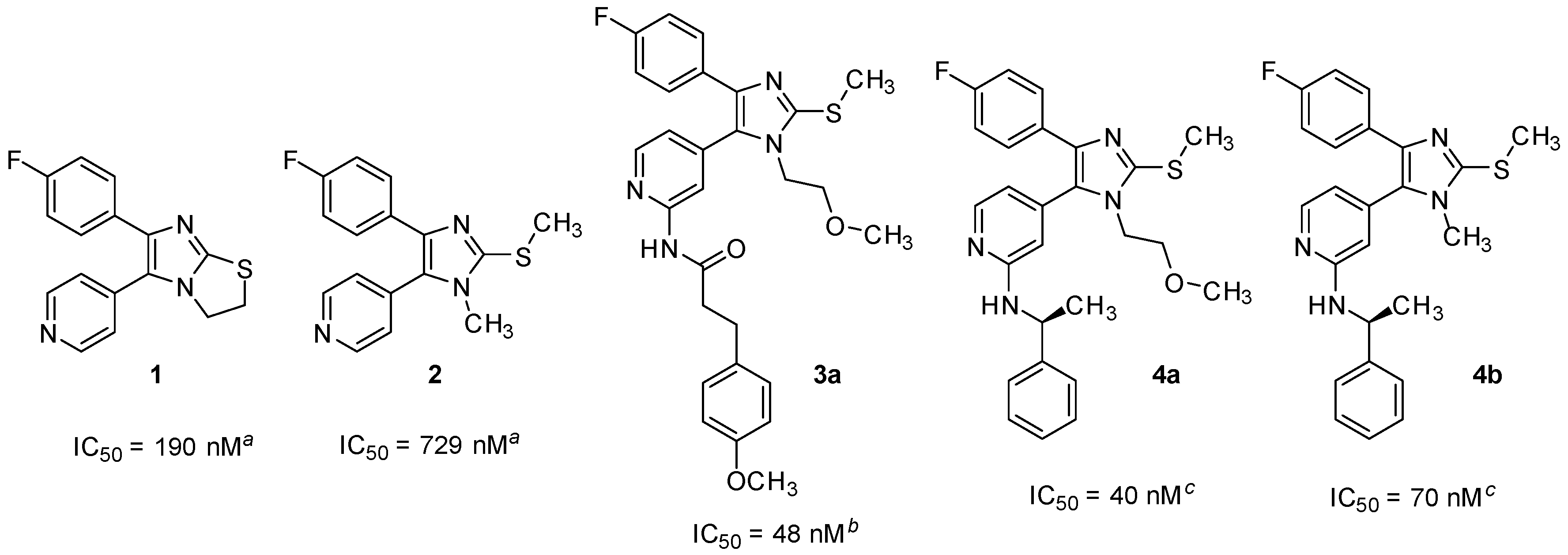
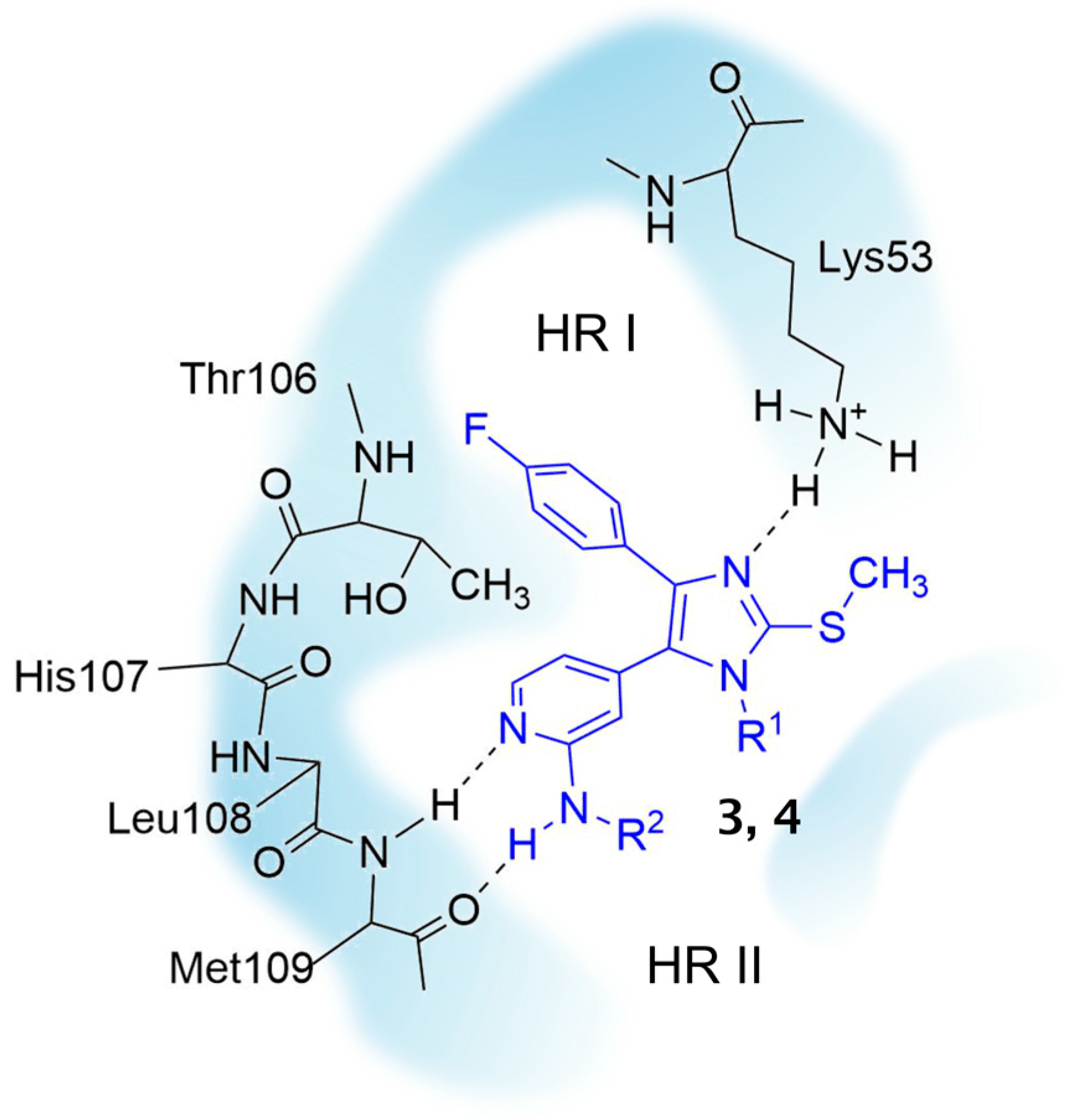
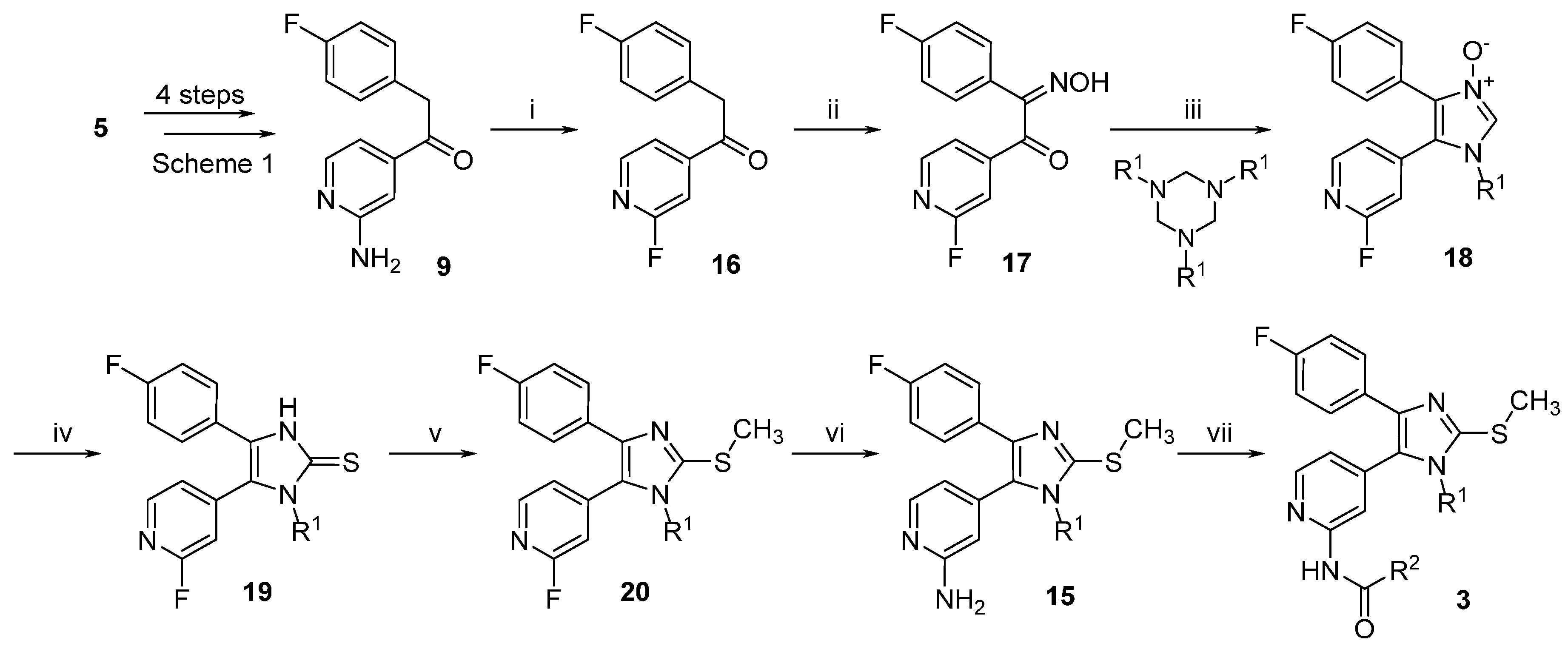
3.2.5. Detailed Procedures for the Preparation of Synthesized Compounds
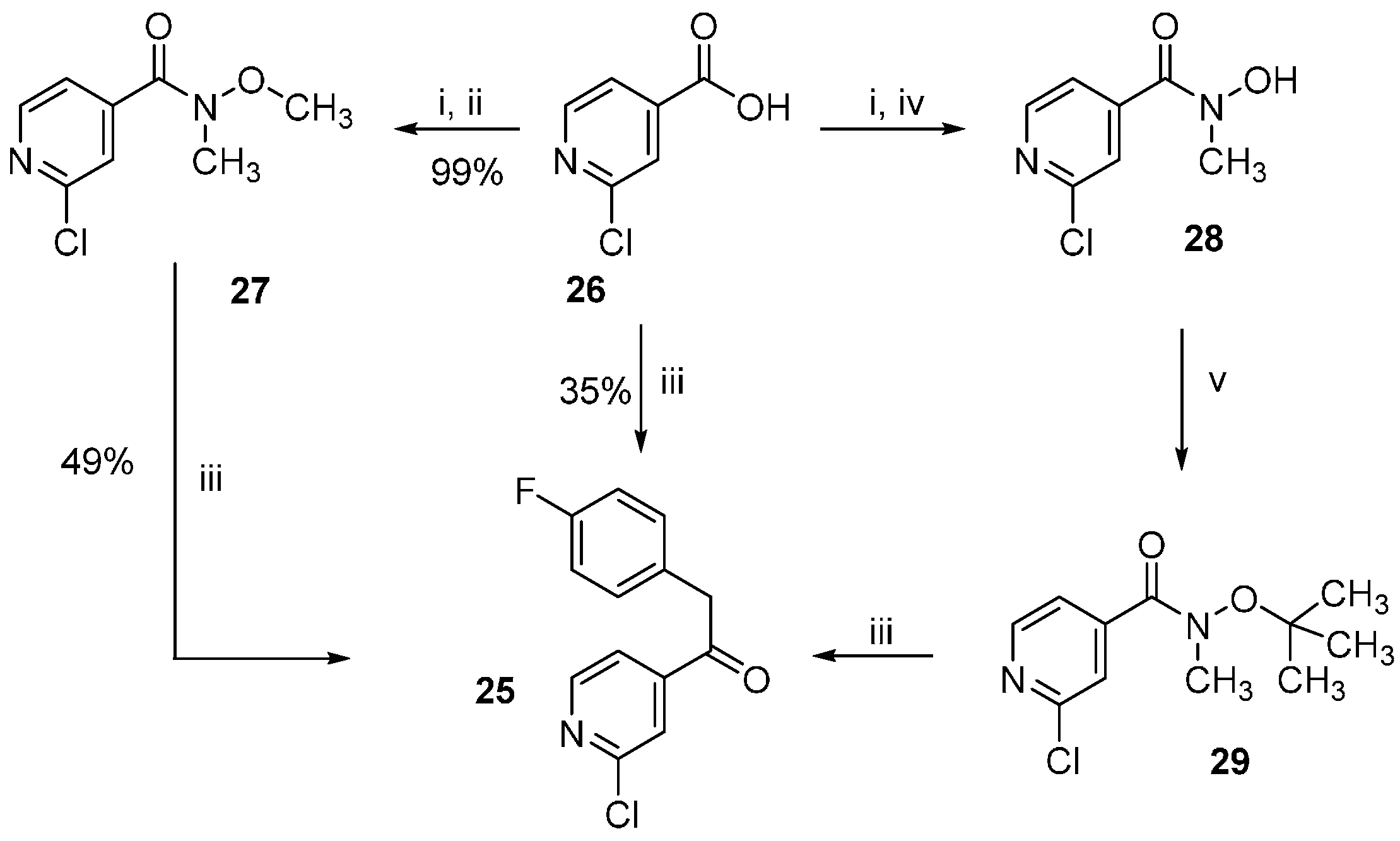
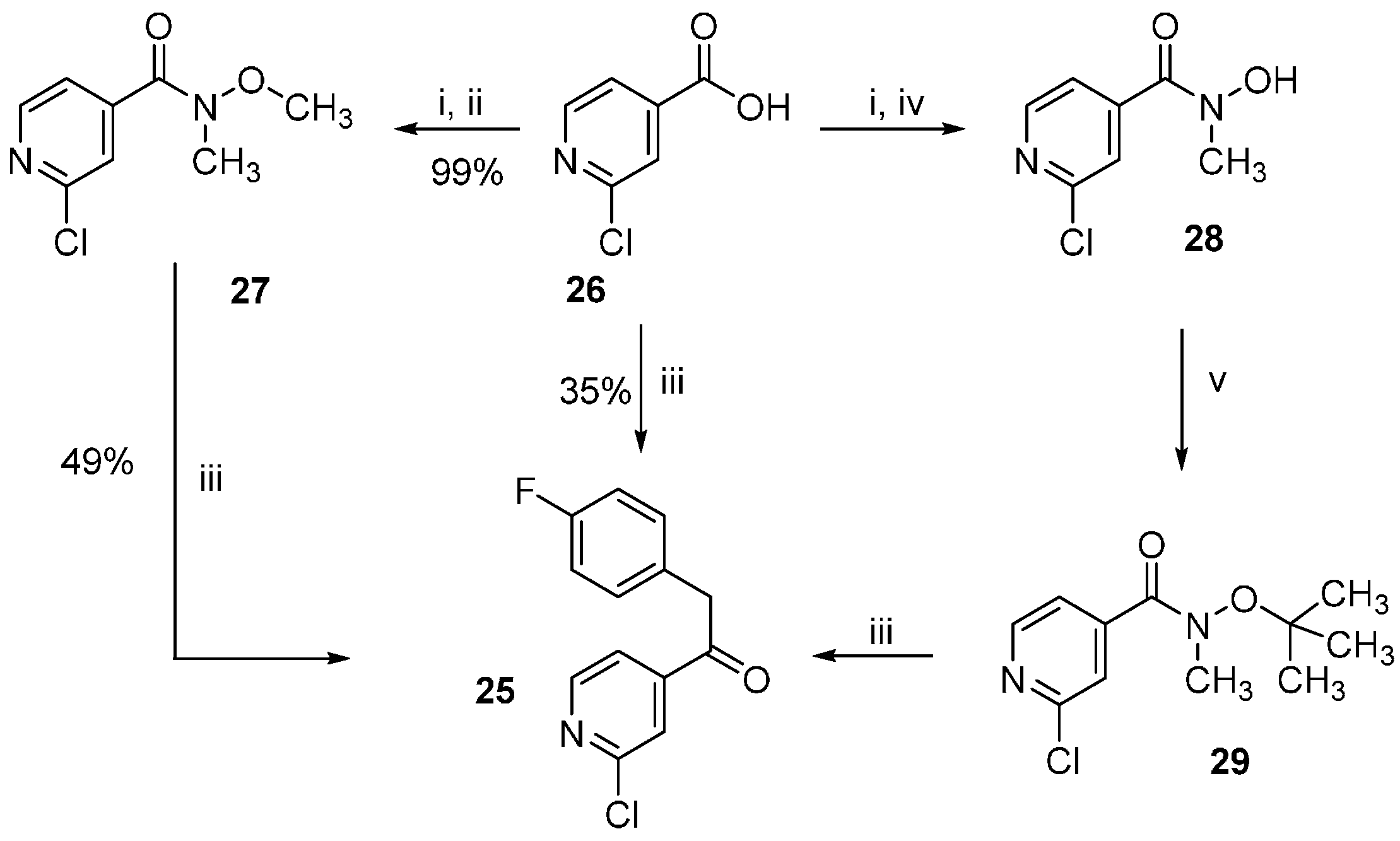
2-Chloro-N-methoxy-N-methylisonicotinamide (27). 2-Chloroisonicotinic acid (26, 21.0 g, 133.3 mmol) was suspended in SOCl2 (60 mL) and the mixture was stirred at reflux temperature for 5 h. After removing the excess of solvent, the residue was taken up in dry DCM and added dropwise to an ice-cooled previously prepared suspension of N,O-dimethylhydroxylamine hydrochloride (15.6 g, 160 mmol) and NEt3 (45.0 mL, 320 mmol) in dry DCM (50 mL). After completion of the addition the mixture was let heating at rt and stirred overnight. After evaporating the solvent at reduced pressure H2O was added and the aqueous phase was extracted with DCM. The combined organic layers were then washed with NaCl saturated solution, dried over anhydrous Na2SO4, and concentrated at reduced pressure giving 26.0 g of product as a light brown solid, which was directly used for the following step without further purification (97% yield); 1H-NMR (300 MHz, CDCl3) δ 3.35 (s, 3H), 3.53 (s, 3H), 7.43 (dd, J = 5.0, 1.2 Hz, 1H), 7.54 (s, 1H), 8.45 (d, J = 5.1 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ 32.9, 61.5, 120.7, 122.8, 144.5, 149.8, 151.6, 166.0; ESI-MS: (m/z) 201.1 [M + H]+; HPLC: tr = 2.051 min.
2-Chloro-N-hydroxy-N-methylisonicotinamide (28). 2-Chloroisonicotinic acid (26, 5.0 g, 31.7 mmol) was suspended in SOCl2 (25 mL) and the mixture was stirred at reflux temperature for 6 h. After removing the excess of solvent, the residue was taken up in dry DCM and added dropwise to an ice-cooled previously prepared suspension of N-methylhydroxylamine hydrochloride (3.2 g, 38.0 mmol) and NEt3 (9.7 mL, 76.1 mmol) in dry DCM (15 mL). After completion of the addition the mixture was let heating at rt and stirred overnight. After evaporating the solvent at reduced pressure H2O was added and the aqueous phase was extracted with EtOAc. The combined organic layers were then washed with NaCl saturated solution, dried over anhydrous Na2SO4, and concentrated at reduced pressure giving 5.6 g of product as a white-pink solid, which was directly used for the following step without further purification (95% yield); 1H-NMR (300 MHz, DMSO-d6) δ 3.27 (s, 3H), 7.53 (dd, J = 5.0, 1.2 Hz, 1H), 7.61 (dd, J = 1.2, 0.5 Hz, 1H), 8.49 (dd, J = 5.0, 0.5 Hz, 1H), 10.40 (br. s, 1H); 13C-NMR (75 MHz, DMSO-d6) δ 36.9, 122.1, 123.1, 146.5, 150.50, 150.56, 165.7; ESI-MS: (m/z) 187.2 [M + H]+, 185.1 [M − H]−; HPLC: tr = 1.568 min.
N-(tert-butoxy)-2-chloro-N-methylisonicotinamide (29). In a pressure vial 2-chloro-N-hydroxy-N-methylisonicotinamide (27, 500 mg, 2.68 mmol) was suspended in dry 1,4-dioxane (5 mL) and t-BuOAc (20 mL). After adding 70% HClO4(aq) (27 mg, 0.27 mmol) the vial was tightly closed and the mixture was stirred at 60 °C for 48 h. After cooling down, the mixture was poured in a K2CO3 saturated solution and the aqueous phase was extracted with EtOAc. The combined organic layers were then dried over anhydrous Na2SO4 and the solvent was evaporated at reduced pressure giving 402 mg of the desired compound, which was used for the following step without further purification; 1H-NMR (300 MHz, CDCl3) δ 1.10 (s, 9H), 3.43 (s, 3H), 7.48 (dd, J = 5.1, 1.2 Hz, 1H), 7.60 (br. s, 1H), 8.43 (dd, J = 5.0, 0.5 Hz, 1H); ESI-MS: (m/z) 243.1 [M + H]+; HPLC: tr = 4.733 min.
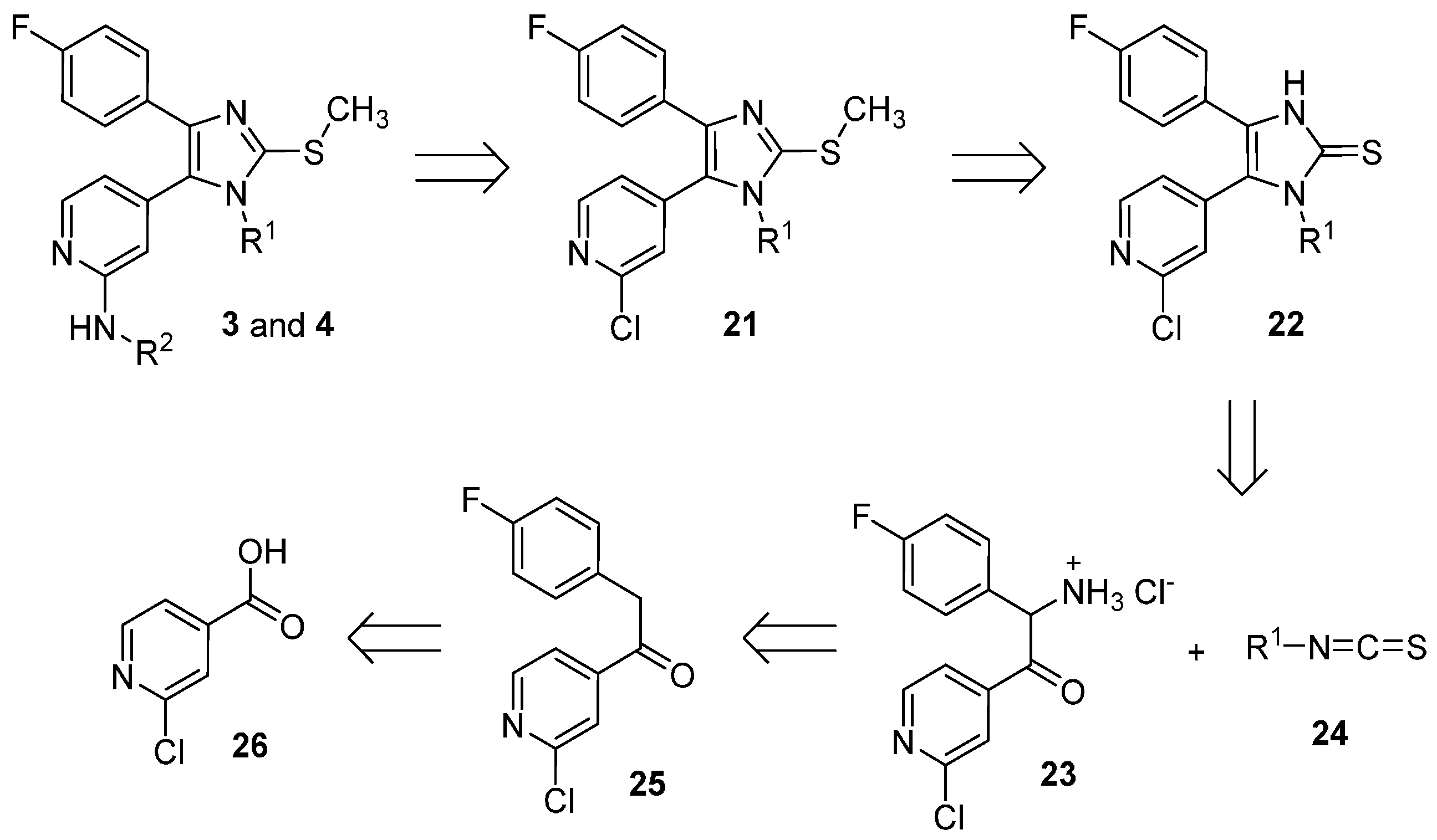
1-(2-Chloropyridin-4-yl)-2-(4-fluorophenyl)ethan-1-one (25). The title compound could be obtained alternatively through the following three procedures:
1) Under argon atmosphere Mg turnings (1.2 g, 50.0 mmol) were suspended in dry THF (120 mL) and after that 4-fluorobenzyl chloride (6.0 g, 41.5 mmol) was added in one portion. After the reaction was initiated, the mixture warmed up and was stirred until it cooled down to rt. The residual Mg was let decanting and the supernatant was added dropwise to a solution of 2-chloro-N-methoxy-N-methylisonicotinamide (27, 3.2 g, 16.0 mmol) in dry THF (35 mL) under argon atmosphere. The mixture was then stirred for 2 h at a temperature of 30–35 °C. The reaction was quenched with NH4Cl saturated solution (100 mL) and stirred overnight at rt. The two formed phases were separated and the aqueous phase was then further extracted by EtOAc. The combined organic layers were then washed with NaCl saturated solution, dried over anhydrous Na2SO4, and concentrated at reduced pressure. Finally, the residue was purified by flash column chromatography (SiO2, petroleum ether 40/60: EtOAc 4:1) giving 1.96 g of pure product as a yellow oil, which solidifies after cooling (49% yield).
2) Under argon atmosphere Mg turnings (1.22 g, 50.1 mmol) were suspended in dry THF (15 mL) and then 4-fluorobenzyl chloride (1.07 g, 7.4 mmol) was added in one portion. When the mixture started to become warm it was immediately cooled with an ice bath and then stirred for 2 h. After letting the residual Mg decanting, the supernatant was added dropwise to a solution of N-(tert-butoxy)-2-chloro-N-methylisonicotinamide (29, 900 mg, 3.7 mmol) in dry THF (10 mL), previously cooled at −10 °C. After completion of the addition the mixture was let slowly heating at rt and stirred for 3 h. NH4Cl saturated solution (80 mL) was added and the two formed phases were separated. The aqueous phase was then further extracted 3 times with EtOAc. The combined organic layers were then washed with NaCl saturated solution, dried over anhydrous Na2SO4, and concentrated at reduced pressure. Finally, the residue was purified by flash column chromatography (SiO2, n-hexane: EtOAc gradient elution from 4:1 to 3:2) giving 747 mg of pure product as a yellow oil, which solidifies after cooling (81% yield).
3) Under argon atmosphere Mg turnings (6.94 g, 285.6 mmol) were suspended in dry THF (35 mL). After that 4-fluorobenzyl chloride (13.6 g, 92.5 mmol) was added in portions: after adding 1 mL and starting the reaction, the mixture was cooled at 0 °C with an ice bath and the addition continued by 0.5 mL/min at the same temperature. After the addition was competed the mixture was let stirring until warming up to rt. After letting the Mg decanting the supernatant was added slowly dropwise to a suspension of 2-chloroisonicotinic acid (26, 6.0 g, 38.1 mmol) in dry THF (24 mL), previously cooled at −30 °C. During the addition the temperature was maintained between −25 and −30 °C. After complete addition the mixture was let heating to rt and stirred overnight. The mixture was poured on NH4Cl saturated solution (100 mL). The aqueous phase was extracted twice with EtOAc and the combined organic layers were dried over anhydrous Na2SO4, and concentrated at reduced pressure. Finally, the residue was purified by flash column chromatography (SiO2, petroleum ether 40/60:EtOAc gradient elution from 3:1 to 1:1) giving 3.37 g of pure compound as a yellow oil, which solidifies after cooling (35% yield); 1H-NMR (300 MHz, CDCl3) δ 4.24 (s, 2H), 6.99–7.10 (m, 2H), 7.15–7.23 (m, 2H), 7.67 (dd, J = 5.1, 1.5 Hz, 1H), 7.79 (dd, J = 1.4, 0.7 Hz, 1H), 8.56 (dd, J = 5.1, 0.5 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ 44.9, 115.9 (d, J = 21.6 Hz), 120.2, 122.7, 128.4 (d, J = 3.3 Hz), 131.1 (d, J = 8.3 Hz), 145.2, 151.0, 153.0, 162.2 (d, J = 246.6 Hz); ESI-MS: (m/z) 249.9 [M + H]+; HPLC: tr = 5.555 min.
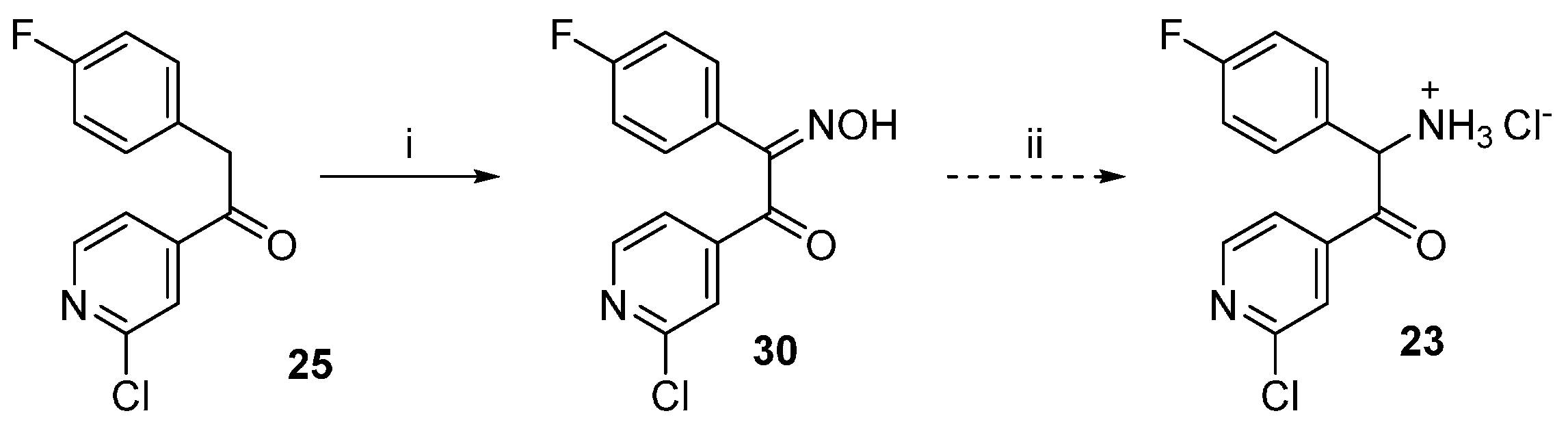
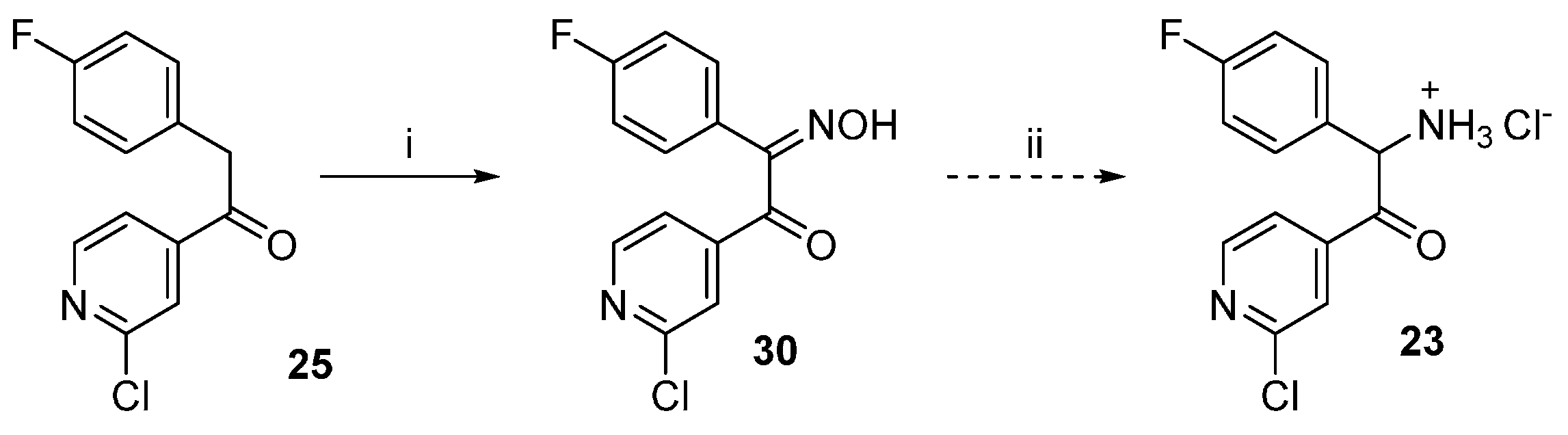
1-(2-Chloropyridin-4-yl)-2-(4-fluorophenyl)-2-(hydroxyimino)ethan-1-one (30). Ethanone derivative 25 (1.96 g, 7.85 mmol) was dissolved in glacial AcOH (15 mL) and after that NaNO2 (1.67 g, 24.2 mmol), previously dissolved in H2O (7 mL), was added dropwise and the reaction mixture was stirred overnight at rt. Afterwards 50 mL H2O were added and the voluminous precipitate formed was filtered off and washed with H2O. The residue was then suspended in H2O and the suspension was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and the solvent was then removed at reduced pressure, affording 1.8 g of the desired product, which was used for the following step without further purification (82% yield); 1H-NMR (300 MHz, DMSO-d6) δ 7.23–7.37 (m, 2H), 7.51–7.62 (m, 2H), 7.73 (dd, J = 5.0, 1.4 Hz, 1H), 7.89 (dd, J = 1.2, 0.7 Hz, 1H), 8.58 (dd, J = 5.0, 0.7 Hz, 1H), 13.14 (s, 1H); 13C-NMR (75 MHz, DMSO-d6) δ 115.3 (d, J = 21.6 Hz), 122.8, 124.3, 125.6 (d, J = 3.9 Hz), 132.5 (d, J = 8.3 Hz), 149.2, 150.5, 150.6, 154.3, 162.8 (d, J = 246.6 Hz), 190.4; ESI-MS: (m/z) 276.9 [M − H]−; HPLC: tr = 7.405 min.
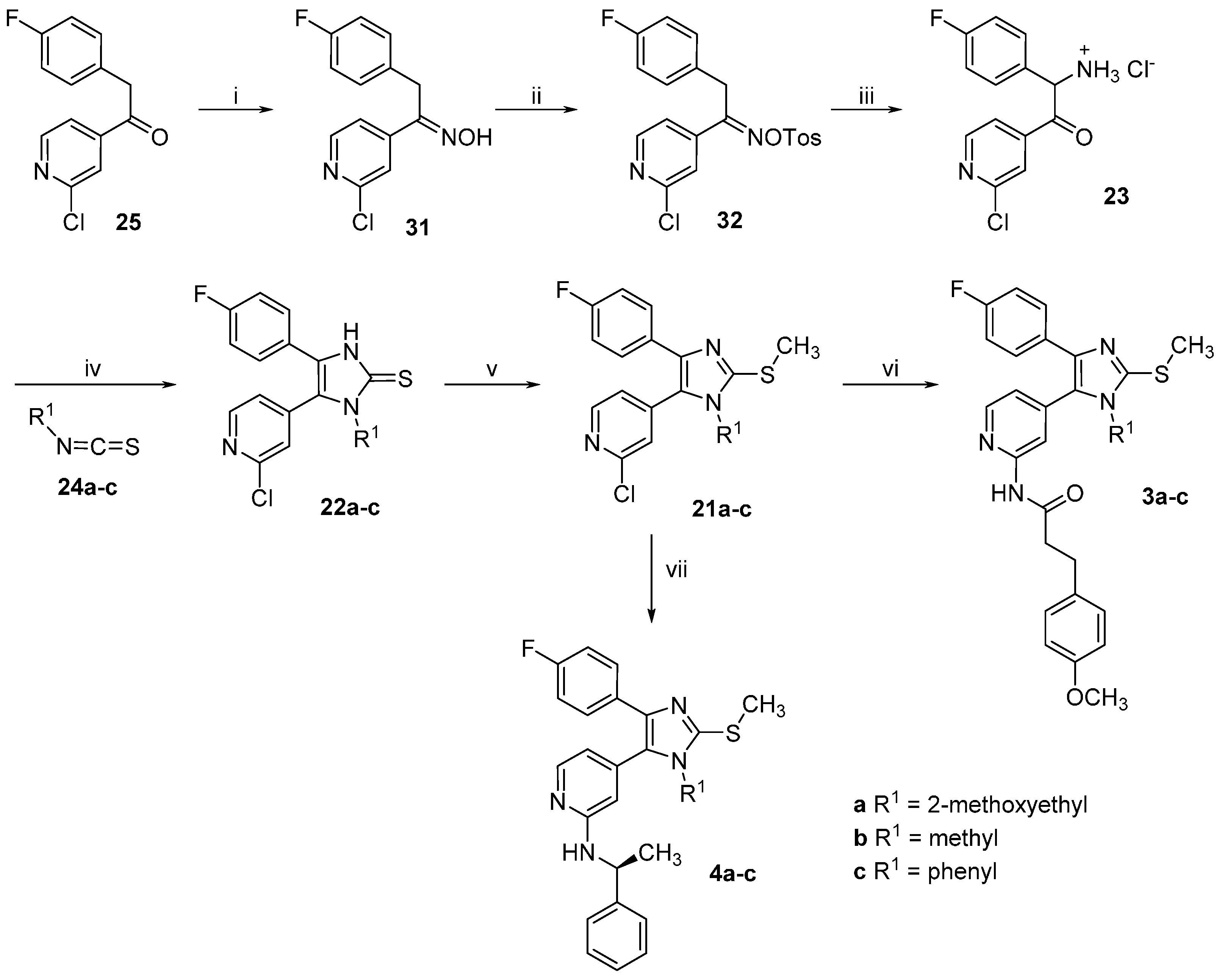
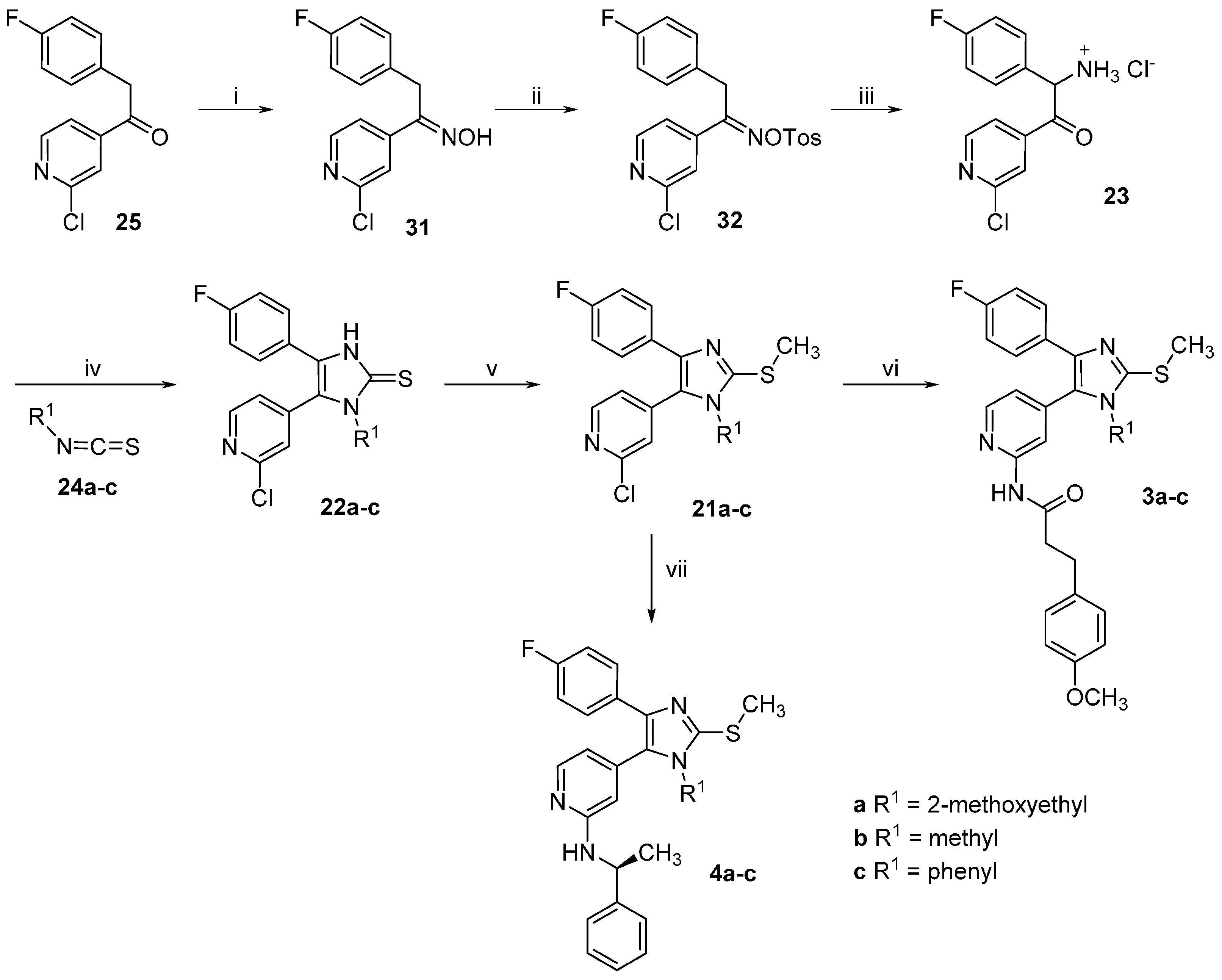
1-(2-Chloropyridin-4-yl)-2-(4-fluorophenyl)ethan-1-one oxime (31). Hydroxylamine hydrochloride (1.52 g, 21.93 mmol) was dissolved in H2O (3.5 mL) and then 20% NaOH(aq) (0.7 mL) and a solution of ethanone derivative 25 (3.65 g, 14.62 mmol) in MeOH (17.5 mL) were added. The reaction mixture was stirred at rt for 48 h while a precipitate formed. Saturated NH4Cl solution (50 mL) was added and the precipitate was filtered off, rinsed with H2O and dried in vacuo over P2O5 affording 3.71 g of the desired product as an off-white solid, which was used for the following step without further purification (96% yield). Extraction with EtOAc represents an alternative work-up procedure giving similar yields; 1H-NMR (300 MHz, DMSO-d6) δ 4.16 (s, 2H), 7.12–7.02 (m, 2H), 7.28–7.20 (m, 2H), 7.64 (dd, J = 5.2, 1.5 Hz, 1H), 7.71–7.68 (m, 1H), 8.37 (d, J = 5.2 Hz, 1H), 12.27 (s, 1H); 13C-NMR (75 MHz, DMSO-d6) δ 29.0, 115.4 (d, J = 21.3 Hz), 119.7, 120.5, 130.2 (d, J = 8.0 Hz), 132.5 (d, J = 3.1 Hz), 146.5, 150.2, 151.0, 152.7, 160.9 (d, J = 242.4 Hz); ESI-MS: 265.0 [M + H]+, 263.0 [M − H]−; HPLC: tr = 6.934 min.
1-(2-Chloropyridin-4-yl)-2-(4-fluorophenyl)ethan-1-one O-tosyl oxime (32). A solution of ketoxime 31 (1.20 g, 4.53 mmol) and p-toluenesulfonyl chloride (3.46 g, 18.14 mmol) in dry pyridine (6 mL) was stirred at rt under N2 atmosphere for 50 h. The mixture was poured into a NaCl saturated solution (30 mL) and extracted three times with EtOAc. The combined organic layers were then washed 3 times with a NaCl saturated solution, dried over anhydrous Na2SO4 and concentrated at reduced pressure. Finally, the residue was purified by flash column chromatography (SiO2, petroleum ether 40/60:EtOAc 2:3) giving 1.68 g of the desired product as a yellow oil (88% yield); 1H-NMR (300 MHz, CDCl3) δ 2.48 (s, 3H), 4.12 (s, 2H), 6.89–7.06 (m, 4H), 7.33 (dd, J = 5.2, 1.5 Hz, 1H), 7.39 (d, J = 8.2 Hz, 2H), 7.42–7.46 (m, 1H), 7.89 (d, J = 8.3 Hz, 2H), 8.39 (d, J = 5.2 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ 21.8, 32.6, 116.1 (d, J = 21.6 Hz), 120.0, 122.1, 128.8 (d, J = 3.3 Hz), 129.0, 129.9 (d, J = 7.7 Hz), 129.9, 131.9, 143.4, 145.9, 150.3, 152.4, 162.5 (d, J = 246.6 Hz), 161.5; ESI-MS: (m/z) 473.2 [M + Na + MeOH]+; HPLC: tr = 9.342 min.
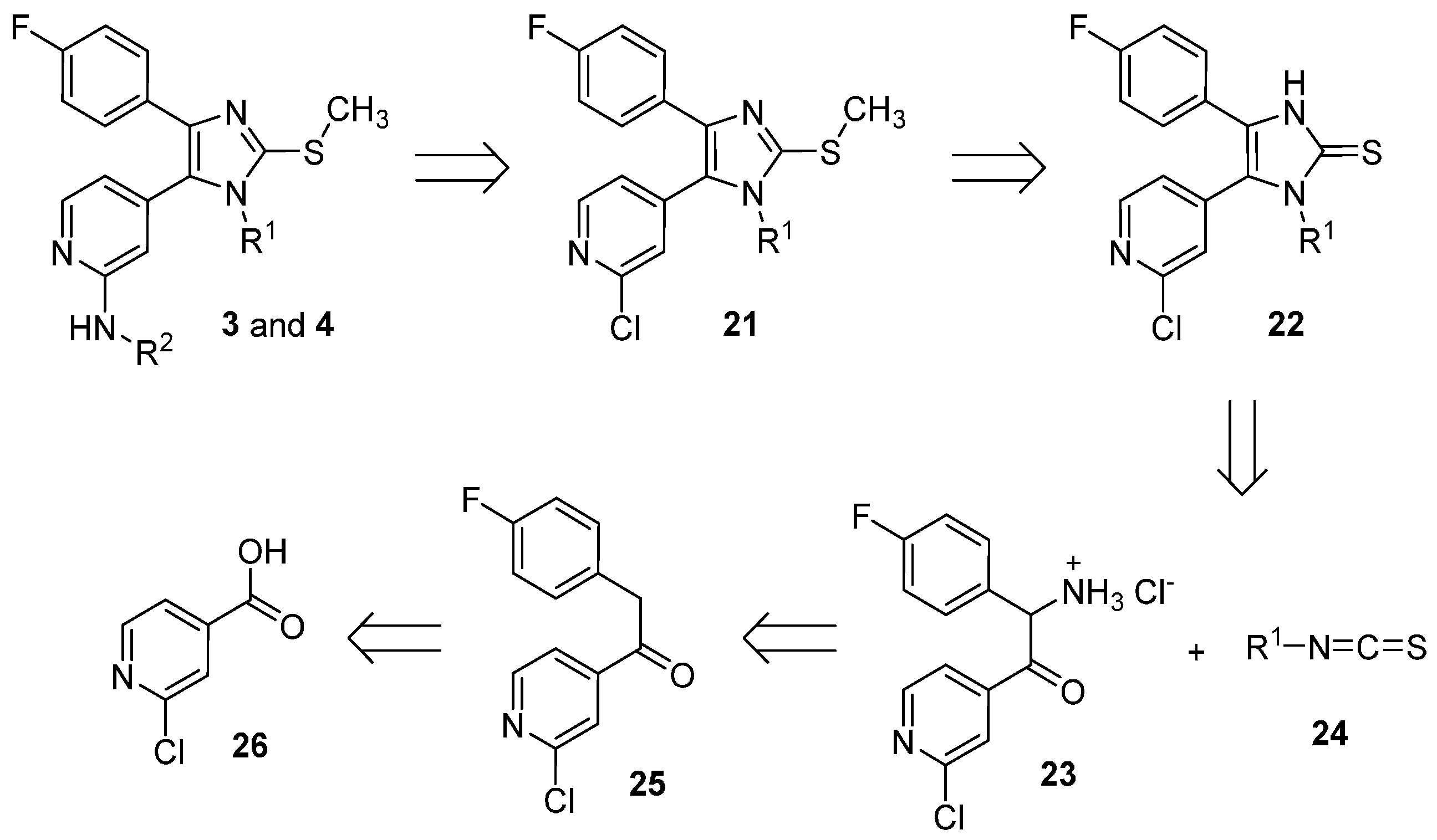
2-Amino-1-(2-chloropyridin-4-yl)-2-(4-fluorophenyl)ethan-1-one hydrochloride (23). In a three-necks round bottom flask under argon atmosphere K chunks (175 mg, 4.4 mmol) were added portionswise to EtOH(abs) (20 mL). After complete dissolution the mixture was cooled at 0 °C and 1-(2-chloropyridin-4-yl)-2-(4-fluorophenyl)ethan-1-one O-tosyl oxime (31, 1.70 g, 4.0 mmol), previously dissolved in EtOH(abs) (50 mL) was added slowly dropwise. The mixture was then stirred at 0 °C for 3 h and after that dry Et2O (250 mL) was added and the mixture was stirred for 30 min at rt. The white precipitate formed was removed by filtration and the filtrate was concentrated at reduced pressure. The residue was taken up in conc. HCl(aq) and the mixture was stirred at 60 °C for 2 h. The residual solvent was removed at reduced pressure and the residue was treated with a mixture of THF:Et2O 1:2. The white precipitate obtained was filtered off and dried affording 725 mg of the desired product, which was used for the following step without further purification (60% yield); ESI-MS: (m/z) 264.9 [M + H]+, 263.0 [M − H]−; HPLC: tr = 2.329 min.
5-(2-Chloropyridin-4-yl)-4-(4-fluorophenyl)-1-(2-methoxyethyl)-1,3-dihydro-2H-imidazole-2-thione (22a). The title compound was prepared following general procedure A starting from compound 23 (500 mg, 1.65 mmol) and 2-methoxyethyl isothiocyanate (24a, 965 mg, 8.25 mmol); The residue was treated with Et2O obtaining a precipitate, which was filtered off and dried. The filtrate was then concentrated at reduced pressure and the residue was purified by flash column chromatography (SiO2, n-hexane:EtOAc gradient elution from 7:3 to 1:1). 293 mg of the desired product were obtained in total (49% yield); 1H-NMR (300 MHz, DMSO-d6) δ 3.06 (s, 3H), 3.52–3.60 (m, 2H), 3.99–4.14 (m, 2H), 7.08–7.32 (m, 4H), 7.42 (d, J = 4.6 Hz, 1H), 7.64 (s, 1H), 8.49 (d, J = 4.9 Hz, 1H), 13.07 (br. s, 1H); ESI-MS: (m/z) 364.2 [M + H]+, 362.2 [M − H]−; HPLC: tr = 5.321 min.
5-(2-Chloropyridin-4-yl)-4-(4-fluorophenyl)-1-methyl-1,3-dihydro-2H-imidazole-2-thione (22b). The title compound was prepared following general procedure A starting from compound 23 (400 mg, 1.32 mmol) and methyl isothiocyanate (24b, 643 mg, 6.62 mmol); After the addition of NaHCO3 saturated solution, a precipitate was formed, which was filtered off and purified twice by flash column chromatography (SiO2, DCM:EtOH gradient elution from 100:0 to 97:03) and (SiO2, DCM:EtOH 99:01). The obtained solid was treated with a mixture of Et2O:n-hexane 1:2 and the yellow precipitate obtained was then filtered off and dried, affording 255 mg of the desired compound (60% yield); 1H-NMR (300 MHz, DMSO-d6) δ 3.43 (s, 3H), 7.16–7.25 (m, 2H), 7.26–7.35 (m, 2H), 7.42 (dd, J = 5.1, 1.3 Hz, 1H), 7.6 (s, 1H), 8.49 (d, J = 5.0 Hz, 1H), 13.01 (br. s, 1H); 13C-NMR (75 MHz, DMSO-d6) δ 32.1, 115.9 (d, J = 22.1 Hz), 123.1, 123.9 (d, J = 2.8 Hz), 124.3, 125.1, 125.6, 129.7 (d, J = 8.3 Hz), 139.9, 150.7, 151.0, 161.9 (d, J = 246.6 Hz), 162.7; ESI-MS: (m/z) 318.3 [M − H]−; HPLC: tr = 4.389 min.
5-(2-Chloropyridin-4-yl)-4-(4-fluorophenyl)-1-phenyl-1,3-dihydro-2H-imidazole-2-thione (22c). The title compound was prepared following general procedure A starting from compound 23 (400 mg, 1.32 mmol) and phenyl isothiocyanate (24c, 535 mg, 3.96 mmol); The residue was purified by flash column chromatography (SiO2, DCM:EtOH gradient elution from 100:0 to 98:02). The solid obtained was treated with a mixture of Et2O:n-hexane 1:2 and the white precipitate formed was then filtered off and dried, affording 260 mg of the desired compound (51% yield);1H-NMR (300 MHz, CDCl3) δ 6.69 (d, J = 4.7 Hz, 1H), 6.81 (s, 1H), 6.89–7.03 (m, 2H), 7.11–7.21 (m, 2H), 7.21–7.30 (m, 2H), 7.32–7.46 (m, 3H), 8.11 (d, J = 5.2 Hz, 1H), 12.63 (br. s, 1H); ESI-MS: (m/z) 380.5 [M − H]−; HPLC: tr = 5.867 min.
2-Chloro-4-(4-(4-fluorophenyl)-1-(2-methoxyethyl)-2-(methylthio)-1H-imidazol-5-yl)pyridine (21a). The title compound was prepared following general procedure B starting from compound 22a (263 mg, 0.72 mmol), t-BuONa (83 mg, 0.86 mmol), and iodomethane (511 mg, 3.6 mmol) obtaining 270 mg of the desired product, which was used for the following step without further purification (99% yield); 1H-NMR (300 MHz, CDCl3) δ 2.73 (s, 3H), 3.25 (s, 3H), 3.55 (t, J = 5.5 Hz, 2H), 4.00 (t, J = 5.5 Hz, 2H), 6.87–6.98 (m, 2H), 7.21 (dd, J = 5.1, 1.4 Hz, 1H), 7.33–7.41 (m, 2H), 7.42 (dd, J = 1.3, 0.6 Hz, 1H), 8.40 (dd, J = 5.1, 0.6 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ 16.0, 44.4, 58.9, 70.5, 115.3 (d, J = 21.6 Hz), 124.1, 125.7, 126.3, 128.9 (d, J = 8.3 Hz), 129.5 (d, J = 3.3 Hz), 139.5, 141.9, 145.4, 150.1, 152.1, 162.0 (d, J = 246.6 Hz); ESI-MS: (m/z) 378.3 [M + H]+; HPLC: tr = 7.524 min.
2-Chloro-4-(4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridine (21b). The title compound was prepared following general procedure B starting from compound 22b (225 mg, 0.70 mmol), t-BuONa (81 mg, 0.84 mmol), and iodomethane (497 mg, 3.5 mmol). The crude product was purified by flash column chromatography (SiO2, DCM:EtOH gradient elution from 100:0 to 99:01) giving 189 mg of the desired compound (80% yield); 1H-NMR (300 MHz, CDCl3) δ 2.73 (s, 3H), 3.49 (s, 3H), 6.90–7.02 (m, 2H), 7.13 (dd, J = 5.1, 1.4 Hz, 1H), 7.34–7.45 (m, 2H), 8.42 (d, J = 5.1 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ 15.8, 32.0, 115.4 (d, J = 21.6 Hz), 123.4, 124.8, 126.1, 129.1 (d, J = 7.7 Hz), 129.5 (d, J = 3.3 Hz), 139.8, 141.6, 146.1, 150.4, 152.4, 162.2 (d, J = 246.6 Hz); ESI-MS: (m/z) 334.3 [M + H]+; HPLC: tr = 6.695 min.
2-Chloro-4-(4-(4-fluorophenyl)-2-(methylthio)-1-phenyl-1H-imidazol-5-yl)pyridine (21c). The title compound was prepared following general procedure B starting from compound 22c (219 mg, 0.57 mmol), t-BuONa (55 mg, 0.57 mmol), and iodomethane (404 mg, 2.85 mmol). After adding H2O a precipitate was formed, which was filtered off and dried, giving 185 mg of the desired product that was used for the following step without further purification (82% yield); 1H-NMR (300 MHz, CDCl3) δ 2.68 (s, 3H), 6.82 (d, J = 4.7 Hz, 1H), 6.93 (s, 1H), 6.96–7.09 (m, 2H), 7.11–7.25 (m, 2H), 7.37–7.58 (m, 5H), 8.15 (d, J = 4.9 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ 15.1, 115.5 (d, J = 21.6 Hz), 122.6, 124.2, 126.2, 127.6, 129.5 (d, J = 3.3 Hz), 129.6, 129.7, 135.1, 140.8 (d, J = 7.7 Hz), 147.6, 149.6, 151.7, 162.4 (d, J = 247.1 Hz); ESI-MS: (m/z) 396.6 [M + H]+; HPLC: tr = 9.718 min.
N-(4-(4-(4-Fluorophenyl)-1-(2-methoxyethyl)-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)-3-(4-methoxyphenyl)propanamide (
3a) [
14]. The title compound was prepared according to general procedure C starting from imidazole
21a (100 mg, 0.26 mmol), 3-(4-methoxyphenyl)propanamide (72 mg, 0.40 mmol), Pd
2(dba)
3 (12 mg, 0.013 mmol), XantPhos (15 mg, 0.026 mmol), and Cs
2CO
3 (254 mg, 0.78 mmol). The residue was purified twice by flash column chromatography (SiO
2, DCM:EtOH gradient elution from 100:0 to 95:05) and (SiO
2, DCM:EtOH gradient elution from 100:0 to 97:03) giving 50 mg of the desired product (37% yield);
1H-NMR (300 MHz, CDCl
3) δ 2.64–2.77 (m, 5H), 3.00 (t,
J = 7.6 Hz, 2H), 3.24 (s, 3H), 3.51 (t,
J = 6.0 Hz, 2H), 3.79 (s, 3H), 4.10 (t,
J = 6.0 Hz, 2H), 6.79–6.88 (m, 2H), 6.88–6.99 (m, 3H), 7.11–7.20 (m, 2H), 7.36–7.47 (m, 2H), 8.16 (br. s, 1H), 8.24 (d,
J = 5.1 Hz, 1H), 8.29 (s, 1H); ESI-MS: (
m/
z) 521.5 [M + H]
+, 543.4 [M + Na]
+, 519.5 [M − H]
−; HPLC: t
r = 8.476 min.
N-(4-(4-(4-Fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)-3-(4-methoxyphenyl)-propanamide (3b). The title compound was prepared according to general procedure C starting from imidazole 21b (70 mg, 0.21 mmol), 3-(4-methoxyphenyl)propanamide (75 mg, 0.41 mmol), Pd2(dba)3 (10 mg, 0.01 mmol), XantPhos (12 mg, 0.021 mmol), and Cs2CO3 (205 mg, 0.63 mmol). The residue was purified twice by flash column chromatography (SiO2, DCM:EtOH gradient elution from 99:01 to 95:05) and (SiO2, n-hexane:EtOAc gradient elution from 4:1 to 1:1) giving 44 mg of the desired product (45% yield); 1H-NMR (300 MHz, CDCl3) δ 2.62–2.81 (m, 5H), 3.00 (t, J = 7.5 Hz, 2H), 3.54 (s, 3H), 3.79 (s, 3H), 6.84 (d, J = 8.6 Hz, 2H), 6.88–7.00 (m, 3H), 7.14 (d, J = 8.6 Hz, 2H), 7.36–7.50 (m, 2H), 8.21 (d, J = 5.1 Hz, 1H), 8.27 (s, 1H), 8.36 (br. s, 1H); 13C-NMR (75 MHz, CDCl3) δ 16.1, 30.3, 32.1, 39.6, 55.3, 114.0, 114.6, 115.3 (d, J = 21.6 Hz), 121.1, 127.6, 129.1 (d, J = 7.7 Hz), 129.3, 130.0 (d, J = 3.3 Hz), 132.3, 139.3, 141.1, 145.3, 148.1, 151.9, 158.2, 162.1 (d, J = 246.6 Hz), 171.0; ESI-TOF-HRMS: (m/z) [M + H]+ calcd. for C26H25FN4O2S 477.1755, found 477.1762; HPLC: tr = 8.855 min.
N-(4-(4-(4-Fluorophenyl)-2-(methylthio)-1-phenyl-1H-imidazol-5-yl)pyridin-2-yl)-3-(4-methoxyphenyl)-propanamide (3c). The title compound was prepared according to general procedure C starting from imidazole 21c (70 mg, 0.18 mmol), 3-(4-methoxyphenyl)propanamide (64 mg, 0.36 mmol), Pd2(dba)3 (8 mg, 0.009 mmol), XantPhos (10 mg, 0.018 mmol), and Cs2CO3 (173 mg, 0.53 mmol). The residue was purified twice by flash column chromatography (SiO2, DCM:EtOH gradient elution from 99:01 to 95:05) and (SiO2, n-hexane:EtOAc gradient elution from 4:1 to 1:1) giving 24 mg of the desired product (25% yield); 1H-NMR (300 MHz, DMSO-d6) δ 2.54–2.68 (m, 5H), 2.70–2.84 (m, 2H), 3.71 (s, 3H), 6.77–6.90 (m, 3H), 7.05–7.22 (m, 4H), 7.25–7.36 (m, 2H), 7.39–7.60 (m, 5H), 7.93 (s, 1H), 8.16 (dd, J = 5.1, 0.7 Hz, 1H), 10.44 (s, 1H); 13C-NMR (75 MHz, CDCl3) δ 15.3, 30.3, 39.7, 55.2, 113.9, 114.5, 115.3 (d, J = 21.6 Hz), 120.5, 127.9, 129.1, 129.2, 129.4, 129.5 (d, J = 7.7 Hz), 129.8 (d, J = 2.8 Hz), 132.3, 135.4, 140.0, 140.8, 146.8, 147.2, 151.4, 158.1, 162.2 (d, J = 246.6 Hz), 170.4; ESI-TOF-HRMS: (m/z) [M + H]+ calcd. for C31H27FN4O2S 539.1911, found 539.1915; HPLC: tr = 10.160 min.
(S)-4-(4-(4-Fluorophenyl)-1-(2-methoxyethyl)-2-(methylthio)-1H-imidazol-5-yl)-N-(1-phenylethyl)pyridin-2-amine (
4a) [
15]. The title compound was prepared according to general procedure D starting from compound
21a (100 mg, 0.26 mmol). The reaction was stopped after 24 h. The residue was then purified twice by flash column chromatography (SiO
2, DCM:EtOH gradient elution from 100:0 to 95:05) and (SiO
2, DCM:EtOH gradient elution from 99:01 to 97:03) giving 50 mg of the desired product (41% yield);
1H-NMR (300 MHz, DMSO-
d6) δ 1.42 (d,
J = 6.8 Hz, 3H), 2.63 (s, 3H), 3.06 (s, 3H), 3.22–3.32 (m, 2H, partially overlapping with H
2O signal), 3.78–3.95 (m, 2H), 4.90–5.09 (m, 1H), 6.35–6.52 (m, 2H), 7.01–7.13 (m, 2H), 7.13–7.24 (m, 2H), 7.24–7.46 (m, 6H), 8.03 (d,
J = 5.1 Hz, 1H); ESI-MS: (
m/
z) 463.5 [M + H]
+, 461.5 [M − H]
−; HPLC: t
r = 6.398 min.
(S)-4-(4-(4-Fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)-N-(1-phenylethyl)pyridin-2-amine (
4b) [
15]. The title compound was prepared according to general procedure D starting from compound
21b (35 mg, 0.105 mmol). The reaction was stopped after 32 h although not completed. The residue was then purified by preparative TLC (SiO
2, DCM:EtOH 95:05) and by flash column chromatography (SiO
2,
n-hexane:EtOAc gradient elution from 7:3 to 1:1) giving 17 mg of the desired product (39% yield);
1H-NMR (300 MHz, CDCl
3) δ 1.54 (d,
J = 6.7 Hz, 3H), 2.64 (s, 3H), 3.08 (s, 3H), 4.47–4.69 (m, 1H), 5.51 (br. s, 1H), 6.05 (s, 1H), 6.42 (d,
J = 4.7 Hz, 1 H), 6.76–6.95 (m, 2H), 7.14–7.53 (m, 7H, overlapping with the solvent peak), 8.05 (d,
J = 5.1 Hz, 1 H); ESI-MS: (
m/
z) 419.3 [M + H]
+, 417.2 [M − H]
−; HPLC: t
r = 7.063 min.
(S)-4-(4-(4-Fluorophenyl)-2-(methylthio)-1-phenyl-1H-imidazol-5-yl)-N-(1-phenylethyl)pyridin-2-amine (4c). The title compound was prepared according to general procedure D starting from compound 21b (42 mg, 0.106 mmol). The reaction was stopped after 26 h. The residue was purified by flash column chromatography (SiO2, n-hexane:EtOAc gradient elution from 9:1 to 1:1) giving 42 mg of the desired product (82% yield); 1H-NMR (300 MHz, CDCl3) δ 1.41 (d, J = 6.8 Hz, 3H), 2.63 (s, 3H), 4.16–4.34 (m, 1H), 5.18–5.36 (m, 1H), 5.90 (s, 1H), 6.21 (dd, J = 5.3, 1.2 Hz, 1H), 6.86–6.98 (m, 2H), 7.00–7.10 (m, 2H), 7.11–7.31 (m, 5H, overlapping with the solvent peak), 7.31–7.41 (m, 3H), 7.41–7.51 (m, 2H), 7.83 (d, J = 5.2 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ 15.3, 24.4, 52.2, 107.4, 114.1, 115.2 (d, J = 21.6 Hz), 125.6, 127.1, 127.6, 128.4, 128.7, 128.9, 129.2 (d, J = 7.7 Hz), 129.3, 129.9 (d, J = 3.3 Hz), 135.5, 139.2, 139.8, 144.0, 146.0, 147.4, 157.8, 162.0 (d, J = 246.0 Hz); ESI-TOF-HRMS: (m/z) [M + H]+ calcd. for C29H25FN4S 481.1857, found 481.1865; HPLC: tr = 9.148 min.
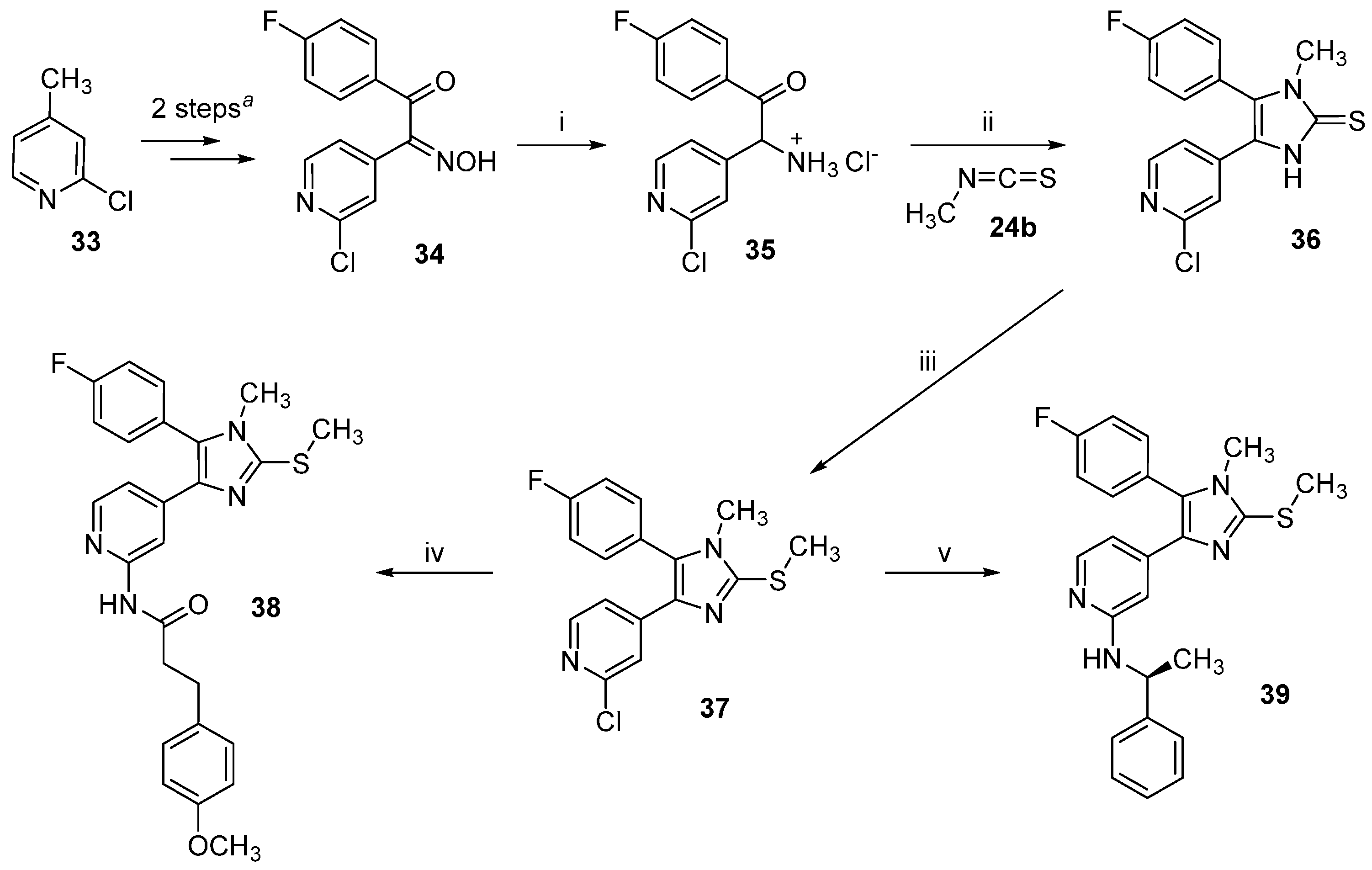
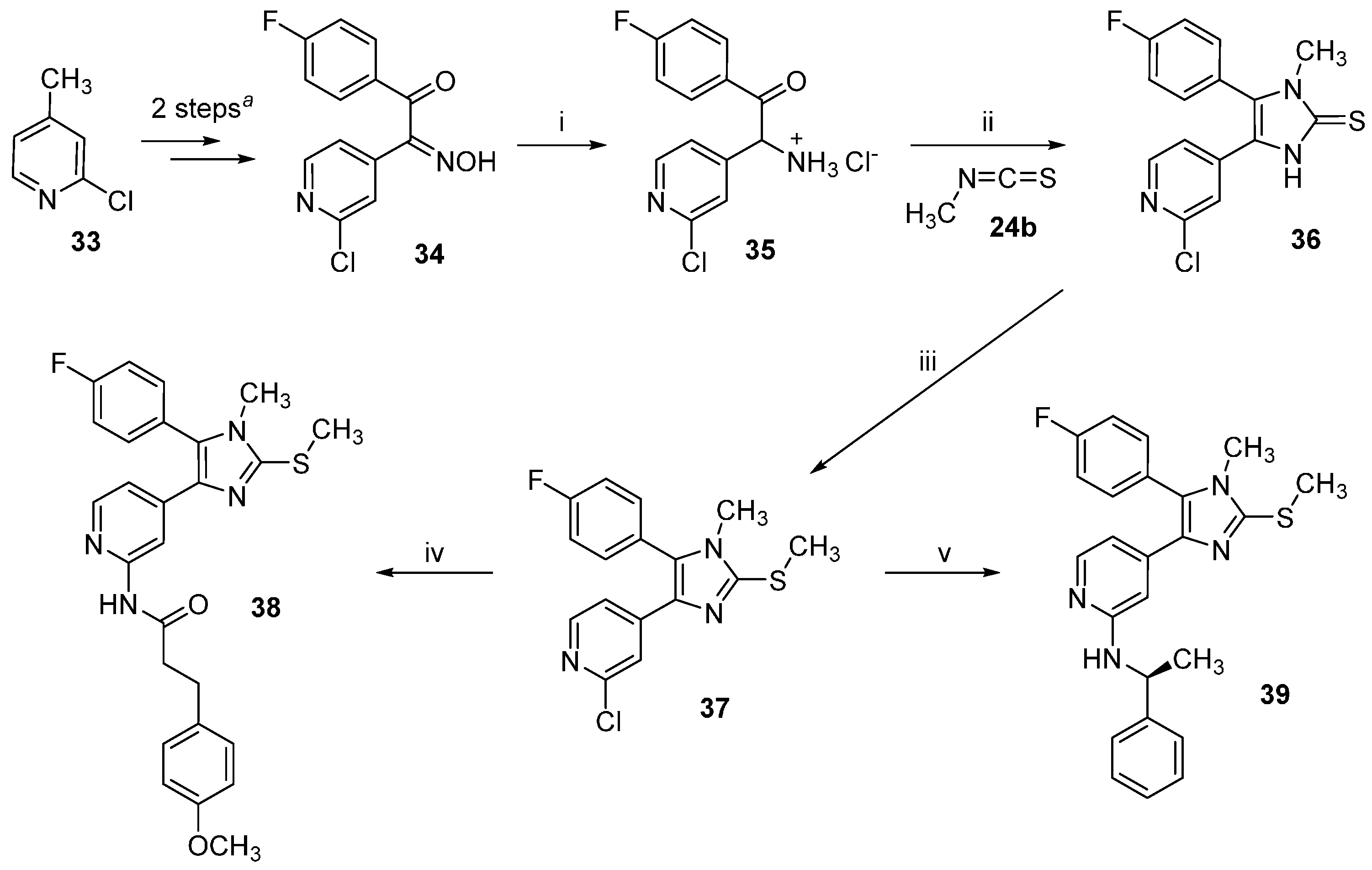
2-Amino-2-(2-chloropyridin-4-yl)-1-(4-fluorophenyl)ethan-1-one hydrochloride (
35) [
13,
30]. Oxime
34 [
13,
30] (500 mg, 1.8 mmol) and 10% Pd on charcoal (100 mg, 0.095 mmol) were placed in a Schlenk reaction tube, which was then evacuated and filled with a H
2 atmosphere. Isopropanolic HCl (10 mL) was then added and the mixture was vigorously stirred at rt under a constant supply of H
2 for 1 h. The solvent was evaporated at reduced pressure and then MeOH was added. The Pd catalyst was removed by filtration and the filtrate was concentrated at reduced pressure. Finally, the residue was treated with EtOAc and the white solid obtained was filtered off and dried, affording 520 mg of the desired product, which were used for the following step without further purification (92% yield); ESI-MS: (
m/
z) 264.9 [M + H]
+, 262.9 [M − H]
−; HPLC: t
r = 2.486 min.
4-(2-Chloropyridin-4-yl)-5-(4-fluorophenyl)-1-methyl-1,3-dihydro-2H-imidazole-2-thione (36). The title compound was prepared following general procedure A starting from compound 35 (550 mg, 1.82 mmol) and methyl isothiocyanate (24b, 665 mg, 9.1 mmol); the residue was then purified by flash column chromatography (SiO2, DCM:EtOH gradient elution from 100:0 to 95:05) obtaining 267 mg of the desired product (46% yield); 1H-NMR (300 MHz, DMSO-d6) δ 3.27 (s, 3H), 7.03 (dd, J = 5.3, 1.6 Hz, 1H), 7.32 (d, J = 1.1 Hz, 1H), 7.37–7.48 (m, 2H), 7.51–7.63 (m, 2H), 8.22 (d, J = 5.3 Hz, 1H), 13.09 (br. s, 1H); ESI-MS: (m/z) 318.0 [M − H]−; HPLC: tr = 5.864 min.
2-Chloro-4-(5-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-4-yl)pyridine (37). The title compound was prepared following general procedure B starting from compound 36 (267 mg, 0.83 mmol), t-BuONa (96.1 mg, 1.0 mmol), and iodomethane (639 mg, 4.15 mmol) obtaining 255 mg of the desired product, which was used for the following step without further purification (92% yield); 1H-NMR (300 MHz, CDCl3) δ 2.65 (s, 3H), 3.28 (s, 3H), 7.03 (d, J = 5.0 Hz, 1H), 7.08–7.31 (m, 4H), 7.44 (s, 1H), 8.02 (d, J = 5.2 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ 15.6, 31.3, 116.7 (d, J = 21.6 Hz), 118.7, 120.4, 125.6 (d, J = 3.3 Hz), 132.3 (d, J = 8.3 Hz), 132.5, 134.3, 144.7, 144.9, 149.2, 151.7, 163.3 (d, J = 251.0 Hz); ESI-MS: (m/z) 334.0 [M + H]+; HPLC: tr = 8.065 min.
N-(4-(5-(4-Fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(4-methoxyphenyl)-propanamide (38). The title compound was prepared according to general procedure C starting from imidazole 37 (90 mg, 0.27 mmol), 3-(4-methoxyphenyl)propanamide (73 mg, 0.41 mmol), Pd2(dba)3 (12 mg, 0.013 mmol), XantPhos (16 mg, 0.027 mmol), and Cs2CO3 (264 mg, 0.81 mmol). The residue was purified by flash column chromatography (SiO2, DCM:EtOH gradient elution from 100:0 to 95:05) affording 82 mg of the desired product (64% yield); 1H-NMR (300 MHz, CDCl3) δ 2.59 (t, J = 7.7 Hz, 2H), 2.76 (s, 3H), 2.94 (t, J = 7.6 Hz, 2H), 3.37 (s, 3H), 3.78 (s, 3H), 6.82 (d, J = 8.6 Hz, 2H), 7.05–7.15 (m, 3H), 7.18–7.27 (m, 2H), 7.29–7.38 (m, 2H), 8.00 (d, J = 5.4 Hz, 1H), 8.21–8.42 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 15.7, 30.3, 31.3, 39.6, 55.2, 110.8, 113.9, 116.5 (d, J = 21.6 Hz), 116.8, 125.9 (d, J = 3.3 Hz), 129.3, 132.2, 132.4 (d, J = 8.3 Hz), 132.6, 135.7, 144.4, 144.5, 146.9, 151.5, 158.0, 163.3 (d, J = 246.6 Hz), 170.4; ESI-TOF-HRMS: (m/z) [M + H]+ calcd. for C26H25FN4O2S 477.1755, found 477.1763; HPLC: tr = 7.745 min.
(S)-4-(5-(4-Fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-4-yl)-N-(1-phenylethyl)pyridin-2-amine (39). The title compound was prepared according to general procedure D starting from compound 21a (55 mg, 0.16 mmol). The reaction was stopped after 40 h although not fully completed. The residue was then purified by flash column chromatography (SiO2, n-hexane:EtOAc gradient elution from 4:1 to 1:1) obtaining 39 mg of the desired product (58% yield); 1H-NMR (300 MHz, CDCl3) δ 1.39 (d, J = 6.8 Hz, 3H), 2.60 (s, 3H), 3.22 (s, 3H), 4.37–4.50 (m, 1 H), 4.91 (d, J = 6.4 Hz, 1H), 6.30 (s, 1H), 6.57 (dd, J = 5.5, 1.4 Hz, 1H), 6.98–7.31 (m, 9H overlapping with the solvent peak), 7.80 (d, J = 5.4 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ 15.8, 24.2, 31.2, 51.7, 103.4, 110.9, 116.3 (d, J = 21.6 Hz), 125.8, 126.3 (d, J = 3.9 Hz), 126.8, 128.4, 131.3, 132.4 (d, J = 8.3 Hz), 136.1, 143.1, 143.8, 144.6, 147.6, 158.0, 163.0 (d, J = 249.9 Hz); ESI-TOF-HRMS: (m/z) [M + H]+ calcd. for C24H23FN4S 419.1700, found 419.1706; HPLC: tr = 6.899 min.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






