Synthesis and Reactions of α-Hydroxyphosphonates



Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(6), 1493; https://doi.org/10.3390/molecules23061493
Submission received: 8 May 2018
/
Revised: 11 June 2018
/
Accepted: 16 June 2018
/
Published: 20 June 2018
(This article belongs to the Special Issue Organophosphorus Chemistry 2018)
Abstract
:This review summarizes the main synthetic routes towards α-hydroxyphosphonates that are known as enzyme inhibitors, herbicides and antioxidants, moreover, a number of representatives express antibacterial or antifungal effect. Special attention is devoted to green chemical aspects. α-Hydroxyphosphonates are also versatile intermediates for other valuable derivatives. O-Alkylation and O-acylation are typical reactions to afford α-alkoxy-, or α-acyloxyphosphonates, respectively. The oxidation of hydroxyphosphonates leads to ketophosphonates. The hydroxy function at the α carbon atom of hydroxyphosphonates may be replaced by a halogen atom. α-Aminophosphonates formed in the nucleophilic substitution reaction of α-hydroxyphosphonates with primary or secondary amines are also potentially bioactive compounds. Another typical reaction is the base-catalyzed rearrangement of α-hydroxy-phosphonates to phosphates. Hydrolysis of the ester function of hydroxyphosphonates leads to the corresponding phosphonic acids.
1. Synthetic Routes towards α-Hydroxyphosphonates
Due to the bioactivity of α-hydroxyphosphonates 1, the synthesis of these derivatives is an evergreen topic in organophosphorus chemistry [1,2,3,4,5,6,7,8]. The main approaches to obtain α-hydroxyphosphonates 1 are shown in Scheme 1. The most commonly studied method is the addition of a dialkyl phosphite to an oxo compound (method “A”) [9]. The good atom economy makes this method the most appealing way to synthesize α-hydroxyphosphonates 1. In the majority of cases, the addition is carried out in the presence of a base catalyst, but a few acid-catalyzed variations are also known. An alternative route is the condensation of an oxo compound and a trialkyl phosphite (method “B”) [10]. In contrast to method “A”, this reaction is usually catalyzed by different acids. In the literature, the denomination of the reactions is not consistent, as both methods “A” and “B” are referred to as the Pudovik and Abramov reaction, and also as the phospha-aldol reaction. The third main approach towards α-hydroxyphosphonates 1 involves the reaction of α-ketophosphonates 2 with Grignard reagents [11] or other organometallic compounds [11,12], followed by hydrolysis (method “C”). α-Alkyl phosphonates 3 may also be converted to the corresponding α-hydroxy derivatives by oxidation (method “D”) [13,14]. In this review, methods “A” and “B” are discussed in detail. Although the asymmetric synthesis of α-hydroxyphosphonates 1 is of special importance [4,5,6,7,8], in this review the discussion is limited to racemic derivatives.
1.1. Synthesis of α-Hydroxyphosphonates by the Reaction of Aldehydes/Ketones and Dialkyl Phosphites
The reaction of oxo compounds and dialkyl phosphites catalyzed by alkali alcoholates (Scheme 1, method “A”) was first reported by Pudovik [15]. Since then, a number of variants involving different catalysts and conditions were elaborated to obtain α-hydroxyphosphonates by the reaction under discussion (Table 1). Recent methods have targeted the use of inexpensive and simple catalysts, and mild reaction conditions in the spirit of green chemistry. In the great majority of the cases, the addition was carried out without using any solvent. It is worth mentioning that starting from ketones, the accomplishment of the reaction is more challenging than in the cases applying aldehydes. The synthesis of α-alkyl-α-hydroxyphosphonates often requires the use of “exotic” catalysts [16,17,18,19,20], or an excess of a base. Procedures also applicable for the conversion of ketones are marked by an asterisk in Table 1.
A base-catalyzed variation of the Pudovik reaction was carried out in the presence of 5 mol % potassium phosphate as the catalyst (Table 1/Entry 1) [21]. This method has the advantage of using an inexpensive catalyst, requiring short reaction times and an easy work-up procedure that comprises simple extraction, and, in most cases, provides the products in high yields. Barium hydroxide was also an efficient catalyst in the addition [22,23]. One method employed 10 mol % Ba(OH)2 (Table 1/Entry 2) [22], while according to another protocol, Ba(OH)2∙8H2O was used (Table 1/Entry 3) [23]. The first Ba(OH)2-catalyzed variation suffers from the drawback of a tedious work-up procedure involving extraction, and washing, followed by crystallization [22], while in the latter case, the use of THF as the solvent is a disadvantage [23]. One equivalent of magnesium oxide efficiently catalyzed the addition of diethyl phosphite to substituted benzaldehydes (Table 1/Entry 4) [24]. The method could be extended to the conversion of γ-cyanoketones by applying the base in a twofold quantity (Table 1/Entry 5) [25]. The reaction of dimethyl phosphite with oxo compounds including ketones afforded the corresponding α-hydroxyphosphonates 1 in the presence of three equivalents of aluminum oxide after stirring for 72 h (Table 1/Entry 6) [26]. The reaction time could be shortened dramatically by using potassium fluoride with only one equivalent of Al2O3. However, this latter method was efficient only in case of aldehydes as the starting compounds (Table 1/Entry 7) [27]. The addition was also performed in the presence of one equivalent of triethylamine as the catalyst (Table 1/Entry 8) [28]. A simple crystallization afforded the desired products in good to excellent yields. Another method employed three equivalents of triethylamine together with one equivalent of magnesium chloride (Table 1/Entry 9) [29]. In the latter case, an extraction was inserted before the crystallization step in the work-up procedure [29]. In agreement with the requirements of environmentally friendly approaches, green activation methods, such as microwave (MW) irradiation [30,31] and grinding [32,33] were also employed in the synthesis of α-hydroxyphosphonates 1. According to a MW-assisted procedure, the reaction of benzaldehydes and diethyl phosphite was carried out without the use of any catalyst or solvent (Table 1/Entry 10) [30], while another method combined MW irradiation with sodium carbonate as the base (Table 1/Entry 11) [31]. Na2CO3 was also used efficiently in another protocol, when the contact among the reaction components was promoted by grinding (Table 1/Entry 12) [32]. The drawback of the method is the complicated work-up procedure comprising washing, extraction, and finally recrystallization. A similar protocol utilizes the piperazine-catalyzed reaction of aldehydes and diethyl phosphite in a mill (Table 1/Entry 13) [33]. In the latter case, the work-up involved washing, extraction and column chromatography. The synthesis of α-hydroxyphosphonates 1 was also carried out in the presence of 10 mol % choline hydroxide as an IL catalyst (Table 1/Entry 14) [34]. According to a plausible mechanism suggested by the authors, the IL promotes the reaction by activation through H-bridges. An interesting protocol is a special fluoroapatite-catalyzed Pudovik reaction (Table 1/Entry 15) [35]. A mixture of the starting dialkyl phosphite and oxo compound was stirred with a spatula, and then left standing on the catalyst over 1–1.5 min. The work-up involved extraction with dichloromethane, and recrystallization of the crude product. Acid-catalyzed variations of the Pudovik reaction occur rarely in the literature [36,37]. Potassium hydrogensulfate proved to be an efficient catalyst in an amount of 20 mol % in addition of diethyl phosphite to substituted aldehydes (Table 1/Entry 16) [36]. According to another method, silica-supported tungstic acid was applied as the catalyst (Table 1/Entry 17) [37]. Organometallic compounds may also play the role of the catalyst during the synthesis of α-hydroxyphosphonates 1 [38,39,40]. Butyllithium was applied in the amount of 0.1 mol % in hexane as the solvent (Table 1/Entry 18) [38]. The method not only bears the disadvantages of using an exotic catalyst in an inert atmosphere, but also the need for a complicated work-up involving quenching, washing, and finally column chromatography. As for the advantages, mild reaction conditions and wide substrate scope including ketones can be mentioned. Titanium tetraisopropylate was also applied to catalyze the reaction of ketones and dimethyl phosphite (Table 1/Entry 19) [39]. Column chromatography of the crude product afforded α-alkyl α-hydroxyphosphonates 1 in good to excellent yields. Molybdenum dioxide dichloride (MoO2Cl2) was also tested as the catalyst in the Pudovik reaction (Table 1/Entry 20) [40]. Using it in an amount of 5 mol % under solvent-free conditions at 80 °C, the hydroxyphosphonates 1 were obtained good yields.
1.2. Synthesis of α-Hydroxyphosphonates by the Reaction of Aldehydes/Ketones and Trialkyl Phosphites
Besides the reaction of oxo compounds with dialkyl phosphites discussed in Section 1.1 (Scheme 1, method “A”), the other widespread method for the synthesis of α-hydroxyphosphonates is the condensation of aldehydes or ketones with trialkyl phosphites (Scheme 1, method “B”) [10]. This way is of somewhat less importance due to the reduced atom economy, as compared to the major route outlined. While the reaction involving dialkyl phosphites is usually catalyzed by different bases, the typical catalysts for the condensation of oxo compounds with trialkyl phosphites exhibit acidic character (Table 2).
A method of choice is a solvent- and catalyst-free accomplishment under ultrasonic irradiation (Table 2/Entry 1) [41]. The reaction of aldehydes and trialkyl phosphites at 25 °C was complete after 10–35 min and provided the α-hydroxyphosphonates 1 in excellent yields after crystallization (Table 2/Entry 1). The method was successfully applied to ketones as starting materials as well. Another ultrasound-assisted variation is the potassium dihydrogen phosphate-catalyzed reaction (Table 2/Entry 2) [42]. According to a plausible mechanism, the role of the acid catalyst is the protonation of the carbonyl function of the aldehyde making the C=O bond more electrophilic, and hence facilitating the nucleophilic attack of the (RO)3P reagent. A similar procedure employed sulphamic acid as the catalyst (Table 2/Entry 3) [43]. Again, the catalytic effect of the sulphamic acid was attributed to its capability to protonate the oxo compound. The camphorsulfonic acid-catalyzed syntheses of α-hydroxyphosphonates 1 from aldehydes and triethyl phosphite was carried out under ultrasonic irradiation on stirring (Table 2/Entry 4) [44]. Applying the ultrasound technique, the reaction times could be shortened from 30–75 min to 8–15 min after comparing the results with those obtained without ultrasonication. At the same time, the yields were similar (85–93%) for the two procedures [44]. Oxalic acid also proved to be an efficient catalyst in the condensation under discussion at 80 °C (Table 2/Entry 5) [45]. A series of organic solvents were tested as the reaction medium, but the desired products were obtained in low yields (<30%). Under neat conditions, the α-hydroxyphosphonates 1 were formed efficiently as suggested by the yields of 83–98% obtained after quenching, extraction and column chromatography. In the hope of synthesizing a new class of antibiotics, α-hydroxy-phosphonates 1 containing a β-lactam scaffold were prepared in the presence of tartaric acid as the catalyst (Table 2/Entry 6) [46].
Among a series of acids (e.g., acetic acid, trifluoroacetic acid, p-toluenesulfonic acid, lactic acid, fumaric acid and tartaric acid) screened as potential catalysts of the reaction, fumaric acid and tartaric acid were found to be the most efficient. It was concluded that weak acids (with a pKa value around 4.5) are the best catalysts of the reaction [46]. According to another procedure, pyridine-2,6-dicarboxylic acid was applied as the catalyst (Table 2/Entry 7) [47]. The reaction of oxo compounds including ketones with trimethyl phosphite was carried out in water at 50 °C to furnish hydroxyphosphonates. In another approach, the pyridine-based catalyst was replaced by guanidine hydrochloride (Table 2/Entry 8) [48]. The drawback of this protocol is the rather complicated work-up comprising quenching, extraction, and then chromatography. An interesting finding is that iodine may also be a catalyst in the reaction of aldehydes and triethyl phosphite (Table 2/Entry 9) [49]. The catalytic effect of I2 was explained similarly as that of the acid catalysts: iodine is able to interact with the oxygen atom of the carbonyl group, making the C=O bond more electrophilic. β-Cyclodextrin was found to be a promising and recoverable catalyst of the condensation (Table 2/Entry 10) [50]. However, it was necessary to use this catalyst in a one equivalent quantity, and completion of the reaction required longer times (8–12 h) as compared with other protocols. Ammonium metavanadate was tested as the catalyst in the reaction of benzaldehydes with triethyl phosphite, as well as with diethyl phosphite [51]. In the first case, the reaction was complete after 12–25 min (Table 2/Entry 11). However, in the second case, the desired products were only formed in traces. Metal salts were also applied in the reaction of oxo compounds and trialkyl phosphites with success (Table 2/Entries 12–14) [52,53,54]. The reaction of slightly electron-rich benzaldehydes and triethyl phosphite in the presence of ZnBr2 afforded the corresponding α-hydroxyphosphonates (Table 2/Entry 12) [52]. Bismuth(III) nitrate pentahydrate was applied efficiently as the catalyst in the reaction of aldehydes and triethyl phosphite under MW irradiation at 70 °C (Table 2/Entry 13) [53], while the niobium(V)chloride-trimethylsilyl chloride system was used in an amount of only 0.05 mol % at 25 °C to prepare hydroxyphosphonates (Table 2/Entry 14) [54].
1.3. The “Greenest” Protocol for the Synthesis of α-Hydroxyphosphonates
As it was presented in Section 1.1 and Section 1.2, a great number of procedures have been elaborated for the synthesis of α-hydroxyphosphonates 1 from oxo compounds by reaction with dialkyl or trialkyl phosphites. Although, several methods involved the use of inexpensive and easily available catalysts together with mild and solvent-free reaction conditions, the work-up procedures still suffered from drawbacks. In most cases, the crude product was extracted with organic solvents, and then purified by column chromatography or recrystallization. The combination of the purification steps mentioned is also a frequent option [22,32,33,35,38,48].
We aimed at the elaboration of a new, environmentally-friendly, procedure for the synthesis of α-hydroxyphosphonates 1. As opposed to the procedures already published, we wished to reduce the amount of volatile organic solvents used not only during the reaction, but also in the course of the work-up procedure. According to our method, an equimolar mixture of substituted benzaldehyde and dialkyl phosphite was stirred at reflux in the presence of 10 mol % of triethylamine as the catalyst, in a minimal amount of acetone (1.0 mL/11.0 mmol of the reagents). Then, n-pentane was added to the mixture, which was then cooled to 5 °C. The desired product crystallized from the reaction mixture, and the α-hydroxyphosphonates 1 could be obtained easily by a simple filtration in good to excellent yields (78–99%) in a pure form. The main novelty of our protocol is the absence of further purification steps, a consequence of the “one-pot” synthesis and precipitation from the reaction mixture (Scheme 2) [55].
To reveal the role of triethylamine in the addition, the reaction of benzaldehyde with diethyl phosphite was investigated by quantum chemical calculations [56]. It was found that in the absence of a catalyst, the enthalpy of activation is around 85.9 kJ/mol, and the addition is thermoneutral. The calculations revealed that triethylamine is able to promote the proton transfer from the >P(O)H reagent to the oxo compound. The amine-catalyzed reaction goes with a decreased enthalpy of activation of 68.8 kJ/mol, and follows an exothermic route (∆H = −17.2 kJ/mol). The suggested mechanism involving transition state 4 and intermediate 5 is shown in Scheme 3.
In conclusion, we were successful in the elaboration of another green protocol for the synthesis of α-hydroxyphosphonates 1 from substituted benzaldehydes and a series of dialkyl phosphites in the presence of triethylamine as the catalyst [55]. The new method involves the preparation in boiling acetone followed by crystallization of the product in high purity. There was no need for additional purification. The role of triethylamine in the reaction was justified by quantum chemical calculations [56].
2. Reactions of α-Hydroxyphosphonates
α-Hydroxyphosphonates 1 deserve interest not only as potential enzyme inhibitors [57], herbicides [58], antibiotics [59] and antibacterial or antifungal [60] agents, but also as starting materials for other valuable derivatives [61]. Section 2.1 summarizes the alkylation reactions of α-hydroxyphosphonates (Scheme 4, Route “A”). Acylation of the hydroxy function is one of the most extensively studied transformations within this family of compounds (Scheme 4, Route “B”, Section 2.2). A series of α-acyloxyphosphonates (8) was synthesized applying different acylating agents, like carboxylic and sulfonyl chlorides, as well as carboxylic anhydrides and acids. The oxidation of α-hydroxyphosphonates leads to α-ketophosphonates 9 (Scheme 4, Route “C”, Section 2.3). Another thoroughly studied field is nucleophilic substitution at the α carbon atom of α-hydroxyphosphonates to afford α-halo-, and α-amino or α-alkylphosphonates 10 (Scheme 4, Route “D”, Section 2.4). The rearrangement of hydroxyphosphonates to benzyl phosphates 11 is an interesting field (Scheme 4, Route “E”, Section 2.5). The curiosity of this transformation is the high substrate-dependence. Last but not least, α-hydroxyphosphonic acids 12 may be obtained by the hydrolysis of the ester function of α-hydroxyphosphonates (Scheme 4, Route “F”, Section 2.6).
The derivatives 7–12 synthesized from α-hydroxyphosphonates were summarized in the family tree shown in Scheme 4. Classes 7–12 represent potentially bioactive compounds.
2.1. Alkylations
Alkylation of the hydroxy function of α-hydroxyphosphonates 1 does not belong to their extensively studied reactions, as only a few articles reported the synthesis of α-alkoxyphosphonates 7. A related article targeted antimalarial drugs, and the synthesis involved the O-alkylation of hydroxyphosphonates 1B with benzyl bromoacetate in the presence of silver oxide (Scheme 5) [62].
The protection of the α-hydroxy function may also occur in the literature as another utilization of O-alkylation. A tetrahydropyranyl (THP) protecting group was introduced to an α-hydroxyphosphonate 1B by treatment with 3,4-dihydro-2H-pyran applying p-toluenesulfonic acid as the catalyst [63]. The silylation of α-hydroxyphosphonates 1 is also a useful tool to protect the hydroxy function [64,65,66,67]. The reaction of hydroxyphosphonates 1B with hexamethyldisilazane (HMDS) was studied using a great variety of catalysts including iodine (Scheme 6, Route “a”) [65], copper triflate (Scheme 6, Route “b”) [66] or iron(III) trifluoroacetate (Scheme 6, Route “c”) [67].
A unique protocol has been reported for the cyclization of α-hydroxyphosphonates bearing an alkynyl function in the ortho-position of the aromatic ring (15) [68]. Applying Pd(II) acetate as the catalyst, the nucleophilic attack of the α-O atom occurred on the more distant C-atom of the triple bond of the alkynyl substituent to form the six-membered product 16 selectively. Surprisingly, changing the Pd(OAc)2 catalyst to DBU used in 2 equivalents led to a five-membered cyclic product 17. In the latter case, the attack of the hydroxy group occurred on the nearer C-atom of the triple bond, followed by isomerization (Scheme 7).
2.2. Acylations
The synthesis of α-acyloxyphosphonates 8 is one of the most thoroughly studied transformations of α-hydroxyphosphonates 1. α-Acyloxyphosphonates 8 attracted attention as potential herbicides [69,70,71], fungicides [69], and insecticides [72]. A number of articles reported the acylation of α-hydroxyphosphonates 1 with carboxylic acid chlorides in the presence of triethylamine [73,74,75] or pyridine [76,77] as the hydrochloric acid scavenger. Among the –C(O)Cl reagents, phenoxyacetyl chloride derivatives 18 are frequently applied acylating agents [70,71,76,77,78,79,80,81,82,83,84].
In general, the reaction of α-hydroxyphosphonates 1 with phenoxyacetyl chlorides 18 required a reaction time of 3–7 h, and the corresponding α-acyloxyphosphonates 19 were isolated in yields of 53–89% (Scheme 8) [78]. The compounds 19 thus-obtained are popular target molecules, as a series of related derivatives exhibited herbicidal activity [70,76,77,78,79,80,83,84].
The reaction of α-hydroxy-phosphonates 1A with 2,6-pyridinedicarboxylic acid chloride (20) led to bis[dimethyl phosphonomethyl pyridine-2,6-dicarboxylates] 21 containing two α-acyloxyphosphonate moieties (Scheme 9). The reaction took place under mild conditions, and the products 21 were isolated with variable yields (38–80%). The derivatives so-obtained 21 also showed herbicidal activity [85].
The acylation of α-hydroxyphosphonates 1B has been also performed with acetic anhydride [86,87,88]. This reaction was carried out in the presence of a catalytic amount (1.3–8 mol %) of Cu(OTf)2 at 25 °C to afford the desired α-acetyloxyphosphonates 22 almost quantitatively (Scheme 10, Route “a”) [86]. In another instance, 1 mol % of TiCl3(OTf) was employed as the catalyst (Scheme 10, Route “b”). In this case, completion of the reaction required a somewhat higher temperature (70 °C) [87]. A more benign synthetic route to α-acetyloxyphosphonates 22 is the reaction of α-hydroxyphosphonates (1B) and acetic anhydride under MW irradiation (Scheme 10, Route “c”) [88]. The acylation was complete after a short reaction time of 5 min, and the products were obtained in excellent yields (90–98%). However, a lack of control of the reaction temperatures is a shortcoming that prevents reproduction.
The synthesis of α-acyloxyphosphonates 8 was also carried out starting from substituted benzaldehydes, dimethyl phosphite and acetic anhydride in the presence of magnetic nanoparticle-supported guanidine [89]. In this one-pot procedure, the formation of the α-hydroxyphosphonate 1 in the Pudovik reaction was followed by acylation of the hydroxy function.
Carboxylic acids may also be efficient acylating agents of α-hydroxyphosphonates 1 in the presence of N,N′-dicyclohexylcarbodiimide (DCC) as the coupling reagent, and 4-dimethyl-aminopyridine (DMAP) as the base [27,90,91]. This is exemplified by the reaction of 2-(4,6-dimethoxypyrimidin-2-yloxy)benzoic acid (23) at 25 °C to give α-acyloxyphosphonates 24 possessing herbicidal activity (Scheme 11) [27].
Another method of choice for the acylation of α-hydroxyphosphonates 1B with carboxylic acids, is the Mitsunobu reaction applying 4,4′-azopyridine (25) and triphenylphosphine in acetonitrile as the solvent (Scheme 12) [92]. Completion of the reaction required a reaction time of 9–24 h at 80 °C, and the products 26 were obtained in yields of 65–88%.
The reaction of α-hydroxyphosphonates 1 with isocyanates 27 or isothiocyanates 29 resulted in the formation of α-carbamoyloxy-28 or α-thiocarbamoyloxyphosphonates 30, respectively [93,94]. One study targeted herbicides by the acylation of α-hydroxyphosphonates (1) with pyrimidine-based isocyanates 27 (Scheme 13, Route “A”) [93].
The reaction took place under benign conditions, and the products 28 so-obtained revealed a weak herbicidal activity. On reacting hydroxyphosphonates (1) and isothiocyanates 29 in the absence of a catalyst, the corresponding α-thiocarbamoyl-oxyphosphonates 30 were obtained in modest yields of 20–30%. However, the application of 15 mol % of sodium methoxide promoted the acylation, and the products 30 could be isolated in yields of 74–90% (Scheme 13, Route “B”) [94]. The authors of the article emphasize the need for mild reaction conditions (25 °C, 0.5–3.5 h), as heating of the reaction mixture resulted in the formation of a by-product through the trimerization of the starting isothiocyanate. The α-thiocarbamoyl-oxyphosphonates 30 obtained in this way expressed plant growth regulating activity.
As shown above, the acylation of α-hydroxyphosphonates (1B) was carried out with a wide range of carboxylic acid derivatives. Besides carboxylic chlorides, anhydrides and acids, sulfonic acid derivatives may also function as the acylating agents. By analogy with the reaction of α-hydroxyphosphonates (1B) with carboxylic chlorides, the acylation was also attempted with sulfonyl chlorides [95,96,97]. Methanesulfonyl chloride (Scheme 14, Route “a”) [96] and p-toluenesulfonyl chloride (Scheme 14, Route “b”) [97] worked well as sulfonylating agents in the presence of triethylamine.
In contrast to the wide range of α-acyloxy and α-sulfonyloxy phosphonates, α-phosphoryloxy analogues appear rarely in the literature. A study from the early 1960’s reported the synthesis of a >P(O)OCHP(O)< type compound 33 by a thermo-induced rearrangement (Scheme 15) [98]. However, the phosphorylation of α-hydroxyphosphonates (1) with P-chlorides presented a challenge.
Recognizing the lack of O-phosphorylated α-hydroxyphosphonates 34 in the literature, we aimed at the synthesis of new classes of derivatives by the acylation of α-hydroxyphosphonates 1 with phosphoryl and phosphinic chlorides. It was observed that by stirring the reaction components at ambient temperature, the O-phosphorylated phosphonates 34 were formed with conversions of 55–90% (Scheme 16) [99].
2.3. Oxidations
The oxidation of α-hydroxyphosphonates 1B affords the corresponding α-ketophosphonates (35) that are versatile precursors of a series of organophosphorus compounds [100,101,102]. The application of metal compounds with variable valency as the oxidizing agent is predominating in the literature. Alumina-supported chromium(III) oxide (CrO3/Al2O3) (Scheme 17, Route “a”) [103], and chromium salts, like zinc dichromate trihydrate (ZnCr2O7·3H2O) (Scheme 17, Route “b”) [104] were found to be efficient reagents in the oxidation of α-hydroxyphosphonates 1B. In both cases, the reaction took place at ambient temperature under solvent-free conditions. The procedures provided the desired products 35 in good to excellent yields (up to 96%).
In other variations, pyridinium dichromate [105] or pyridinium chlorochromate [106] were applied successfully to give α-ketophosphonates 35. The reaction under discussion was also reported using KMnO4 as the oxidant (Scheme 18). A shortcoming of this latter protocol is the need for dry benzene as the reaction medium [107]. Another problem of the chemical oxidating agents is the low atom efficiency and the lack of “greenness”.
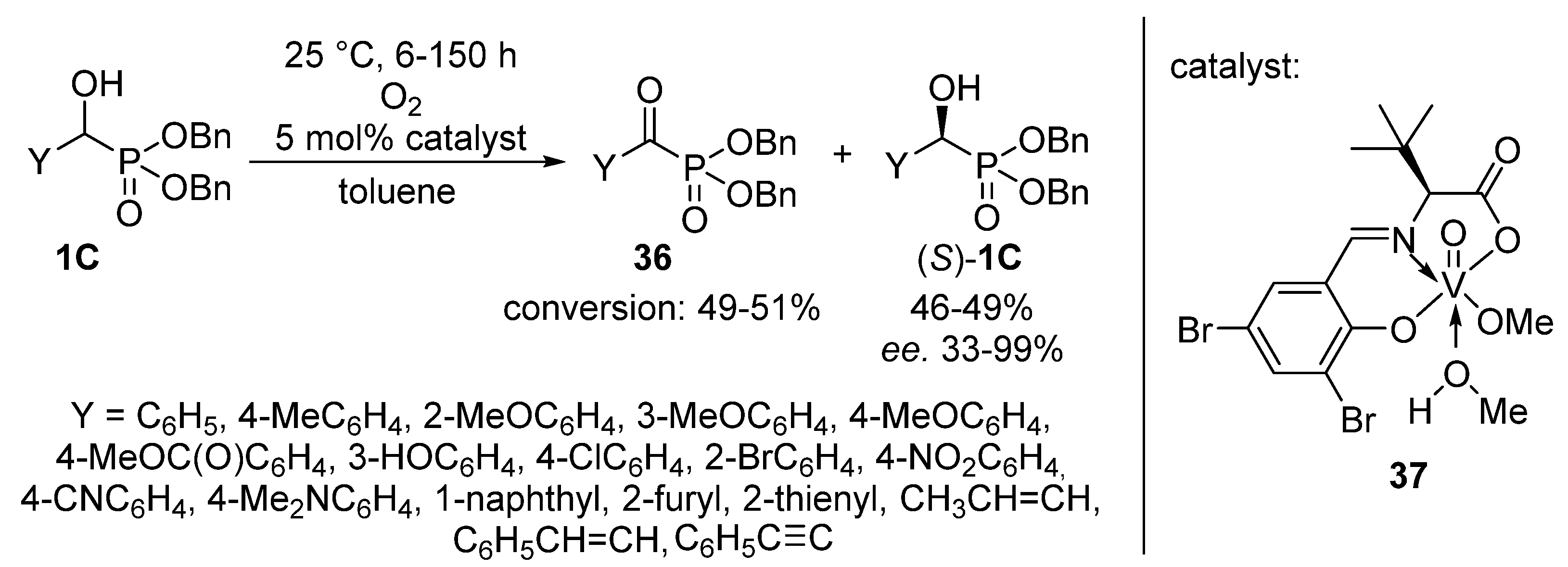
An elegant achievement is the oxidation of dibenzyl α-hydroxyphosphonates 1C with oxygen, employing a chiral vanadyl(V) complex (37) as the catalyst (Scheme 19) [108]. After a reaction time of 6–150 h, the corresponding α-ketophosphonates 36 were obtained in conversions of 49–51%, while due to the presence of the optically active catalyst, the (S) enantiomer of the α-hydroxyphosphonate (S)-1C remained untouched. This protocol offered an efficient way for the resolution of α-hydroxyphosphonates 1C with high enantiomeric excesses up to 99% (with two exceptions), in parallel with the formation of α-ketophosphonates 36.
2.4. Nucleophilic Substitutions
The nucleophilic substitution of the hydroxy group of α-hydroxyphosphonates 1 represents one of the most widely studied reaction types of this class of compounds. A typical transformation is the change of the hydroxy function to a halogen atom. α-Halobenzylphosphonates 38 deserve interest as potential inhibitors of protein tyrosine phosphatases [109,110]. A chloro or bromo substituent may be introduced into an α-hydroxyphosphonate 1 molecule by treatment with thionyl chloride [111] or thionyl bromide (Scheme 20, Route “a”) [110], respectively. Another method of choice to obtain α-halobenzylphosphonates 38 is the joint application of triphenylphosphine and the corresponding carbon tetrahalide [112,113,114]. One procedure applied this synthetic route to obtain α-chlorobenzylphosphonates (Scheme 20, Route “b”) [114], while another paper reported the introduction of a chloro atom to both aliphatic and aromatic hydroxyphosphonates under similar conditions (Scheme 20, Route “c”) [113].
A further variation involves the use of tetrabutylammonium bromide as the nucleophile in the presence of triphenylphosphine—DDQ (2,3-dichloro-5,6-dicyanobenzoquinone) system, affording α-bromophosphonates [115,116].
In another protocol, hydroxyphosphonates 1 were converted to their corresponding α-halo analogues 38 by reaction with molecular halogens (Br2 or I2) applying 4-aminophenyl-diphenylphosphinite 39 as the acid scavenger (Scheme 21) [117]. The amino function of the phosphinite 39 made possible the removal of the forming hydrogen halogenide as an ammonium salt after a simple filtration.
To synthesize α-fluorophosphonates, the most commonly used fluorinating agent is diethylaminosulfur trifluoride (Et2NSF3) [111,114,118,119,120,121]. As a variation of this method, a protocol applying morpholinosulfur trifluoride was elaborated for the OH → F change in α-hydroxy-bisphosphonates [122]. N-fluorobisbenzenesulfonimide was used efficiently to introduce a fluoro function to the α-carbon atom of α-monohalophosphonates 38 in the presence of sodium hexamethyldisilazane [113,114,116].
Halogens are not the sole nucleophiles that the α-hydroxy function of benzylphosphonates 1B may be replaced with. One method operated with the above mentioned triphenylphosphine−DDQ system to catalyze the reaction of α-hydroxyphosphonates 1B with ammonium thiocyanate to obtain α-thiocyanatophosphonates 40 (Scheme 22, Route “A”) [115]. Another study reported the Mitsunobu reaction of α-hydroxyphosphonates 1B with hydrazoic acid as the nucleophile. The treatment of the so-formed α-azidophosphonates 41 with triphenylphosphine and carbon disulfide resulted in the formation of α-isothiocyanatophosphonates 42 (Scheme 22, Route “B”) [123].
α-Aminophosphonates 43 are usually obtained by the three-component condensation (Kabachnik-Fields reaction) of oxo compounds, amines and dialkyl phosphites or secondary phosphine oxides [124,125,126,127]. However, these compounds may also be synthesized via the nucleophilic substitution of α-hydroxyphosphonates 1B with primary or secondary amines. According to a simple protocol, this latter reaction was performed under MW irradiation, in the presence of Al2O3 [128]. After 3–7 min, a complete conversion was attained (Scheme 23). However, reaction temperatures are missing from the articles, as the experiments were carried out in a kitchen MW oven.
We found that α-hydroxyphosphonates 1B may be converted to the corresponding α-aminophosphonates 44–46 under MW conditions without the use of any catalyst or solvent (Scheme 24) [56,129]. The starting hydroxyphosphonates 1B were reacted with three equivalents of a primary amine (propyl-, butyl- or cyclohexyl-amine) applying MW irradiation at 100 °C. Following this method, the substitution took place with surprising ease. Starting from substituted hydroxyphosphonates 1Bb, 1Bc and 1Bg, a short reaction time of 15–30 min was sufficient for complete conversion (Table 3/Entries 4–12). The unsubstituted analogue 1Ba was even more reactive, and in this case the reaction took place after 10–15 min (Table 3/Entries 1–3).
Encouraged by the recognition that the reaction of α-hydroxyphosphonates 1B with primary amines takes place rather easily, we wished to study the mechanism of the substitution by quantum chemical (DFT) calculations [129]. The nucleophilic attack of methylamine on diethyl α-hydroxybenzylphosphonate 1Ba was chosen as the model reaction. It was found that the reaction follows an SN2 mechanism. It was also revealed that a favorable neighboring group effect facilitates the transfer of the oxygen atom of the hydroxy group from the α-carbon atom to the phosphorus (transition state 47) [129]. The proposed mechanism is shown on Scheme 25.
α-Sulfonamidophosphonates were obtained through the reaction of α-hydroxyphosphonates and sulfonamides in the presence of HOTf in dioxane. The reaction took place at ambient temperature within 5 h, and the desired products were obtained in yields of 70–94% [130]. The products so-obtained were found to be as promising corrosion inhibitors for mild steel [131].
The Friedel-Crafts alkylation using α-hydroxyphosphonates 1 in the presence of an acid catalyst (e.g., FeCl3 or HOTf) led to α,α-diarylphosphonates 53 [132,133,134]. Benzene and naphthalene served as the aromatic substrate. According to the proposed mechanism, the first step is the formation of benzylphosphonate carbocation 52 that is followed by its electrophilic attack on the aromatic substrate (Scheme 26) [132].
An interesting reaction is when α-hydroxyphosphonates 1B function as an alkylating agent of 1,3-diketones to afford γ-ketophosphonates 54 (Scheme 27) [135]. The reaction of the starting components in organic solvents in the presence of FeCl3 or Cu(OTf)2 Lewis acids resulted in the formation of compound 54 (Scheme 27, Route “A”). The optimization of the alkylations was a real challenge for the authors, as each hydroxyphosphonate—diketone combination required different reaction conditions regarding the Lewis acid, temperature, time and solvent. However, applying FeCl3·6H2O as the catalyst, a C–C bond cleavage of the desired product 54 also occurred due to the presence of the crystal hydrate of the iron salt. In this case, a mixture of compounds 54 and 55 was attained (Scheme 27, Route “B”).
2.5. Rearrangements
The rearrangement of α-hydroxyphosphonates 1 to benzyl phosphates was first discovered through the example of the widely known insecticide prodrug, trichlorofon (O,O-dimethyl (2,2,2-trichloro-1-hydroxyethyl)phosphonate) that is rearranged to 2,2-dichlorovinyl dimethyl phosphate (DDVP) acting as an acetylcholinesterase inhibitor [136]. Phosphonate-phosphate rearrangements were investigated in the presence of strong bases, such as sodium hydroxide [136], sodium ethoxide [137] and sodium hydride [138]. A number of protocols operated with triethylamine as the catalyst [139,140,141]. However, the Et3N-catalyzed rearrangement is not a versatile method, as it was reported to apply to only six-membered cyclic phosphonates [139,140,141].
A new approach of this transformation is when the Pudovik reaction of benzaldehydes and dialkyl phosphites and the subsequent rearrangement of the so-formed α-hydroxyphosphonate 1 take place in “one-pot”, under the same conditions. The synthesis of benzyl phosphates 56 directly from an oxo compound and a P-reagent was efficiently catalyzed by butyllithium (Scheme 28, Route “a”) [142] as well as by DBU (Scheme 28, Route “b”) [143]. The tandem Pudovik reaction and rearrangement was also reported in the presence of superbases 57 (Scheme 28, Route “c”) [144]. The authors of this latter article emphasized that both the reaction media and the substituent in the aromatic ring had a great impact on the reaction. The rearrangement took place faster in alcohols (EtOH or iPrOH) than in acetonitrile. It was also shown that the presence of electron-withdrawing substituents facilitated the formation of the benzyl phosphate 56 [144].
In this field, our research targets the accomplishment of the rearrangement of α-hydroxyphosphonates 1 using bases under phase transfer catalytic conditions (Scheme 29) [145].
Our preliminary results highlighted that the substituent in the aromatic ring has a significant impact on the reaction. In the presence of electron-withdrawing substituents (Cl and F), the reaction took place at ambient temperature, whereas the rearrangement of α-hydroxyphosphonates bearing electron-releasing substituents required 2–3 h of heating [145].
2.6. Hydrolysis
The hydrolysis of α-hydroxyphosphonates 1 is the most common way for the synthesis of α-hydroxyphosphonic acids 12 that may be inhibitors of a wide variety of enzymes including protein tyrosine phosphatases [110,146,147], mandelate racemase [148], phosphoglycerate kinase [149], plant P5C reductase [150] and plant glutamine synthetase [151]. A series of α-hydroxyphosphonic acid (12) and α-hydroxybisphosphonic acid was recognized as antibiotics against Gram-positive bacteria [152]. α-Hydroxyphosphonic acids 12 bearing an amide function in the para-position of the aromatic ring were found to be in vivo inhibitors of autotaxin [153].
As for the synthesis of α-hydroxyphosphonic acids 12, most of the related articles reported the acidic hydrolysis of the corresponding methyl or ethyl ester [149,150,152,154]. A number of protocols applied hydrochloric acid in an excess at reflux, and the hydrolysis usually required a prolonged reaction time of 6–24 h [149,150,152]. According to another method, beside the excess of 6 M hydrochloric acid, dioxane was also added as a co-solvent [154].
In the hope of selective hydrolysis of one of the ester functions, the hydrolytic reaction of dimethyl 1-hydroxy-1-phenylmethylphosphonate 1Aa was carried out in aqueous sodium hydroxide at reflux for 5 h (Scheme 30, Route “A”) [155]. After pH adjustment, the desired product (57) was obtained in a yield of 85%. This method was suitable only for the synthesis of the racemic target compound 57. In order to maintain the C chirality center of the starting α-hydroxyphosphonate (R)-1Aa, a milder protocol had to be elaborated. On stirring the starting optically active α-hydroxyphosphonate (R)-1Aa in the presence of sodium iodide in acetone at reflux for 22 h, racemization of the hydroxyphosphonate (R)-1Aa could be avoided (Scheme 30, Route “B”) [155].
Another way of synthesizing α-hydroxyphosphonic acids (12) from α-hydroxyphosphonates 1 involves the cleavage of the C–O bond of the ester function by trimethylsilyl chloride [77,138] or trimethylsilyl bromide [110]. This reaction is usually carried out at ambient temperature within 2–18 h, generally in acetonitrile as the solvent.
3. Conclusions
This review summarizes the synthesis and reactions of α-hydroxyphosphonates that are of importance due to their bioactivity. The first half of the review presents the most common synthetic routes towards α-hydroxyphosphonates, with special stress on the addition of dialkyl phosphite to an oxo compound, and the condensation of trialkyl phosphites with aldehydes or ketones. The overview is followed by the discussion of the green synthetic protocols for α-hydroxyphosphonates. Then, the reactions of α-hydroxyphosphonates involving O-alkylation, O-acylation and oxidation of the hydroxy function, substitutions of the hydroxy group by a chloro or amino function, rearrangements to phosphate derivatives and hydrolysis of the ester function are discussed. Beside the literature examples, our own synthetic results are also included.
Acknowledgments
The authors are indebted to David W. Allen for his advice. The above project was supported by the National Research Development and Innovation Fund (K119202). Z. Rádai is grateful for the fellowship provided by the Chinoin-Sanofi Pharmaceuticals and the József Varga Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Olszewski, T.K. Environmentally benign syntheses of α-substituted phosphonates: Preparation of α-amino- and α-hydroxyphosphonates in water, in ionic liquids, and under solvent-free conditions. Synthesis 2014, 46, 403–429. [Google Scholar] [CrossRef]
- Failla, S.; Finocchiaro, P.; Consiglio, G.A. Syntheses, characterization, stereochemistry and complexing properties of acyclic and macrocyclic compounds possessing α-amino- or α-hydroxyphosphonate units: A review article. Heteroatom. Chem. 2000, 11, 493–504. [Google Scholar] [CrossRef]
- Rádai, Z.; Kiss, N.Z.; Keglevich, G. Synthesis of α-Hydroxyphosphonates, an Important Class of Bioactive Compounds; György, K., Ed.; Organophosphorus Chemistry: Novel Developments; Walter de Gruyter: Berlin, Germany; Boston, MA, USA, 2018; pp. 91–107. 315p. [Google Scholar]
- Gröger, H.; Hammer, B. Catalytic concepts for the enantioselective synthesis of α-amino and α-hydroxy phosphonates. Chem. Eur. J. 2000, 6, 943–948. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Chiral hydroxyl phosphonates: Synthesis, configuration and biological properties. Russ. Chem. Rev. 2006, 75, 227–253. [Google Scholar] [CrossRef]
- Merino, P.; Marqués-López, E.; Herrera, R.P. Catalytic enantioselective hydrophosphonylation of aldehydes and imines. Adv. Synth. Catal. 2008, 350, 1195–1208. [Google Scholar] [CrossRef]
- Spilling, C.D.; Malla, R.K. Synthesis of non-racemic α-hydroxyphosphonates via asymmetric phosphor-aldol reaction. Top. Curr. Chem. 2015, 361, 83–136. [Google Scholar] [PubMed]
- Phillips, A.M.F. Organocatalytic asymmetric synthesis of chiral phosphonates. Mini Rev. Org. Chem. 2014, 11, 164–185. [Google Scholar] [CrossRef]
- Pudovik, A.N.; Konovalova, I.V. Addition reactions of esters of phosphorus(III) acids with unsaturated systems. Synthesis 1979, 2, 81–96. [Google Scholar] [CrossRef]
- Abramov, V.S.; Kiryukhina, L.I.; Kudryavtseva, N. Reaction of dialkyl esters of phosphorus acids with aldehydes and ketones. XXV. Esters of α-hydroxy-β-(2,2,3-trimethyl3-cyclopentenyl)ethylphosphonic and α-hydroxynitrobenzylphosphonic acids. Zh. Obshch. Khim. 1964, 34, 2235–2238. [Google Scholar]
- Maeda, H.; Takahashi, K.; Ohmori, H. Reactions of acyl tributylphosphonium chlorides and dialkyl acylphosphonates with Grignard and organolithium reagents. Tetrahedron 1998, 54, 12233–12242. [Google Scholar] [CrossRef]
- Seven, O.; Polat-Cakir, S.; Hossain, M.S.; Emrullahoglu, M.; Demir, A.S. Reactions of acyl phosphonates with organoaluminum reagents: A new method for the synthesis of secondary and tertiary α-hydroxy phosphonates. Tetrahedron 2011, 67, 3464–3469. [Google Scholar] [CrossRef]
- Gu, L.; Jin, C.; Zhang, H. The catalytic aerobic synthesis of quaternary α-hydroxy phosphonates via direct hydroxylation of phosphonate compounds. New J. Chem. 2015, 39, 1579–1582. [Google Scholar] [CrossRef]
- Li, X.; Jin, C.; Gu, L. C–H hydroxylation of phosphonates with oxygen in [bmIm]OH to produce quaternary α-hydroxy phosphonates. J. Org. Chem. 2015, 80, 2443–2447. [Google Scholar] [CrossRef] [PubMed]
- Pudovik, A.N.; Zametaeva, G.A. New synthesis of esters of phosphonic and thiophosphonic acids. XIII. Addition of diethyl thiophosphite to ketones and aldehydes. Izv. Akad. Nauk SSSR Ser. Khim. 1952, 1952, 932–939. [Google Scholar]
- Zhou, S.; Wu, Z.; Rong, J.; Wang, S.; Yang, G.; Zhu, X.; Zhang, L. Highly efficient hydrophosphonylation of aldehydes and unactivated ketones catalyzed by methylene-linked pyrrolyl rare earth metal amido complexes. Chem. Eur. J. 2012, 18, 2653–2659. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ding, H.; Zhao, B.; Lu, C.; Yao, Y. Synthesis and characterization of amidate rare-earth metal amides and their catalytic activities toward hydrophosphonylation of aldehydes and unactivated ketones. Polyhedron 2014, 83, 50–59. [Google Scholar] [CrossRef]
- Yang, S.; Zhu, X.; Zhou, S.; Wang, S.; Feng, Z.; Wei, Y.; Miao, H.; Guo, L.; Wang, F.; Zhang, G.; et al. Synthesis, structure, and catalytic activity of novel trinuclear rare-earth metal amido complexes incorporating μ–η5:η1 bonding indolyl and μ3-oxo groups. Dalton Trans. 2014, 43, 2521–2533. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Zhou, S.L.; Wang, S.W.; Zhang, L.J.; Wei, Y.; Yang, S.; Wang, F.H.; Chen, Z.; Chen, Y.; Yuan, Q.B. Rare-earth metal amido complexes supported by bridged bis(β-diketiminato) ligand as efficient catalysts for hydrophosphonylation of aldehydes and ketones. Sci. China Chem. 2013, 56, 329–336. [Google Scholar] [CrossRef]
- Liu, B.; Carpentier, J.F.; Sarazin, Y. Highly effective alkaline earth catalysts for the sterically governed hydrophosphonylation of aldehydes and nonactivated ketones. Chem. Eur. J. 2012, 18, 13259–13264. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, M.A.; Lad, U.P.; Desai, U.V.; Mitragotri, S.D.; Wadgaonkar, P.P. Mechanistic approach for expeditious and solvent-free synthesis of α-hydroxy phosphonates using potassium phosphate as catalyst. C. R. Chim. 2013, 16, 148–152. [Google Scholar] [CrossRef]
- Nandre, K.P.; Nandre, J.P.; Patil, V.S.; Bhosale, S.V. Barium hydroxide catalyzed greener protocol for the highly efficient and rapid synthesis of α-hydroxyphosphonates under solvent free conditions. Chem. Biol. Interface 2012, 2, 314–321. [Google Scholar]
- Pandi, M.; Chanani, P.K.; Govindasamy, S. An efficient synthesis of α-hydroxy phosphonates and 2-nitroalkanols using Ba(OH)2 as catalyst. Appl. Catal. A 2012, 441–442, 119–123. [Google Scholar] [CrossRef]
- Sardarian, A.R.; Kaboudin, B. Surface-mediated solid phase reactions: Preparation of diethyl 1-hydroxyarylmethylphosphonates on the surface of magnesia. Synth. Commun. 1997, 27, 543–551. [Google Scholar] [CrossRef]
- Aouani, I.; Lahbib, K.; Touil, S. Green synthesis and antioxidant activity of novel γ-cyano-α-hydroxyphosphonate derivatives. Med. Chem. 2015, 11, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Hudson, H.R.; Yusuf, R.O.; Matthews, R.W. The preparation of dimethyl α-hydroxyphosphonates and the chemical shift non-equivalence of their diastereotopic methyl ester groups. Phosphorus Sulfur Silicon Relat. Elem. 2008, 183, 1527–1540. [Google Scholar] [CrossRef]
- Jin, C.; He, H. Synthesis and herbicidal activity of novel dialkoxyphosphoryl aryl methyl 2-(4,6-dimethoxypyrimidin-2-yloxy) benzoate derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2011, 186, 1397–1403. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, J.; Lv, X.; Wen, J.; He, H. Solvent-free synthesis of tertiary α-hydroxyphosphates by the triethylamine-catalyzed hydrophosphonylation of ketones. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 1334–1339. [Google Scholar] [CrossRef]
- Tajbakhsh, M.; Samad, K.; Zahra, T.; Ahmadreza, B. MgCl2/Et3N base system as a new catalyst for the synthesis of α-hydroxyphosphonate. Chin. J. Chem. 2012, 30, 827–829. [Google Scholar] [CrossRef]
- Kumari, S.; Shekhar, A.; Pathak, D.D. A new catalyst and solvent-free green synthesis of α-hydroxy phosphonates and α-aminophosphonates. Chem. Sci. Trans. 2014, 3, 45–54. [Google Scholar]
- Keglevich, G.; Tóth, V.R.; Drahos, L. Microwave-assisted synthesis of α-hydroxy-benzylphosphonates and –benzylphosphine oxides. Heteroatom Chem. 2011, 22, 15–17. [Google Scholar] [CrossRef]
- Kong, D.L.; Liu, R.D.; Li, G.Z.; Zhang, P.W.; Wu, M.S. A rapid, convenient, solventless green approach for the synthesis of α-hydroxyphosphonates by grinding. Asian J. Chem. 2014, 26, 1246–1248. [Google Scholar]
- Kumar, K.S.; Reddy, C.B.; Reddy, M.V.N.; Rani, C.R.; Reddy, C.S. Green chemical synthesis of α-hydroxyphosphonates. Org. Commun. 2012, 5, 50–57. [Google Scholar]
- Kalla, R.M.N.; Zhang, Y.; Kim, I. Highly efficient green synthesis of α-hydroxyphosphonates using a recyclable choline hydroxide catalyst. New J. Chem. 2017, 41, 5373–5379. [Google Scholar] [CrossRef]
- Ramananarivo, H.R.; Solhy, A.; Sebti, J.; Smahi, A.; Zahouily, M.; Clark, J.; Sebti, S. An eco-friendly paradigm for the synthesis of α-hydroxyphosphonates using sodium-modified fluorapatite under solventless conditions. ACS Sustain. Chem. Eng. 2013, 1, 403–409. [Google Scholar] [CrossRef]
- Rao, K.U.M.; Sundar, C.S.; Prasad, S.S.; Rani, C.R.; Reddy, C.S. Neat synthesis and anti-oxidant activity of α-hydroxyphosphonates. Bull. Korean Chem. Soc. 2011, 32, 3343–3347. [Google Scholar] [CrossRef]
- Santhisudha, S.; Sreelakshimi, P.; Jayaprakash, S.H.; Kumar, B.V.; Reddy, C.S. Silica-supported tungstic acid catalyzed synthesis and antioxidant activity of α-hydroxyphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 1479–1488. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Y.; Qian, Q.; Yuan, D.; Yao, Y. n-BuLi as a highly efficient precatalyst for hydrophosphonylation of aldehydes and unactivated ketones. Org. Lett. 2014, 16, 6172–6175. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Y.; Chang, L.; Zhao, J.; Shang, D.; Liu, X.; Lin, L.; Feng, X. Highly efficient synthesis of quaternary α-hydroxy phosphonates via Lewis acid-catalyzed hydrophosphonylation of ketones. Adv. Synth. Catal. 2009, 351, 2567–2572. [Google Scholar] [CrossRef]
- de Noronha, R.G.; Costa, P.J.; Romão, C.C.; Calhorda, M.J.; Fernandes, A.C. MoO2Cl2 as a novel catalyst for C–P bond formation and for hydrophosphonylation of aldehydes. Organometallics 2009, 28, 6206–6212. [Google Scholar] [CrossRef]
- Bouzina, A.; Aouf, N.E.; Berredjem, M. Ultrasound assisted green synthesis of α-hydroxyphosphonates under solvent-free conditions. Res. Chem. Intermed. 2016, 42, 5993–6002. [Google Scholar] [CrossRef]
- Mandhane, P.G.; Joshi, R.S.; Nagargoje, D.R.; Gill, C.H. Ultrasound-promoted greener approach to synthesize α-hydroxy phosphonates catalyzed by potassium dihydrogen phosphate under solvent-free condition. Tetrahedron Lett. 2010, 51, 1490–1492. [Google Scholar] [CrossRef]
- Sadaphal, A.S.; Sonar, S.S.; Pokalwar, R.U.; Shitole, N.V.; Shingare, M.S. Sulphamic acid: An efficient catalyst for the synthesis of α-hydroxy phosphonates using ultrasound irradiation. J. Korean Chem. Soc. 2009, 53, 536–541. [Google Scholar]
- Shinde, P.V.; Kategaonkar, A.H.; Shingate, B.B.; Shingare, M.S. An organocatalyzed facile and rapid access to α-hydroxy and α-amino phosphonates under conventional/ultrasound technique. Tetrahedron Lett. 2011, 52, 2889–2892. [Google Scholar] [CrossRef]
- Vahdat, S.M.; Baharfar, R.; Tajbakhsh, M.; Heydari, A.; Baghbanian, S.M.; Khaksar, S. Organocatalytic synthesis of α-hydroxy and α-aminophosphonates. Tetrahedron Lett. 2008, 49, 6501–6504. [Google Scholar] [CrossRef]
- Vangala, V.B.; Pati, H.N. Efficient synthesis of β-lactam containing α-hydroxy phosphonates using tartaric acid and fumaric acid as mild catalysts. Synth. Commun. 2016, 46, 374–378. [Google Scholar] [CrossRef]
- Jahani, F.; Zamenian, B.; Khaksar, S.; Tajbakhsh, M. Pyridine 2,6-dicarboxylic acid as a bifunctional organocatalyst for hydrophosphonylation of aldehydes and ketones in water. Synthesis 2010, 19, 3315–3318. [Google Scholar] [CrossRef]
- Heydari, A.; Arefi, A.; Khaksar, S.; Tajbakhsh, M. Hydrophosphonylation of aldehydes catalyzed by guanidine hydrochloride in water. Catal. Commun. 2006, 7, 982–984. [Google Scholar] [CrossRef]
- Wang, H.S.; Zeng, J.E. Iodine-catalyzed, efficient synthesis of α-hydroxy phosphonates in water. Phosphorus Sulfur Silicon Relat. Elem. 2010, 185, 1425–1428. [Google Scholar] [CrossRef]
- Ramesh, K.; Madhav, B.; Murthy, S.N.; Nageswar, Y.V.D. Aqueous-phase synthesis of α-hydroxyphosphonates catalyzed by β-cyclodextrin. Synth. Commun. 2012, 42, 258–265. [Google Scholar] [CrossRef]
- Sonar, S.S.; Kategaonkar, A.H.; Ware, M.N.; Gill, C.H.; Shingate, B.B.; Shingare, M.S. Ammonium metavanadate: An effective catalyst for synthesis of α-hydroxyphosphonates. Arkivoc 2009, 2, 138–148. [Google Scholar]
- Raju, P.; Rajeshwaran, G.G.; Nandakumar, M.; Mohanakrishnan, A.K. Unusual reactivity of aryl aldehydes with triethyl phosphite and zinc bromide: A facile preparation of epoxides, benzisoxazoles, and α-hydroxy phosphonate esters. Eur. J. Org. Chem. 2015, 2015, 3513–3523. [Google Scholar] [CrossRef]
- Kumar, A.; Jamwal, S.; Khan, S.; Singh, N.; Rai, V.K. Bi(NO3)3·5H2O catalyzed phosphonylation of aldehydes: An efficient route to α-hydroxyphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2016, 192, 381–385. [Google Scholar] [CrossRef]
- Thottempudi, V.; Chung, K.H. Niobium(V) chloride catalyzed Abramov reaction: An efficient protocol for the preparation of α-hydroxy phosphonates. Bull. Korean Chem. Soc. 2008, 29, 1781–1783. [Google Scholar] [CrossRef]
- Keglevich, G.; Rádai, Z.; Kiss, N.Z. To date the greenest method for the preparation of α-hydroxyphosphonates from substituted benzaldehydes and dialkyl phosphites. Green Process Synth. 2017, 6, 197–201. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Rádai, Z.; Mucsi, Z.; Keglevich, G. Synthesis of α-aminophosphonates from α-hydroxyphosphonates; a theoretical study. Heteroat. Chem. 2016, 27, 260–268. [Google Scholar] [CrossRef]
- Lorenz, W.; Henglein, A.; Schrader, G. The new insecticide O,O-dimethyl 2,2,2-trichloro-1-hydroxyethylphosphonate. J. Am. Chem. Soc. 1955, 77, 2554–2556. [Google Scholar] [CrossRef]
- Song, H.; Mao, H.; Shi, D. Synthesis and herbicidal activity of α-hydroxy phosphonate derivatives containing pyrimidine moiety. Chin. J. Chem. 2010, 28, 2020–2024. [Google Scholar] [CrossRef]
- Pokalwar, R.U.; Hangarge, R.V.; Maske, P.V.; Shingare, M.S. Synthesis and antibacterial activities of α-hydroxyphosphonates and α-acetyloxyphosphonates derived from 2-chloroquinoline-3-carbaldehyde. Arkivoc 2006, 11, 196–204. [Google Scholar]
- Kategaonkar, A.H.; Pokalwar, R.U.; Sonar, S.S.; Gawali, V.U.; Shingate, B.B.; Shingare, M.S. Synthesis, in vitro antibacterial and antifungal evaluations of new α-hydroxyphosphonate and new α-acetoxyphosphonate derivatives of tetrazolo [1, 5-a] quinolone. Eur. J. Med. Chem. 2010, 45, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, S.; Tashrifi, Z. Synthesis of α-functionalized phosphonates from α-hydroxyphosphonates. Tetrahedron 2010, 66, 1429–1439. [Google Scholar] [CrossRef]
- Brücher, K.; Illarionov, B.; Held, J.; Tschan, S.; Kunfermann, A.; Pein, M.K.; Bacher, A.; Gräwert, T.; Maes, L.; Mordmüller, B.; et al. α-Substituted β-oxa isosteres of fosmidomycin: Synthesis and biological evaluation. J. Med. Chem. 2012, 55, 6566–6575. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Yang, F.; Guo, D.; Xu, J.; Jiang, M.; Liu, C.; Bao, K.; Wu, Y.; Zhang, W. Synthesis and biological evaluation of novel 3,4-diaryl-1,2,5-selenadiazol analogues of combretastatin A-4. Eur. J. Med. Chem. 2014, 87, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sekine, M.; Nakajima, M.; Kume, A.; Hashizume, A.; Hata, T. Silyl phosphites. XVIII. Versatile utility of α-(trimethylsilyloxy)-alkylphosphonates as key intermediates for transformation of aldehydes into several carbonyl derivatives. Bull. Chem. Soc. Jpn. 1982, 55, 224–238. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Sobhani, S. A high yielding preparation of α-trimethylsilyloxyphosphonates by silylation of α-hydroxyphosphonates with HMDS catalyzed by iodine. Tetrahedron Lett. 2002, 43, 3653–3655. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Sobhani, S.; Ghassamipour, S.; Amoozgar, Z. Copper triflate [Cu(OTf)2] is an efficient and mild catalyst for the silylation of α-hydroxyphosphonates to α-trimethylsilyloxyphosphonates with HMDS at room temperature. Tetrahedron Lett. 2003, 44, 891–893. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Jafari, A.A.; Jafari, M.R. Iron(III) trifluoroacetate [Fe(F3CCO2)3] as an easily available, non-hygroscopic, non-corrosive, highly stable and a reusable Lewis acis catalyst: Efficient O-silylation of α-hydroxyphosphonates, alcohols and phenols by hexamethyldisilazane (HMDS) under solvent-free conditions. J. Organomet. Chem. 2008, 693, 2711–2714. [Google Scholar]
- Wang, F.; Miao, Z.; Chen, R. Efficient syntheses of phosphonylated isochromenes by regioselective 6-endo-dig addition to carbon-carbon triple bond catalyzed by Pd(OAc)2. Org. Biomol. Chem. 2009, 7, 2848–2850. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.B.; Shi, D.Q. Synthesis and biological activity of novel phosphonate derivatives containing of pyridyl and 1,2,3-triazole rings. Phosphorus Sulfur Silicon Relat. Elem. 2008, 183, 1134–1144. [Google Scholar] [CrossRef]
- Chen, T.; Shen, P.; Li, Y.; He, H. Synthesis and herbicidal activity of O,O-dialkyl phenoxyacetoxyalkylphosphonates containing fluorine. J. Fluorine Chem. 2006, 127, 291–295. [Google Scholar] [CrossRef]
- Peng, H.; Wang, T.; Xie, P.; Chen, T.; He, H.W.; Wan, J. Molecular docking and three-dimensional quantitative structure-activity relationship studied on the binding modes of herbicidal 1-(substituted phenoxyacetoxy)alkylphosphonates to the E1 component of pyruvate dehydrogenase. J. Agric. Food Chem. 2007, 55, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.P.; Ning, B.K.; Mao, M.Z.; Xue, C.; Wang, H.Y. Synthesis and insecticidal activities of O,O-dialkyl-2-[3-bromo-1-(3-chloropyridin-2-yl)-1H-pyrazole-5-carbonyloxy] (aryl) methylphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1362–1367. [Google Scholar] [CrossRef]
- Fang, H.; Wang, D.; Chen, W.; Zhao, Y.; Fang, M. Synthesis, crystal structure and fragmentation pathway of arylcarboxy ester of α-hydroxyphosphonate. Chin. J. Org. Chem. 2010, 30, 1377–1382. [Google Scholar]
- Hudson, H.R.; Jászay, Z.M.; Pianka, M. The preparation and properties of some α-acyloxy- and α-carbamoyloxy-phosphonothionates. Phosphorus Sulfur Silicon Relat. Elem. 2003, 178, 1571–1582. [Google Scholar] [CrossRef]
- Shi, D.Q.; Li, X.J.; Wei, J. 5-Fluorouracil derivatives containing α-hydroxy phosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 405–412. [Google Scholar] [CrossRef]
- Peng, H.; Long, Q.; Deng, X.; He, H. Synthesis and herbicidal activities of lithium or potassium hydrogen 1-(substituted phenoxyacetoxy)alkylphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 1868–1874. [Google Scholar] [CrossRef]
- Long, Q.; Deng, X.; Gao, Y.; Xie, H.; Peng, H.; He, H. Synthesis and herbicidal activities of sodium hydrogen 1-(substituted phenoxyacetoxy)alkylphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 819–825. [Google Scholar] [CrossRef]
- He, H.W.; Yuan, J.L.; Peng, H.; Chen, T.; Shen, P.; Wan, S.Q.; Li, Y.; Tan, H.L.; He, Y.H.; He, J.B.; et al. Studies of O,O-dimethyl α-(2,4-dichlorophenoxyacetoxy)-ethylphosphonate (HW02) as a new herbicide. 1. Synthesis and herbicidal activity of HW02 and analogues as novel inhibitors of pyruvate dehydrogenase complex. J. Agric. Food Chem. 2011, 59, 4801–4813. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Joshi, R.; Li, M.; He, H. Synthesis and biological activity of novel 1-(substituted phenoxyacetoxy) alkyl phosphonates and phosphinates. J. Nepal Chem. Soc. 2009, 23, 11–20. [Google Scholar]
- Soung, M.G.; Hwang, T.Y.; Sung, N.D. Synthesis and 3D-QSARs analyses of herbicidal O,O-dialkyl-1-phenoxy-1-acetoxy-1-methylphosphonate analogues as a new class of potent inhibitors of pyruvate dehydrogenase. Bull. Korean Chem. Soc. 2010, 31, 1361–1367. [Google Scholar] [CrossRef]
- Wang, T.; He, H.W. An efficient synthesis of α-(2,4-dichloro-phenoxyacetoxy)aryl methyl phosphonate monosodium salts. Synth. Commun. 2004, 34, 1415–1423. [Google Scholar] [CrossRef]
- Wang, T.; He, H.W. Simple and improved preparation of α-oxophosphonate monolithium salts. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 2081–2089. [Google Scholar] [CrossRef]
- Wang, W.; He, H.W.; Zuo, N.; Zhang, X.; Lin, J.S.; Chen, W.; Peng, H. Synthesis and herbicidal activity of 2-(substituted phenoxyacetoxy)alkyl-5,5-dimethyl-1,3,2-dioxaphosphinan-2-one containing fluorine. J. Fluorine Chem. 2012, 142, 24–28. [Google Scholar] [CrossRef]
- Wang, W.; He, H.W.; Zuo, N.; He, H.F.; Peng, H.; Tan, X.S. Synthesis and herbicidal activity of 2-(substituted phenoxyacetoxy)alkyl-5,5-dimethyl-1,3,2-dioxaphosphinan-2-one. J. Agric. Food Chem. 2012, 60, 7581–7587. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lei, D.Y.; Huang, Y.; Ao, L.H. Synthesis and biological activity of O,O-dimethyl-2,6-pyridinyl diformyloxy alkyl phosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 2777–2785. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Sobhani, S.; Amoozgar, Z. Copper triflate as a useful catalyst for the high-yielding preparation of α-acetyloxyphosphonates under solvent-free conditions. Synthesis 2004, 2, 295–297. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Farahi, S. Solid trichlorotitanium(IV) trifluoromethanesulfonate TiCl3(OTf) catalyzed efficient acylation of –OH and –SH: Direct esterification of alcohols with carboxylic acids and transesterification of alcohols with esters under neat conditions. J. Mol. Catal. A: Chem. 2008, 289, 61–68. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Sobhani, S.; Amoozgar, Z. Facile and high-yielding preparation of α-hydroxyphosphonates assisted by microwave irradiation. Synthesis 2004, 11, 1771–1774. [Google Scholar] [CrossRef]
- Rostami, A.; Atashkar, B.; Moradi, D. Synthesis, characterization and catalytic properties of magnetic nanoparticle supported guanidine in base catalyzed synthesis of α-hydroxyphosphonates and α-acetoxyphosphonates. Appl. Catal. A 2013, 467, 7–16. [Google Scholar] [CrossRef]
- Green, D.; Elgendy, S.; Patel, G.; Skordalakes, E.; Goodwin, C.A.; Scully, M.F.; Kakkar, V.V.; Deadman, J.J. Substrate related O,O-dialkyldipeptidyly ψ carboxybenzylphosphonates, a new type of thrombin inhibitor. Phosphorus Sulfur Silicon Relat. Elem. 2000, 156, 151–155. [Google Scholar] [CrossRef]
- Yang, J.; Ma, J.; Che, W.; Li, M.; Li, G.; Song, B. Microwave-assisted synthesis and antitumor activity of salicyl acyloxy phosphonate derivatives. Chin. J. Org. Chem. 2014, 34, 2566–2571. [Google Scholar] [CrossRef]
- Iranpoor, N.; Firouzabadi, H.; Khalili, D. The first Mitsunobu protocol for efficient synthesis of α-acyloxyphosphonates using 4,4′-azopyridine. Phosphorus Sulfur Silicon Relat. Elem. 2011, 186, 2166–2171. [Google Scholar] [CrossRef]
- Xu, L.; You, G.; Peng, H.; He, H. Synthesis and biological activities of O,O-dialkyl 1-((4,6-dichloropyrimidin-2-yl)carbamyloxy) alkylphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2014, 189, 812–818. [Google Scholar] [CrossRef]
- Li, J.P.; Zhu, J.G.; Liu, R.J.; Cui, F.L.; Liu, P.; Liu, G.S. Straightforward synthesis of a new series of α-(arylamino thiocarbonyloxy) hydrocarbylphosphonates. S. Afr. J. Chem. 2008, 61, 5–8. [Google Scholar]
- Li, Z.G.; Sun, H.K.; Wang, Q.M.; Huang, R.Q. An α-hydrazinoalkylphosphonate as building block for novel N-phosphonoalkylheterocycles. Heteroatom Chem. 2003, 14, 384–386. [Google Scholar] [CrossRef]
- Creary, X.; Geiger, C.C.; Hilton, K. Mesylate derivatives of α-hydroxy phosphonates. Formation of carbocations adjacent to the diethyl phosphonate group. J. Am. Chem. Soc. 1983, 105, 2851–2858. [Google Scholar] [CrossRef]
- Kong, D.L.; Li, G.Z.; Liu, R.D. Synthesis and crystal structure of diethyl tosyloxybenzylphosphonate. Asian J. Chem. 2014, 26, 2138–2140. [Google Scholar]
- Davidson, R.S.; Sheldon, R.A.; Trippett, S. The reaction of tetraphenyldiphosphine with aromatic carboxylic acids. J. Chem. Soc. 1967, 1547–1552. [Google Scholar] [CrossRef]
- Rádai, Z.; Hodula, V.; Kiss, N.Z.; Kegelevich, G. Unpublished results.
- Samanta, S.; Zhao, C.G. Organocatalyzed nitroaldol reaction of α-ketophosphonates and nitromethane revisited. Arkivoc 2007, 8, 218–226. [Google Scholar]
- Guang, J.; Zhao, C.G. Organocatalyzed asymmetric Michael reaction of β-aryl-α-ketophosphonates and nitroalkenes. Tetrahedron Lett. 2013, 54, 5703–5706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telan, L.A.; Poon, C.D.; Evans, S.A. Diastereoselectivity in the Mukaiyama–Michael reaction employing α-acyl β,γ-unsaturated phosphonates. J. Org. Chem. 1996, 61, 7455–7462. [Google Scholar] [CrossRef] [PubMed]
- Kaboudin, B. Surface-mediated solid-phase reactions: The preparation of acyl phosphonates by oxidation of 1-hydroxyphosphonates on the solid surface. Tetrahedron Lett. 2000, 41, 3169–3171. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Sobhani, S.; Sardarian, A.R. High yields preparation of α-ketophosphonates by oxidation of α-hydroxyphosphonates with zinc dichromate trihydrate (ZnCr2O7·3H2O) under solvent-free conditions. Tetrahedron Lett. 2001, 42, 4369–4371. [Google Scholar] [CrossRef]
- Guliaiko, I.; Nesterov, V.; Sheiko, S.; Kolodiazhnyi, O.I.; Freytag, M.; Jones, P.G.; Schmutzler, R. Synthesis of optically active hydroxyphosphonates. Heteroatom Chem. 2008, 19, 133–139. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Sobhani, S. Preparation of α-ketophosphonates by oxidation of α-hydroxyphosphonates with pyridinium chlorochromate (PCC). Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 1483–1491. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Sobhani, S. Preparation of α-ketophosphonates by oxidation of α-hydroxyphosphonates with neutral alumina supported potassium permanganate (NASPP) under solvent-free conditions and potassium permanganate in dry benzene. Tetrahedron Lett. 2002, 43, 477–480. [Google Scholar] [CrossRef]
- Pawar, V.D.; Bettigeri, S.; Weng, S.S.; Kao, J.Q.; Chen, C.T. Highly enantioselective aerobic oxidation of α-hydroxyphosphonates catalyzed by chiral vanadyl(V) methoxides bearing N-salicylidene-α-aminocarboxylates. J. Am. Chem. Soc. 2006, 128, 6308–6309. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.P.; Zhang, Z.Y.; Widlanski, T.S. Quiescent affinity inactivators of protein tyrosine phosphatases. Bioorg. Med. Chem. 1996, 4, 1515–1520. [Google Scholar] [CrossRef]
- Tulsi, N.S.; Downey, A.M.; Cairo, C.W. A protected l-bromophosphonomethylphenylalanine amino acid derivative (BrPmp) for synthesis of irreversible protein tyrosine phosphatase inhibitors. Bioorg. Med. Chem. 2010, 18, 8679–8686. [Google Scholar] [CrossRef] [PubMed]
- Caplan, N.A.; Pogson, C.I.; Hayes, D.J.; Blackburn, G.M. The synthesis of novel bisphosphonates as inhibitors of phosphoglycerate kinase (3-PGK). J. Chem. Soc. Perkin Trans. 1 2000, 421–437. [Google Scholar] [CrossRef]
- Gajda, T. Preparation of diethyl 1-bromoalkylphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 1990, 53, 327–331. [Google Scholar] [CrossRef]
- Guan, Z.; Wu, D.; Fu, J.P.; He, Y.H. A facile and efficient synthesis of diethyl α,α-chlorofluoroalkanephosphonates. Heteroatom Chem. 2010, 21, 250–255. [Google Scholar] [CrossRef]
- Wu, D.; He, Y.; Tang, R.; Guan, Z. The first synthesis of diethyl α,α-chlorofluorobenzylphosphonates. Synlett 2009, 13, 2180–2182. [Google Scholar]
- Firouzabadi, H.; Iranpoor, N.; Sobhani, S. PPh3/DDQ as a neutral system for the facile preparation of diethyl α-bromo, α-iodo and α-azidophosphonates from diethyl α-hydroxyphosphonates. Tetrahedron 2004, 60, 203–210. [Google Scholar] [CrossRef]
- Fu, J.P.; He, Y.H.; Zhong, J.; Yang, Y.; Deng, X.; Guan, Z. An efficient and general route to the synthesis of diethyl α,α-bromofluorophosphonates. J. Fluorine Chem. 2011, 132, 636–640. [Google Scholar] [CrossRef]
- Iranpoor, N.; Firouzabadi, H.; Gholinejad, M. 4-Aminophenyldiphenylphosphinite (APDPP), a new heterogeneous and acid scavenger phosphinite—Conversion of alcohols, trimethylsilyl, and tetrahydropyranyl ethers to alkyl halides with halogens or N-halosuccinimides. Can. J. Chem. 2006, 84, 1006–1012. [Google Scholar] [CrossRef]
- Allmendinger, T.; Fujimoto, R.; Gasparini, F.; Schilling, W.; Satoh, Y. α-Fluoro-benzylphosphonates as reagents for the preparation of 1-fluoro-1-aryl alkenes and α-fluorostilbenes. Chimia 2004, 58, 133–137. [Google Scholar] [CrossRef]
- Blackburn, G.M.; Kent, D.E. A novel synthesis of α- and γ-fluoroalkylphosphonates. J. Chem. Soc. Chem. Commun. 1981, 511–513. [Google Scholar] [CrossRef]
- Blackburn, G.M.; Kent, D.E. Synthesis of α- and γ-fluoroalkylphosphonates. J. Chem. Soc. Perkin Trans. 1986, 913–917. [Google Scholar] [CrossRef]
- Yokomatsu, T.; Yamagishi, T.; Matsumoto, K.; Shibuya, S. Stereocontrolled synthesis of hydroxymethylene phosphonate analogues of phosphorylated tyrosine and their conversion to monofluoromethylene phosphonate analogues. Tetrahedron 1996, 52, 11725–11738. [Google Scholar] [CrossRef]
- Guzyr, O.I.; Zasukha, S.V.; Vlasenko, Y.G.; Chernega, A.N.; Rozhenko, A.B.; Shermolovich, Y.G. Simple route to adducts of (amino)(aryl)carbene with phosphorus pentafluoride. Eur. J. Inorg. Chem. 2013, 2013, 4154–4158. [Google Scholar] [CrossRef]
- Psurski, M.; Błażewska, K.; Gajda, A.; Gajda, T.; Wietrzyk, J.; Oleksyszyn, J. Synthesis and antiproliferative activity of novel α- and β-dialkoxyphosphoryl isothiocyanates. Bioorg. Med. Chem. Lett. 2011, 21, 4572–4576. [Google Scholar] [CrossRef] [PubMed]
- Keglevich, G.; Bálint, E. The Kabachnik–Fields reaction: Mechanism and synthetic use. Molecules 2012, 17, 12821–12835. [Google Scholar] [CrossRef] [PubMed]
- Bálint, E.; Fazekas, E.; Tripolszky, A.; Tajti, Á.; Kangyal, R.; Milen, M.; Keglevich, G. Synthesis of aminophosphonate derivatives by microwave-assisted Kabachnik–Fields reaction. Organomet. Chem. 2012, 717, 655–659. [Google Scholar] [CrossRef]
- Zefirov, N.S.; Matveeva, E.D. Catalytic Kabachnik–Fields reaction: New horizons for old reaction. Arkivoc 2008, 1, 1–17. [Google Scholar]
- Kafarski, P.; Gorny vel Gorniak, M.; Andrasiak, I. Kabachnik–Fields reaction under green conditions—A critical overview. Curr. Green Chem. 2015, 2, 218–222. [Google Scholar] [CrossRef]
- Kaboudin, B. A convenient synthesis of 1-aminophosphonates from 1-hydroxyphosphonates. Tetrahedron Lett. 2003, 44, 1051–1053. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Kaszás, A.; Drahos, L.; Mucsi, Z.; Keglevich, G. A neighbouring group effect leading to enhanced nucleophilic substitution of amines at the hindered α-carbon atom of an α-hydroxyphosphonate. Tetrahedron Lett. 2012, 53, 207–209. [Google Scholar] [CrossRef]
- Pallikonda, G.; Chakravarty, M. Triflic acid mediated functionalization of α-hydroxyphosphonates: Route for sulfonamide phosphates. RSC Adv. 2013, 3, 20503–20511. [Google Scholar] [CrossRef]
- Verma, C.; Singh, A.; Pallikonda, G.; Chakravarty, M.; Quraishi, M.A.; Bahadur, I.; Ebenso, E.E. Aryl sulfonamidomethylphosphonates as new class of green corrosion inhibitors for mild steel in 1 M HCl: Electrochemical, surface and quantum chemical investigation. J. Mol. Liq. 2015, 209, 306–319. [Google Scholar] [CrossRef]
- Chen, L.; Zou, Y.X.; Fang, X.Y.; Yin, X. Convenient synthesis of α-diarylmethylphosphonates by HOTf catalyzed Friedel–Crafts arylation of α-aryl α-hydroxyphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2018, 193, 168–177. [Google Scholar] [CrossRef]
- Khalid, M.B.Z.; Pallikonda, G.; Tulichala, R.N.P.; Chakravarty, M. Oxy-Wittig reactions of 1-naphthyl(aryl)methylphosphonates: A new approach to naphthylarylketones. Tetrahedron 2016, 72, 2094–2101. [Google Scholar] [CrossRef]
- Pallikonda, G.; Chakravarty, M. FeCl3-mediated arylation of α-hydroxyphosphonates with unactivated arenes: Pseudo-umpolung in allylic phosphonates. Eur. J. Org. Chem. 2013, 2013, 944–951. [Google Scholar] [CrossRef]
- Pallikonda, G.; Chakravarty, M.; Sahoo, M. An easy access to α-aryl substituted γ-ketophosphonates: Lewis acid mediated reactions of 1,3-diketones with α-hydroxyphosphonates and tandem regioselective C–C bond cleavage. Org. Biomol. Chem. 2014, 12, 7140–7149. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.A.R.; Stephens, C.W.; Drysdale, J.J. A rearrangement to from diethyl 1-cyanoethyl phosphate. J. Am. Chem. Soc. 1957, 79, 1768–1769. [Google Scholar] [CrossRef]
- Yoshino, K.; Kohno, T.; Morita, T.; Tsukamoto, G. Organic phosphorus compounds. 2. Synthesis and coronary vasodilator activity of (benzothiazolylbenzyl)phosphonate derivatives. J. Med. Chem. 1989, 32, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- McGeary, R.P.; Vella, P.; Mak, J.Y.W.; Guddat, L.W.; Schenk, G. Inhibition of purple acid phosphatase with α-alkoxynaphthylmethylphosphonic acids. Bioorg. Med. Chem. Lett. 2009, 19, 163–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumaraswamy, S.; Selvi, R.S.; Swamy, K.C.K. Synthesis of new α-hydroxy-, α-halogeno- and vinylphosphonates derived from 5,5-dimethyl-1,3,2-dioxaphosphinan-2-one. Synthesis 1997, 1997, 207–212. [Google Scholar] [CrossRef]
- Jankowski, S.; Marczak, J.; Olczak, A.; Główka, M.L. Stereochemistry of 1-hydroxyphosphonate–phosphate rearrangement. Retention of configuration at the phosphorus atom. Tetrahedron Lett. 2006, 47, 3341–3344. [Google Scholar] [CrossRef]
- Pallitsch, K.; Roller, A.; Hammerschmidt, F. The stereochemical course of the α-hydroxyphosphonate–phosphate rearrangement. Chem. Eur. J. 2015, 21, 10200–10206. [Google Scholar] [CrossRef] [PubMed]
- Pallikonda, G.; Santosh, R.; Ghosal, S.; Chakravarty, M. BuLi-triggered phospha-Brook rearrangement: Efficient synthesis of organophosphates from ketones and aldehydes. Tetrahedron Lett. 2015, 56, 3796–3798. [Google Scholar] [CrossRef]
- El Kaïm, L.; Gaultier, L.; Grimaud, L.; Santos, A.D. Formation of new phosphates from aldehydes by a DBU-catalysed phospha-brook rearrangement in a polar solvent. Synlett 2005, 15, 2335–2336. [Google Scholar] [CrossRef]
- Galeta, J.; Potáček, M. Applications of caged-designed proton sponges in base-catalyzed transformations. J. Mol. Catal. A: Chem. 2014, 395, 87–92. [Google Scholar] [CrossRef]
- Rádai, Z.; Szabó, R.; Kiss, N.Z.; Keglevich, G. Unpublished results.
- Baguley, T.D.; Xu, H.C.; Chatterjee, M.; Nairn, A.C.; Lombroso, P.J.; Ellman, J.A. Substrate-based fragment identification for the development of selective, nonpeptidic inhibitors of striatal-enriched protein tyrosine phosphatase. J. Med. Chem. 2013, 56, 7636–7650. [Google Scholar] [CrossRef] [PubMed]
- Beers, S.A.; Malloy, E.A.; Wu, W.; Wachter, M.P.; Gunnia, U.; Cavender, D.; Harris, C.; Davis, J.; Brosius, R.; Pellegrino-Gensey, J.L.; et al. Nitroarylhydroxymethylphosphonic acids as inhibitors of CD45. Bioorg. Med. Chem. 1997, 5, 2203–2211. [Google Scholar] [CrossRef]
- Burley, R.K.M.; Bearne, S.L. Inhibition of mandelate racemase by the substrate–intermediate–product analogue 1,1-diphenyl-1-hydroxymethylphosphonate. Bioorg. Med. Chem. 2005, 15, 4342–4344. [Google Scholar] [CrossRef] [PubMed]
- Caplan, N.A.; Pogson, C.I.; Hayes, D.J.; Blackburn, G.M. Novel bisphosphonate inhibitors of phosphoglycerate kinase. Bioorg. Med. Chem. Lett. 1998, 8, 515–520. [Google Scholar] [CrossRef]
- Forlani, G.; Occhipinti, A.; Berlicki, Ł.; Dziedzioła, G.; Wieczorek, A.; Kafarski, P. Tailoring the structure of aminobisphosphonates to target plant P5C reductase. J. Agric. Food Chem. 2008, 56, 3193–3199. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, A.; Berlicki, Ł.; Giberti, S.; Dziedzioła, G.; Kafarski, P.; Forlani, G. Effectiveness and mode of action of phosphonate inhibitors of plant glutamine synthetase. Pest. Manag. Sci. 2010, 66, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, J.; Wang, Y.; Wang, K.; Malwal, S.R.; Oldfield, E. Isoprenoid biosynthesis inhibitors targeting bacterial cell growth. Chem. Med. Chem. 2016, 11, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Madan, D.; Prestwich, G.D. Aromatic phosphonates inhibit the lysophospholipase D activity of autotaxin. Bioorg. Med. Chem. 2011, 21, 5098–5101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesterov, V.V.; Kolodiazhnyi, O.I. Efficient method for the asymmetric reduction of α- and β-ketophosphonates. Tetrahedron 2007, 63, 6720–6731. [Google Scholar] [CrossRef]
- Colton, I.J.; Yin, D.T.; Grochulski, P.; Kazlauskas, R.J. Molecular basis of chiral acid recognition by candida rugosa lipase: X-ray structure of transition state analog and modeling of the hydrolysis of methyl 2-methoxy-2-phenylacetate. Adv. Synth. Catal. 2011, 353, 2529–2544. [Google Scholar] [CrossRef]
Scheme 1.
Synthetic pathways towards α-hydroxyphosphonates 1.

Scheme 2.
A green, solvent-economical synthetic method towards α-hydroxyphosphonates (1).

Scheme 3.
Calculated mechanism of the triethylamine-catalyzed Pudovik reaction.

Scheme 4.
Family tree of compounds 7–12 derived from α-hydroxyphosphonates (1).

Scheme 5.
O-alkylation of hydroxyphosphonates 1B with benzyl bromoacetate.

Scheme 6.
Possible ways of introducing a silyl protecting group to α-hydroxyphosphonates 1B.

Scheme 7.
Selective cyclization of ortho-alkynyl α-hydroxyphosphonates 15.

Scheme 8.
Synthesis of α-acyloxyphosphonates (19) from α-hydroxyphosphonates (1).

Scheme 9.
Acylation of α-hydroxyphosphonates (1A) with 2,6-pyridinedicarboxylic acid chloride (20).

Scheme 10.
Possible ways for the synthesis of α-acetyloxyphosphonates 22.

Scheme 11.
Coupling reaction of α-hydroxyphosphonates (1) with carboxylic acid 23.

Scheme 12.
Synthesis of α-acyloxyphosphonates (26) via Mitsunobu reaction.

Scheme 13.
The reaction of α-hydroxyphosphonates (1) with isocyanates 27 and isothiocyanates 29.

Scheme 14.
Sulfonylation of α-hydroxyphosphonates 1B.

Scheme 15.
Synthesis of O-phosphoryloxy phosphonate 33 by rearrangement.

Scheme 16.
Phosphorylation of α-hydroxyphosphonates 1 with phosphoryl and phosphinic chlorides.

Scheme 17.
Oxidation of α-hydroxyphosphonates 1B using metal compounds with variable valency.

Scheme 18.
Synthesis of α-ketophosphonates 35 from α-hydroxyphosphonates 1B with KMnO4 oxidant.

Scheme 19.
Tandem resolution and oxidation of α-hydroxyphosphonates 1C.
Scheme 20.
Synthetic routes to obtain α-halophosphonates 38.
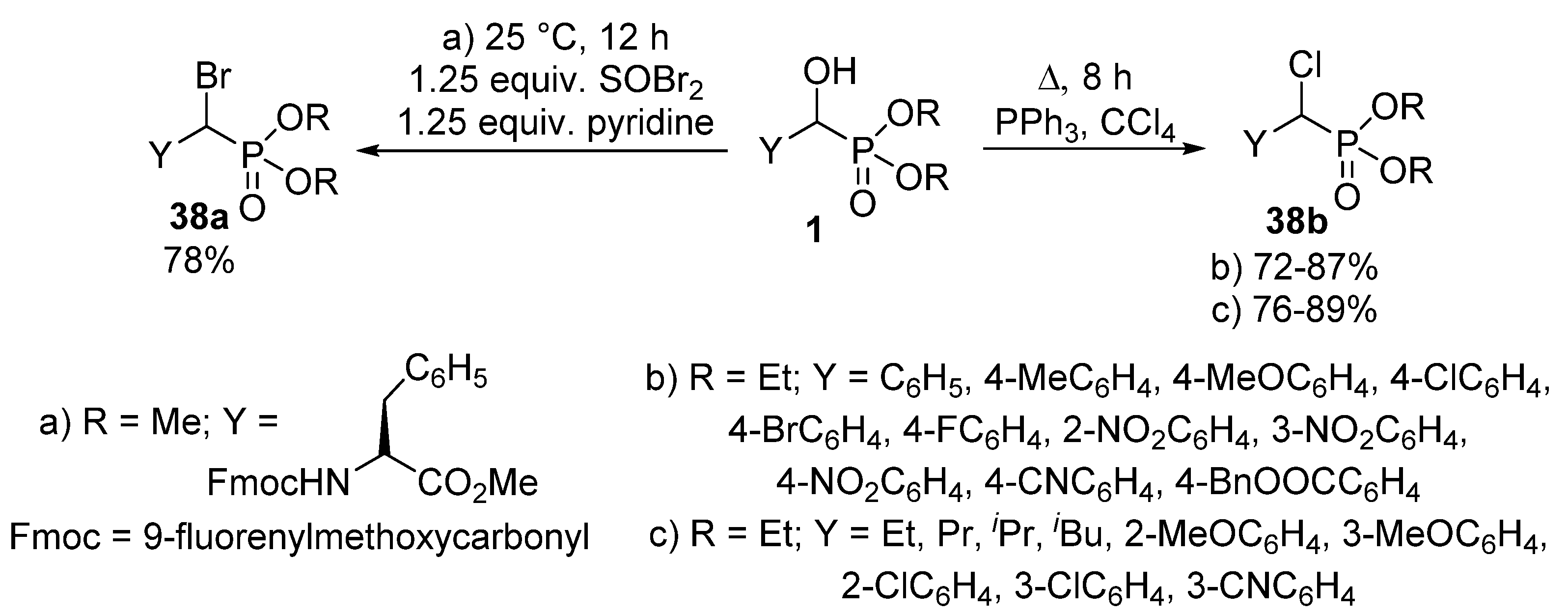
Scheme 21.
Synthesis of α-halophosphonates 38 from α-hydroxyphosphonates 1 with molecular halogens.

Scheme 22.
Nucleophilic substitution of α-hydroxyphosphonates 1B with NH4SCN and hydrazoic acid.

Scheme 23.
Synthesis of α-aminophosphonates 43 by nucleophilic substitution of 1B with amines.

Scheme 24.
MW-Assisted synthesis of α-aminophosphonates 44–46 through nucleophilic substitution.

Scheme 25.
Calculated mechanism of the substitution of hydroxyphosphonate 1Ba with methylamine.

Scheme 26.
Friedel-Crafts alkylation of α-hydroxyphosphonates 1 with arene nucleophiles.

Scheme 27.
Alkylation of 1,3-diketones with α-hydroxyphosphonates 1B.

Scheme 28.
Methods of the tandem Pudovik reaction and rearrangement affording benzyl phosphates 56.

Scheme 29.
Rearrangement of α-hydroxyphosphonates 1 under phase-transfer catalytic conditions.

Scheme 30.
Selective hydrolysis of one ester function of hydroxyphosphonate (R)-1Aa.


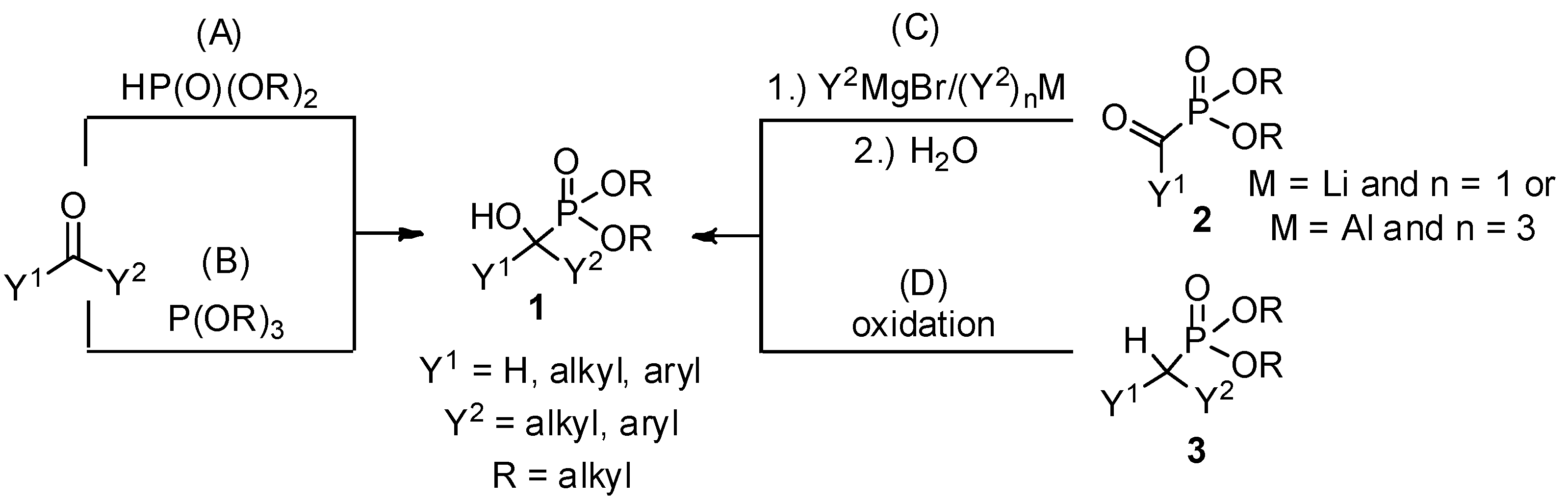{kind=link}
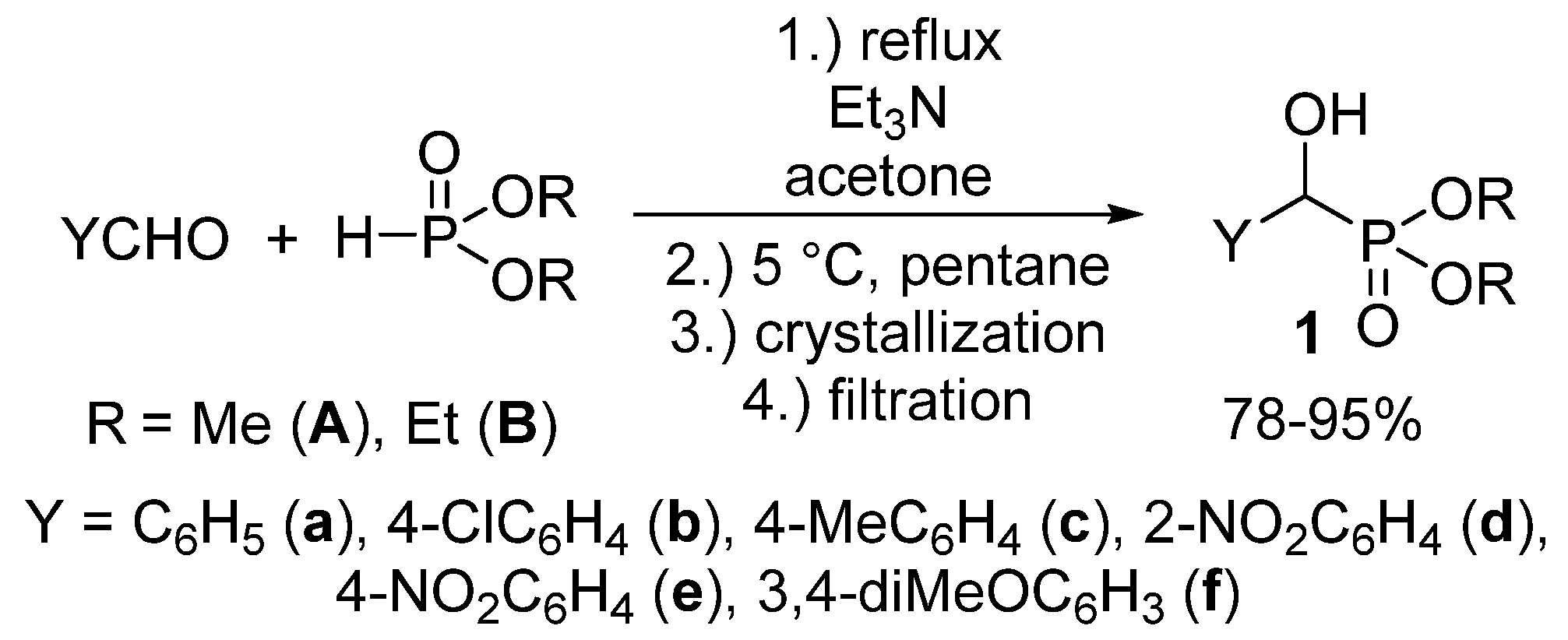{kind=link}
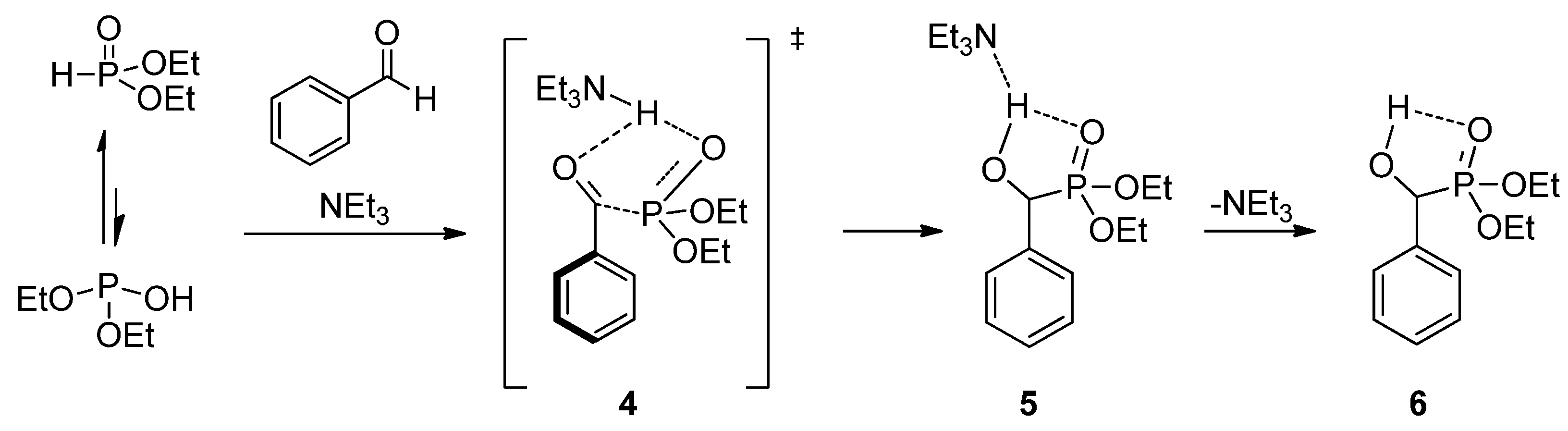{kind=link}
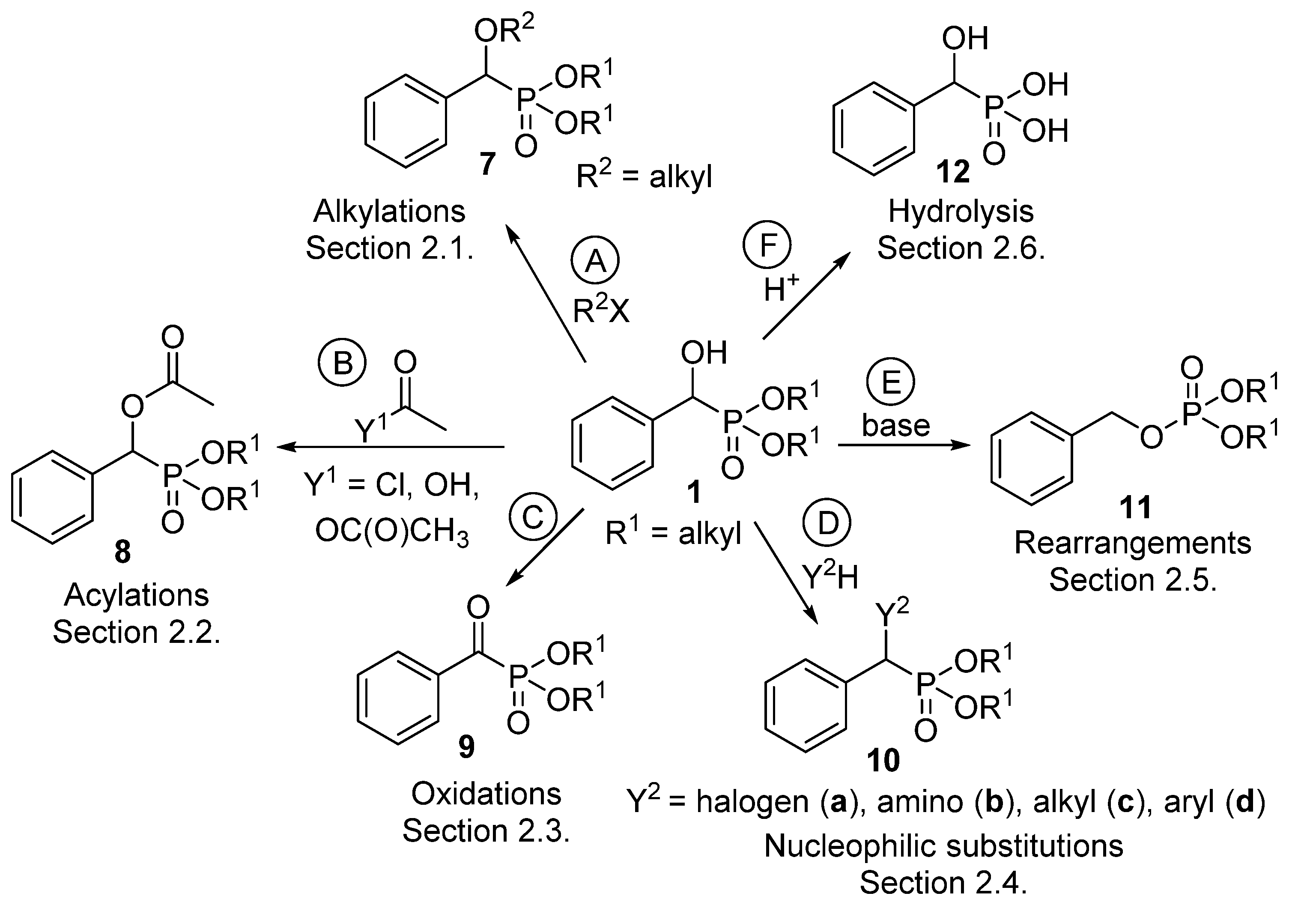{kind=link}
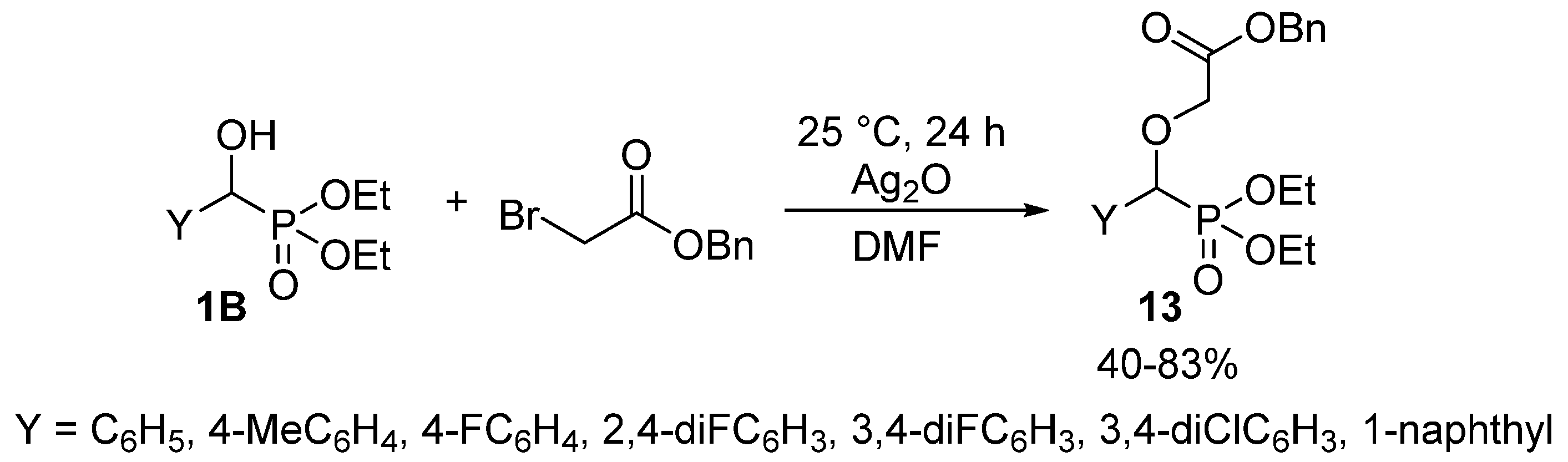{kind=link}
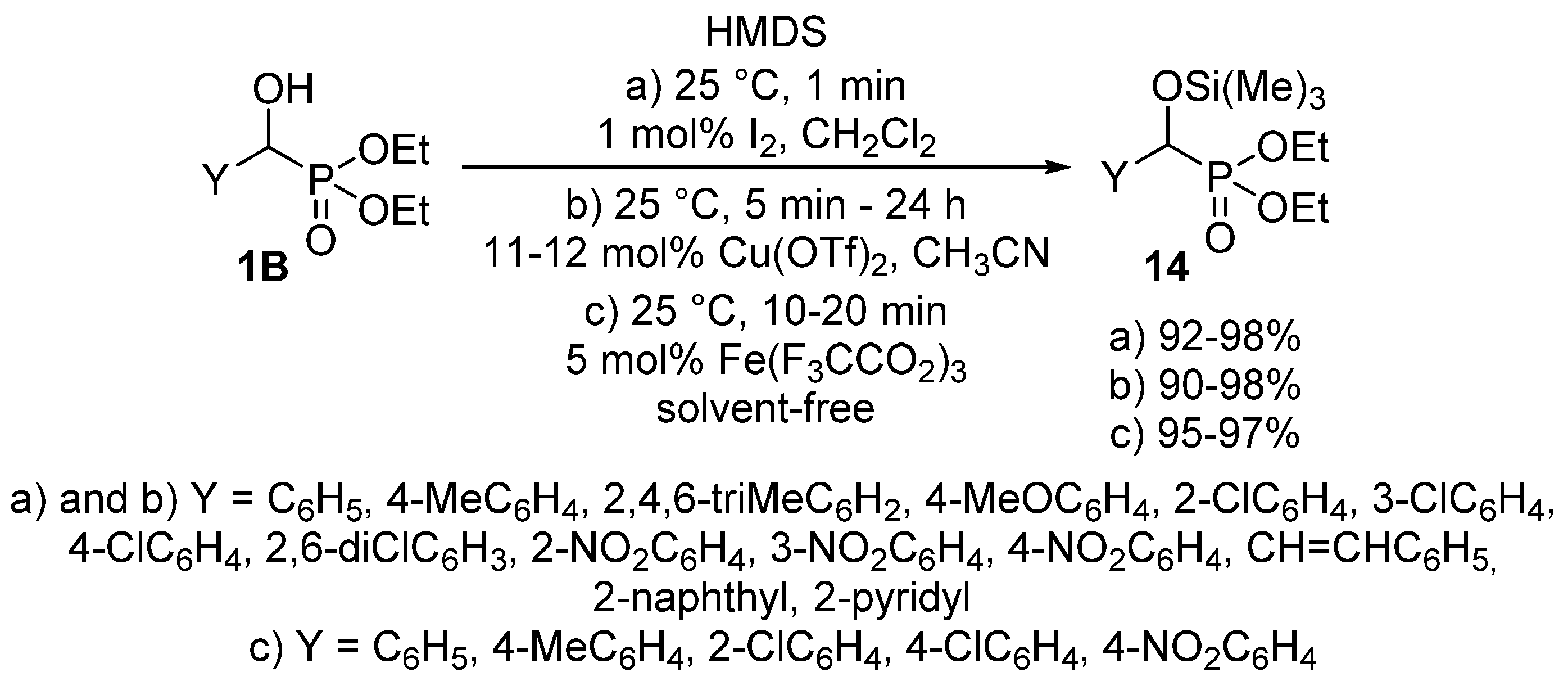{kind=link}
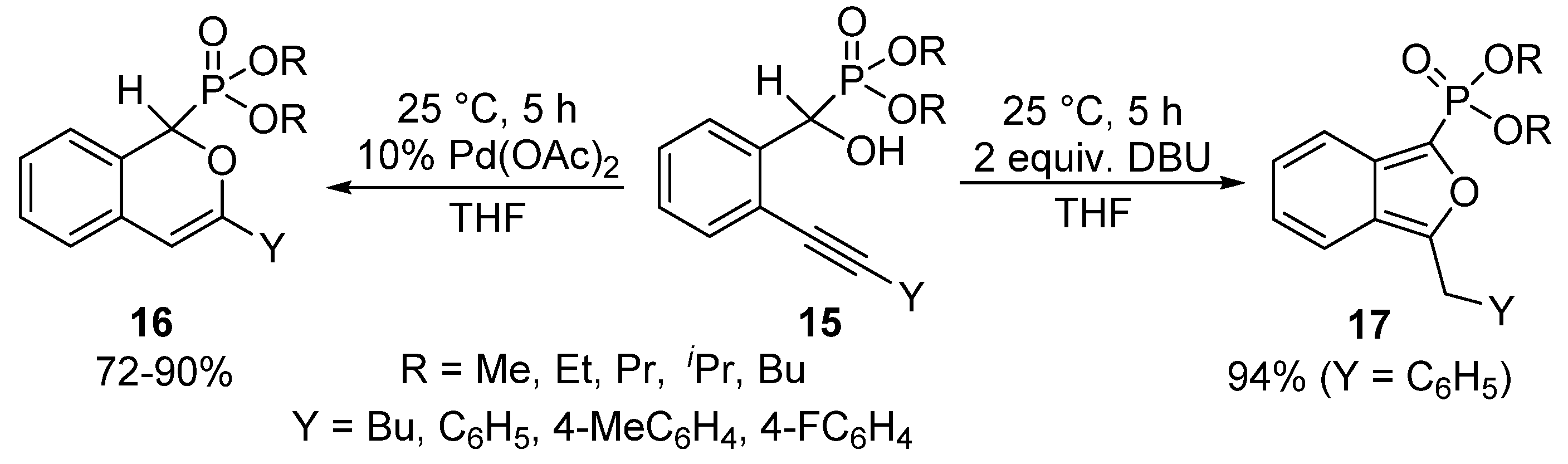{kind=link}
{kind=link}
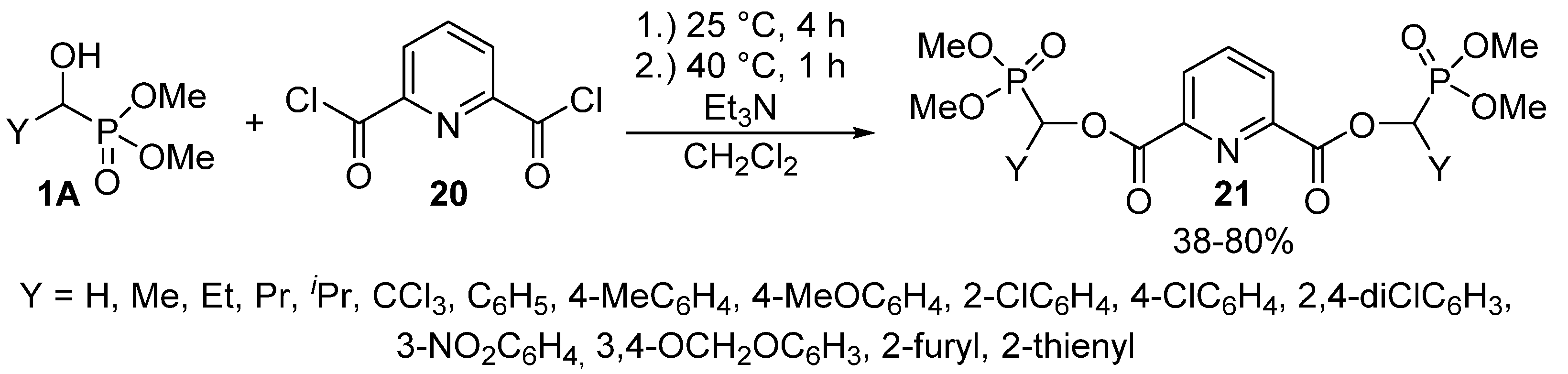{kind=link}
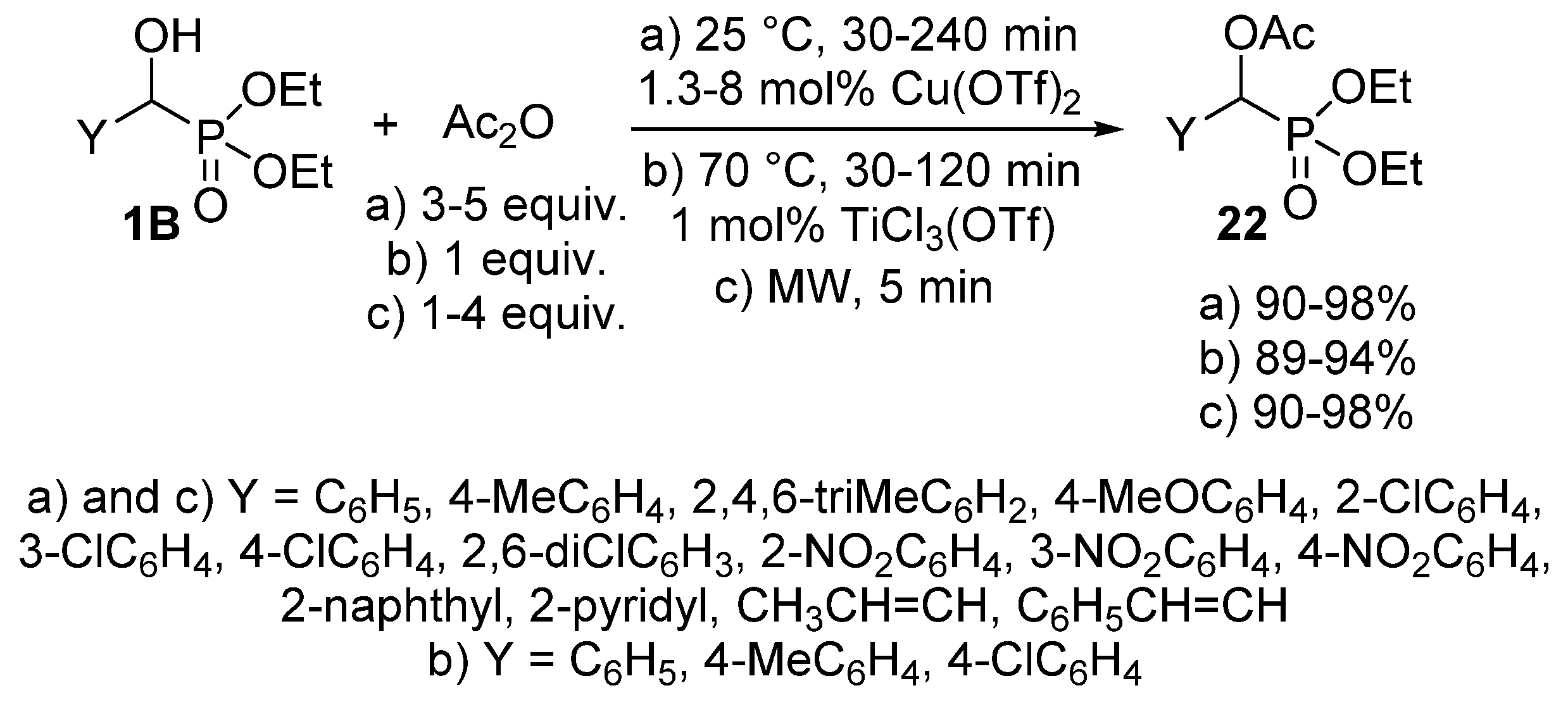{kind=link}
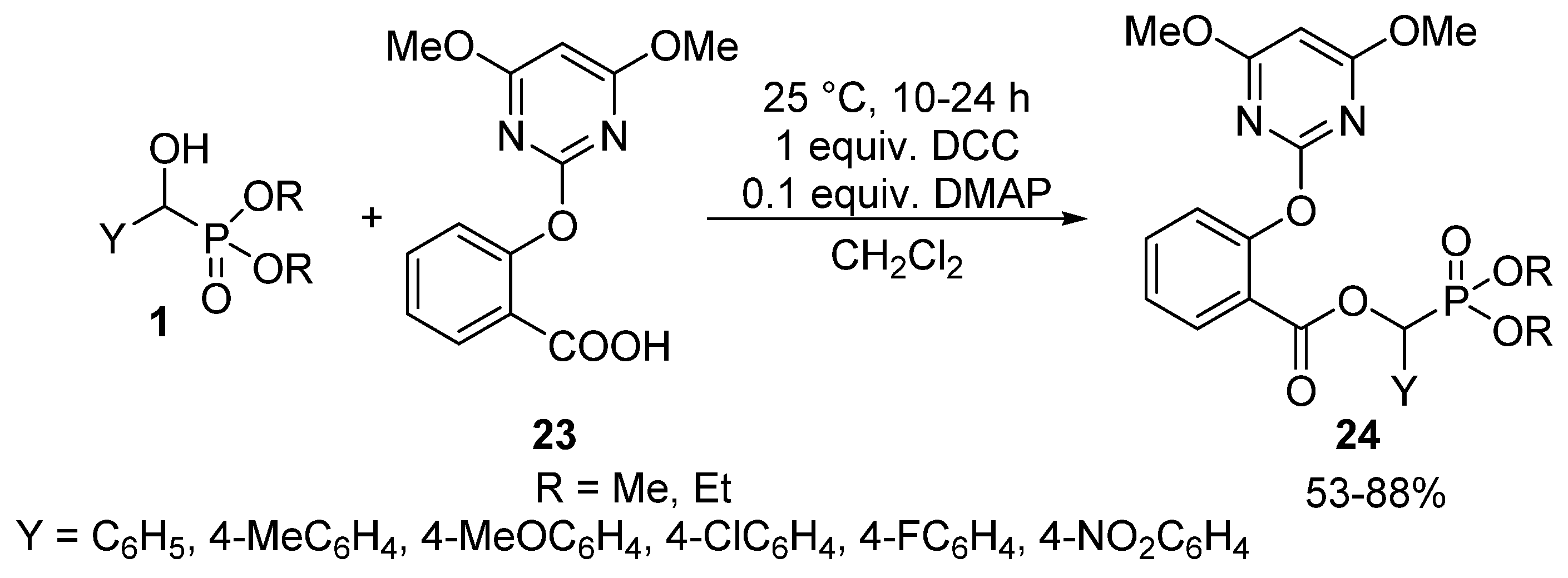{kind=link}
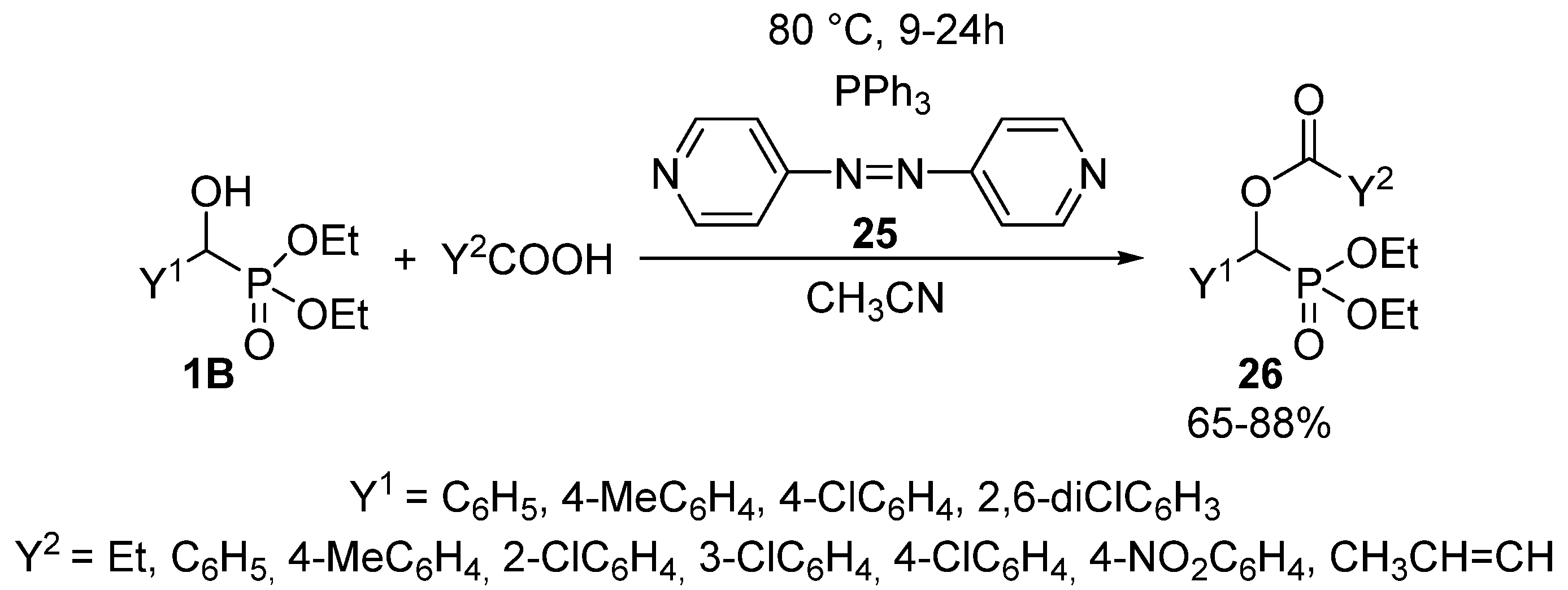{kind=link}
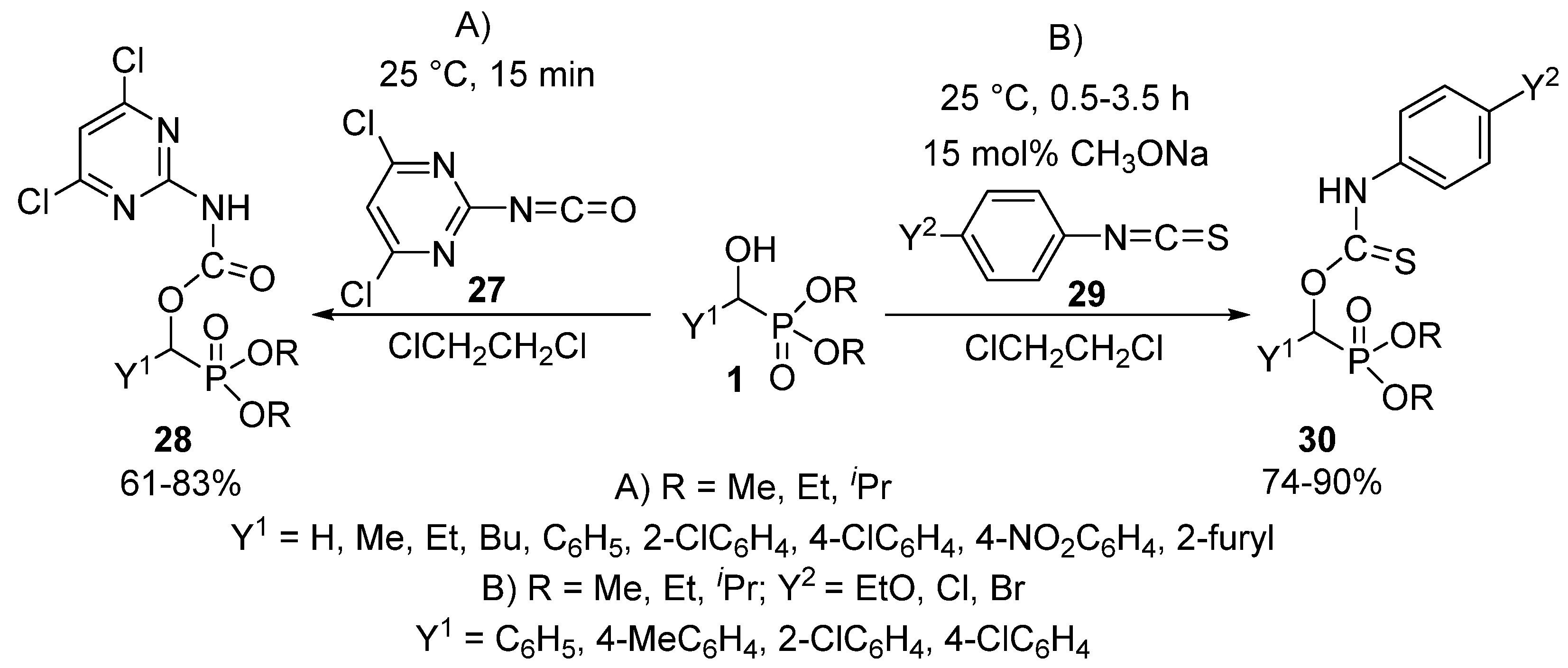{kind=link}
{kind=link}
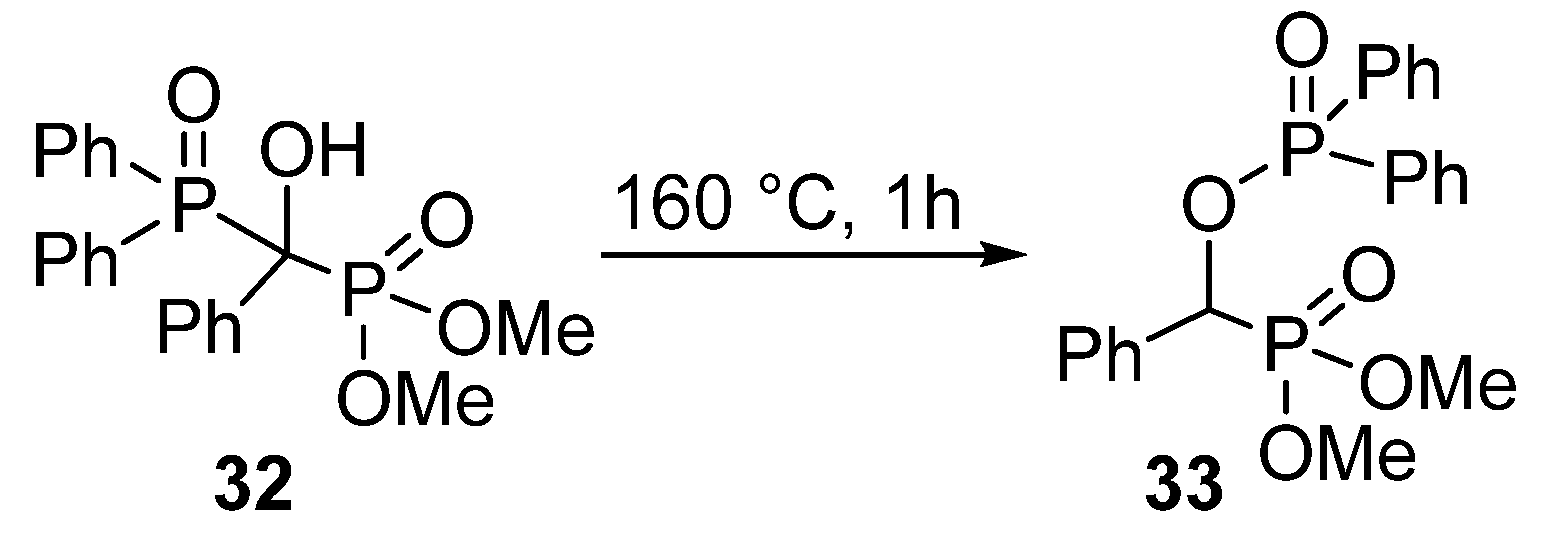{kind=link}
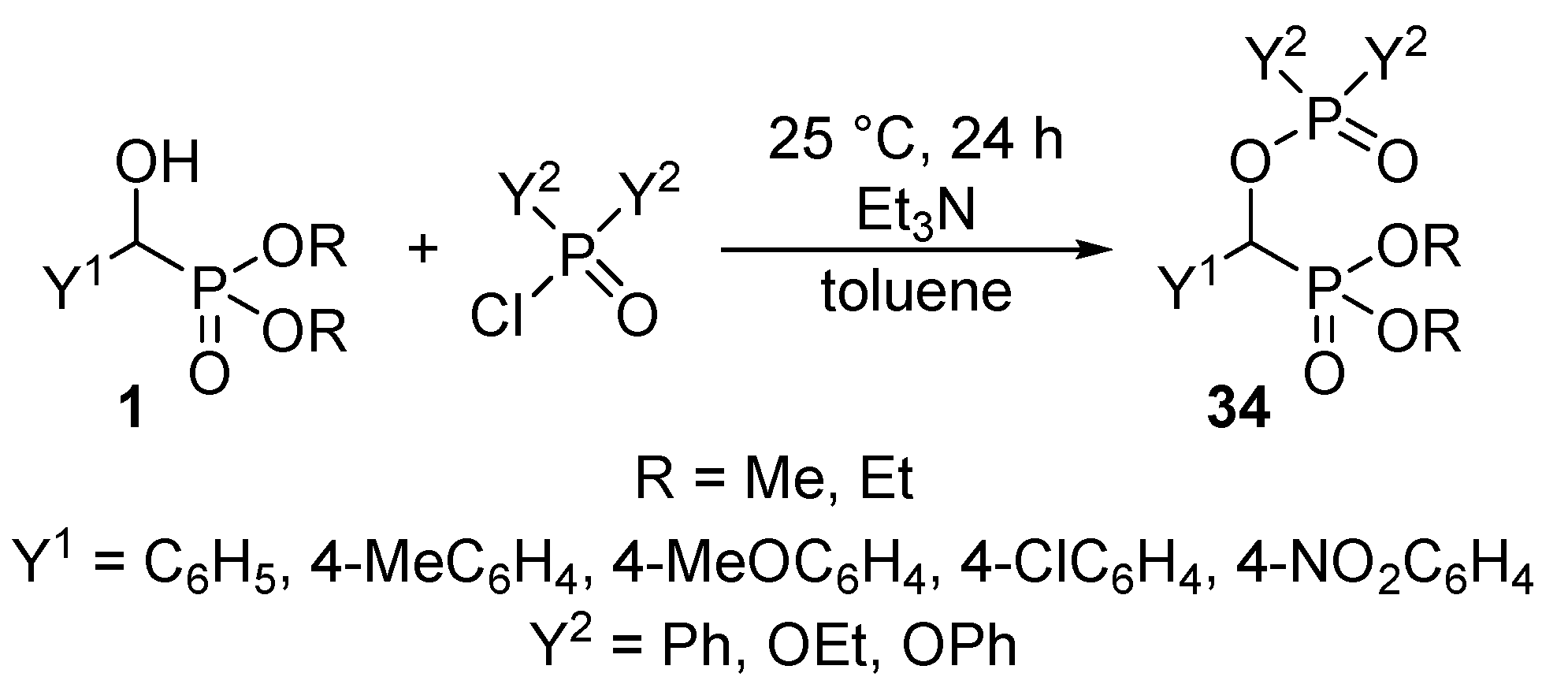{kind=link}
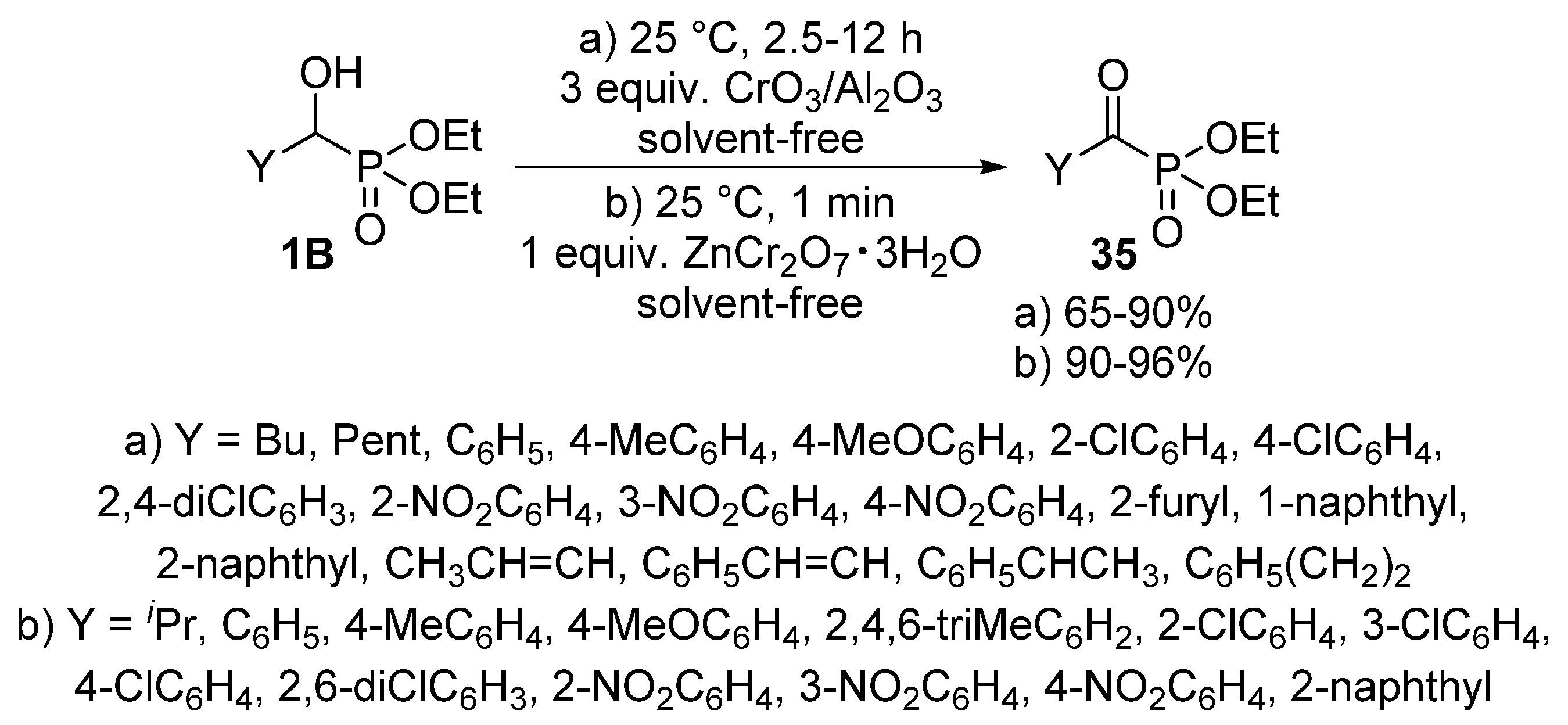{kind=link}
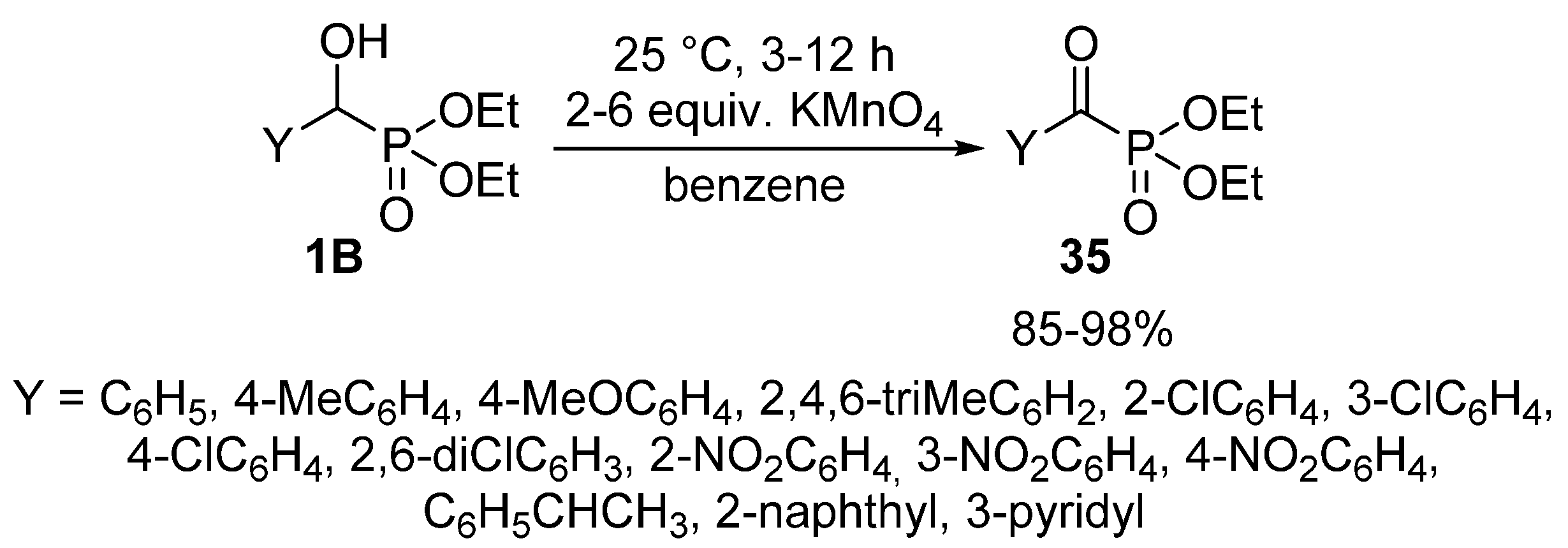{kind=link}
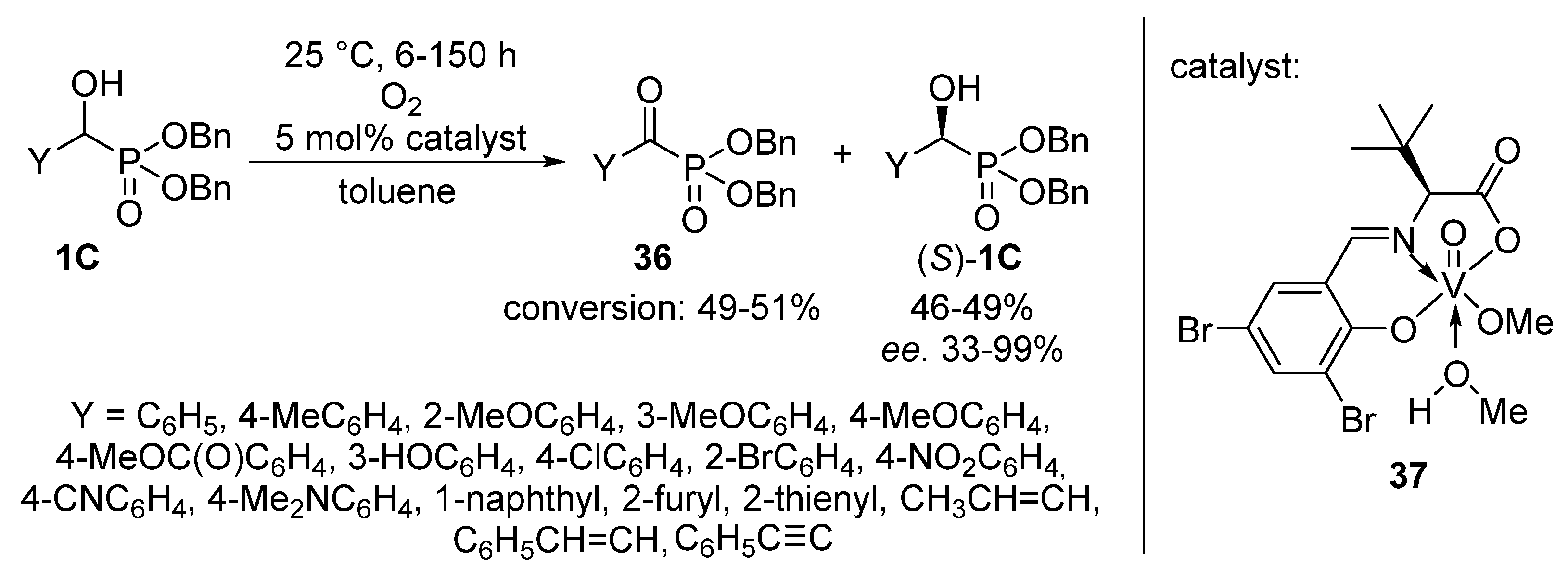{kind=link}
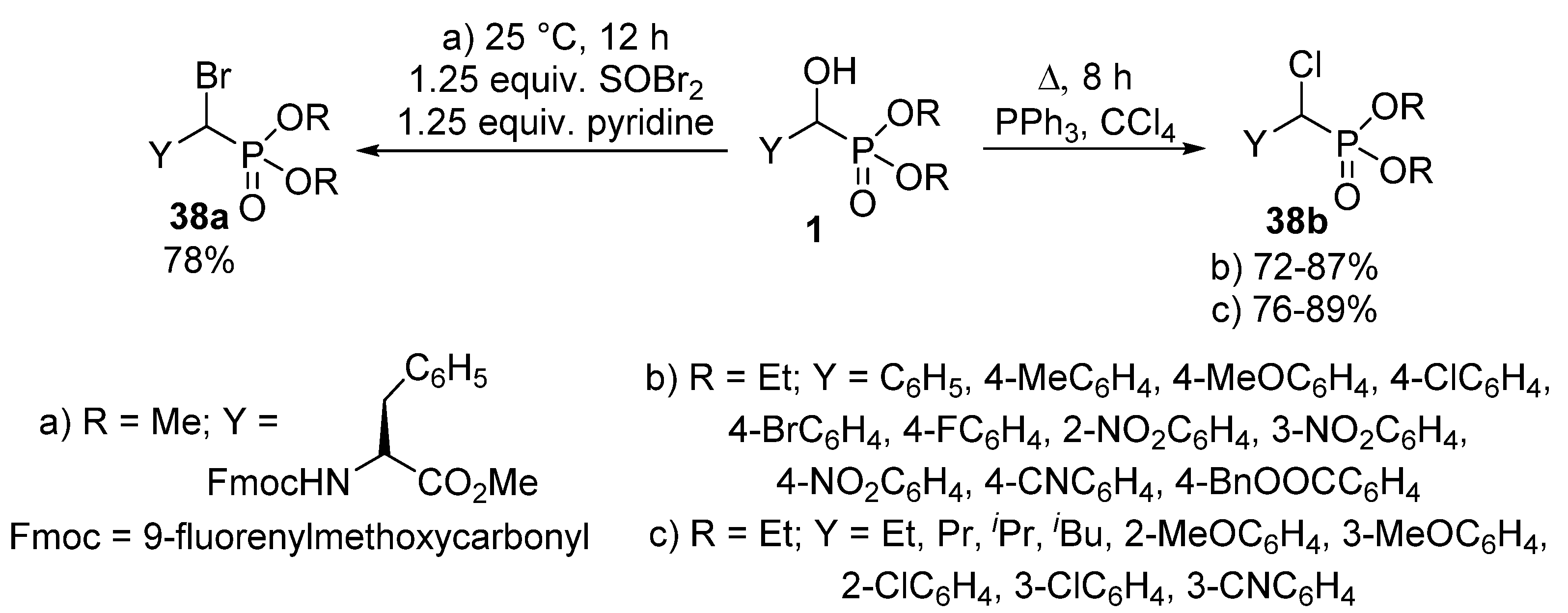{kind=link}
{kind=link}
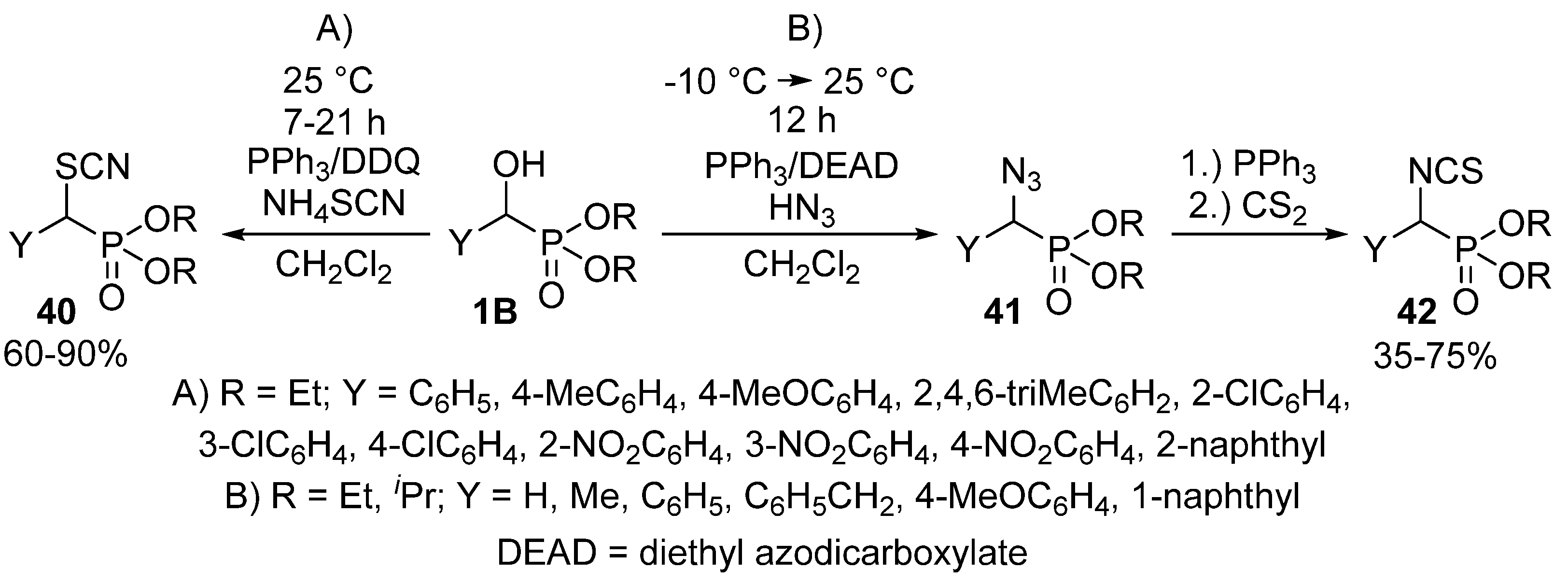{kind=link}
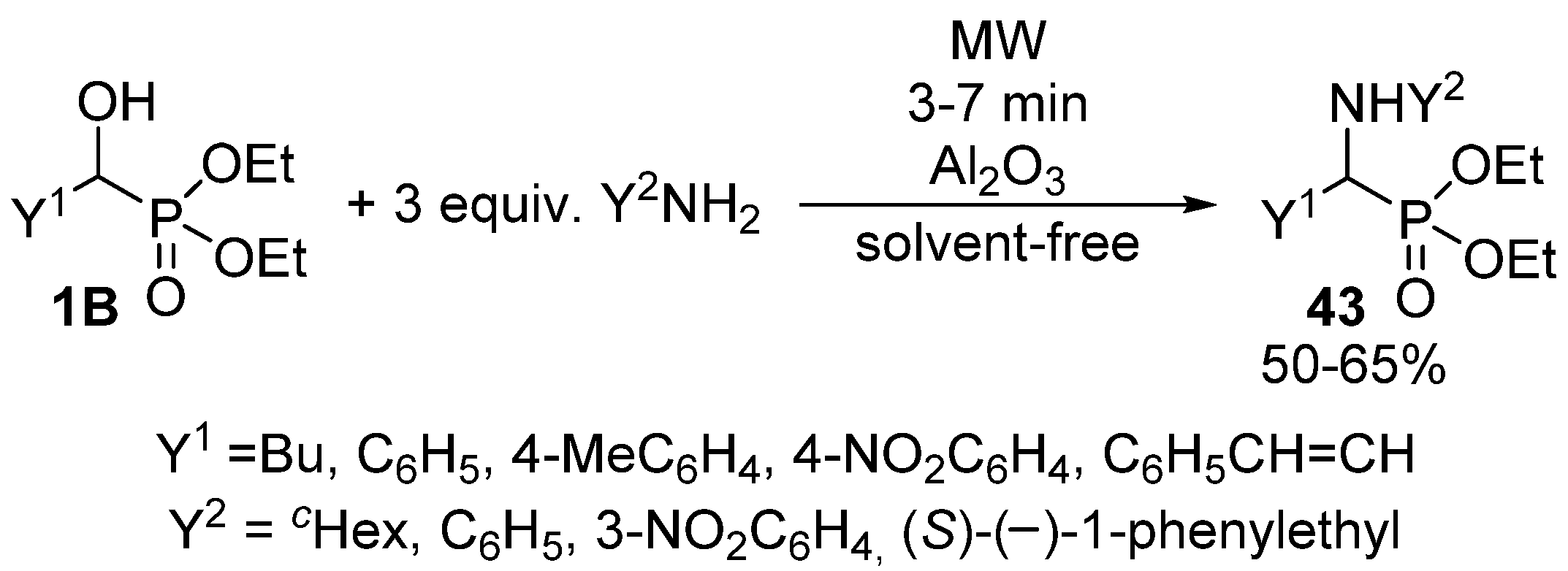{kind=link}
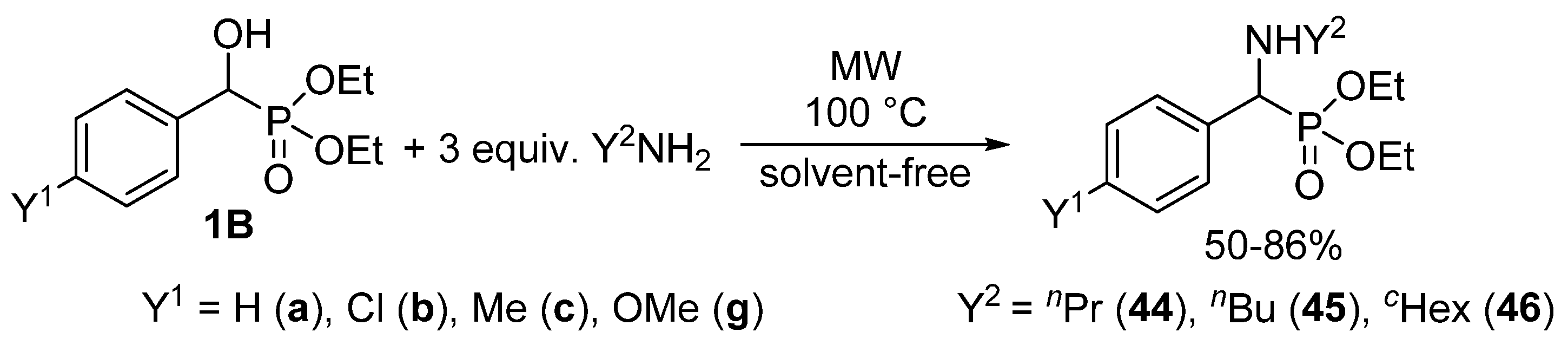{kind=link}
{kind=link}
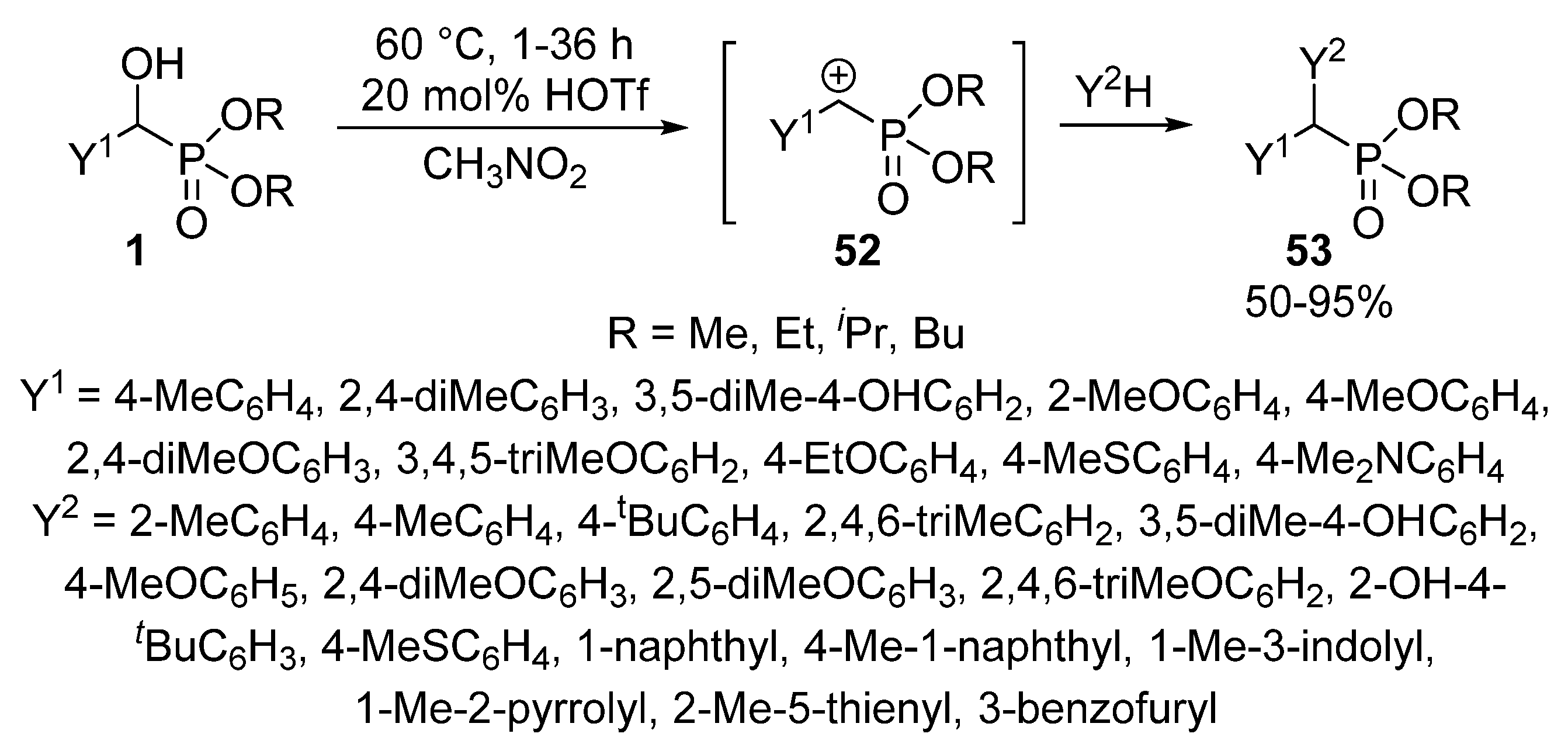{kind=link}
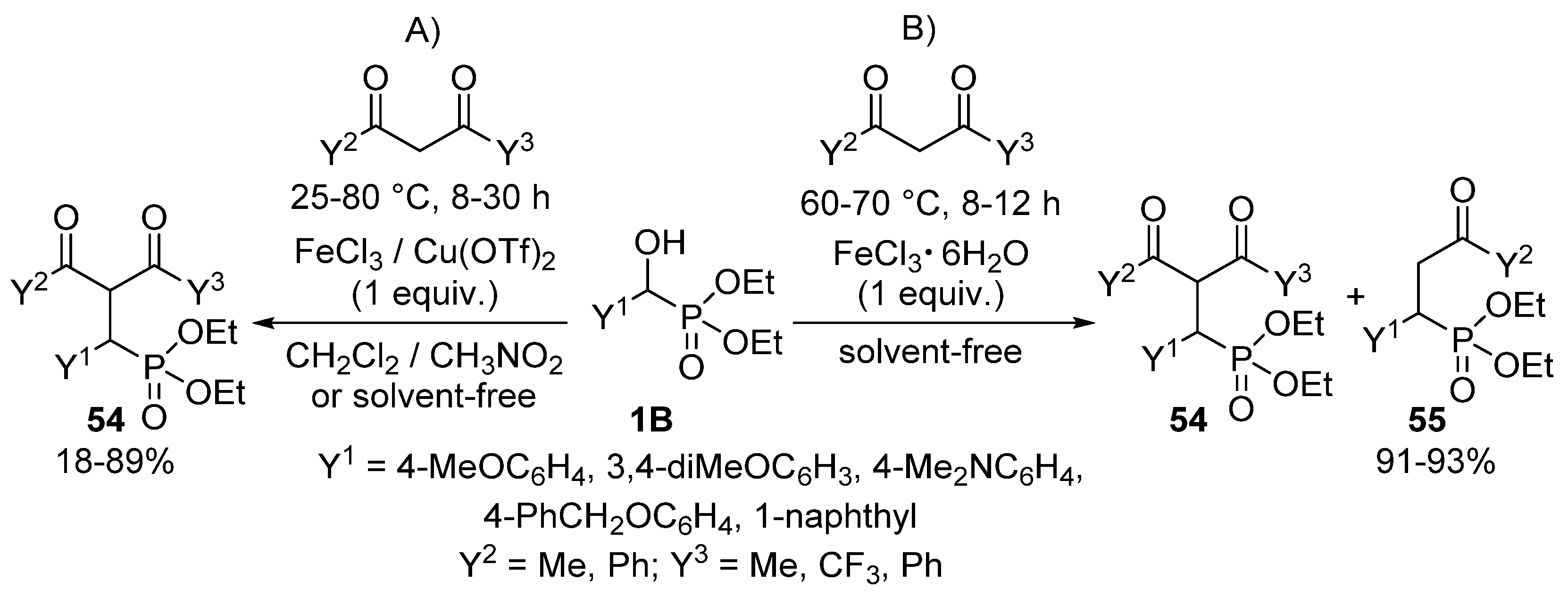{kind=link}
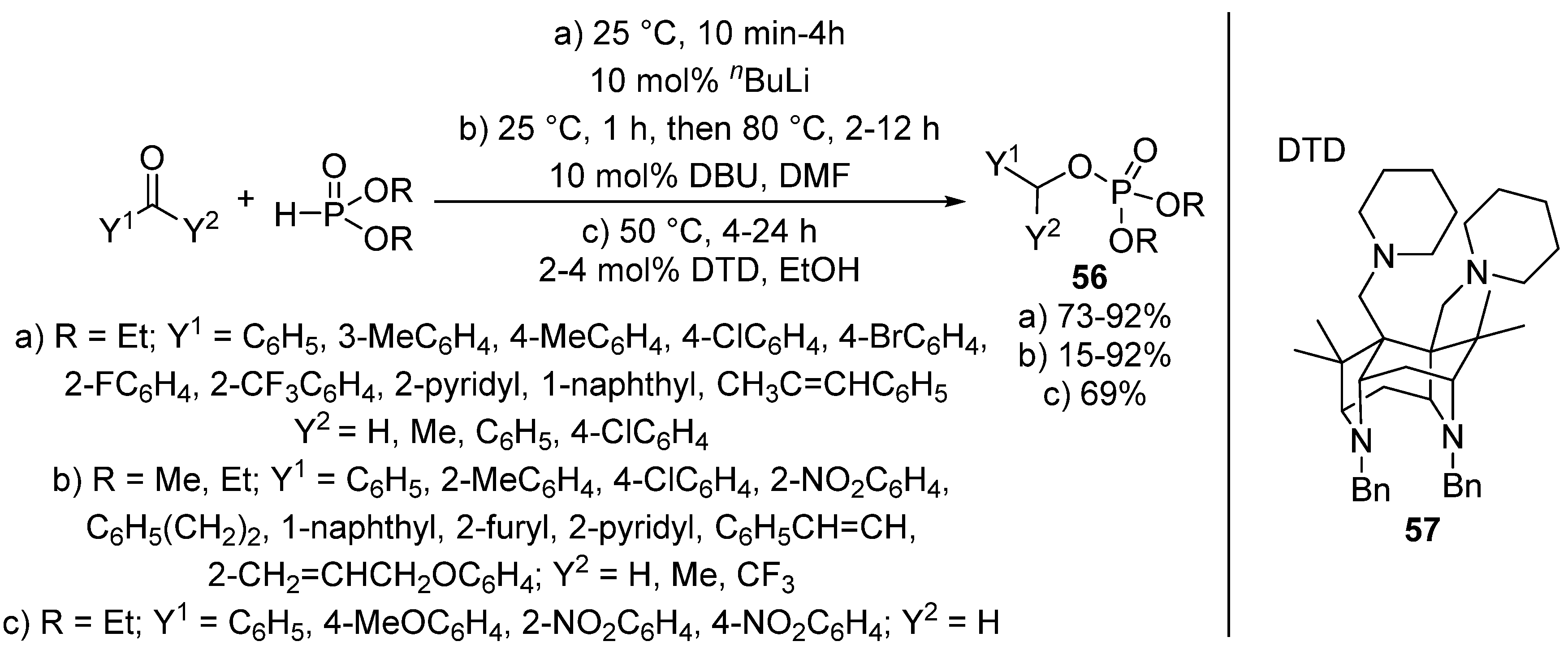{kind=link}
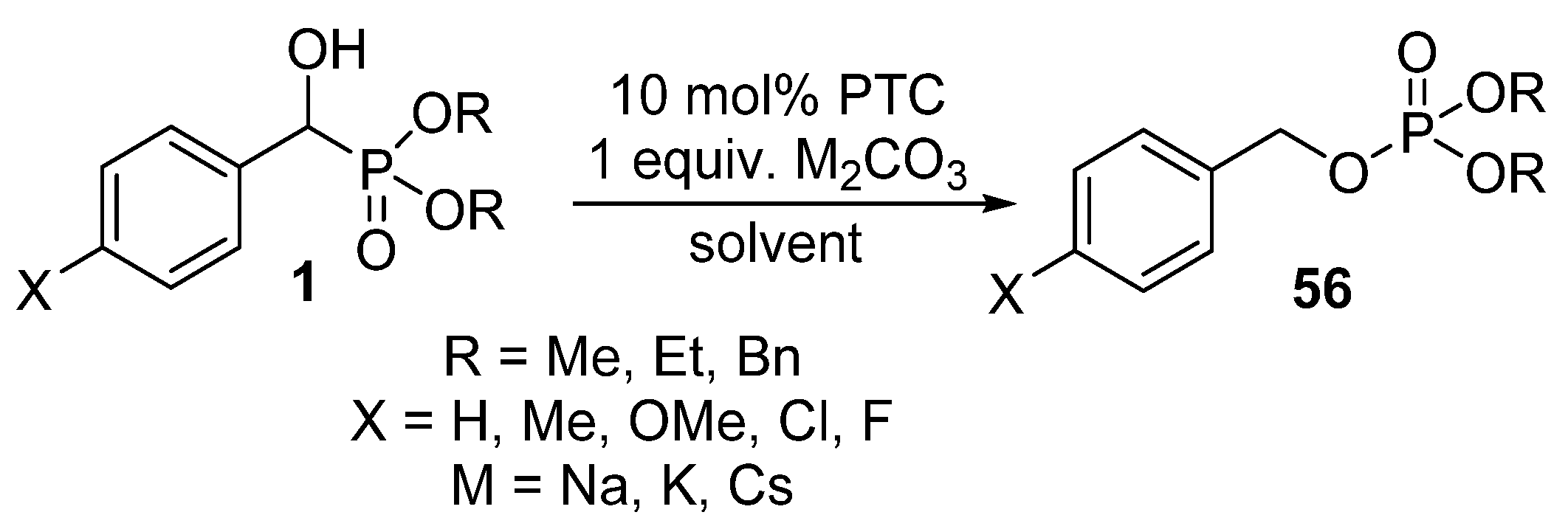{kind=link}
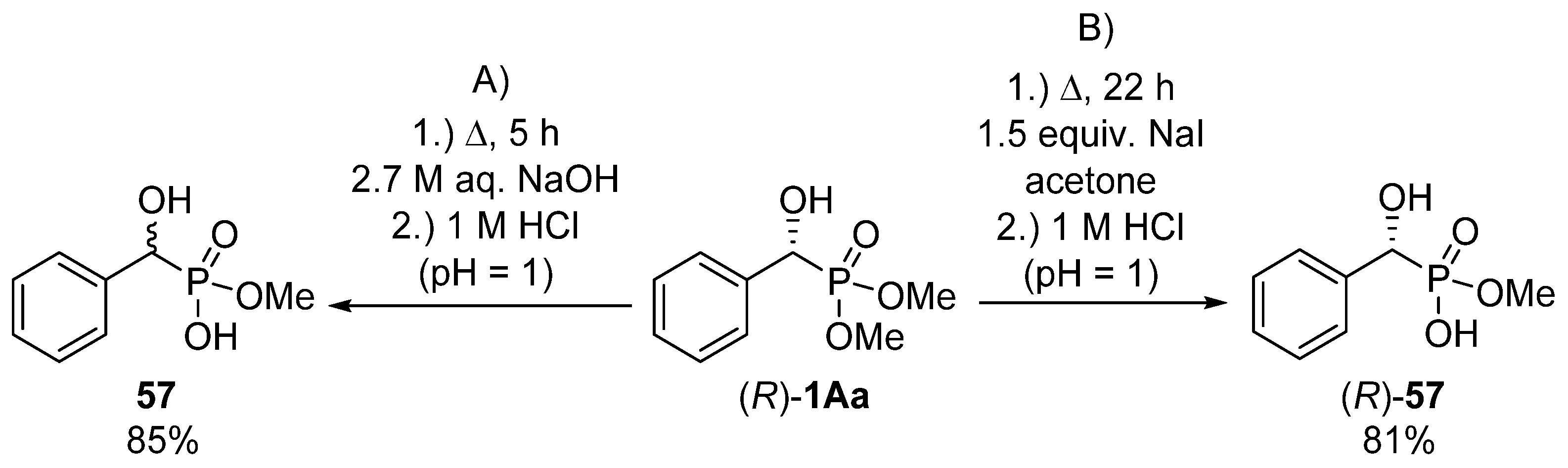{kind=link}
Table 1.
Selected examples for the synthesis of α-hydroxyphosphonates (1) from aldehydes or ketones and dialkyl phosphites (method “A”).
Table 1.
Selected examples for the synthesis of α-hydroxyphosphonates (1) from aldehydes or ketones and dialkyl phosphites (method “A”).
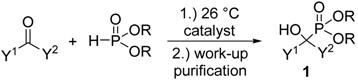
| Entry | Y1 | Y2 | R | Catalyst | Amount of Catalyst | Conditions | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | iPr, cHex, C6H5, 4-MeC6H4, 4-iPrC6H4, 4-MeOC6H4, 3,4-diMeOC6H3, 4-ClC6H4, 2-NO2C6H4, 4-CNC6H4, C6H5CH=CH, 3,4-OCH2OC6H3, 4-CH2=CHCH2OC6H4, 4-PhCH2OC6H4, 2-furyl, 2-thienyl | H | Me, Et, iPr | K3PO4 | 5 mol % | 25 °C, 4–8 min for aromatic aldehydes, 24 h for aliphatic aldehydes | 20–98 | [21] |
| 2 | C6H5, 4-MeOC6H4, 3,4-diMeOC6H3, 2-ClC6H4, 4-ClC6H4, 2,3-diClC6H3, 2,4-diClC6H3, 2,6-diClC6H3, 2-BrC6H4, 4-BrC6H4, 3-FC6H4, 4-FC6H4, 2-NO2C6H4, 3-NO2C6H4, 4-NO2C6H4, 4-CNC6H4, C6H5CH=CH, 2-thienyl, 4-pyridyl | H | Et | Ba(OH)2 | 10 mol % | 25 °C, 4–10 min | 70–98 | [22] |
| 3 | iBu, C6H5, 2-MeC6H4, 3-MeC6H4, 4-MeC6H4, 4-EtC6H4, 4-MeOC6H4, 2,3,4-triMeOC6H2, 2-ClC6H4, 4-ClC6H4, 2,6-diClC6H3, 3-BrC6H4, 4-BrC6H4, 3-FC6H4, 4-FC6H4, 2-NO2C6H4, 3-NO2C6H4, 4-NO2C6H4, 3-CNC6H4, 4-CNC6H4, 2-furyl, 2-thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 1-naphthyl, 2-naphthyl, 9-anthryl, C6H5CH=CH | H | Et, Bu, Bn | Ba(OH)2·8H2O | 2–7 mol % | 25 °C, 15 min, THF | 72–99 | [23] |
| 4 | C6H5, 4-MeC6H4, 3-MeOC6H4, 4-MeOC6H4, 3-HOC6H4, 2-ClC6H4, 4-ClC6H4, 2,6-diClC6H3, 2-NO2C6H4, 3-NO2C6H4, 4-NO2C6H4, 4-Me2NC6H4, C6H5CH=CH, 2-furyl, 1-naphthyl, 2-naphthyl | H | Et | MgO | 1 equiv. | 25 °C, 2 min–4 h | 80–100 | [24] |
| 5 * | C6H5, 4-MeC6H4, 4-MeOC6H4 | (CH2)2CN, MeCHCH2CN | Me, Et | MgO | 2 equiv. | 25 °C, 1–6 h | 70–82 | [25] |
| 6 * | Pr, Bu, C6H5, 4-MeC6H4, 4-MeOC6H4, 4-iPrC6H4, 2,4,6-triMeC6H2, 2-OHC6H4, 4-ClC6H4, 4-BrC6H4, 3-FC6H4, 4-FC6H4, 4-Br-2-OHC6H3, 3,4-diMeOC6H3, 3,4-OCH2OC6H3, 2-NO2C6H4, 4-NO2C6H4, 2-furyl, 2-thienyl, 3-thienyl, 1-naphthyl, 2-naphthyl | H, Me | Me | Al2O3 | 3 equiv. | 25 °C, 72 h | 52–98 | [26] |
| 7 | C6H5, 4-MeC6H4, 4-MeOC6H4, 4-ClC6H4, 4-FC6H4, 4-NO2C6H4 | H | Me, Et | Al2O3 + KF | 1 + 2 equiv. | 25 °C, 30 min | 53–88 | [27] |
| 8 * | Me, ClCH2, C6H5, 4-MeC6H4, 4-MeOC6H4, 2-ClC6H4, 3-ClC6H4, 4-ClC6H4, 3-BrC6H4, 4-BrC6H4, 2-FC6H4, 4-FC6H4, 2,4-diClC6H3, 2-furyl, 2-thienyl | Me, Pr, Ph | Me | Et3N | 1 equiv. | 40 °C, 2 h | 63–89 | [28] |
| 9 * | Pr, C6H5, 4-MeC6H4, 4-MeOC6H4, 2-ClC6H4, 4-ClC6H4, 3-NO2C6H4, 4-NO2C6H4, 4-CNC6H4, (CH2)2C6H5, C6H5CH=CH, 2-furyl, 2-naphthyl, 9-anthryl | H, Me | Me | Et3N + MgCl2 | 3 + 1 equiv. | 50 °C, 1–2 h | 85–98 | [29] |
| 10 | C6H5, 4-MeC6H4, 4-MeOC6H4, 4-OHC6H4, 2-ClC6H4, 4-ClC6H4, 4-BrC6H4, 2-NO2C6H4, 3-NO2C6H4, 4-NO2C6H4 | H | Et | – | – | MW, 90–100 °C, 10–20 min | 79–95 | [30] |
| 11 | C6H5, 4-MeC6H4, 4-MeOC6H4, 4-ClC6H4, 4-NO2C6H4 | H | Me, Et | Na2CO3 | 0.75 equiv. | MW, 110 °C, 20 min | 62–88 | [31] |
| 12 | C6H5, 4-MeC6H4, 4-MeOC6H4, 4-ClC6H4, 3-NO2C6H4, 4-NO2C6H4 | H | Et | Na2CO3 | 1 equiv. | Grinding, 25 °C, 10 min | 75–83 | [32] |
| 13 | C6H5, 4-MeC6H4, 4-iPrC6H4, 4-MeOC6H4, 3,4-diMeOC6H3, 2-ClC6H4, 4-ClC6H4, 2-NO2C6H4, 3-NO2C6H4, 4-NO2C6H4, 4-Me2NC6H4, 4-PhCH2OC6H4, 4-C5H4NC6H4, 9-anthryl | H | Et | 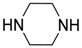 | 1 equiv. | Grinding, 25 °C, 2–10 min | 78–96 | [33] |
| 14 | C6H5, 2-MeC6H4, 4-MeC6H4, 2-iPrC6H4, 3,5-diMeC6H3, 2-MeOC6H4, 4-MeOC6H4, 3,5-diMeOC6H3, 3,4,5-triMeOC6H2, 4-EtOC6H4, 2-ClC6H4, 2-BrC6H4, 3-BrC6H4, 4-BrC6H4, 2-Cl-6-furylC6H3, 2-F-4-BrC6H3, 2-NO2C6H4, 4-NO2C6H4, 9-anthryl, 1-pyrenyl, 2-furyl, 1-C6H5-4-pyrazolyl | H | Et | 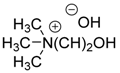 | 10 mol % | 25 °C, 5–10 min | 90–98 | [34] |
| 15 * | C6H5, 3-MeOC6H4, 4-MeOC6H4, 4-ClC6H4, 3-NO2C6H4, 4-NO2C6H4, C6H5CH=CH | H, Me | Me, Et | Na-modified fluoroapatite | 1 g/1.25 mol acetophenone | 20–25 °C, 1–1.5 min | 75–98 | [35] |
| 16 | 4-iPrC6H4, 4-MeOC6H4, 2,6-diMeOC6H3, 3,5-diMeOC6H3, 4-OHC6H4, 3-NO2C6H4, 4-Me2NC6H4, 2-PhCH2OC6H4, 2-imidazyl, 3-indolyl | H | Et | KHSO4 | 20 mol % | 25 °C, 2–4 h | 82–91 | [36] |
| 17 | 3-FC6H4, 4-NO2C6H4, 3,4-OCH2OC6H3, 4-MeSC6H4, C5H10N, 3-MeO-4-OHC6H3, 4-C4H8NC6H4, 2-furyl, 2-thienyl, 4-imidazyl, 2-pyrrolyl, 4-pyridyl | H | Me | silica-supported tungstic acid | 20 mol % | 25 °C, 30 min | 85–96 | [37] |
| 18 * | Me, Pr, iBu, Pent, C6H5, 2-MeC6H4, 4-MeC6H4, 2-MeOC6H4, 3-MeOC6H4, 4-MeOC6H4, 2-ClC6H4, 3-ClC6H4, 4-ClC6H4, 2-BrC6H4, 4-BrC6H4, 4-FC6H4, 3-NO2C6H4, 4-NO2C6H4, 1-naphthyl, 2-naphthyl, 2-furyl, 2-thienyl, 3-pyridyl | H, Me, Ph, CF3, (CH2)10CH3, C(O)Ph, CH2C(O)Ph | Et, iPr, Ph | BuLi | 0.1 mol % | 10–25 °C, 5 min, hexane | 35–99 | [38] |
| 19 * | Et, cHex, C6H5, 3-MeC6H4, 4-MeC6H4, 3-MeOC6H4, 4-ClC6H4, 3,4-diClC6H3, 2,3,4-triClC6H2, 2-FC6H4, 4-FC6H4, 4-NO2C6H4, 3-CF3C6H4CH2=CH, 3-CF3C6H4CH(OH)CH2, 2-thienyl, 2-naphthyl, EtOC(O)CHBn | Me, Et, Ph, CH(OEt)2, COOMe, (CH2)2Cl | Me | Ti(OiPr)4 | 5 mol % | 30 °C, 15 min | 74–98 | [39] |
| 20 | C6H5, 4-MeOC6H4, 4-FC6H4, 4-NO2C6H4, 4-CF3C6H4, 4-CNC6H4, 4-MeOC(O)C6H4, (CH2)2C6H5, C6H5CH=CH | H | Et | MoO2Cl2 | 5 mol % | 80 °C, 1–24 h | 70–96 | [40] |
* The procedure is also suitable starting from ketones.
Table 2.
Selected procedures to synthesize α-hydroxyphosphonates (1) from aldehydes or ketones and trialkyl phosphites (method “B”).
Table 2.
Selected procedures to synthesize α-hydroxyphosphonates (1) from aldehydes or ketones and trialkyl phosphites (method “B”).
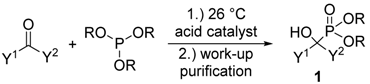
| Entry | Y1 | Y2 | R | Catalyst | Amount of Catalyst | Conditions | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 * | C6H5, 4-MeC6H4, 4-iPrC6H4, 4-MeOC6H4, 2-ClC6H4, 4-ClC6H4, 4-BrC6H4, 4-FC6H4, 4-NO2C6H4, 4-CF3C6H4, 4-Me2NC6H4, CH2C6H5, (CH2)2C6H5 | H, Me, Et, Ph | Me, Et | – | – | Sonication, 25 °C, 10–35 min | 84–94 | [41] |
| 2 | Pr, iBu, C6H5, 4-MeC6H4, 4-MeOC6H4, 3-OHC6H4, 2-ClC6H4, 4-ClC6H4, C6H5CH=CH, 3-chromonyl, 6-Cl-7-Me-3-chromonyl, 2-Cl-3-quinolinyl | H | Et | KH2PO4 | 5 mol % | Sonication, 25 °C, 5–45 min | 48–92 | [42] |
| 3 | C6H5, 4-MeC6H4, 4-MeOC6H4, 4-ClC6H4, 3-pyridyl, 3-chromonyl, 6,8-diMe-3-chromonyl, 2-Cl-3-quinolinyl, 6-Cl-3-chromonyl, 6,7-diCl-3-chromonyl, 6,8-diCl-3-chromonyl, 6-Br-3-chromonyl, 6-Cl-7-Me-3-chromonyl, 2-Cl-6-Me-3-quinolinyl, 2-Cl-7-Me-3-quinolinyl, 2-Cl-8-Me-3-quinolinyl, 2-Cl-6-MeO-3-quinolinyl, 2-Cl-6-EtO-3-quinolinyl, 2-Cl-8-Et-3-quinolinyl | H | Me, Et |  | 25 mol % | Sonication, 25 °C, 1–60 min | 78–98 | [43] |
| 4 | C6H5, 4-MeC6H4, 4-MeOC6H4, 4-OHC6H4, 4-ClC6H4, 4-NO2C6H4, 3,4-OCH2OC6H3, C6H5CH=CH, 2-furyl, 2-thienyl, 2-Cl-3-quinolinyl, 4-tetrazolo[1,5-a]quinolinyl | H | Et | 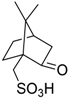 | 10 mol % | Sonication, 25 °C, 8–20 min | 85–93 | [44] |
| 5 | Pr, iPr, Pent, cHex, C6H5, 4-MeC6H4, 4-MeOC6H4, 4-OHC6H4, 2-ClC6H4, 4-ClC6H4, 4-CNC6H4, C6H5CH=CH, 2-furyl | H | Me | (COOH)2 | 10 mol % | 80 °C, 3 h | 83–98 | [45] |
| 6 | 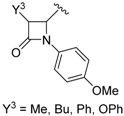 | H | Me, Et |  | 10 mol % | ∆, 30 min, acetonitrile | 41–69 | [46] |
| 7 * | Pr, C6H5, 4-MeC6H4, 4-MeOC6H4, 2-ClC6H4, 4-ClC6H4, 3-NO2C6H4, 4-NO2C6H4, 4-CNC6H4, (CH2)2C6H5, C6H5CH=CH, 2-furyl | H, Me | Me | 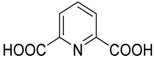 | 10 mol % | 50 °C, 1.3–3 h, H2O | 60–95 | [47] |
| 8 | Pr, iPr, Bu, tBu, Hex, cHex, C6H5, 4-ClC6H4, 2-furyl, 2-pyridyl | H | Me | 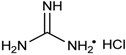 | 0.5 mol % | 50 °C, 2 h, H2O | 60–95 | [48] |
| 9 | Me, Et, iPr, C6H5, 4-MeC6H4, 4-MeOC6H4, 2-ClC6H4, 4-ClC6H4, 2,4-diClC6H3, 2-NO2C6H4, 3-NO2C6H4, 4-NO2C6H4, C6H5CH=CH, 2-furyl, 2-thienyl | H | Et | I2 | 10 mol % | 80 °C, 15–120 min, H2O | 83–97 | [49] |
| 10 | C6H5, 4-MeC6H4, 4-MeOC6H4, 2-OHC6H4, 4-ClC6H4, 4-BrC6H4, 4-FC6H4, 3-NO2C6H4, 4-NO2C6H4, 2-pyridyl, 2-naphthyl, 2-furyl, 2-thienyl | H | Et | β-cyclodextrin | 1 equiv. | 60–70 °C, 8–12 h, H2O | 80–93 | [50] |
| 11 | C6H5, 4-MeC6H4, 4-MeOC6H4, 3-OHC6H4, 2-ClC6H4, 4-ClC6H4, 4-NO2C6H4, C6H5CH=CH, 3-chromonyl, 6-Cl-3-chromonyl, 6,7-diCl-3-chromonyl, 6,8-diCl-3-chromonyl, 6-Cl-7-Me-3-chromonyl, 2-Cl-3-quinolinyl, 2-Cl-6-Me-3-quinolinyl, 2-Cl-7-MeO-3-quinolinyl, 2-Cl-8-Et-3-quinolinyl, 2-Cl-6-EtO-3-quinolinyl | H | Et | NH4VO3 | 10 mol % | 25 °C, 5–40 min | 80–94 | [51] |
| 12 | 4-ClC6H4, 2,4-diClC6H3, 4-BrC6H4, 4-MeOC(O)C6H4, 4-CF3C6H4, 2-NO2-3,6-diMeOC6H2, 1-naphthyl | H | Et | ZnBr2 | 10 mol % | 25 °C, 10–30 min | 68–91 | [52] |
| 13 | C6H5, 4-MeC6H4, 4-MeOC6H4, 4-HOC6H4, 2-ClC6H4, 4-ClC6H4, 4-NO2C6H4, 4-Me2NC6H4, C6H5CH=CH, 2-Cl-3-quinolinyl, 2-Cl-6-Me-3-quinolinyl | H | Et | Bi(NO3)3·5H2O | 10 mol % | MW, 70 °C, 10–15 min | 88–95 | [53] |
| 14 * | Et, Pr, iPr, tBu, cHex, CH3CH=CH, C6H5, 4-MeOC6H4, 4-ClC6H4, (CH2)2C6H5, C6H5CH=CH | H, Me | Me, Et | NbCl5, TMSCl | 0.05 mol % | 25 °C, 20–90 min | 44–96 | [54] |
* The procedure is also suitable starting from ketones.
Table 3.
Details of the synthesis of α-aminophosphonates 44–46 from α-hydroxyphosphonates 1B.
| Entry | Y1 | Y2 | Reaction Time (min) | Yield (%) | Product | Ref. |
|---|---|---|---|---|---|---|
| 1 | H | nPr | 10 | 78 | 44a | [129] |
| 2 | H | nBu | 15 | 86 | 45a | [129] |
| 3 | H | cHex | 10 | 84 | 46a | [129] |
| 4 | Cl | nPr | 15 | 60 | 44b | [50] |
| 5 | Cl | nBu | 20 | 50 | 45b | [56] |
| 6 | Cl | cHex | 30 | 54 | 46b | [56] |
| 7 | Me | nPr | 15 | 58 | 44c | [56] |
| 8 | Me | nBu | 15 | 79 | 45c | [56] |
| 9 | Me | cHex | 30 | 73 | 46c | [56] |
| 10 | OMe | nPr | 15 | 72 | 44g | [56] |
| 11 | OMe | nBu | 30 | 66 | 45g | [56] |
| 12 | OMe | cHex | 30 | 70 | 46g | [56] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rádai, Z.; Keglevich, G. Synthesis and Reactions of α-Hydroxyphosphonates. Molecules 2018, 23, 1493. https://doi.org/10.3390/molecules23061493
AMA Style
Rádai Z, Keglevich G. Synthesis and Reactions of α-Hydroxyphosphonates. Molecules. 2018; 23(6):1493. https://doi.org/10.3390/molecules23061493
Chicago/Turabian StyleRádai, Zita, and György Keglevich. 2018. "Synthesis and Reactions of α-Hydroxyphosphonates" Molecules 23, no. 6: 1493. https://doi.org/10.3390/molecules23061493