Structure Related Inhibition of Enzyme Systems in Cholinesterases and BACE1 In Vitro by Naturally Occurring Naphthopyrone and Its Glycosides Isolated from Cassia obtusifolia



Abstract
:1. Introduction
2. Results
2.1. Inhibitory Activities of Rubrofusarin and Its Derivatives against AChE, BChE, and BACE1
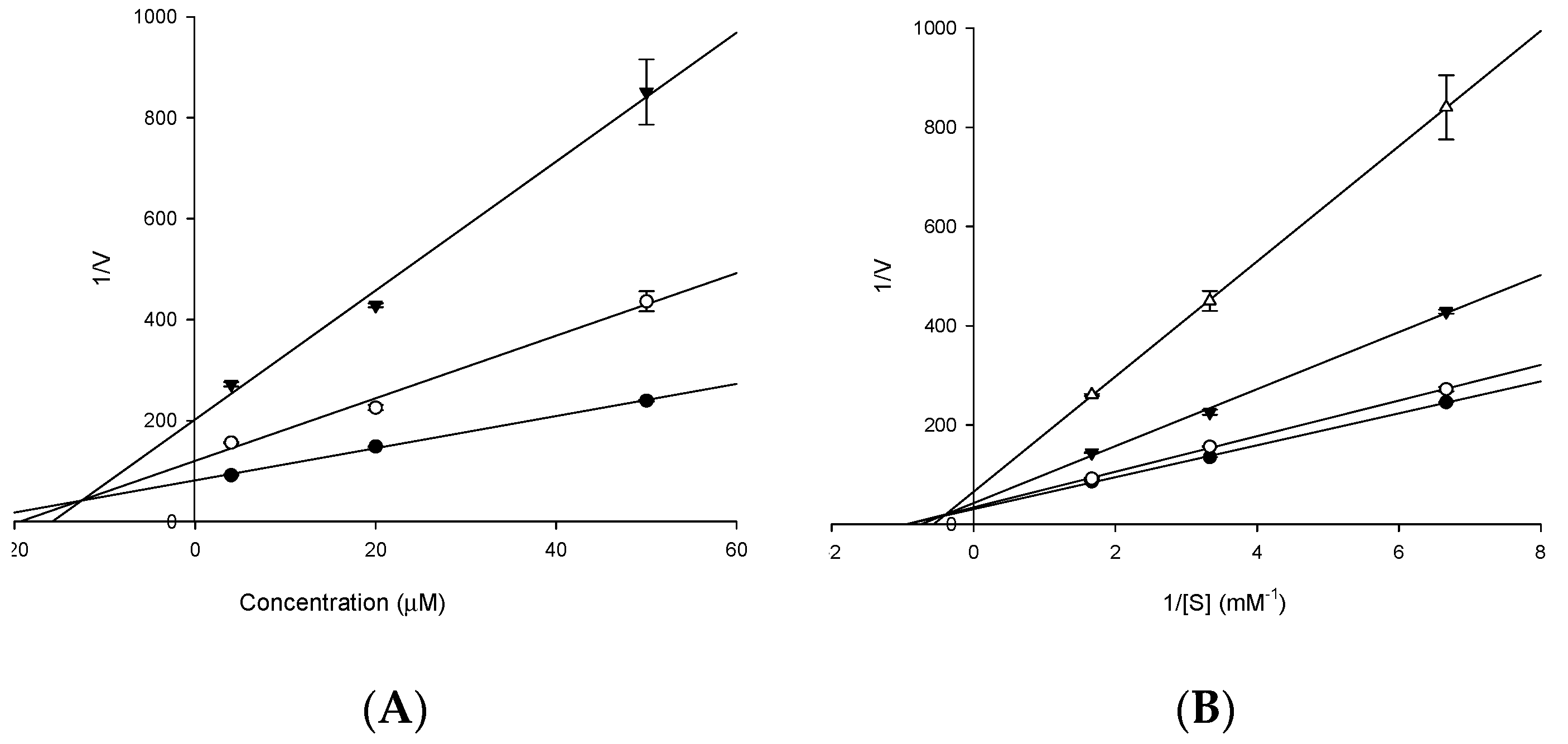
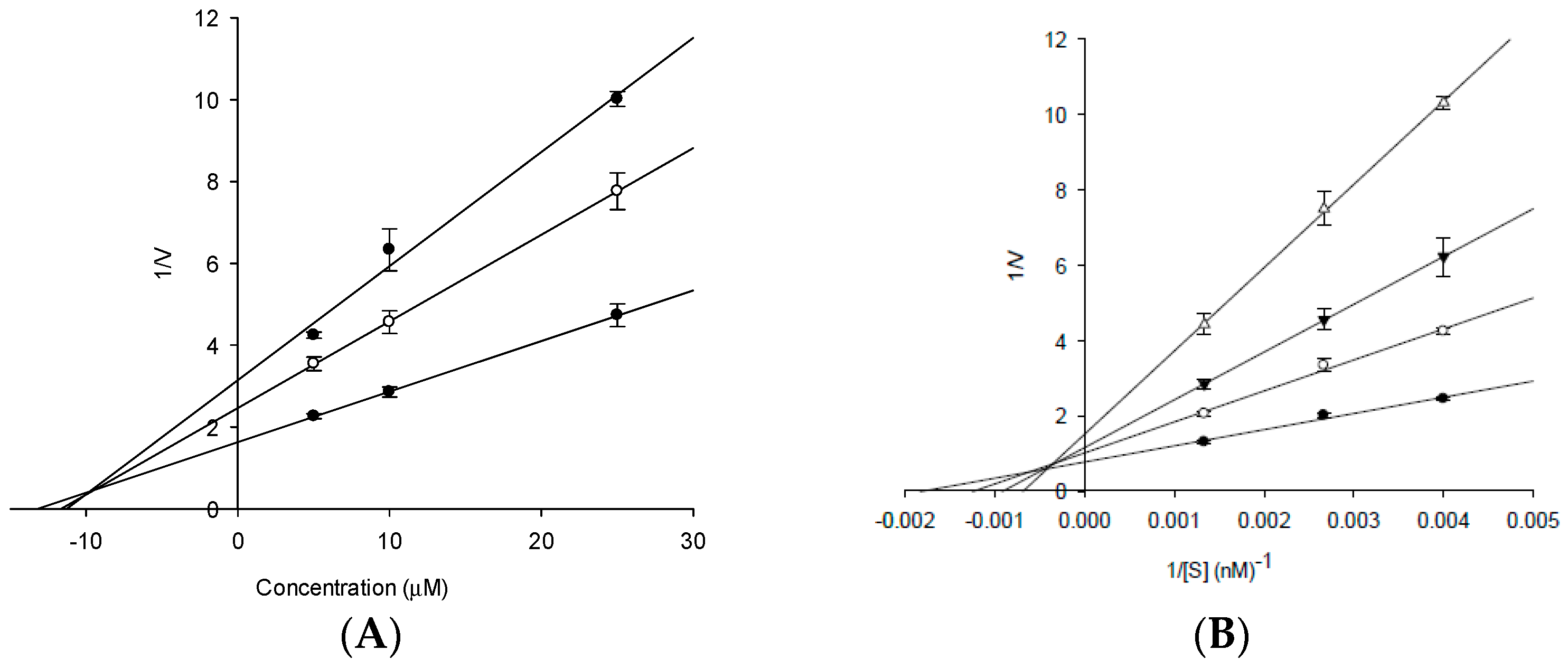
2.2. Enzyme Kinetic Analysis with AChE and BACE1
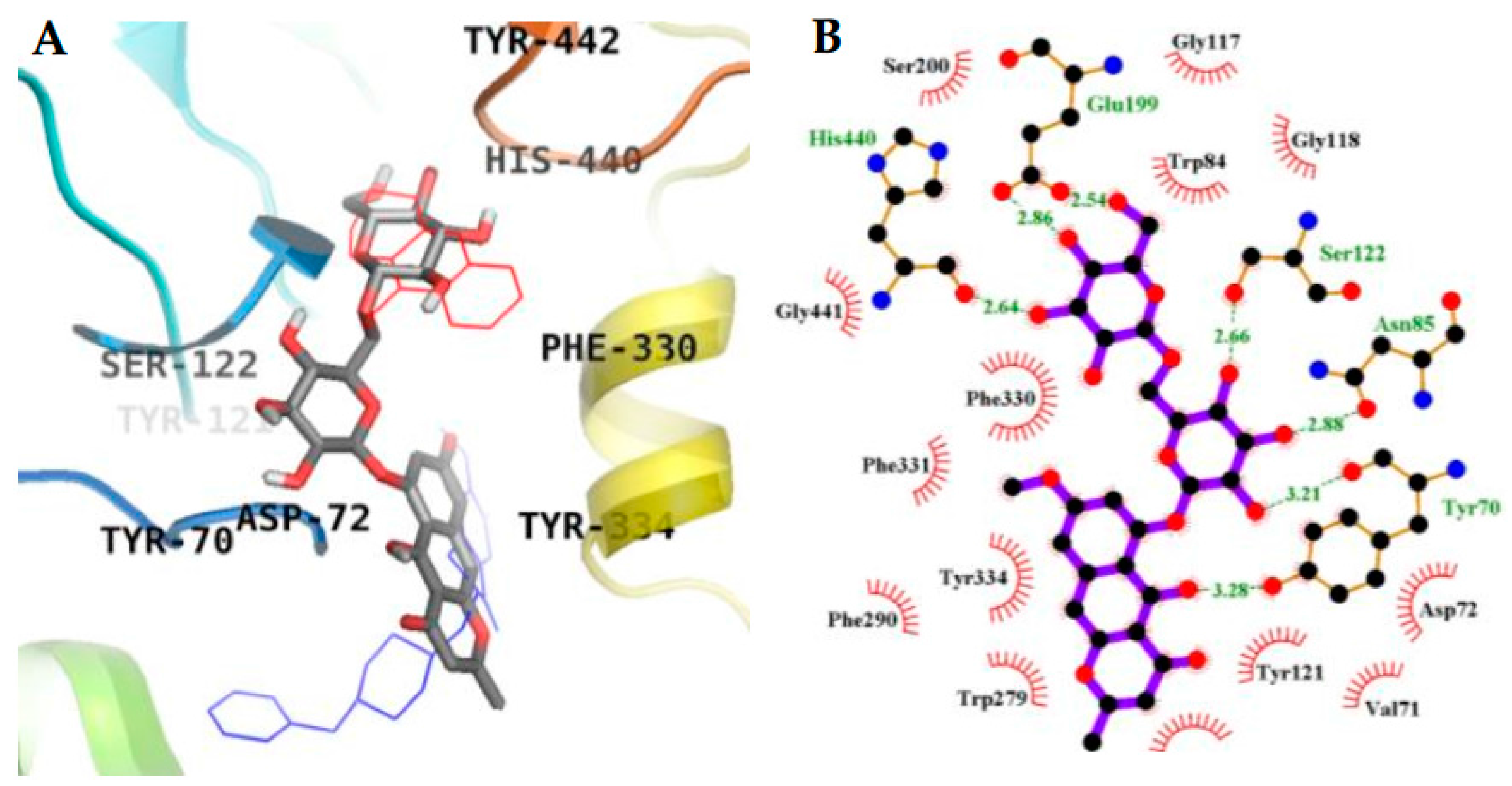
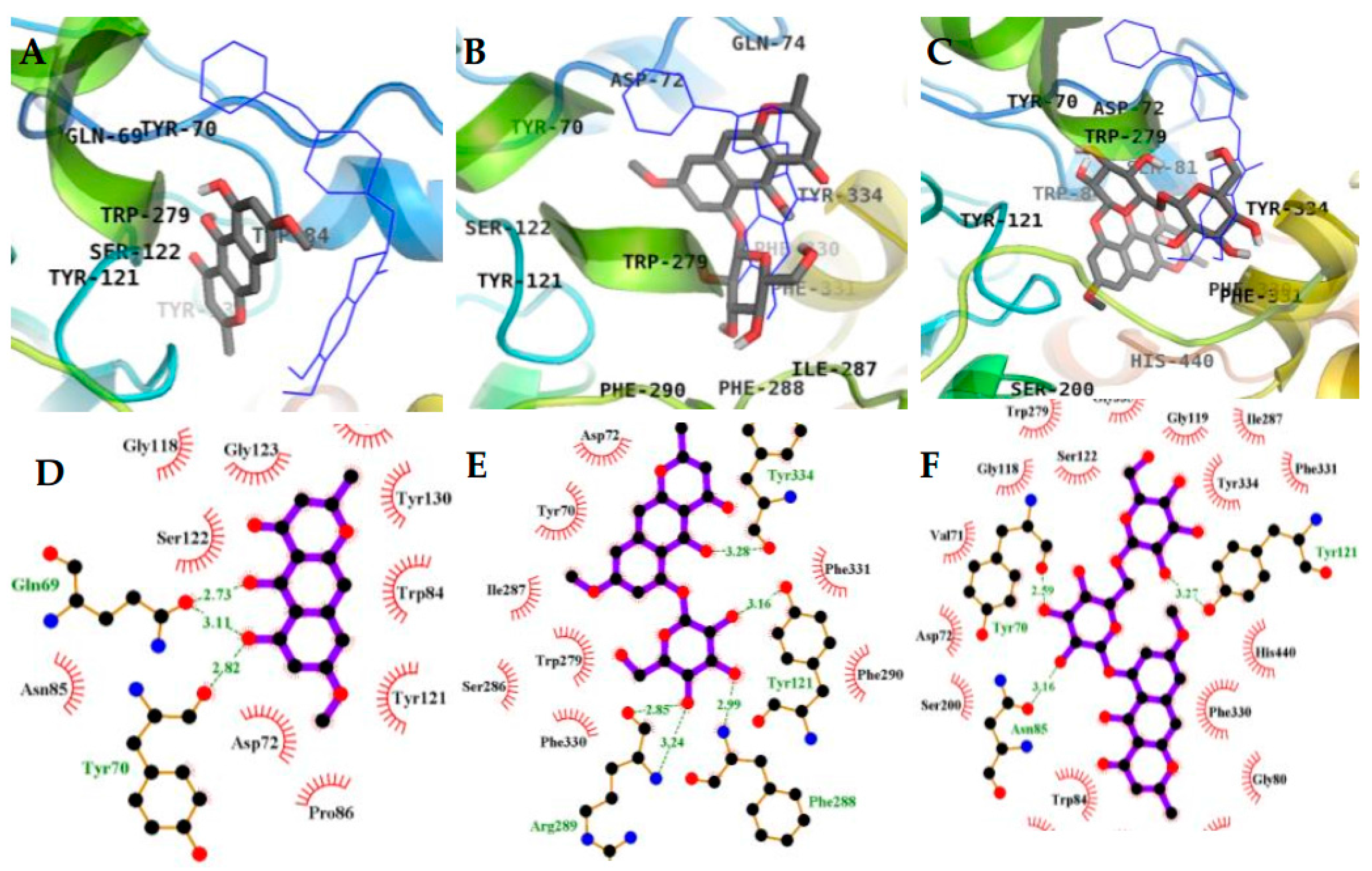
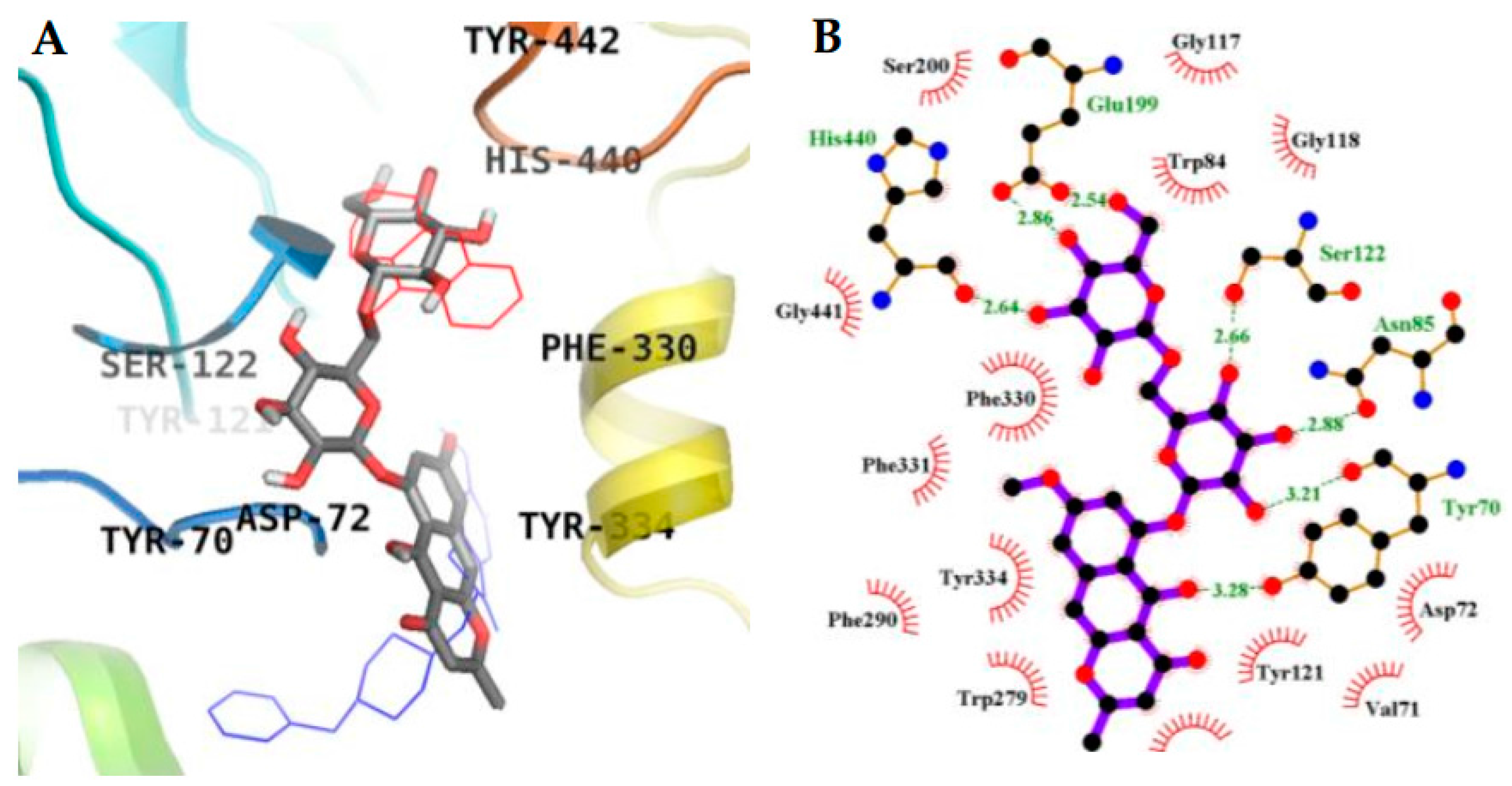
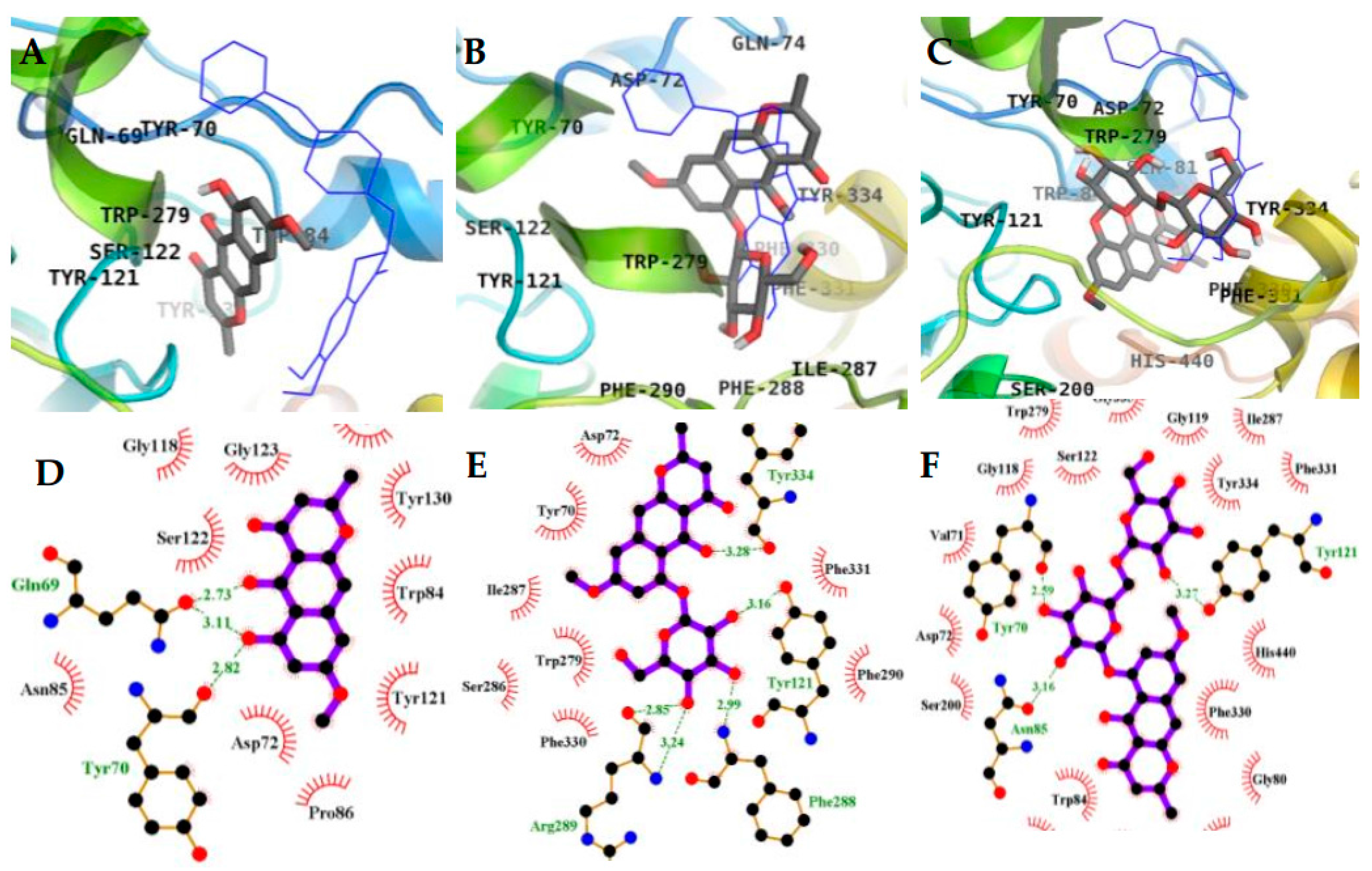
2.3. Molecular Docking Simulation in AChE inhibition
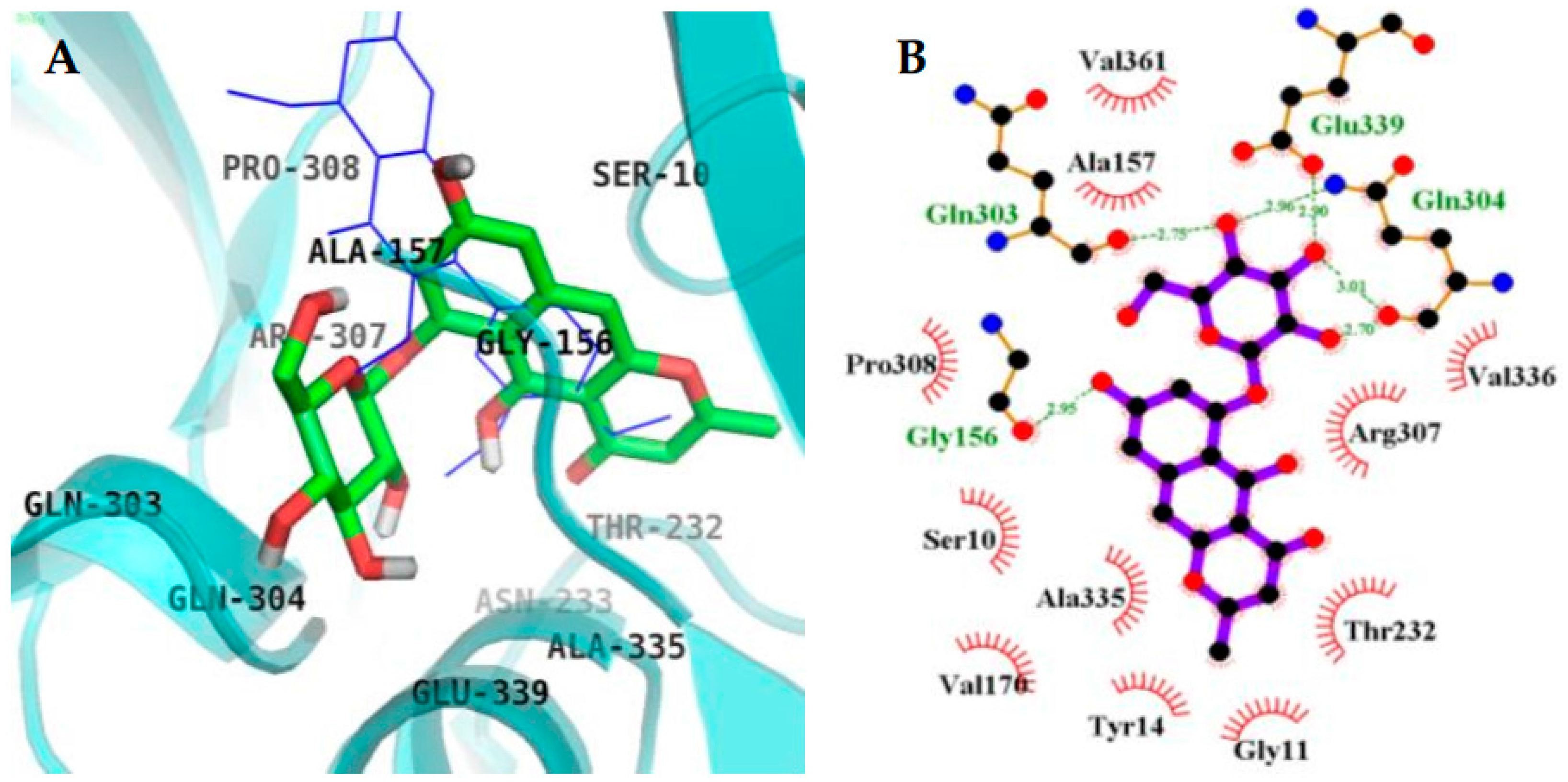
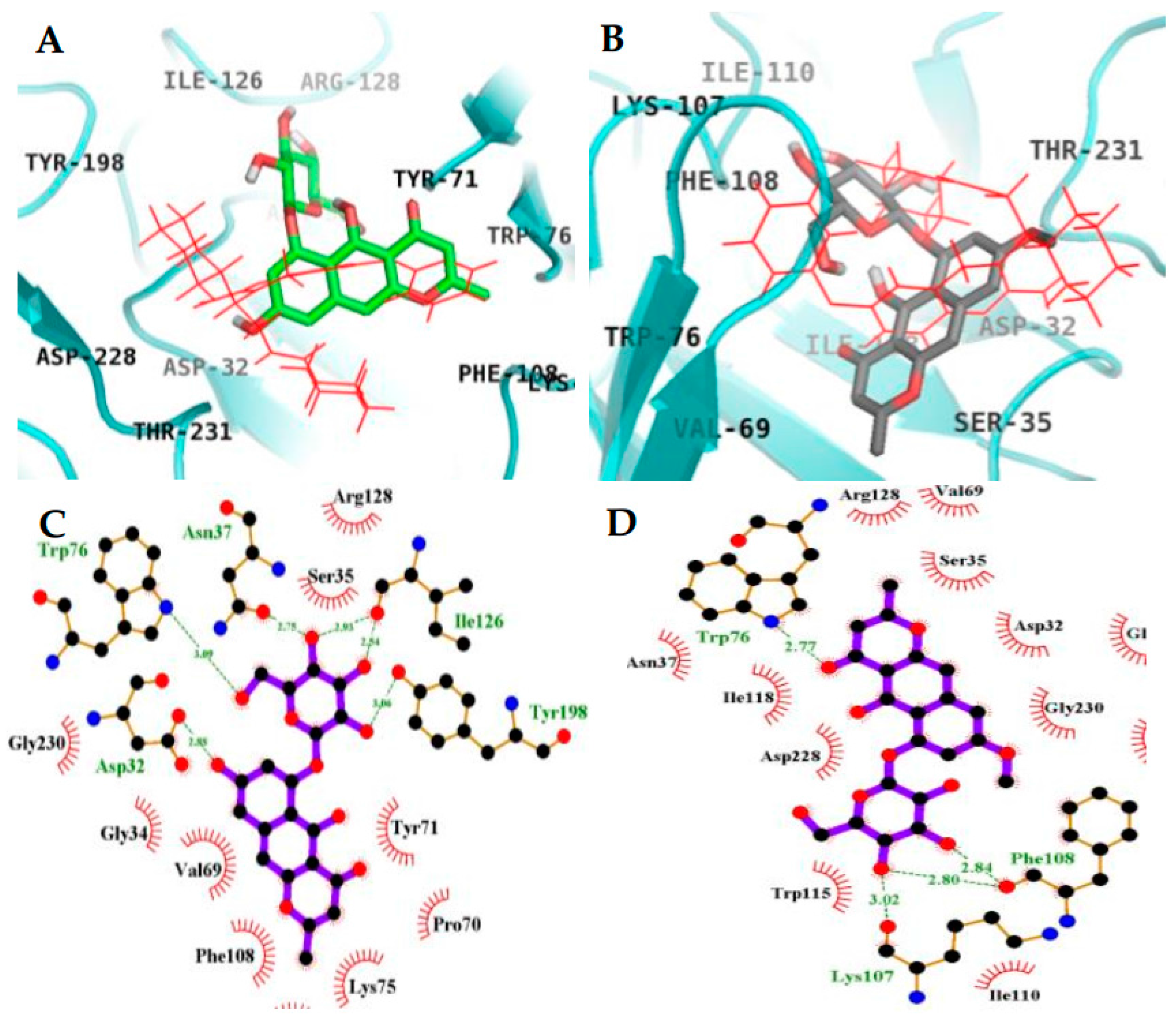
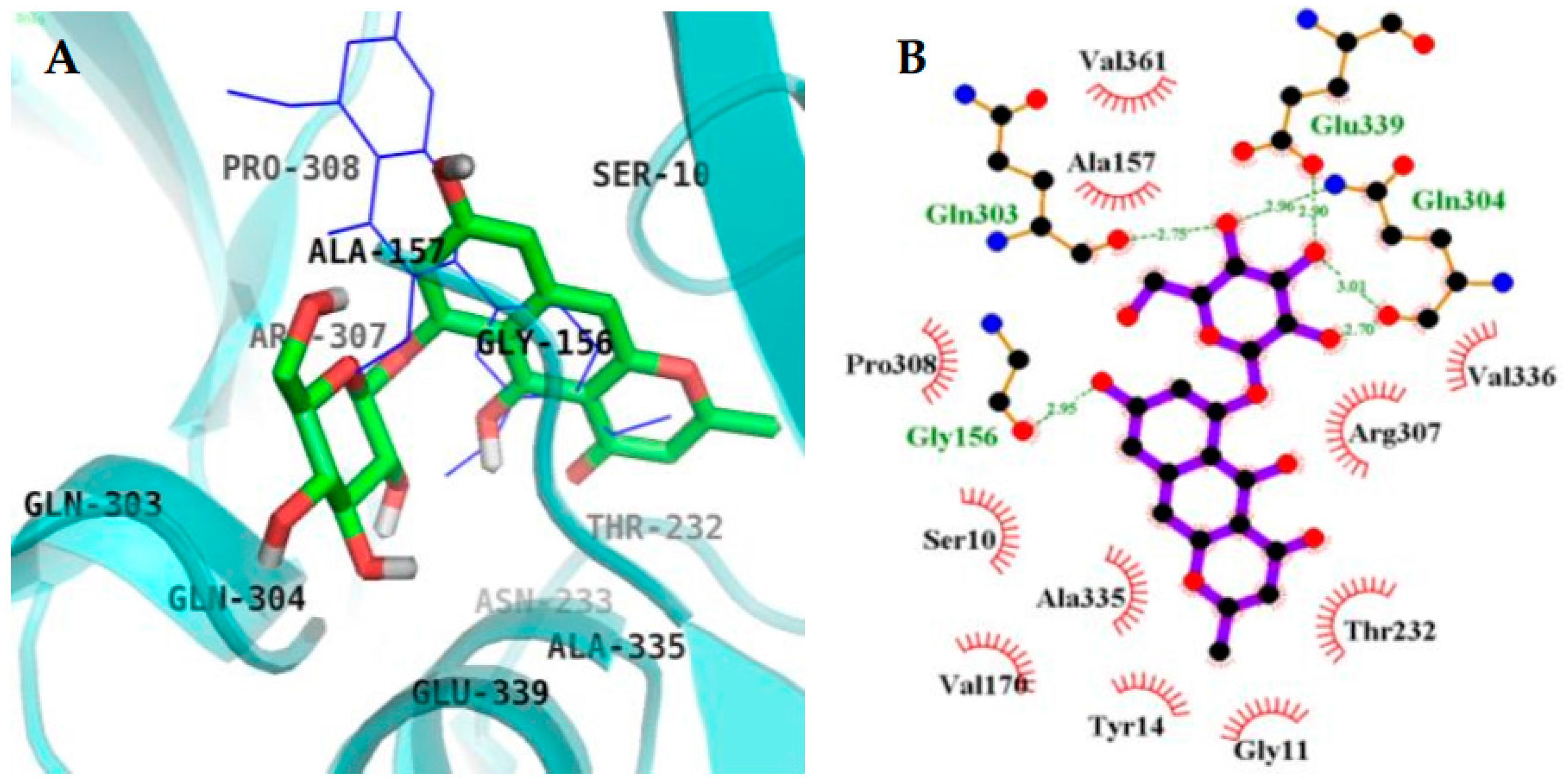
2.4. Molecular Docking Simulation in BACE1 Inhibition
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Chemicals and Reagents
4.3. Plant Material
4.4. Extraction, Fractionation, and Isolation
4.5. In Vitro Ches Enzyme Assay
4.6. In Vitro BACE1 Enzyme Assay
4.7. Kinetic Parameters of 3 towards AChE and 4 towards BACE1 Inhibition
4.8. AChE and BACE1 Molecular Docking Simulations
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alzheimer’s Disease International. World Alzheimer Report 2015. Available online: https://www.alz.co.uk/research/worldAlzheimerReport2015.pdf (accessed on 1 September 2017).
- Chertkow, H. Diagnosis and treatment of dementia: Introduction. Introducing a series based on the third Canadian consensus conference on the diagnosis and treatment of dementia. Can. Med. Assoc. J. 2008, 178, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R. BACE1: The beta-secretase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Chen, J.K.; Chen, T.T. Liver-calming and wind-extinguishing herbs. In Chinese Medical Herbology and Pharmacology; Art of Medicine Press: City of Industry, CA, USA, 2004; pp. 803–804. [Google Scholar]
- Yen, G.C.; Chuang, D.Y. Antioxidant properties of water extracts from Cassia tora L. in relation to the degree of roasting. J. Agric. Food Chem. 2010, 48, 2760–2765. [Google Scholar] [CrossRef]
- Ju, M.S.; Kim, H.G.; Choi, J.G.; Ryu, J.H.; Hur, J.; Kim, Y.J.; Oh, M.S. Cassiae semen, a seed of Cassia obtusifolia, has neuroprotective effects in Parkinson’s disease models. Food Chem. Toxicol. 2010, 48, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Yoon, B.H.; Kim, Y.W.; Lee, S.; Shin, B.Y.; Jung, J.W.; Kim, H.J.; Lee, Y.S.; Choi, J.S.; Kim, S.Y.; et al. The seed extract of Cassia obtusifolia ameliorates learning and memory impairments induced by scopolamine or transient cerebral hypoperfusion in mice. J. Pharmacol. Sci. 2007, 105, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Park, H.J.; Lee, S.; Jung, J.W.; Kim, B.C.; Lee, Y.C.; Ryu, J.H.; Kim, D.H. Cassia obtusifolia seed ameliorates amyloid β-induced synaptic dysfunction through anti-inflammatory and Akt/GSK-3β pathways. J. Ethnopharmacol. 2016, 178, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Drever, B.D.; Anderson, W.G.; Riedel, G.; Kim, D.H.; Ryu, J.H.; Choi, D.Y.; Platt, B. The seed extract of Cassia obtusifolia offers neuroprotection to mouse hippocampal cultures. J. Pharmacol. Sci. 2008, 107, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Kitanaka, S.; Takido, M. Studies on the constituents in the roots of Cassia obtusifolia L. and the antimicrobial activities of constituents of the roots and the seeds. Yakugaku Zasshi (J. Pharm. Soc. Jpn.) 1986, 106, 302–306. [Google Scholar] [CrossRef]
- Jang, D.S.; Lee, G.Y.; Kim, Y.S.; Lee, Y.M.; Kim, C.S.; Yoo, J.L.; Kim, J.S. Anthraquinones from the seeds of Cassia tora with inhibitory activity on protein glycation and aldose reductase. Biol. Pharm. Bull. 2007, 30, 2207–2210. [Google Scholar] [CrossRef] [PubMed]
- Megawati; Dewi, R.T.; Mulyani, H.; Maryani, F.; Lotullung, P.N.D.; Minarti. Rubrofusarin from Aspergillus niger GTS01-4 and its biological activity. In AIP Conference Proceedings; American Institute of Physics: College Park, MD, USA, 2017; Volume 1803, p. 020026. [Google Scholar]
- Wong, S.M.; Wong, M.M.; Seligmann, O.; Wagner, H. New antihepatotoxic naphtho-pyrone glycosides from the seeds of Cassia tora. Planta Med. 1989, 55, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Jang, D.S.; Lee, Y.M.; Kim, J.M.; Kim, J.S. Naphthopyrone glucosides from the seeds of Cassia tora with inhibitory activity on advanced glycation end products (AGEs) formation. Arch. Pharm. Res. 2006, 29, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Lee, H.J.; Kang, S.S. Alaternin, cassiaside and rubrofusarin gentiobioside, radical scavenging principles from the seeds of Cassia tora on 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical. Arch. Pharm. Res. 1994, 17, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Park, T.H.; Kim, D.H.; Kim, C.H.; Jung, H.A.; Choi, J.S.; Lee, J.W.; Chung, H.Y. Peroxynitrite scavenging mode of alaternin isolated from Cassia tora. J. Pharm. Pharmacol. 2004, 56, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Ali, M.Y.; Jung, H.J.; Jeong, H.O.; Chung, H.Y.; Choi, J.S. Inhibitory activities of major anthraquinones and other constituents from Cassia obtusifolia against β-secretase and cholinesterases. J. Ethnopharmacol. 2016, 191, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Ali, M.Y.; Choi, J.S. Promising inhibitory effects of anthraquinones, naphthopyrone, and naphthalene glycosides from Cassia obtusifolia on α-glucosidase and human protein tyrosine phosphatases 1B. Molecules 2017, 22, 28. [Google Scholar] [CrossRef] [PubMed]
- El-Halawany, A.M.; Chung, M.H.; Nakamura, N.; Ma, C.M.; Nishihara, T.; Hattori, M. Estrogenic and anti-estrogenic activities of Cassia tora phenolic constituents. Chem. Pharm. Bull. 2007, 55, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.G.; Zhang, H.; Pendland, S.L.; Santarsiero, B.D.; Mesecar, A.D.; Cabieses, F.; Farnsworth, N.R. Antimycobacterial naphthopyrones from Senna oblique. J. Nat. Prod. 2004, 67, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Paudel, P.; Seong, S.H.; Min, B.S.; Choi, J.S. Structure-related protein tyrosine phosphatase 1B inhibition by naringenin derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- McKinney, J.D.; Richard, A.; Waller, C.; Newman, M.C.; Gerberick, F. The practice of structure-activity relationships (SAR) in toxicology. Toxicol. Sci. 2000, 56, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Guha, R. On exploring structure–activity relationships. Methods Mol. Biol. 2013, 993, 81–94. [Google Scholar] [PubMed]
- Schultz, T.W.; Dumont, J.N.; Sankey, F.D.; Schmoyer, R.L. Structure activity relationships of selected naphthalene derivatives. Ecotoxicol. Environ. Saf. 1983, 7, 191–203. [Google Scholar] [CrossRef]
- Ahmed, M.; Rocha, J.B.; Correa, M.; Mazzanti, C.M.; Zanin, R.F.; Morsch, V.M.; Schetinger, M.R. Inhibition of two different cholinesterases by tacrine. Chem. Biol. Interact. 2006, 162, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.W.; Ryu, G.; Park, S.H.; Kim, E.S.; Shin, J.; Roh, S.S.; Shin, H.C.; Lee, B.H. Anticholinesterase activity of plastoquinones from Sargassum sagamianum: Lead compounds for Alzheimer’s disease therapy. Phytother. Res. 2007, 21, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef]
- Jin, H.; Nguyen, T.; Go, M.L. Acetylcholinesterase and butyrylcholinesterase inhibitory properties of functionalized tetrahydroacridines and related analogs. Med. Chem. 2014, 4, 688–696. [Google Scholar] [CrossRef]
- Youn, K.; Lee, J.; Ho, C.T.; Jun, M. Discovery of polymethoxyflavones from black ginger (Kaempferia parviflora) as potential β-secretase (BACE1) inhibitors. J. Funct. Foods 2016, 20, 567–574. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Perez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase accelerates assembly of amyloid-β-peptides into Alzheimer’s fibrils: Possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef]
- Tricco, A.C.; Soobiah, C.; Lillie, E.; Perrier, L.; Chen, M.H.; Hemmelgarn, B.; Majumdar, S.R.; Straus, S.E. Efficacy of cognitive enhancers for Alzheimer’s disease: Protocol for a systematic review and network meta-analysis. Syst. Rev. 2012, 1, 31. [Google Scholar] [CrossRef] [PubMed]
- Schulz, V. Ginkgo extract or cholinesterase inhibitors in patients with dementia: What clinical trials and guidelines fail to consider. Phytomedicine 2003, 10, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Cerella, C.; Teiten, M.H.; Radogna, F.; Dicato, M.; Diederich, M. From nature to bedside: Pro-survival and cell death mechanisms as therapeutic targets in cancer treatment. Biotechnol. Adv. 2014, 32, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Gechev, T.S.; Hille, J.; Woerdenbag, H.J.; Benina, M.; Mehterov, N.; Toneva, V.; Fernie, A.R.; Mueller-Roeber, B. Natural products from resurrection plants: Potential for medical applications. Biotechnol. Adv. 2014, 32, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, M.I. From plants to pharmacy shelf: Natural products revival. Phytochem. Rev. 2016, 15, 511–513. [Google Scholar] [CrossRef]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Plant-derived epigenetic modulators for cancer treatment and prevention. Biotechnol. Adv. 2014, 32, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Weymouth-Wilson, A.C. The role of carbohydrates in biologically active natural products. Nat. Prod. Rep. 1997, 14, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Metwally, K.; Khalil, A.; Sallam, A.; Pratsinis, H.; Kletsas, D.; El, S.K. Structure–activity relationship investigation of methoxy substitution on anticancer pyrimido[4,5-c]quinolin-1(2H)-ones. Med. Chem. Res. 2013, 22, 4481–4491. [Google Scholar] [CrossRef]
- Sato, H.; Chuang, V.T.; Yamasaki, K.; Yamaotsu, N.; Watanabe, H.; Nagumo, K.; Anraku, M.; Kadowaki, D.; Ishima, Y.; Hirono, S.; et al. Differential effects of methoxy group on the interaction of curcuminoids with two major ligand binding sites of human serum albumin. PLoS ONE 2014, 9, e87919. [Google Scholar] [CrossRef] [PubMed]
- Yanagita, R.C.; Kamachi, H.; Kikumori, M.; Tokuda, H.; Suzuki, N.; Suenaga, K.; Nagai, H.; Irie, K. Effects of the methoxy group in the side chain of debromoaplysiatoxin on its tumor-promoting and anti-proliferative activities. Bioorg. Med. Chem. Lett. 2013, 23, 4319–4323. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Jannat, S.; Jung, H.A.; Choi, R.J.; Roy, A.; Choi, J.S. Anti-Alzheimer’s disease potential of coumarins from Angelica decursiva and Artemisia capillaris and structure-activity analysis. Asian Pac. J. Trop. Med. 2016, 9, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Hu, Y.; Kong, W.; Yang, M. Evaluation and structure-activity relationship analysis of a new series of arylnaphthalene lignans as potential anti-tumor agents. PLoS ONE 2014, 9, e93516. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. Methods Mol. Biol. 2008, 443, 365–382. [Google Scholar] [PubMed]
- Ma, D.L.; Chan, D.S.H.; Leung, C.H. Molecular docking for virtual screening of natural product databases. Chem. Sci. 2011, 2, 1656–1665. [Google Scholar] [CrossRef]
- Choi, J.S.; Islam, M.N.; Ali, M.Y.; Kim, E.J.; Kim, Y.M.; Jung, H.A. Effects of C-glycosylation on anti-diabetic, anti-Alzheimer’s disease and anti-inflammatory potential of apigenin. Food Chem. Toxicol. 2014, 64, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Barman, A.; Prabhakar, R. Elucidating the catalytic mechanism of β-secretase (BACE1): A quantum mechanics/molecular mechanics (QM/MM) approach. J. Mol. Graph. Model. 2013, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Rodríguez, M.; Correa-Basurto, J.; Gutiérrez, A.; Vitorica, J.; Rosales-Hernández, M.C. Asp32 and Asp228 determine the selective inhibition of BACE1 as shown by docking and molecular dynamics simulations. Eur. J. Med. Chem. 2016, 124, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Messana, I.; Ferrari, F.; Cavalcanti, M.S.; Morace, G. An anthraquinone and three naphthopyrone derivatives from Cassia pudibunda. Phytochemistry 1991, 30, 708–710. [Google Scholar] [CrossRef]
- Lee, H.J.; Jung, J.H.; Kang, S.S.; Choi, J.S. A rubrofusarin gentiobioside isomer from roasted Cassia tora. Arch. Pharm. Res. 1997, 20, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-stone, R.M. A new and rapid colorimetric determination of acetylcholineserase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Jung, H.A.; Jung, Y.J.; Hyun, S.K.; Min, B.S.; Kim, D.W.; Jung, J.H.; Choi, J.S. Selective cholinesterase inhibitory activities of a new monoterpene diglycoside and other constituents from Nelumbo nucifera stamens. Biol. Pharm. Bull. 2010, 33, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef] [PubMed]
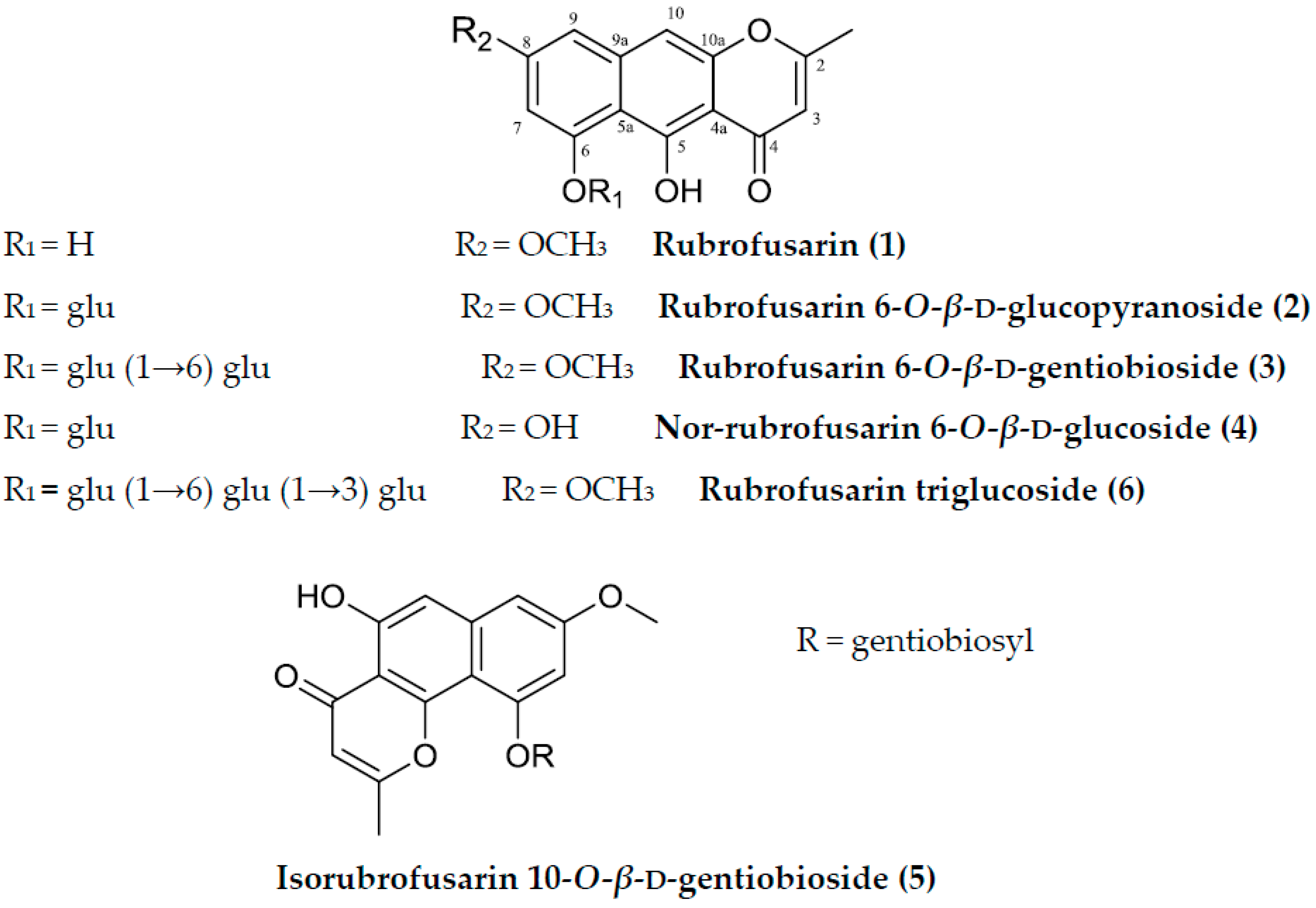
Sample Availability: Samples of the compounds (rubrofusarin, rubrofusarin 6-O-β-d-glucopyranoside, rubrofusarin 6-O-β-d-gentiobioside, nor-rubrofusarin 6-O-β-d-glucoside, isorubrofusarin 10-O-β-d-gentiobioside, and rubrofusarin 6-O-β-d-triglucoside) are available from the authors. |

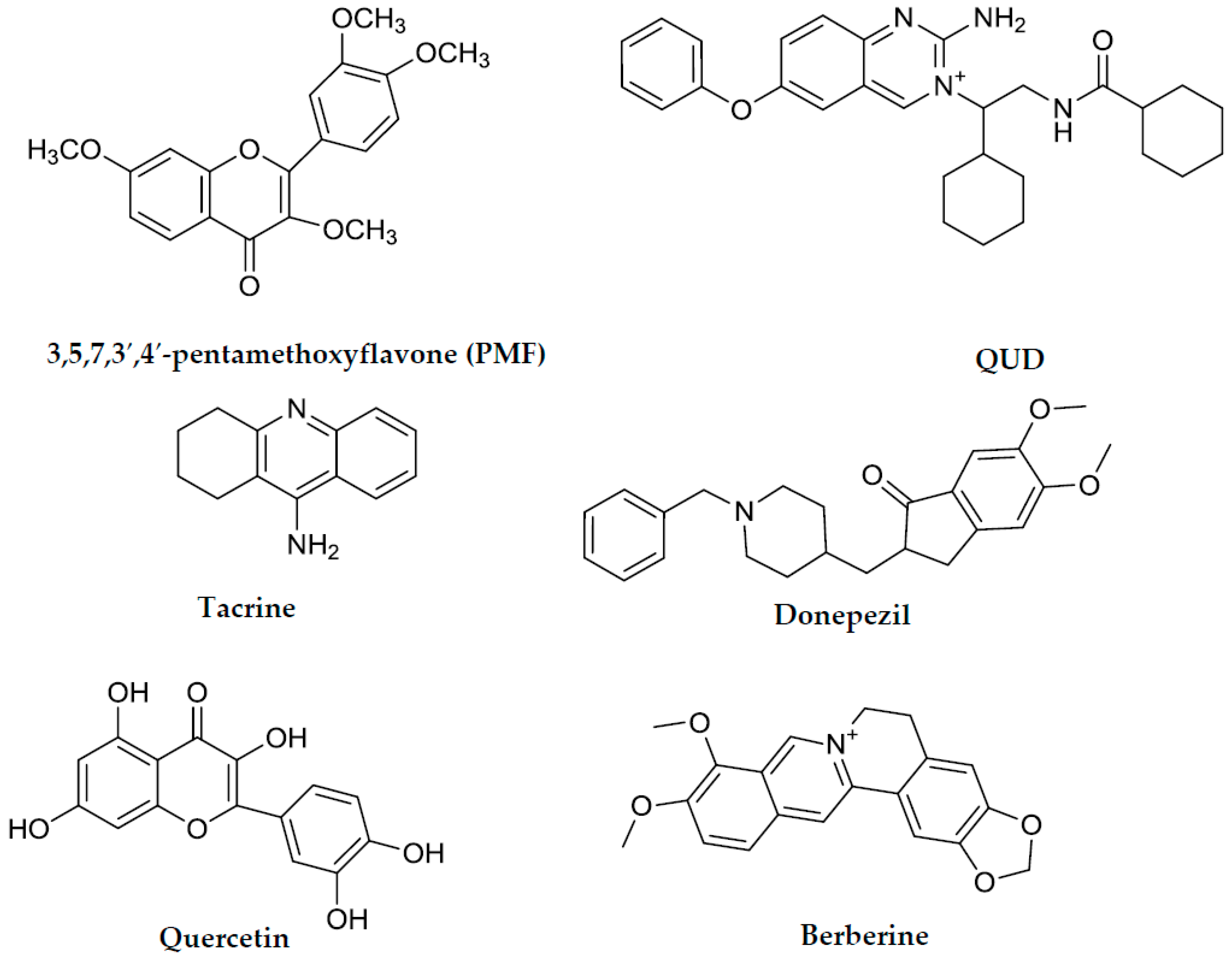




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | AChE | BChE | BACE1 | Ki Value | Inhibition Type |
|---|---|---|---|---|---|
| IC50 (µM) a | |||||
| 1 | 124.13 ± 1.39 | >200 | 90.01 ± 2.38 | NT | NT |
| 2 | 148.08 ± 2.09 | >200 | 190.63 ± 4.68 | NT | NT |
| 3 | 15.94 ± 0.32 | 141.15 ± 1.23 | 85. 66 ± 3.98 | 12.83 b | Mixed type c |
| 4 | 86.05 ± 2.01 | >200 | 14.41 ± 2.87 | 10.01 d | Mixed type e |
| 5 | 83.52 ± 1.56 | >200 | >200 | NT | NT |
| 6 | 82.31 ± 1.63 | >200 | >200 | NT | NT |
| Berberine g | 0.68 ± 0.01 | 25.77 ± 0.26 | NT | NT | NT |
| Tacrine g | 0.25 h | 0.01 h | NT | NT | Mixed type f |
| Donepezil g | 0.005 h | 1.74 h | NT | NT | NT |
| Querceting | NT | NT | 21.42 ± 1.04 | NT | NT |
| PMF g,i | NT | NT | 59.8 h | NT | NT |
| Compounds | Binding Energy (Kcal/mol) a | No. of H-Bonds | H-Bonds Interacting Residues | Van Der Waals Interacting Residues |
|---|---|---|---|---|
| 1 (Allosteric inhibition mode) | −7.95 | 3 | Gln69, Tyr70 | Asp72, Trp84, Asn85, Pro86, Gly117, Gly118, Tyr121, Ser122, Gly123, Leu127, Tyr130 |
| 2 (Allosteric inhibition mode) | −7.51 | 5 | Tyr121, Arg289, Tyr334, Phe288 | Tyr70, Asp72, Trp279, Ser286, Ile287, Phe290, Phe330, Phe331 |
| 3 (Mixed inhibition mode) | −9.06 | 7 | Tyr70, Asn85, Ser122, Glu199, His440 | Val71, Asp72, Gln74, Trp84, Gly117, Gly118, Tyr121, Ser200, Phe290, Phe330, Phe331, Tyr334, Gly441 |
| 3 (Allosteric inhibition mode) | −9.57 | 3 | Tyr70, Asn85, Tyr121 | Val71, Asp72, Gly80, Ser81, Trp84, Gly118, Gly119, Ser122, Ser200, Trp279, Ile287, Phe330, Phe331, Tyr334, Gly335, Trp432, Ile439, His440, Tyr442 |
| Tacrine b (Catalytic inhibitor) | −9.8 c | 1 | His440 | Tyr442, Phe330, Trp84, Trp432, Gly441, Glu199, Ile439 |
| Donepezil b (Allosteric inhibitor) | −10.6 | - | - | Tyr70, Ile275, Asp276, Trp279, Ile287, Phe288, Arg289, Tyr334, Tyr121, Ser286, Phe290, Phe330, Phe331 |
| Compounds | Binding Energy (Kcal/mol) a | No. Of H-Bonds | H-Bonds Interacting Residues | Van Der Waals Interacting Residues |
|---|---|---|---|---|
| 4 (Catalytic inhibition mode) | −6.61 | 6 | Asp32, Trp76, Asn37, Ile126, Tyr198 | Gly230, Gly34, Val69, Phe108, Asp106, Lys75, Pro70, Tyr71, Ser35, Arg128 |
| 4 (Allosteric inhibition mode) | −8.34 | 6 | Gln303, Gln304, Glu339, Gly156 | Ser10, Ala335, Val170, Tyr14, Gly13, Gly11, Thr232, Arg307, Val336, Val361, Ala157, Pro308 |
| 2 (Catalytic inhibition mode) | −5.38 | 4 | Trp76, Lys107, Phe108 | Asp32, Gly34, Ser35, Asn37, Val69, Ile110, Trp115, Ile118, Arg128, Asp228, Gly230, Thr231 |
| QUD b (Catalytic inhibitor) | −11.19c | 4 | Asp228, Asp32, Gly230 | Lys107, Lys75, Gly76, Leu30, Thr231, Val69, Tyr198, Ile226, Phe108, Gly34, Arg235, Ser35, Tyr71, Ile118 |
| PMF b (Allosteric inhibitor) | −6.5 | 1 | Ser10 | Gly11, Ala157, Ala168, Val170, Thr232, Gln304, Arg307, Pro308, Tyr320, Ala335, Glu339 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrestha, S.; Seong, S.H.; Paudel, P.; Jung, H.A.; Choi, J.S. Structure Related Inhibition of Enzyme Systems in Cholinesterases and BACE1 In Vitro by Naturally Occurring Naphthopyrone and Its Glycosides Isolated from Cassia obtusifolia. Molecules 2018, 23, 69. https://doi.org/10.3390/molecules23010069
Shrestha S, Seong SH, Paudel P, Jung HA, Choi JS. Structure Related Inhibition of Enzyme Systems in Cholinesterases and BACE1 In Vitro by Naturally Occurring Naphthopyrone and Its Glycosides Isolated from Cassia obtusifolia. Molecules. 2018; 23(1):69. https://doi.org/10.3390/molecules23010069
Chicago/Turabian StyleShrestha, Srijan, Su Hui Seong, Pradeep Paudel, Hyun Ah Jung, and Jae Sue Choi. 2018. "Structure Related Inhibition of Enzyme Systems in Cholinesterases and BACE1 In Vitro by Naturally Occurring Naphthopyrone and Its Glycosides Isolated from Cassia obtusifolia" Molecules 23, no. 1: 69. https://doi.org/10.3390/molecules23010069