Convergent Synthesis of N,S-bis Glycosylquinolin-2-ones via a Pd-G3-XantPhos Precatalyst Catalysis


1
Laboratoire de Physico-Chimie des Matériaux et Catalyse, Faculté des Sciences Exactes, Université de Bejaia, 0600 Bejaia, Algeria
2
BioCIS, Université Paris-Sud, CNRS, University Paris-Saclay, 92296 Châtenay-Malabry, France
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(3), 519; https://doi.org/10.3390/molecules23030519
Submission received: 29 January 2018
/
Revised: 8 February 2018
/
Accepted: 10 February 2018
/
Published: 26 February 2018
(This article belongs to the Special Issue Glycomimetics: Design, Synthesis and Therapeutic Applications)
Abstract
:Buchwald-Hartwig-Migita cross-coupling of 1-thiosugars with α- or β-3-iodo-N-glycosylquinolin-2-ones has been accomplished under mild and operationally simple reaction conditions through the use of a Pd-G3 XantPhos palladacycle precatalyst. This new methodology has been successfully applied to a variety of α- or β-mono-, di-, and poly-thiosugar derivatives to efficiently synthesize a series of α- or β-N,S-bis-glycosyl quinolin-2-ones, which are difficult to synthesize by classical methods.
1. Introduction
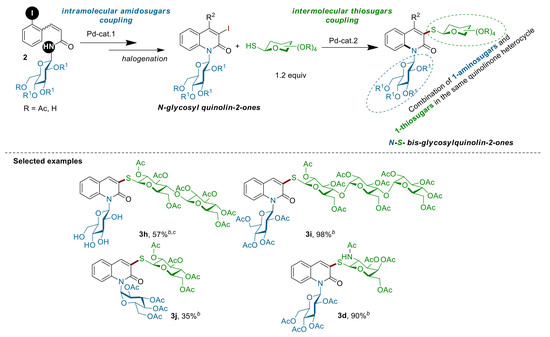
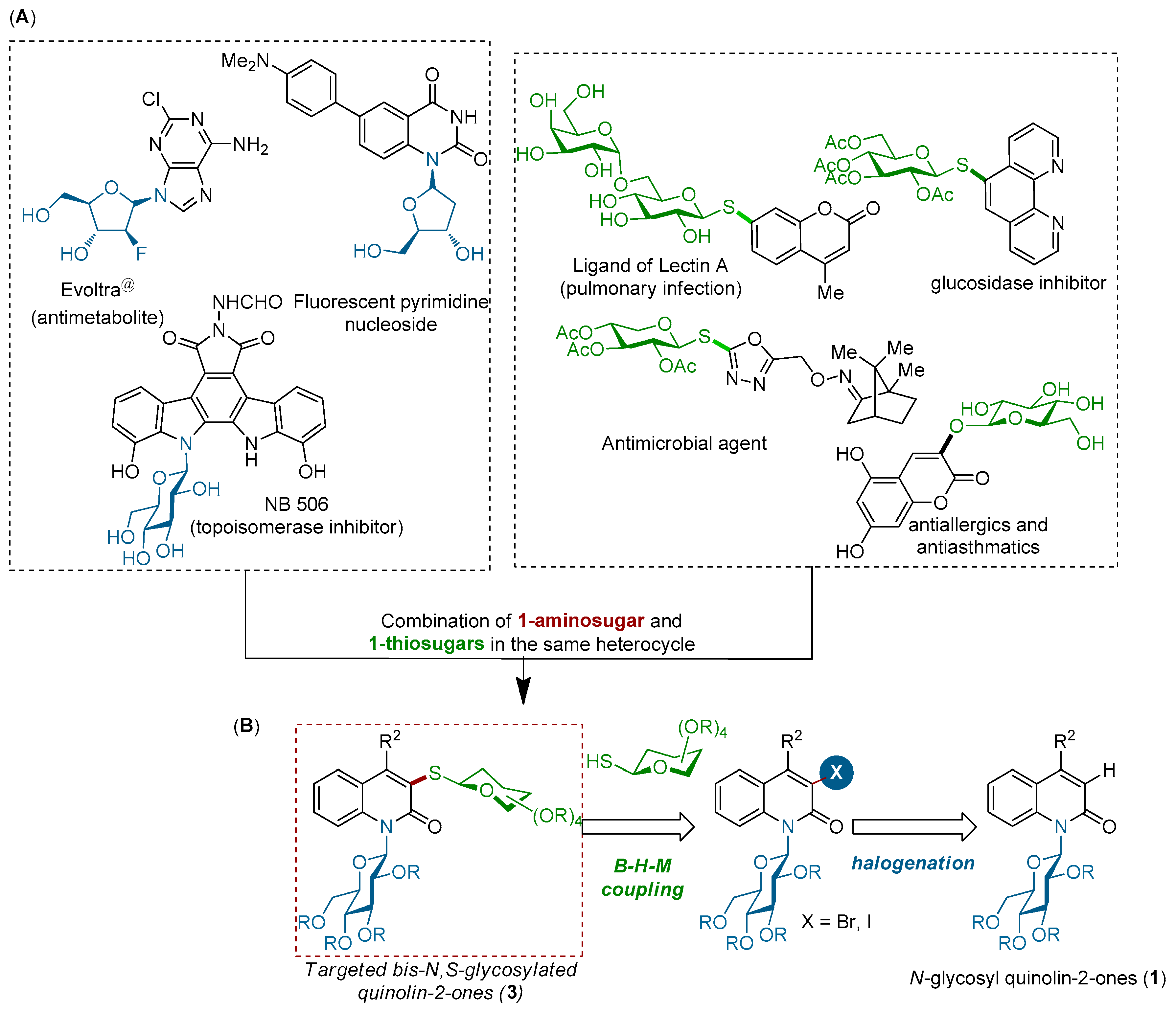
Heteroaryl-glycosides are of high importance in medicinal chemistry and commonly found in many compounds of enormous practical importance, ranging from natural compounds to pharmaceutical agents [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19] (Figure 1). While these derivatives clearly hold great potential in medicinal chemistry, relatively little attention has been devoted to the synthesis of heteroaryl-bis-glycosides bearing two different sugar moieties, such as thiosugars and aminosugars, in the same heterocycle. One of the most important subfamilies of heteroaryl-N-glycosides is N-glycosyl quinolin-2-ones, in which a glycosyl unit is attached to a quinolin-2-one core. Quinolin-2-(1H)-ones are present in many biologically active compounds and pharmaceutical agents [20,21,22,23,24,25,26,27]. Thus, the attachment of S- and N-glycosyl units to a quinolin-2-one nucleus can cause several changes in their features, including their chemical, physical, biochemical, and biological properties. Thus, developing a simple method for the synthesis of bis N,S-glycosyl quinolin-2-ones would be of great interest for the preparation of large libraries of new potentially active compounds. Recently, our group reported an efficient protocol for the synthesis of N-glycosyl quinolin-2-ones (Figure 1) via a palladium-catalyzed intramolecular N-arylation of various substituted (2-iodophenyl)-acrylamidosugars [28,29]. As part of our continued efforts to functionalize sugars under transition-metal catalysis to access complex glycosides [30,31,32,33,34,35,36], we envisioned whether N-glycosyl quinolin-2-ones of type (1) could be utilized as a building block in the synthesis of 3-thioglycosyl N1-glycosyl quinolin-2-ones (3) through C3-halogenation followed by the Pd(0)-catalyzed Buchwald-Hartwig-Migita (B-H-M) coupling reaction with various thiosugars (Figure 1). This modular strategy is conceptually attractive in terms of diversifying the N-glycosyl quinolinone frameworks with the aim of identifying novel scaffolds of biological interest. Herein, we report our success in the development of such a strategy.
2. Results and Discussion
2.1. Synthesis of Starting Materials
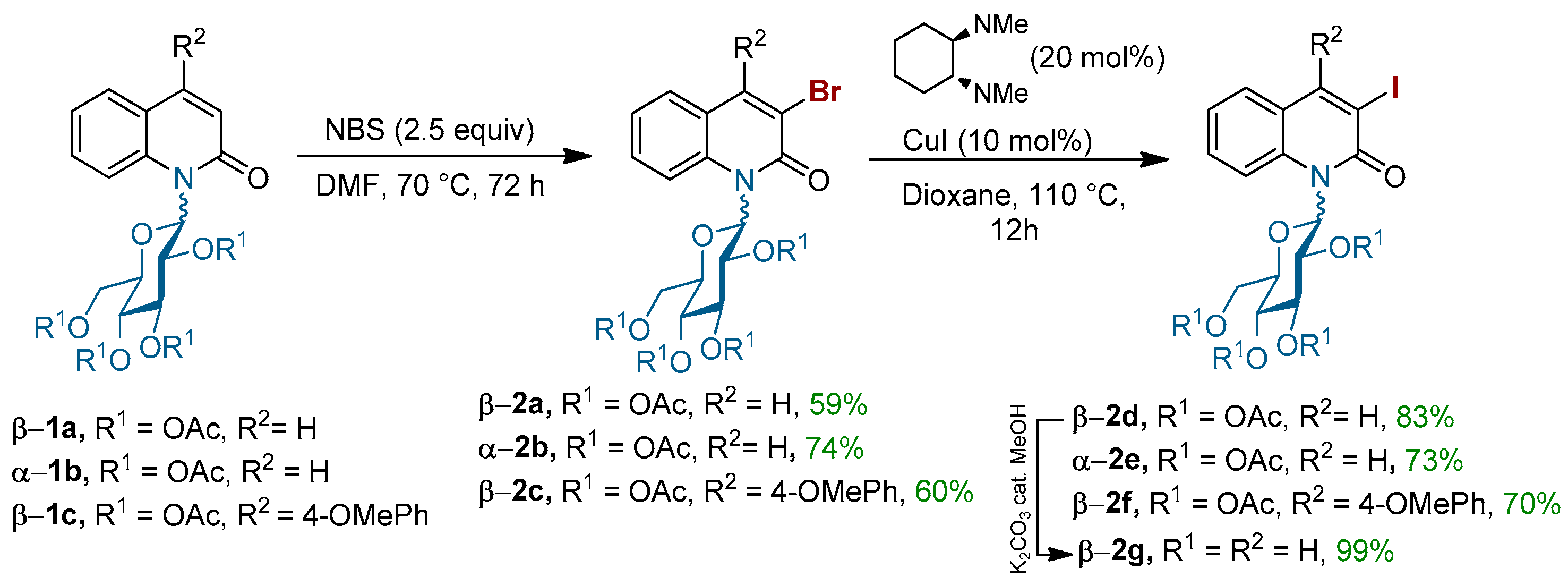
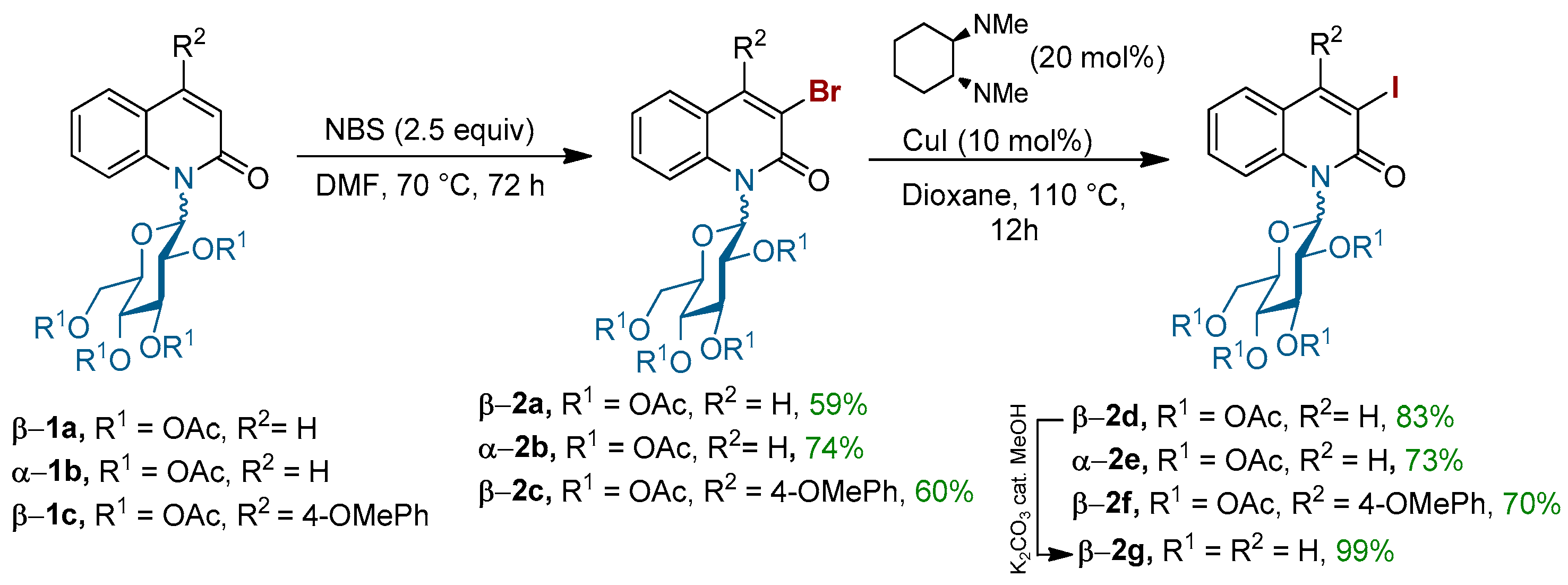
To establish the appropriate conditions for the coupling of 3-halo-N-glycosyl quinolin-2-ones with various thiosugars, we initially started our chemistry by the synthesis of the appropriate α- or β-3-halo-N-glycosyl quinolin-2-ones 2a–g (Scheme 1). The compounds 2a–c were prepared by the electrophilic regioselective aromatic bromination of N-glycosyl quinolin-2-one 1a–c using N-bromosuccinimide in anhydrous dimethylformamide (DMF). Under these conditions, 3-bromo-N-glucopyranosylquinolin-2-ones 2a–c were isolated in good yields (Scheme 1). To compare the reactivity of brominated quinolinones 2a–c with its analogue, in which the bromine atom is replaced by the iodine one, derivatives 2d–f were synthesized from 2a–c through a halogen exchange by a Cu-catalyzed Finkelstein reaction [37]. Finally, compound β-2g bearing an unprotected sugar was also prepared in order to study the influence of a free hydroxyls group on the outcome of the coupling.
2.2. Optimization of the Reaction Conditions on the Model Study
With these starting materials in hand, we turned our attention to explore the feasibility of the coupling of the quinolones β-2a and β-2d with tetra-O-acetylated 1-thio-β-d-galactopyranose 1a under various reaction conditions (Scheme 2). When β-2a and 1a were mixed under our previously reported conditions [38] (G3-XantPhos (5 mol %), Et3N (1.5 equiv.) in tetrahydrofuran (THF) at room temperature), only the starting material was recovered unchanged; however, when the reaction mixture was heated at 60 °C, product 3a was detected by NMR of the crude reaction mixture and the conversion rate was calculated to be around 35% (Table 1, entry 2). The conversion rate has never exceeded 50%, even when the amount of the thiogalactose 1a was increased until 2.5 equiv. and the reaction temperature was at 100 °C, probably due to the fact that the formation of disulfide dimer was faster than the coupling of product 3a. In the next set of experiences, we decided to use the iodinated quinolinone β-2d instead of β-2a. Delightfully, the coupling of β-2d with 1a in the presence of Pd-G3-XantPhos (5 mol %), with Et3N (1.2 equiv.) as the base in THF at room temperature, led to N-β-glycosyl S-β-galactosyl quinolin-2-one 3a (J1,2 = 9.9 Hz) in 70% yield (entry 3, Table 1). Decreasing the amount of thiogalactose 1a into 1.5 equiv. led to a lower conversion rate (40%, entry 4), indicating that the thiosugar concentration plays a critical role in the outcome of the reaction. It should be noted that the palladium catalyst is necessary to achieve this transformation, since no reaction occurs when the coupling is conducted in the absence of the Pd-G3-precatalyst.
2.3. Scope and Limitation of the Cross-Coupling
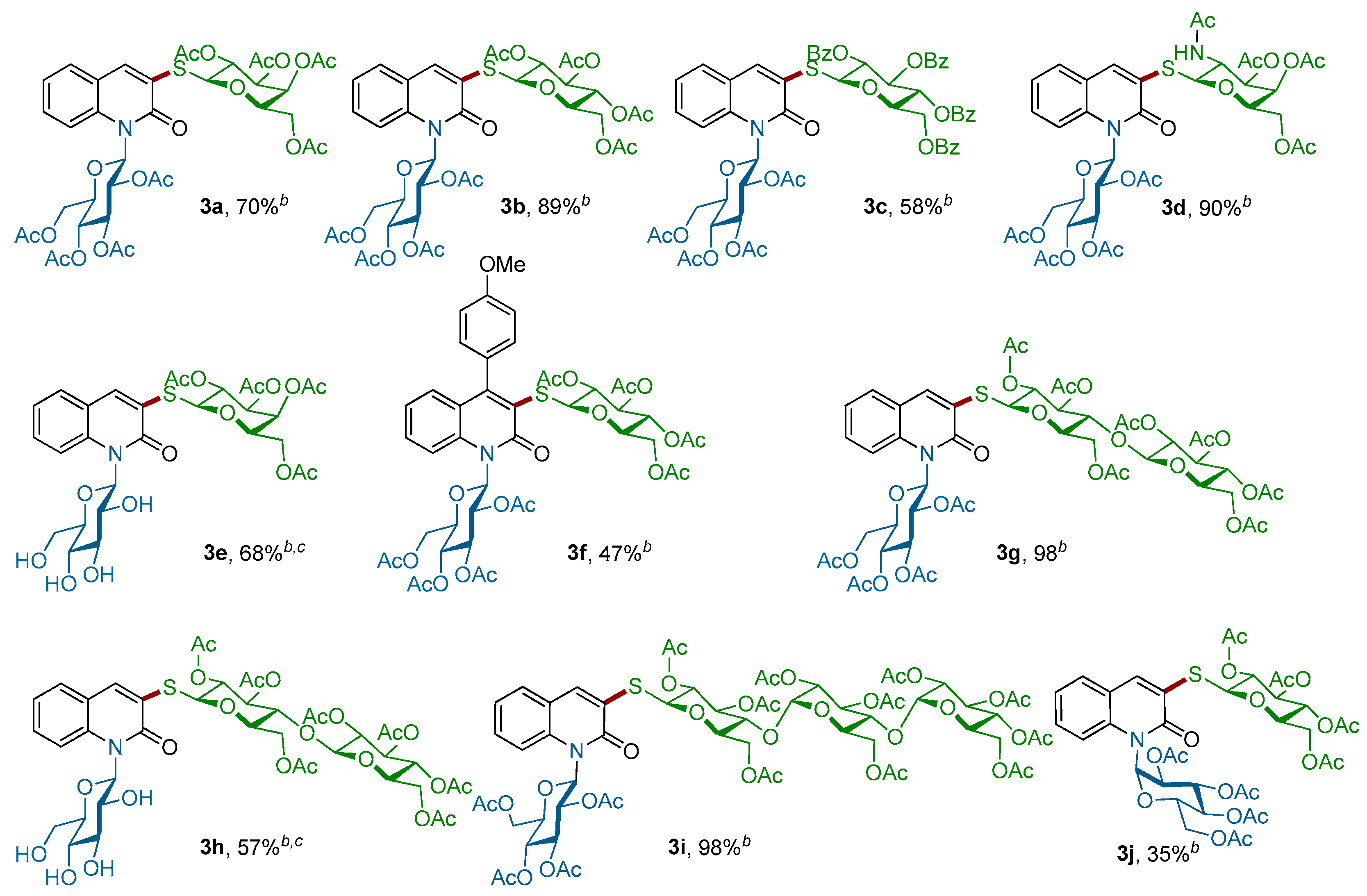
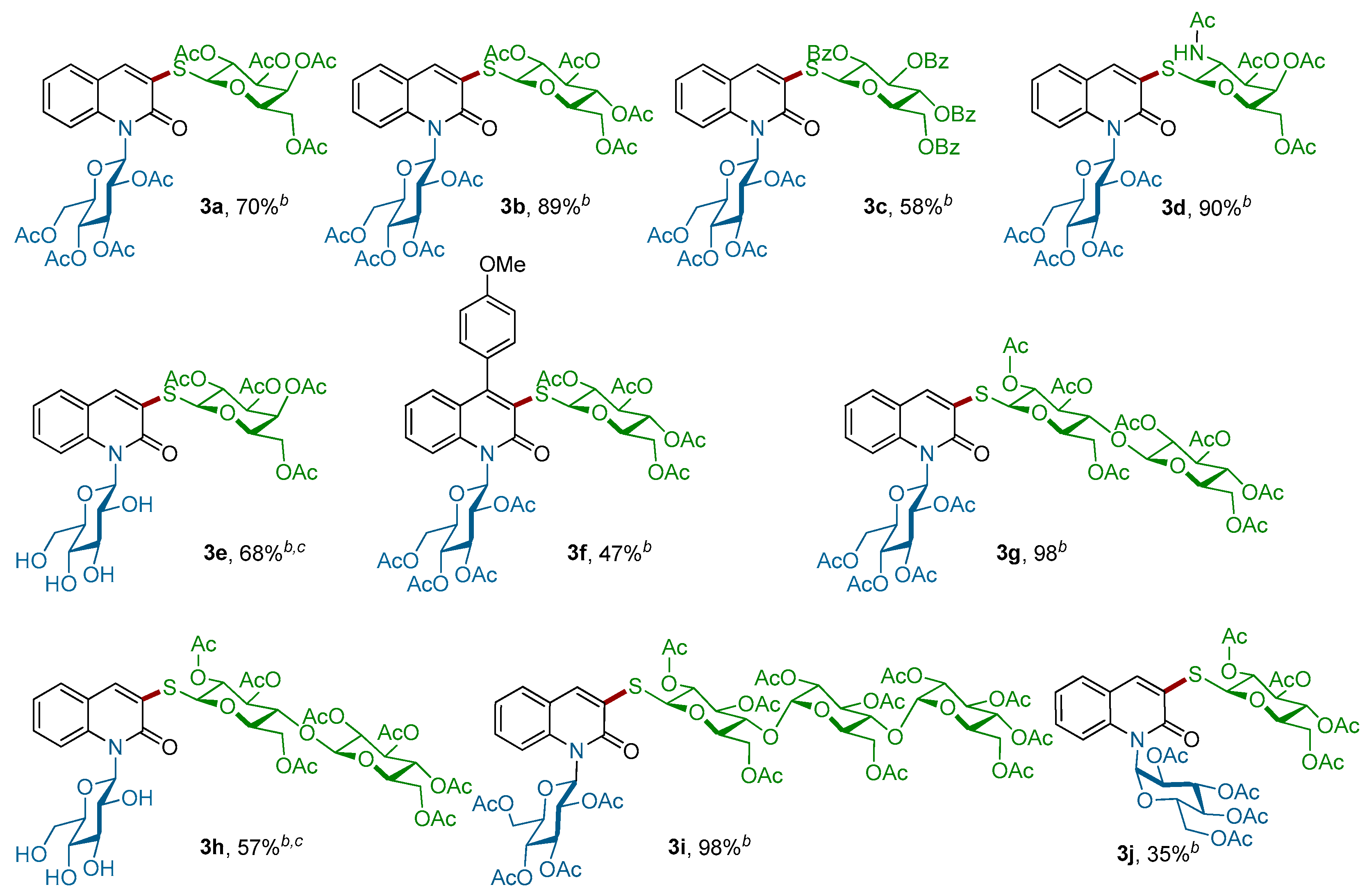
Motivated by these results, we next explored the scope of the coupling reaction of structurally diverse mono-, di-, and tri-thiosugar derivatives 1a–f with various α- or β-N-glucosylquinolinones 2d–g (Scheme 3). Gratifyingly, all of the couplings proceeded in good yields as well as with a retention of the anomeric configuration. The nature of the N-β-glucosylquinolinone partner does not interfere with the outcome of the reaction, since both O-acetylated β-glucosylquinolinone β-2d and unprotected β-glucosylquinolinone β-2g were successfully coupled. Regarding the thio-nucleophilic partners, this coupling reaction tolerates a large variety of thiosugars 1a–f: O-acetylated 1-thio-β-d-galactopyranose 1a, O-acetylated 1-thio-β-d-glycopyranose 1b, O-acetylated N-Ac-1-thio-β-d-glucopyranose 1c, and O-benzoylated 1-thio-β-d-glucopyranose 1d were coupled with both glucosylquinolinones β-2d and β-2g to give the β-N,S-bis-glycosyl quinolin-2-ones 3a–f without any loss of reactivity, except for the O-acetylated β-glucosylquinolinone β-2f due to the steric effects (Figure 2).
Importantly, this procedure is not limited to only β-glucosyl quinolin-2-ones, but it also worked successfully with 1-N-glucosylquinolin-2-one α-2e, which had an anomeric α-configuration. In this case, the corresponding α-N,S-bis-glycosyl quinolin-2-one 3j was obtained with a slightly lower yield of 35%. Finally, the efficiency of this C–S bond-forming reaction was well-demonstrated by the coupling of more complex di- and trisaccharide derivatives. Thus, 1-thio-β-d-cellobiose 1e as well as 1-thio-β-d-maltotriose 1f were readily reacted with β-2d and β-2g to give the corresponding thioglycosides 3g–i in 97%, 57% and 98% yields, respectively. More importantly, the stereochemistry of the β-1,4′-O-glycosidic bond in the di-saccharides 3g,h and the α-1,4′ in β-tri-saccharide 3i remained intact. It is worth noting that all our attempts to react an unprotected thiogalactose with β-2d or β-2g under our optimized conditions failed. Alternatively, in order to produce completely unprotected β-N,S-bis-glycosyl quinolin-2-ones and show that their purification and characterization may be achieved easily, the deprotection of representative β-N,S-bis-glycosyl quinolin-2-one was performed (Scheme 4). Thus, acetyl protecting groups of 3b could be removed through the Zemplen reaction [39,40,41] by using a catalytic amount of potassium carbonate as the base in methanol. Under these conditions, unprotected β-N,S-bis-glycosyl quinolin-2-one 4a was isolated in a quantitative yield.
3. Materials and Methods
3.1. General Experimental Methods
The compounds were all identified by the usual physical methods, that is, 1H-NMR, 13C-NMR, IR, and MS (ESI). 1H- and 13C-NMR spectra were measured in CDCl3 or DMSO-d6, Acetone-d6 or MeOH-d4 with NMR 300 and 400. 1H chemical shifts are reported in ppm from an internal standard trimethylsilane (TMS). The following abbreviations are used: m (multiplet), s (singlet), bs (broad singlet), d (doublet), t (triplet), dd (doublet of doublet), td (triplet of doublet), q (quadruplet), qui (quintuplet), sex (sextuplet). 13C chemical shifts are reported in ppm from the central peak of deuterochloroform (77.14), acetone d6 (29.84 and 206.26), MeOH (49.00), and DMSO (39.52). High resolution mass spectra (HR-MS) were recorded on a Micromass spectrometer, using ESI. IR spectra were measured and are reported in wave numbers (cm−1).
3.2. Typical Procedure A for the Synthesis of β or α 3-iodo N-glucosylquinolinones 2a–c
A 50-mL round tube flash was charged with 1a–c (1 equiv.), freshly crystallized N-bromosuccinimide (NBS) (2.5 equiv.). Under an argon atmosphere, anhydrous DMF was added. The mixture was heated to 70 °C and stirred until reaction completeness (72 h) ascertained by thin layer chromatography (TLC). The crude was diluted with EtOAc and extracted with saturated NH4Cl (50 mL × 3). The organic layer was washed with water, dried by MgSO4, and concentrated under vacuum. The residue was purified by silica gel column chromatography.
3.3. Typical Procedure B for the Synthesis of β or α 3-iodo N-glucosylquinolinones 2d–g
A reactor tube was charged with CuI (10 mol %), trans-N,N′-dimethylcyclohexane-1,2-diamine (20 mol %), NaI (2 equiv.), and β- or α-3-bromo N-glucosylquinolinones 2a–c (1 equiv., 0.721 mmol) followed by the addition of 1,4-dioxane (12 mL). The reaction under argon atmosphere was then stirred at 110 °C in an oil bath overnight. The mixture was cooled to room temperature. The crude product was purified by silica gel flash chromatography.
3.4. Typical Procedure C for Pd-Catalyzed Coupling of Thiosugars (1a–f) with β- or α-3-iodo N-glucosylquinolin-2-ones (2d–g)
A resealable and dry tube (5 mL) was charged with XantPhos Pd-G3 (5.0 mol %), thiosugar 1a–f (2.5 equiv.), and 3-iodo N-glucosylquinolin-2-ones 2d–g (0.083 mmol, 1.0 equiv.). The tube was capped with a rubber septum, evacuated, and backfilled with argon. Then, THF (1 mL) or THF/H2O (0.8 mL THF, 0.2 mL H2O) for unprotected compounds and Et3N (1.2 equiv.) were added. The tube was sealed and the mixture was stirred at room temperature for 2–3 h. After evaporation of the THF or THF/H2O, the residue was then purified by flash chromatography over silica gel. This first purification was followed by HPLC preparative for products 3e, 3g, 3h, 3i and 3j. The column used was XSELECT 4.6 × 150 mm–5 μm.
3.5. Typical Procedure D for the Synthesis of Unprotected 3-iodo Glucosylquinolinone β-2g
A mixture of 3-iodo β-N-glucosylquinolinones (100 mg, 1.0 equiv.) and K2CO3 (12 mg, 0.5 equiv.) in methanol (3 mL) was placed in a small balloon and the mixture was stirred under argon at room temperature for 30 min to 1 h. The crude mixture was then filtered through celite, washed with 10 mL of methanol, and filtered for only 1 min. The filtrate was concentrated under reduced pressure at 25 °C for 1–2 h.
(2R,3R,4S,5R,6R)-2-(acetoxymethyl)-6-(3-bromo-2-oxoquinolin-1(2H)-yl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2a: Following procedure A, A 50-mL round tube flash was charged with 1a N-(2,3,4,6-tetra-O-acetyl-1-deoxy-α-d-glucopyranosyl)-quinolin-2-one (500 mg, 1 equiv.) and freshly crystallized NBS (469 mg, 2.5 equiv.). Under an argon atmosphere, 30 mL of anhydrous DMF was added. The mixture was heated to 70 °C and stirred until reaction completeness (72 h), ascertained by TLC. The crude was diluted with EtOAc and extracted with saturated NH4Cl (50 mL × 3). The organic layer was washed with water, dried by MgSO4, and concentrated under vacuum. The residue was purified by silica gel column chromatography (cyclohexane/EtOAc 6/4) to afford the desired product 2a as white powder (345 mg, 59%); m.p. = 213–215 °C. + 103.0 (c, 1.0 in CHCl3). 1H-NMR (300 MHz, CDCl3) δ 8.08 (s, 1H), 7.95 (d, J = 8.8 Hz, 1H), 7.58 (dd, J = 7.3 Hz, 8.6 Hz, 1H), 7.48 (d, J = 7.4 Hz, 1H), 7.29 (t, J = 7.9 Hz, 1H), 6.89 (d, J = 10.0 Hz, 1H), 5.89 (t, J = 9.3 Hz, 1H), 5.46 (t, J = 9.3 Hz, 1H), 5.36 (t, J = 9.8 Hz, 1H), 4.30 (dd, J = 12.4, 4.4 Hz, 1H), 4.23 (dd, J = 12.5, 2.0 Hz, 1H), 4.08–4.00 (m, 1H), 2.09 (s, 6H), 2.00 (s, 3H), 1.81 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 170.5 (C), 169.9 (C), 169.8 (C), 169.3 (C), 158.8 (C), 142.8 (CH), 137.1 (C), 130.8 (CH), 128.9 (CH), 123.9 (CH), 121.1 (C), 116.9 (CH), 82.4 (CH), 75.3 (CH), 73.8 (CH), 68.0 (CH), 67.9 (CH), 61.8 (CH2), 20.9 (CH3), 20.7 (CH3), 20.7 (CH3), 20.3 (CH3). FT-IR (neat, cm−1) 1757, 1652, 1366, 1229, 1032, 905, 837. HRMS (ESI, m/z) calcd. for C23H24NO10BrNa [M + Na]+: 576.0481 found 576.0485.
(2S,3S,4R,5S,6R)-2-(acetoxymethyl)-6-(3-bromo-2-oxoquinolin-1(2H)-yl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2b: Following procedure A, a 30-mL round tube flash was charged with 1b N-(2,3,4,6-tetra-O-acetyl-1-deoxy-α-d-glucopyranosyl)-quinolin-2-one (200 mg, 1 equiv.) and freshly crystallized NBS (188 mg, 2.5 equiv.). Under an argon atmosphere, 12 mL of anhydrous DMF was added. The mixture was heated to 70 °C and stirred until reaction completeness (72 h), ascertained by TLC. The crude was diluted with EtOAc and extracted with saturated NH4Cl (50 mL × 3). The organic layer was washed with water, dried by MgSO4, and concentrated under vacuum. The residue was purified by silica gel column chromatography (heptane/EtOAc 6/4) to afford the desired product 2b as white powder (173 mg, 74%); 1H-NMR (300 MHz, CDCl3) δ 8.11 (s, 1H), 7.76 (d, J = 8.6 Hz, 1H), 7.54 (dd, J = 8.6 Hz, 7.4, 1H), 7.47 (d, J = 7.6 Hz, 1H), 7.26 (t, J = 7.4 Hz, 1H), 5.37 (t, J = 6.4 Hz, 1H), 5.24 (t, J = 7.8 Hz, 1H), 4.71–4.62 (m, 1H), 4.39 (dd, J = 12.4, 4.8 Hz, 1H), 4.16 (dd, J = 12.4, 2.7 Hz, 1H), 2.10 (s, 3H), 2.08 (s, 3H), 2.05 (s, 3H), 1.73 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 170.7 (C), 169.9 (C), 169.8 (C), 169.8 (C), 159.5 (C), 142.4 (CH), 139.4 (C), 130.7 (CH), 128.1 (CH), 123.7 (CH), 121.3 (C), 117.1 (C), 116.6 (CH), 80.8 (CH), 73.4 (CH), 72.5 (CH), 70.2 (CH), 68.0 (CH), 61.7 (CH2), 21.0 (CH3), 20.9 (CH3), 20.9 (CH3), 20.4 (CH3).
(2R,3R,4S,5R,6R)-2-(acetoxymethyl)-6-(3-bromo-4-(4-methoxyphenyl)-2-oxoquinolin-1(2H)-yl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2c: Following procedure A, a 10-mL round tube flash was charged with 1c (50 mg, 1 equiv.) and freshly crystallized NBS (39 mg, 2.5 equiv.). Under an argon atmosphere, 3 mL of anhydrous DMF was added. The mixture was heated to 70 °C and stirred until reaction completeness (4 h), ascertained by TLC. The crude was diluted with EtOAc and extracted with saturated NH4Cl (50 mL × 3). The organic layer was washed with water, dried by MgSO4, and concentrated under vacuum. The residue was purified by silica gel column chromatography (cyclohexane/EtOAc 5/5) to afford the desired product 2c as pale yellow solid (34 mg, 60%); m.p.: 105–107 °C; Rf = 0.48 (Ethyl/Cyclohexane: 5/5); + 35.55 (c, 1.0 in CHCl3); IR (neat): 2963, 1756, 1650, 1604, 1554, 1511, 1455, 1366, 1260, 1248, 1033,1013,802, 763 cm−1; 1H-NMR (300 MHz, CDCl3) δ 8.00 (d, J = 8.5 Hz, 1H), 7.59–7.53 (m, 1H), 7.22–7.13 (m, 4H), 7.02 (dd, J = 17.8, 9.3 Hz, 3H), 5.97 (t, J = 9.3 Hz, 1H), 5.44 (dt, J = 30.0, 9.5 Hz, 2H), 4.33–4.24 (m, 2H), 4.17–4.09 (m, 1H), 3.90 (s, 3H), 2.11 (s, 6H), 2.02 (s, 4H), 1.85 (s, 2H); 13C-NMR (75 MHz, CDCl3) δ 170.56, (C) 169.95 (C), 169.77 (C), 169.65 (C), 169.28 (C), 160.00 (C), 158.88 (C), 152.51 (C), 136.55 (C), 130.55 (CH), 130.08 (CH), 129.90 (CH), 129.42 (C), 129.25 (CH), 123.56 (CH), 122.35 (C), 116.76 (CH), 114.23 (2CH), 82.55 (CH), 75.33 (CH), 73.96 (CH), 68.05 (CH), 61.85 (CH2), 55.49 (CH) 20.86 (CH3), 20.75 (CH3), 20.73 (2CH3), 20.37 (CH3); HR-MS (ESI positive, m/z): found 682.0894 ([M + Na]+), calc. for C30H30NO11NaBr (M + Na): 682.0900.
(2R,3R,4S,5R,6R)-2-(acetoxymethyl)-6-(3-iodo-2-oxoquinolin-1(2H)-yl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2d: Following procedure B, a solution of β-3-bromo N-glycosylquinolinone 2a (0.722 mmol, 400 mg), NaI (1.443 mmol, 217 mg), trans-N,N′-dimethylcyclohexane-1,2-diamine (21 mg, 0,14 mmol), and CuI (14 mg, 0.072 mmol) was stirred at 110 °C overnight. The crude product was purified by silica gel flash chromatography (Cyclohexane/EtOAc 5/5), and the product 2d was isolated as a pale yellow solid (362 mg, 83%); m.p.: 222.7–223.8 °C; Rf = 0.56 (Cyclohexane/EtOAc: 5/5); + 96.29 (c, 1.0 in CHCl3); IR (neat): 1746, 1648, 1596, 1365, 1217, 1077, 914, 751 cm−1; 1H-NMR (300 MHz, CDCl3) δ 8.35 (s, 1H), 7.95 (d, J = 8.6 Hz, 1H), 7.58 (t, J = 7.9 Hz, 1H), 7.45 (d, J = 7.7 Hz, 1H), 7.31–7.24 (m, 1H), 6.89 (d, J = 9.8 Hz, 1H), 5.88 (t, J = 9.3 Hz, 1H), 5.49–5.29 (m, 2H), 4.33–4.19 (m, 2H), 4.07–3.97 (m, 1H), 2.10 (s, 6H), 2.00 (s, 3H), 1.80 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 170.55, 169.90, 169.76, 169.24, 159.22, 150.33, 137.80, 131.02, 128.61, 123.69, 122.32, 116.94, 92.17, 82.92, 75.30, 73.77, 68.03, 67.94, 61.82, 20.85, 20.74, 20.70, 20.27; HR-MS (ESI positive, m/z): found 624.0339 ([M + Na]+), calc. for C23H24NO10NaI (M + Na): 624.0343.
(2S,3S,4R,5S,6R)-2-(acetoxymethyl)-6-(3-iodo-2-oxoquinolin-1(2H)-yl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2e: Following procedure B, α-3-bromo N-glycosylquinolinone 2b (0.288 mmol, 160 mg), NaI (0.577 mmol, 87 mg), trans-N,N′-dimethylcyclohexane-1,2-diamine (8.2 mg, 0,057 mmol), and CuI (5.5 mg, 0.028 mmol) were stirred at 110 °C overnight. The crude product was purified by silica gel flash chromatography (EtOAc/heptan: 5/5), and the product 2e was isolated as a pale yellow solid (124 mg, 73%): m.p.: 111.4–123.4 °C; Rf = 0.26 (EtOAc/heptan: 5/5); + 370 (c, 1.0 in CHCl3); IR (neat): 1739, 1641, 1596, 1367, 1260, 1205, 1031, 817, 798 cm−1; 1H-NMR (300 MHz, CDCl3) δ 8.37 (s, 1H), 7.77 (d, J = 8.7 Hz, 1H), 7.53 (t, J = 7.9 Hz, 1H), 7.44 (d, J = 7.8 Hz, 1H), 7.23 (d, J = 7.6 Hz, 1H), 6.70 (d, J = 6.2 Hz, 1H), 6.01 (t, J = 6.7 Hz, 1H), 5.36 (t, J = 6.3 Hz, 1H), 5.23 (t, J = 7.7 Hz, 1H), 4.70–4.63 (m, 1H), 4.39 (dd, J = 12.4, 4.9 Hz, 1H), 4.17 (dd, J = 12.4, 3.0 Hz, 1H), 2.13–2.04 (m, 9H), 1.73 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 170.73 (C), 169.93 (C), 169.76 (2C), 160.03 (C), 149.95 (CH), 140.12 (C), 130.86 (CH), 127.84 (CH), 123.48 (CH), 122.48 (C), 116.70(CH), 93.58 (C), 80.99 (CH), 73.35 (CH), 72.45 (CH), 70.17 (CH), 68.06 (CH), 61.67 (CH2), 21.06 (CH3), 20.94 (CH3), 20.89 (CH3), 20.46 (CH3); HR-MS (ESI positive, m/z): found 624.0344 ([M + Na]+), calc. for C23H24NO10NaI (M + Na): 624.0343.
(2R,3R,4S,5R,6R)-2-(acetoxymethyl)-6-(3-iodo-4-(4-methoxyphenyl)-2-oxoquinolin-1(2H)-yl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2f: Following procedure B, N-glycosylquinolinone β-2c (0.136 mmol, 90 mg), NaI (0.272 mmol, 41 mg), trans-N,N′-dimethylcyclohexane-1,2-diamine (4 mg, 0,027 mmol), and CuI (3 mg, 0.0135 mmol) were stirred at 110 °C overnight. The crude product was purified by silica gel flash chromatography (Cyclohexane/EtOAc 6/4), and the product 2f was isolated as a yellow solid (67 mg, yield 70%); m.p.: 106.6–121.8 °C; Rf = 0.48 (Cyclohexane/EtOAc 6/4); + 22.35 (c, 1.0 in CHCl3); IR (neat): 1756, 1652, 1604, 1511, 1367, 1249, 1217, 1113, 1097, 1035, 765 cm−1; 1H-NMR (300 MHz, Acetone) δ 8.34 (d, J = 8.6 Hz, 1H), 7.70–7.63 (m, 1H), 7.26–7.06 (m, 5H), 7.02 (d, J = 9.9 Hz, 1H), 5.98 (t, J = 10.6 Hz, 1H), 5.59 (t, J = 9.5, 1H), 5.47 (t, J = 9.2 Hz, 1H), 4.37–4,28 (m, 3H), 4.06 (d, J = 7.1 Hz, 1H), 3.92 (s, 3H), 2.11–2.01 (m, 9H), 1.97 (s, 3H); 13C-NMR (75 MHz, Acetone) δ 170.84, 170.25, 169.48, 167.30, 161.09, 154.96, 137.19, 131.79, 130.85, 130.54, 130.05, 124.11, 117.14, 115.16 (3), 83.93, 76.03, 74.54, 69.00 (2), 62.72, 55.89, 20.84 (2), 20.72, 20.30. HR-MS (ESI positive, m/z): found 730.0739 ([M + Na]+), calc. for C30H30NO11NaI (M + Na): 730.0761.
3-iodo-1-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)quinolin-2(1H)-one 2g: Following procedure D, a mixture of β-2d (100 mg, 1.0 equiv.) and K2CO3 (12 mg, 0.5 equiv.) in methanol (3 mL) was stirred under argon at room temperature for 30 min to 1 h. The crude mixture was then filtered through celite, washed with 10 mL of methanol, and filtered for only 1 min. The filtrate was concentrated under reduced pressure at 25 °C for 1–2 h. The product 2g was isolated as a pale brown solid (100 mg, yield 99%); m.p.: 155.2–171.8 °C; Rf = 0.4 (Ethyl/MeOH: 9/1); + 45 (c, 1.0 in MeOH); IR (neat): 3360, 1629, 1589, 1451, 1314, 1277, 1082, 766, 749 cm−1; 1H-NMR (300 MHz, MeOD-d4) δ 8.59 (s, 1H), 8.10 (d, J = 8.9 Hz, 1H), 7.58 (t, J = 9.7 Hz, 2H), 7.27 (t, J = 7.4 Hz, 1H), 6.57 (d, J = 9.8 Hz, 1H), 4.27 (t, J = 9.0 Hz, 1H), 3.94 (d, J = 11.8 Hz, 1H), 3.81 (dd, J = 12.5, 4.6 Hz, 1H), 3.62–3.53 (m, 3H); 3C-NMR (75 MHz, MeOD) δ 161.39, 151.53, 132.47, 131.55, 129.90, 129.46, 124.33, 124.02, 119.17, 86.94, 81.86, 79.54, 71.30, 70.75, 62.62; HR-MS (ESI positive, m/z): found 455.9928 ([M + Na]+), calc. for C15H16NO6NaI (M + Na): 455.9920.
(2R,3S,4S,5R,6S)-2-(acetoxymethyl)-6-((2-oxo-1-((2R,3R,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)-1,2-dihydroquinolin-3-yl)thio)tetrahydro-2H-pyran-3,4,5-triyl triacetate 3a: Following procedure C, 3-iodo N-β-glycosylquinolinone β-2d (0.0831 mmol, 50 mg) and β-thiogalactose 1a (0.2077 mmol, 76 mg) were stirred at room temperature for 3 h. The crude product was purified by silica gel flash chromatography (Cyclohexane/EtOAc 4/6). The product 3a was isolated as pale yellow solid (46 mg, yield 70%); m.p.: 222.7–223.8 °C; Rf = 0.4 (Cyclohexan/EtOAc: 4/6); + 16.21 (c, 1.0 in CHCl3); IR (neat): 1745, 1648, 1595, 1367, 1208, 1034, 794, 733,702 cm−1; 1H-NMR (300 MHz, Acetone) δ 8.28 (d, J = 8.7 Hz, 1H), 7.98 (s, 1H), 7.69–7.58 (m, 2H), 7.33 (t, J = 7.5 Hz, 1H), 6.94 (d, J = 9.7 Hz, 1H), 5.94 (t, J = 9.3 Hz, 1H), 5.60–5.26 (m, 6H), 4.50 (t, J = 6.3 Hz, 1H), 4.41–4.26 (m, 3H), 4.18 (d, J = 6.3 Hz, 2H), 2.19 (s, 3H), 2.10–1.92 (m, 21H); 13C-NMR (75 MHz, Acetone-d6) δ 169.87(C), 169.75 (C), 169.67 (C), 169.25 (C), 169.21 (C), 169.15 (C), 169.04 (C), 168.38 (C), 159.72 (C), 135.91 (C), 135.60 (CH), 129.62 (CH), 128.41 (CH), 127.91 (C), 123.16 (CH), 121.09 (C), 117.49 (CH), 81.87 (CH), 81.25 (CH), 74.84 (CH), 74.51 (CH), 73.38 (CH), 71.56 (CH), 67.88 (CH), 67.81 (CH), 67.74 (CH), 66.74 (CH), 61.96 (CH2), 61.55 (CH2), 19.74 (6CH3), 19.61 (CH3), 19.20 (CH3). HR-MS (ESI positive, m/z): found 860.2037 ([M + Na]+), calc. for C37H43NO19NaS (M + Na): 860.2048.
(2R,3R,4S,5R,6S)-2-(acetoxymethyl)-6-((2-oxo-1-((2R,3R,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)-1,2-dihydroquinolin-3-yl)thio)tetrahydro-2H-pyran-3,4,5-triyl triacetate 3b: Following procedure C, 3-iodo N-β-glycosylquinolinone β-2d (0.0831 mmol, 50 mg) and β-thioglucose 1b (0.2077 mmol, 76 mg) were stirred at room temperature for 3 h. The crude product was purified by silica gel flash chromatography (diethylether/Pentan: 9/1), and the product 3b was isolated as a pale yellow solid (62 mg, yield 89%); m.p.: 221.8–223 °C; Rf = 0.33 (diethylether/Pentan: 9/1); + 23.07 (c, 1.0 in CHCl3); IR (neat): 1756, 1649, 1595, 1366, 1206, 1032, 914, 798,764 cm−1; 1H-NMR (300 MHz, Acetone) δ 8.27 (d, J = 8.8 Hz, 1H), 7.92 (s, 1H), 7.70–7.58 (m, 2H), 7.33 (t, J = 7.5 Hz, 1H), 6.94 (d, J = 9.8 Hz, 1H), 5.94 (t, J = 9.4 Hz, 1H), 5.58 (dd, J = 18.7, 9.2 Hz, 1H), 5.51–5.38 (m, 3H), 5.21–5.09 (m, 2H), 4.40–4.16 (m, 6H), 2.08–1.96 (m, 24H); 13C-NMR (75 MHz, Acetone) δ 169.75 (C), 169.37 (C), 169.20 (C), 169.14 (2C), 168.97 (C), 168.91 (C), 168.38 (C), 159.68 (C), 135.86 (C), 135.31 (CH), 129.63 (CH), 128.46 (CH), 127.84 (C), 123.16 (CH), 121.08 (C), 117.48 (CH), 81.30 (2CH), 75.47 (CH), 74.85 (CH), 73.42 (2CH), 69.69 (CH), 68.49 (CH), 67.88 (CH), 67.81 (CH), 62.34 (CH2), 61.55 (CH2), 19.73 (3CH3), 19.68 (2CH3), 19.61 (2CH3), 19.20 (CH3); HR-MS (ESI positive, m/z): found 860.2056 ([M + Na]+), calc. for C37H43NO19NaS (M + Na): 860.2048.
(2R,3R,4S,5R,6S)-2-((benzoyloxy)methyl)-6-((2-oxo-1-((2R,3R,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)-1,2-dihydroquinolin-3-yl)thio)tetrahydro-2H-pyran-3,4,5-triyl tribenzoate 3c: Following procedure C, 3-iodo N-β-glycosylquinolinone β-2d (0.0831 mmol, 50 mg) and O-benzoylated 1-thio-β-d-glucopyranose 1d (0.2077 mmol, 128 mg) were stirred at room temperature for 3 h. The crude product was purified by silica gel flash chromatography (Cyclohexan/EtOAc: 5/5), and the product 3c was isolated as a pale yellow solid (52 mg, yield 58%); m.p.: 221–249 °C; Rf = 0.4 (Cyclohexan/EtOAc: 5/5); + 86.95 (c, 1.0 in CHCl3); IR (neat): 1728, 1648, 1597, 1452, 1367, 1277,1215, 1111,1086, 1068,801,766,708 cm−1; 1H-NMR (300 MHz, Acetone-d6) δ 8.24 (d, J = 8.4 Hz, 1H), 8.05–7.92 (m, 7H), 7.88–7.83 (m, 2H), 7.63–7.38 (m, 14H), 7.19 (t, J = 7.3 Hz, 1H), 6.88 (d, J = 9.7 Hz, 1H), 6.21 (t, J = 9.3 Hz, 1H), 5.93–5.78 (m, 4H), 5.57–5.38 (m, 2H), 4.89–4.72 (m, 2H), 4.59 (dd, J = 12.3, 6.2 Hz, 1H), 4.39–4.25 (m, 3H), 2.09–2.02 (m, 9H), 1.93 (s, 3H);13C-NMR (75 MHz, Acetone-d6) δ 169.73, 169.40, 169.14, 168.23, 168.84, 165.58, 165.17, 164.97, 164.77, 159.64, 135.85, 135.75, 135.04, 133.59, 133.54, 133.45, 133.18, 129.79, 129.5–128.53 (m), 123.06, 120.95, 117.36, 81.94, 81.30, 75.86, 74.81, 74.20, 73.27, 70.44, 69.59, 67.85, 67.77, 63.41, 61.55, 19.72, 19.58, 19.01; HR-MS (ESI positive, m/z): found 1086.2856 ([M + H]+), calc. for C57H52NO19S (M + H): 1086.2854.
(2R,3R,5R,6R)-2-(3-(((2S,3R,4R,5S,6R)-3-acetamido-4,5-diacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)thio)-2-oxoquinolin-1(2H)-yl)-6-(acetoxymethyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 3d: Following procedure C, 3-iodo N-β-glycosylquinolinone β-2d (0.083 mmol, 50 mg) and N-acetyl-O-acetylated 1-thio-β-d-glucopyranose 1c (0.2077mmol, 76 mg) were stirred at room temperature for 3 h. The crude product was purified by silica gel flash chromatography (dichloromethan/EtOAc: 2/8). The product 3d was isolated as a white solid (62 mg, yield 90%); m.p.: 258.9–261.5 °C; Rf = 0.28 (dichloromethan/EtOAc: 2/8); + 20 (c, 1.0 in CHCl3); IR (neat): 1740, 1649, 1596, 1367, 1260, 1213,1076, 796,748 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.97 (s, 1H), 7.89 (d, J = 8.7 Hz, 1H), 7.51 (t, J = 7.5 Hz, 2H), 7.27–7.21 (m, 1H), 6.86 (d, J = 9.9 Hz, 1H), 6.75 (s, 1H), 5.88 (t, J = 9.4 Hz, 1H), 5.47–5.30 (m, 3H), 5.22–5.17 (m, 1H), 5.06 (t, J = 9.7 Hz, 1H), 4.25–4.03 (m, 6H), 3.81–3.74 (m, 1H), 2.09–1.85 (m, 24H); 13C-NMR (101 MHz, CDCl3) δ 170.77 (2C), 170.62 (C), 170.47 (C), 169.85 (C), 169.56 (C), 169.43 (C), 168.66, (C) 161.38 (C), 143.41 (CH), 136.63(C), 130.77 (CH), 129.30 (CH), 124.77 (C), 123.84 (CH), 120.99 (C), 116.84 (CH), 90.76 (CH), 82.98 (CH), 81.62 (CH), 76.03 (CH), 75.13 (CH), 73.85 (CH), 73.65 (CH), 70.79 (CH), 68.81 (CH), 68.04 (CH), 67.80 (CH), 62.44 (CH2), 61.62 (CH2), 23.51 (CH3), 23.10 (CH3), 21.00 (CH3), 20.77 (CH3), 20.65 (CH3), 20.63 (CH3), 20.59 (CH3), 19.98 (CH3); HR-MS (ESI positive, m/z): found 859.2211 ([M + Na]+), calc. for C37H44N2O18S (M + Na): 859.2208.
(2R,4S,5R,6S)-2-(acetoxymethyl)-6-((2-oxo-1-((2R,3R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)-1,2-dihydroquinolin-3-yl)thio)tetrahydro-2H-pyran-3,4,5-triyl triacetate 3e: Following procedure C, 3-iodo N-β-glycosylquinolinone β-2g (0.115 mmol, 50 mg) and β-thiogalactose 1a (0.2885 mmol, 106 mg) were stirred at room temperature for 3 h. After purification of the crude reaction by HPLC preparative (conditions: H2O + 0.1% AF/ACN gradient from 20% to 50% in 20 min), the product 3e was isolated as a pale yellow solid (52 mg, yield 68%); m.p.: 244–252.8 °C; Rf = 0.4 (Cyclohexane/EtOAc: 5/5); − 16 (c, 1.0 in CHCl3); IR (neat): 1750, 1633, 1561, 1367, 1211, 1046, 732 cm−1; 1H-NMR (300 MHz, Acetone-d6) δ 8.10 (d, J = 8.1 Hz, 1H), 7.92 (s, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.48 (t, J = 7.7 Hz, 1H), 7.28 (t, J = 7.5 Hz, 1H), 6.59 (d, J = 9.7 Hz, 1H), 5.55 (d, J = 2.9 Hz, 1H), 5.47–5.27 (m, 4H), 4.54 (t, J = 6.1 Hz, 1H), 4,33 (t, J=8 Hz,1H), 4.24–4.12 (m, 3H), 3.97–3.56 (m, 7H), 2.08–2.03 (m, 12H);13C-NMR (75 MHz, Acetone-d6) δ 169.95 (C), 169.79 (C), 169.36 (C), 159.99 (C), 135.96 (C), 133.46 (CH), 129.20 (C), 128.81 (CH), 128.02 (CH), 122.70 (CH), 121.35 (C), 118.83 (C), 117.83 (CH), 84.04 (CH), 81.38 (CH), 80.44 (CH), 78.57 (CH), 74.45 (CH), 71.65 (CH), 70.16 (CH), 69.28 (CH), 67.67(CH), 66.68(CH), 62.05 (CH2), 61.57 (CH2), 19.80 (2CH3), 19.74 (CH3), 19.63 (CH3); HR-MS (ESI positive, m/z): found 692.1636 ([M + Na]+), calc. for C29H35NO15NaS (M + Na): 692.1625.
(2R,3R,4S,6S)-2-(acetoxymethyl)-6-((4-(4-methoxyphenyl)-2-oxo-1-((2R,3R,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)-1,2-dihydroquinolin-3-yl)thio)tetrahydro-2H-pyran-3,4,5-triyl triacetate 3f: Following procedure C, 3-iodo-N-β-glycosylquinolinone β-2f (0.071 mmol, 50 mg) and β-thioglucose 1a (0.1767 mmol, 65 mg) were stirred at room temperature for 3 h. The crude product was purified by silica gel flash chromatography (EtOAc/Cyclohexane: 5/5), and the product 3f was isolated as a pale brown solid (31 mg, yield 47%); m.p.: 143.4–150.3 °C; Rf = 0.36 (EtOAc/Cyclohexane: 5/5); − 1.42 (c, 1.0 in CHCl3); IR (neat): 1756, 1367, 1277, 1260, 1035, 766, 749 cm−1; 1H-NMR (400 MHz, Acetone d6) δ 8.31 (d, J = 8.7 Hz, 1H), 7.63 (t, J = 8.4 Hz, 1H), 7.27 (dd, J = 9.1, 1.9 Hz, 1H), 7.19–7.11 (m, 3H), 7.07–7.01 (m, 3H), 6.04 (t, J = 9.5 Hz, 1H), 5.59 (t, J = 9.4 Hz, 1H), 5.49 (t, J = 10.3 Hz, 1H), 5.38–5.32 (m, 2H), 5.16 (t, J = 9.6 Hz, 1H), 5.06 (t, J = 9.8 Hz, 1H), 4.98–4.92 (m, 2H), 4.85 (t, J = 9.7 Hz, 1H), 4.35–4.30 (m, 2H), 4.17 (dd, J = 12.5, 2.3 Hz, 1H), 3.89 (s, 3H), 2.07 (d, J = 4.5 Hz, 11H), 2.00 (s, 3H), 1.96 (d, J = 8.2 Hz, 10H); 13C-NMR (101 MHz, Acetone) δ 170.88, 170.66, 170.62, 170.24, 170.18, 170.06, 169.99, 169.94, 169.90, 169.69, 169.47, 131.62, 131.57, 131.24, 129.86, 129.63, 123.66, 122.91, 118.33, 114.67, 114.16, 87.75, 82.88, 82.18, 76.70, 75.95, 75.85, 74.49, 74.36, 74.19, 71.92, 70.34, 69.45, 68.98, 68.77, 68.59, 62.97, 62.53, 55.59, 20.79, 20.65, 20.55, 20.50, 20.40; HR-MS (ESI positive, m/z): found 966.2470 ([M + Na]+), calc. for C44H49NO20NaS (M + Na): 966.2466.
(2R,3R,4S,5R,6S)-2-(acetoxymethyl)-6-(((2R,3R,4S,5R,6S)-4,5-diacetoxy-2-(acetoxymethyl)-6-((2-oxo-1-((2R,3R,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)-1,2-dihydroquinolin-3-yl)thio)tetrahydro-2H-pyran-3-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate 3g: Following procedure C, 3-iodo N-β-glycosylquinolinone β-2d (0.083 mmol, 50 mg) and β-thiocellebiose 1e (0.2077 mmol, 136 mg) were stirred at room temperature for 3 h. After purification of the crude reaction by HPLC preparative (conditions: H2O + 0.1% AF/ACN gradient from 40% to 100% in 15 min), the product 3g was isolated as a pale yellow solid (92 mg, yield 98%): m.p.: 154.3–157.8 °C; Rf = 0.53 (dichloromethane/EtOAc: 6/4); − 15 (c, 1.0 in CHCl3); IR (neat): 1755, 1649, 1595, 1397, 1229, 1035,910 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.90 (d, J = 8.6 Hz, 1H), 7.63 (s, 1H), 7.52–7.43 (m, 2H), 7.26–7.22 (m, 1H), 6.81 (d, J = 9.9 Hz, 1H), 5.86 (t, J = 9.4 Hz, 1H), 5.41 (t, J = 9.3 Hz, 1H), 5.33 (t, J = 9.8 Hz, 1H), 5.24 (t, J = 8.7 Hz, 1H), 5.12–5.03 (m, 3H), 4.96 (d, J = 10.0 Hz, 1H), 4.90 (t, J = 8.6 Hz, 1H), 4.64–4.52 (m, 1H), 4.49 (d, J = 7.8 Hz, 1H), 4.42 (d, J = 11.3 Hz, 1H), 4.33 (dd, J = 12.5, 4.5 Hz, 1H), 4.26–4.19 (m, 1H), 4.09 (dd, J = 11.9, 5.9 Hz, 1H), 4.05–3.96 (m, 2H), 3.79–3.71 (m, 2H), 3.68–3.61 (m, 1H), 2.08–2.03 (m, 10H), 2.01–1.93 (m, 23H). 13C-NMR (101 MHz, CDCl3) δ 170.57 (C), 170.49 (C), 170.33 (C), 170.27 (C), 169.86 (C), 169.81 (C), 169.73 (C), 169.48 (C), 169.40 (C), 169.14 (C), 169.08 (C), 160.26 (C), 137.79 (CH), 136.19 (C), 130.16 (CH), 128.73 (CH), 126.93 (C), 123.62 (CH), 121.04 (C), 116.84 (CH), 100.93 (CH), 82.07 (CH), 81.52 (CH), 76.94 (CH), 76.69 (CH), 75.21 (CH), 73.79 (CH), 73.62 (CH), 73.01 (CH), 72.15 (CH),71.76 (CH), 70.32 (CH), 67.95 (2CH), 67.84 (CH), 62.52 (CH2), 61.75 (CH2), 61.61 (CH2), 20.83 (CH3), 20.78 (CH3), 20.75 (CH3), 20.73 (CH3), 20.69 (CH3), 20.62 (4CH3), 20.23 (CH3), 1.10 (CH3); HR-MS (ESI positive, m/z): found 1148.2888 ([M + Na]+), calc. for C49H59NO27NaS (M + Na): 1148.2893.
(2R,3R,4S,5R,6S)-2-(acetoxymethyl)-6-(((2R,3R,4S,5R,6S)-4,5-diacetoxy-2-(acetoxymethyl)-6-((2-oxo-1-((2R,3R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)-1,2-dihydroquinolin-3-yl)thio)tetrahydro-2H-pyran-3-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate 3h: Following procedure C, 3-iodo N-β-glycosylquinolinone β-2g (0.115 mmol, 50 mg) and β-thiocellobiose 1e (0.2885 mmol, 189 mg) were stirred at room temperature for 3 h. After purification of the crude reaction by HPLC preparative (conditions: H2O + 0.1% AF/ACN gradient from 20% to 50% in 15 min), the product 3h was isolated as a white solid (63 mg, yield 57%); m.p.: 249.2–251.9 °C; Rf = 0.52 (EtOAc/MeOH: 9/1); + 7.14 (c, 1.0 in CHCl3); IR (neat): 1740, 1629, 1590, 1366, 1212, 1033, 907, 751 cm−1; 1H-NMR (300 MHz, Acetone-d6) δ 8.09 (d, J = 8.0 Hz, 1H), 7.82 (s, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.48 (t, J = 7.90 Hz, 1H), 7.28 (t, J = 7.3 Hz, 1H), 6.58 (d, J = 9.7 Hz, 1H), 5.43–5.23 (m, 4H), 5.14–5,03 (m, 2H), 4.94–4.84 (m, 2H), 4.60–4.30 (m, 6H), 4.25–4.14 (m, 3H), 3.96–3.56 (m, 8H), 2.05–1.92 (m, 16H); 13C-NMR (75 MHz, Acetone-d6) δ 169.97 (C), 169.83 (C), 169.41 (C), 169.31 (C), 169.23 (C), 169.03 (C), 168.82 (C), 159.87 (C), 135.88 (C), 132.79 (CH), 130.77 (C), 128.77 (CH), 128.00 (CH), 122.63 (CH), 121.34 (C), 117.80 (CH), 100.53 (CH), 84.05 (CH), 80.55 (CH), 80.46 (CH), 78.56 (CH), 76.73 (CH), 76.45 (CH), 73.33 (CH), 72.78 (CH), 71.57 (CH), 71.53(CH), 70.25 (CH), 69.80 (CH), 69.35 (CH), 68.03 (CH), 62.69 (CH2), 61.67 (CH2), 61.57 (CH2), 19.93 (CH3), 19.84 (CH3), 19.76 (CH3), 19.71 (CH3), 19.69 (CH3), 19.61 (CH3), 19.52 (CH3); HR-MS (ESI positive, m/z): found 980.2461 ([M + Na]+), calc. for C41H51NO23NaS (M + Na): 980.2470.
(2R,3R,4S,5R,6R)-2-(acetoxymethyl)-6-(((2R,3S,4S,5R,6R)-4,5-diacetoxy-2-(acetoxymethyl)-6-(((2R,3S,4S,5R,6S)-4,5-diacetoxy-2-(acetoxymethyl)-6-((2-oxo-1-((2R,3R,5R,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)-1,2-dihydroquinolin-3-yl)thio)tetrahydro-2H-pyran-3-yl)oxy)tetrahydro-2H-pyran-3-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate 3i: Following procedure C, 3-iodo N-β-glycosylquinolinone β-2d (0.083 mmol, 50 mg) and β-thiomalthotriose 1f (0.2077mmol, 196 mg) were stirred at room temperature for 3 h. After purification of the crude reaction by HPLC preparative (conditions: H2O + 0.1% AF/MeOH gradient from 50% to 100% in 15 min), the product 3i was isolated as a white solid (116.25 mg, yield 98%); m.p.: 110.8–134.3 °C; Rf = 0.19 (Cyclohexane/EtOAc: 4/6); + 40 (c, 1.0 in CHCl3); IR (neat): 1756, 1649, 1367, 1260, 1208, 1011, 794, 708, 702 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.93 (d, J = 8.6 Hz, 1H), 7.73 (s, 1H), 7.54 (t, J = 9.1 Hz, 2H), 7.31 (d, J = 7.4 Hz, 1H), 6.85 (d, J = 9.9 Hz, 1H), 5.91 (t, J = 9.5 Hz, 1H), 5.45–5.27 (m, 8H), 5.12–4.94 (m, 4H), 4.86 (dd, J = 10.5, 4.0 Hz, 1H), 4.75 (dd, J = 10.3, 4.0 Hz, 1H), 4.49–4.18 (m, 8H), 3.99–3.89 (m, 4H), 2.12–1.96 (m, 42H); 13C-NMR (75 MHz, CDCl3) δ 170.68, (C) 170.59 (C), 170.52 (C), 170.39 (C), 170.18 (C), 170.12 (C), 170.00 (C), 169.86 (C), 169.75 (C), 169.69 (C), 169.61 (C), 169.51 (C), 169.39 (C), 168.97 (C), 144.13 (C), 136.14 (C), 130.09 (CH), 126.52 (C), 123.61 (CH), 120.99 (C), 116.67 (CH), 95.90 (CH), 95.75 (CH), 81.76 (CH), 81.43 (CH), 76.15 (CH), 75.98 (CH), 75.76 (CH), 75.13 (CH), 74.17 (CH), 73.72 (CH), 72.67 (CH), 71.71 (CH), 70.81 (CH), 70.36 (CH), 70.06 (CH), 69.38 (CH), 69.02 (CH), 68.59 (CH), 67.89 (CH), 68.19 (CH), 67.78 (CH), 67.56 (CH), 63.50 (CH2), 62.46 (CH2), 61.66 (CH2), 61.42 (CH2), 20.84 (2CH3), 20.81 (CH3), 20.77 (CH3), 20.56 (9CH3), 20.17 (CH3); HR-MS (ESI positive, m/z): found 1436.3730 ([M + Na]+), calc. for C61H75NO35NaS (M + Na): 1436.3738.
(2R,3R,4S,5R,6S)-2-(acetoxymethyl)-6-((2-oxo-1-(((2R,3S,4R,5S,6S)-3,4,5-triacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)-1,2-dihydroquinolin-3-yl)thio)tetrahydro-2H-pyran-3,4,5-triyl triacetate 3j: Following procedure C, 3-iodo N-α-glycosylquinolinone α-2e (0.083 mmol, 50 mg) and β-thioglucose 1a (0.2077 mmol, 76 mg) were stirred at room temperature for 3 h. After purification of the crude reaction by HPLC preparative (conditions: H2O + 0.1% AF/ACN gradient from 40 to 100% in 15 min), the product 3j was isolated as a pale yellow solid (26 mg, yield 35%); m.p.: 224–225 °C; Rf = 0.35 (EtOAc/heptan: 7/3); + 4.44 (c, 1.0 in CHCl3); IR (neat): 1755, 1641, 1367, 1259, 1031, 913, 799, 748 cm−1; 1H-NMR (400 MHz, Acetone-d6) δ 7.87–7.77 (m, 2H), 7.54 (d, J = 7.8 Hz, 1H), 7.44 (t, J = 8.07 Hz, 1H), 7.19 (t, J = 7.5 Hz, 1H), 6.71 (d, J = 5.9 Hz, 1H), 5.84 (t, J = 6.12 Hz,1H), 5.33–5.22 (m, 3H), 5.08 (t, J = 7.3 Hz, 1H), 5.01 (dd, J = 18.4, 9.4 Hz, 2H), 4.57–4.52 (m, 1H), 4.34 (dd, J = 12.5, 5.8 Hz, 1H), 4.18–4.05 (m, 4H), 1.97 (s, 2H), 1.93–1.88 (m, 13H), 1.86–1.82 (m, 9H); 13C-NMR (101 MHz, Acetone-d6) δ 170.71 (C), 170.70 (C), 170.29 (C), 170.19 (C), 170.03 (C), 170.01 (C), 169.96 (C), 169.92 (C), 161.31 (C), 138.86 (C), 136.24 (CH), 130.31 (CH), 129.88 (C), 128.96 (CH), 124.01 (CH), 122.29 (C), 117.77 (CH),82.25 (CH), 80.36 (CH), 76.37 (CH), 74.67 (CH), 74.37 (CH), 70.52 (CH), 70.41, 69.38, 68.58, 63.26 (CH2), 62.23 (CH2), 20.83 (CH3), 20.79 (CH3), 20.70 (CH3), 20.64 (CH3), 20.62 (CH3), 20.60 (CH3), 20.53 (CH3), 20.30 (CH3); HR-MS (ESI positive, m/z): found 860.2042 ([M + Na]+), calc. for C37H43NO19NaS (M + Na): 860.2048.
1-((2R,3R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)-3-(((2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)thio)quinolin-2(1H)-one 4a: Following procedure D, a mixture of 3b (20 mg, 1.0 equiv.) and K2CO3 (2 mg, 0.5 equiv.) in methanol (2 mL) was stirred under argon at room temperature for 1 h. The crude mixture was then filtered through celite, washed with 10 mL of methanol, and filtered again. The filtrate was concentrated under reduced pressure at 25. The product 4a was isolated as a white solid (12 mg, yield 98%); m.p.: 187–197.8 °C; Rf = 0.52 (EtOAc/MeOH: 9/1); − 63.15 (c, 1.0 in CHCl3); IR (neat): 1277, 1281, 798, 766, 749 cm−1; 1H-NMR (400 MHz, DMSO-d6) δ 7.95 (d, J = 8.6 Hz, 1H), 7.87 (s, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.45 (t, J = 8.0 Hz, 1H), 7.24 (t, J = 7.5 Hz, 1H), 6.31 (d, J = 9.7 Hz, 1H), 4.75 (d, J = 9.6 Hz, 1H), 4.04 (t, J = 9 Hz; 2H), 3.74 (d, J = 10.6 Hz, 3H), 3.24–3.13 (m, 15H); 13C-NMR (101 MHz, DMSO-d6) δ 160.08 (C), 135.24 (C), 133.12 (CH), 130.37 (C), 128.51 (CH), 128.23 (CH), 122.82 (CH), 121.52 (C), 117.63 (CH), 84.15 (CH), 84.02 (CH), 81.38 (CH), 80.89 (CH), 78.52 (CH), 78.18 (CH), 72.78 (CH), 70.22 (CH), 69.82 (CH), 68.91 (CH), 61.20 (CH2), 61.08 (CH2); HR-MS (ESI positive, m/z): found 524.1205 ([M + Na]+), calc. for C21H27NO11NaS (M + Na): 524.1203.
4. Conclusions
In summary, we have successfully developed an efficient method to synthesize various bis β-N,S-glycosyl quinolin-2-ones via the palladium-catalyzed coupling of α- or β-mono-, di-, and poly-thiosugar derivatives with α- or β-3-iodo-N-glycosylquinolin-2-ones. Efforts are now in progress to synthesize a large library of analogues through this strategy to study their biological activity. We expect this simple and general methodology to be of broad utility for the synthesis and development of new medicinal agents.
Supplementary Materials
The supplementary materials are available online. Spectra for all synthesized compounds are available online.
Acknowledgments
Authors acknowledge the support of this project by CNRS, University Paris Sud, ANR (ANR-15-CE29-0002), and by la Ligue Contre le Cancer through an Equipe Labellisée 2014 grant. We also thank the Algerian Ministry of Education and Research for a fellowship (PNE) to Wafa Redjdal. Our laboratory is a member of the Laboratory of Excellence LERMIT supported by a grant (ANR-10-LABX-33).
Author Contributions
Samir Messaoudi and Mouad Alami conceived and designed the experiments; Wafa Redjdal and Nada Ibrahim performed the experiments; Wafa Redjdal, Nada Ibrahim, Belkacem Benmerad, Mouad Alami, and Samir Messaoudi analyzed the data. Samir Messaoudi wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dwek, R.A. Glycobiology: Toward understanding the function of sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, A.A.-H.; El Ashry, S.H.; Richard, R.S. Synthesis of C-(d-glycopyranosyl)ethylamines and C-(d-glycofuranosyl)methylamines as potential glycosidase inhibitors. Carbohydr. Res. 1999, 315, 106–116. [Google Scholar] [CrossRef]
- Huryn, D.M.; Okabe, M. AIDS-driven nucleoside chemistry. Chem. Rev. 1992, 92, 1745–1768. [Google Scholar] [CrossRef]
- Knölker, H.J.; Reddy, K.R. Isolation and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2002, 102, 4303–4428. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, S.; Anizon, F.; Léonce, S.; Pierré, A.; Pfeiffer, B.; Prudhomme, M. Synthesis and in vitro cytotoxicities of 7-aza rebeccamycin analogues bearing various substituents on the sugar moiety, on the imide nitrogen and on the carbazole framework. Eur. J. Med. Chem. 2005, 40, 961–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messaoudi, S.; Anizon, F.; Léonce, S.; Pierré, A.; Pfeiffer, B.; Prudhomme, M. Synthesis and in vitro cytotoxicities of bridged aza-rebeccamycin derivatives. Tetrahedron 2005, 61, 7304–7316. [Google Scholar] [CrossRef] [Green Version]
- Henon, H.; Messaoudi, S.; Hugon, B.; Anizon, F.; Pfeiffer, B.; Prudhomme, M. Synthesis of granulatimide bis-imide analogues. Tetrahedron 2005, 61, 5599–5614. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Bailly, C.; Chaires, J.-B. NB-506, an indolocarbazole topoisomerase I inhibitor, binds preferentially to triplex DNA. FEBS Lett. 2000, 470, 355–359. [Google Scholar] [CrossRef]
- Ahmed Belmokhtar, C.; Hillion, J.; Ségal-Bendirdjian, E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 2001, 20, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, M.; Pengei, J.; Rohit, S.; Dmitri, S.; Ila, B.; Natsuko, N.; Ying, C.; Sandra, P. High-efficiency liposomal encapsulation of a tyrosine kinase inhibitor leads to improved in vivo toxicity and tumor response profile. Int. J. Nanomed. 2013, 8, 3991–4006. [Google Scholar]
- Huguet, F.; Leguay, T.; Raffoux, E.; Rousselot, P.; Vey, N.; Pigneux, A.; Ifrah, N.; Dombret, H. Clofarabine for the treatment of adult acute lymphoid leukemia: The Group for Research on Adult Acute Lymphoblastic Leukemia intergroup. Leuk. Lymphoma 2015, 56, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Mata, G.; Luedtke, N.W. Synthesis and solvatochromic fluorescence of biaryl pyrimidine nucleosides. Org. Lett. 2013, 15, 2462–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaneda, F.; Burse, A.; Boland, W.; Kinne, R.K.H. Thioglycosides as inhibitors of hSGLT1 and hSGLT2: Potential therapeutic agents for the control of hyperglycemia in diabetes. Int. J. Med. Sci. 2007, 4, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Samoshin, A.V.; Dotsenko, I.A.; Samoshina, N.M.; Franz, A.H.; Samoshin, V.V. Thio-β-d-glucosides: Synthesis and evaluation as glycosidase inhibitors and activators. Int. J. Carbohydr. Chem. 2014, 2014, 8. [Google Scholar] [CrossRef]
- Elgemeie, G.H.; Farag, A.B.; Amin, K.M.; El-Badry, O.M.; Hassan, G.S. Design, synthesis and cytotoxic evaluation of novel heterocyclic thioglycosides. Med. Chem. 2014, 4, 814–820. [Google Scholar] [CrossRef]
- Rodrigue, J.; Ganne, G.; Blanchard, B.; Saucier, C.; Giguere, D.; Shiao, T.C.; Varrot, A.; Imberty, A.; Roy, R. Aromatic thioglycoside inhibitors against the virulence factor LecA from Pseudomonas aeruginosa. Org. Biomol. Chem. 2013, 11, 6906–6918. [Google Scholar] [CrossRef] [PubMed]
- Kato, E.; Nagano, H.; Yamamura, S.; Ueda, M. Synthetic inhibitor of leaf-closure that reveals the biological importance of leaf-movement for the survival of leguminous plants. Tetrahedron 2003, 59, 5909–5917. [Google Scholar] [CrossRef]
- Schnabelrauch, M.; Vasella, A.; Withers, S.G. Synthesis and evaluation as irreversible glycosidase inhibitors of mono and oligo (glycosy1thio) benzoquinones. Helv. Chim. Acta 1994, 77, 778–799. [Google Scholar] [CrossRef]
- Adinolfi, M.; d’Ischia, M.; Iadonisi, A.; Leone, L.; Pezzella, A.; Valerio, S. Glycosylated eumelanin building blocks by thioglycosylation of 5,6-diacetoxyindole with an expedient selenium-based dynamic-mixture methodology. Eur. J. Org. Chem. 2012, 2012, 4333–4338. [Google Scholar] [CrossRef]
- Audisio, D.; Messaoudi, S.; Cegielkowski, L.; Peyrat, J.-F.; Brion, J.-D.; Methy-Gonnot, D.; Radanyi, C.; Renoir, J.-M.; Alami, M. Discovery and biological activity of 6BrCaQ as an inhibitor of the Hsp90 protein folding machinery. ChemMedChem 2011, 11, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Hewawasam, P.; Chen, N.; Ding, M.; Natale, J.T.; Boissard, C.G.; Yeola, S.; Gribkoff, V.K.; Starrett, J.; Dworetzky, S.I. The synthesis and structure-activity relationships of 3-amino-4-benzylquinolin-2-ones; discovery of novel KCNQ2 channel openers. Bioorg. Med. Chem. Lett. 2004, 14, 1615–1618. [Google Scholar] [CrossRef] [PubMed]
- Raitio, K.H.; Savinainen, J.R.; Vepsäläinen, J.; Laitinen, J.T.; Poso, A.; Järvinen, T.; Nevalainen, T. Synthesis and SAR studies of 2-oxoquinoline derivatives as CB2 receptor inverse agonists. J. Med. Chem. 2006, 49, 2022–2027. [Google Scholar] [CrossRef] [PubMed]
- Hewawasam, P.; Fan, W.; Ding, M.; Flint, K.; Cook, D.; Goggings, G.D.; Myers, R.A.; Gribkoff, V.K.; Boissard, C.G.; Dworetzky, S.I.; et al. 4-aryl-3-(hydroxyalkyl)quinolin-2-ones: Novel maxi-K channel opening relaxants of corporal smooth muscle targeted for erectile dysfunction. J. Med. Chem. 2003, 46, 2819–2822. [Google Scholar] [CrossRef] [PubMed]
- Hewawasam, P.; Fan, W.; Knipe, J.; Moon, S.L.; Boissard, C.G.; Gribkoff, V.K.; Starret, J.E. The synthesis and structure-activity relationships of 4-aryl-3-aminoquinolin-2-ones: A new class of calcium-dependent, large conductance, potassium (maxi-K) channel openers targeted for post-stroke neuroprotection. Bioorg. Med. Chem. Lett. 2002, 12, 1779–1783. [Google Scholar] [CrossRef]
- Cordi, A.A.; Desos, P.; Randle, J.C.; Lepagnol, J. Structure-activity relationships in a series of 3-sulfonylamino-2-(1H)-quinolones, as new AMPA/kainate and glycine antagonists. Bioorg. Med. Chem. 1995, 3, 129–141. [Google Scholar] [CrossRef]
- Desos, P.; Lepagnol, J.M.; Morain, P.; Lestage, P.; Cordi, A.A. Structure-activity relationships in a series of 2(1H)-quinolones bearing different acidic function in the 3-position: 6,7-dichloro-2(1H)-oxoquinoline-3-phosphonic acid, a new potent and selective AMPA/kainate antagonist with neuroprotective properties. J. Med. Chem. 1996, 39, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Carling, R.W.; Leeson, P.D.; Moore, K.W.; Smith, J.D.; Moyes, C.R.; Mawer, I.M.; Thomas, S.; Chan, T.; Baker, R.; Foster, A.; et al. 3-Nitro-3,4-dihydro-2(1H)-quinolones. Excitatory amino acid antagonists acting at glycine-site NMDA and (RS)-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. J. Med. Chem. 1993, 36, 3397–3408. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.T.H.; Brion, J.-D.; Lescop, E.; Alami, M.; Messaoudi, S. Intramolecular Pd-catalyzed arylation of 1-amidosugars: A new route to n-glycosyl quinolin-2-ones. Org. Lett. 2016, 18, 2126–2129. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.T.H.; Touchet, S.; Alami, M.; Messaoudi, S. Selective palladium-catalyzed domino Heck/Buchwald-Hartwig arylations of N-glycosylcinnamamides: An efficient route to 4-aryl N-glycosyl quinolin-2-ones. Adv. Synth. Catal. 2017, 359, 1320–1330. [Google Scholar] [CrossRef]
- Bruneau, A.; Brion, J.-D.; Alami, M.; Messaoudi, S. Stereoselective copper-catalyzed Chan-Lam-Evans N-arylation of glucosamines with arylboronic acids at room temperature. Chem. Commun. 2013, 49, 8359–8361. [Google Scholar] [CrossRef] [PubMed]
- Brachet, E.; Brion, J.-D.; Messaoudi, S.; Alami, M. Stereoselective palladium-catalyzed alkenylation and alkynylation of thioglycosides. Adv. Synth. Catal. 2013, 355, 2627–2636. [Google Scholar] [CrossRef]
- Brachet, E.; Brion, J.-D.; Alami, M.; Messaoudi, S. Nickel catalyzed arylation, alkenylation and alkynylation of unprotected thioglycosides at room temperature. Chem. Eur. J. 2013, 19, 15276–15280. [Google Scholar] [CrossRef] [PubMed]
- Chabrier, A.; Bruneau, A.; Benmahdjoub, S.; Benmerad, B.; Belaid, S.; Brion, J.-D.; Alami, M.; Messaoudi, S. Stereoretentive copper catalyzed directed thioglycosylation of C(sp2)–H bonds of benzamides. Chem. Eur. J. 2016, 22, 15006–15010. [Google Scholar] [CrossRef] [PubMed]
- Probst, N.; Grelier, G.; Ghermani, N.; Gandon, V.; Alami, M.; Messaoudi, S. Intramolecular Pd-catalyzed anomeric C(sp3)–H activation of glycosyl carboxamides. Org. Lett. 2017, 19, 5038–5041. [Google Scholar] [CrossRef] [PubMed]
- AL-Shuaeeb, R.-A.-A.; Montoir, D.; Alami, M.; Messaoudi, S. Synthesis of (1→2)-S-linked saccharides and S-linked glycoconjugates via a Palladium-G3-XantPhos precatalyst catalysis. J. Org. Chem. 2017, 82, 6720–6728. [Google Scholar] [CrossRef] [PubMed]
- Probst, N.; Lartia, R.; Théry, O.; Alami, M.; Defrancq, E.; Messaoudi, S. Efficient Buchwald-Hartwig-Migita cross-coupling for DNA thioglycoconjugation. Chem. Eur. J. 2018, 24, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Klapars, A.; Buchwald, S.L. Copper-catalyzed halogen exchange in aryl halides: An aromatic Finkelstein reaction. J. Am. Chem. Soc. 2002, 124, 14844–14845. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, A.; Roche, M.; Hamze, A.; Brion, J.-D.; Alami, M.; Messaoudi, S. Stereoretentive palladium-catalyzed arylation, alkenylation and alkynylation of 1-thiosugars and thiols using aminobiphenyl palladacycle precatalyst at room temperature. Chem. Eur. J. 2015, 21, 8375–8379. [Google Scholar] [CrossRef] [PubMed]
- Zemplen, G.; Kunz, A. Studien über amygdalin, IV: Synthese des natürlichen l-Amygdalins. Eur. J. Inorg. Chem. 1924, 57, 1357–1359. [Google Scholar] [CrossRef]
- Ren, B.; Wang, M.; Liu, J.; Ge, J.; Zhang, X.; Dong, H. Zemplén transesterification: A name reaction that has misled us for 90 years. Green Chem. 2015, 17, 1390–1394. [Google Scholar] [CrossRef]
- Wang, Z. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |
Figure 1.
(A) Heteroaryl glycoside-based bioactive molecules; (B) General structure of the targeted -N,S-bis-glycosylated quinolinones (3).
Figure 1.
(A) Heteroaryl glycoside-based bioactive molecules; (B) General structure of the targeted -N,S-bis-glycosylated quinolinones (3).
Scheme 1.
Halogenation of N-(β-glucopyranosyl)quinolin-2-ones.
Scheme 2.
Coupling of the quinolones β-2a and β-2d with tetra-O-acetylated 1-thio-β-d-galactopyranose 1a under various reaction conditions.
Scheme 2.
Coupling of the quinolones β-2a and β-2d with tetra-O-acetylated 1-thio-β-d-galactopyranose 1a under various reaction conditions.
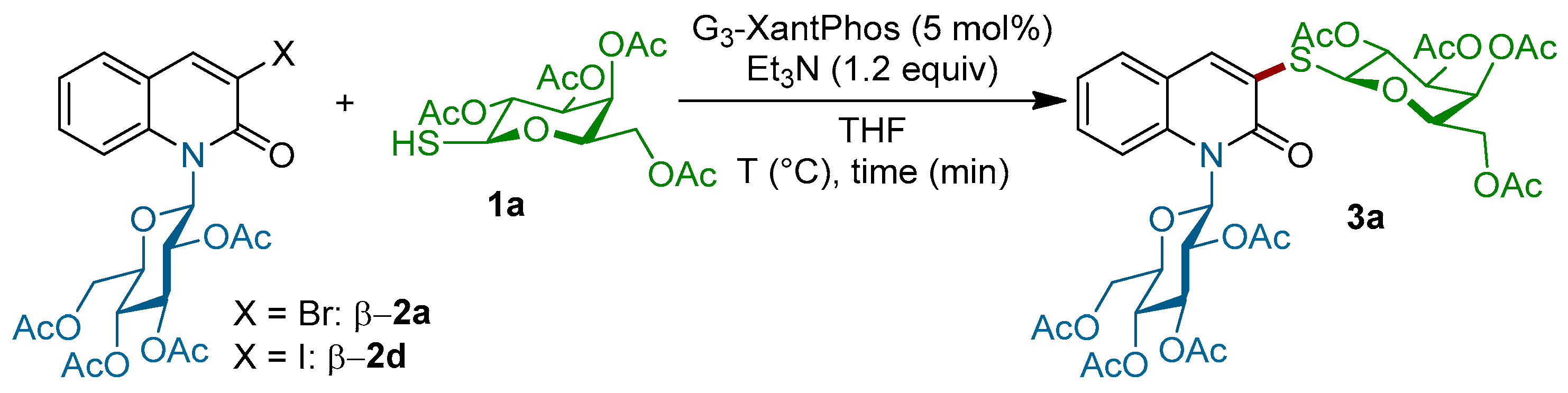
Scheme 3.
Coupling of the quinolones 2d–g with thiosugars 1a–f under the optimized conditions.
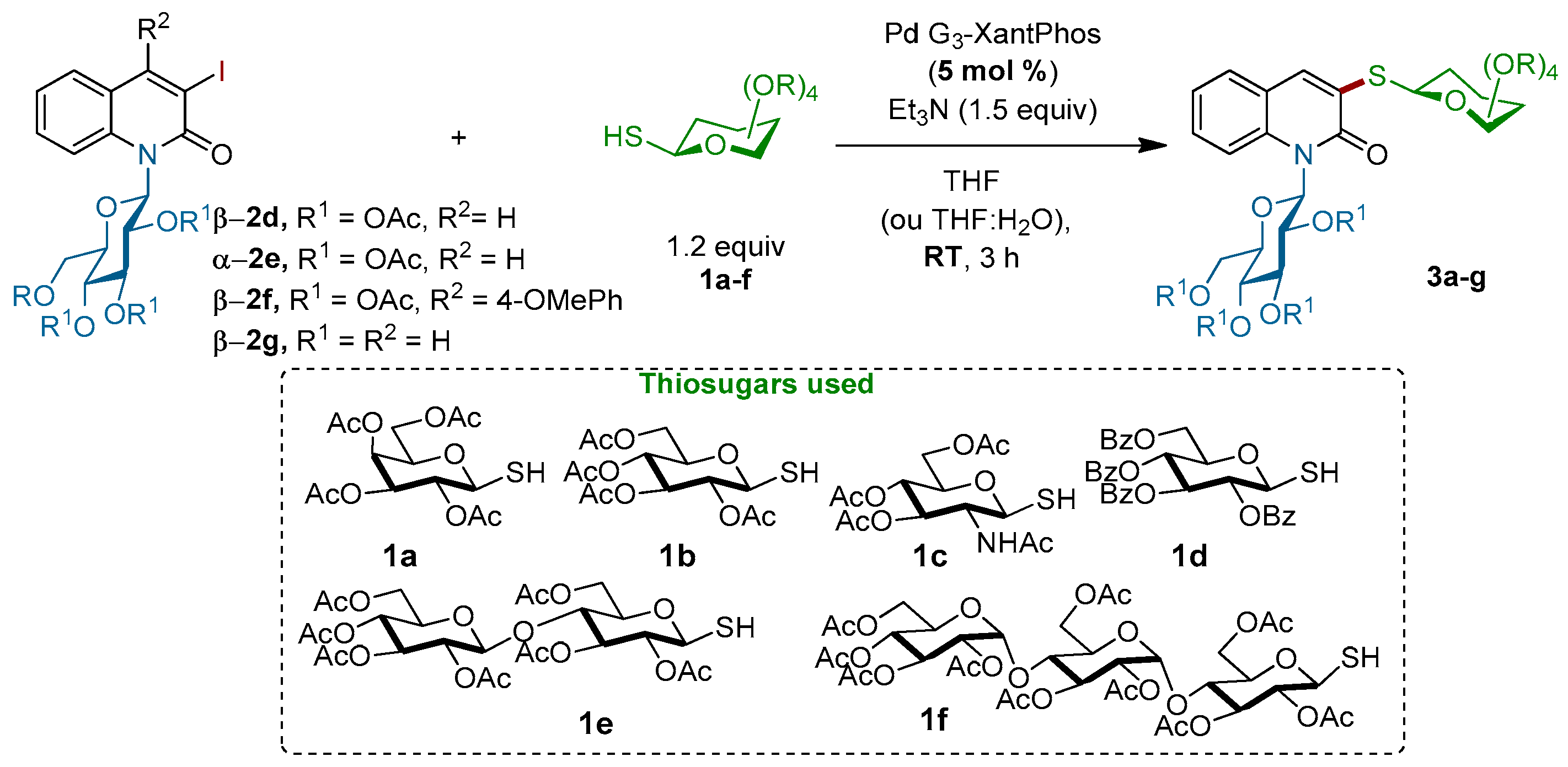
Figure 2.
Scope of thiosugars 1a–f for the Pd-catalyzed coupling with N-glucosylquinolinones 2d–g a. a Conditions: Reactions of 1 (2.5 equiv.) with 2 (1.0 equiv.) were performed in a resealable tube by using Pd-G3-XantPhos (5 mol %) and Et3N (1.5 equiv.) in 1,4-dioxane (0.1 M) at room temperature for 3 h; b Yield of isolated; c THF:H2O (8:2) was used as a solvent.
Figure 2.
Scope of thiosugars 1a–f for the Pd-catalyzed coupling with N-glucosylquinolinones 2d–g a. a Conditions: Reactions of 1 (2.5 equiv.) with 2 (1.0 equiv.) were performed in a resealable tube by using Pd-G3-XantPhos (5 mol %) and Et3N (1.5 equiv.) in 1,4-dioxane (0.1 M) at room temperature for 3 h; b Yield of isolated; c THF:H2O (8:2) was used as a solvent.
Scheme 4.
Deprotection of β-N,S-bis glycosyl quinolin-2-one 3b. a isolated yield.
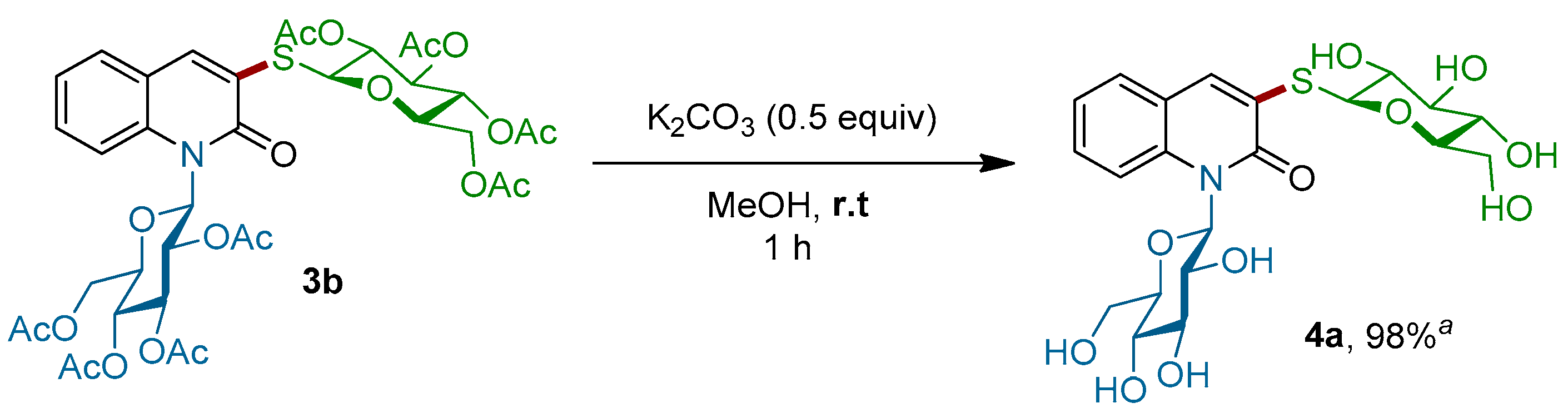

{kind=link}
{kind=link}
{kind=link}
{kind=link}
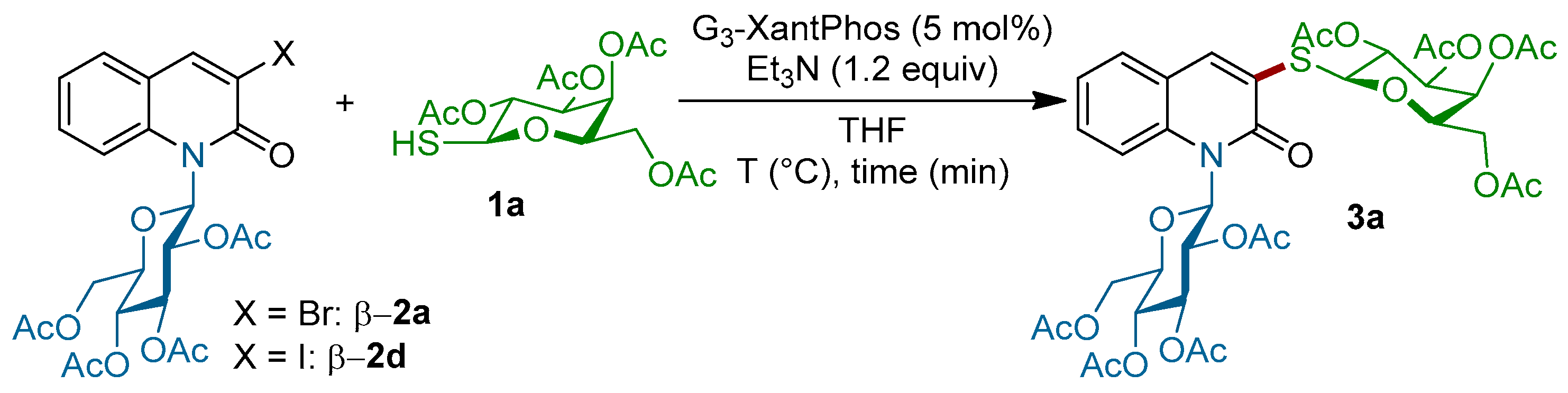{kind=link}
{kind=link}
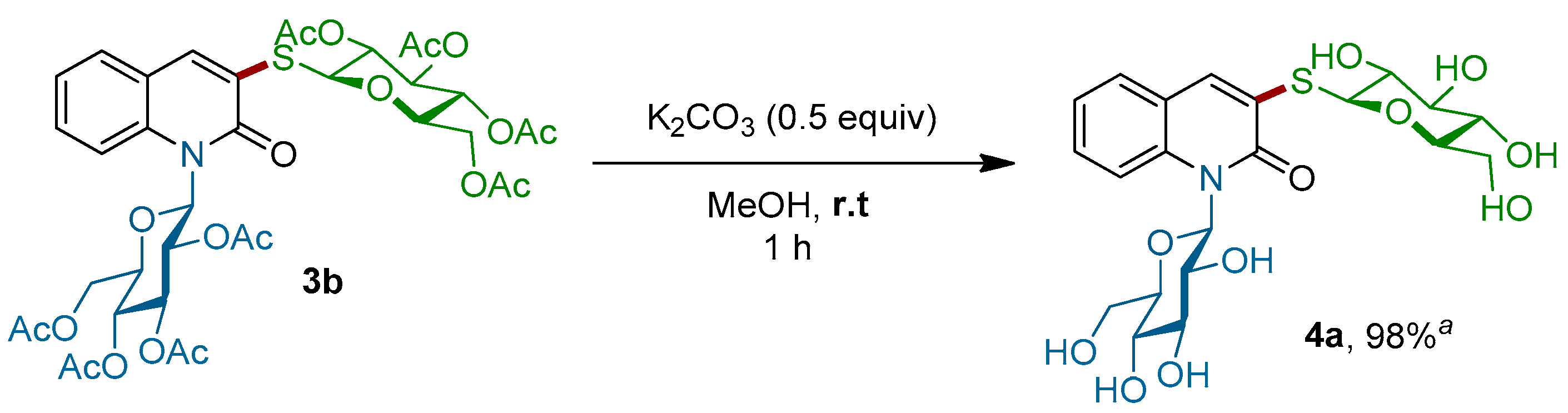{kind=link}
Table 1.
Survey of reaction conditions for the coupling of tetra-O-acetylated 1-thio-β-d-galactopyranose 1a with 3-halo N-glucosylquinolinone β-2a,d.
Table 1.
Survey of reaction conditions for the coupling of tetra-O-acetylated 1-thio-β-d-galactopyranose 1a with 3-halo N-glucosylquinolinone β-2a,d.
| Entry | Comp. β-2 | 1a (equiv.) | Time (h) | Temperature (°C) | Conversion Rate a | Yield (%) b |
|---|---|---|---|---|---|---|
| 1 | 2a | 1.5 | 5 | r.t. | 0 | - |
| 2 | 2a | 1.5 | 5 | 60 | 36% | - |
| 3 | 2a | 2.5 | 1 | 100 | 42% | - |
| 4 | 2d | 2.5 | 3 | r.t. | 100% | 70% |
| 5 | 2d | 1.5 | 3 | r.t. | 40% | - |
a Conversion rate was determined by 1H-NMR in the crude reaction mixture based on the chemical shift (ppm) of the proton signal H4 for haloquinolinone β-2a,b (δ = 8.35) and 3a (δ = 8.27); b Yield of isolated 3a.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Redjdal, W.; Ibrahim, N.; Benmerad, B.; Alami, M.; Messaoudi, S. Convergent Synthesis of N,S-bis Glycosylquinolin-2-ones via a Pd-G3-XantPhos Precatalyst Catalysis. Molecules 2018, 23, 519. https://doi.org/10.3390/molecules23030519
AMA Style
Redjdal W, Ibrahim N, Benmerad B, Alami M, Messaoudi S. Convergent Synthesis of N,S-bis Glycosylquinolin-2-ones via a Pd-G3-XantPhos Precatalyst Catalysis. Molecules. 2018; 23(3):519. https://doi.org/10.3390/molecules23030519
Chicago/Turabian StyleRedjdal, Wafa, Nada Ibrahim, Belkacem Benmerad, Mouad Alami, and Samir Messaoudi. 2018. "Convergent Synthesis of N,S-bis Glycosylquinolin-2-ones via a Pd-G3-XantPhos Precatalyst Catalysis" Molecules 23, no. 3: 519. https://doi.org/10.3390/molecules23030519