Design, Synthesis and Biological Evaluation of Novel N-Pyridyl-Hydrazone Derivatives as Potential Monoamine Oxidase (MAO) Inhibitors

Abstract
:1. Introduction
2. Results and Discussion
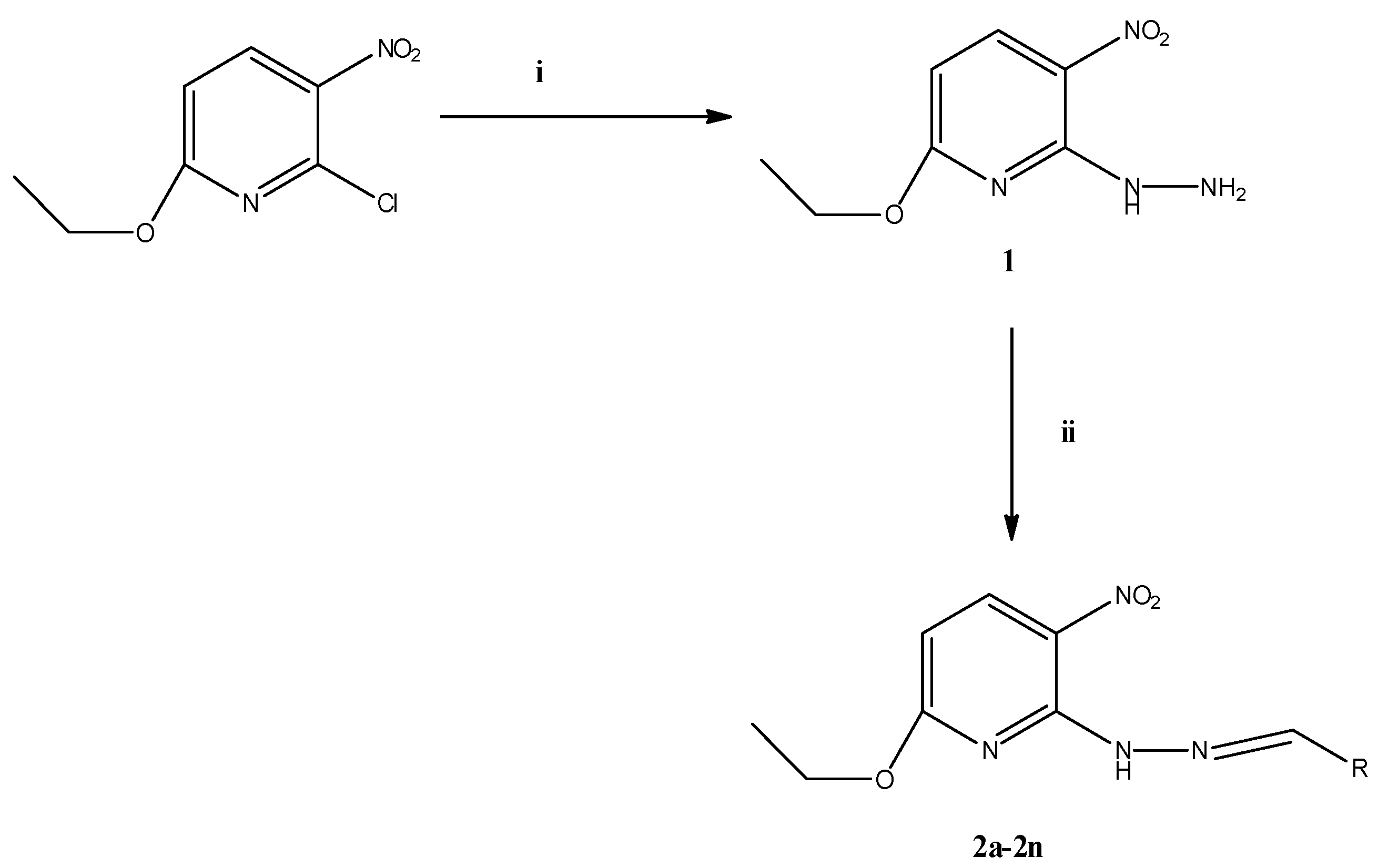
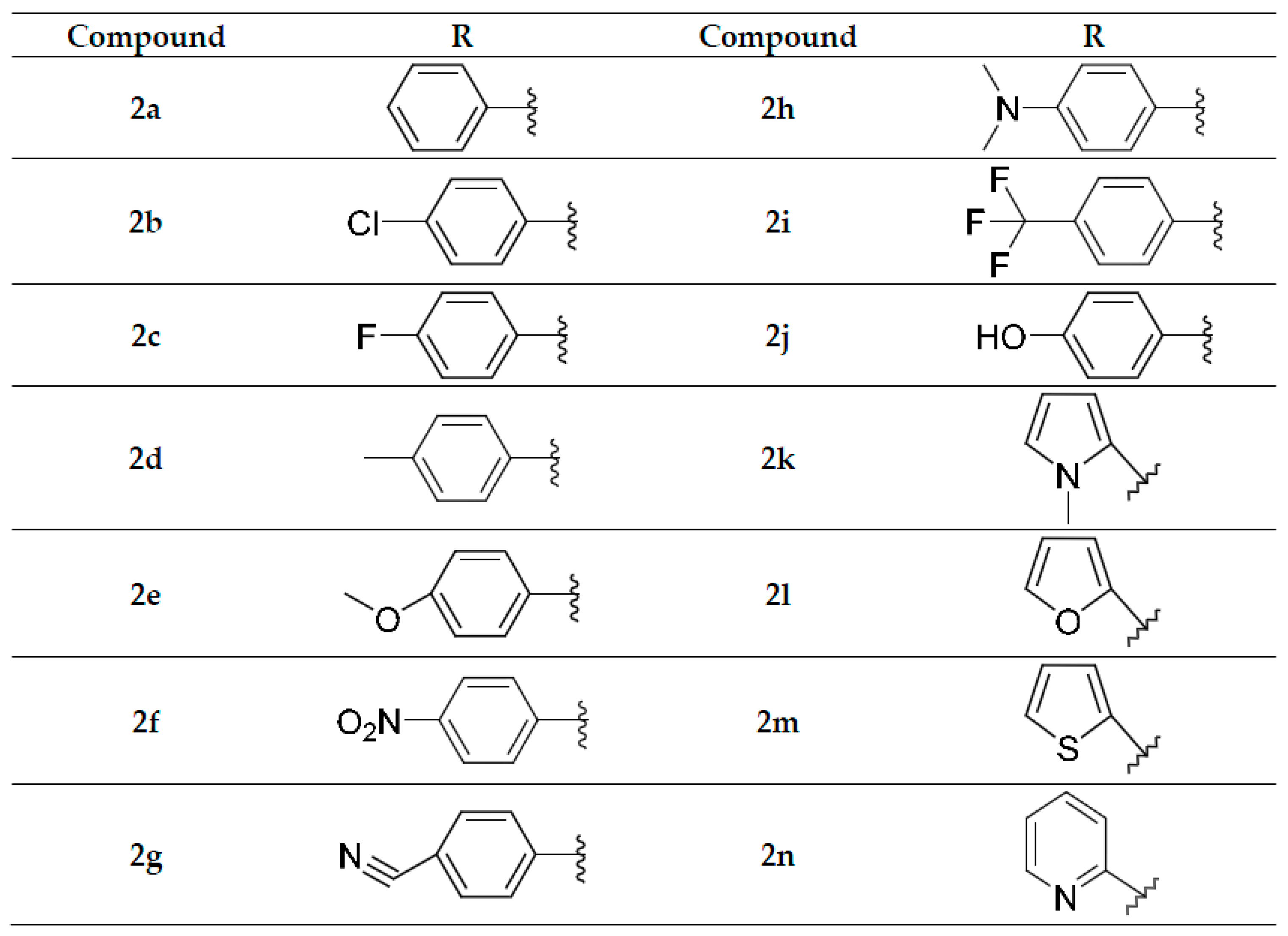
2.1. Chemistry
2.2. Inhibition Potency of the Compounds
2.3. Cytotoxicity Test
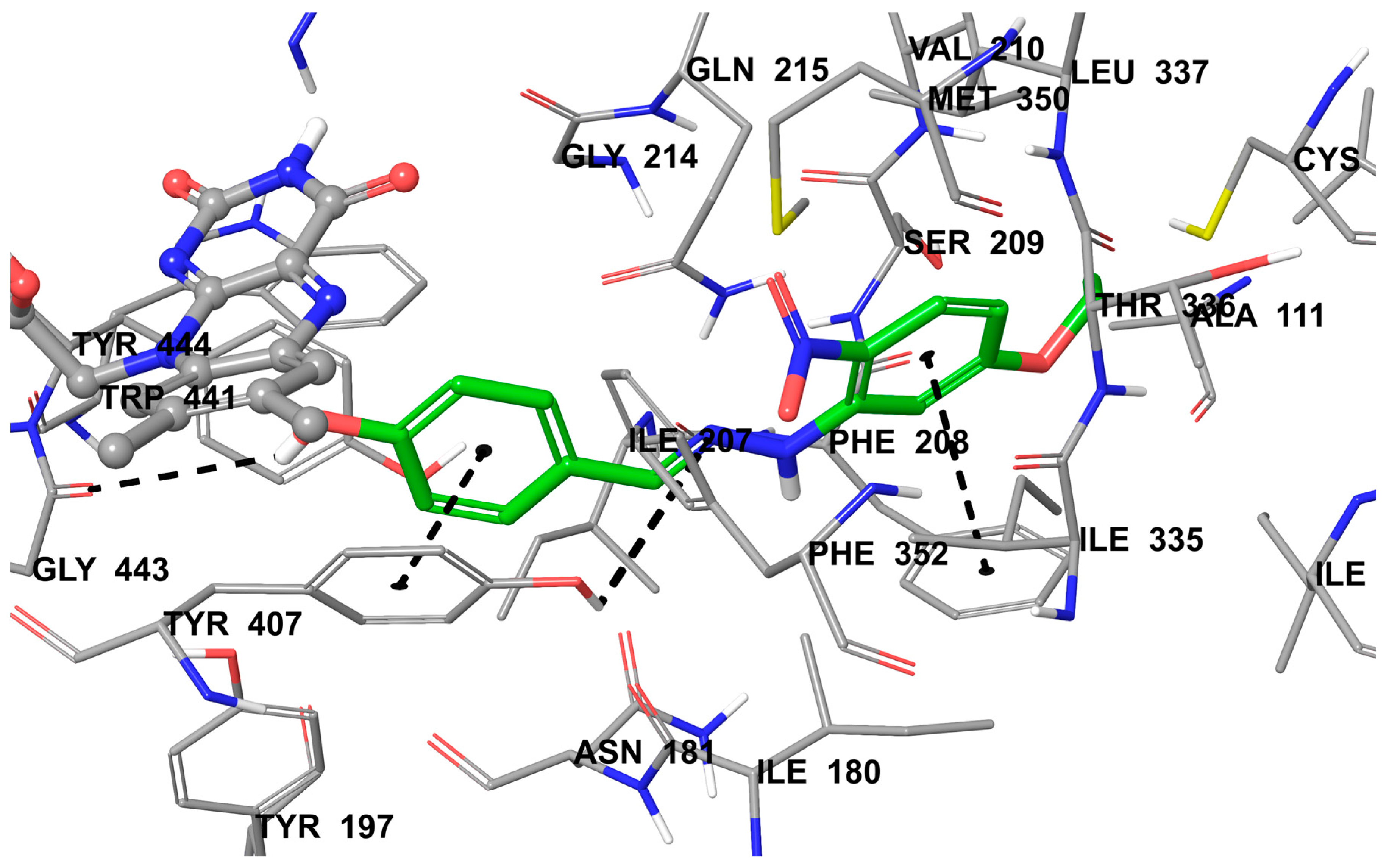
2.4. Molecular Docking Studies
3. Materials and Methods
3.1. Chemistry
3.2. Synthesis of the Compounds
3.2.1. Preparation of 6-Ethoxy-2-hydrazinyl-3-nitropyridine (1)
3.2.2. Preparation of 2-[(2-Substitutedbenzylidene)hydrazinyl]-6-ethoxy-3-nitropyridines 2a–2n
3.3. Biological Activity
3.3.1. MAO Activity Assay
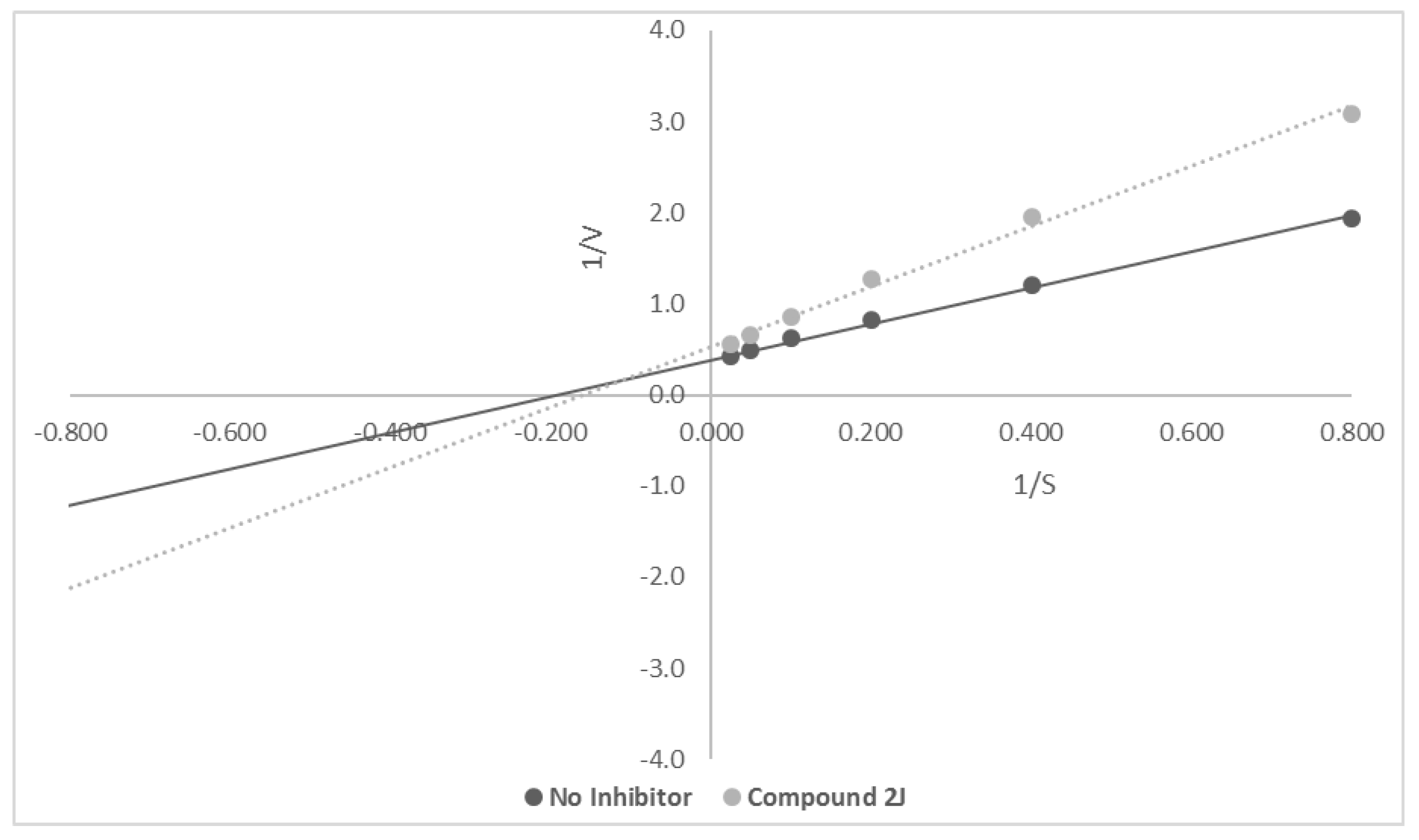
3.3.2. Enzyme Kinetic Studies
3.4. Cytotoxicity Test
3.5. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, A.; Carotti, A.; Barreca, M.L.; Altomare, C. Binding models of reversible inhibitors to type-B monoamine oxidase. J. Comput. Aided Mol. Des. 2002, 16, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Gentili, F.; Pizzinat, N.; Ordener, C.; Marchal-Victorion, S.; Maurel, A.; Hofmann, R.; Renard, P.; Delagrange, P.; Pigini, M.; Parini, A.; et al. 3-[5-(4,5-Dihydro-1H-imidazol-2-yl)-furan-2-yl]phenylamine (Amifuraline), a promising reversible and selective peripheral MAO-A inhibitor. J. Med. Chem. 2006, 49, 5578–5586. [Google Scholar] [CrossRef] [PubMed]
- Sant’ Anna, G.S.; Machado, P.; Sauzem, P.D.; Rosa, F.A.; Rubin, M.A.; Ferreira, J.; Bonacorso, H.G.; Zanatta, N.; Martins, M.A. Ultrasound promoted synthesis of 2-imidazolines in water: A greener approach toward monoamine oxidase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R. Monoamine Oxidases: The biochemistry of the proteins as targets in medicinal chemistry and drug discovery. Curr. Top. Med. Chem. 2012, 12, 2189–2209. [Google Scholar] [CrossRef] [PubMed]
- Secci, D.; Bolasco, A.; Chimenti, P.; Carradori, S. The state of the art of pyrazole derivatives as monoamine oxidase inhibitors and antidepressant/anticonvulsant agents. Curr. Med. Chem. 2011, 18, 5114–5144. [Google Scholar] [CrossRef] [PubMed]
- Christophe, J.; Kutzner, R.; Hguyen-Bui, N.D.; Damien, C.; Chatelain, P.; Gillet, L. Conversion of orally administered 2-n.pentylaminoacetamide into glycinamide and glycine in the rat brain. Life Sci. 1983, 33, 533–541. [Google Scholar] [CrossRef]
- Chimenti, F.; Bolasco, A.; Secci, D.; Chimenti, P.; Granese, A.; Carradori, S.; Yanez, M.; Orallo, F.; Ortuso, F.; Alcaro, S. Investigations on the 2-thiazolylhydrazyne scaffold: Synthesis and molecular modeling of selective human monoamine oxidase inhibitors. Bioorg. Med. Chem. 2010, 18, 5715–5723. [Google Scholar] [CrossRef] [PubMed]
- Secci, D.; Bolasco, A.; Carradori, S.; Ascenzio, M.D.; Nescatelli, R.; Yanez, M. Recent Advances in the Development of Selective Human MAO-B Inhibitors: (Hetero)arlylidene-(4-substituted-thiazol-2-yl) hydrazines. Eur. J. Med. Chem. 2012, 58, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Krejčová, P.; Kučerová, P.; Stafford, G.; Jäger, A.K.; Kubec, R. Antiinflammatory and neurological activity of pyrithione and related sulfur-containing pyridine N-oxides from Persian shallot (Allium stipitatum). J. Ethnopharmacol. 2014, 154, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Park, H.R.; Kim, J.; Kim, T.; Jo, S.; Yeom, M.; Moon, B.; Choo, I.H.; Lee, J.; Lim, E.J.; Park, K.D.; et al. Oxazolopyridines and thiazolopyridines as monoamine oxidase B inhibitors for the treatment of Parkinson’s disease. Bioorg. Med. Chem. 2013, 21, 5480–5487. [Google Scholar] [CrossRef] [PubMed]
- Turan-Zitouni, G.; Yurttaş, L.; Kaplancıklı, Z.A. Synthesis and anti-nociceptive, anti-inflammatory activities of new aroyl propionic acid derivatives including N-acylhydrazone motif. Med. Chem. Res. 2015, 24, 2406–2416. [Google Scholar] [CrossRef]
- Turan-Zitouni, G.; Mehlika Dilek, A.; Özdemir, A.; Demirci, F.; Abu Mohsend, U.; Asım Kaplancıklı, Z. Synthesis and antifungal activity of new hydrazide derivatives. J. Enzyme Inhib. Med. Chem. 2013, 28, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Patila, P.O.; Bari, S.B. Nitrogen heterocycles as potential monoamine oxidase inhibitors: Synthetic aspects. Arab. J. Chem. 2014, 7, 857–884. [Google Scholar] [CrossRef]
- Matsumoto, T.; Suzuki, O.; Furuta, T.; Asai, M.; Kurokawa, Y.; Nimura, Y.; Katsumata, Y.; Takahashi, I. A sensitive fluorometric assay for serum monoamine oxidase with kynuramine as substrate. Clin. Biochem. 1985, 18, 126–129. [Google Scholar] [CrossRef]
- International Organization for Standardization. Biological Evaluation of Medical Devices-Part 5: Tests for in vitro Cytotoxicity ISO-10993-5, 3rd ed.; International Organization for Standardization: Geneva, Switzerland, 2009. [Google Scholar]
- Son, S.-Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed]
- Jhonson, J.A.; Thomas, H.J.; Schaeffer, H.J. Synthesis of potantial Anticancers Agents. XIIL Ribosides of 6-Substituted Purines. J. Am. Chem. Soc. 1958, 80, 699–702. [Google Scholar] [CrossRef]
- Turan-Zitouni, G.; Altıntop, M.D.; Özdemir, A.; Kaplaqncıklı, Z.A.; Dikmen, M. Synthesis of some hydrazine derivatives bearing purine moiety as anticancer agents. Turk. J. Pharm. Sci. 2014, 11, 55–66. [Google Scholar]
- Chaurasiya, N.D.; Ibrahim, M.A.; Muhammad, I.; Walker, L.A.; Tekwani, B.L. Monoamine oxidase inhibitory constituents of propolis: Kinetics and mechanism of inhibition of recombinant human MAO-A and MAO-B. Molecules 2014, 19, 18936–18952. [Google Scholar] [CrossRef] [PubMed]
- Sağlık, B.N.; Ilgın, S.; Özkay, Y. Synthesis of new donepezil analogues and investigation of their effects on cholinesterase enzymes. Eur. J. Med. Chem. 2016, 124, 1026–1040. [Google Scholar] [CrossRef] [PubMed]
- Demir Özkay, Ü.; Can, Ö.D.; Sağlık, B.N.; Acar Çevik, U.; Levent, S.; Özkay, Y.; Ilgın, S.; Atlı, Ö. Design, synthesis, and AChE inhibitory activity of new benzothiazole-piperazines. Bioorg. Med. Chem. Lett. 2016, 26, 5387–5394. [Google Scholar] [CrossRef] [PubMed]
- Can, Ö.D.; Osmaniye, D.; Demir Özkay, Ü.; Sağlık, B.N.; Levent, S.; Ilgın, S.; Baysal, M.; Özkay, Y.; Kaplancıklı, Z.A. MAO enzymes inhibitory activity of new benzimidazole derivatives including hydrazone and propargyl side chains. Eur. J. Med. Chem. 2016, 131, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Can, N.Ö.; Osmaniye, D.; Levent, S.; Sağlık, B.N.; İnci, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Hydrazone Derivatives for MAO Enzymes Inhibitory Activity. Molecules 2017, 22, 1381. [Google Scholar] [CrossRef] [PubMed]
- Karaca Gençer, H.; Acar Çevik, U.; Kaya Çavuşoğlu, B.; Sağlık, B.N.; Levent, S.; Atlı, Ö.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis, and evaluation of novel 2-phenylpropionic acid derivatives as dual COX inhibitory-antibacterial agents. J. Enzyme Inhib. Med. Chem. 2017, 32, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Karaca Gençer, H.; Acar Çevik, U.; Levent, S.; Sağlık, B.N.; Korkut, B.; Özkay, Y.; Ilgın, S.; Öztürk, Y. New Benzimidazole-1,2,4-Triazole Hybrid Compounds: Synthesis, Anticandidal Activity and Cytotoxicity Evaluation. Molecules 2017, 22, 507. [Google Scholar] [CrossRef] [PubMed]
- Maestro, Version 10.6; Schrödinger, LLC: New York, NY, USA, 2016.
- MaestroProtein Preparation WizardSchrödinger, LLC: New York, NY, USA, 2016.
- LigPrep, Version 3.8; Schrödinger, LLC: New York, NY, USA, 2016.
- Glide, Version 7.1; Schrödinger, LLC: New York, NY, USA, 2016.
- Mustafa Toprakçı, Kemal Yelekçi. Docking studies on monoamine oxidase-B inhibitors: Estimation of inhibition constants (Ki) of a series of experimentally tested compounds. Bioorg. Med. Chem. Lett. 2005, 15, 4438–4446. [Google Scholar]
- Gökhan-Kelekçi, N.; Özgün Şimşek, Ö.; Ercan, A.; Yelekçi, K.; Sibel Şahin, Z.; Işık, S.; Uçar, G.; Altan Bilgin, A. Synthesis and molecular modeling of some novel hexahydroindazole derivatives as potent monoamine oxidase inhibitors. Bioorg. Med. Chem. 2009, 17, 6761–6772. [Google Scholar] [CrossRef] [PubMed]
- Evranos-Aksöz, B.; Yabanoğlu-Çiftçi, S.; Uçar, G.; Yelekçi, K.; Ertan, R. Synthesis of some novel hydrazone and 2-pyrazoline derivatives: Monoamine oxidase inhibitory activities and docking studies. Bioorg. Med. Chem. Lett. 2014, 24, 3278–3284. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 2a–2n are available from the authors. Or not available. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MAO-A Inhibition % | MAO-B Inhibition % | ||
|---|---|---|---|---|
| 10−3 M | 10−4 M | 10−3 M | 10−4 M | |
| 2a | 25.63 ± 0.88 | 17.85 ± 0.68 | 0.76 ± 0.03 | 0.58 ± 0.01 |
| 2b | 48.97 ± 1.22 | 0.46 ± 0.01 | 0.43 ± 0.02 | 0.11 ± 0.004 |
| 2c | 51.95 ± 1.02 | 7.09 ± 0.18 | 60.21 ± 1.18 | 0.69 ± 0.03 |
| 2d | 20.14 ± 0.41 | 11.41 ± 0.28 | 0.52 ± 0.02 | 0.18 ± 0.006 |
| 2e | 55.61 ± 1.05 | 13.96 ± 0.41 | 33.09 ± 1.14 | 0.22 ± 0.008 |
| 2f | 64.98 ± 1.23 | 16.02 ± 0.56 | 7.39 ± 0.31 | 0.55 ± 0.02 |
| 2g | 25.86 ± 0.87 | 14.42 ± 0.54 | 0.67 ± 0.03 | 0.27 ± 0.01 |
| 2h | 80.78 ± 1.31 | 16.93 ± 0.49 | 86.28 ± 1.25 | 3.52 ± 0.15 |
| 2i | 55.72 ± 1.26 | 51.70 ± 1.18 | 38.28 ± 1.19 | 29.12 ± 0.99 |
| 2j | 75.67 ± 1.85 | 69.76 ± 1.43 | 63.77 ± 1.28 | 52.69 ± 1.29 |
| 2k | 76.56 ± 1.38 | 66.08 ± 1.47 | 40.91 ± 1.02 | 39.73 ± 1.15 |
| 2l | 67.80 ± 1.44 | 64.43 ± 1.30 | 51.00 ± 1.08 | 43.05 ± 1.06 |
| 2m | 41.42 ± 1.13 | 36.05 ± 1.15 | 34.42 ± 0.88 | 21.17 ± 0.80 |
| 2n | 72.07 ± 1.56 | 56.90 ± 1.50 | 42.00 ± 1.21 | 25.29 ±1 .08 |
| Moclobemide | 94.12 ± 2.76 | 82.14 ± 2.69 | - | - |
| Selegiline | - | - | 98.94 ± 2.06 | 94.73 ± 1.97 |
| Comp. | MAO-A Inhibition % | MAO-A IC50 (µM) | ||||||
|---|---|---|---|---|---|---|---|---|
| 10−3 M | 10−4 M | 10−5 M | 10−6 M | 10−7 M | 10−8 M | 10−9 M | ||
| 2i | 55.72 ± 1.26 | 51.70 ± 1.18 | 31.19 ± 1.20 | 21.08 ± 0.84 | 15.45 ± 0.62 | 12.66 ± 0.43 | 11.57 ± 0.38 | 13.06 |
| 2j | 75.67 ± 1.85 | 69.76 ± 1.43 | 56.12 ± 1.27 | 43.18 ± 1.06 | 38.77 ± 1.07 | 32.68 ± 0.82 | 26.74 ± 0.96 | 6.25 |
| 2k | 76.56 ± 1.38 | 66.08 ± 1.47 | 54.72 ± 1.20 | 36.58 ± 1.08 | 33.23 ± 0.92 | 25.20 ± 0.88 | 22.99 ± 0.89 | 6.12 |
| 2l | 67.80 ± 1.44 | 64.43 ± 1.30 | 42.97 ± 1.02 | 31.01 ± 0.98 | 27.17 ± 0.89 | 19.75 ± 0.41 | 17.88 ± 0.17 | 10.64 |
| 2n | 72.07 ± 1.56 | 56.90 ± 1.50 | 42.13 ± 1.22 | 35.11 ± 1.18 | 24.96 ± 0.83 | 18.02 ± 0.77 | 10.40 ± 0.29 | 9.52 |
| Moclobemide | 94.12 ± 2.76 | 82.14 ± 2.69 | 60.45 ± 2.55 | 36.15 ± 1.98 | 22.13 ± 1.33 | 18.16 ± 0.81 | 14.12 ± 0.72 | 6.06 |
| Comp. | MAO-B Inhibition % | MAO-B IC50 (µM) | ||||||
| 10−3 M | 10−4 M | 10−5 M | 10−6 M | 10−7 M | 10−8 M | 10−9 M | ||
| 2j | 63.77 ± 1.28 | 52.69 ± 1.29 | 40.22 ± 1.12 | 31.60 ± 1.08 | 25.00 ± 0.95 | 21.93 ± 0.82 | 12.48 ± 0.51 | 9.30 |
| Selegiline | 98.94 ± 2.06 | 94.73 ± 1.97 | 86.96 ± 1.82 | 79.22 ± 1.71 | 65.36 ± 1.12 | 43.27 ± 1.02 | 14.70 ± 0.25 | 0.038 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turan-Zitouni, G.; Hussein, W.; Sağlık, B.N.; Tabbi, A.; Korkut, B. Design, Synthesis and Biological Evaluation of Novel N-Pyridyl-Hydrazone Derivatives as Potential Monoamine Oxidase (MAO) Inhibitors. Molecules 2018, 23, 113. https://doi.org/10.3390/molecules23010113
Turan-Zitouni G, Hussein W, Sağlık BN, Tabbi A, Korkut B. Design, Synthesis and Biological Evaluation of Novel N-Pyridyl-Hydrazone Derivatives as Potential Monoamine Oxidase (MAO) Inhibitors. Molecules. 2018; 23(1):113. https://doi.org/10.3390/molecules23010113
Chicago/Turabian StyleTuran-Zitouni, Gülhan, Weiam Hussein, Begüm Nurpelin Sağlık, Aouatef Tabbi, and Büşra Korkut. 2018. "Design, Synthesis and Biological Evaluation of Novel N-Pyridyl-Hydrazone Derivatives as Potential Monoamine Oxidase (MAO) Inhibitors" Molecules 23, no. 1: 113. https://doi.org/10.3390/molecules23010113