Development and Validation of HPLC-DAD and UHPLC-DAD Methods for the Simultaneous Determination of Guanylhydrazone Derivatives Employing a Factorial Design


 and
and
Abstract
:1. Introduction
2. Results
2.1. Development and Validation of the HPLC Method
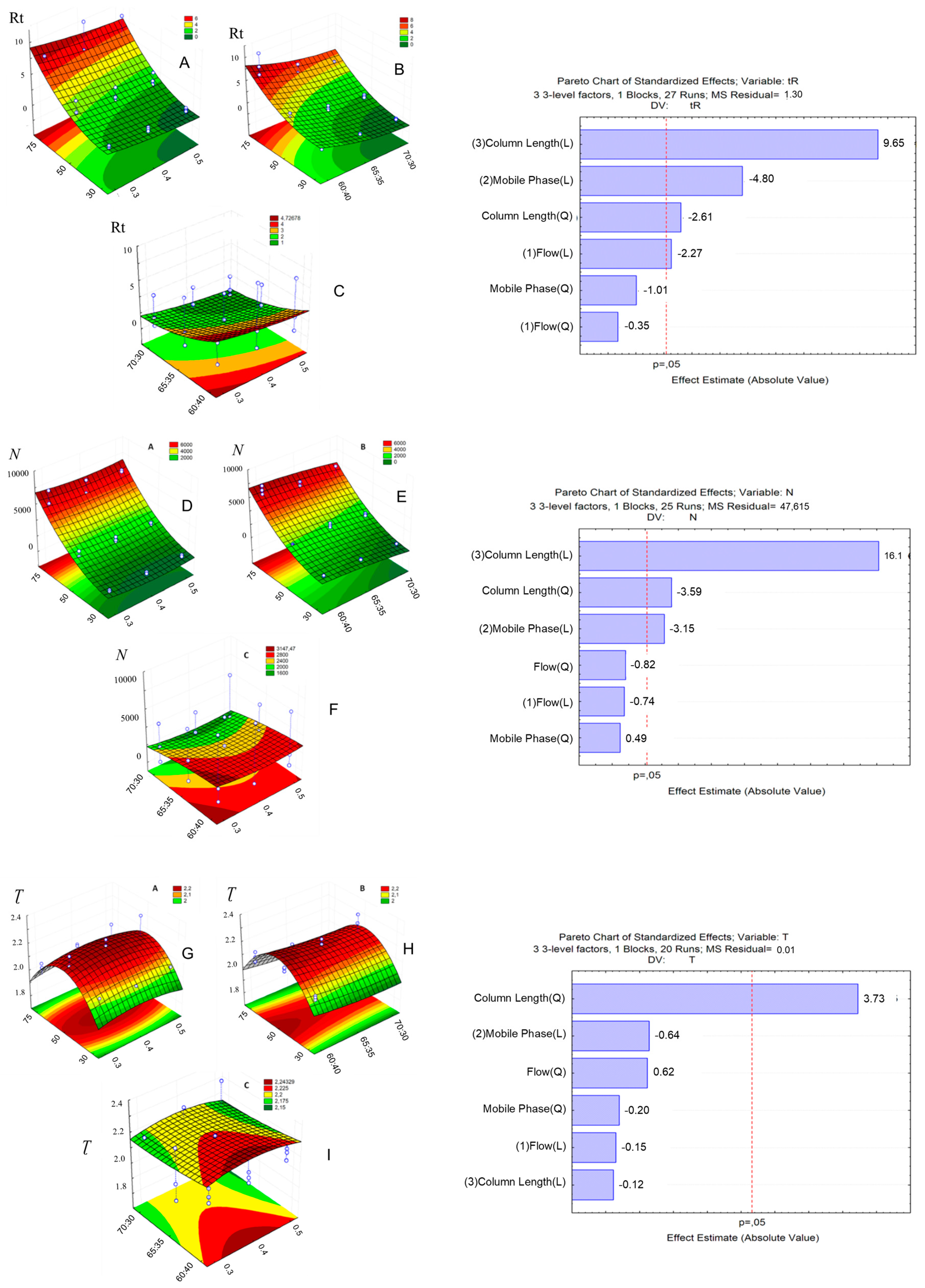
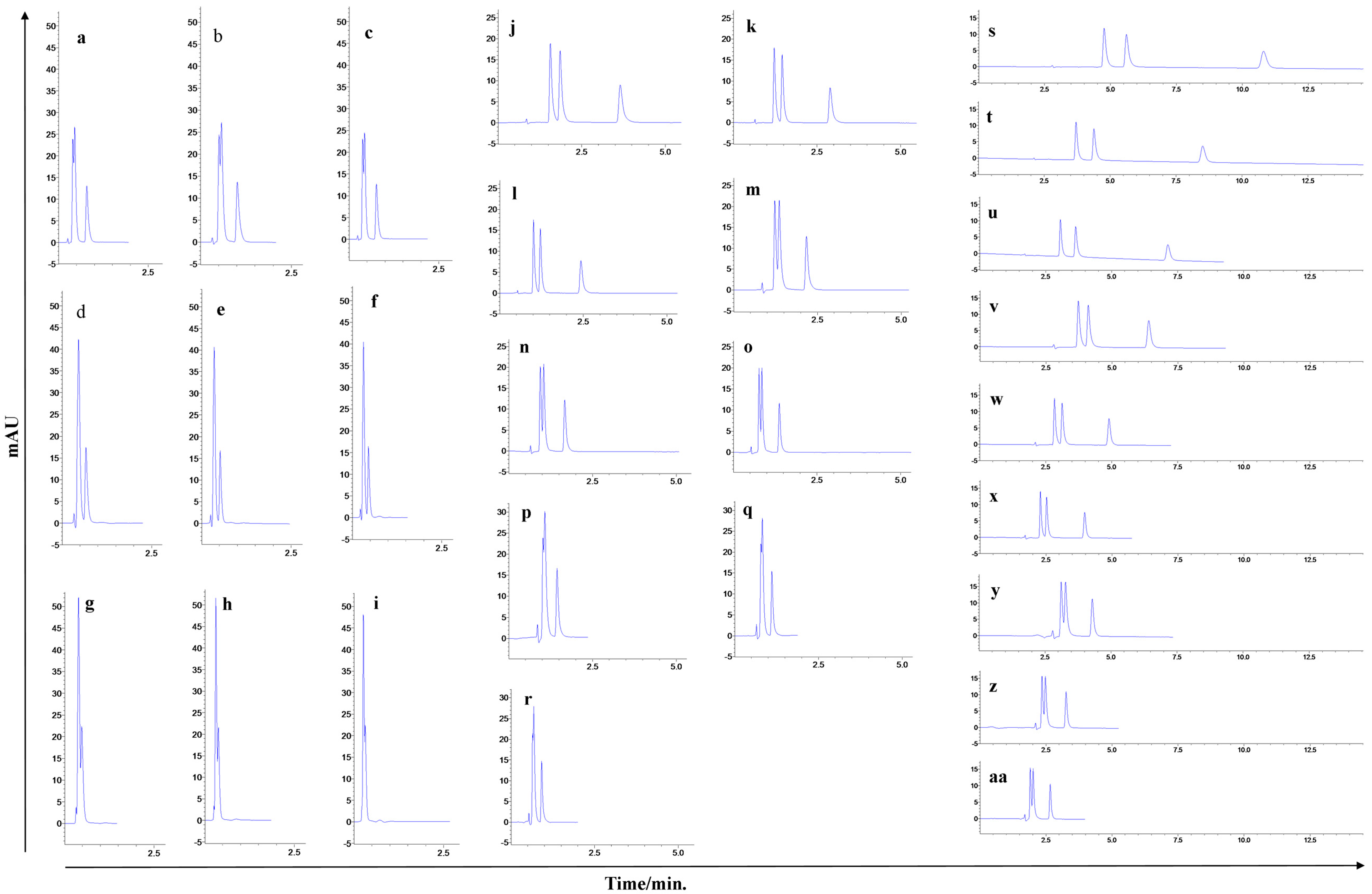
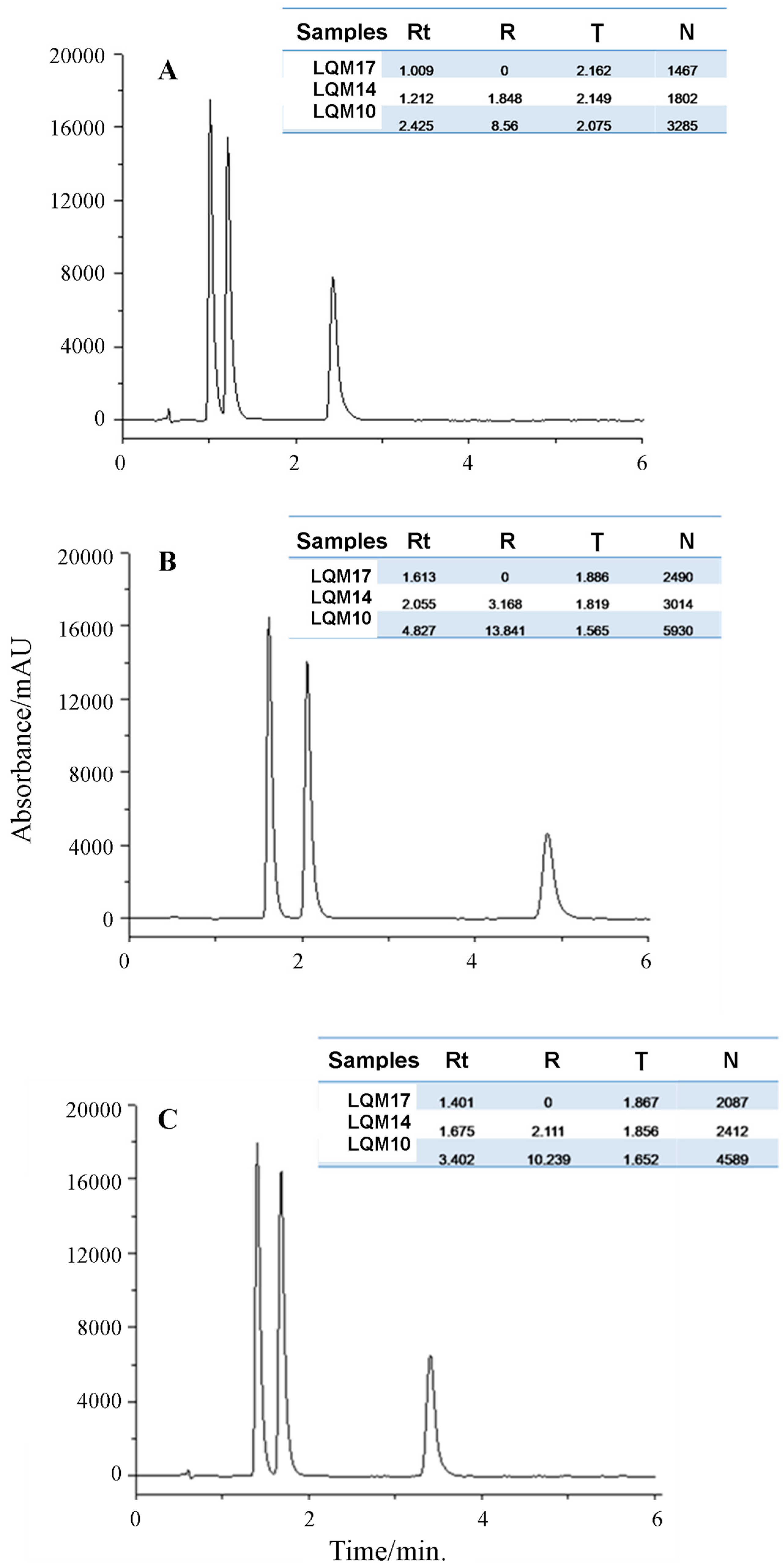
2.1.1. Optimization of the Chromatographic Conditions
2.1.2. HPLC Method Validation
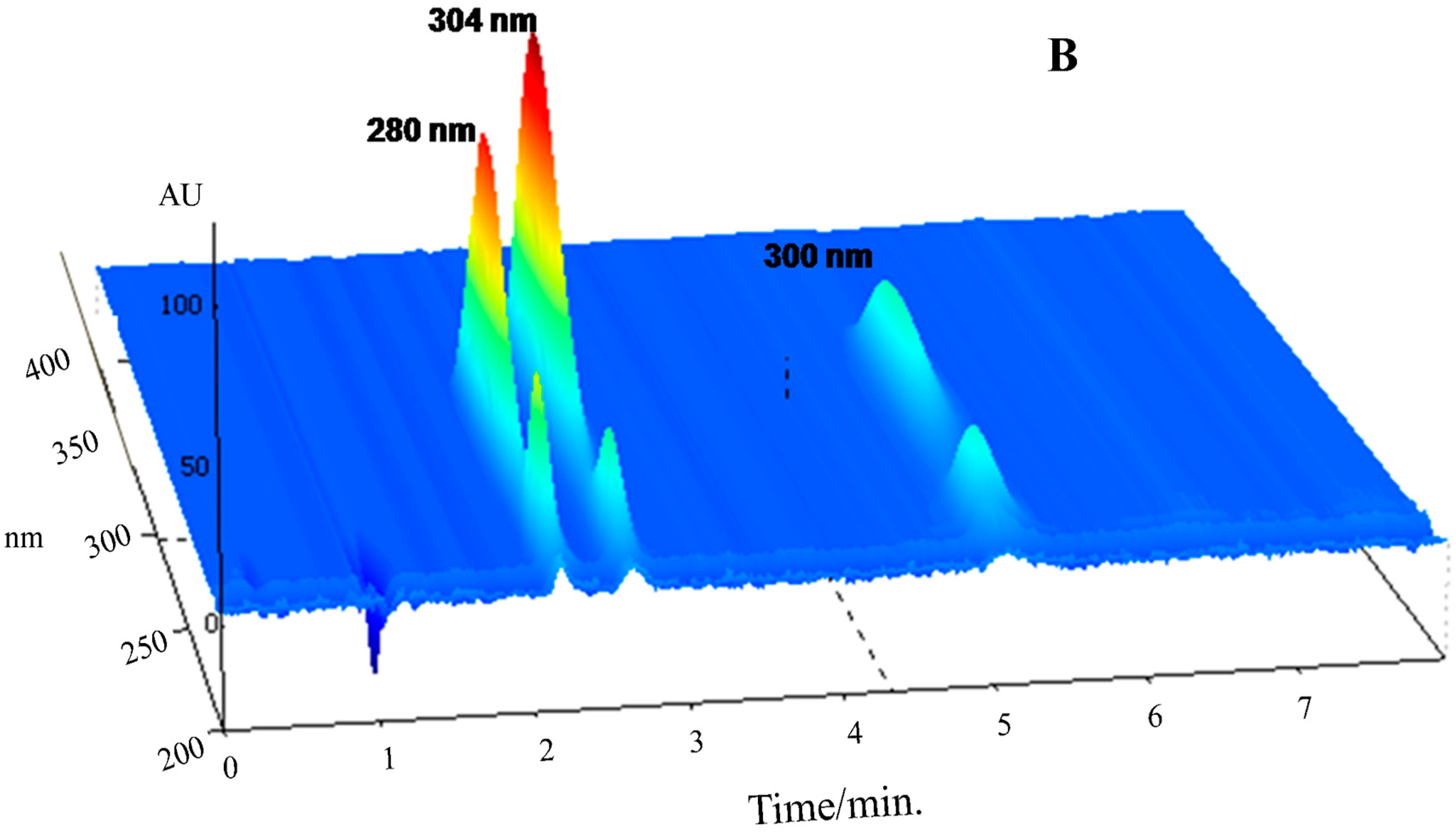
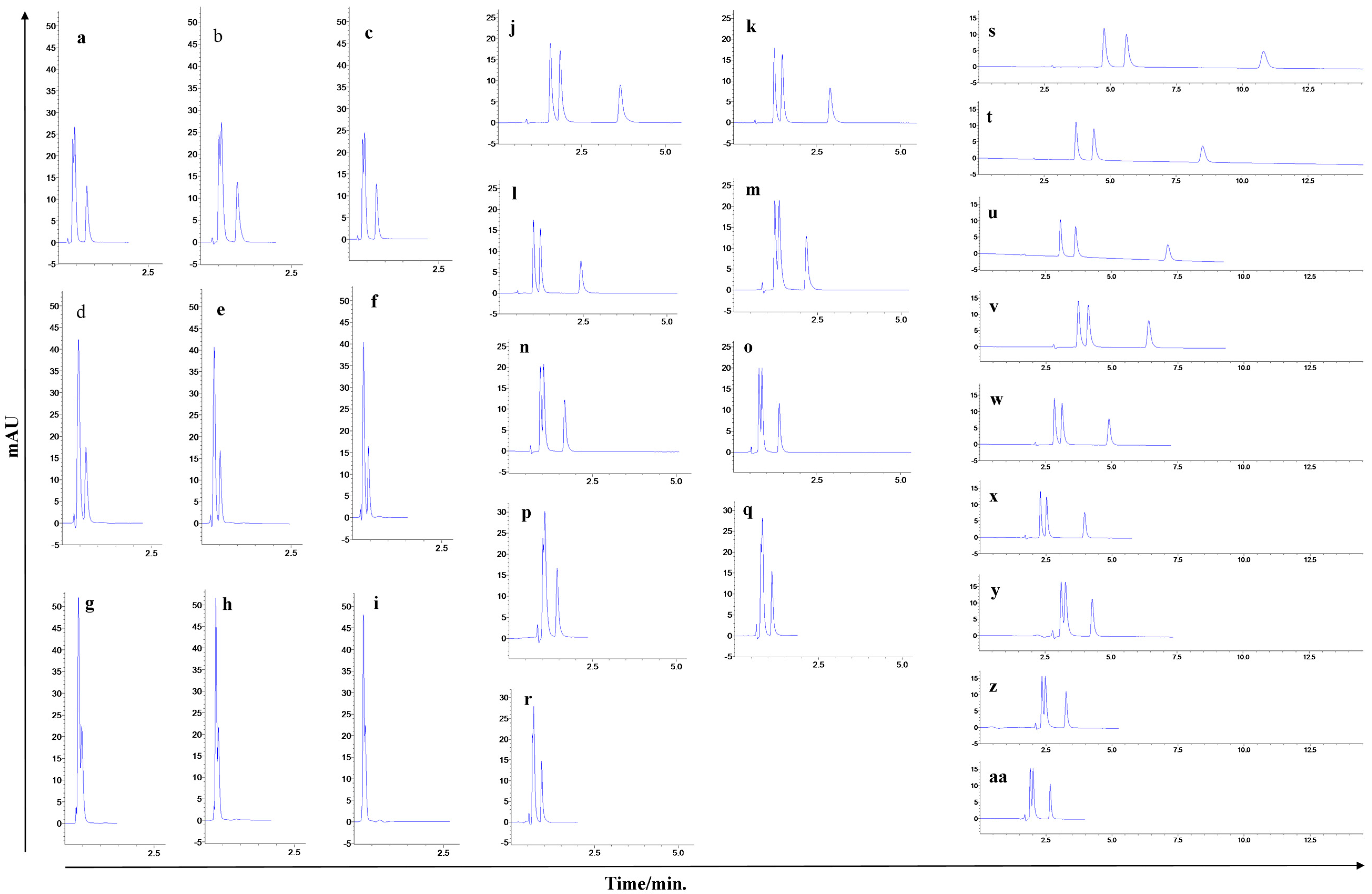
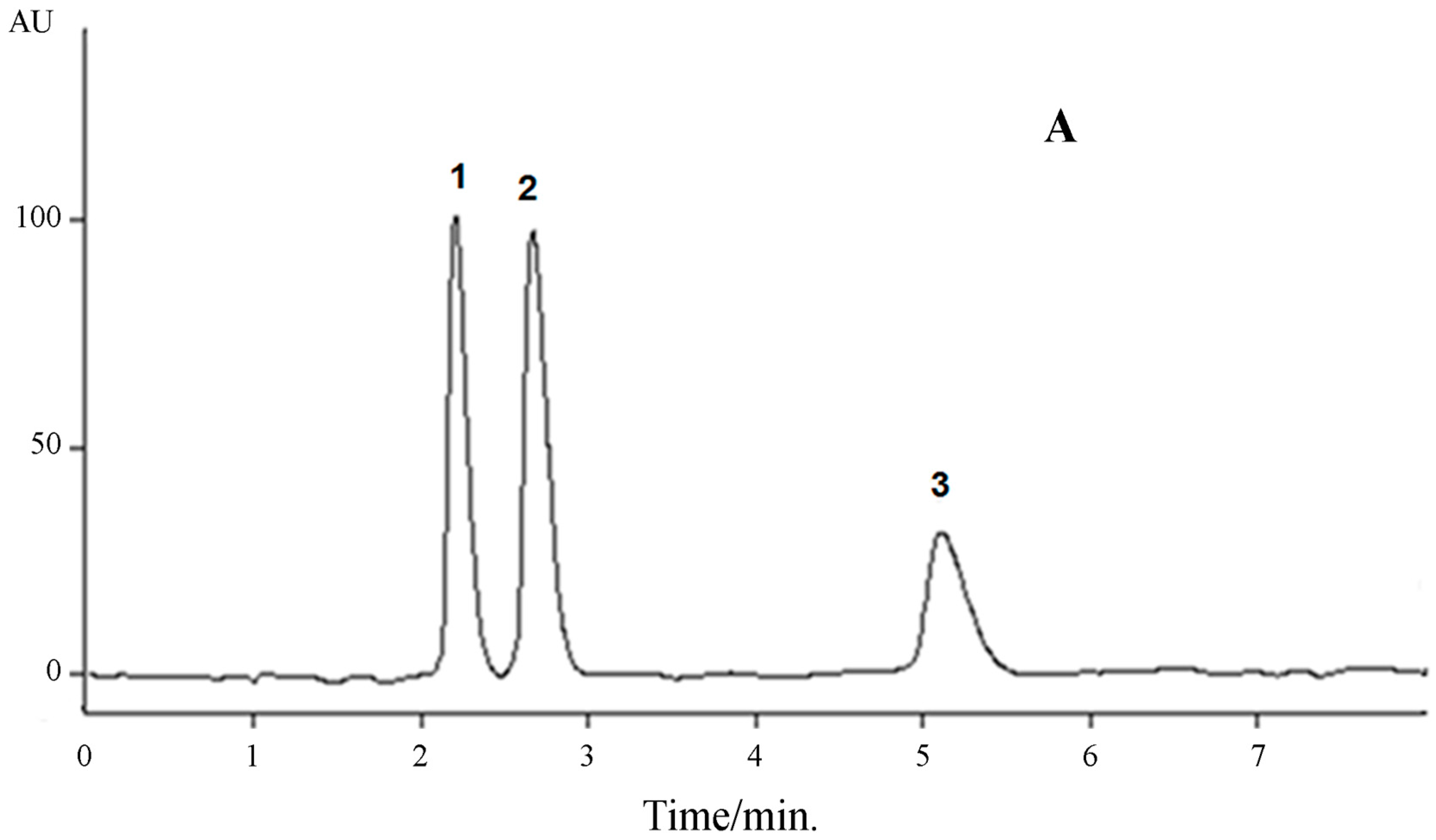
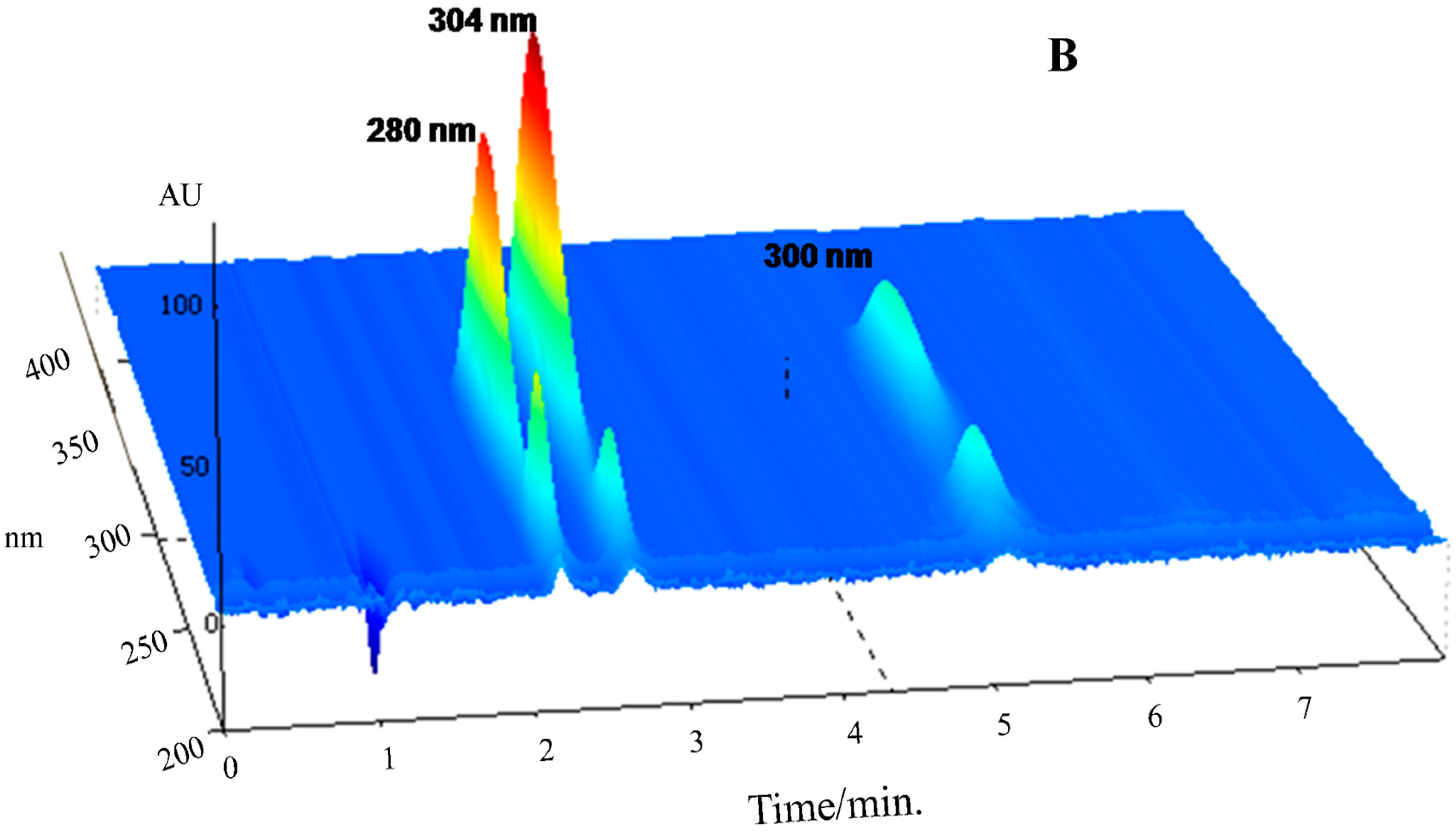
Selectivity
Linearity
LOD and LOQ
Precision
Accuracy
Robustness
2.2. Experimental Design and Validation of UHPLC Method
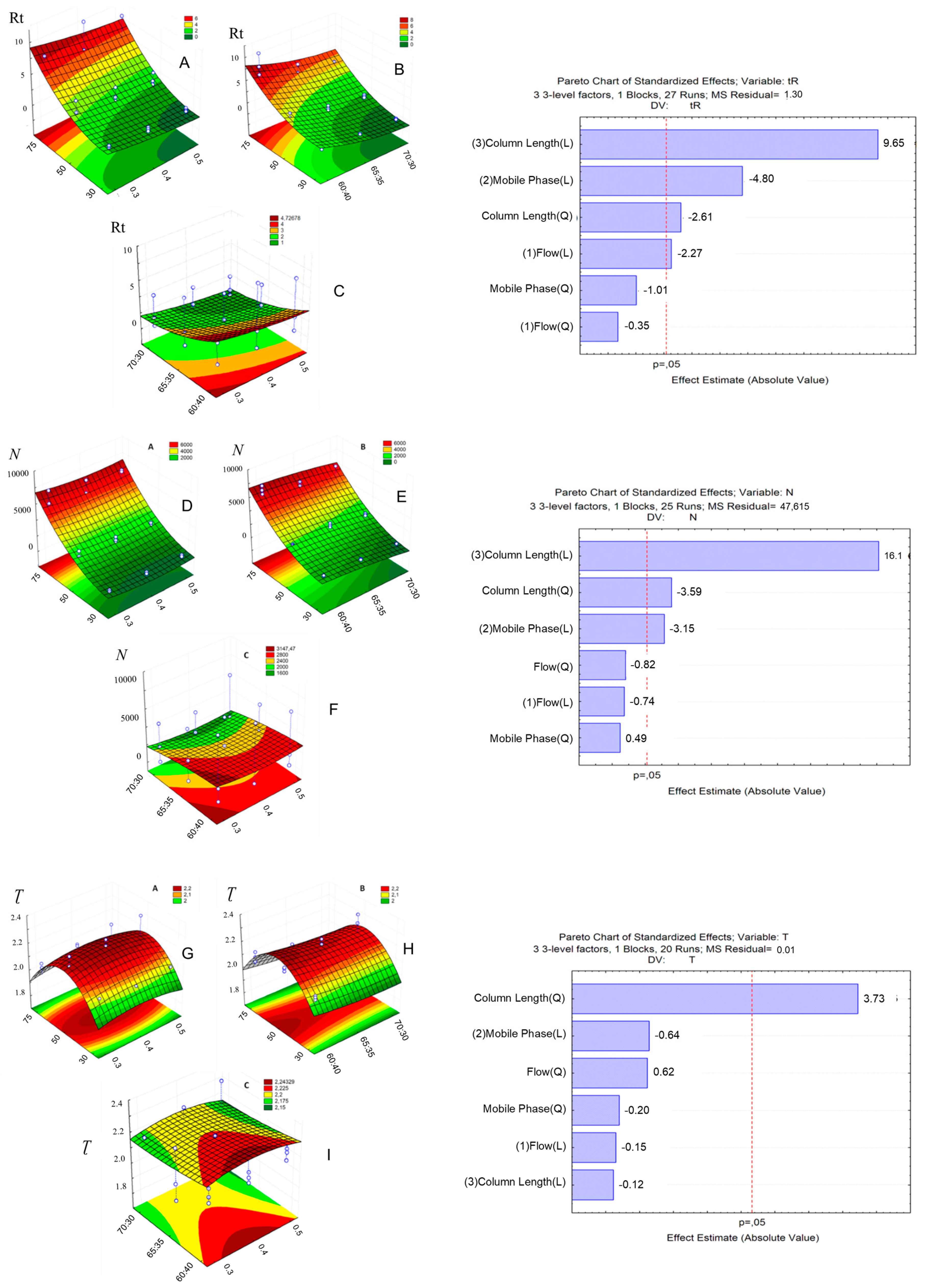
2.2.1. Response Surface Methodology (RSM) and UHPLC Method Development
2.2.2. UHPLC Method Validation
Selectivity
Linearity
LOD and LOQ
Precision
Accuracy
Robustness
3. Material and Methods
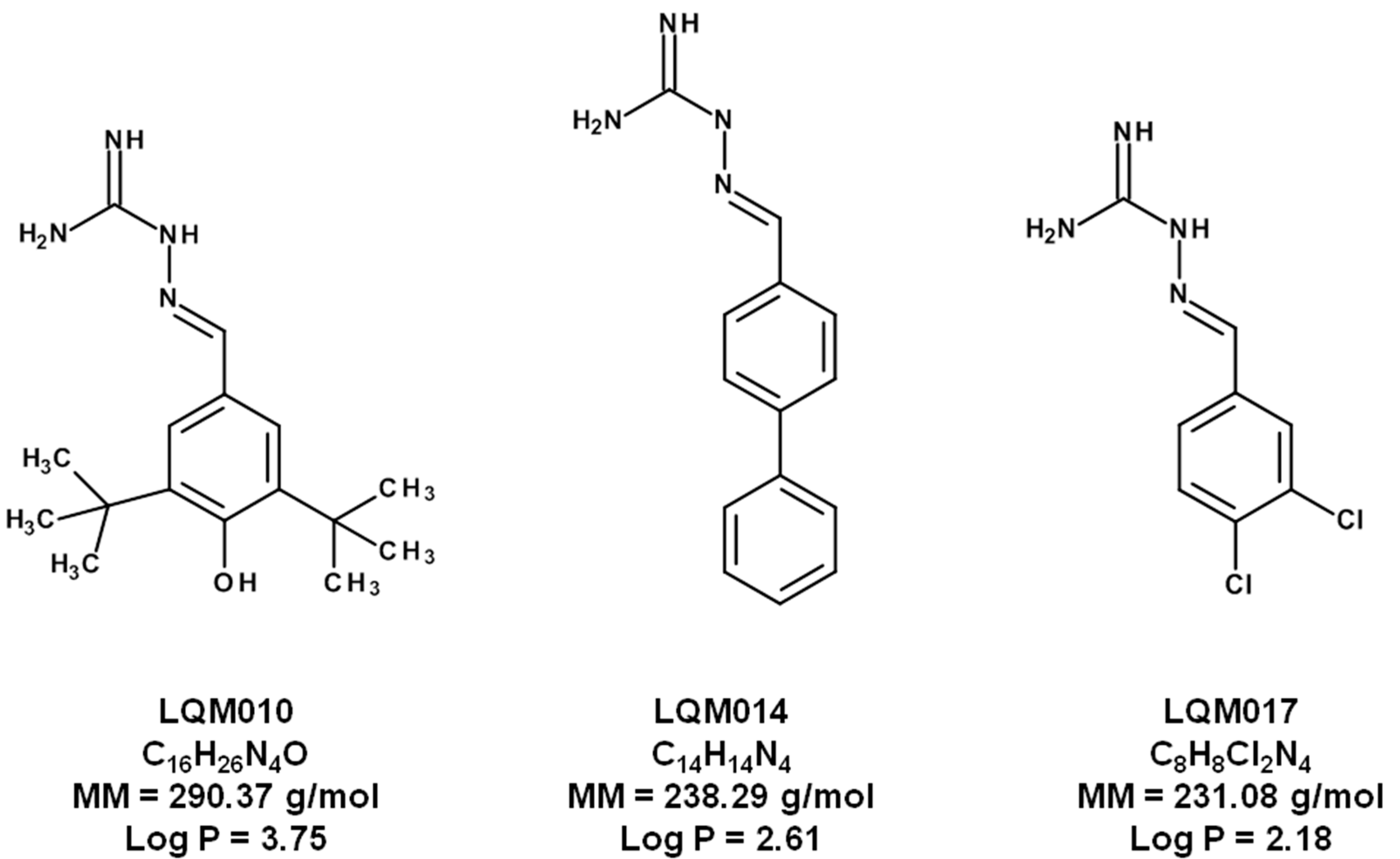
3.1. Chemicals
3.2. Instrumental and Analytical Conditions
3.3. Preparation of Sample Solution
3.4. Methods Development
3.4.1. HPLC Method Development
3.4.2. UHPLC Method Development
3.5 Method Validation
3.5.1. Specificity/Selectivity
3.5.2. Linearity
3.5.3. Limit of Detection (LOD) Limit of Quantification (LOQ)
3.5.4. Precision
3.5.5. Accuracy
3.5.6. Robustness
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Edmont, D.; Rocher, R.; Plisson, C.; Chenault, J. Synthesis and evaluation of quinoline carboxyguanidines as antidiabetic agents. Bioorg. Med. Chem. Lett. 2000, 10, 1831–1834. [Google Scholar] [CrossRef]
- Messeder, J.C.; Tinoco, L.W.; Figueroa-Villar, J.D.; Souza, E.M.; Santa Rita, R.; de Castro, S.L. Aromatic guanyl hydrazones: Synthesis, structural studies and in vitro activity against Trypanosoma cruzi. Bioorg. Med. Chem. Lett. 1995, 5, 3079–3084. [Google Scholar] [CrossRef]
- Santos-Filho, O.A.; Figueroa-Villar, J.D.; Araujo, M.T. Molecular modeling of the interaction of trypanocide guanyl hydrazones with B-DNA. Bioorg. Med. Chem. Lett. 1997, 7, 1797–1802. [Google Scholar] [CrossRef]
- Gadad, A.K.; Mahajanshetti, C.S.; Nimbalkar, S.; Raichurkar, A. Synthesis and antibacterial activity of some 5-guanylhydrazone/thiocyanato-6-arylimidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide derivatives. Eur. J. Med. Chem. 2000, 35, 853–857. [Google Scholar] [CrossRef]
- Foye, W.O.; Almassian, B.; Eisenberg, M.S.; Maher, T.J. Synthesis and biological activity of guanylhydrazones of 2- and 4-pyridine and 4-quinoline carboxaldehydes. J. Pharm. Sci. 2017, 79, 527–530. [Google Scholar] [CrossRef]
- Knight, W.A.T.; Fabian, C.; Costanzi, J.J.; Jones, S.E.; Coltman, C.A. Methyl-glyoxal bis guanyl hydrazone (methyl-GAG, MGBG) in lymphoma and Hodgkin’s disease. Investig. New Drugs 1983, 1, 235–237. [Google Scholar] [CrossRef]
- Pinhatti, V.R.; da Silva, J.; Martins, T.L.C.; Moura, D.J.; Rosa, R.M.; Villela, I.; Stopiglia, C.D.O.; da Silva Santos, S.; Scroferneker, M.L.; Machado, C.R.; et al. Cytotoxic, mutagenicity, and genotoxicity effects of guanylhydrazone derivatives. Mutat. Res. Toxicol. Environ. Mutagen. 2016, 806, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Güzin Aslan, H.; Karacan, N.; Aslan, E. Synthesis, characterization and antimicrobial activity of a new aromatic sulfonyl hydrazone derivative and its transition metal complexes. J. Chin. Chem. Soc. 2013, 60, 212–217. [Google Scholar] [CrossRef]
- Koskinen, M.; Elo, H.; Lukkari, P.; Riekkola, M.-L. Determination of the antileukemic drug mitoguazone and seven other closely related bis(amidinohydrazones) in human blood serum by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1996, 685, 141–149. [Google Scholar] [CrossRef]
- Clement, B.; Demesmaeker, M.; Linne, S. Microsomal catalyzed N-hydroxylation of guanabenz and reduction of the N-hydroxylated metabolite: Characterization of the two reactions and genotoxic potential of guanoxabenz1. Chem. Res. Toxicol. 1996, 9, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Andreani, A.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Giorgi, G.; Garaliene, V. Potential antitumor agents. 34. (1) Synthesis and antitumor activity of guanylhydrazones from imidazo[2,1-b]thiazoles and from diimidazo[1,2-a:1,2-c]pyrimidine. Anticancer Res. 2004, 24, 203–212. [Google Scholar] [PubMed]
- Franca, P.H.B.; Da Silva-Junior, E.F.; Aquino, P.G.V.; Santana, A.E.G.; Ferro, J.N.S.; De Oliveira Barreto, E.; Pessoa, C.D.Ó.; Meira, A.S.; De Aquino, T.M.; Alexandre-Moreira, M.S.; et al. Preliminary in vitro evaluation of the anti-proliferative activity of guanylhydrazone derivatives. Acta Pharm. 2016, 66, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Galvão, F.P.; De Brito, W.A.; Dantas, M.G.; Pereira, M.A.V.; Araújo, J.X.; França, P.H.B.; Santana, A.E.G.; Macêdo, R.O.; Ribeiro, E.A.N.; Barbosa, E.G.; et al. Thermal characterization of some guanylhydrazones by thermal and nonthermal techniques. J. Therm. Anal. Calorim. 2015, 120, 879–887. [Google Scholar] [CrossRef]
- El-Houssini, O.M.; Zawilla, N.H. Chromatographic methods for the determination of fluphenazine, nortriptyline and its impurity amitriptyline in bulk and pharmaceutical formulations. J. Anal. Chem. 2014, 69, 1187–1192. [Google Scholar] [CrossRef]
- Zhang, H.; Wood, T.; Young, S.; Millington, D. A straightforward, quantitative ultra-performance liquid chromatography tandem mass spectrometric method for heparan sulfate, dermatan sulfate and chondroitin. Mol. Genet. 2014, 114, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, X. Mixed-mode chromatography in pharmaceutical and biopharmaceutical applications. J. Pharm. Biomed. Anal. 2016, 128, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Solon, L.G.; de Barros Lima, I.P.; Nogueira, F.H.A.; de Araújo, J.P.; Vivacqua, C.A.; Aragão, C.F.S. Development and validation of an UHPLC method for the determination of betamethasone valerate in cream, gel, ointment and lotion. Steroids 2016, 106, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Do Nascimento, T.G.; Aragão, C.F.S.; de Medeiros, F.D.; Oliveira, E.D.J.; Macêdo, R.O. Validation of a method for determination of ampicillin in human plasma using LC-DAD. J. Chromatogr. Sci. 2009, 47, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Gumustas, M.; Kurbanoglu, S.; Uslu, B.; Ozkan, S.A. UPLC versus HPLC on drug analysis: Advantageous, applications and their validation parameters. Chromatographia 2013, 76, 1365–1427. [Google Scholar] [CrossRef]
- Hu, L.; Liu, Y.; Cheng, S. Simultaneous determination of six analytes by HPLC-UV for high throughput analysis in permeability assessment. J. Chromatogr. Sci. 2011, 49, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Cerami, C.; Xini, Z.; Ulrich, P.; Bianchi, M.; Tracey, K.J.; Berger, B.J. High-performance liquid chromatographic method for guanylhydrazone compounds. J. Chromatogr. B Biomed. Sci. Appl. 1996, 675, 71–75. [Google Scholar] [CrossRef]
- Ferreira, S.L.C.; Bruns, R.E.; da Silva, E.G.P.; Dos Santos, W.N.L.; Quintella, C.M.; David, J.M.; de Andrade, J.B.; Breitkreitz, M.C.; FontesJardim, I.C.S.; Neto, B.B. Statistical designs and response surface techniques for the optimization of chromatographic systems. J. Chromatogr. A 2007, 1158, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Hibbert, D.B. Experimental design in chromatography: A tutorial review. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 910, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.W. Modern HPLC for Practicing Scientists; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- ICH International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. Q2 (R1) Validation of Analytical Procedures: Text and Methodolog. 2005. Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (accessed on 21 August 2017).
- Samanidou, V.F.; Nika, M.K.; Papadoyannis, I.N. Development of an HPLC method for the monitoring of tricyclic antidepressants in biofluids. J. Sep. Sci. 2007, 30, 2391–2400. [Google Scholar] [CrossRef] [PubMed]
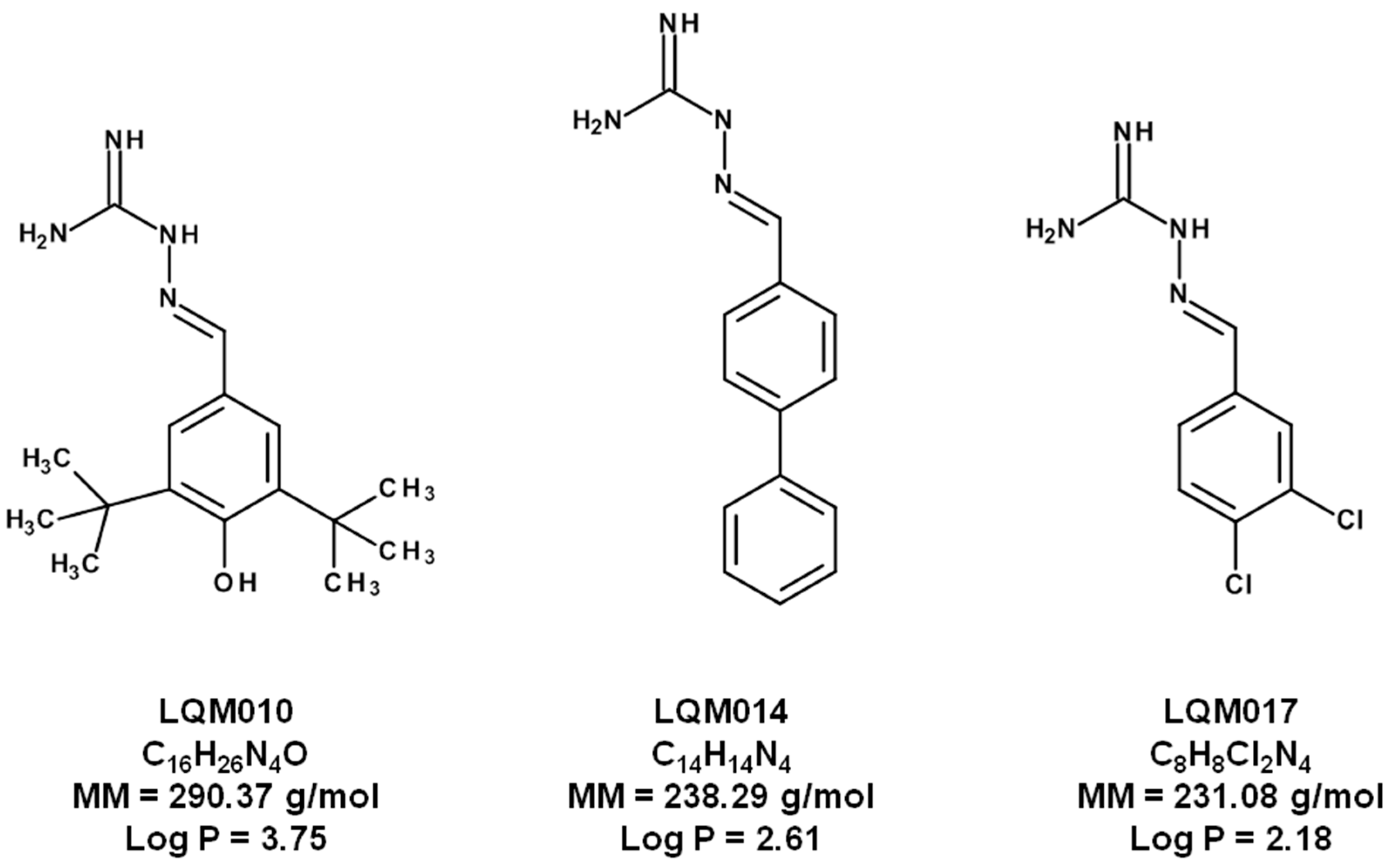
Sample Availability: Samples of the compounds LQM10, LQM14, and LQM17 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromatographic Method | Validation Parameters | LQM010 | LQM014 | LQM017 | ||||
|---|---|---|---|---|---|---|---|---|
| Linearity (n = 3) | r2: 0.9995 y = 42.04453x + 6.54327 | r2: 0.9999 y = 74.20108x + 0.66868 | r2: 0.9994 y = 62.60385x + 0.43507 | |||||
| Specificity, SI | 979 | 973 | 959 | |||||
| % | % | % | ||||||
| Accuracy (n = 5), µg·mL−1 | 8 | 99.71 ± 1.67 | 98.69 ± 1.85 | 100.22 ± 1.86 | ||||
| 10 | 100.46 ± 1.34 | 101.47 ± 0.24 | 99.71 ± 1.36 | |||||
| 12 | 99.49 ± 1.79 | 98.71 ± 1.50 | 100.15 ± 1.25 | |||||
| HPLC | Mean Area ± RSD | Mean Area ± RSD | Mean Area ± RSD | |||||
| Precision (n = 6), 10 µg·mL−1 | Intra-day | 58046 ± 1.48 | 101134 ± 2.00 | 79412 ± 1.24 | ||||
| Inter-day | 56976 ± 2.81 | 101459 ± 1.56 | 78202 ± 2.20 | |||||
| Mean Area ± RSD | RT | Area | RT | Area | RT | |||
| Robustness Flow (mL·min−1) | 1.50 ± 0.05 | 556.53 ± 2.07 | 5.08 | 1019.33 ± 2.34 | 2.64 | 765.33 ± 2.54 | 2.18 | |
| % | RT | % | RT | % | RT | |||
| Robustness pH | 3.50 ± 0.05 | 561.04 ± 1.76 | 5.10 | 1027.50 ± 1.64 | 2.60 | 772.81 ± 1.61 | 2.20 | |
| Linearity (n = 3) | r2: 0.9994 y = 5092.94x − 180.59 | r2: 0.9997 y = 8464.98x − 533.43 | r2: 0.9997 y= 8215.21x − 522.16 | |||||
| Specificity, SI | 999 | 999 | 1000 | |||||
| % | % | % | ||||||
| Accuracy (n = 5), µg·mL−1 | 8 | 101.62 ± 1.92 | 99.12 ± 1.35 | 100.48 ± 1.42 | ||||
| 10 | 99.32 ± 0.24 | 99.07 ± 0.97 | 99.48 ± 1.34 | |||||
| 12 | 100.23 ± 1.45 | 100.30 ± 0.76 | 100.33 ± 1.33 | |||||
| UHPLC | Mean Area ± RSD | Mean Area ± RSD | Mean Area ± RSD | |||||
| Precision (n = 6), 10 µg·mL−1 | Intra-day | 50,277 ± 0.53 | 80,625 ± 0.84 | 80,742 ±1.27 | ||||
| Inter-day | 50,134 ± 0.43 | 80,321 ± 0.68 | 80,894 ± 0.63 | |||||
| Area | RT | Area | RT | Area | RT | |||
| Robustness Flow (mL·min−1) | 0.50 ± 0.05 | 46,917.61 ± 1.66 | 3.40 | 76,600.00 ± 1.29 | 1.67 | 75,945.22 ± 1.63 | 1.40 | |
| Area | RT | Area | RT | Area | RT | |||
| Robustness pH | 3.50 ± 0.05 | 46,709.83 ± 1.93 | 3.40 | 76,835.67 ± 1.05 | 1.67 | 75,542.72 ± 1.53 | 1.40 | |
| Factor | Levels | ||
|---|---|---|---|
| −1 | 0 | +1 | |
| Column length, mm | 30 | 50 | 75 |
| Flow rate, mL min−1 | 0.3 | 0.4 | 0.5 |
| Mobile phase composition, MeOH:H2O | 60:40 | 65:35 | 70:30 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azevedo de Brito, W.; Gomes Dantas, M.; Andrade Nogueira, F.H.; Ferreira da Silva-Júnior, E.; Xavier de Araújo-Júnior, J.; Aquino, T.M.d.; Adélia Nogueira Ribeiro, Ê.; Da Silva Solon, L.G.; Soares Aragão, C.F.; Barreto Gomes, A.P. Development and Validation of HPLC-DAD and UHPLC-DAD Methods for the Simultaneous Determination of Guanylhydrazone Derivatives Employing a Factorial Design. Molecules 2017, 22, 1394. https://doi.org/10.3390/molecules22091394
Azevedo de Brito W, Gomes Dantas M, Andrade Nogueira FH, Ferreira da Silva-Júnior E, Xavier de Araújo-Júnior J, Aquino TMd, Adélia Nogueira Ribeiro Ê, Da Silva Solon LG, Soares Aragão CF, Barreto Gomes AP. Development and Validation of HPLC-DAD and UHPLC-DAD Methods for the Simultaneous Determination of Guanylhydrazone Derivatives Employing a Factorial Design. Molecules. 2017; 22(9):1394. https://doi.org/10.3390/molecules22091394
Chicago/Turabian StyleAzevedo de Brito, Wanessa, Monique Gomes Dantas, Fernando Henrique Andrade Nogueira, Edeildo Ferreira da Silva-Júnior, João Xavier de Araújo-Júnior, Thiago Mendonça de Aquino, Êurica Adélia Nogueira Ribeiro, Lilian Grace Da Silva Solon, Cícero Flávio Soares Aragão, and Ana Paula Barreto Gomes. 2017. "Development and Validation of HPLC-DAD and UHPLC-DAD Methods for the Simultaneous Determination of Guanylhydrazone Derivatives Employing a Factorial Design" Molecules 22, no. 9: 1394. https://doi.org/10.3390/molecules22091394