Structure–Activity Relationship and Molecular Docking of Natural Product Library Reveal Chrysin as a Novel Dipeptidyl Peptidase-4 (DPP-4) Inhibitor: An Integrated In Silico and In Vitro Study

 , ,
, ,
Abstract
:
1. Introduction
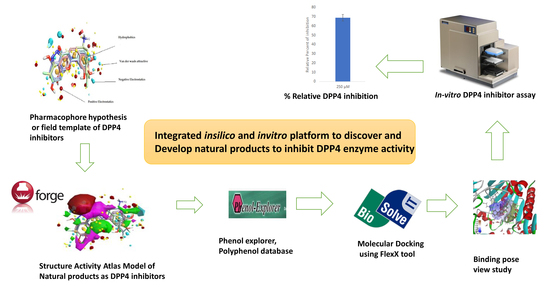
2. Results and Discussion
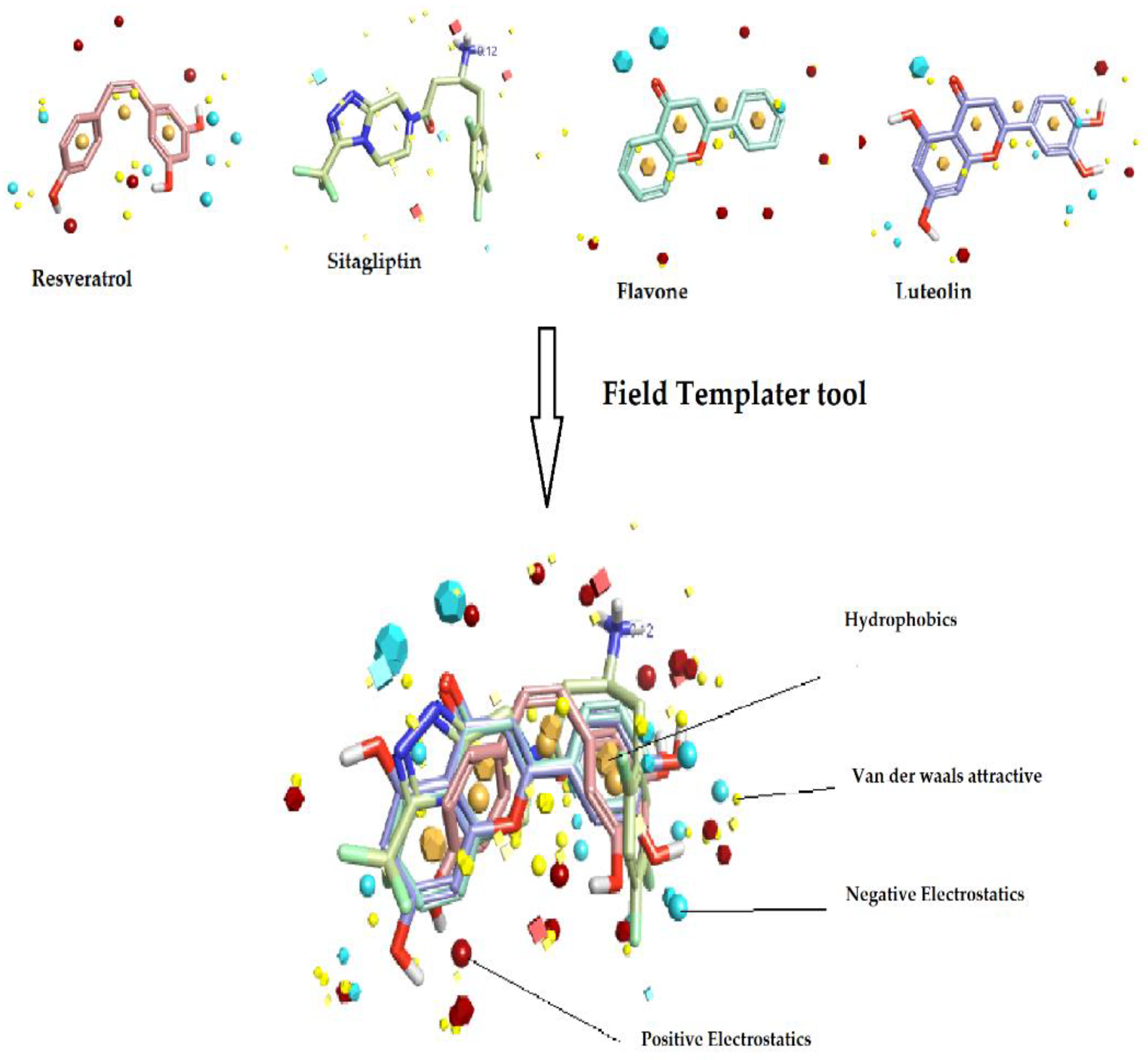
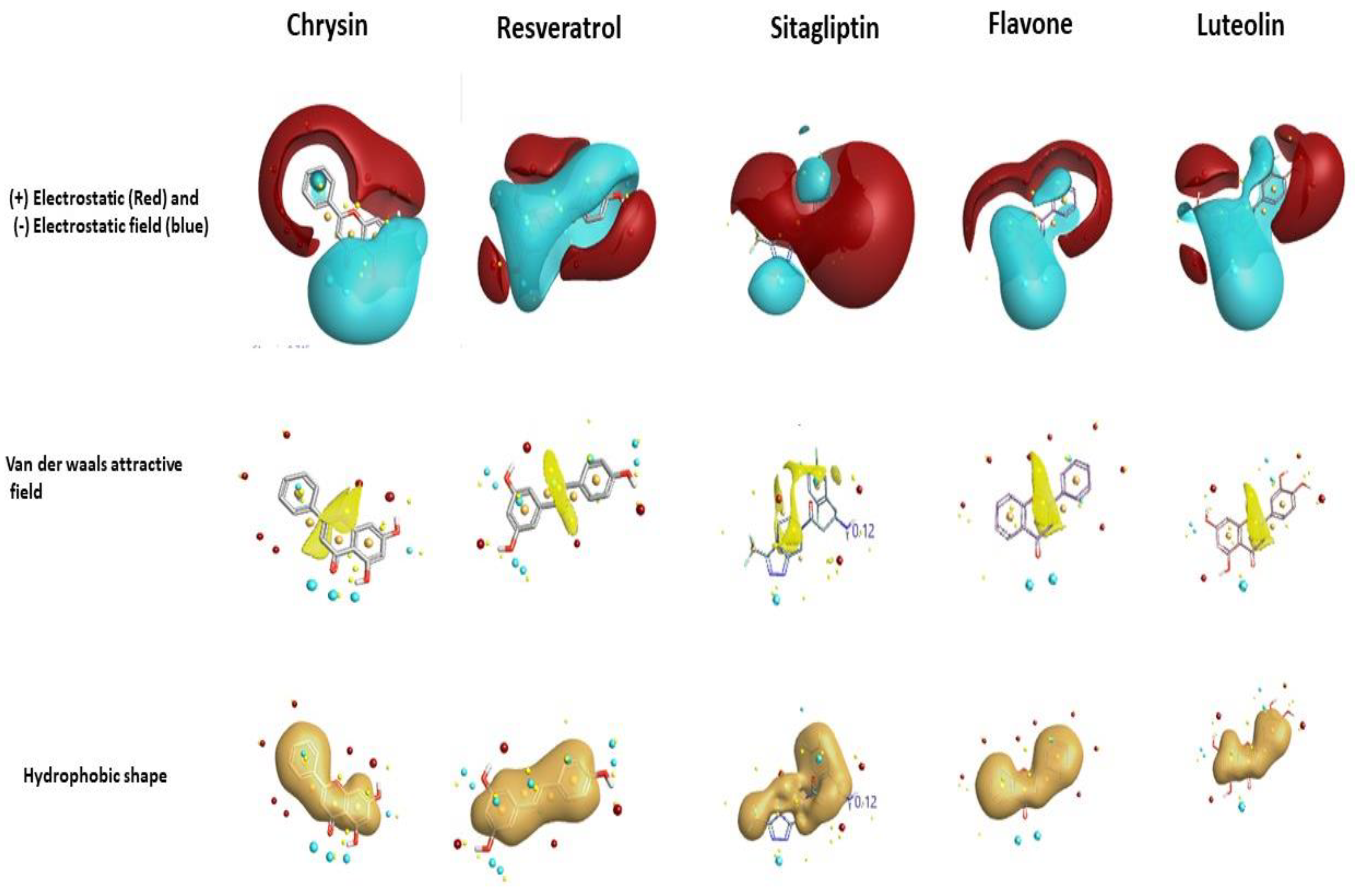
2.1. Field-Based Virtual Screening
2.1.1. Field Template Generation
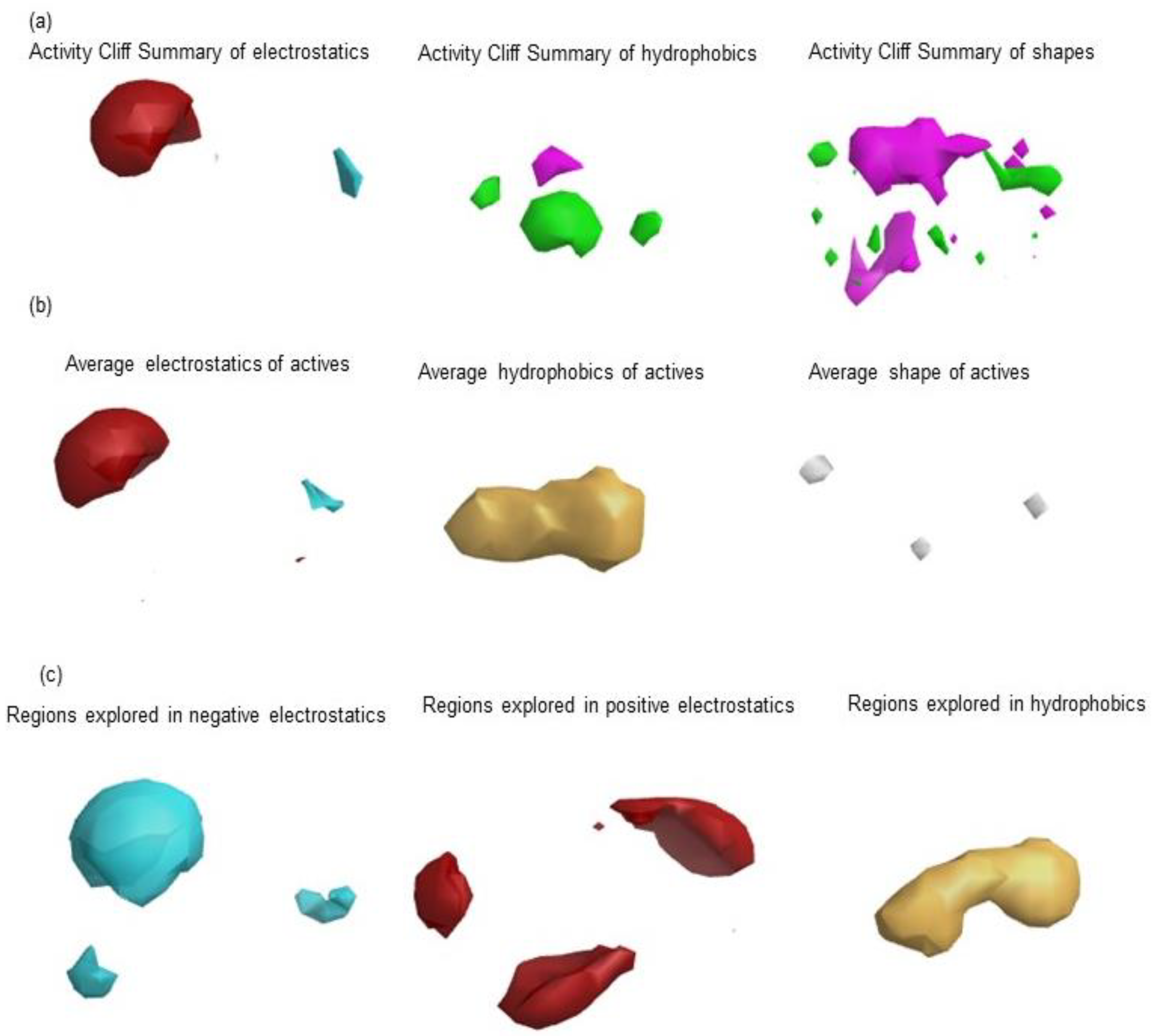
2.1.2. Activity Atlas Model Generation
2.1.3. Screening of Molecules Based on Field Template Alignment and Novelty Score
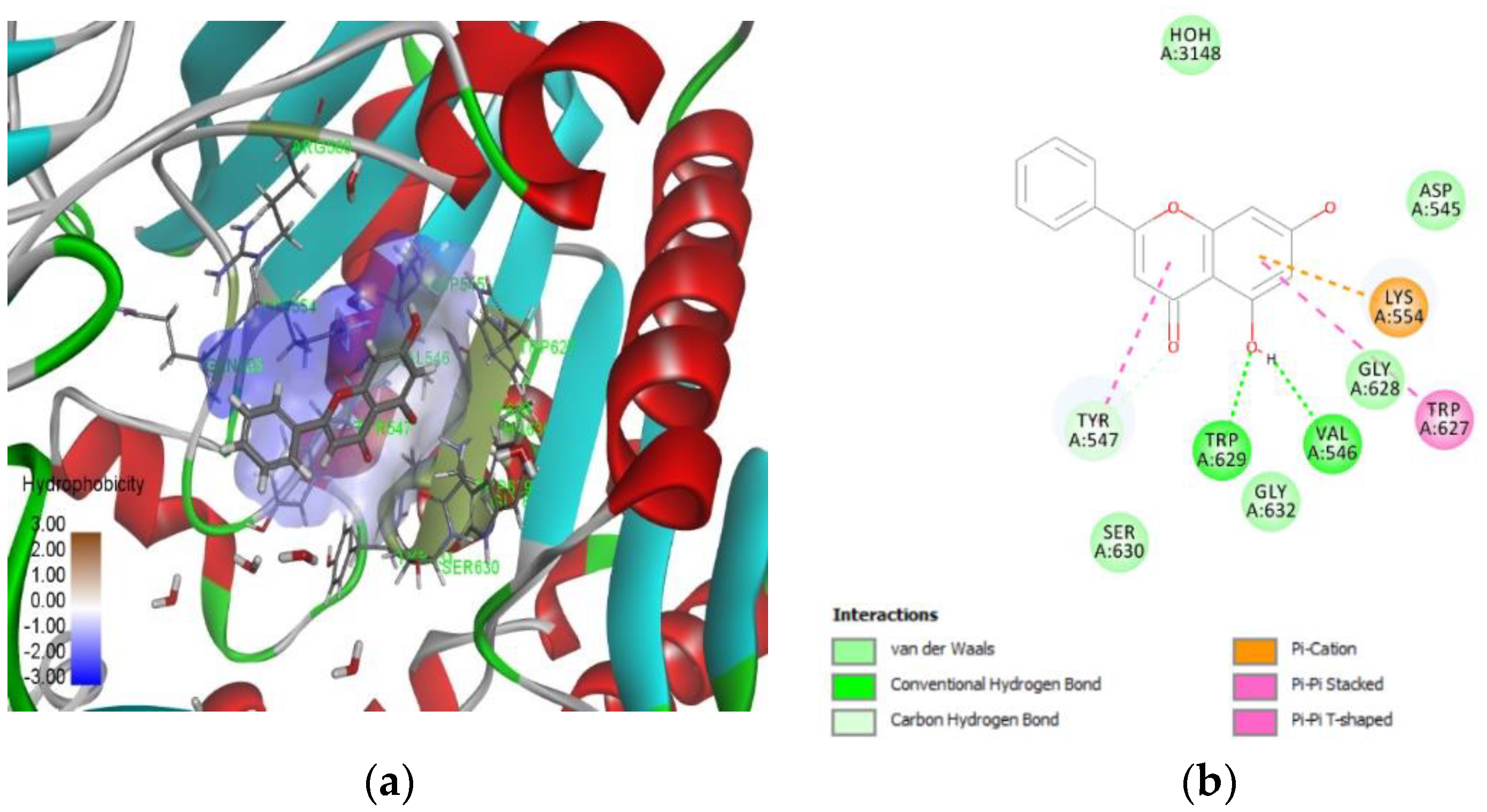
2.2. Molecular Docking
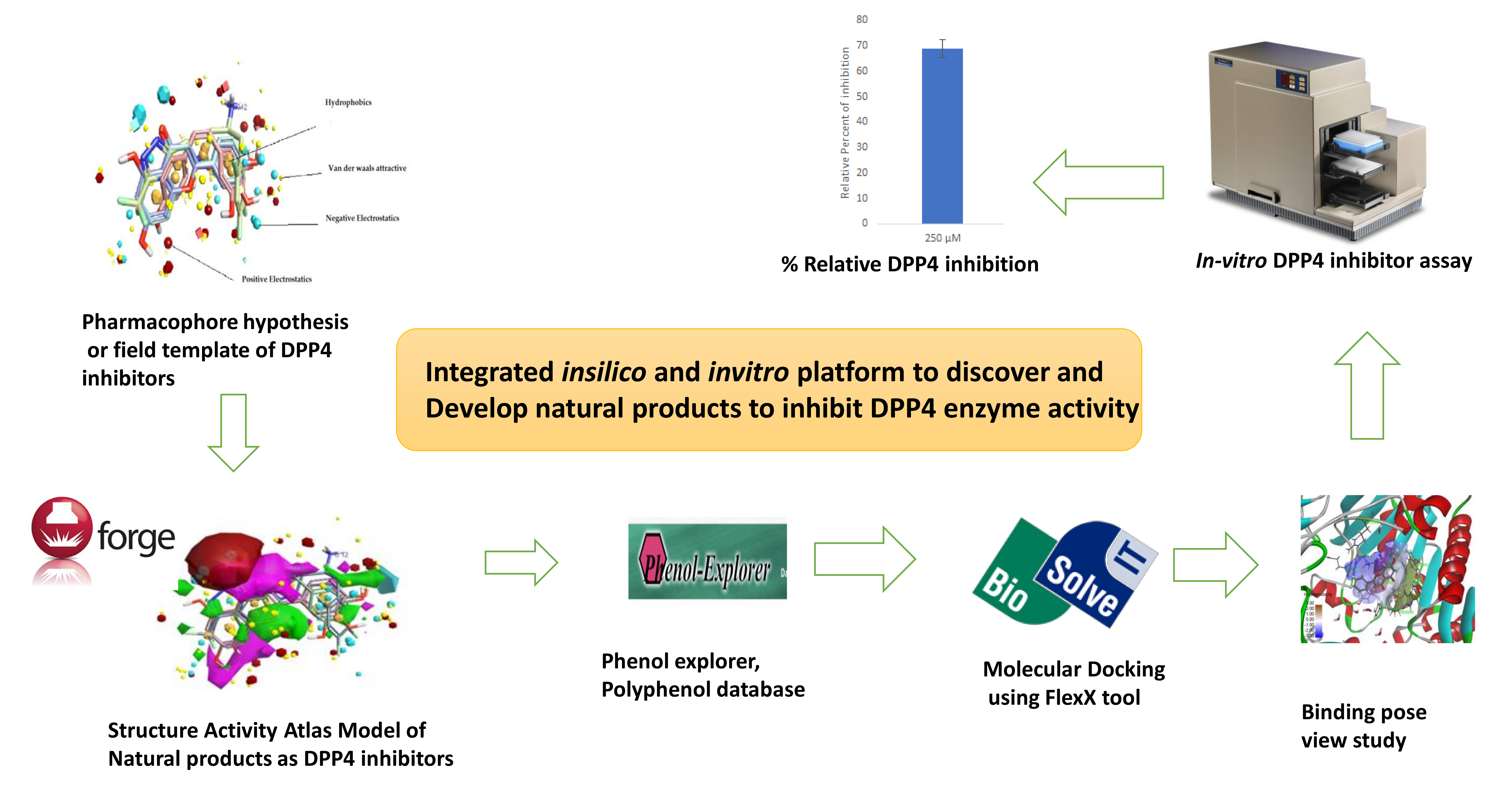
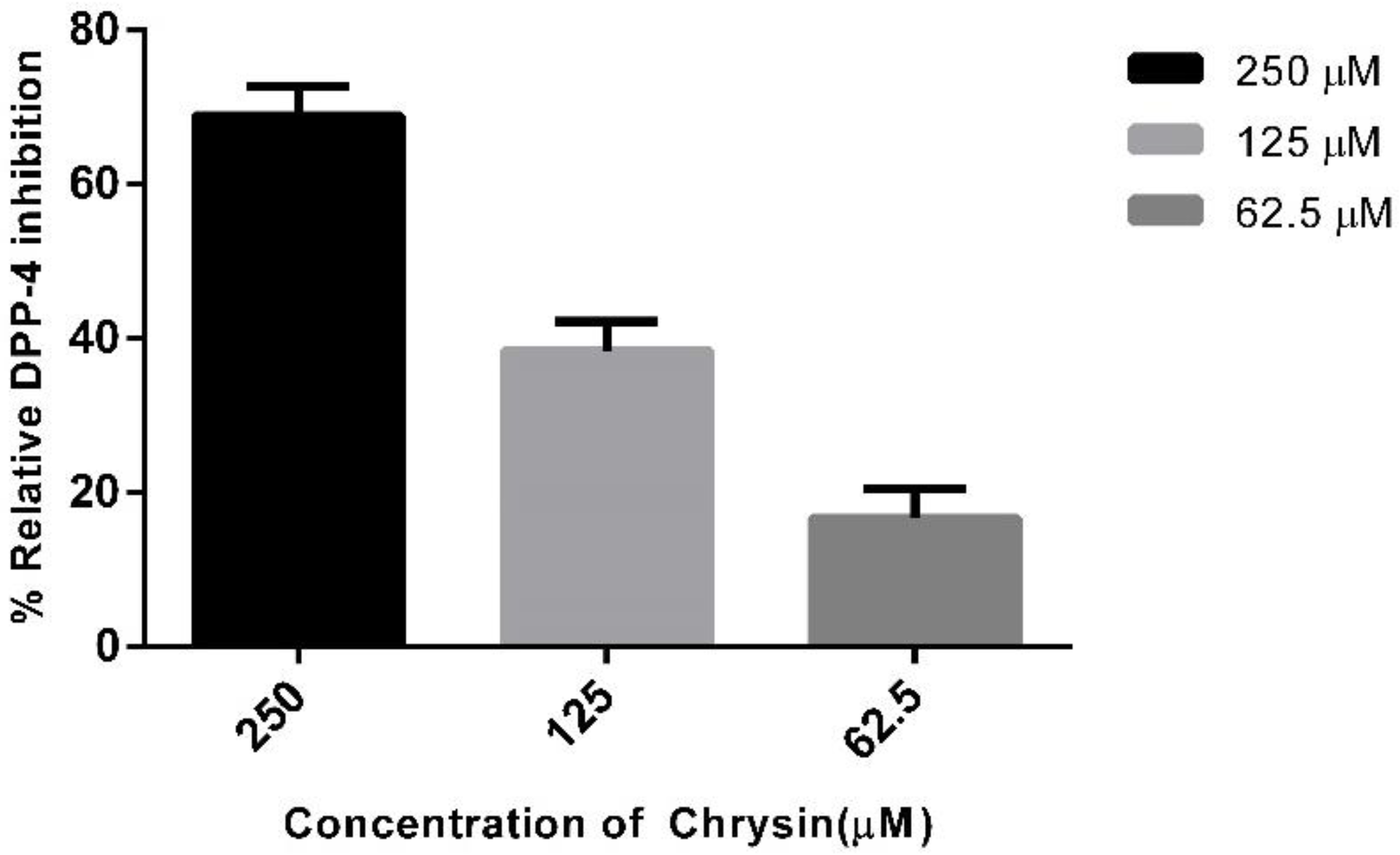
2.3. In Vitro Assay to Validate Chrysin as Dipeptidyl Peptidase-4 Inhibitor
2.4. In Silico Absorption, Distribution, Metabolism, and Toxicity of Chrysin
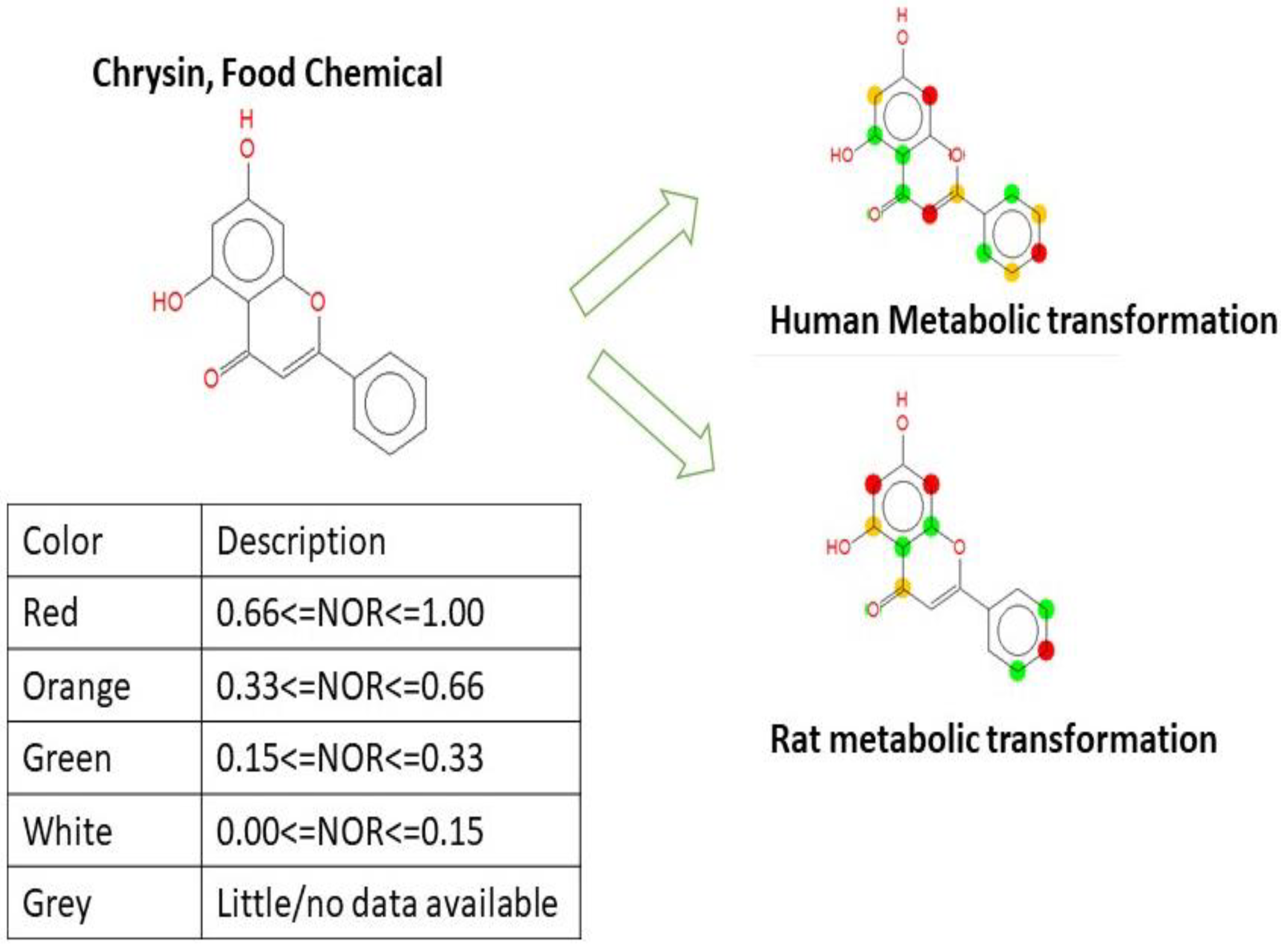
2.5. Metabolic Transformation of Chrysin
3. Materials and Methods
3.1. Generation of Field Template Hypothesis and Activity Atlas Model
3.2. Field-Based and Activity Atlas Model-Based Virtual Screening
3.3. Molecular Docking
3.4. Chemicals and Reagents
3.5. In Vitro DPP-4 Assay
3.6. In Silico Absorption, Distribution, Metabolism, and Toxicity
3.7. Metabolic Biotransformation of Chrysin
4. Conclusions
Supplementary Material
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Barbosa-Filho, J.M.; Vasconcelos, T.H.C.; Alencar, A.A.; Batista, L.M.; Oliveira, R.A.G.; Guedes, D.N.; Falcão, H.D.S.; Moura, M.D.; Diniz, M.F.F.M.; Modesto-Filho, J. Plants and their active constituents from South, Central, and North America with hypoglycemic activity. Rev. Bras. Farmacogn. 2005, 15, 392–413. [Google Scholar] [CrossRef] [Green Version]
- Rao, E.V. Drug discovery from plants. Curr. Sci. 2007. [Google Scholar] [CrossRef]
- Chin, Y.-W.; Balunas, M.J.; Chai, H.B.; Kinghorn, A.D. Drug discovery from natural sources. AAPS J. 2006, 8, E239–E253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saklani, A.; Kutty, S.K. Plant-derived compounds in clinical trials. Drug Discov. Today 2008, 13, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, R.R.; Preedy, V. Bioactive Food as Dietary Interventions for Diabetes; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Lee, H.; Lee, I.S.; Choue, R. Obesity, Inflammation and Diet. Pediatr. Gastroenterol. Hepatol. Nutr. 2013, 16, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Cancer Research Fund; American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective; American Institute for Cancer Research: Washington, DC, USA, 2007. [Google Scholar]
- Fujiwara, K.; Inoue, T.; Yorifuji, N.; Iguchi, M.; Sakanaka, T.; Narabayashi, K.; Kakimoto, K.; Nouda, S.; Okada, T.; Ishida, K.; et al. Combined treatment with dipeptidyl peptidase 4 (DPP4) inhibitor sitagliptin and elemental diets reduced indomethacin-induced intestinal injury in rats via the increase of mucosal glucagon-like peptide-2 concentration. J. Clin. Biochem. Nutr. 2015, 56, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nistala, R.; Habibi, J.; Aroor, A.; Sowers, J.R.; Hayden, M.R.; Meuth, A.; Knight, W.; Hancock, T.; Klein, T.; DeMarco, V.G.; et al. DPP4 inhibition attenuates filtration barrier injury and oxidant stress in the zucker obese rat. Obesity 2014, 22, 2172–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, R.B.; Laird, M.E.; Yatim, N.; Fiette, L.; Ingersoll, M.A.; Albert, M.L. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat. Immunol. 2015, 16, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Rohrborn, D.; Wronkowitz, N.; Eckel, J. DPP4 in diabetes. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, B.; Hennig, M.; Mattei, P. Molecular recognition of ligands in dipeptidyl peptidase IV. Curr. Top. Med. Chem. 2007, 7, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Zettl, H.; Schubert-Zsilavecz, M.; Steinhilber, D. Medicinal chemistry of incretin mimetics and DPP-4 inhibitors. ChemMedChem 2010, 5, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Aertgeerts, K.; Ye, S.; Tennant, M.G.; Kraus, M.L.; Rogers, J.O.E.; Sang, B.; Skene, R.J.; Webb, D.R.; Prasad, G.S. Crystal structure of human dipeptidyl peptidase IV in complex with a decapeptide reveals details on substrate specificity and tetrahedral intermediate formation. Protein Sci. 2004, 1, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.; Boura, P.; Tsapas, A. Safety of dipeptidyl peptidase 4 inhibitors: A perspective review. Ther. Adv. Drug Saf. 2014, 5, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Wadood, A.; Ahmed, N.; Shah, L.; Ahmad, A.; Hassan, H.; Shams, S. In-silico drug design: An approach which revolutionarised the drug discovery process. Open Access Drug Des. Deliv. 2013, 1, 1–4. [Google Scholar] [CrossRef]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-art in ligand-based virtual screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Polgár, T.; Keserue, G.M. Structure-Based Virtual Screening. Front. Drug Des. Discov. 2007, 2007, 477–502. [Google Scholar]
- Wichapong, K.; Rohe, A.; Platzer, C.; Slynko, I.; Erdmann, F.; Schmidt, M.; Sippl, W. Application of docking and QM/MM-GBSA rescoring to screen for novel Myt1 kinase inhibitors. J. Chem. Inf. Model. 2014, 54, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Füllbeck, M.; Michalsky, E.; Dunkel, M.; Preissner, R. Natural products: Sources and databases. Nat. Prod. Rep. 2006, 23, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.-W. Public Databases of Plant Natural Products for Computational Drug Discovery. Curr. Comput. Aided-Drug Des. 2015, 10, 191–196. [Google Scholar] [CrossRef]
- Ma, D.-L.; Chan, D.S.-H.; Leung, C.-H. Molecular docking for virtual screening of natural product databases. Chem. Sci. 2011, 2, 1656–1665. [Google Scholar] [CrossRef]
- Veeramachaneni, G.K.; Raj, K.K.; Chalasani, L.M.; Annamraju, S.K.; Js, B.; Talluri, V.R. Shape based virtual screening and molecular docking towards designing novel pancreatic lipase inhibitors. Bioinformation 2015, 11, 535–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheeseright, T.J.; Mackey, M.D.; Melville, J.L.; Vinter, J.G. FieldScreen: Virtual screening using molecular fields. Application to the DUD data set. J. Chem. Inf. Model. 2008, 48, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, Y.; Zhu, J.; Li, B.; Li, Z.; Zhu, W.; Shi, J.; Jia, Q.; Li, Y. Recent progress in natural products as DPP-4 inhibitors. Future Med. Chem. 2015, 7, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.J.; Mackey, M.D.; Scoffin, R.A. High Content Pharmacophores from Molecular Fields: A Biologically Relevant Method for Comparing and Understanding Ligands. Curr. Comput. Aided-Drug Des. 2011, 7, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Espinosa, J.J.; Saldaña-Ríos, J.; García-Jiménez, S.; Villalobos-Molina, R.; Ávila-Villarreal, G.; Rodríguez-Ocampo, A.N.; Bernal-Fernández, G.; Estrada-Soto, S. Chrysin induces antidiabetic, antidyslipidemic and anti-inflammatory effects in athymic nude diabetic mice. Molecules 2018, 23, 67. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Otake, Y.; Brubaker, J.A.; Walle, U.K.; Halushka, P.V. Disposition and metabolism of the flavonoid chrysin in normal volunteers. Br. J. Clin. Pharmacol. 2001, 51, 143–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, L.; Spjuth, O.; Adams, S.; Glen, R.C.; Boyer, S. Use of historic metabolic biotransformation data as a means of anticipating metabolic sites using MetaPrint2D and Bioclipse. BMC Bioinf. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Spjuth, O.; Helmus, T.; Willighagen, E.L.; Kuhn, S.; Eklund, M.; Wagener, J.; Murray-Rust, P.; Steinbeck, C.; Wikberg, J.E.S. Bioclipse: An open source workbench for chemo- and bioinformatics. BMC Bioinf. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Inhibitory Concentration (μM) |
|---|---|
| Hispidulin | 0.49 ± 0.1 |
| Crisimaritin | 0.43 ± 0.07 |
| Luteolin | 0.12 ± 0.01 |
| Apigenin | 0.14 ± 0.02 |
| Kaempferol | 0.49 ± 0.1 |
| Flavone | 0.17 ± 0.01 |
| Hesperetin | 0.28 ± 0.07 |
| Naringenin | 2.5 ± 0.3 |
| Genistein | 0.48 ± 0.04 |
| Cyanidin | 1.41 ± 0.25 |
| Cyanidin-3-glucoside | 0.42 ± 0.09 |
| Malvidin | 1.41 ± 0.44 |
| Resveratrol | 0.0006 ± 0.0004 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalhotra, P.; Chittepu, V.C.S.R.; Osorio-Revilla, G.; Gallardo-Velázquez, T. Structure–Activity Relationship and Molecular Docking of Natural Product Library Reveal Chrysin as a Novel Dipeptidyl Peptidase-4 (DPP-4) Inhibitor: An Integrated In Silico and In Vitro Study. Molecules 2018, 23, 1368. https://doi.org/10.3390/molecules23061368
Kalhotra P, Chittepu VCSR, Osorio-Revilla G, Gallardo-Velázquez T. Structure–Activity Relationship and Molecular Docking of Natural Product Library Reveal Chrysin as a Novel Dipeptidyl Peptidase-4 (DPP-4) Inhibitor: An Integrated In Silico and In Vitro Study. Molecules. 2018; 23(6):1368. https://doi.org/10.3390/molecules23061368
Chicago/Turabian StyleKalhotra, Poonam, Veera C. S. R. Chittepu, Guillermo Osorio-Revilla, and Tzayhri Gallardo-Velázquez. 2018. "Structure–Activity Relationship and Molecular Docking of Natural Product Library Reveal Chrysin as a Novel Dipeptidyl Peptidase-4 (DPP-4) Inhibitor: An Integrated In Silico and In Vitro Study" Molecules 23, no. 6: 1368. https://doi.org/10.3390/molecules23061368