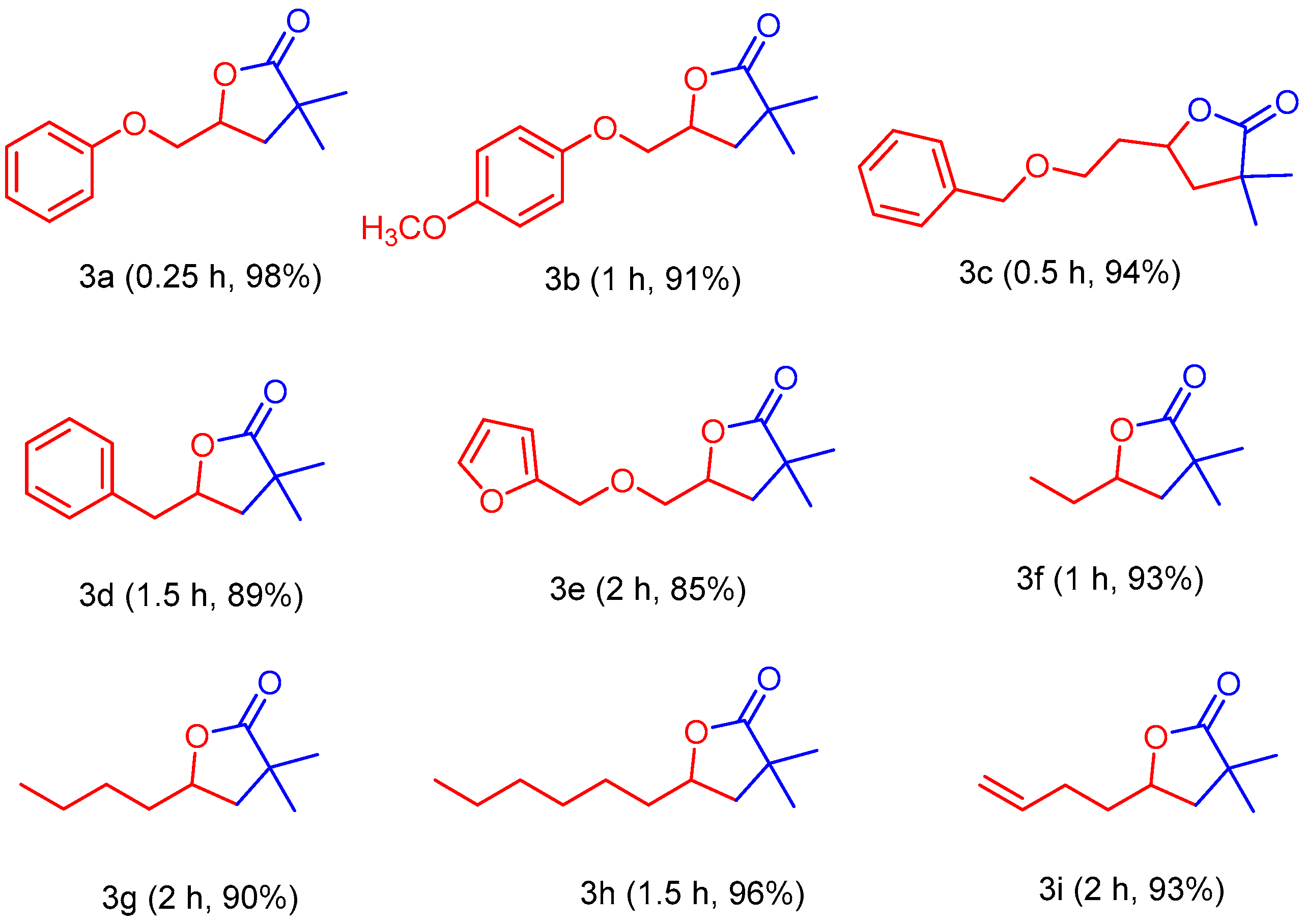
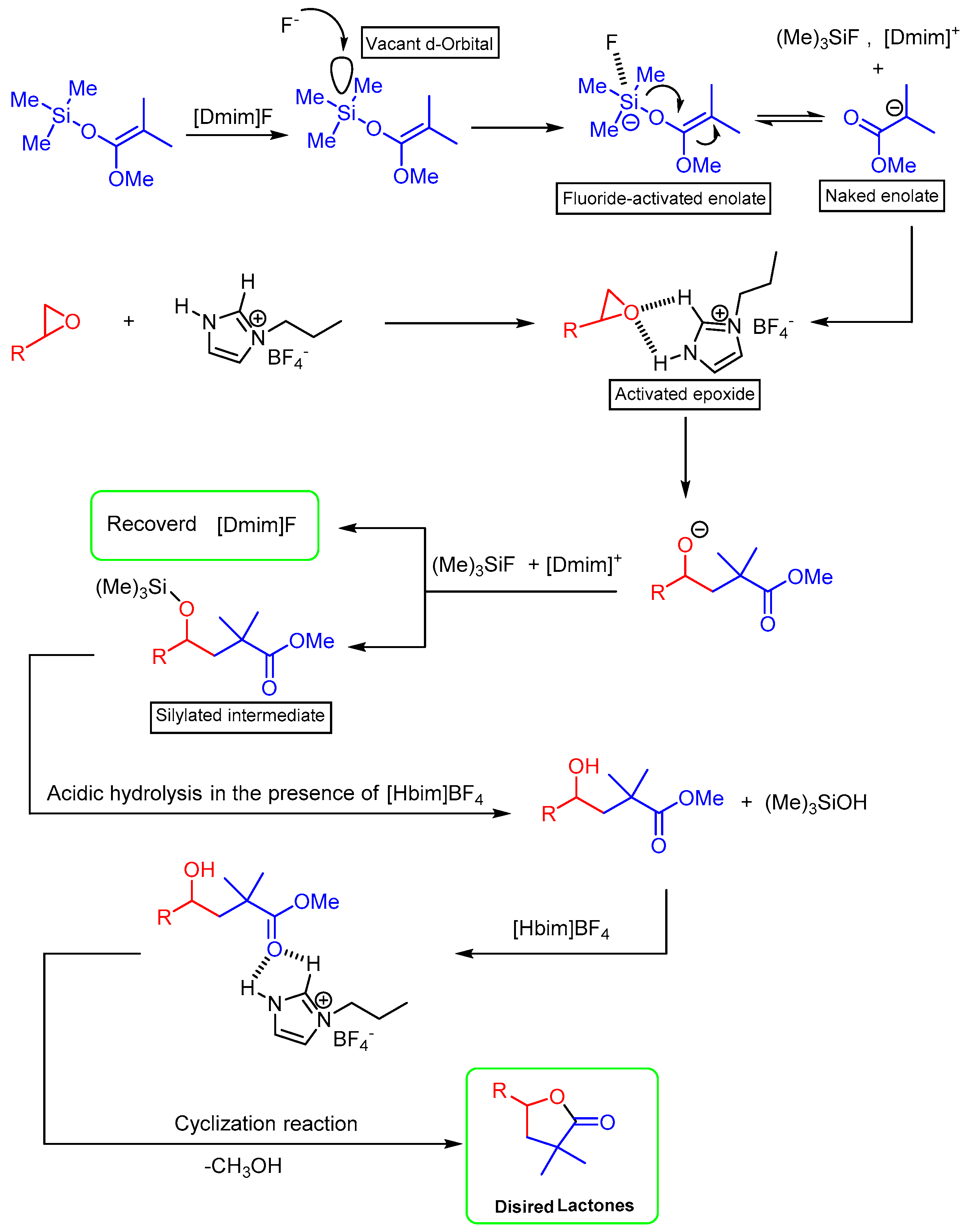
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Preparation of 1,3-Dimethylimidazolium Fluoride ([Dmim]F)
3.3. General Procedure for the Synthesis of γ-Lactones Using 1,3Dimethylimidazolium Fluoride as Catalyst
3.4. Selected Spectral Data
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schneider, C. Synthesis of 1,2-difunctionalized fine chemicals through catalytic, enantioselective ring-opening reactions of epoxides. Synthesis 2006, 3919–3944. [Google Scholar] [CrossRef]
- Pineschi, M. Asymmetric ring-opening of epoxides and aziridines with carbon nucleophiles. Eur. J. Org. Chem. 2006, 4979–4988. [Google Scholar] [CrossRef]
- Taylor, S.K. Reactions of epoxides with ester, ketone and amide enolates. Tetrahedron 2000, 56, 1149–1163. [Google Scholar] [CrossRef]
- Sturm, T.J.; Marolewski, A.E.; Rezenka, D.S.; Taylor, S.K. Diastereoselective reactions of ester enolates with epoxides. J. Org. Chem. 1989, 54, 2039–2040. [Google Scholar] [CrossRef]
- Yura, T.; Iwasawa, N.; Clark, R.; Mukaiyama, T. Asymmetric sulfenylation of tin (ii) enolates of ketones and 3-acyl-2-oxazolidones. Application to the synthesis of optically active epoxides. Chem. Lett. 1986, 15, 1809–1812. [Google Scholar] [CrossRef]
- Danishefsky, S.; Kitahara, T.; Tsai, M.; Dynak, J. Functionalized alanes for the conversion of epoxides to trans-fused. Gamma.-lactones. J. Org. Chem. 1976, 41, 1669–1671. [Google Scholar] [CrossRef]
- Movassaghi, M.; Jacobsen, E.N. A direct method for the conversion of terminal epoxides into γ-butanolides. J. Am. Chem. Soc. 2002, 124, 2456–2457. [Google Scholar] [CrossRef] [PubMed]
- Ugurchieva, T.M.; Veselovsky, V.V. Advances in the synthesis of natural butano-and butenolides. Russ. Chem. Rev. 2009, 78, 337–373. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Serra, S. Enantioselective perception of chiral odorants. Tetrahedron Asymmetr. 2003, 14, 1–42. [Google Scholar] [CrossRef]
- Kraft, P.; Bajgrowicz, J.A.; Denis, C.; Fráter, G. Odds and trends: Recent developments in the chemistry of odorants. Angew. Chem. Int. Ed. 2000, 39, 2980–3010. [Google Scholar] [CrossRef]
- Yoshimura, F.; Takahashi, M.; Tanino, K.; Miyashita, M. Stereospecific epoxide-opening reactions of 1,1-dibromo-3,4-epoxy-1-alkenes with carbon nucleophiles. Tetrahedron Lett. 2008, 49, 6991–6994. [Google Scholar] [CrossRef]
- Maslak, V.; Matović, R.; Saičić, R.N. Reaction of silyl ketene acetals with epoxides: A new method for the synthesis of γ-butanolides. Tetrahedron 2004, 60, 8957–8966. [Google Scholar] [CrossRef]
- Maslak, V.; Matović, R.; Saičić, R.N. Titanium tetrachloride promoted reaction of silyl ketene acetals with epoxides: A new method for the synthesis of γ-butanolides. Tetrahedron Lett. 2002, 43, 5411–5413. [Google Scholar] [CrossRef]
- Ipaktschi, J.; Heydari, A. Liclo4-katalysierte nucleophile addition an α-chirale aldehyde, aldimine und oxirane. Eur. J. Inorg. Chem. 1993, 126, 1905–1912. [Google Scholar] [CrossRef]
- Fringuelli, F.; Lanari, D.; Pizzo, F.; Vaccaro, L. An e-factor minimized protocol for the preparation of methyl β-hydroxy esters. Green Chem. 2010, 12, 1301–1305. [Google Scholar] [CrossRef]
- Fringuelli, F.; Lanari, D.; Pizzo, F.; Vaccaro, L. Solid-supported ammonium fluorides in organic synthesis. Curr. Org. Synth. 2009, 6, 203–218. [Google Scholar] [CrossRef]
- Fringuelli, F.; Lanari, D.; Pizzo, F.; Vaccaro, L. Amberlite ira900f as a solid fluoride source for a variety of organic transformations under solvent-free conditions. Eur. J. Org. Chem. 2008, 3928–3932. [Google Scholar] [CrossRef]
- Amantini, D.; Beleggia, R.; Fringuelli, F.; Pizzo, F.; Vaccaro, L. Tbaf-catalyzed synthesis of 5-substituted 1 h-tetrazoles under solventless conditions. J. Org. Chem. 2004, 69, 2896–2898. [Google Scholar] [CrossRef] [PubMed]
- Bonollo, S.; Ahmady, A.Z.; Petrucci, C.; Marrocchi, A.; Pizzo, F.; Vaccaro, L. A catalytic approach to the metal-free reaction of epoxides with ketene silyl acetals for accessing γ-lactones. Org. Lett. 2014, 16, 5721–5723. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-Q.; Jiang, M.-Y.; Zheng, C.-G.; Xiao, J.-C. Efficient synthesis of 1-alkyl-3-methylimidazolium fluorides and possibility of the existence of hydrogen bonding between fluoride anion and c (sp3)–h. J. Fluor. Chem. 2012, 133, 160–162. [Google Scholar] [CrossRef]
- Keshavarz, M.; Karami, B.; Ahmady, A.Z.; Ghaedi, A.; Vafaei, H. [Bmim]BF4/[Cu(im12)2]CuCl2 as a novel catalytic reaction medium for click cyclization. C. R. Chim. 2014, 17, 570–576. [Google Scholar] [CrossRef]
- Eckert, H. Synergy effects in the chemical synthesis and extensions of multicomponent reactions (mcrs)—The low energy way to ultra-short syntheses of tailor-made molecules. Molecules 2017, 22, 349. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Zhou, G.; Gong, Y.; Wei, Q.-D.; Tian, M.-Y.; Liu, X.-L.; Feng, T.-T.; Zhou, Y.; Yuan, W.-C. Molecular hybridization-guided one-pot multicomponent synthesis of turmerone motif-fused 3,3′-pyrrolidinyl-dispirooxindoles via a 1,3-dipolar cycloaddition reaction. Molecules 2017, 22, 645. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, J.; Shin, K.J.; Oh, E.; Seo, J.H. Consecutive one-pot versus domino multicomponent approaches to 3-(diarylmethylene) oxindoles. Molecules 2017, 22, 503. [Google Scholar] [CrossRef] [PubMed]
- Heidarizadeh, F.; Zarei, A. Bronsted acidic ionic liquid 1-n-butylimidazolium tetrafluoroborate ([hbim] bf4): A green catalyst and recyclable medium for the azidolysis of epoxides. J. Chem. Soc. Pak. 2012, 34, 593. [Google Scholar]
- Ahmady, A.Z.; Heidarizadeh, F.; Keshavarz, M. Ionic liquid containing copper (i): A new, green, homogeneous, and reusable catalyst for click cyclization. Synth. Commun. 2013, 43, 2100–2109. [Google Scholar] [CrossRef]
- Baudequin, C.; Brégeon, D.; Levillain, J.; Guillen, F.; Plaquevent, J.-C.; Gaumont, A.-C. Chiral ionic liquids, a renewal for the chemistry of chiral solvents? Design, synthesis and applications for chiral recognition and asymmetric synthesis. Tetrahedron Asymmetr. 2005, 16, 3921–3945. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, S.; Shi, F.; Zhang, Q.; Ma, X.; Lu, L.; Deng, Y. The influence of the acidity of ionic liquids on catalysis. ChemSusChem 2010, 3, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Kölle, P.; Dronskowski, R. Hydrogen bonding in the crystal structures of the ionic liquid compounds butyldimethylimidazolium hydrogen sulfate, chloride, and chloroferrate (ii, iii). Inorg. Chem. 2004, 43, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst (mol %) | Solvent | 1a:2 (mmol:mmol) | T (°C) | Time (h) | Yield (%) a |
|---|---|---|---|---|---|---|
| 1 | - | - | 1:1 | 60 | 72 | 0 |
| 2 | - | [Hbim]BF4 | 1:1.5 | 60 | 72 | 0 |
| 3 | [Dmim]F(2%) | - | 1:1 | 60 | 5 | 15 |
| 4 | [Dmim]F(2%) | - | 1:1.5 | 60 | 5 | 30 |
| 5 | [Dmim]F(2%) | - | 1.5:1 | 60 | 5 | 20 |
| 6 | [Dmim]F(5%) | - | 1:1 | 60 | 5 | 40 |
| 7 | [Dmim]F(5%) | - | 1.5:1 | 60 | 5 | 45 |
| 8 | [Dmim]F(5%) | - | 1:1.5 | 60 | 2 | 90 |
| 9 | [Dmim]F(5%) | [Hbim]BF4 | 1:1.5 | 60 | 0.25 | 96 |
| 10 | [Dmim]F(5%) | [Hbim]BF4 | 1:1.5 | 80 | 0.25 | 98 |
| 11 | [Dmim]F(10%) | [Hbim]BF4 | 1:1.5 | 60 | 0.25 | 97 |

| Entry | Catalyst/Solvent System | Run | Tim (h) | Yield(%) b |
|---|---|---|---|---|
| 1 | [Dmim]F (5%)/[Hbim]BF4 | 1 | 0.25 | 96 |
| 2 | Recovered from 1st Run | 2 | 0.25 | 95 |
| 3 | Recovered from 2nd Run | 3 | 0.25 | 90 |
| 4 | Recovered from 3rd Run | 4 | 0.25 | 73 |
| 5 | [Dmim]Cl (5%)/[Hbim]BF4 | 1 | 72 | 0 |
| 6 | [Dmim]Br (5%)/[Hbim]BF4 | 1 | 72 | 0 |
| 7 | [Dmim]I (5%)/[Hbim]BF4 | 1 | 72 | 0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keshavarz, M.; Zarei Ahmady, A.; Mostoufi, A.; Mohtasham, N. One-Pot Green Regioselesctive Synthesis of γ-Lactones from Epoxides and Ketene Silyl Acetals Using 1,3-Dimethylimidazolium Fluoride as a Recoverable Metal-Free Catalyst. Molecules 2017, 22, 1385. https://doi.org/10.3390/molecules22091385
Keshavarz M, Zarei Ahmady A, Mostoufi A, Mohtasham N. One-Pot Green Regioselesctive Synthesis of γ-Lactones from Epoxides and Ketene Silyl Acetals Using 1,3-Dimethylimidazolium Fluoride as a Recoverable Metal-Free Catalyst. Molecules. 2017; 22(9):1385. https://doi.org/10.3390/molecules22091385
Chicago/Turabian StyleKeshavarz, Mosadegh, Amanollah Zarei Ahmady, Azar Mostoufi, and Neda Mohtasham. 2017. "One-Pot Green Regioselesctive Synthesis of γ-Lactones from Epoxides and Ketene Silyl Acetals Using 1,3-Dimethylimidazolium Fluoride as a Recoverable Metal-Free Catalyst" Molecules 22, no. 9: 1385. https://doi.org/10.3390/molecules22091385