Speeding Up the Identification of Cystic Fibrosis Transmembrane Conductance Regulator-Targeted Drugs: An Approach Based on Bioinformatics Strategies and Surface Plasmon Resonance

 ,
,  , , , and
, , , and
Abstract
:
1. Introduction
2. Results
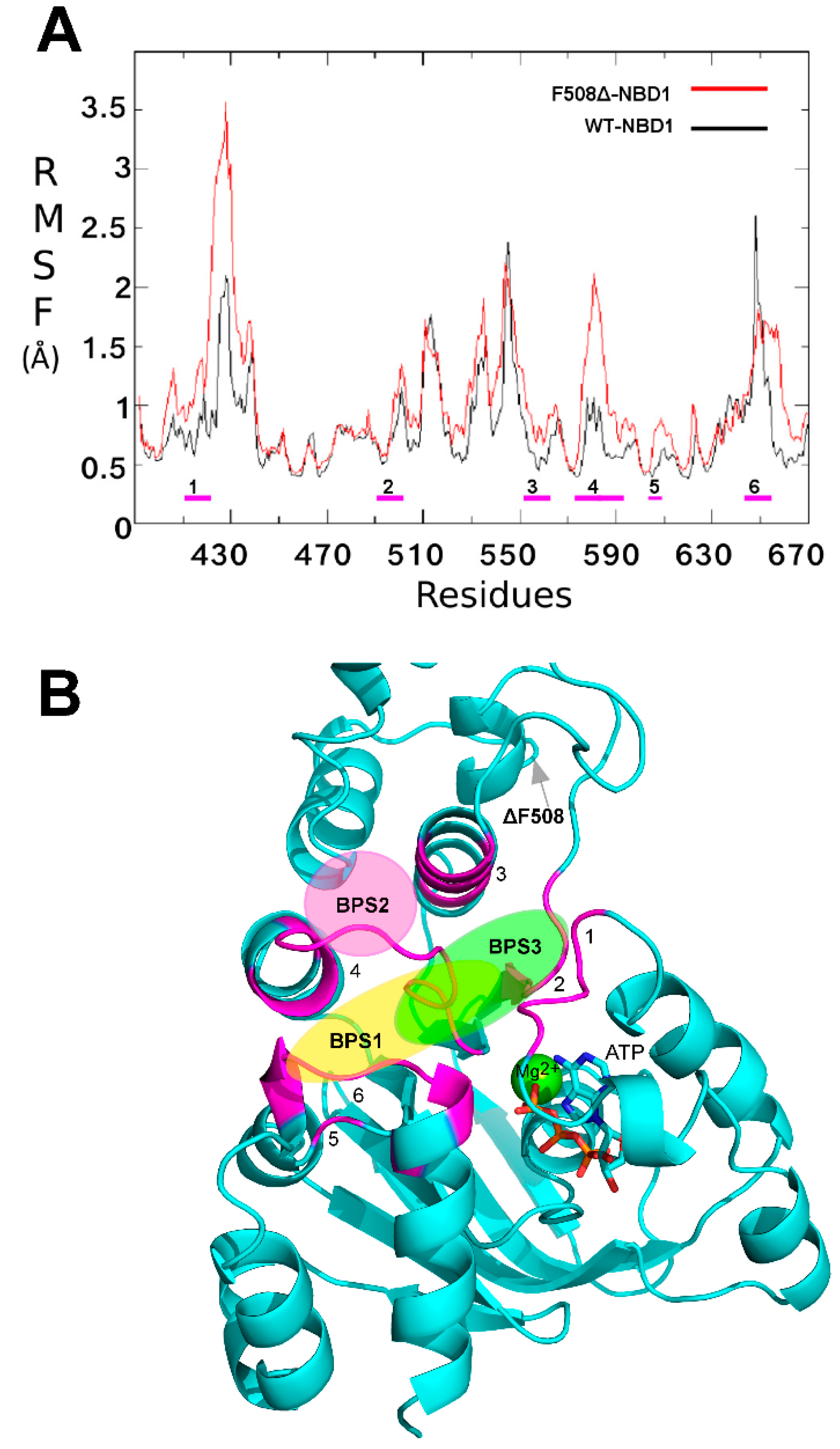
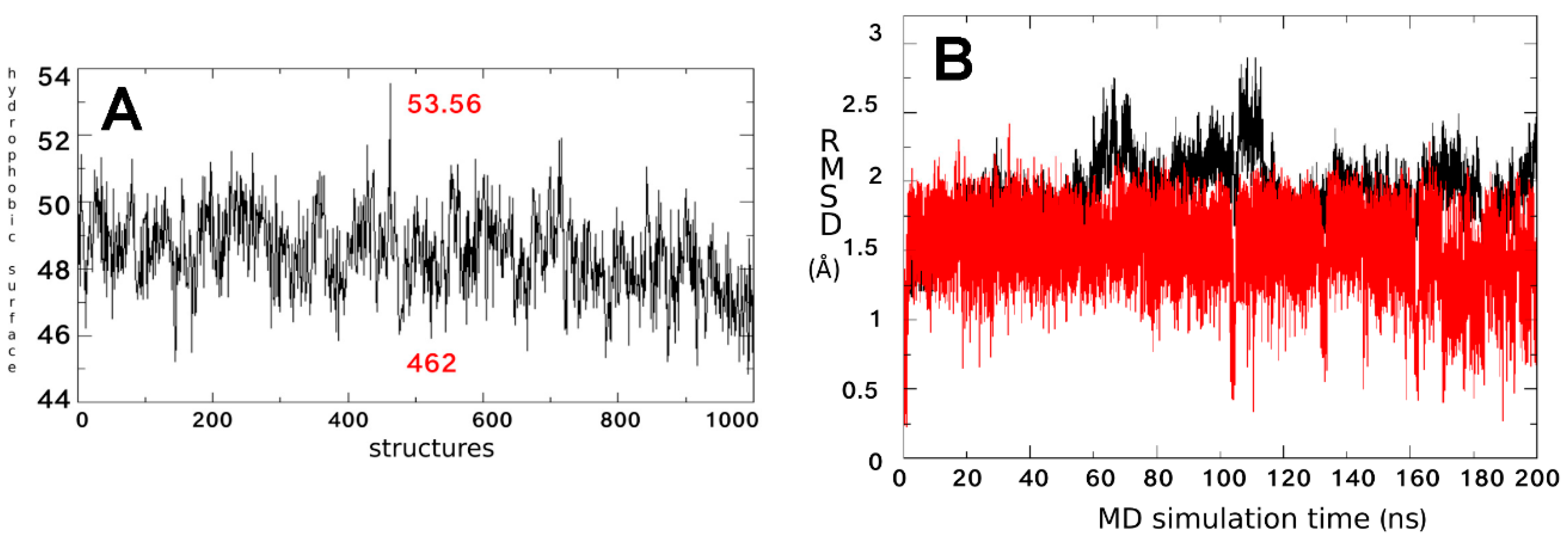
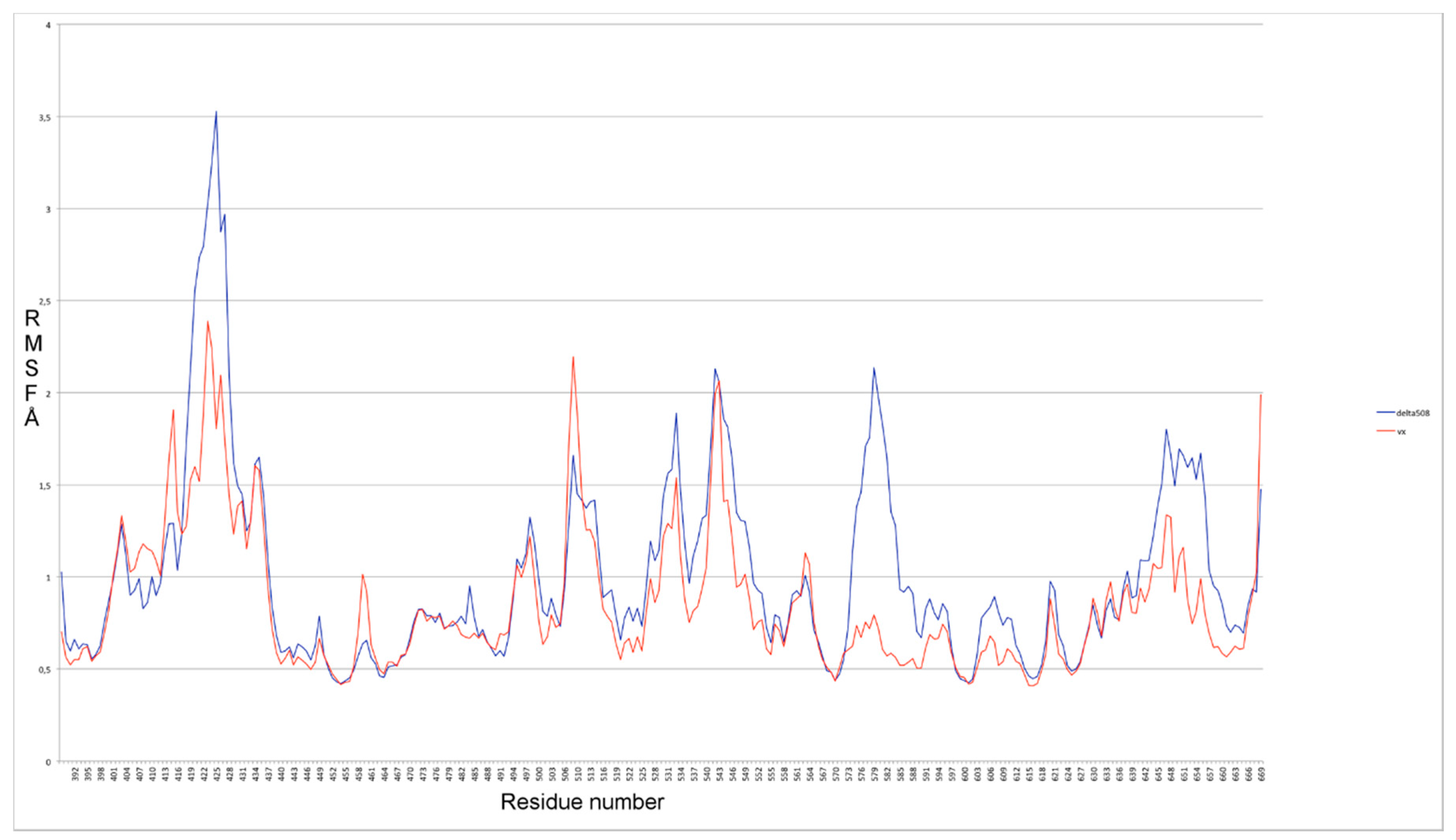
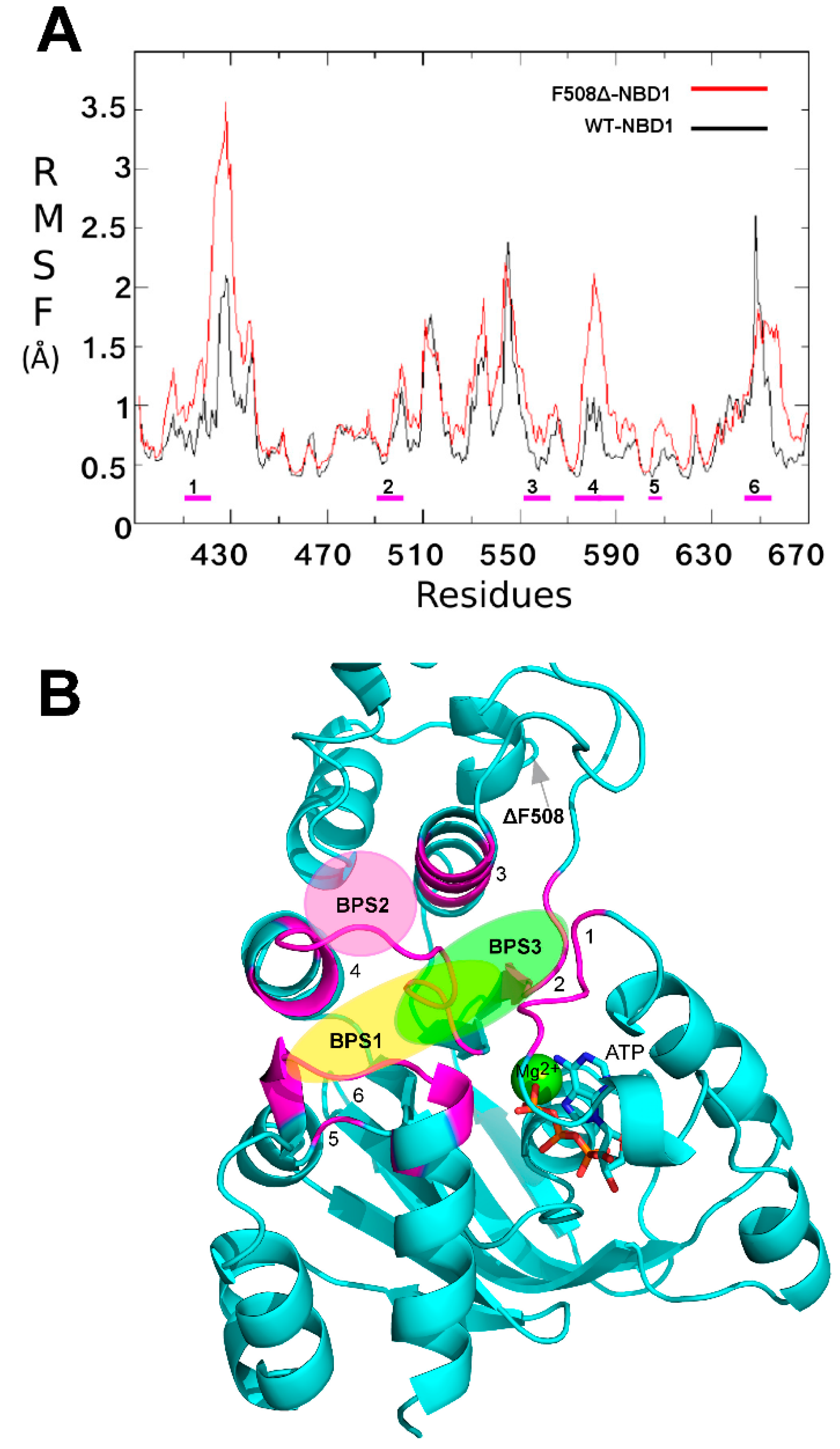
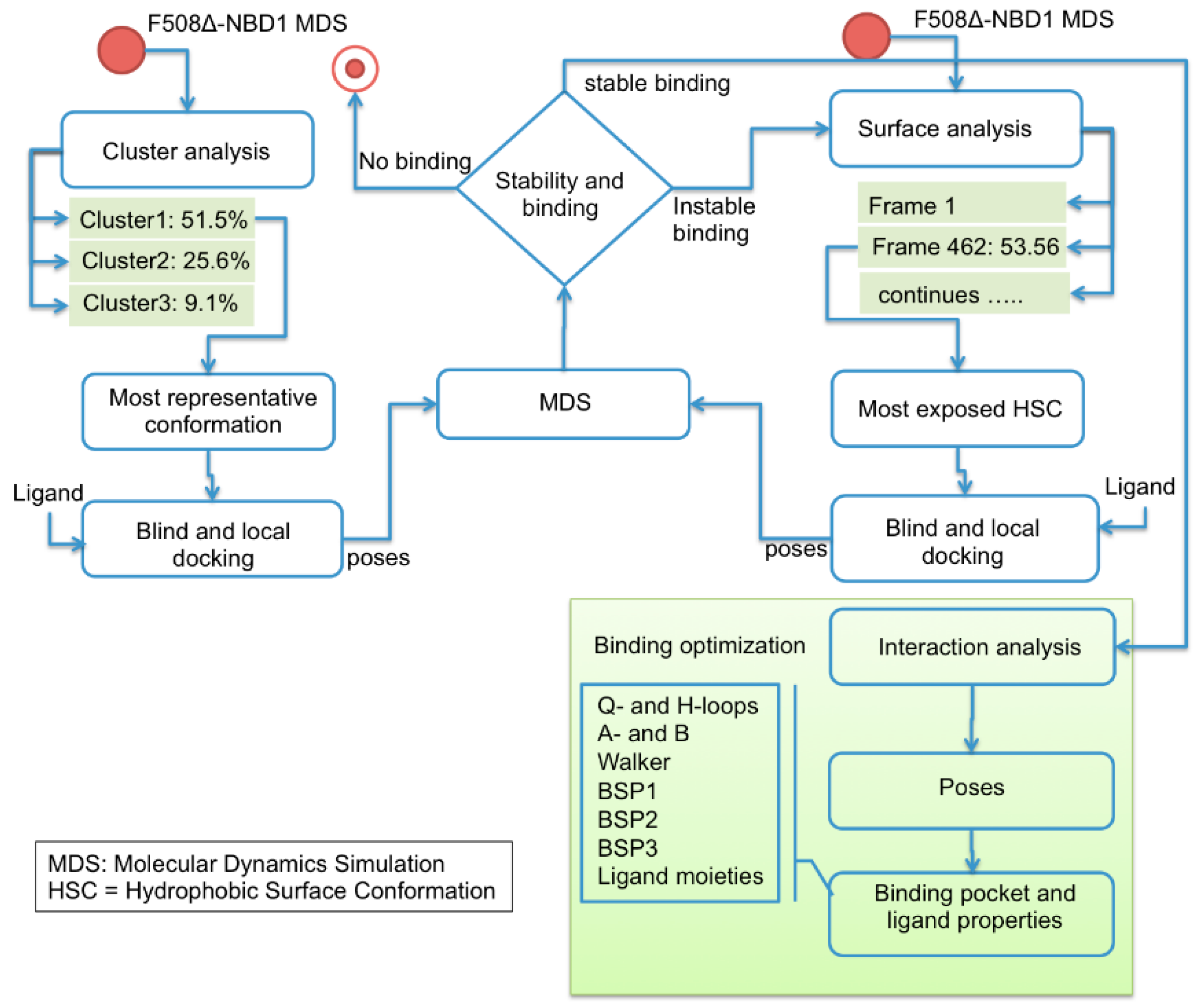
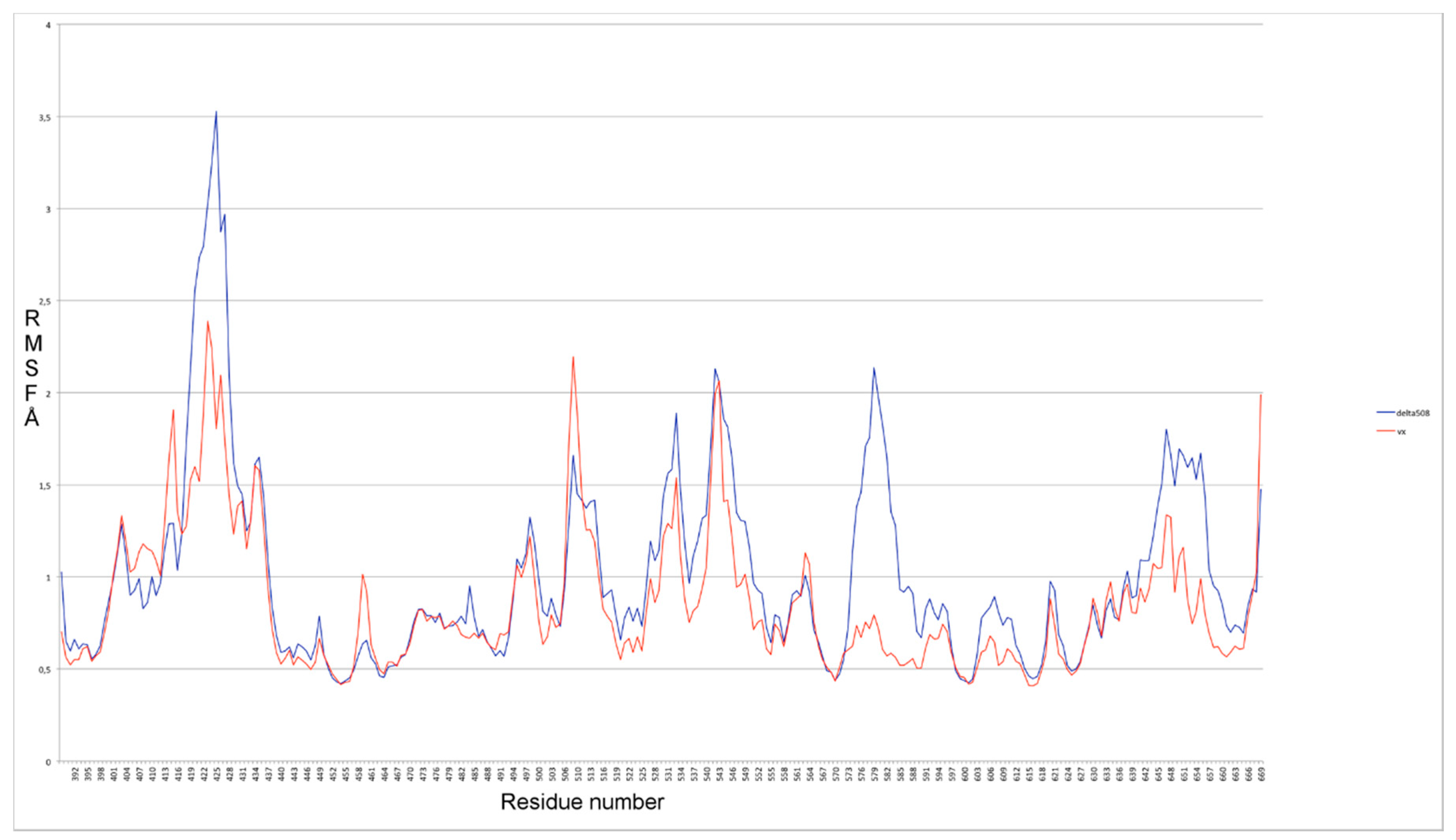
2.1. Characterization of the ΔF508-NBD1 Flexibility by Molecular Dynamics Simulations (MDS)
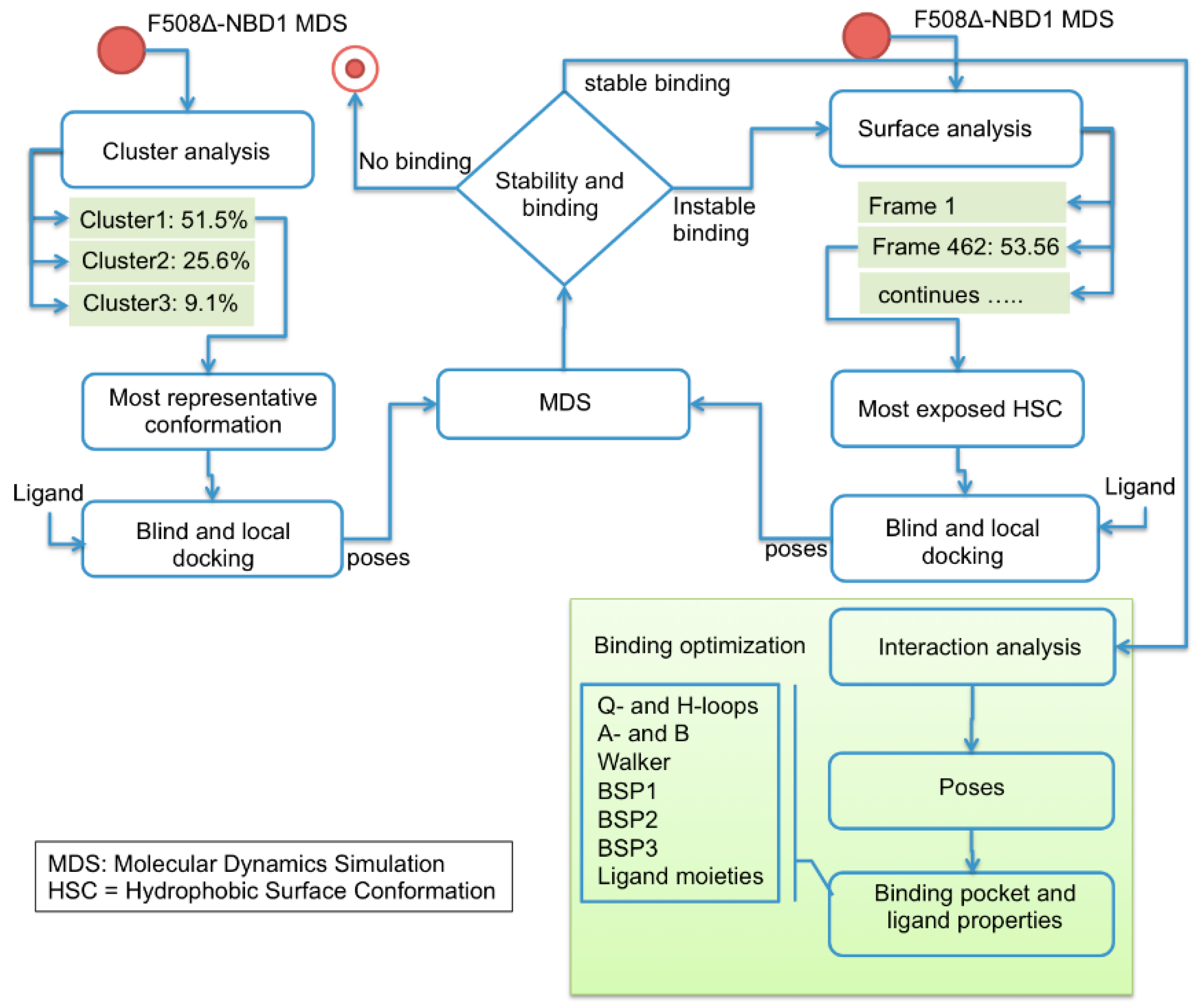
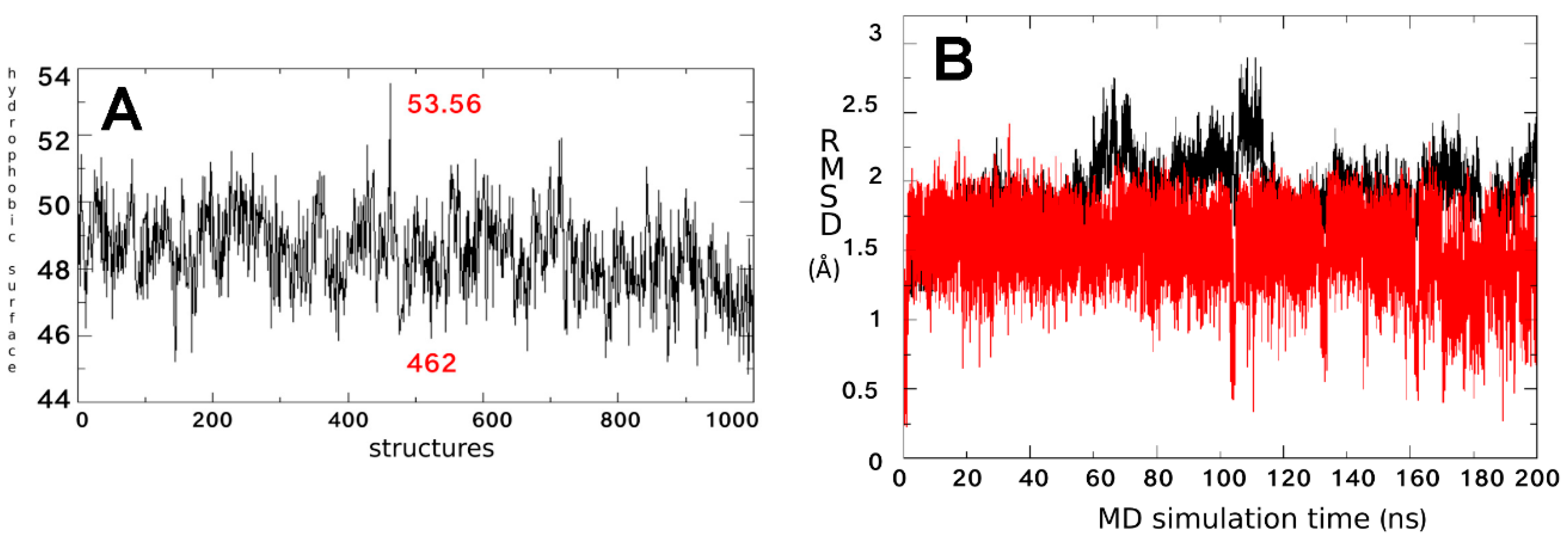
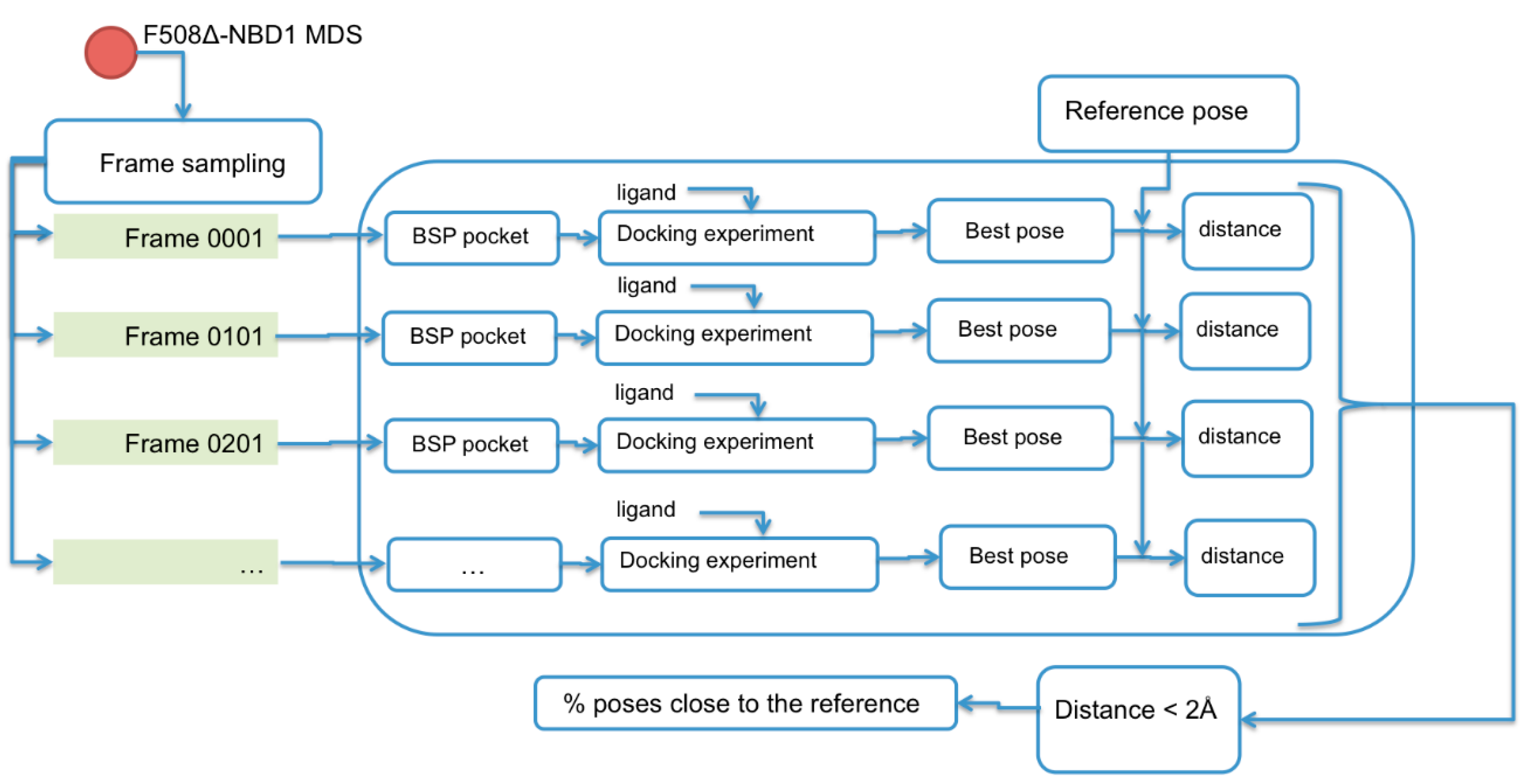
2.2. Binding Sites Prediction
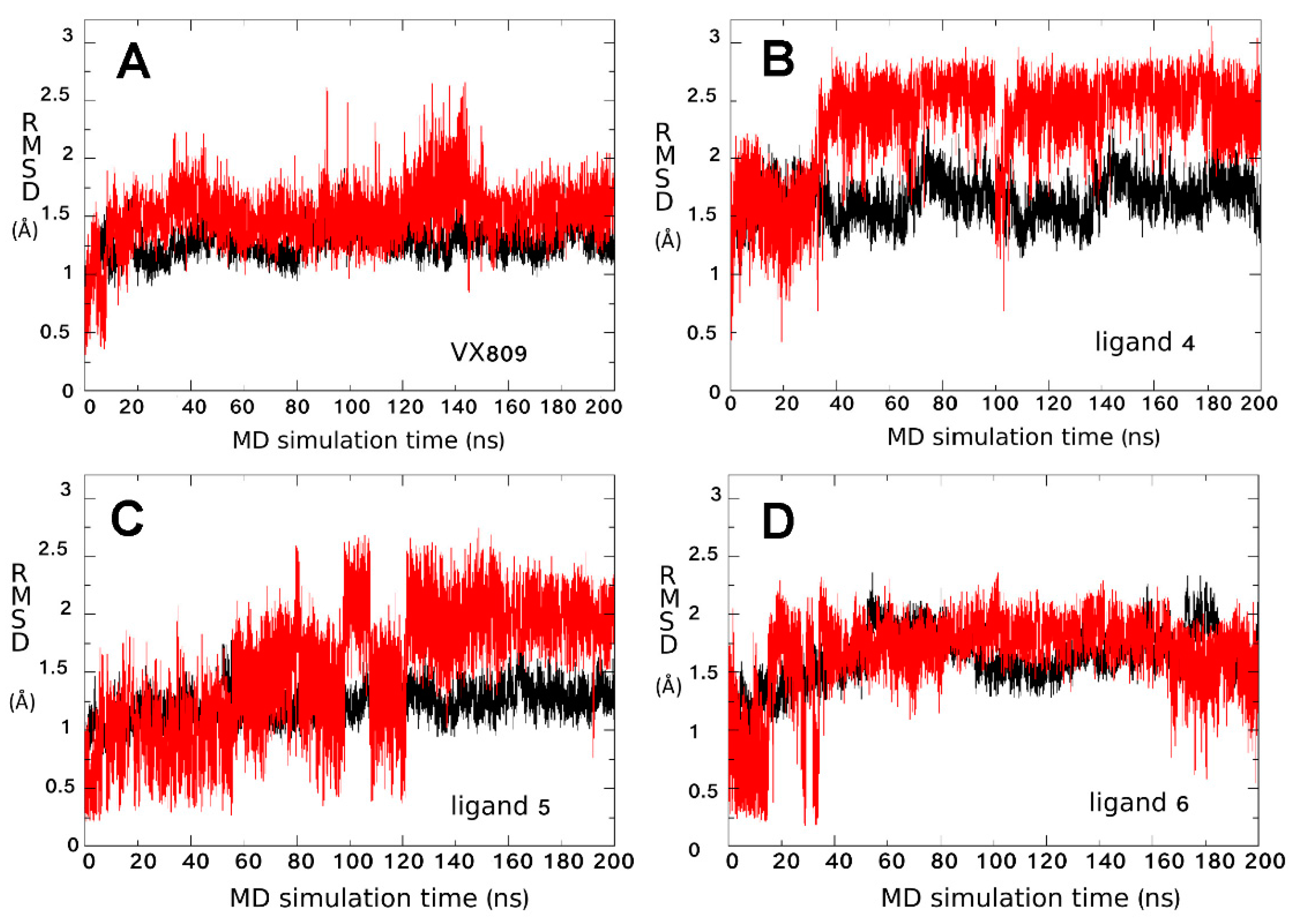
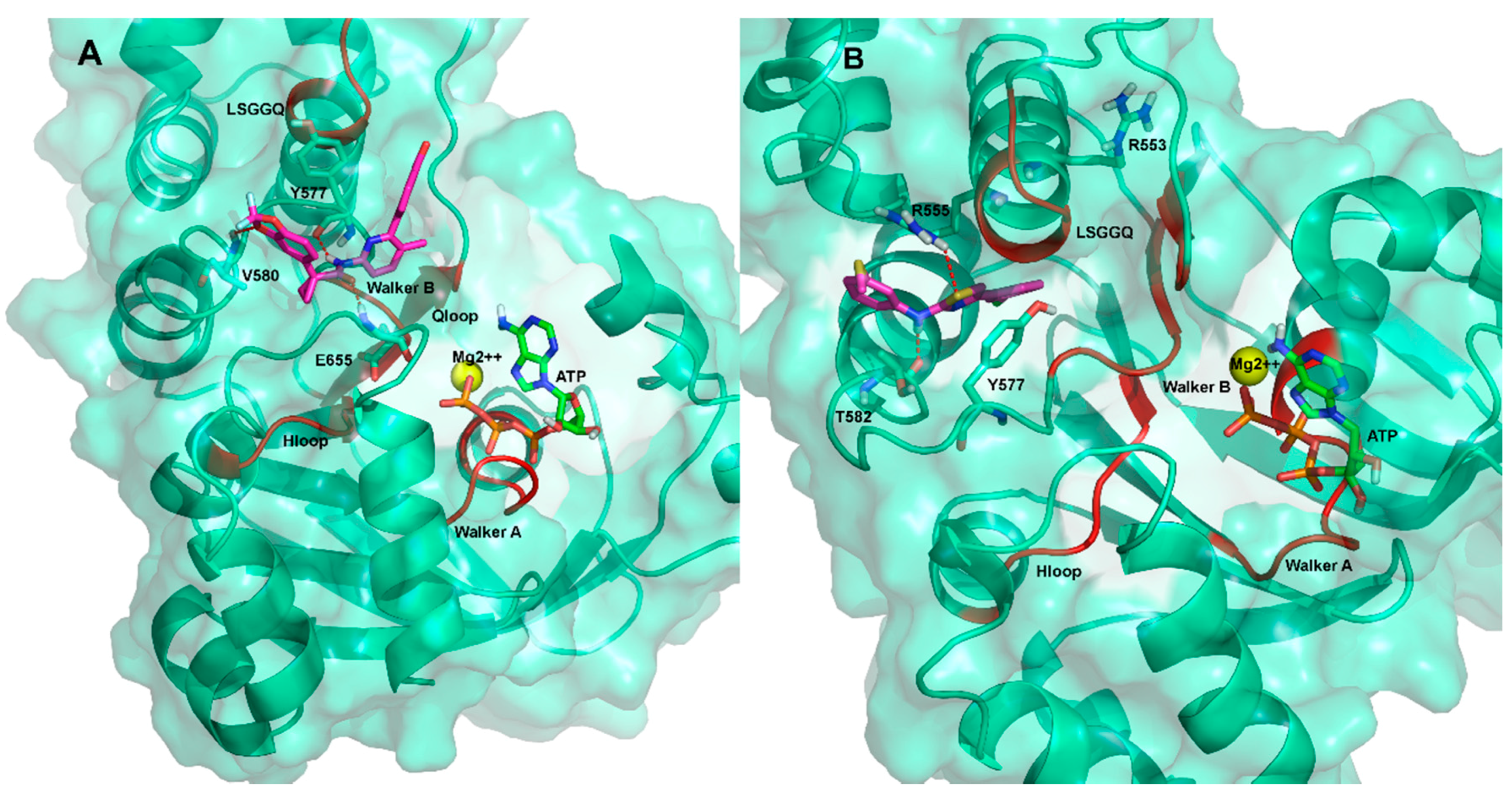
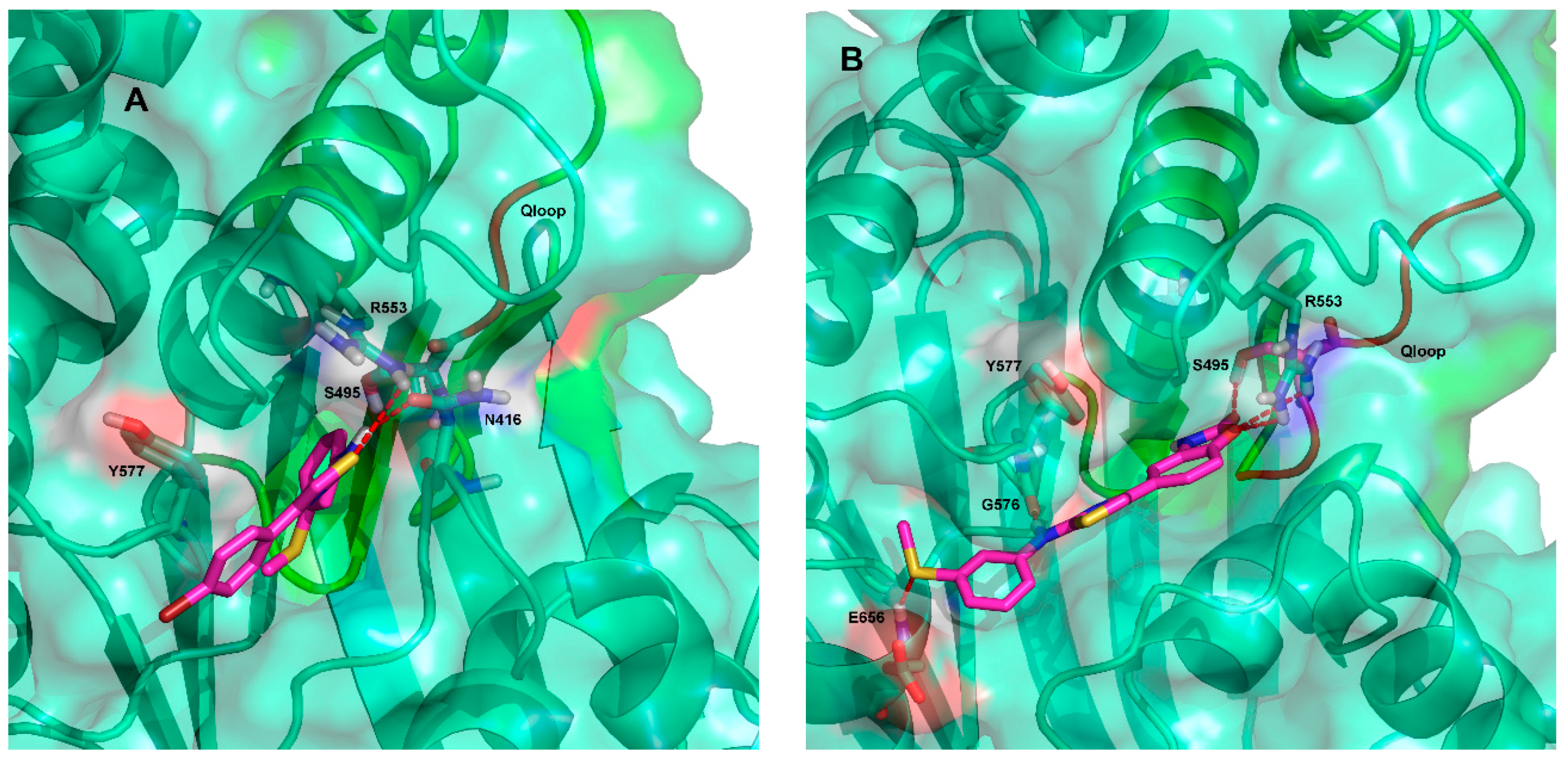
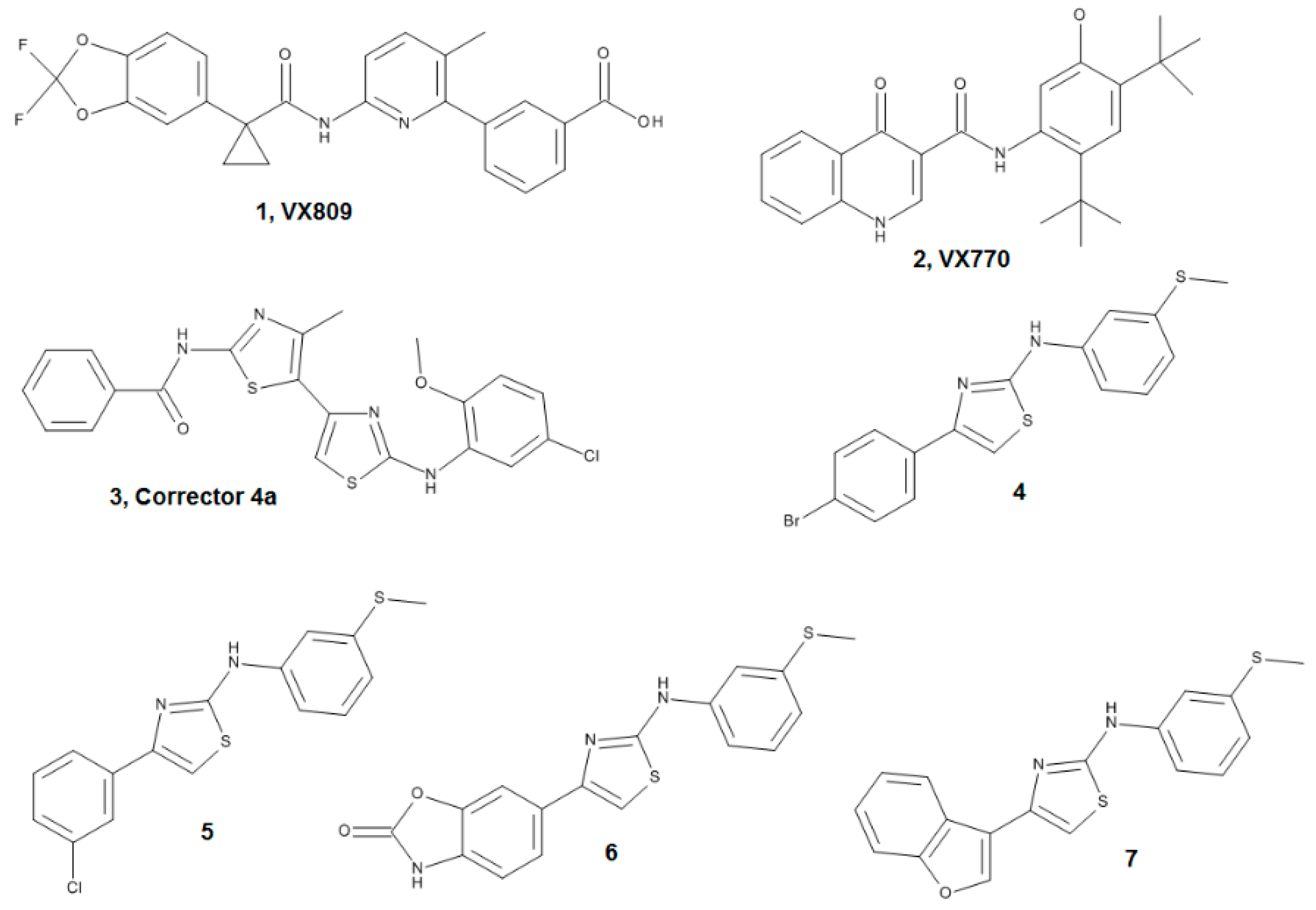
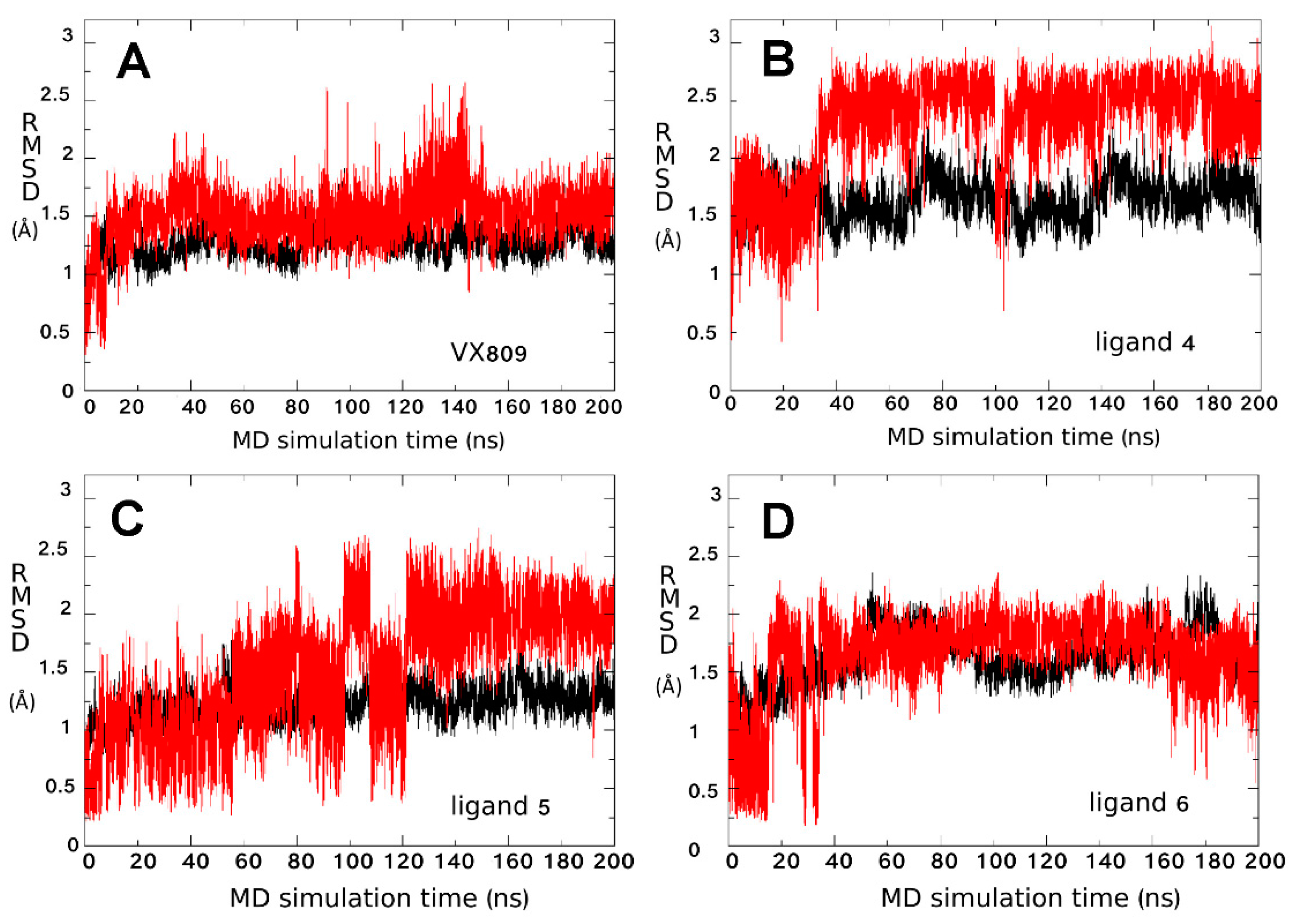
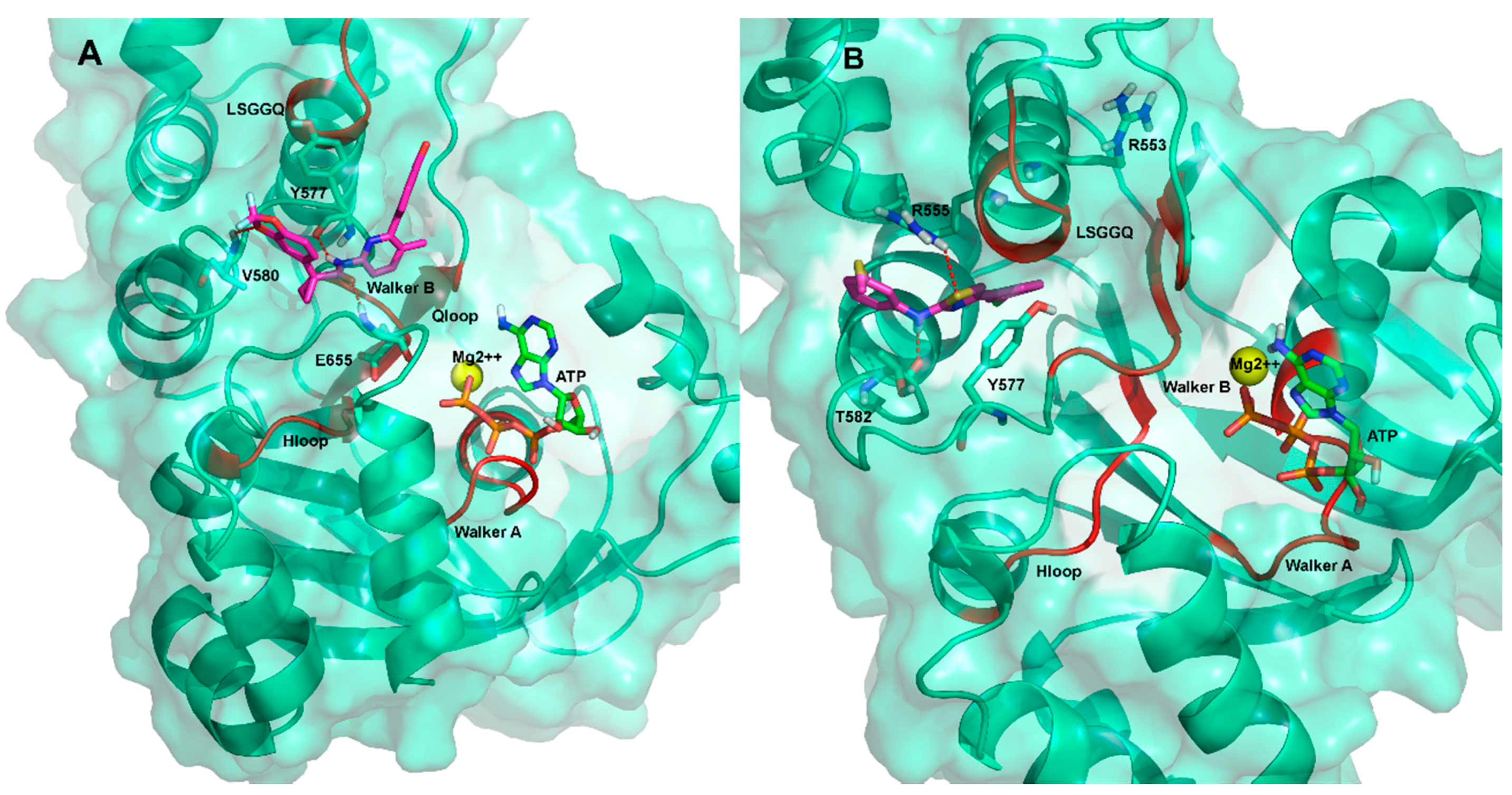
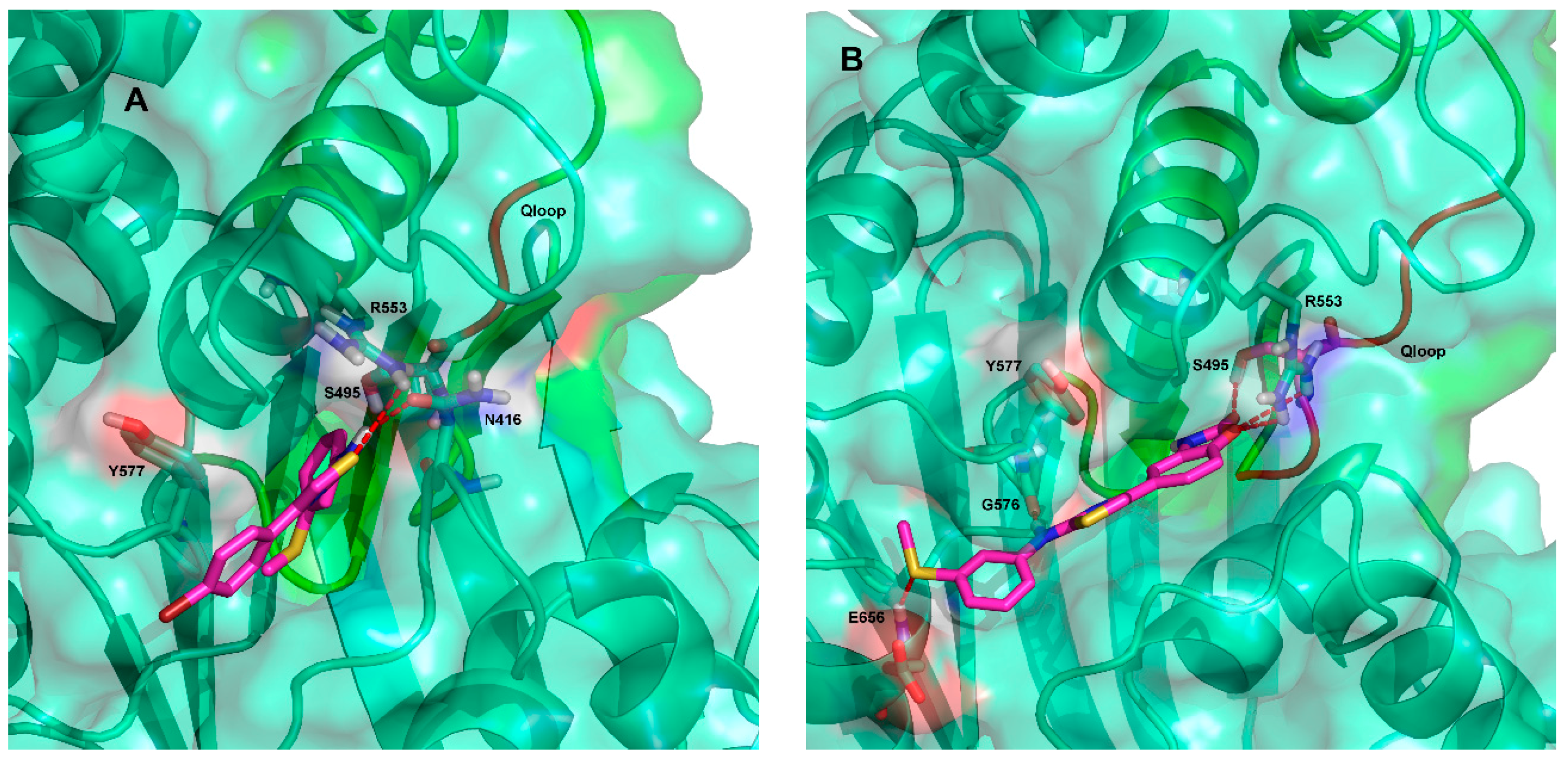
2.3. Ligand Interactions on ΔF508-NBD1
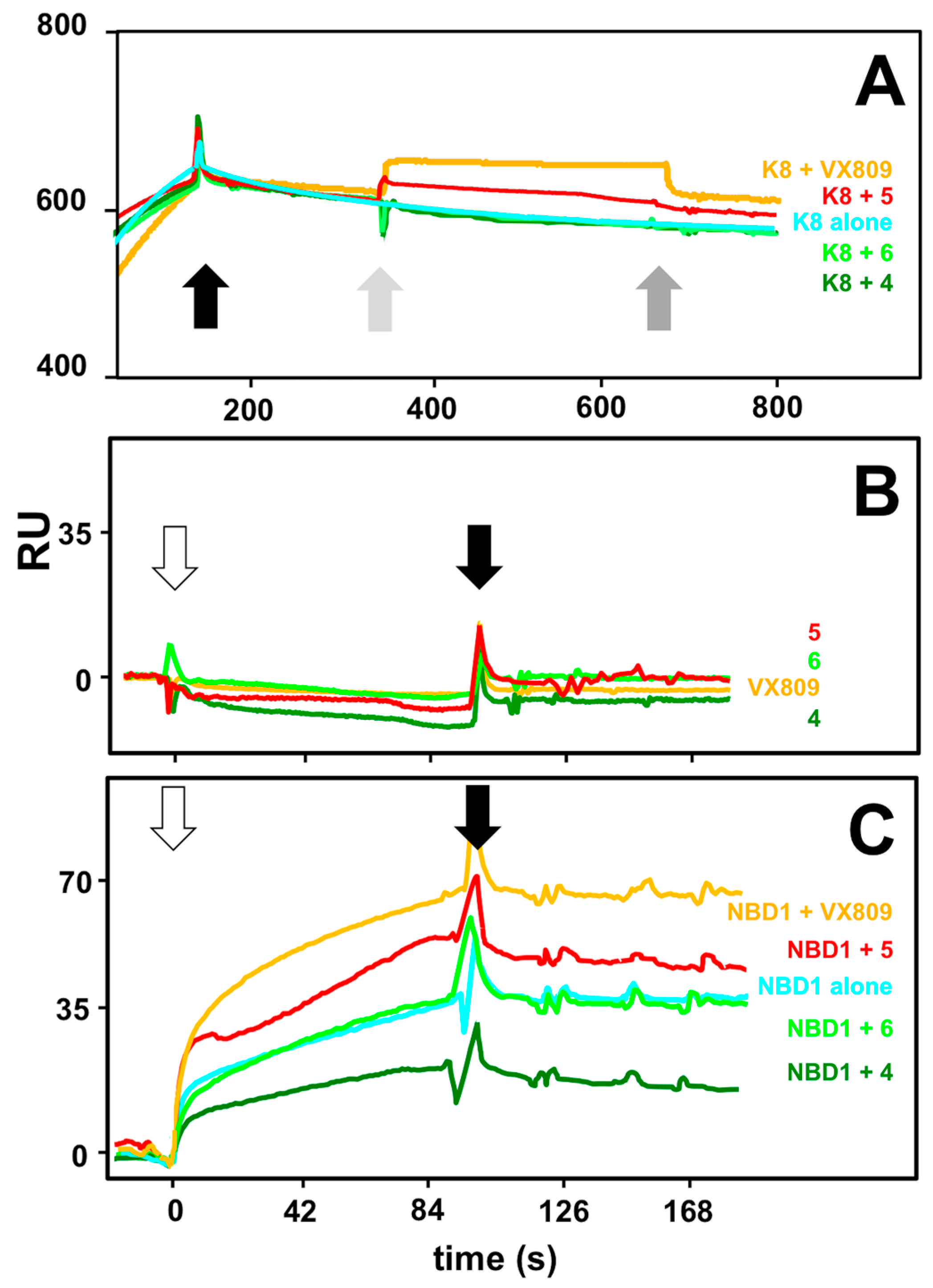
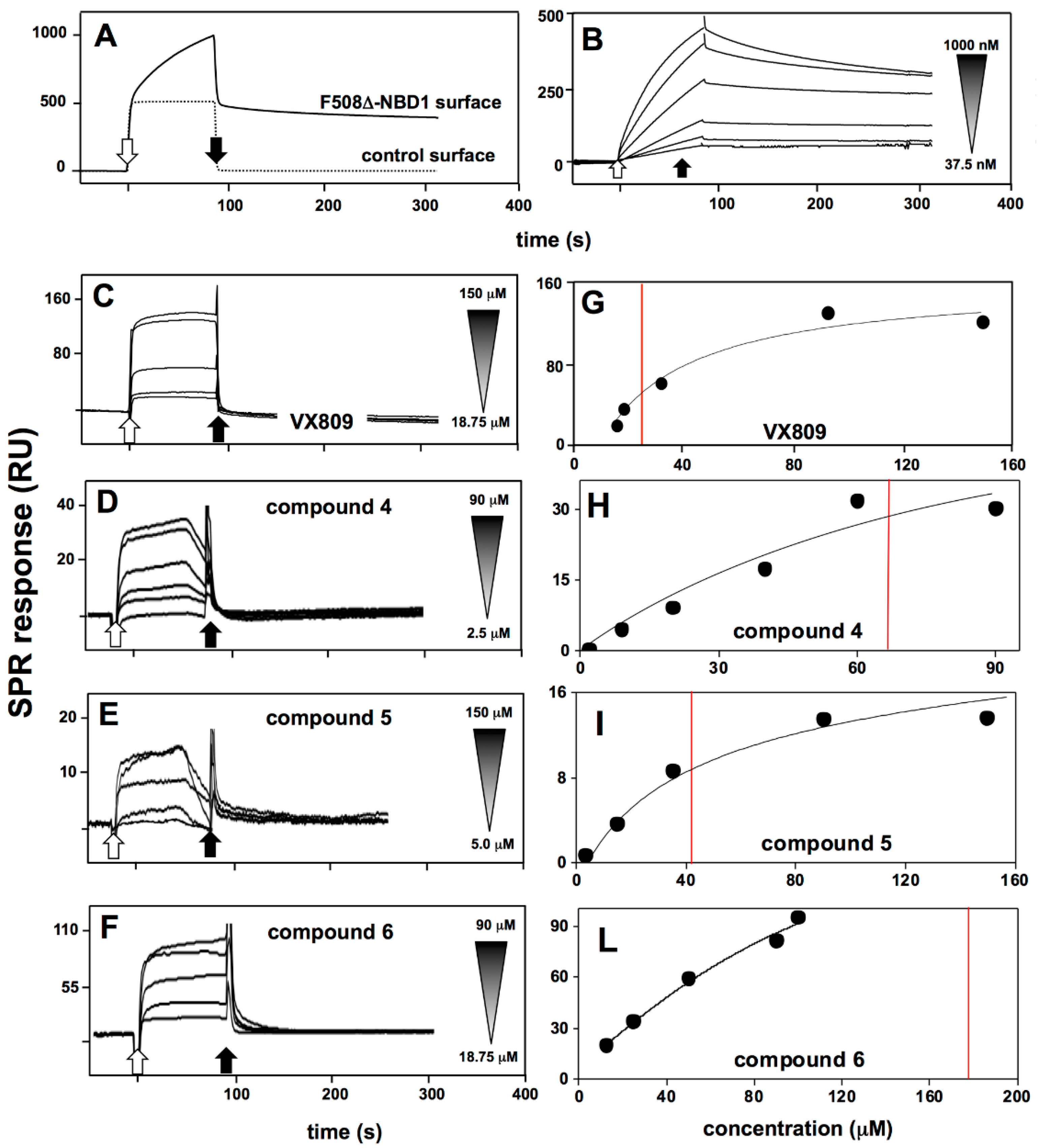
2.4. Interaction Analysis by Surface Plasmon Resonance
3. Discussion
4. Materials and Methods
4.1. Computational Studies
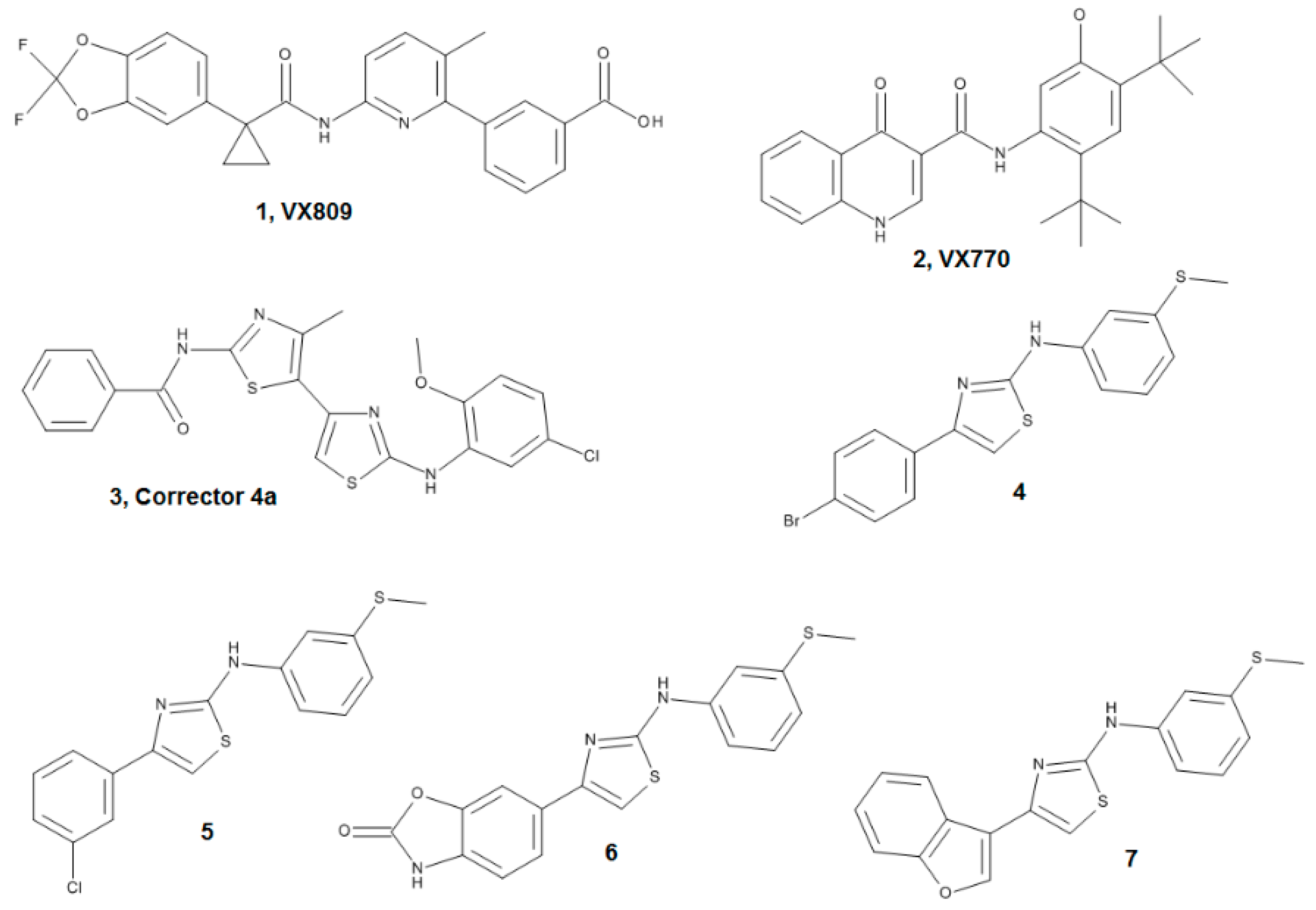
4.1.1. Ligand Dataset
4.1.2. Model Refinement
4.1.3. MDS Simulations
4.1.4. Docking Studies, Ligand-Protein Docking
4.1.5. Docking Studies, Inverse Docking Technique to Calculate Binding Propensity
4.1.6. Trajectory Analysis
4.2. Reagents and Recombinant Proteins
4.3. Surface Plasmon Resonance (SPR) Binding Assay
4.3.1. Direct Binding Analysis
4.3.2. Competition Assays
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rogan, M.P.; Stoltz, D.A.; Hornick, D.B. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest 2011, 139, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Fanen, P.; Wohlhuter-Haddad, A.; Hinzpeter, A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int. J. Biochem. Cell Biol. 2014, 52, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Mendes, F.; Farinha, C.M.; Felicio, V.; Alves, P.C.; Vieira, I.; Amaral, M.D. BAG-1 stabilizes mutant F508del-CFTR in a ubiquitin-like-domain-dependent manner. Cell. Physiol. Biochem. 2012, 30, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Colas, J.; Faure, G.; Saussereau, E.; Trudel, S.; Rabeh, W.M.; Bitam, S.; Guerrera, I.C.; Fritsch, J.; Sermet-Gaudelus, I.; Davezac, N.; et al. Disruption of cytokeratin-8 interaction with F508del-CFTR corrects its functional defect. Hum. Mol. Genet. 2012, 21, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Le Henaff, C.; Faria Da Cunha, M.; Hatton, A.; Tondelier, D.; Marty, C.; Collet, C.; Zarka, M.; Geoffroy, V.; Zatloukal, K.; Laplantine, E.; et al. Genetic deletion of keratin 8 corrects the altered bone formation and osteopenia in a mouse model of cystic fibrosis. Hum. Mol. Genet. 2016, 25, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Rivas Caldas, R.; Boisrame, S. Upper aero-digestive contamination by Pseudomonas aeruginosa and implications in Cystic Fibrosis. J. Cyst. Fibros. 2015, 14, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Scott-Ward, T.S.; Amaral, M.D. Deletion of Phe508 in the first nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator increases its affinity for the heat shock cognate 70 chaperone. FEBS J. 2009, 276, 7097–7109. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; Farinha, C.M. Rescuing mutant CFTR: A multi-task approach to a better outcome in treating cystic fibrosis. Curr. Pharm. Des. 2013, 19, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef] [PubMed]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Aleksandrov, A.A.; An, J.; Cui, L.; Yang, Z.; Brouillette, C.G.; Riordan, J.R. Restoration of NBD1 thermal stability is necessary and sufficient to correct F508 CFTR folding and assembly. J. Mol. Biol. 2015, 427, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Csanady, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.P.; Dawson, J.E.; Chong, P.A.; Yang, Z.; Millen, L.; Thomas, P.J.; Brouillette, C.G.; Forman-Kay, J.D. Direct Binding of the Corrector VX-809 to Human CFTR NBD1: Evidence of an Allosteric Coupling between the Binding Site and the NBD1:CL4 Interface. Mol. Pharmacol. 2017, 92, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Sampson, H.M.; Robert, R.; Liao, J.; Matthes, E.; Carlile, G.W.; Hanrahan, J.W.; Thomas, D.Y. Identification of a NBD1-binding pharmacological chaperone that corrects the trafficking defect of F508del-CFTR. Chem. Biol. 2011, 18, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.A.; Wang, C.; Zhao, X.; Hamuro, Y.; Conners, K.; Kearins, M.C.; Lu, F.; Sauder, J.M.; Molnar, K.S.; Coales, S.J.; et al. Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J. Mol. Biol. 2010, 396, 406–430. [Google Scholar] [CrossRef] [PubMed]
- Tordai, H.; Leveles, I.; Hegedus, T. Molecular dynamics of the cryo-EM CFTR structure. Biochem. Biophys. Res. Commun. 2017, 491, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.M.; Weiwer, M.; Koehler, A.N. Unbiased binding assays for discovering small-molecule probes and drugs. Bioorg. Med. Chem. 2012, 20, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Presta, M. Angiogenic growth factors interactome and drug discovery: The contribution of surface plasmon resonance. Cytokine Growth Factor Rev. 2015, 26, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Bugatti, A.; Mitola, S.; Leali, D.; Bergese, P.; Depero, L.E.; Presta, M. Exploiting Surface Plasmon Resonance (SPR) Technology for the Identification of Fibroblast Growth Factor-2 (FGF2) Antagonists Endowed with Antiangiogenic Activity. Sensors 2009, 9, 6471–6503. [Google Scholar] [CrossRef] [PubMed]
- Eckford, P.D.; Li, C.; Ramjeesingh, M.; Bear, C.E. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 2012, 287, 36639–36649. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Caci, E.; Millo, E.; Armirotti, A.; Damonte, G.; Zegarra-Moran, O.; Galietta, L.J. Dual activity of aminoarylthiazoles on the trafficking and gating defects of the cystic fibrosis transmembrane conductance regulator chloride channel caused by cystic fibrosis mutations. J. Biol. Chem. 2011, 286, 15215–15226. [Google Scholar] [CrossRef] [PubMed]
- Kim Chiaw, P.; Eckford, P.D.; Bear, C.E. Insights into the mechanisms underlying CFTR channel activity, the molecular basis for cystic fibrosis and strategies for therapy. Essays Biochem. 2011, 50, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Lukacs, G.L.; Du, K.; Caci, E.; Zegarra-Moran, O.; Galietta, L.J.; Verkman, A.S. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Investig. 2005, 115, 2564–2571. [Google Scholar] [CrossRef] [PubMed]
- Pesce, E.; Bellotti, M.; Liessi, N.; Guariento, S.; Damonte, G.; Cichero, E.; Galatini, A.; Salis, A.; Gianotti, A.; Pedemonte, N.; et al. Synthesis and structure-activity relationship of aminoarylthiazole derivatives as correctors of the chloride transport defect in cystic fibrosis. Eur. J. Med. Chem. 2015, 99, 14–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Protasevich, I.; Yang, Z.; Seehausen, D.; Skalak, T.; Zhao, X.; Atwell, S.; Spencer Emtage, J.; Wetmore, D.R.; Brouillette, C.G.; et al. Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci. 2010, 19, 1932–1947. [Google Scholar] [CrossRef] [PubMed]
- Protasevich, I.; Yang, Z.; Wang, C.; Atwell, S.; Zhao, X.; Emtage, S.; Wetmore, D.; Hunt, J.F.; Brouillette, C.G. Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 2010, 19, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Rabeh, W.M.; Bossard, F.; Xu, H.; Okiyoneda, T.; Bagdany, M.; Mulvihill, C.M.; Du, K.; di Bernardo, S.; Liu, Y.; Konermann, L.; et al. Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell 2012, 148, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.P.; Chong, P.A.; Protasevich, I.I.; Vernon, R.; Noy, E.; Bihler, H.; An, J.L.; Kalid, O.; Sela-Culang, I.; Mense, M.; et al. Conformational changes relevant to channel activity and folding within the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2012, 287, 28480–28494. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, G.; Zielenkiewicz, P. DeltaF508 mutation increases conformational flexibility of CFTR protein. J. Cyst. Fibros. 2008, 7, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, I.; Hoffmann, B.; Lehn, P.; Mornon, J.P. Molecular modelling and molecular dynamics of CFTR. Cell. Mol. Life Sci. 2017, 74, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Kim Chiaw, P.; Huan, L.J.; Gagnon, S.; Ly, D.; Sweezey, N.; Rotin, D.; Deber, C.M.; Bear, C.E. Functional rescue of DeltaF508-CFTR by peptides designed to mimic sorting motifs. Chem. Biol. 2009, 16, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Odolczyk, N.; Fritsch, J.; Norez, C.; Servel, N.; da Cunha, M.F.; Bitam, S.; Kupniewska, A.; Wiszniewski, L.; Colas, J.; Tarnowski, K.; et al. Discovery of novel potent DeltaF508-CFTR correctors that target the nucleotide binding domain. EMBO Mol. Med. 2013, 5, 1484–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhenin, M.; Noy, E.; Senderowitz, H. REMD Simulations Reveal the Dynamic Profile and Mechanism of Action of Deleterious, Rescuing, and Stabilizing Perturbations to NBD1 from CFTR. J. Chem. Inf. Model. 2015, 55, 2349–2364. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, C.; Russo, S.; Incerti, M.; Bugatti, A.; Vacondio, F.; Barocelli, E.; Mor, M.; Pala, D.; Hassan-Mohamed, I.; Gioiello, A.; et al. Biochemical characterization of EphA2 antagonists with improved physico-chemical properties by cell-based assays and surface plasmon resonance analysis. Biochem. Pharmacol. 2016, 99, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.W.; Okiyoneda, T.; Balch, W.E.; Braakman, I.; Brodsky, J.L.; Guggino, W.B.; Penland, C.M.; Pollard, H.B.; Sorscher, E.J.; Skach, W.R.; et al. CFTR Folding Consortium: Methods available for studies of CFTR folding and correction. Methods Mol. Biol. 2011, 742, 335–353. [Google Scholar] [PubMed]
- Moran, O.; Galietta, L.J.; Zegarra-Moran, O. Binding site of activators of the cystic fibrosis transmembrane conductance regulator in the nucleotide binding domains. Cell. Mol. Life Sci. 2005, 62, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.A.; Farber, P.J.; Vernon, R.M.; Hudson, R.P.; Mittermaier, A.K.; Forman-Kay, J.D. Deletion of Phenylalanine 508 in the First Nucleotide-binding Domain of the Cystic Fibrosis Transmembrane Conductance Regulator Increases Conformational Exchange and Inhibits Dimerization. J. Biol. Chem. 2015, 290, 22862–22878. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.A.; Buchanan, S.G.; Burley, S.K.; Conners, K.; Dickey, M.; Dorwart, M.; Fowler, R.; Gao, X.; Guggino, W.B.; Hendrickson, W.A.; et al. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004, 23, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.L.; Schmidt, A.; Li, Q.; Nuvaga, E.; Barrett, T.; Bridges, R.J.; Feranchak, A.P.; Brautigam, C.A.; Thomas, P.J. Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell 2012, 148, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. In Functional Genomics; Kaufmann, M., Klinger, C., Savelsbergh, A., Eds.; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; Volume 1654, pp. 39–54. [Google Scholar] [CrossRef]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Shrake, A.; Rupley, J.A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef]
- Premchandar, A.; Kupniewska, A.; Bonna, A.; Faure, G.; Fraczyk, T.; Roldan, A.; Hoffmann, B.; Faria da Cunha, M.; Herrmann, H.; Lukacs, G.L.; et al. New insights into interactions between the nucleotide-binding domain of CFTR and keratin 8. Protein Sci. 2017, 26, 343–354. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of compounds 4–7 can be available from the author Enrico Millo. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction | Kd (μM) |
|---|---|
| VX770/ΔF508-NBD1 | n.d. |
| C4a/ΔF508-NBD1 | n.d. |
| VX809/ΔF508-NBD1 | 24.2 ± 6.2 |
| 4/ΔF508-NBD1 | 99.3 ± 14.5 |
| 5/ΔF508-NBD1 | 40.3 ± 2.5 |
| 6/ΔF508-NBD1 | 197.9 ± 4.5 |
| 7/ΔF508-NBD1 | n.d. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusnati, M.; Sala, D.; Orro, A.; Bugatti, A.; Trombetti, G.; Cichero, E.; Urbinati, C.; Di Somma, M.; Millo, E.; Galietta, L.J.V.; et al. Speeding Up the Identification of Cystic Fibrosis Transmembrane Conductance Regulator-Targeted Drugs: An Approach Based on Bioinformatics Strategies and Surface Plasmon Resonance. Molecules 2018, 23, 120. https://doi.org/10.3390/molecules23010120
Rusnati M, Sala D, Orro A, Bugatti A, Trombetti G, Cichero E, Urbinati C, Di Somma M, Millo E, Galietta LJV, et al. Speeding Up the Identification of Cystic Fibrosis Transmembrane Conductance Regulator-Targeted Drugs: An Approach Based on Bioinformatics Strategies and Surface Plasmon Resonance. Molecules. 2018; 23(1):120. https://doi.org/10.3390/molecules23010120
Chicago/Turabian StyleRusnati, Marco, Davide Sala, Alessandro Orro, Antonella Bugatti, Gabriele Trombetti, Elena Cichero, Chiara Urbinati, Margherita Di Somma, Enrico Millo, Luis J. V. Galietta, and et al. 2018. "Speeding Up the Identification of Cystic Fibrosis Transmembrane Conductance Regulator-Targeted Drugs: An Approach Based on Bioinformatics Strategies and Surface Plasmon Resonance" Molecules 23, no. 1: 120. https://doi.org/10.3390/molecules23010120