Substrate Binding Switches the Conformation at the Lynchpin Site in the Substrate-Binding Domain of Human Hsp70 to Enable Allosteric Interdomain Communication


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
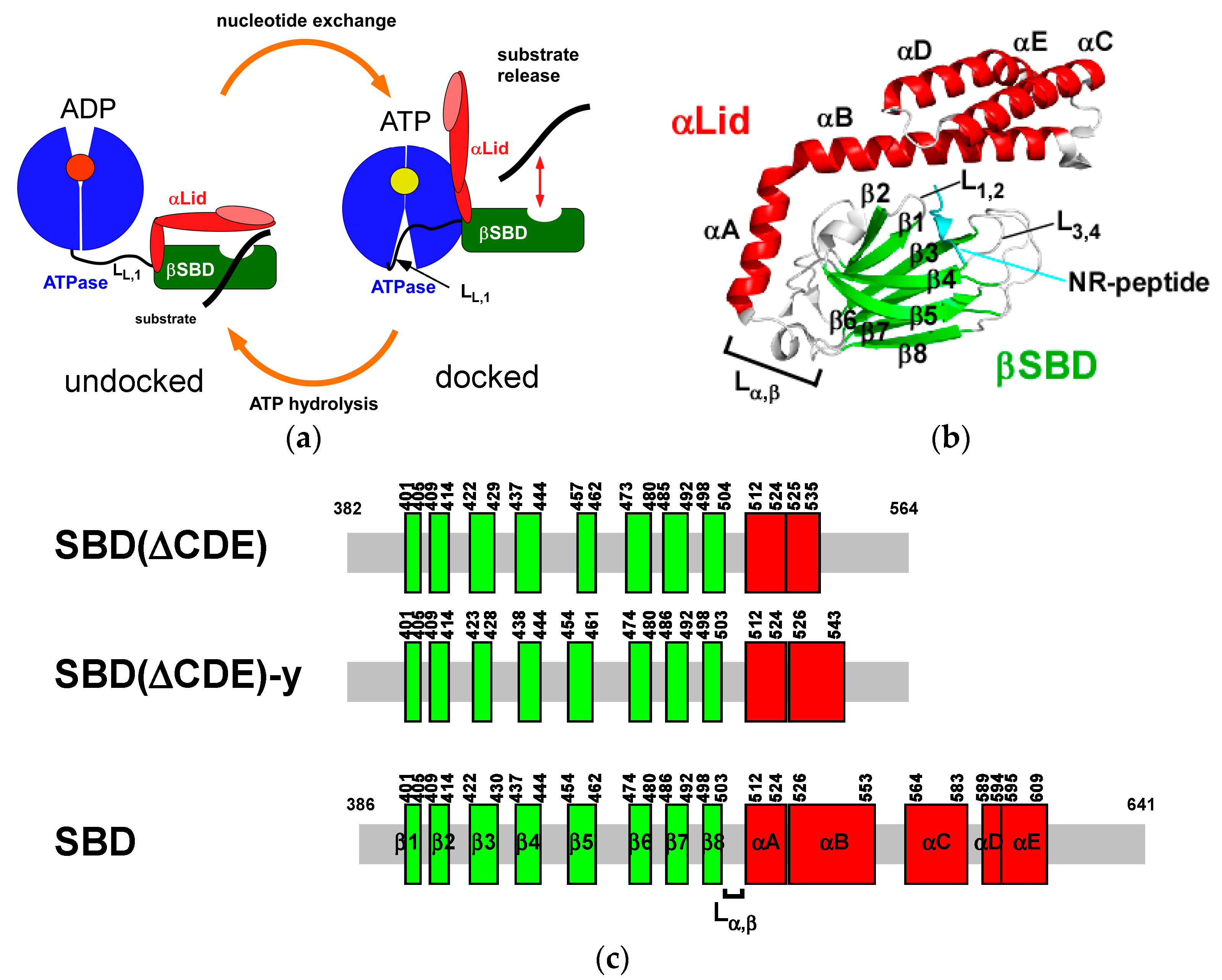
1. Introduction
2. Results
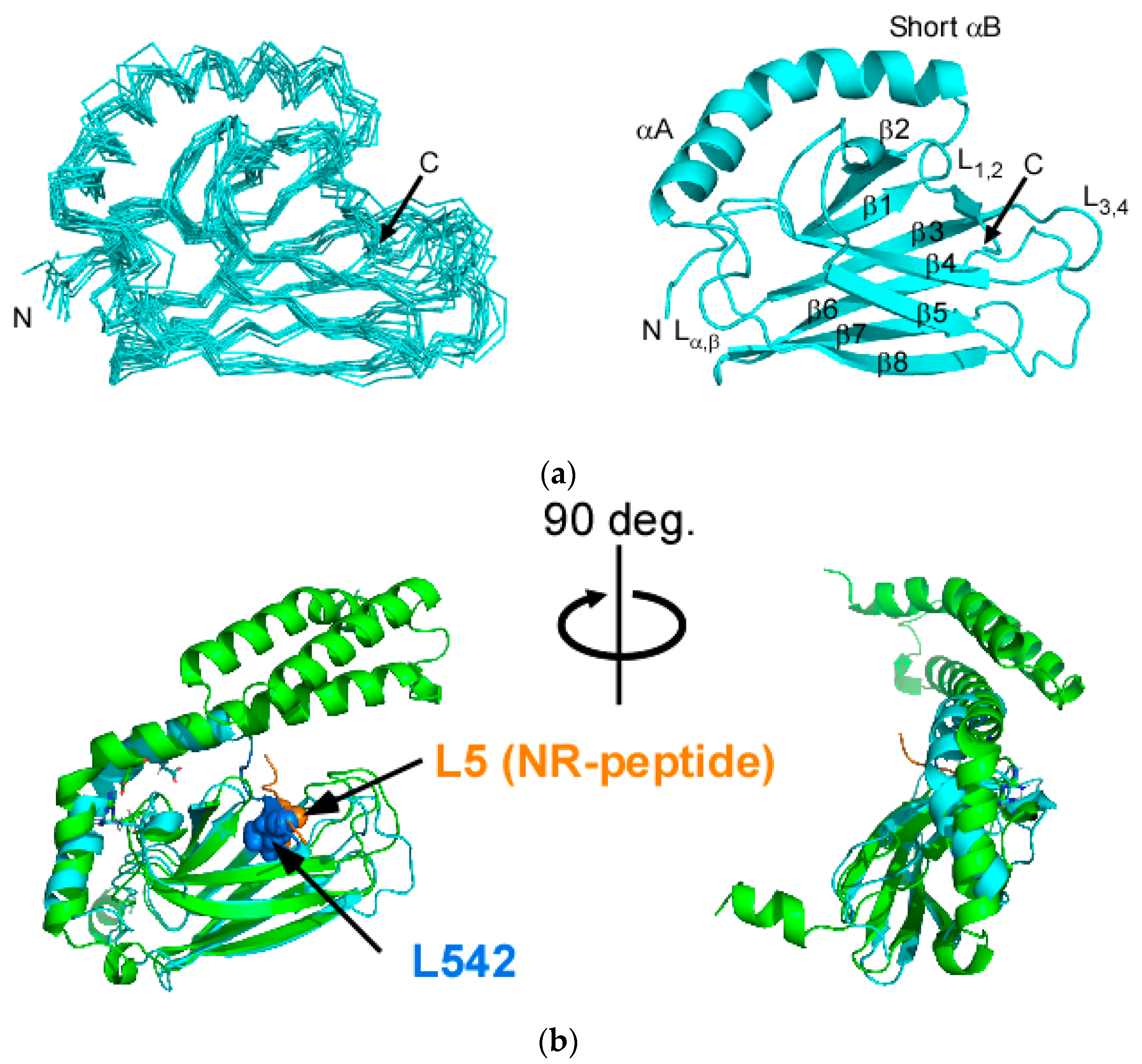
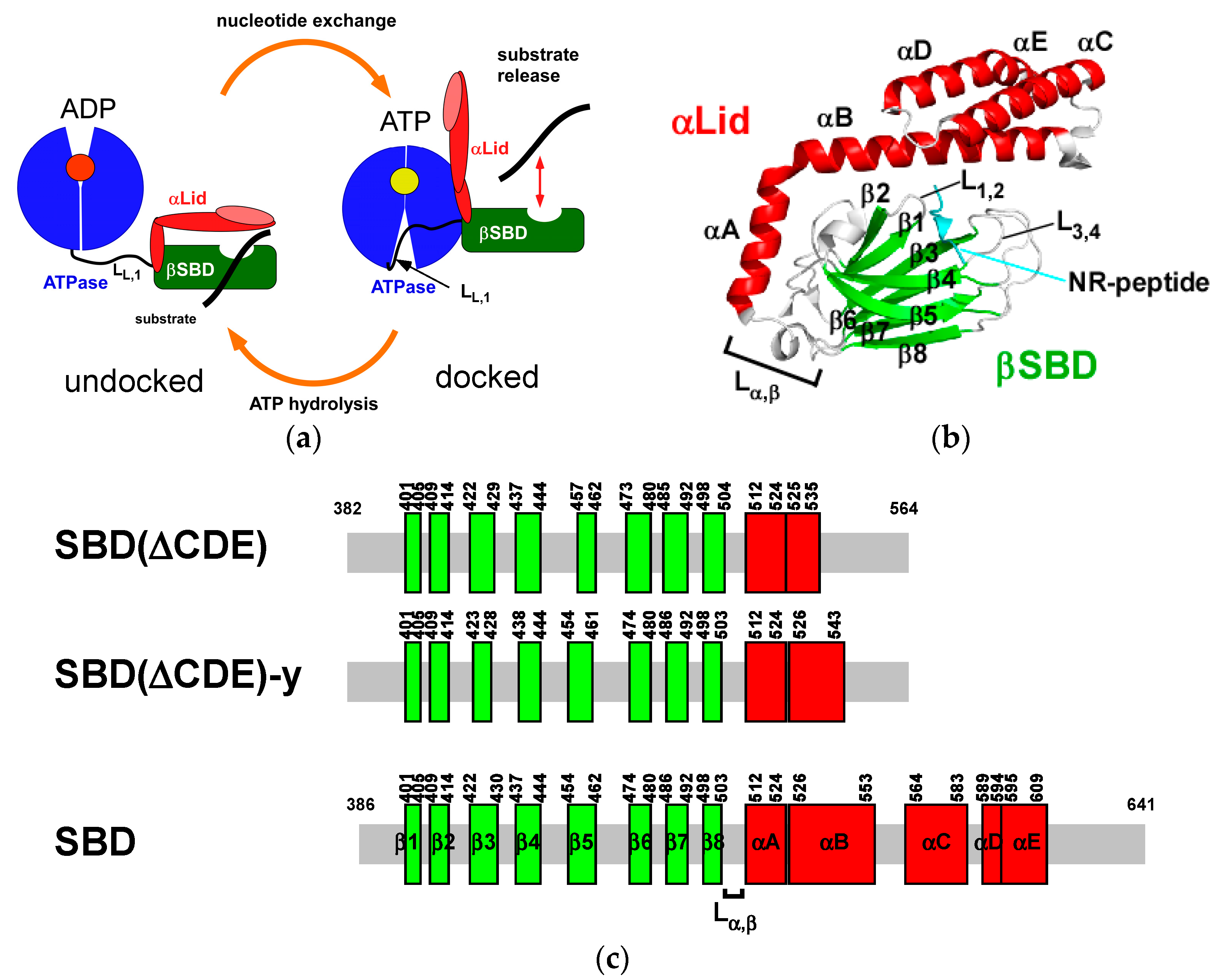
2.1. Solution Structure of Human Hsp70 SBD(∆CDE)
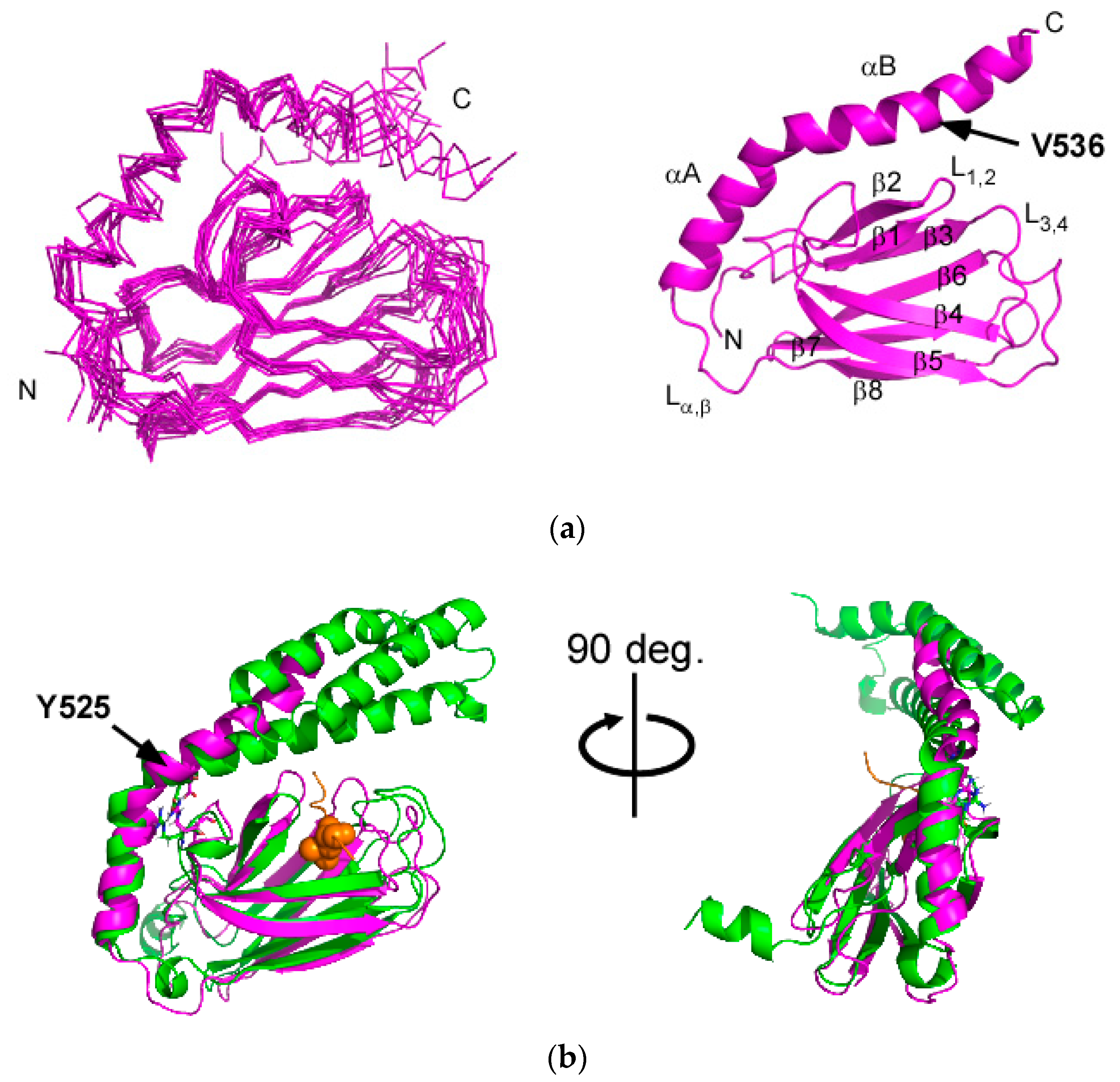
2.2. Solution Structure of the Hsp70 SBD that Lacks the Intramolecular αB Interaction, SBD(∆CDE)-y
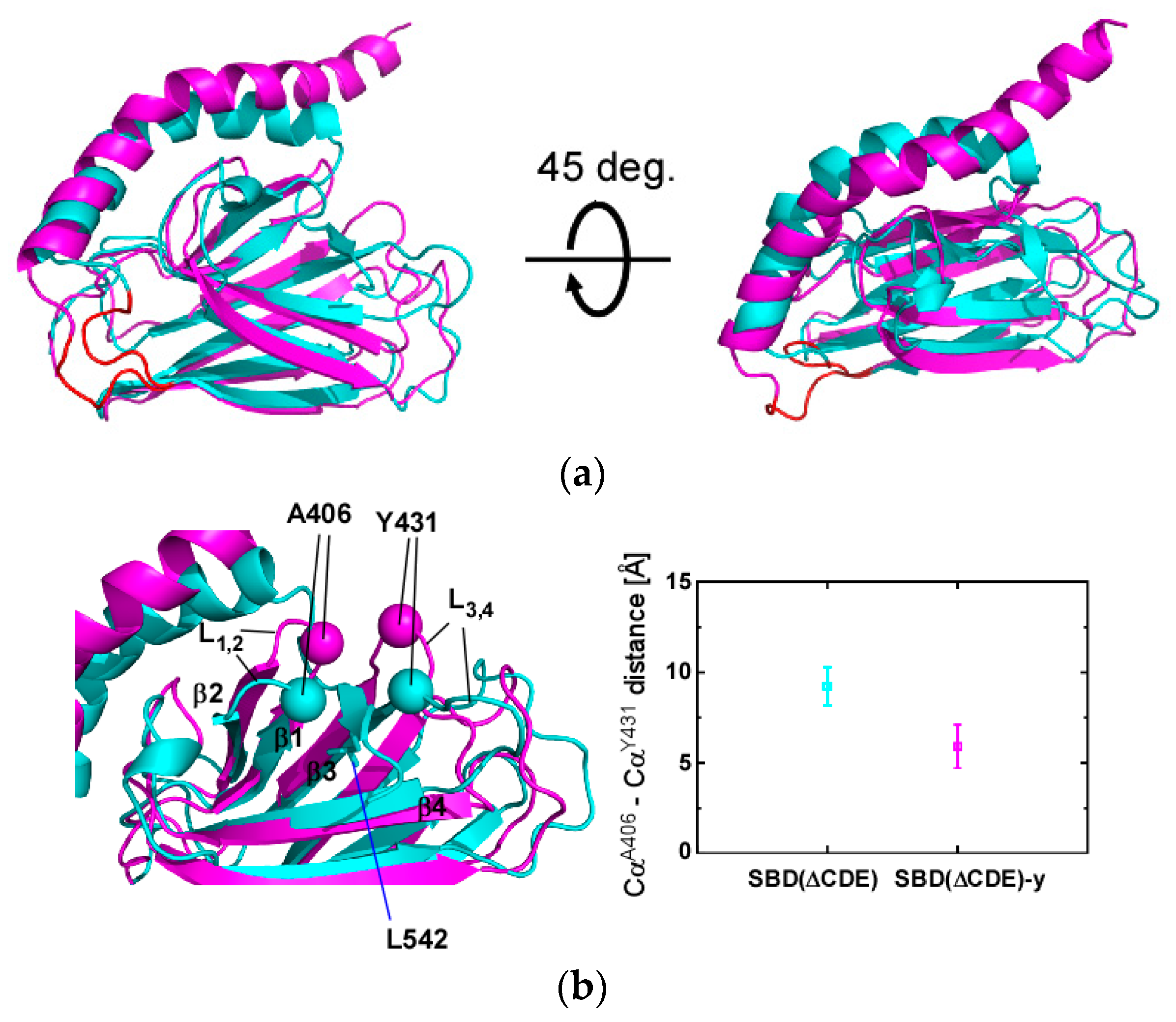
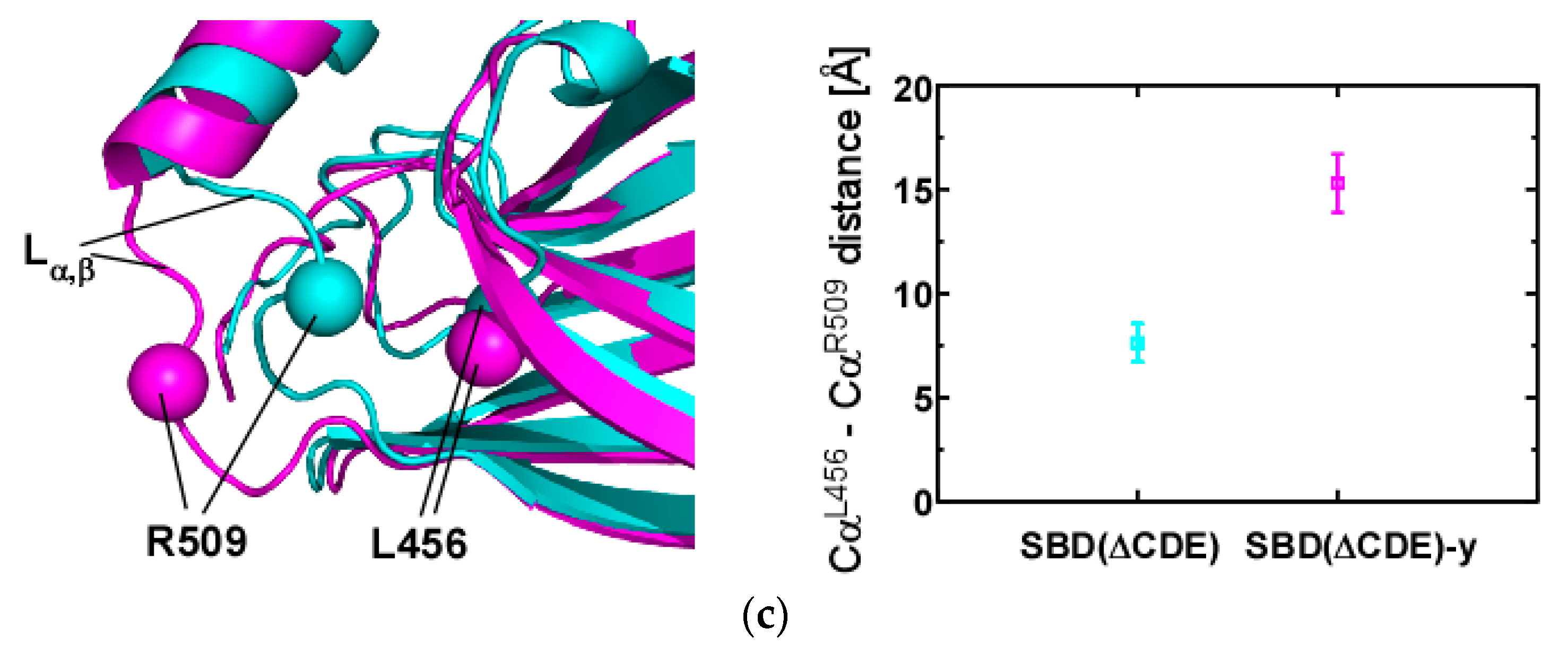
2.3. Structural Comparison of SBD(∆CDE) and SBD(∆CDE)-y
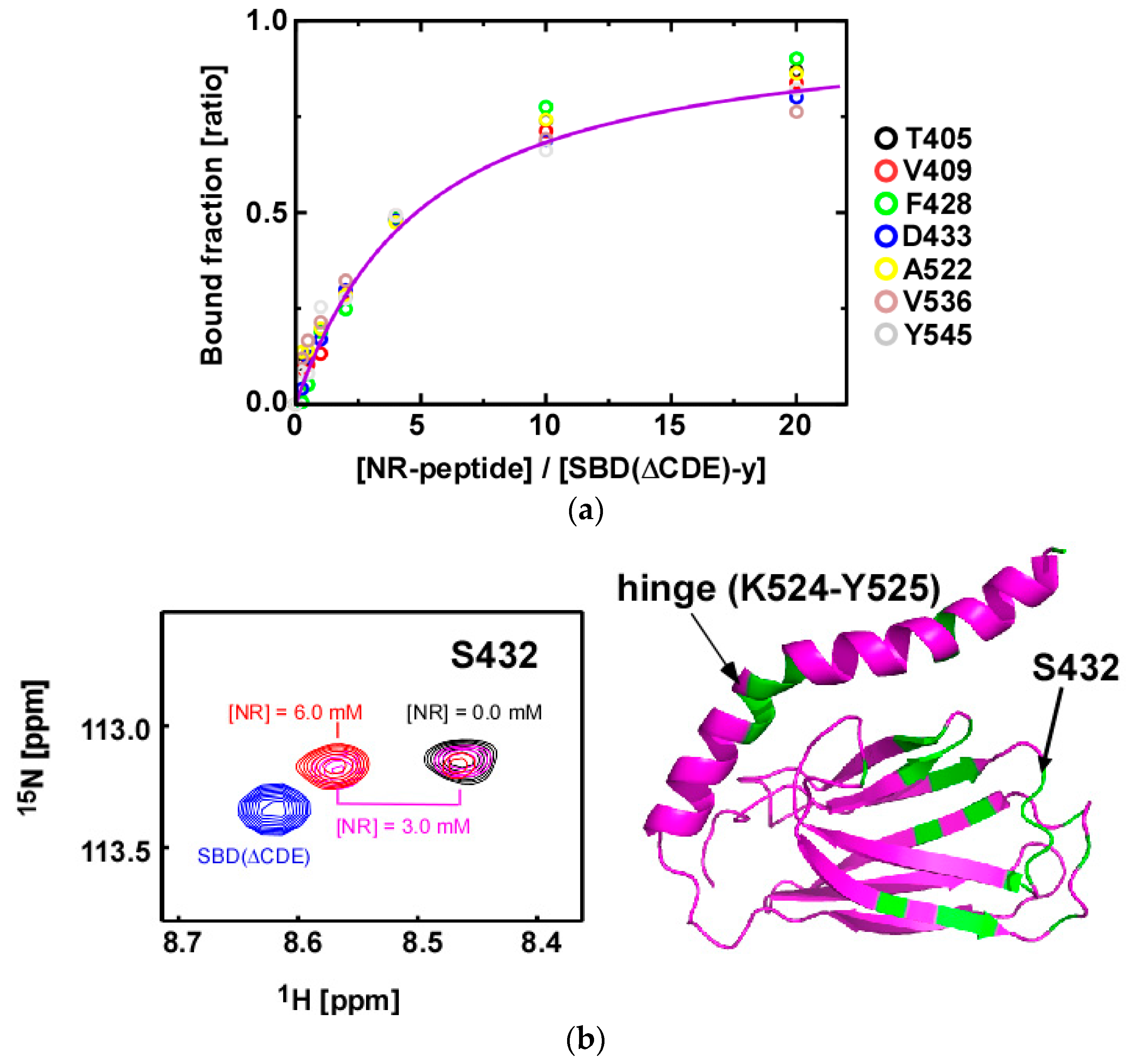
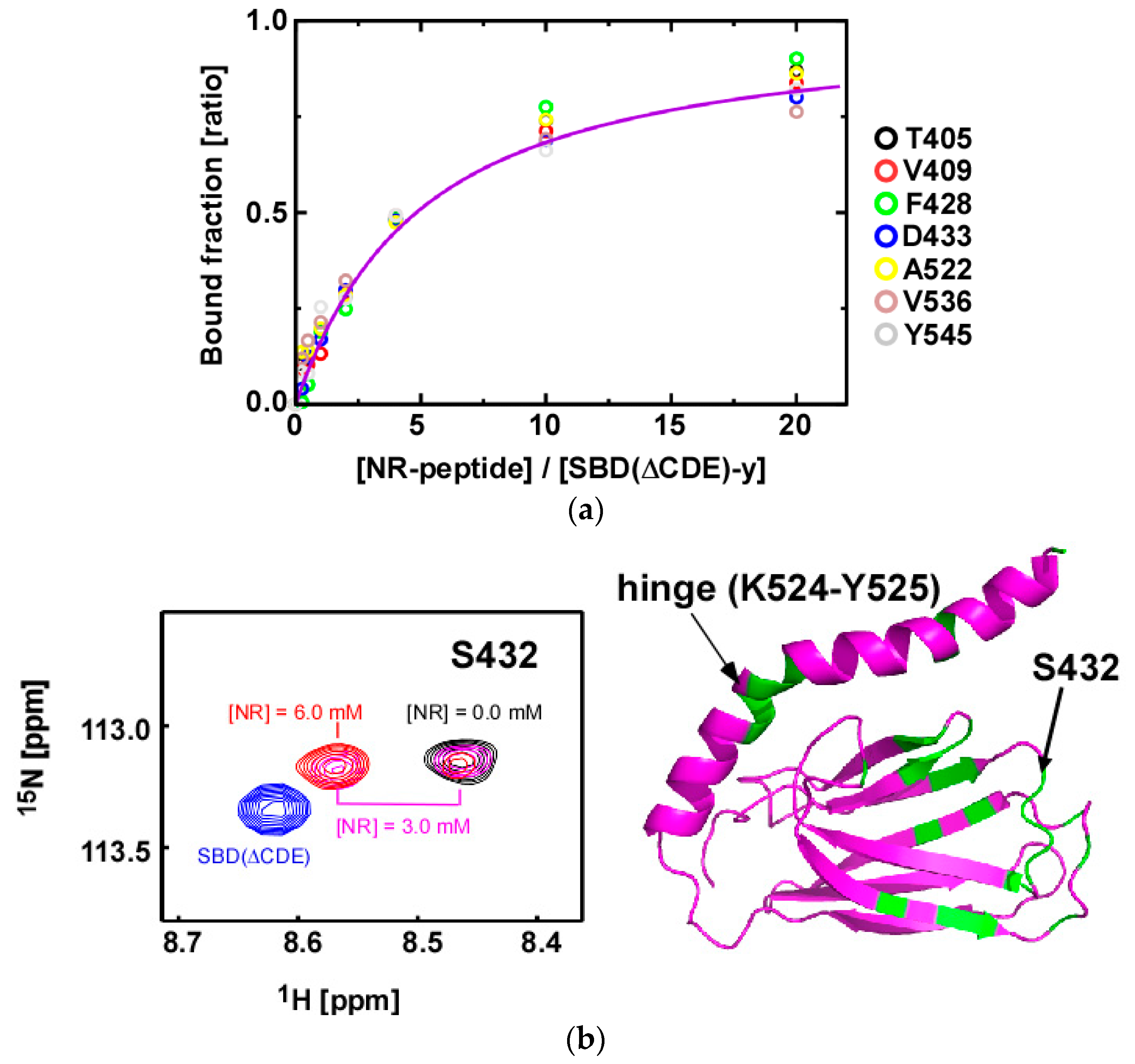
2.4. Substrate Binding Causes Chemical Shift Changes at the Hinge Between αA and αB in the αLid
3. Discussion
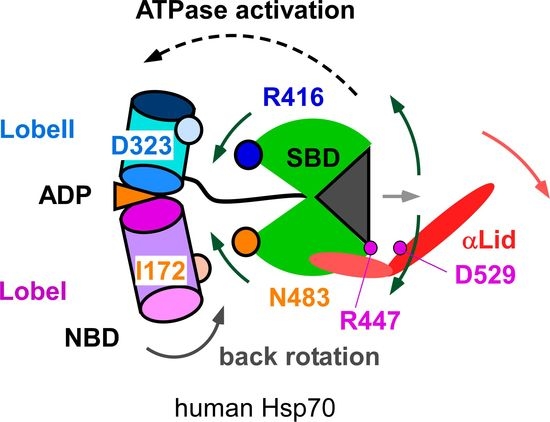
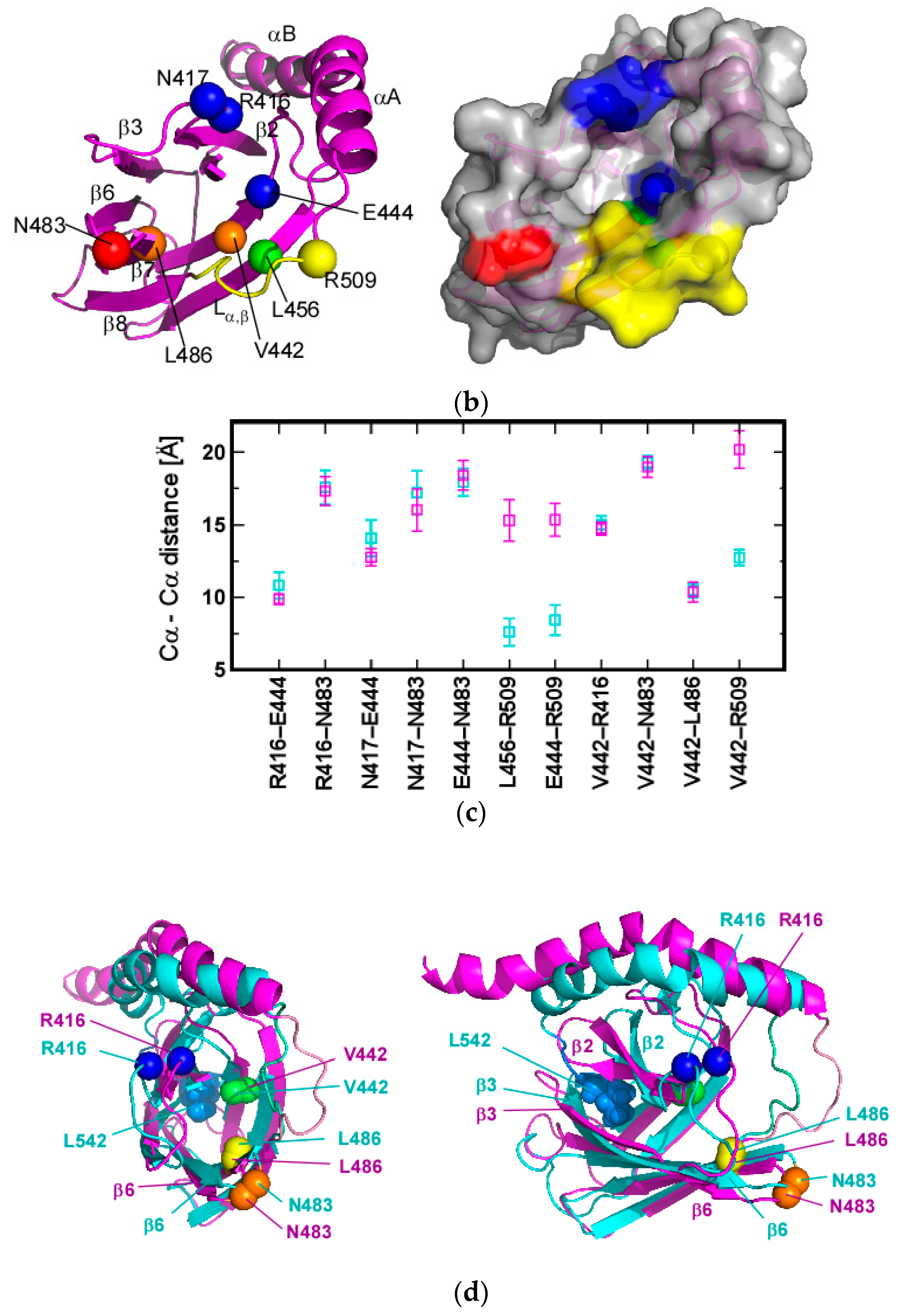
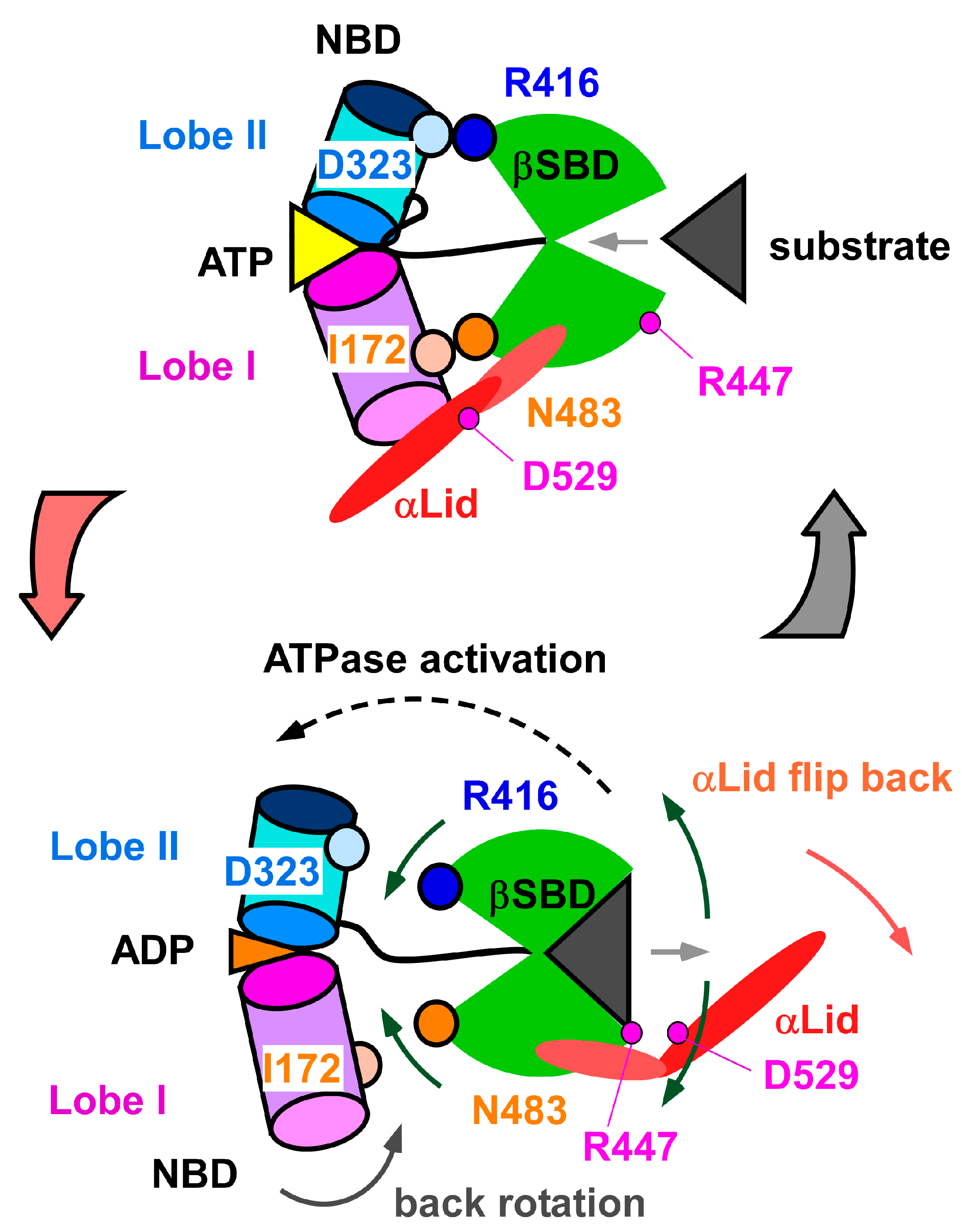
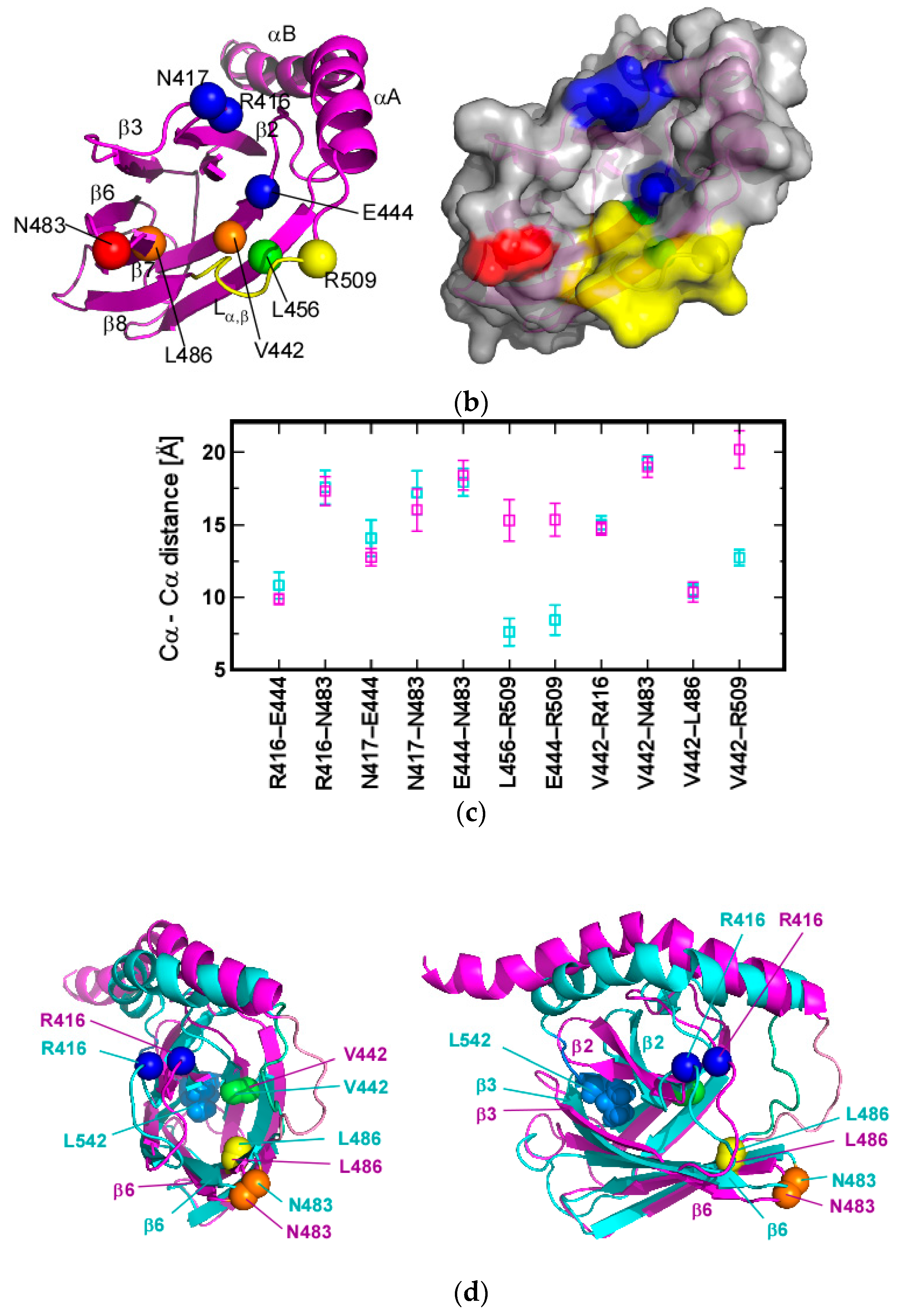
3.1. Client-Peptide Binding Changes the Structure at the Lynchpin Site for the Interdomain Contact
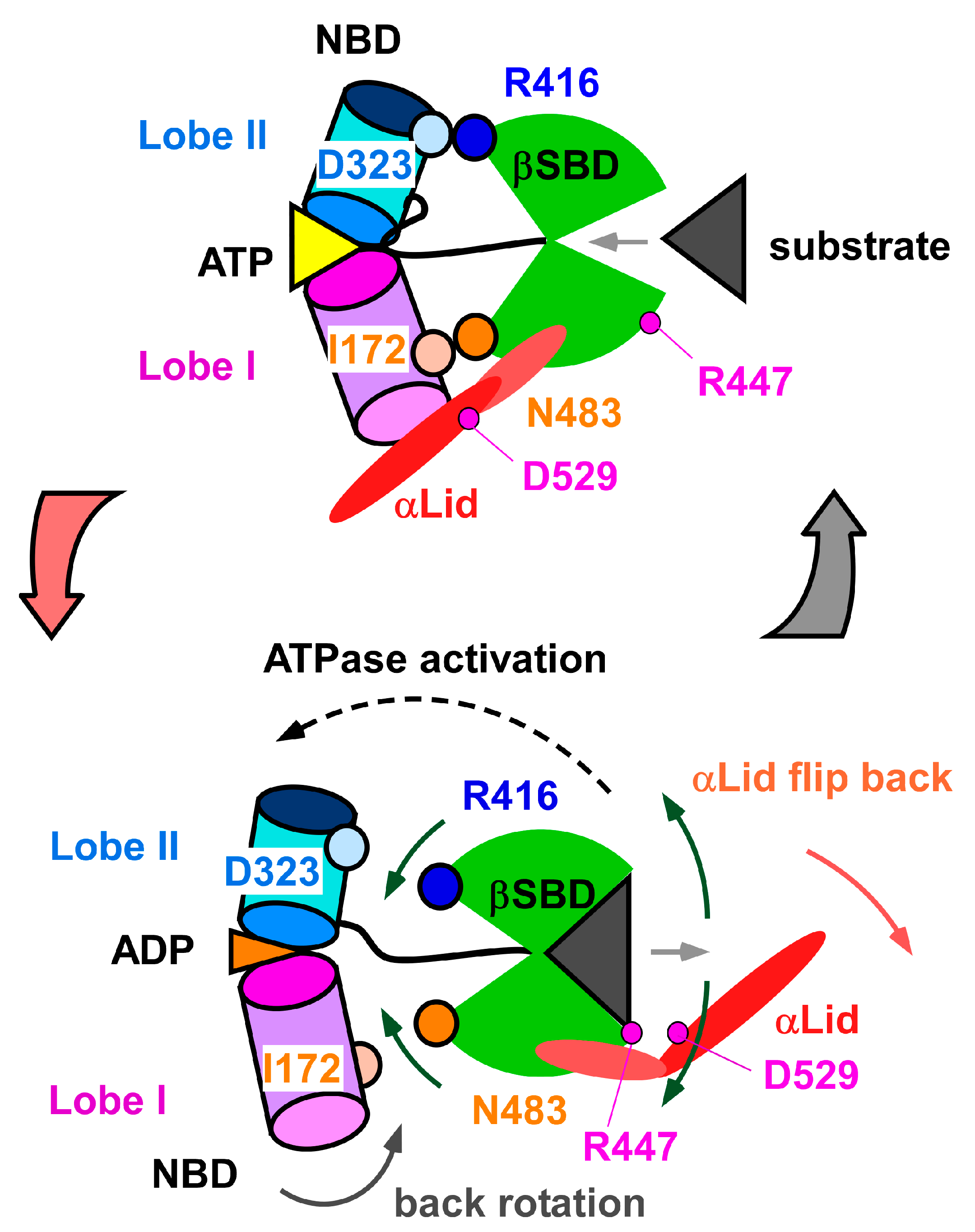
3.2. Change in αA Orientation upon Substrate Binding May Pave the Other Way for Interdomain Communication between SBD and NBD
4. Materials and Methods
4.1. Sample Preparation
4.2. NMR Spectroscopy and Structure Determination
4.3. NMR Spin Relaxation Experiments
4.4. Isothermal Titration Calorimetry (ITC) Experiments
4.5. NMR Titration Experiments
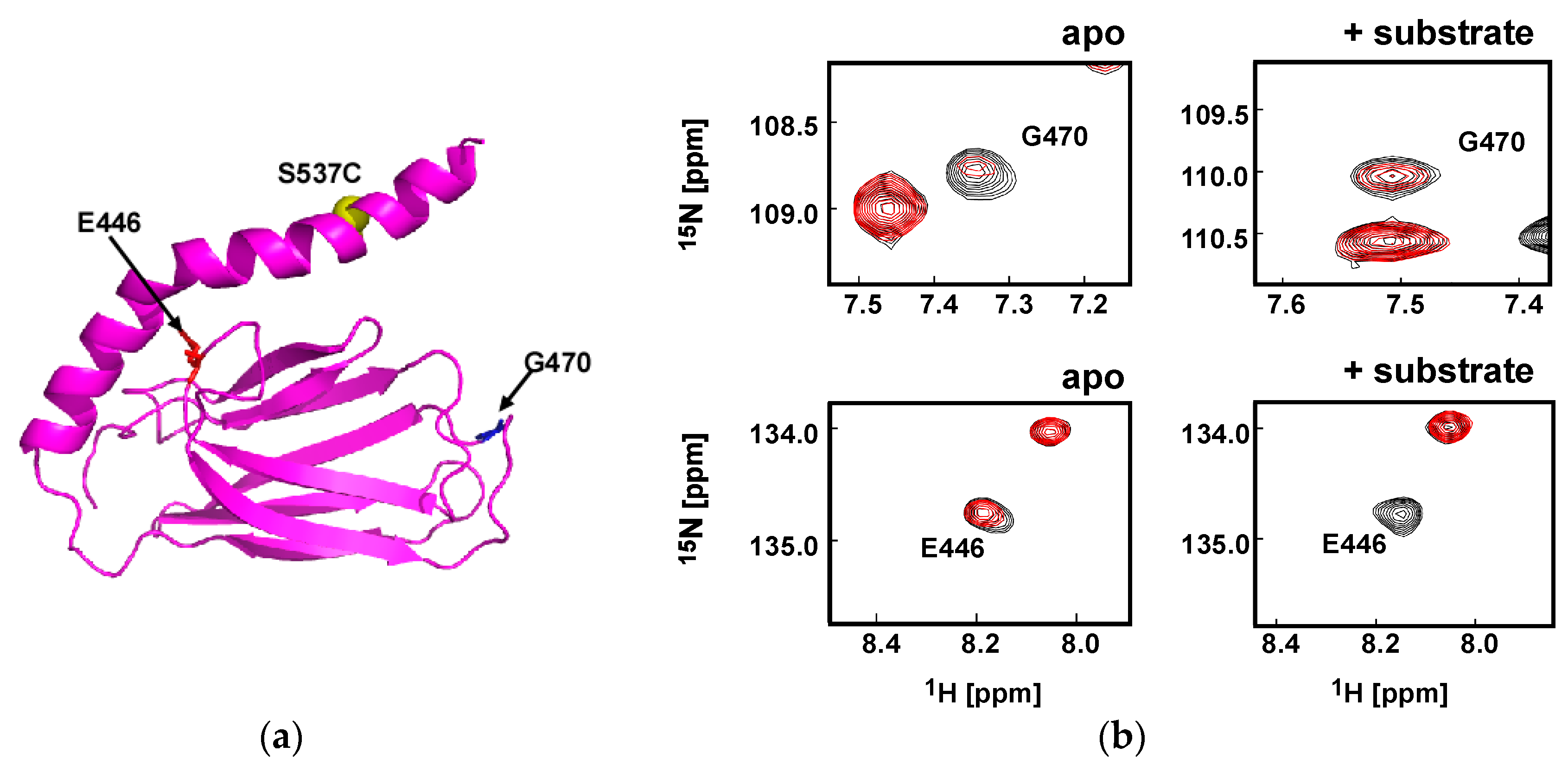
4.6. NMR Paramagnetic Relaxation Enhancement Experiments
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mayer, M.P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013, 38, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Meimaridou, E.; Gooljar, S.B.; Chapple, J.P. From hatching to dispatching: The multiple cellular roles of the hsp70 molecular chaperone machinery. J. Mol. Endocrinol. 2009, 42, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Bukau, B. Molecular chaperones: Structure of a protein disaggregase. Curr. Biol. 2003, 14, R78–R80. [Google Scholar] [CrossRef]
- Daugaard, M.; Rohde, M.; Jaattela, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Rohde, M.; Daugaard, M.; Jensen, M.H.; Helin, K.; Nylandsted, J.; Jäättelä, M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005, 19, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Nylandsted, J.; Jäättelä, M. Heat shock protein 70 promotes cancer cell viability by safeguarding lysosomal integrity. Cell Cycle 2004, 3, 1484–1485. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.E. The hsp70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Zhang, P.; Murphy, M.E.; Marmorstein, R.; George, D.L. Structural basis for the inhibition of hsp70 and dnak chaperones by small-molecule targeting of a c-terminal allosteric pocket. ACS Chem. Biol. 2014, 9, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Patury, S.; Miyata, Y.; Gestwicki, J.E. Pharmacological targeting of the hsp70 chaperone. Curr. Top. Med. Chem. 2009, 9, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L.; Chiosis, G. Hsp70 molecular chaperones: Emerging roles in human disease and identification of small molecule modulators. Curr. Top. Med. Chem. 2006, 6, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.J.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Brocchieri, L.; Conway de Macario, E.; Macario, A.J. Hsp70 genes in the human genome: Conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC Evol. Biol. 2008, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Leu, J.I.; Murphy, M.E.; George, D.L.; Marmorstein, R. Crystal structure of the stress-inducible human heat shock protein 70 substrate-binding domain in complex with peptide substrate. PLoS ONE 2014, 9, e103518. [Google Scholar] [CrossRef] [PubMed]
- Zuiderweg, E.R.P.; Bertelsen, E.B.; Rousaki, A.; Mayer, M.P.; Gestwicki, J.E.; Ahmad, A. Allostery in the hsp70 chaperone proteins. In Molecular Chaperones; Jackson, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 99–153. [Google Scholar]
- Bertelsen, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R. Solution conformation of wild-type E. coli hsp70 (dnak) chaperone complexed with adp and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef] [PubMed]
- Kityk, R.; Kopp, J.; Sinning, I.; Mayer, M.P. Structure and dynamics of the atp-bound open conformation of hsp70 chaperones. Mol. Cell 2012, 48, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Sarbeng, E.B.; Liu, Q.; Le, K.Q.; Xu, X.; Xu, H.; Yang, J.; Wong, J.L.; Vorvis, C.; Hendrickson, W.A.; et al. Allosteric opening of the polypeptide-binding site when an hsp70 binds atp. Nat. Struct. Mol. Biol. 2013, 20, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.F.; Dinler, G.; Sivendran, R.; Montgomery, D.L.; Stotz, M.; Gierasch, L.M. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell 2007, 26, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, A.; Clerico, E.M.; Gierasch, L.M. An interdomain energetic tug-of-war creates the allosterically active state in hsp70 molecular chaperones. Cell 2012, 151, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhao, X.; Burkholder, W.F.; Gragerov, A.; Ogata, C.M.; Gottesman, M.E.; Hendrickson, W.A. Structural analysis of substrate binding by the molecular chaperone dnak. Science 1996, 272, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Schroder, H.; Rudiger, S.; Paal, K.; Laufen, T.; Bukau, B. Multistep mechanism of substrate binding determines chaperone activity of hsp70. Nat. Struct. Biol. 2000, 7, 586–593. [Google Scholar] [PubMed]
- Buczynski, G.; Slepenkov, S.V.; Sehorn, M.G.; Witt, S.N. Characterization of a lidless form of the molecular chaperone dnak: Deletion of the lid increases peptide on- and off-rate constants. J. Biol. Chem. 2001, 276, 27231–27236. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.; Montgomery, D.L.; Stevens, S.Y.; Vander Kooi, C.W.; Feng, H.P.; Gierasch, L.M.; Zuiderweg, E.R. Structural insights into substrate binding by the molecular chaperone dnak. Nat. Struct. Biol. 2000, 7, 298–303. [Google Scholar] [PubMed]
- Schlecht, R.; Erbse, A.H.; Bukau, B.; Mayer, M.P. Mechanics of hsp70 chaperones enables differential interaction with client proteins. Nat. Struct. Mol. Biol. 2011, 18, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, A.; Gierasch, L.M. Substrate-binding domain conformational dynamics mediate hsp70 allostery. Proc. Natl. Acad. Sci. USA 2015, 112, E2865–E2873. [Google Scholar] [CrossRef] [PubMed]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in hsp70 chaperones. Nat. Commun. 2015, 6, 8308. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.Q.; Kirby, C.A.; Zhou, W.; Schuhmann, T.; Kityk, R.; Kipp, D.R.; Baird, J.; Chen, J.; Chen, Y.; Chung, F.; et al. The novolactone natural product disrupts the allosteric regulation of hsp70. Chem. Biol. 2015, 22, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kurochkin, A.V.; Pang, Y.; Hu, W.; Flynn, G.C.; Zuiderweg, E.R.P. Nmr solution structure of the 21 kda chaperone protein dnak substrate binding domain: A preview of chaperone−protein interaction. Biochemistry 1998, 37, 7929–7940. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.F.; Schulz, E.G.; Gierasch, L.M. Direct comparison of a stable isolated hsp70 substrate-binding domain in the empty and substrate-bound states. J. Biol. Chem. 2006, 281, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Morshauser, R.C.; Hu, W.; Wang, H.; Pang, Y.; Flynn, G.C.; Zuiderweg, E.R.P. High-resolution solution structure of the 18 kda substrate-binding domain of the mammalian chaperone protein hsc7011edited by p. E. Wright. J. Mol. Biol. 1999, 289, 1387–1403. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Prasad, K.; Lafer, E.M.; Sousa, R. Structural basis of interdomain communication in the hsc70 chaperone. Mol. Cell 2005, 20, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Rist, W.; Graf, C.; Bukau, B.; Mayer, M.P. Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. J. Biol. Chem. 2006, 281, 16493–16501. [Google Scholar] [CrossRef] [PubMed]
- Nageswara Rao, B.D. Nuclear magnetic resonance line-shape analysis and determination of exchange rates. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1989; Volume 176, pp. 279–311. [Google Scholar]
- Gillespie, J.R.; Shortle, D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. I. Paramagnetic relaxation enhancement by nitroxide spin labels11edited by p. E. Wright. J. Mol. Biol. 1997, 268, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, D.L.; Morimoto, R.I.; Gierasch, L.M. Mutations in the substrate binding domain of the escherichia coli 70 kda molecular chaperone, dnak, which alter substrate affinity or interdomain coupling11edited by m. Gottesman. J. Mol. Biol. 1999, 286, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sáiz, V.; Moro, F.; Arizmendi, J.M.; Acebrón, S.P.; Muga, A. Ionic contacts at dnak substrate binding domain involved in the allosteric regulation of lid dynamics. J. Biol. Chem. 2006, 281, 7479–7488. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, J.; Fairbrother, W.; Palmer, A.; Skelton, N. Heteronuclear NMR experiments. In Protein NMR Spectroscopy, 1st ed.; Academic Press: Cambridge, MA, USA, 1996; pp. 410–453. [Google Scholar]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. Nmrpipe: A multidimensional spectral processing system based on unix pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Iwahara, J.; Koshiba, S.; Tomizawa, T.; Tochio, N.; Guntert, P.; Kigawa, T.; Yokoyama, S. Kujira, a package of integrated modules for systematic and interactive analysis of NMR data directed to high-throughput NMR structure studies. J. Biomol. NMR 2007, 39, 31–52. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A. Using nmrview to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 2004, 278, 313–352. [Google Scholar] [PubMed]
- Guntert, P. Automated NMR structure calculation with cyana. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar] [PubMed]
- Guntert, P. Automated structure determination from NMR spectra. Eur. Biophys. J. 2009, 38, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. Talos+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Schwieters, C.D.; Kuszewski, J.J.; Tjandra, N.; Clore, G.M. The xplor-nih NMR molecular structure determination package. J. Magn. Reson. 2003, 160, 65–73. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. Aqua and procheck-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Tejero, R.; Montelione, G.T. Evaluating protein structures determined by structural genomics consortia. Proteins 2007, 66, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Koradi, R.; Billeter, M.; Wuthrich, K. Molmol: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 29–32, 51–55. [Google Scholar] [CrossRef]
- Tellinghuisen, J. Isothermal titration calorimetry at very low c. Anal. Biochem. 2008, 373, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, W.B.; Daranas, A.H. On the value of c: Can low affinity systems be studied by isothermal titration calorimetry? J. Am. Chem. Soc. 2003, 125, 14859–14866. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: The plasmids to express the proteins in E. coli used in this work are available from the authors. |





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umehara, K.; Hoshikawa, M.; Tochio, N.; Tate, S.-i. Substrate Binding Switches the Conformation at the Lynchpin Site in the Substrate-Binding Domain of Human Hsp70 to Enable Allosteric Interdomain Communication. Molecules 2018, 23, 528. https://doi.org/10.3390/molecules23030528
Umehara K, Hoshikawa M, Tochio N, Tate S-i. Substrate Binding Switches the Conformation at the Lynchpin Site in the Substrate-Binding Domain of Human Hsp70 to Enable Allosteric Interdomain Communication. Molecules. 2018; 23(3):528. https://doi.org/10.3390/molecules23030528
Chicago/Turabian StyleUmehara, Kohei, Miho Hoshikawa, Naoya Tochio, and Shin-ichi Tate. 2018. "Substrate Binding Switches the Conformation at the Lynchpin Site in the Substrate-Binding Domain of Human Hsp70 to Enable Allosteric Interdomain Communication" Molecules 23, no. 3: 528. https://doi.org/10.3390/molecules23030528