Investigation of an 18F-labelled Imidazopyridotriazine for Molecular Imaging of Cyclic Nucleotide Phosphodiesterase 2A

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
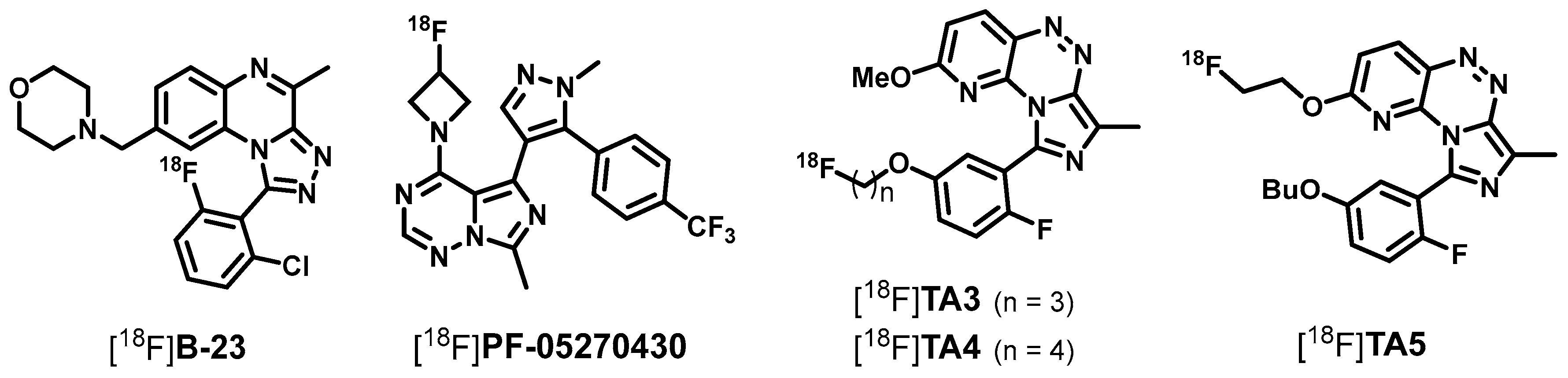
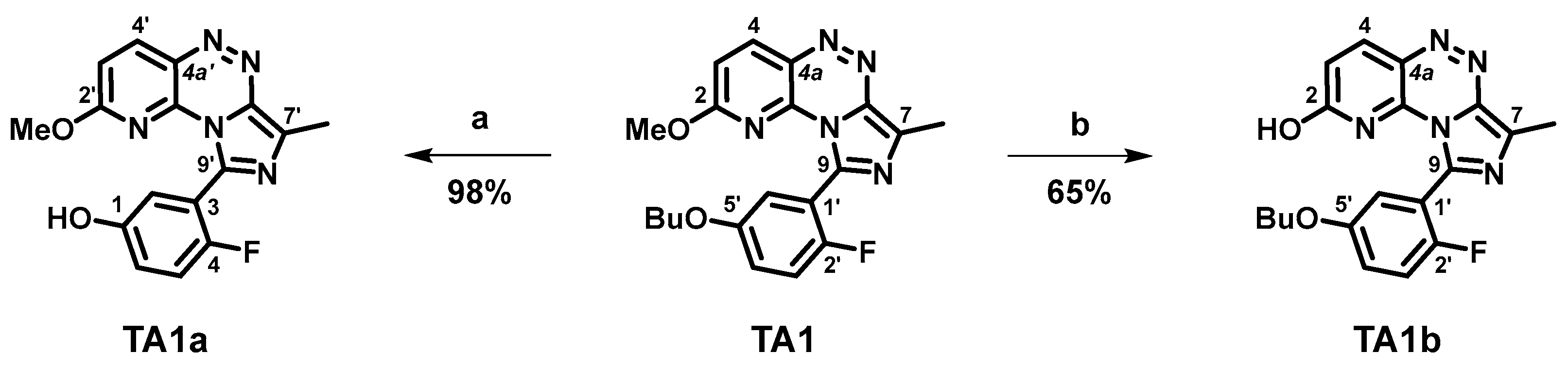
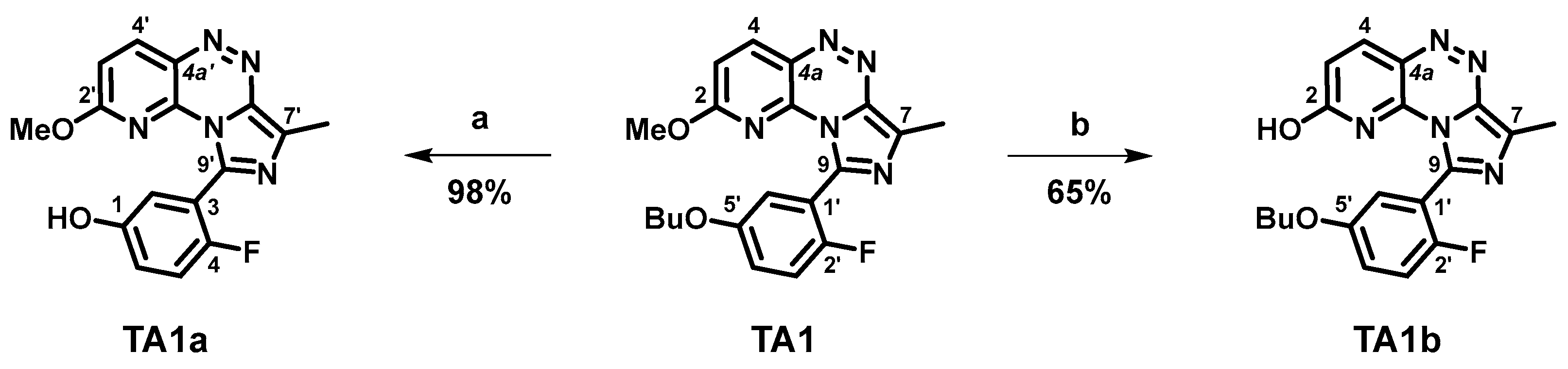
2.1. Organic Syntheses and Inhibitory Potency
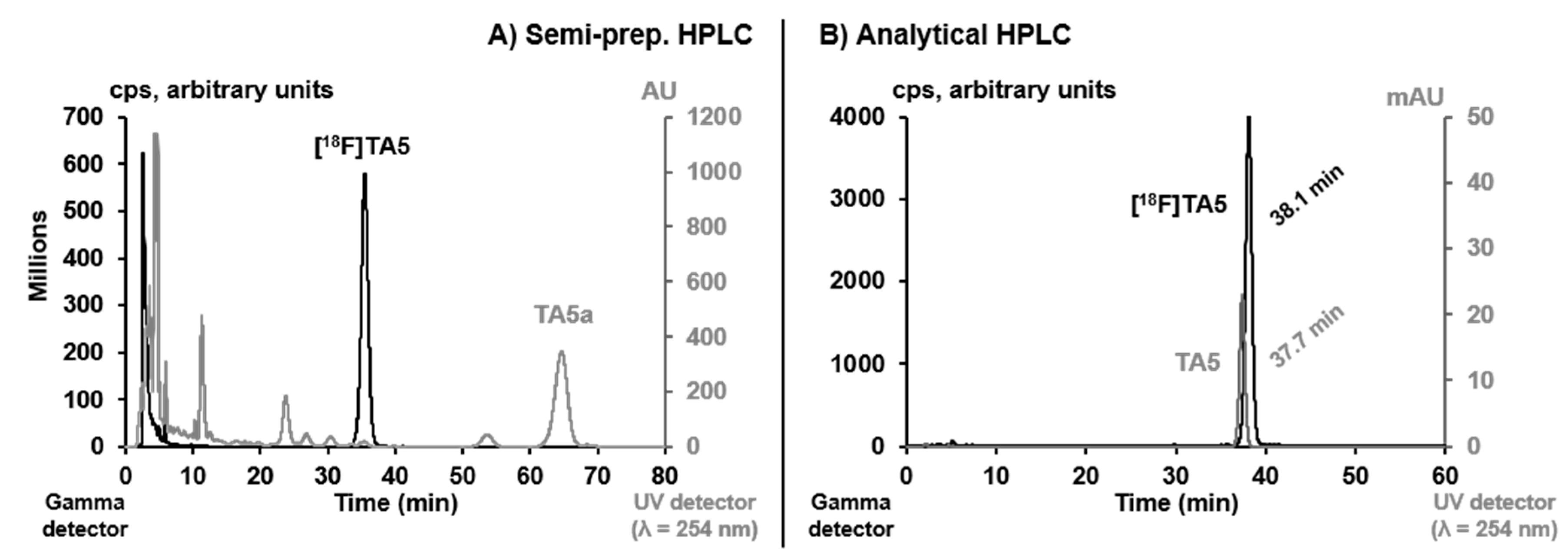
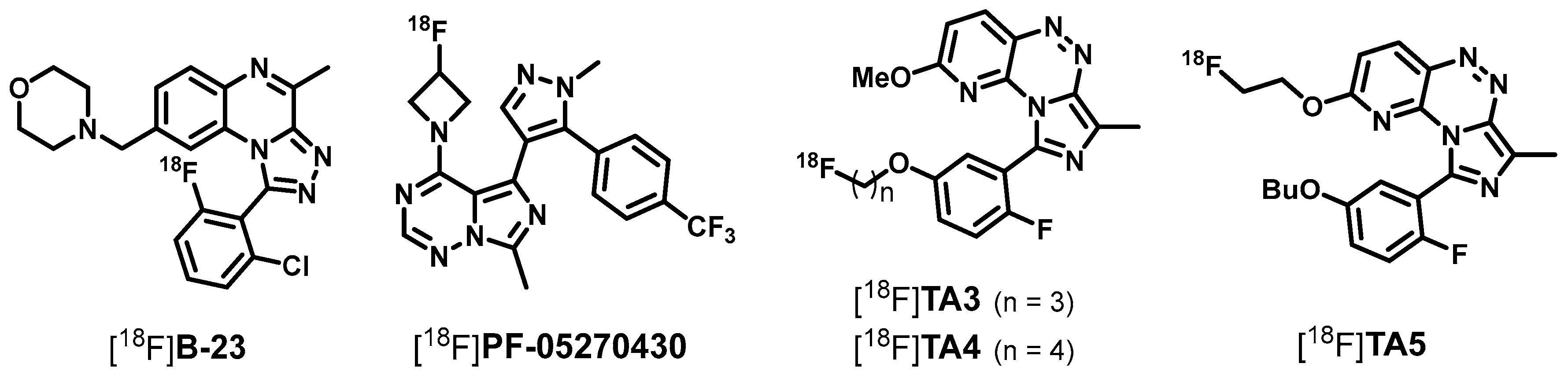
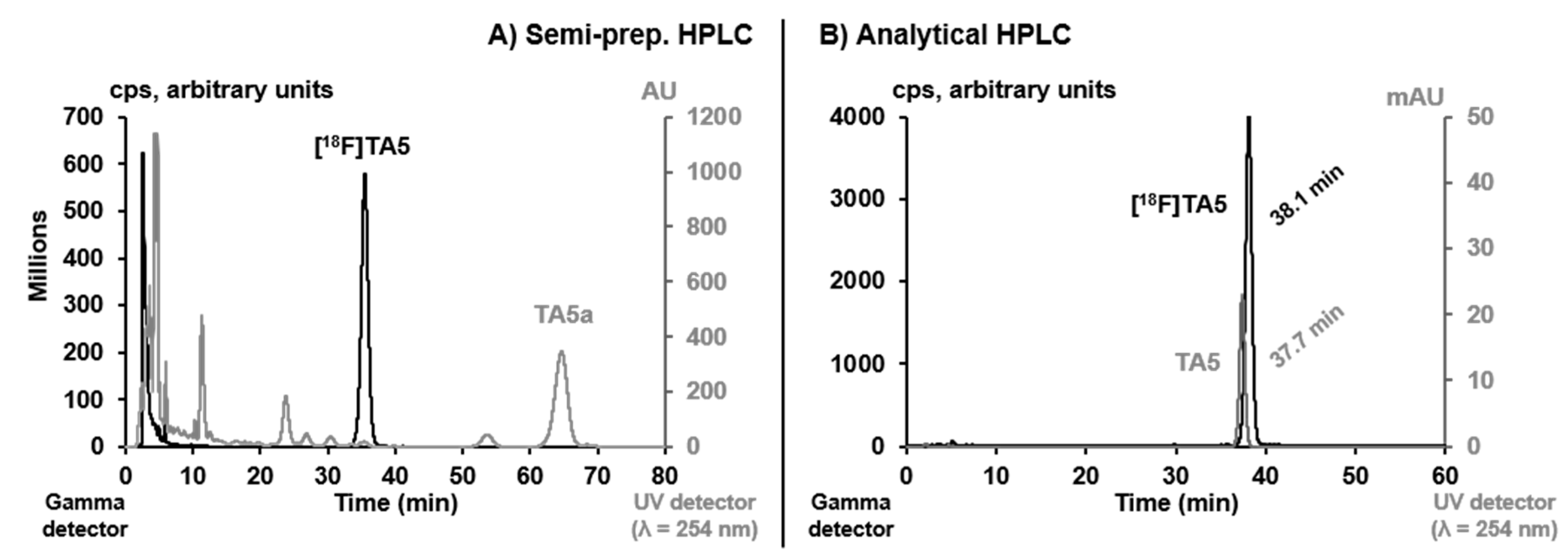
2.2. Radiosynthesis, In Vitro Stability and Lipophilicity
2.3. Biological Evaluation—In Vitro Autoradiography and In Vivo Metabolism
3. Materials and Methods
3.1. General Information
3.2. Organic Syntheses
3.2.1. 9-(5-Butoxy-2-fluorophenyl)-7-methylimidazo[5,1-c]pyrido[2,3-e][1,2,4]triazin-2-ol (TA1b)
3.2.2. 9-(5-Butoxy-2-fluorophenyl)-2-(2-fluoroethoxy)-7-methylimidazo[5,1-c]pyrido[2,3-e]-[1,2,4]triazine (TA5)
3.2.3. 2-((9-(5-Butoxy-2-fluorophenyl)-7-methylimidazo[5,1-c]pyrido[2,3-e][1,2,4]-triazin-2-yl)oxy)ethyl-4-methylbenzenesulfonate (TA5a)
3.3. In Vitro Affinity Assay
3.4. Radiochemistry
3.5. Investigation of In Vitro Stability and Lipophilicity (logD7.4)
3.6. Animal Studies
3.6.1. In Vitro Autoradiographic Studies
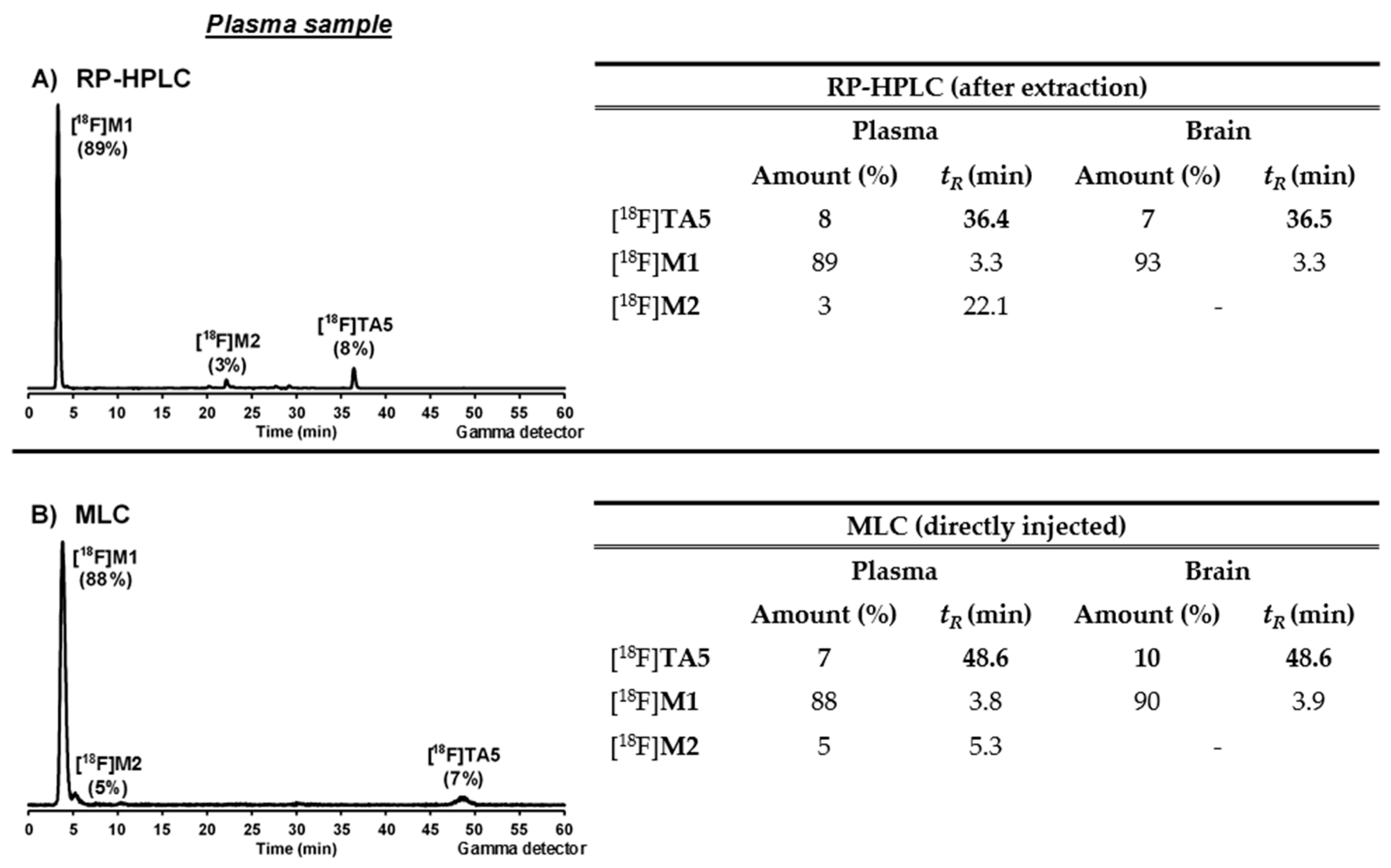
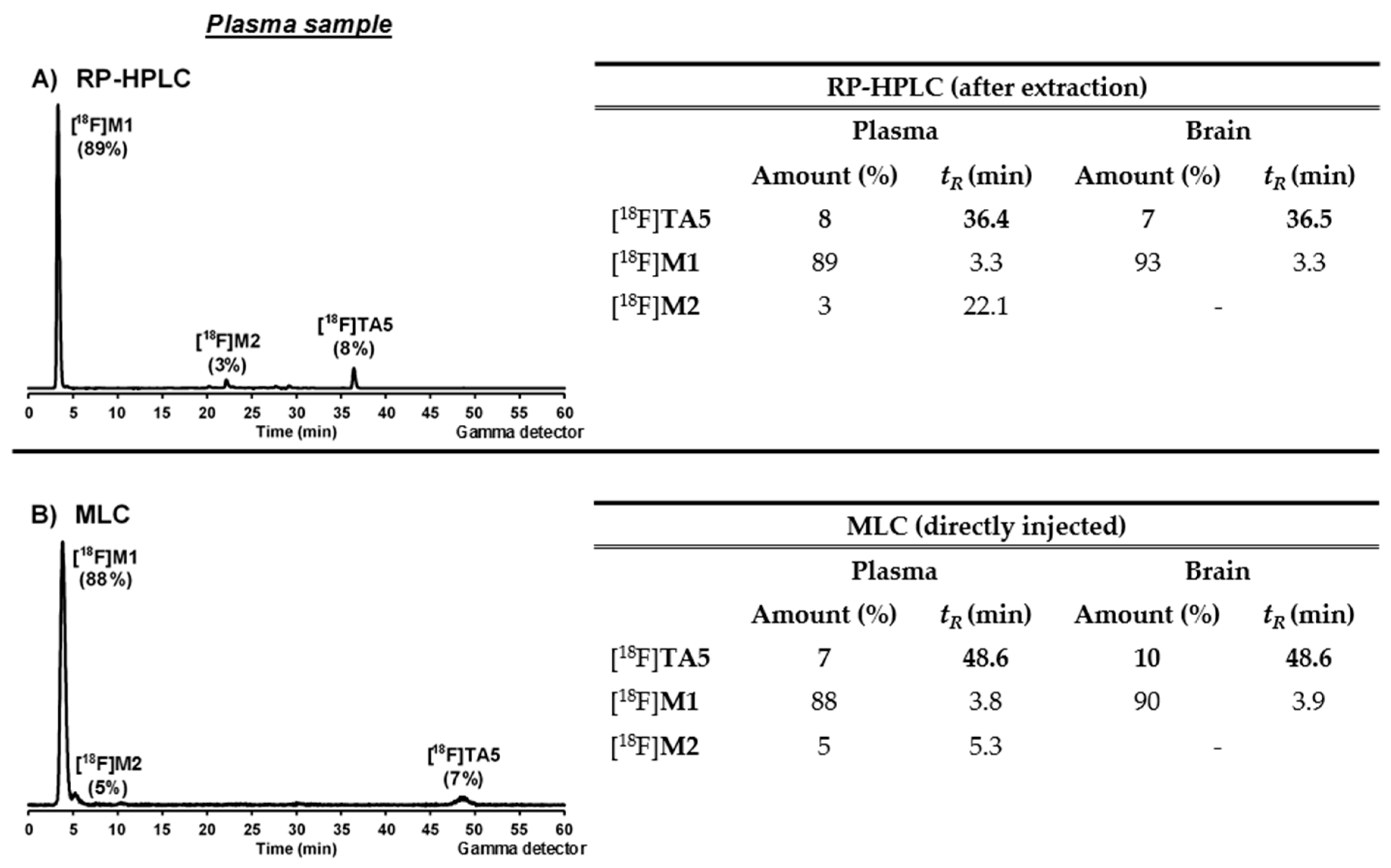
3.6.2. In Vivo Metabolism Studies
Conventional Extraction Method
Micellar Liquid Chromatography (MLC)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Brit. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, Y.; Ruan, L.; Wang, C.; Pan, J.; Klabnik, J.; Lueptow, L.; Zhang, H.-T.; O’Donnell, J.M.; Xu, Y. The roles of phosphodiesterase 2 in the central nervous and peripheral systems. Curr. Pharm. Design 2015, 21, 274–290. [Google Scholar] [CrossRef]
- Trabanco, A.A.; Buijnsters, P.; Rombouts, F.J. Towards selective phosphodiesterase 2A (PDE2A) inhibitors: A patent review (2010–present). Expert Opin. Ther. Pat. 2016, 26, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Coskran, T.M.; Wilhelms, M.B.; Adamowicz, W.O.; O’Donnell, M.M.; Muravnick, K.B.; Menniti, F.S.; Kleiman, R.J.; Morton, D. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J. Histochem. Cytochem. 2009, 57, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Coskran, T.M.; Kelly, M.P.; Kleiman, R.J.; Morton, D.; O’Neill, S.M.; Schmidt, C.J.; Weinberg, R.J.; Menniti, F.S. The distribution of phosphodiesterase 2A in the rat brain. Neuroscience 2012, 226, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Breitenbucher, J.G. PDE2 inhibition: Potential for the treatment of cognitive disorders. Bioorg. Med. Chem. Lett. 2013, 23, 6522–6527. [Google Scholar] [CrossRef] [PubMed]
- Stange, H.; Langen, B.; Egerland, U.; Hoefgen, N.; Priebs, M.; Malamas, M.S.; Erdel, J.J.; Ni, Y. Triazine Derivatives as Inhibitors of Phosphodiesterases. Patent No. WO 2010/054253 A1, 14 May 2010. [Google Scholar]
- Masood, A.; Huang, Y.; Hajjhussein, H.; Xiao, L.; Li, H.; Wang, W.; Hamza, A.; Zhan, C.-G.; O’Donnell, J.M. Anxiolytic effects of phosphodiesterase-2 inhibitors associated with increased cGMP signaling. J. Pharmacol. Exp. Ther. 2009, 331, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, D.; Rosenbrock, H.; Kroker, K.S. Inhibition of PDE2A, but not PDE9A, modulates presynaptic short-term plasticity measured by paired-pulse facilitation in the Ca1 region of the hippocampus. Synapse 2015, 69, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Boess, F.G.; Hendrix, M.; van der Staay, F.J.; Erb, C.; Schreiber, R.; van Staveren, W.; de Vente, J.; Prickaerts, J.; Blokland, A.; Koenig, G. Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology 2004, 47, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Reneerkens, O.A.H.; Rutten, K.; Bollen, E.; Hage, T.; Blokland, A.; Steinbusch, H.W.M.; Prickaerts, J. Inhibition of phoshodiesterase type 2 or type 10 reverses object memory deficits induced by scopolamine or MK-801. Behav. Brain Res. 2013, 236, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Bales, K.; Plath, N.; Svenstrup, N.; Menniti, F. Phosphodiesterase inhibition to target the synaptic dysfunction in Alzheimer’s disease. In Neurodegenerative Diseases; Dominguez, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 6, pp. 57–90. [Google Scholar]
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Scheunemann, M.; Brust, P. Novel radioligands for cyclic nucleotide phosphodiesterase imaging with positron emission tomography: An update on developments since 2012. Molecules 2016, 21, 650. [Google Scholar] [CrossRef]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Andrés, J.I.; Rombouts, F.J.R.; Trabanco, A.A.; Vanhoof, G.C.P.; De Angelis, M.; Buijnsters, P.J.J.A.; Guillemont, J.E.G.; Bormans, G.M.R.; Celen, S.J.L. 1-Aryl-4-methyl-[1,2,4]triazolo[4,3-a]quinoxaline derivatives. Patent No. WO 2013/000924 A1, 3 January 2013. [Google Scholar]
- Zhang, L.; Villalobos, A.; Beck, E.M.; Bocan, T.; Chappie, T.A.; Chen, L.; Grimwood, S.; Heck, S.D.; Helal, C.J.; Hou, X.; et al. Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J. Med. Chem. 2013, 56, 4568–4579. [Google Scholar] [CrossRef] [PubMed]
- Naganawa, M.; Nabulsi, N.; Waterhouse, R.; Lin, S.-F.; Zhang, L.; Cass, T.; Ropchan, J.; McCarthy, T.; Huang, Y.; Carson, R. Human PET studies with [18F]PF-05270430, a PET radiotracer for imaging phosphodiesterase-2A. J. Nucl. Med. 2013, 54, 201. [Google Scholar]
- Naganawa, M.; Waterhouse, R.N.; Nabulsi, N.; Lin, S.F.; Labaree, D.; Ropchan, J.; Tarabar, S.; DeMartinis, N.; Ogden, A.; Banerjee, A.; et al. First-in-human assessment of the novel PDE2A PET radiotracer 18F-PF-05270430. J. Nucl. Med. 2016, 57, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Egerland, U.; Kranz, M.; Scheunemann, M.; Höfgen, N.; Steinbach, J.; Brust, P. Synthesis, 18F-radiolabelling and biological characterization of novel fluoroalkylated triazine derivatives for in vivo imaging of phosphodiesterase 2A in brain via positron emission tomography. Molecules 2015, 20, 9591–9615. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Egerland, U.; Kranz, M.; Fischer, S.; Höfgen, N.; Steinbach, J.; Brust, P. Novel 18F-labelled triazine derivatives for PET imaging of phosphodiesterase 2A. J. Labelled Compd. Rad. 2015, 58, S221. [Google Scholar]
- Schröder, S.; Wenzel, B.; Kranz, M.; Egerland, U.; Teodoro, R.; Deuther-Conrad, W.; Fischer, S.; Höfgen, N.; Steinbach, J.; Brust, P. Development, synthesis and F-18 labelling of a fluoroalkylated triazine derivative for PET imaging of phosphodiesterase 2A. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, S197. [Google Scholar]
- Zoghbi, S.S.; Shetty, H.U.; Ichise, M.; Fujita, M.; Imaizumi, M.; Liow, J.-S.; Shah, J.; Musachio, J.L.; Pike, V.W.; Innis, R.B. PET imaging of the dopamine transporter with [18F]FECNT: A polar radiometabolite confounds brain radioligand measurements. J. Nucl. Med. 2006, 47, 520–527. [Google Scholar] [PubMed]
- Evens, N.; Vandeputte, C.; Muccioli, G.G.; Lambert, D.M.; Baekelandt, V.; Verbruggen, A.M.; Debyser, Z.; Van Laere, K.; Bormans, G.M. Synthesis, in vitro and in vivo evaluation of fluorine-18 labelled FE-GW405833 as a PET tracer for type 2 cannabinoid receptor imaging. Bioorg. Med. Chem. 2011, 19, 4499–4505. [Google Scholar] [CrossRef] [PubMed]
- Langen, K.-J.; Hamacher, K.; Weckesser, M.; Floeth, F.; Stoffels, G.; Bauer, D.; Coenen, H.H.; Pauleit, D. O-(2-[18F]fluoroethyl)-L-tyrosine: Uptake mechanisms and clinical applications. Nucl. Med. Biol. 2006, 33, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, M.; Nowosielski, M.; Putzer, D.; Jansen, N.L.; Seiz, M.; Schocke, M.; McCoy, M.; Göbel, G.; la Fougère, C.; Virgolini, I.J.; et al. [18F]-Fluoro-ethyl-L-tyrosine PET: A valuable diagnostic tool in neuro-oncology, but not all that glitters is glioma. Neuro-Oncology 2013, 15, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Galldiks, N.; Stoffels, G.; Filss, C.; Rapp, M.; Blau, T.; Tscherpel, C.; Ceccon, G.; Dunkl, V.; Weinzierl, M.; Stoffel, M.; et al. The use of dynamic O-(2-18F-fluoroethyl)-L-tyrosine PET in the diagnosis of patients with progressive and recurrent glioma. Neuro. Oncol. 2015, 17, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, H.; Sato, K.; Fukumoto, D.; Kakiuchi, T. Evaluation of D-isomers of O-18F-fluoromethyl, O-18F-fluoroethyl and O-18F-fluoropropyl tyrosine as tumour imaging agents in mice. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Pauleit, D.; Floeth, F.; Herzog, H.; Hamacher, K.; Tellmann, L.; Müller, H.-W.; Coenen, H.; Langen, K.-J. Whole-body distribution and dosimetry of O-(2-[18F]fluoroethyl)-L-tyrosine. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Barret, O.; Thomae, D.; Tavares, A.; Alagille, D.; Papin, C.; Waterhouse, R.; McCarthy, T.; Jennings, D.; Marek, K.; Russell, D.; et al. In vivo assessment and dosimetry of 2 novel PDE10A PET radiotracers in humans: 18F-MNI-659 and 18F-MNI-654. J. Nucl. Med. 2014, 55, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.S.; Barret, O.; Jennings, D.L.; Friedman, J.H.; Tamagnan, G.D.; Thomae, D.; Alagille, D.; Morley, T.J.; Papin, C.; Papapetropoulos, S.; et al. The phosphodiesterase 10 positron emission tomography tracer, [18F]MNI-659, as a novel biomarker for early huntington disease. J. Am. Med. Assoc. Neurol. 2014, 71, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.S.; Jennings, D.L.; Barret, O.; Tamagnan, G.D.; Carroll, V.M.; Caille, F.; Alagille, D.; Morley, T.J.; Papin, C.; Seibyl, J.P.; et al. Change in PDE10 across early huntington disease assessed by [18F]MNI-659 and PET imaging. Neurology 2016, 86, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Nakada, Y.; Aicher, T.D.; Huerou, Y.L.; Turner, T.; Pratt, S.A.; Gonzales, S.S.; Boyd, S.A.; Miki, H.; Yamamoto, T.; Yamaguchi, H.; et al. Novel acyl coenzyme A (CoA): Diacylglycerol acyltransferase-1 inhibitors: Synthesis and biological activities of diacylethylenediamine derivatives. Bioorg. Med. Chem. 2010, 18, 2785–2795. [Google Scholar] [CrossRef] [PubMed]
- Benton, F.L.; Dillon, T.E. The cleavage of ethers with boron bromide. I. Some common ethers. J. Am. Chem. Soc. 1942, 64, 1128–1129. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Greene, T.W. Protection for phenols and catechols. In Greene’s Protective Groups in Organic Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 367–430. [Google Scholar]
- Vickery, E.H.; Pahler, L.F.; Eisenbraun, E.J. Selective O-demethylation of catechol ethers. Comparison of boron tribromide and iodotrimethylsilane. J. Org. Chem. 1979, 44, 4444–4446. [Google Scholar] [CrossRef]
- Bhatt, M.V.; Kulkarni, S.U. Cleavage of ethers. Synthesis 1983, 1983, 249–282. [Google Scholar] [CrossRef]
- Pasquini, C.; Coniglio, A.; Bassetti, M. Controlled dealkylation by BBr3: Efficient synthesis of para-alkoxy-phenols. Tetrahedron Lett. 2012, 53, 6191–6194. [Google Scholar] [CrossRef]
- Robinson, P.D.; Groziak, M.P. A boron-containing estrogen mimic. Acta Crystallogr. C 1999, 55, 1701–1704. [Google Scholar] [CrossRef] [PubMed]
- Shiao, M.J.; Lai, L.L.; Ku, W.S.; Lin, P.Y.; Hwu, J.R. Chlorotrimethylsilane in combination with sodium sulfide as the equivalent of sodium trimethylsilanethiolate in organic reactions. J. Org. Chem. 1993, 58, 4742–4744. [Google Scholar] [CrossRef]
- Testaferri, L.; Tiecco, M.; Tingoli, M.; Bartoli, D.; Massoli, A. The reactions of some halogenated pyridines with methoxide and methanethiolate ions in dimethylformamide. Tetrahedron 1985, 41, 1373–1384. [Google Scholar] [CrossRef]
- Beak, P.; Fry, F.S.; Lee, J.; Steele, F. Equilibration studies. Protomeric equilibria of 2- and 4-hydroxypyridines, 2- and 4-hydroxypyrimidines, 2- and 4-mercaptopyridines, and structurally related compounds in the gas phase. J. Am. Chem. Soc. 1976, 98, 171–179. [Google Scholar] [CrossRef]
- Albert, A.; Phillips, J.N. Ionization constants of heterocyclic substances. Part II. Hydroxy-derivatives of nitrogenous six-membered ring-compounds. J. Chem. Soc. 1956, 1294–1304. [Google Scholar] [CrossRef]
- Hatherley, L.D.; Brown, R.D.; Godfrey, P.D.; Pierlot, A.P.; Caminati, W.; Damiani, D.; Melandri, S.; Favero, L.B. Gas-phase tautomeric equilibrium of 2-pyridinone and 2-hydroxypyridine by microwave spectroscopy. J. Phys. Chem. 1993, 97, 46–51. [Google Scholar] [CrossRef]
- Comins, D.L.; Jianhua, G. N- vs. O-Alkylation in the mitsunobu reaction of 2-pyridone. Tetrahedron Lett. 1994, 35, 2819–2822. [Google Scholar] [CrossRef]
- Chung, N.M.; Tieckelmann, H. Alkylations of heterocyclic ambident anions. Iv. Alkylation of 5-carbethoxy- and 5-nitro-2-pyridone salts. J. Org. Chem. 1970, 35, 2517–2520. [Google Scholar] [CrossRef]
- Hopkins, G.; Jonak, J.; Minnemeyer, H.; Tieckelmann, H. Alkylations of heterocyclic ambident anions ii. Alkylation of 2-pyridone salts. J. Org. Chem. 1967, 32, 4040–4044. [Google Scholar] [CrossRef]
- Waterhouse, R.N. Determination of lipophilicity and its use as a predictor of blood–brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2003, 5, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Nakao, R.; Schou, M.; Halldin, C. Direct plasma metabolite analysis of positron emission tomography radioligands by micellar liquid chromatography with radiometric detection. Anal. Chem. 2012, 84, 3222–3230. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wenzel, B.; Dukic-Stefanovic, S.; Teodoro, R.; Ludwig, F.-A.; Deuther-Conrad, W.; Schröder, S.; Chezal, J.-M.; Moreau, E.; Brust, P.; et al. Development of a new radiofluorinated quinoline analog for PET imaging of phosphodiesterase 5 (PDE5) in brain. Pharmaceuticals 2016, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.L.; Ackermann, R.F. Evaluation of radiolabeled acetate and fluoroacetate as potential tracers of cerebral oxidative metabolism. Metab. Brain Dis. 1990, 5, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Sun, L.-Q.; Kobayashi, M.; Kiyono, Y.; Okazawa, H.; Furukawa, T.; Kawashima, H.; Welch, M.J.; Fujibayashi, Y. Preparation and evaluation of ethyl [18F]fluoroacetate as a proradiotracer of [18F]fluoroacetate for the measurement of glial metabolism by PET. Nucl. Med. Biol. 2009, 36, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Muir, D.; Berl, S.; Clarke, D.D. Acetate and fluoroacetate as possible markers for glial metabolism in vivo. Brain Res. 1986, 380, 336–340. [Google Scholar] [CrossRef]
- Ponde, D.E.; Dence, C.S.; Oyama, N.; Kim, J.; Tai, Y.-C.; Laforest, R.; Siegel, B.A.; Welch, M.J. 18F-Fluoroacetate: A potential acetate analog for prostate tumor imaging—In vivo evaluation of 18F-fluoroacetate versus 11C-acetate. J. Nucl. Med. 2007, 48, 420–428. [Google Scholar] [PubMed]
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of fluorine-containing drugs. Annu. Rev. Pharmacol. 2001, 41, 443–470. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.-Z.; Johnson, B.M.; Yang, T.J. Role of biotransformation studies in minimizing metabolism-related liabilities in drug discovery. AAPS J. 2008, 10, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. Pet radiotracers: Crossing the blood–brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.H.; Chan, H.K.; Miller, J.D.; Jayne, C.L.; Eichhorn, D.M. Synthesis, modification, and characterization of a family of homologues of exo-calix[4]arene: Exo-[n.m.n.m]metacyclophanes, n,m ≥ 3. J. Org. Chem. 2000, 65, 5185–5196. [Google Scholar] [CrossRef] [PubMed]

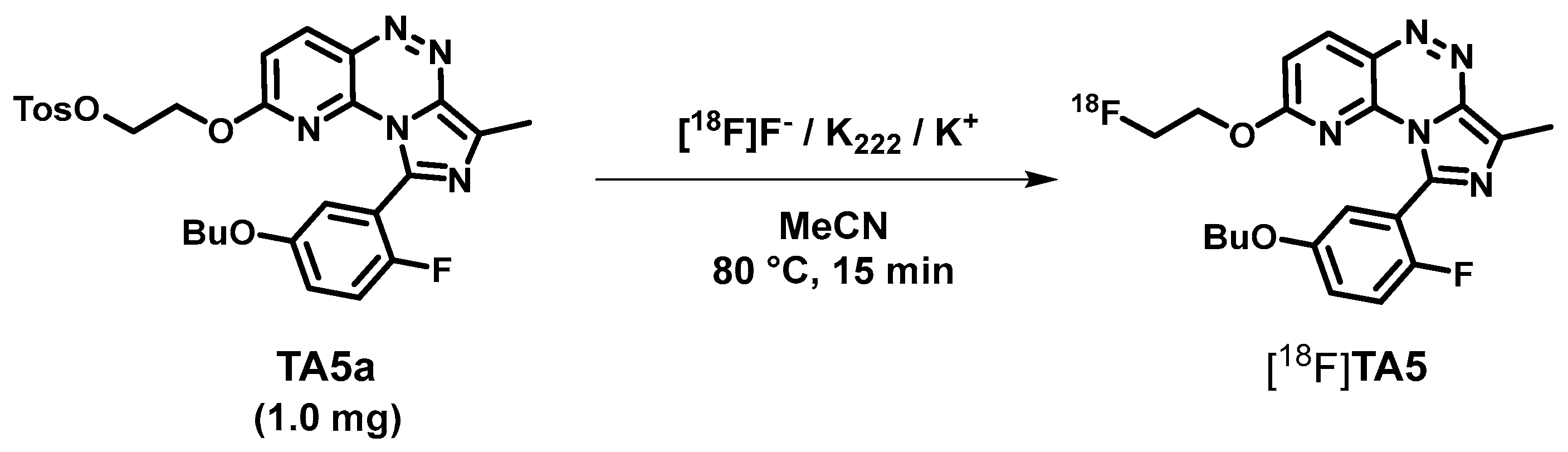

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
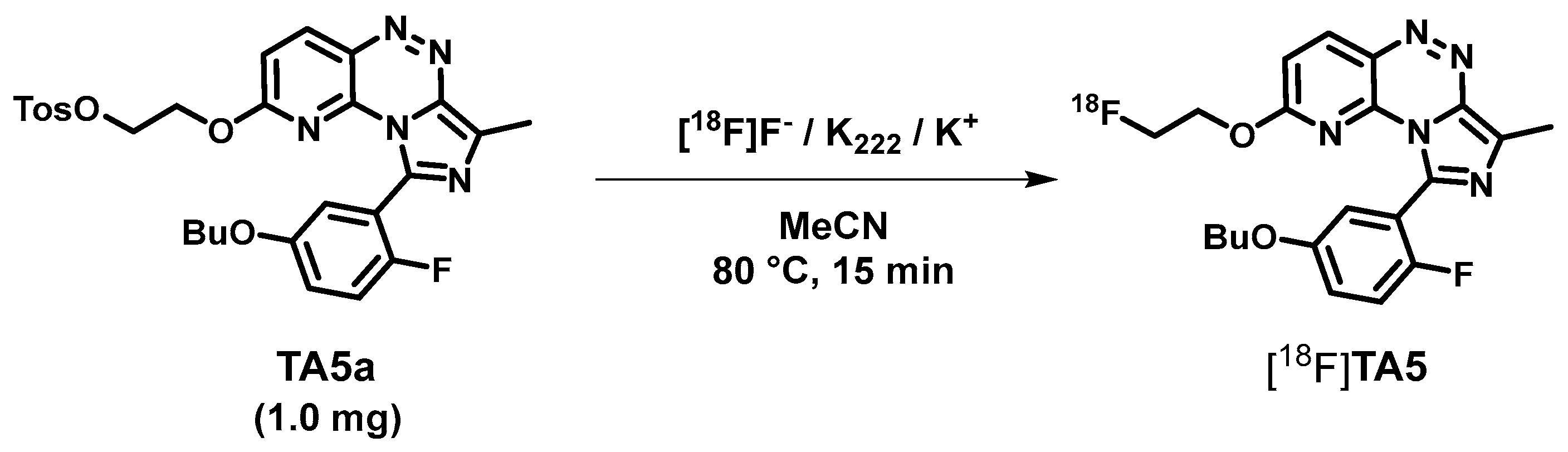{kind=link}
| Ligand | IC50 hPDE2A | IC50 hPDE10A | Selectivity Ratio PDE10A/PDE2A |
|---|---|---|---|
| TA5 (2-fluoroethoxy) | 3.0 nM | >1000 nM | >330 |
| TA1 (lead) | 4.5 nM | 670 nM | 149 |
| TA2 (5′-fluoroethoxy) | 10.4 nM | 77 nM | 7 |
| TA3 (5′-fluoropropoxy) | 11.4 nM | 318 nM | 28 |
| TA4 (5′-fluorobutoxy) | 7.3 nM | 913 nM | 125 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Kranz, M.; Scheunemann, M.; Egerland, U.; Höfgen, N.; Briel, D.; Steinbach, J.; et al. Investigation of an 18F-labelled Imidazopyridotriazine for Molecular Imaging of Cyclic Nucleotide Phosphodiesterase 2A. Molecules 2018, 23, 556. https://doi.org/10.3390/molecules23030556
Schröder S, Wenzel B, Deuther-Conrad W, Teodoro R, Kranz M, Scheunemann M, Egerland U, Höfgen N, Briel D, Steinbach J, et al. Investigation of an 18F-labelled Imidazopyridotriazine for Molecular Imaging of Cyclic Nucleotide Phosphodiesterase 2A. Molecules. 2018; 23(3):556. https://doi.org/10.3390/molecules23030556
Chicago/Turabian StyleSchröder, Susann, Barbara Wenzel, Winnie Deuther-Conrad, Rodrigo Teodoro, Mathias Kranz, Matthias Scheunemann, Ute Egerland, Norbert Höfgen, Detlef Briel, Jörg Steinbach, and et al. 2018. "Investigation of an 18F-labelled Imidazopyridotriazine for Molecular Imaging of Cyclic Nucleotide Phosphodiesterase 2A" Molecules 23, no. 3: 556. https://doi.org/10.3390/molecules23030556