
A3, a Scorpion Venom Derived Peptide Analogue with Potent Antimicrobial and Potential Antibiofilm Activity against Clinical Isolates of Multi-Drug Resistant Gram Positive Bacteria


Abstract
:1. Introduction
2. Materials
2.1. Bacterial Strains
2.2. Antimicrobial Substances
2.3. Cell Lines
3. Methods
3.1. Bioinformatics Analysis and Design of the Modified Peptide
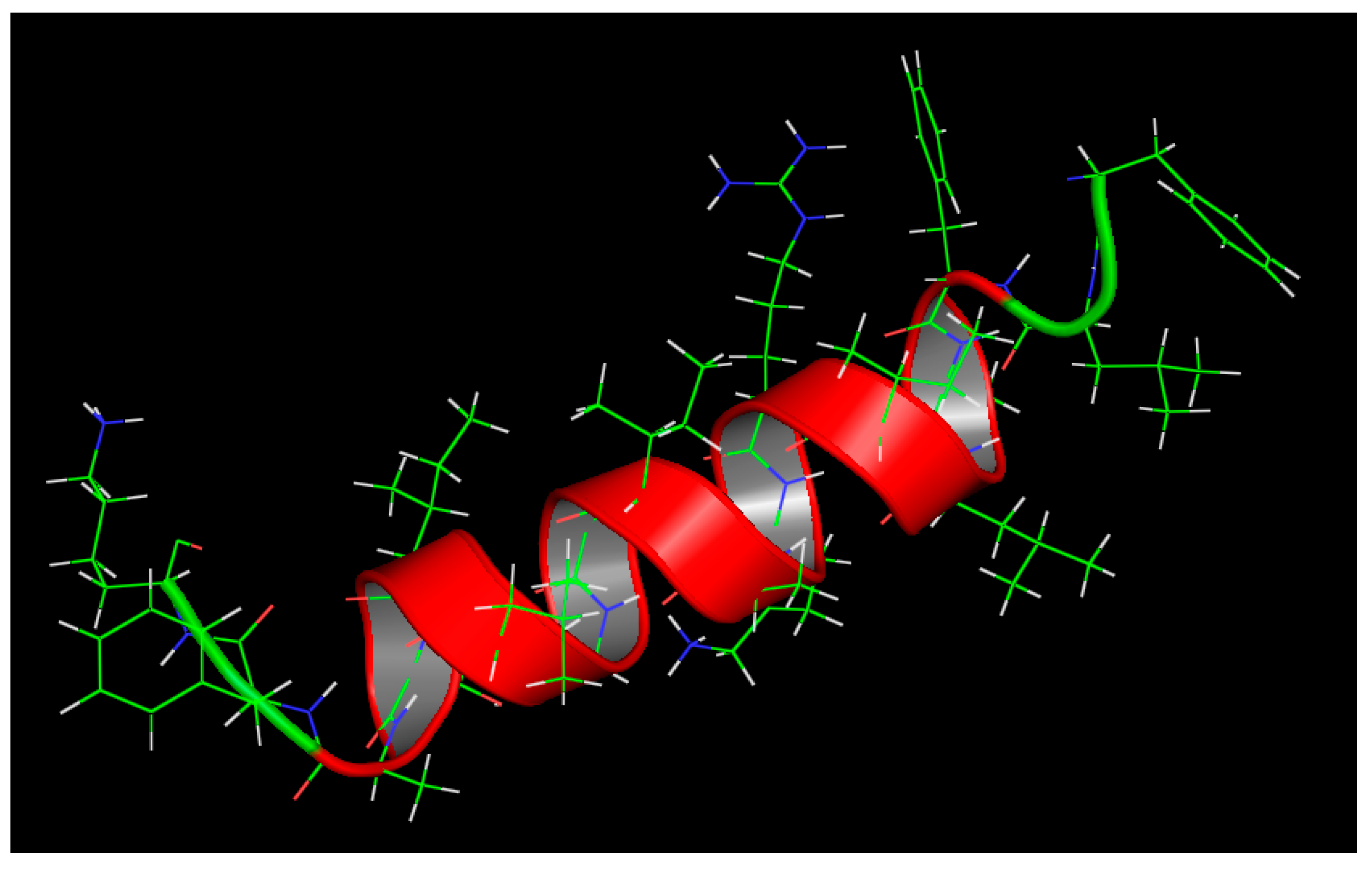
3.2. Molecular Modeling and In Silico Analysis of AamAP1 and Its Modified Peptide Analogue A3
3.3. Minimum Inhibitory Concentrations (MIC) and Minimum Bactericidal Concentrations (MBC)
3.4. MIC and MBC Determination of Individual Antibiotics and Checkerboard Assay
3.5. Synergistic Studies Evaluation and the Fractional Inhibitory Concentration (FIC)
3.6. Antibiofilm Activity
3.7. Mammalian Cytotoxicity Assays
3.8. A3’s Hemolytic Activity
4. Results
4.1. Bioinformatics Analysis of AamAP1 and the Design of A3
4.2. Peptide Synthesis and Purification
4.3. Bacterial Susceptibility Assay
4.4. Synergistic Activity of A3 and the Antibiotics in Combination
4.5. The FIC Index
4.6. Antibiofilm Activity
4.7. MTT Cell Proliferation Assay
4.8. Hemolysis Assay
5. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roca, I.; Akova, M.; Baquero, F.; Carlet, J.; Cavaleri, M.; Coenen, S.; Cohen, J.; Findlay, D.; Gyssens, I.; Heure, O.E.; et al. The global threat of antimicrobial resistance: Science for intervention. New Microbes New Infect. 2015, 6, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Hu, F.P.; Guo, Y.; Zhu, D.M.; Wang, F.; Jiang, X.F.; Xu, Y.C.; Zhang, X.J.; Zhang, C.X.; Ji, P.; Xie, Y.; et al. Resistance trends among clinical isolates in China reported from CHINET surveillance of bacterial resistance, 2005–2014. Clin. Microbiol. Infect. 2016, 22, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Bartlett, J.; Wunderink, R.; Gilbert, D.N. Novel approaches are needed to develop tomorrow’s antibacterial therapies. Am. J. Respir. Crit. Care Med. 2015, 191, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Woodford, N.; Livermore, D.M. Infections caused by Gram-positive bacteria: A review of the global challenge. J. Infect. 2009, 59, S4–S16. [Google Scholar] [CrossRef]
- Bechinger, B.; Lohner, K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 2006, 1758, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Hemshekhar, M.; Anaparti, V.; Mookherjee, N. Functions of cationic host defense peptides in immunity. Pharmaceuticals 2016, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.C.; Pena, O.M.; Hancock, R.E. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Omardien, S.; Brul, S.; Zaat, S.A. Antimicrobial activity of cationic antimicrobial peptides against gram-positives: Current progress made in understanding the mode of action and the response of bacteria. Front. Cell Dev. Biol. 2016, 4, 111. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Gorr, S.U. Antimicrobial peptides: Mechanisms of action and resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Reiling, S.; Zarena, D.; Wang, G. Host defense antimicrobial peptides as antibiotics: Design and application strategies. Curr. Opin. Chem. Biol. 2017, 38, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Almaaytah, A.; Zhou, M.; Wang, L.; Chen, T.; Walker, B.; Shaw, C. Antimicrobial/cytolytic peptides from the venom of the North African scorpion, Androctonus amoreuxi: Biochemical and functional characterization of natural peptides and a single site-substituted analog. Peptides 2012, 35, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deleage, G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Komatsuzawa, H.; Ohta, K.; Sugai, M.; Fujiwara, T.; Glanzmann, P.; Berger-Bächi, B.; Suginaka, H. Tn 551-mediated insertional inactivation of the fmtB gene encoding a cell wall-associated protein abolishes methicillin resistance in Staphylococcus aureus. J. Antimicrob. Chemother. 2000, 45, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Bussmann, R.W.; Malca-García, G.; Glenn, A.; Sharon, D.; Chait, G.; Díaz, D.; Pourmand, K.; Jonat, B.; Somogy, S.; Guardado, G.; Aguirre, C. Minimum inhibitory concentrations of medicinal plants used in Northern Peru as antibacterial remedies. J. Ethnopharmacol. 2010, 132, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sueke, H.; Kaye, S.B.; Neal, T.; Hall, A.; Tuft, S.; Parry, C.M. An in vitro investigation of synergy or antagonism between antimicrobial combinations against isolates from bacterial keratitis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4151–4155. [Google Scholar] [CrossRef] [PubMed]
- Luca, V.; Stringaro, A.; Colone, M.; Pini, A.; Mangoni, M.L. Esculentin (1-21), an amphibian skin membrane-active peptide with potent activity on both planktonic and biofilm cells of the bacterial pathogen Pseudomonas aeruginosa. Cell. Mol. Life Sci. 2013, 70, 2773–2786. [Google Scholar] [CrossRef] [PubMed]
- Rolain, J.M.; Canton, R.; Cornaglia, G. Emergence of antibiotic resistance: Need for a new paradigm. Clin. Microbiol. Infect. 2012, 18, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Sun, Y.; Liu, Q.; Wang, X.; Li, Z.; Hao, J. In vitro synergistic activities of antimicrobial peptide brevinin-2CE with five kinds of antibiotics against multidrug-resistant clinical isolates. Curr. Microbiol. 2014, 68, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Almaaytah, A.; Mohammed, G.K.; Abualhaijaa, A.; Al-Balas, Q. Development of novel ultrashort antimicrobial peptide nanoparticles with potent antimicrobial and antibiofilm activities against multidrug-resistant bacteria. Drug Des. Dev. Ther. 2017, 11, 3159–3170. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ma, Y.; Dai, C.; Zhao, R.; Li, S.; Wu, Y.; Cao, Z.; Li, W. Imcroporin, a new cationic antimicrobial peptide from the venom of the scorpion Isometrus maculates. Antimicrob. Agents Chemother. 2009, 53, 3472–3477. [Google Scholar] [CrossRef] [PubMed]
- Almaaytah, A.; Albalas, Q. Scorpion venom peptides with no disulfide bridges: A review. Peptides 2014, 51, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 2008, 1778, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Almaaytah, A.; Tarazi, S.; Abu-Alhaijaa, A.; Altall, Y.; Alshar’i, N.; Bodoor, K.; Al-Balas, Q. Enhanced antimicrobial activity of AamAP1-Lysine, a novel synthetic peptide analog derived from the scorpion venom peptide AamAP1. Pharmaceuticals 2014, 7, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C. Antibiotics: Actions, Origins, Resistance; American Society for Microbiology (ASM): Washington, DC, USA, 2003. [Google Scholar]
- Huang, H.W. Peptide-Lipid Interactions and Mechanisms of Antimicrobial Peptides. In Gramicidin and Related Ion Channel-Forming Peptides; John Wiley & Sons: Chichester, UK, 2008; Volume 225, pp. 188–200. [Google Scholar]
- Giacometti, A.; Cirioni, O.; Barchiesi, F.; Scalise, G. In-vitro activity and killing effect of polycationic peptides on methicillin-resistant Staphylococcus aureus and interactions with clinically used antibiotics. Diagn. Microbiol. Infect. Dis. 2000, 38, 115–118. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
| Peptide Name | Amin Acid Sequence | Hydrophobicity (H) | Hydrophobic Moment (µH) | % Helicity | Net Charge z |
|---|---|---|---|---|---|
| AamAP1 | FLFSLIPHAIGGLISAFK | 0.904 | 0.435 | 55.56% | +1 |
| A3 | FLFSLIRKAIGGLISAFK | 0.746 | 0.517 | 100% | +3 |
| Control Gram-Positive Strains | ATCC | MIC Value (µM) | MBC Value (µM) |
| Staphylococcus aureus | 29213 | 5 | 5 |
| Enterococcus faecalis | 29212 | 10 | 10 |
| Enterococcus faecalis | 19433 | 12.5 | 12.5 |
| Staphylococcus epidermis | 12228 | 2.5 | 2.5 |
| MDR Gram-Positive Strains | ATCC | MIC Value (µM) | MBC Value (µM) |
| Staphylococcus aureus | 43300 | 5 | 5 |
| Staphylococcus aureus | BAA-41 | 5 | 5 |
| Staphylococcus aureus | 33591 | 5 | 5 |
| Enterococcus faecalis | BAA-2356 | 5 | 5 |
| Enterococcus faecium | BAA-2316 | 15 | 15 |
| Antibiotic | S. aureus (29213) | S. aureus (33591) | S. aureus (BAA-41) | E. faecalis (BAA-2356) | E. faecium (BAA-2316) |
|---|---|---|---|---|---|
| Levofloxacin | 0.5 | 10 | 10 | 27.5 | 12.5 |
| Chloramphenicol | 20 | 130 | 25 | 30 | 20 |
| Rifampicin | 0.025 | 0.04 | 0.005 | 0.03 | 7.5 |
| Erythromycin | 0.5 | 8 | 350 | 35 | 40 |
| MIC in Combination against Gram-Positive Bacteria MIC in Combination/(Individual MIC) | |||
|---|---|---|---|
| Bacterial Strain | Antibiotics (µM) | A3 (µM) | |
| S. aureus (29213) | Levofloxacin | 0.03215/(0.5) | 2/(5) |
| Chloramphenicol | 1.25/(20) | 2/(5) | |
| Rifampicin | 0.01/(0.025) | 0.125/(5) | |
| Erythromycin | 0.0625/(0.5) | 0.125/(5) | |
| S. aureus (33591) | Levofloxacin | 0.75/(10) | 1.5/(5) |
| Chloramphenicol | 30/(130) | 2.5/(5) | |
| Rifampicin | 0.00125/(0.04) | 2/(5) | |
| Erythromycin | 3/(8) | 2/(5) | |
| S. aureus (BAA-41) | Levofloxacin | 0.7/(10) | 3/(5) |
| Chloramphenicol | 2.5/(25) | 2/(5) | |
| Rifampicin | 0.0002/(0.005) | 3/(5) | |
| Erythromycin | 50/(350) | 2/(5) | |
| E. faecium (BAA-2316) | Levofloxacin | 1.25/(12.5) | 2/(15) |
| Chloramphenicol | 10/(20) | 2/(15) | |
| Rifampicin | 6.25/(7.5) | 2.5/(15) | |
| Erythromycin | 30/(40) | 1/(15) | |
| FIC Index | ||||
|---|---|---|---|---|
| Antimicrobial Combinations | S. aureus (29213) | S. aureus (33591) | S. aureus (BAA-41) | E. faecium (BAA-2316) |
| A3-kevofloxacin | 0.46 | 0.38 | 0.68 | 0.23 |
| A3-chloramphenicol | 0.46 | 0.73 | 0.5 | 0.63 |
| A3-rifampicin | 0.43 | 0.43 | 0.64 | 1.0 |
| A3-erythromycin | 0.15 | 0.78 | 0.54 | 0.82 |
| Gram-Positive Strains | A3 | Parent Peptide |
|---|---|---|
| S. aureus (29213) | 25 | No activity |
| S. aureus (BAA-41) | 30 | No activity |
| Peptide Conc. (µM) | 80 | 60 | 40 | 35 | 30 | 25 | 20 | 15 |
|---|---|---|---|---|---|---|---|---|
| S. aureus (29213) | 0.028% | 0.083% | 0.13% | 0.24% | 0.46% | 5.7% | 11.6% | 25.3% |
| S. aureus (BAA-41) | 0.013% | 0.098% | 1.2% | 2.6% | 4.7% | 13.2% | 27.4% | 35.2% |
| A3 | Mammalian IC50 (µM) | |
| HEK | Vero | |
| 33.2 | 26.1 | |
| Peptide Concentration (µM) | Hemolysis (%) |
|---|---|
| 1 | 0 |
| 5 | 0 |
| 10 | 5.1 |
| 20 | 16.8 |
| 40 | 36.1 |
| 60 | 47.9 |
| 80 | 49.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almaaytah, A.; Farajallah, A.; Abualhaijaa, A.; Al-Balas, Q. A3, a Scorpion Venom Derived Peptide Analogue with Potent Antimicrobial and Potential Antibiofilm Activity against Clinical Isolates of Multi-Drug Resistant Gram Positive Bacteria. Molecules 2018, 23, 1603. https://doi.org/10.3390/molecules23071603
Almaaytah A, Farajallah A, Abualhaijaa A, Al-Balas Q. A3, a Scorpion Venom Derived Peptide Analogue with Potent Antimicrobial and Potential Antibiofilm Activity against Clinical Isolates of Multi-Drug Resistant Gram Positive Bacteria. Molecules. 2018; 23(7):1603. https://doi.org/10.3390/molecules23071603
Chicago/Turabian StyleAlmaaytah, Ammar, Ahmad Farajallah, Ahmad Abualhaijaa, and Qosay Al-Balas. 2018. "A3, a Scorpion Venom Derived Peptide Analogue with Potent Antimicrobial and Potential Antibiofilm Activity against Clinical Isolates of Multi-Drug Resistant Gram Positive Bacteria" Molecules 23, no. 7: 1603. https://doi.org/10.3390/molecules23071603