
Glycoside Mimics from Glycosylamines: Recent Progress



Institute of Organic and Analytical Chemistry, UMR 7311, University of Orleans and CNRS, Rue de Chartres, BP 6759, 45067 Orleans CEDEX 2, France
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(7), 1612; https://doi.org/10.3390/molecules23071612
Submission received: 31 May 2018
/
Revised: 26 June 2018
/
Accepted: 28 June 2018
/
Published: 2 July 2018
(This article belongs to the Special Issue Glycomimetics: Design, Synthesis and Therapeutic Applications)
Abstract
:Glycosylamines are valuable sugar derivatives that have attracted much attention as synthetic intermediates en route to iminosugar-C-glycosyl compounds. Iminosugars are among the most important glycomimetics reported to date due to their powerful activities as inhibitors of a wide variety of glycosidases and glycosyltransferases, as well as for their use as pharmacological chaperones. As they provide ready access to these important glycoside mimics, we have reviewed the most significant glycosylamine-based methodologies developed to date, with a special emphasis on the literature reported after 2006. The groups of substrates covered include N-alkyl- and N-benzyl-glycosylamines, N-glycosylhydroxylamines, N-(alkoxycarbonyl)-, and N-tert-butanesulfinyl-glycosylamines.

1. Introduction
Carbohydrates are essential ubiquitous molecules that are involved in many fundamental biological events, such as cell-cell recognition or cell adhesion, glycolysis, gluconeogenesis, and signal transduction [1]. They are the most abundant biomolecules on Earth, thus providing very high incentive for the design of glycomimetics as prospective therapeutics [2,3,4,5,6]. Indeed, these analogues may interfere in biochemical pathways wherein carbohydrates play key roles and are associated with pathological disorders [7,8].
N-linked glycoconjugates in which the anomeric oxygen of glycosides has been replaced by nitrogen are also natural and valuable sugar-related derivatives [9]. These enclose N-glycosyl-amino acids and N-glycopeptide derivatives [10,11,12,13,14] (erythropoietin (EPO) is a well-known example), nucleosides, and nucleotides [6,14]. As an aside, N-glycoside linkages may also be embedded within many other structurally-diverse natural products such as anthraquinone mycorhodin [15,16], anti-carcinogenic N-glycosyl indoles akashines A, B, C [17], staurosporine [18,19,20], and rebeccamycin [21], or ansacarbamitocin antibiotics [22].
Small N-glycosyl mimics of glycosides, also known as “glycosylamines”, are per se attractive targets, as they are capable of inhibiting enzymes acting on glycosides [23,24].
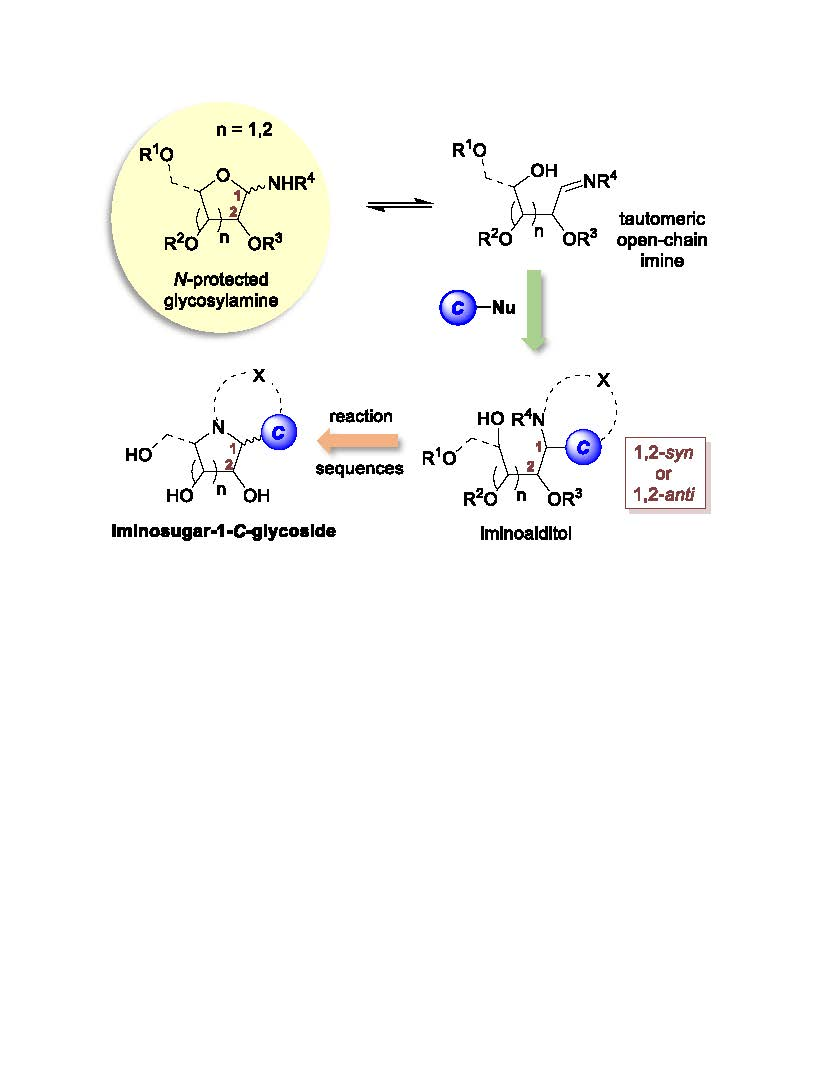
Interestingly, like sugars, some of these N-glycosyl compounds exhibit mutarotation [25]. They rearrange to a tautomeric open-chain imine and are, therefore, capable of reacting with a variety of carbon nucleophiles to provide 1,2-syn or 1,2-anti aminoalditols in good yields and good levels of stereoselectivity. After activation of the pendant alcohol, cyclization, and further deprotections, related iminosugar-C-glycosyl compounds are obtained in good yields (Scheme 1) [26].
Iminosugars are found in a wide variety of microorganisms and plants. These form, probably, the most important class of carbohydrate mimics reported to date [27,28,29,30]. However, one of the major drawbacks associated with imino analogues of glycosides is their instability caused by the lability of the N,O-acetal function, which prevents their use as biological probes or drug candidates. As glycoside mimics, 1-deoxyiminosugars have, thus, gained considerable importance as bioactive molecules. A typical example is 1-deoxygalactonojirimycin (Galafold®), now used to treat Fabry disease [31]. However, unlike this notable example, absence of structural and configurational information of normal (α- or β-linked) glycosides may prevent their use as drugs. This problematic feature could, to some extent, be prevented by relocating pieces of structural information in the nitrogen substituent. For instance, it may be the case of N-[2-hydroxyethyl]-1-deoxynojirimycin (Diastobol®) [32], and N-butyl-1-deoxynojirimycin (Zavesca®) [33] that are used for the treatment of diabetes mellitus type 2 and Gaucher disease, respectively. By analogy, iminosugar-C-glycosides may, thus, be more significant as stable glycoconjugate or oligosaccharide mimetics of biological and therapeutic interest, since a strong and non-hydrolyzable C–C bond has replaced the labile glycosidic linkage of real iminoglycosides [26,27].
Iminosugar-1-C-glycosides are powerful inhibitors of a wide variety of glycosidases [27,28,29,30], and glycosyltransferases down to a femtomolar range [34,35,36]. They may also be employed as pharmacological chaperones to treat deficiencies characterized by improperly folded proteins [37].
Their chemical syntheses and therapeutic applications are well documented [26]. Although recent progress in their de novo preparation through the diversity oriented synthetic approach by elegant asymmetric organocatalyzed processes [38,39,40] have been reported, one of the best methods remains the addition of C-Nu to N-glycosylamines.
In this review, we have compiled the most significant glycosylamine-based -methodologies developed to date, with emphasis on the literature reported after 2006. They involve N-alkyl- and N-benzyl-glycosylamines, N-glycosylhydroxylamines, N-(alkoxycarbonyl)-, and N-tert-butanesulfinyl-glycosylamines.
2. N-(Benzyl)- and Other N-(Alkyl)-N-Glycosides
Some aspects of the chemistry of N-benzylglycosylamines have been reviewed by Behr and Plantier-Royon in 2006 [41], and recent progress in this area will be outlined here.
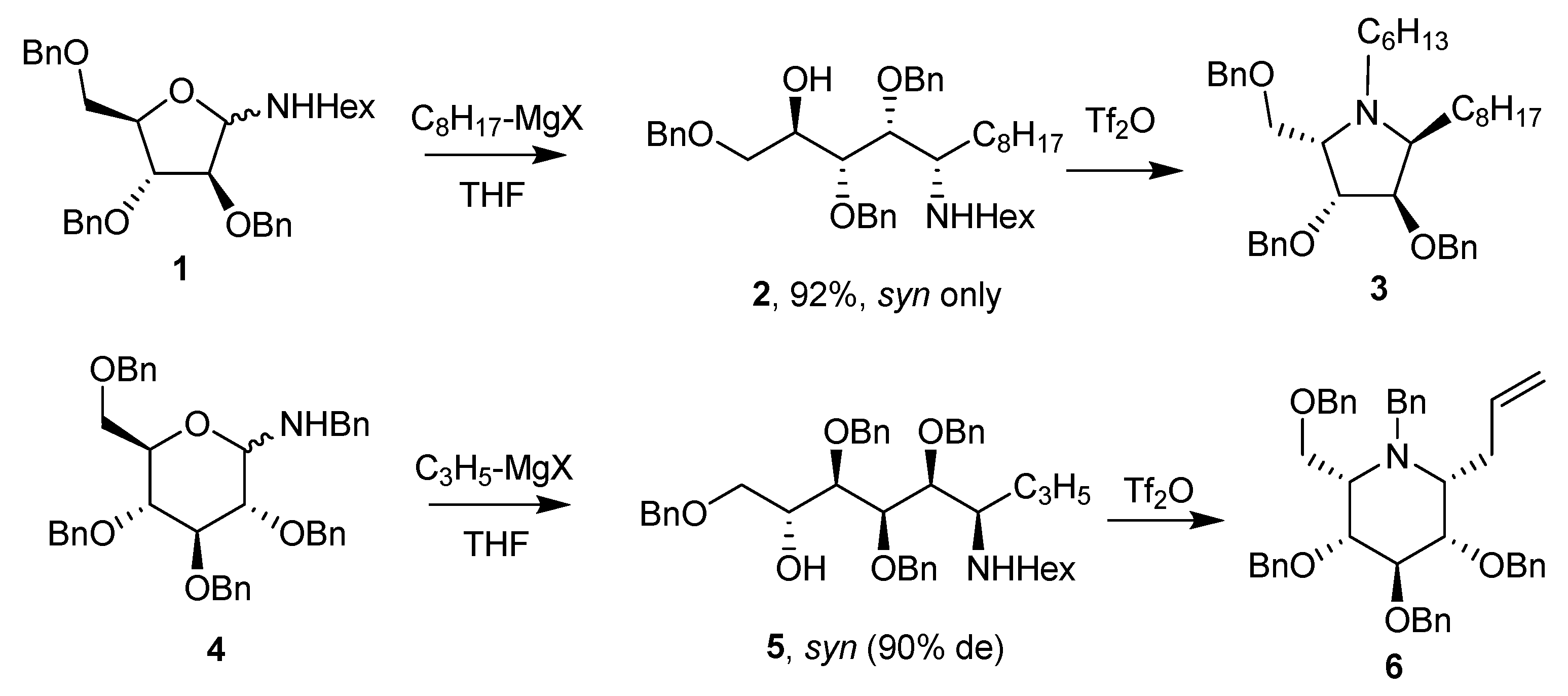
Pioneering studies on glycosylamines have been reported by Nicotra and coworkers since 1989 [42]. These authors have shown, for the first time, that the addition of Grignard reagents to N-benzyl and N-alkyl glycosylamines derived from perbenzylated pentofuranoses or hexopyranoses followed by a simple cyclization procedure afforded a short and convenient approach to imino-C-glycosides in the pyrrolidine and piperidine series [43,44]. The procedure is illustrated in Scheme 2 (see compounds 1–6) from a d-arabinofuranosylamine, using octylmagnesium bromide, and from a d-glucopyranosylamine using allylmagnesium bromide, and cyclization promoted by reacting the intermediate amino alditol with triflic anhydride.
In the gluco series, the final product is a mimic of an α-glycoside (as in 6), whereas in the manno series, the cyclized product is a mimic of a β-glycoside [43,44]. Also it is important to note that cyclization at a secondary position, as in 2 and 5, leads to an inversion at this position, d-arabinof substrates leading to l-xylof products (as in 3) and d-glucop substrates leading to l-idop products (as in 6).
The Nicotra group further investigated this process to prepare the significant DNJ derivatives (d-gluco epimer of 6) [45,46]. This requires the oxidation at C-5 of the addition intermediate (as in 5) and cyclization by a reductive amination, a reaction known to favor axial hydride delivery and, hence, formation of the “d” stereoisomer from 5 [47]. This sequence was made possible providing the nitrogen atom was protected by a Fmoc group during oxidation.
The Nicotra procedure was also used more recently by other groups. 1-C-allyl iminosugar derivatives in the α-d-gluco, β-l-ido, and α-d-xylo series were prepared by Overkleeft et al. [48]. While the synthetic sequences are similar to those already described, the Leiden group chose to use N-p-methoxybenzyl glycosylamines as substrates in order to facilitate the selective cleavage of the N-alkyl substituent and replace it by a carbamate for further functionalization of the allyl group mainly by cross-metathesis. Allylation of the N-p-methoxybenzyl glucosylamine was also exploited by Vankar et al. [49] in order to reach an advanced synthetic intermediate in their synthesis of novel hydroxylated indolizidines and pyrrolizidines. The addition of vinyl-magnesium bromide to N-benzyl pentopyranosylamines (d-xylo, l-arabino) was a key step in recent work of the Fleet’s group leading to the total synthesis of calystegines B2 and B3 [50], as well as of the Peczuh work aiming at amino septanosyl conjugates [51,52]. An improvement of the formation of glycosylamines (i.e., faster reaction times and better yields) using iodine in the presence of imidazole was reported by Chagnault et al. [53].
From a stereochemical viewpoint, the addition of the organometallic reagent appears to be controlled by the group at C-2 (usually an O-benzyl group) of the substrate (Cram chelate) leading to the 1,2-syn diastereoisomer, predominantly or exclusively and, hence to a 1,2-cis configuration after cyclization to an iminosugar. On the other hand, the anti-configuration was only observed in glycosylamines derived from 2,3-O-isopropylidene ribofuranose derivatives and related scaffolds, which afforded iminosugars with a 1,2-trans configuration (Rao et al. [54,55,56,57] (see for example Scheme 3, compounds 7−10), Behr et al. [58,59,60]). For a rationale, see [54].
As an exception, the anti addition product was observed by Zhuang et al. [61] in the reaction of the glycosylamine derived from 2,3-O-isopropylidene-d-erythrofuranose with a Grignard reagent (Scheme 4). It was suggested that the reaction takes place by way of a seven-membered-ring complex (e.g., A).
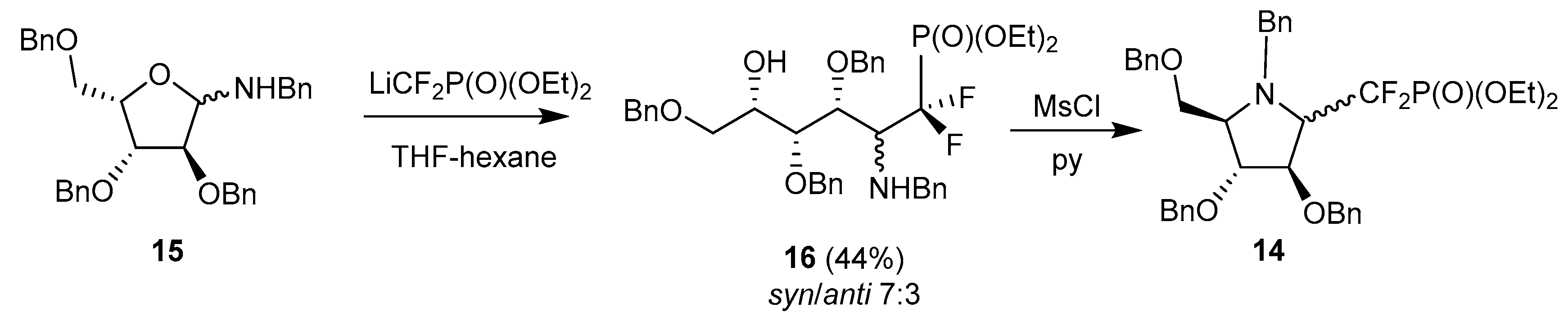
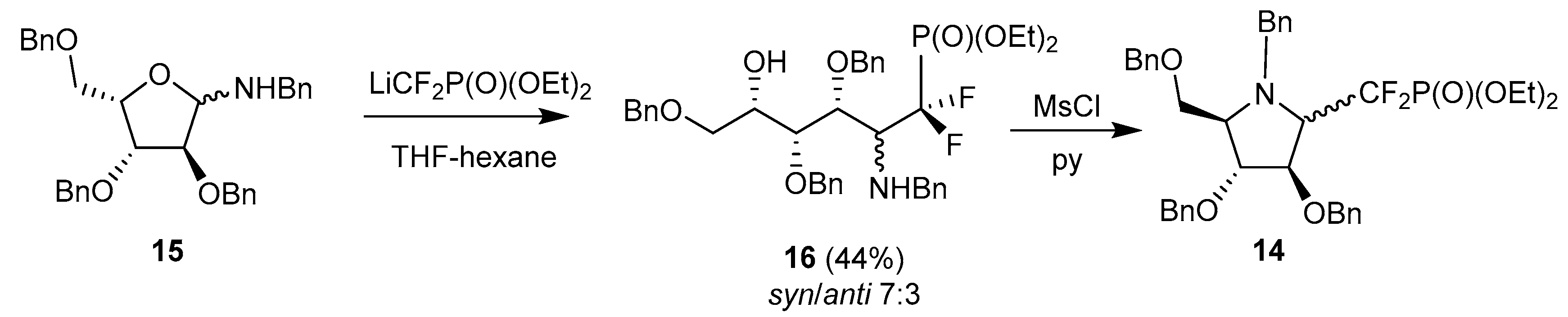
Several research groups have adopted this strategy to reach biologically-significant iminosugar derivatives. In early studies, Behr, Guillerm and coworkers took advantage of this methodology to prepare potential glycosyl transferase inhibitors; in particular they investigated the addition of lithium difluoromethyldiethylphosphonate to N-benzylpentofuranosylamines, as an approach to glycosylphosphate analogs, such as 14, from L-xylofuranosylamine 15 (Scheme 5) [62].
These phosphonates were later deprotected and tested as antifungal agents, together with other pyrrolidines related to DMDP and obtained by the Nicotra’s procedure [63]. The same group also took advantage of the addition of allyl Grignard reagent to glycosylamine 15 and to a related 5-deoxy-l-lyxofuranosylamine to prepare 6-deoxy-homoDMDP and iminosugar-ferrocene conjugates, respectively [59,64]. Interestingly, Behr et al. demonstrated in 2012 that the allylation of free glycosylamines could be achieved using indium metal in MeOH, with excellent syn stereoselectivity, and the two steps could be achieved in one pot (Scheme 6) [65]. Application of this protocol to glycosylamines derived from (R)- and (S)-α-methylbenzylamine revealed that the chiral group did not mediate the stereoselectivity of the reaction.
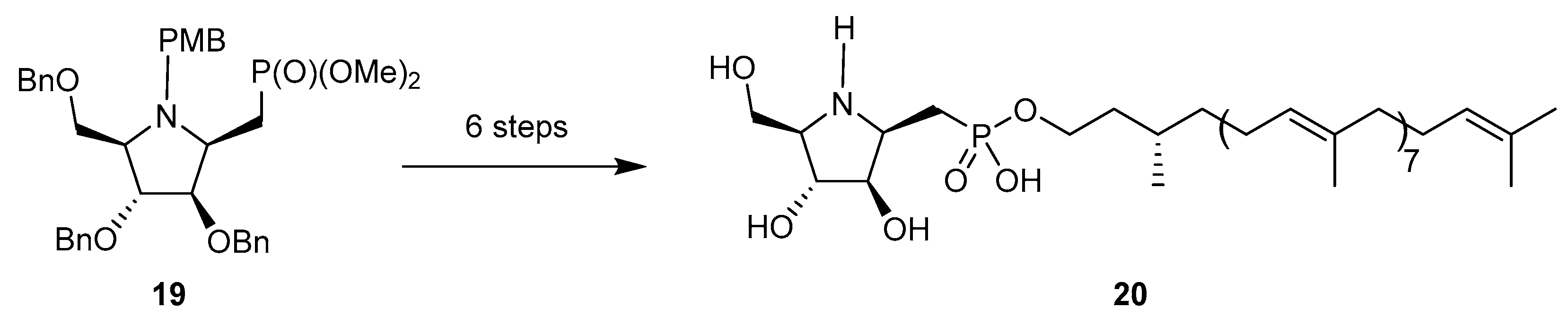
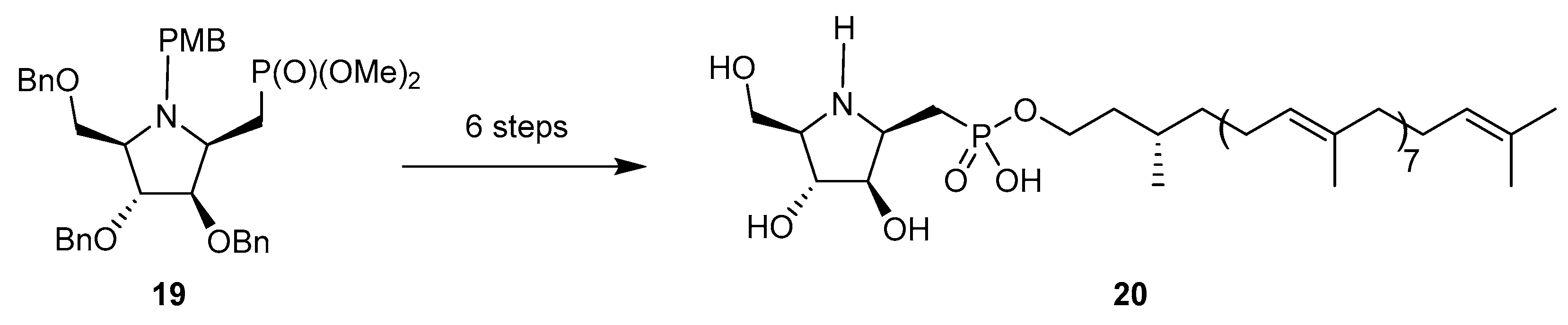
The methylenephosphonate analogs of 14 (in both configurations) were prepared by Eustache et al. starting from the N-p-methoxybenzyl glycosylamine equivalent of 15 [66]. Interestingly, the addition of the lithium methylphosphonate was greatly facilitated when the glycosylamine was first treated with BF3·Et2O. The ‘β’anomer 19 was then converted in six steps into compound 20 (Scheme 7), a remarkable mimic of DPA (β-d-arabinofuranosyl-1-monophosphoryl decaprenol), the glycosyl donor involved in the biosynthesis of arabinans in mycobacteria. This compound is endowed with good MIC values toward mycobacteria, comparable to ethambutol.
In our own work, we have prepared a series of 1-C-alkylated imino-l-iditols using Nicotra’s procedure, in order to compare the activity of these compounds as β-glucocerebrosidase inhibitors with the α-d-gluco epimers [67]. Furthermore, the synthesis of new glucosylceramide mimics based on an iminoxylitol core (e.g., 21, Scheme 8) was achieved form N-benzyl-d-xylopyranosylamine 22 by the stereoselective addition of allylMgBr, cyclization (to give 23) and elaboration of the allyl group into a 2,3-di-O-acyl or 2,3-di-O-alkylglyceryl residue [68]. Compound 21, a potent inhibitor of this enzyme (Ki = 1.8 nM), was found to exhibit a significant activity as chaperone of the mutant form of β-glucocerebrosidase carrying the L444P mutation. This mutation is responsible for the neuronopathic form of Gaucher disease, for which there is currently no treatment.
Compounds such as 21 which act as pharmacological chaperones, constitute new leads for the treatment of this severe form of Gaucher disease, which cannot be treated by Enzyme Replacement Therapy. In more recent work, with the goal of preparing iminosugar derivatives carrying a 1-C-propargyl group for further functionalization, we have investigated the addition of TMS-propargyl bromide to the N-benzyl-d-xylofuranosylamine (ent-15); best conditions consisted in using Zn dust and performing the reaction under ultrasound activation [69]. The reaction gave the expected product (syn relative configuration, 60% d.e.) in 62% yield after cyclization, but the conditions were found to be difficult to reproduce, and better results were obtained from the corresponding N-sulfinyl glycosylamines (vide infra).
A number of interesting methodologies involving in situ formation of the glycosylamines have been reported. In particular, Baskaran and coworkers have developed elegant methodologies in which the glycosylamine is trapped by various nucleophiles:
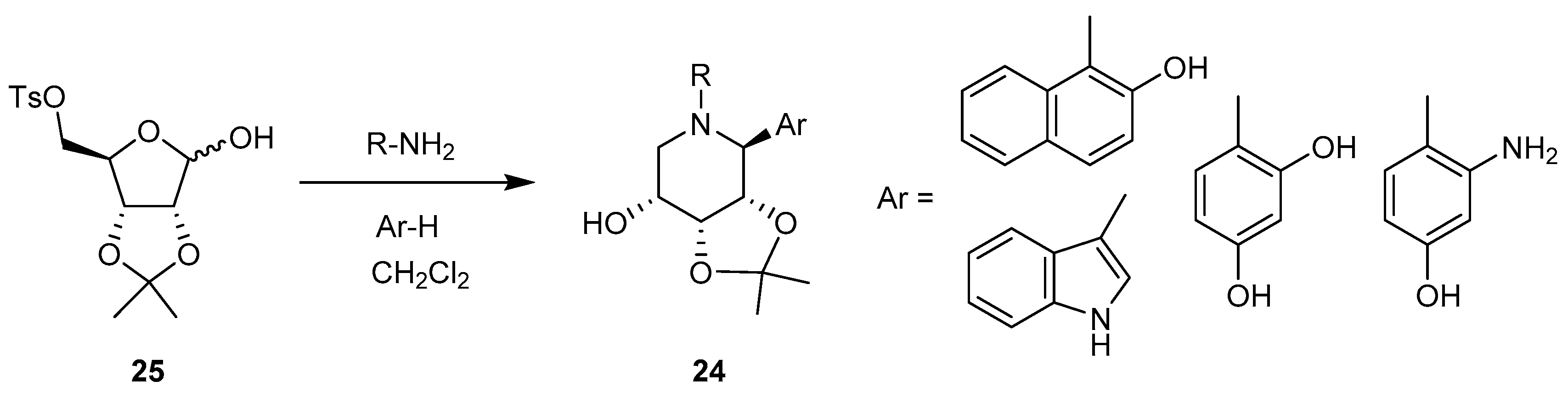
Electron-rich aromatic groups [70]: a great diversity of C-aryl iminosugars 24 have been generated from 25 by the general methodology outlined in Scheme 9.
In addition, using an amine carrying an electon-rich aromatic substituent, the procedure led, in one step, to innovative polycyclic systems, such as 26 (Scheme 10).
In these reactions, the in situ-generated imine 27 undergoes intramolecular N-alkylation by the tosylate leading to a cyclic iminium cation 28, which is sufficiently reactive to promote an electrophilic substitution of the electron-rich aromatic compounds. All reactions occur in high yield (68–92%), and high stereoselectivity, the 1,2-trans stereoisomer being exclusively formed.
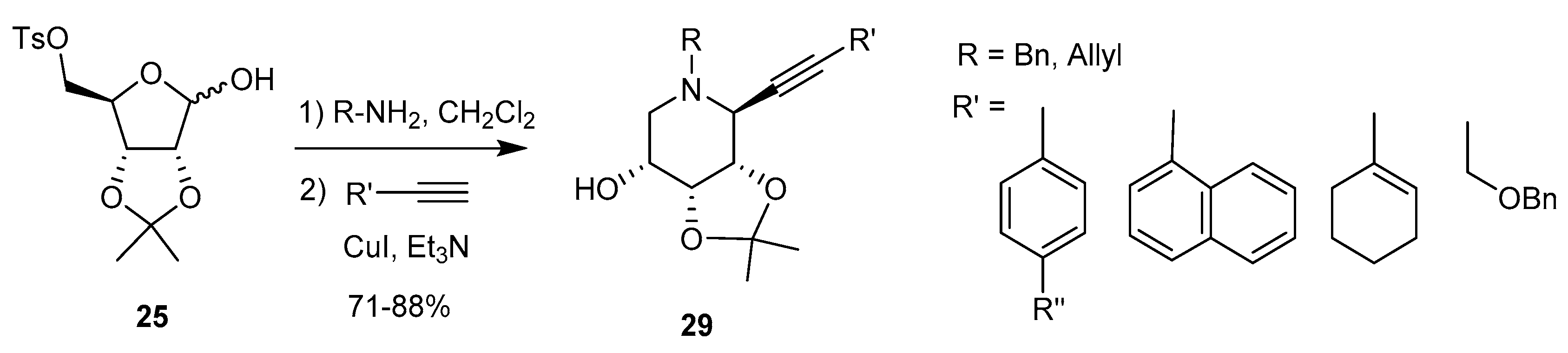
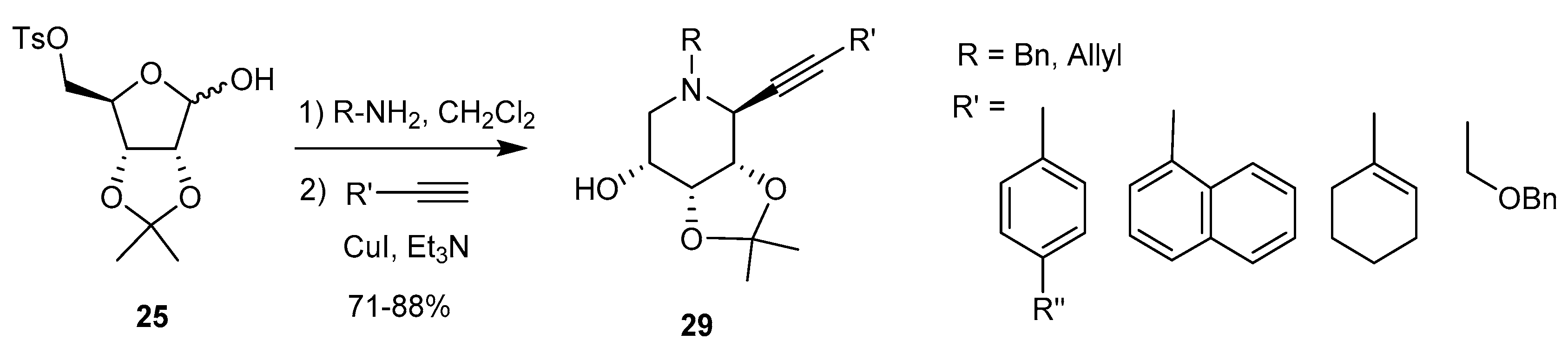
Alkynyl anions [71]: A further extension of this work allowed the introduction of an alkynyl group at the ‘pseudoanomeric’ position: in situ formation of the cyclic iminium ion as before followed by reaction with a terminal alkyne in the presence of a Cu(I) salt gives access to 1-C-alkynyl piperidine iminosugar derivatives 29, in high yield and complete stereoselectivity (Scheme 11).
Various polycyclic systems were obtained from reactions between the nitrogen substituent (allyl, o-bromobenzyl) and the alkynyl group. The authors also showed that pyrrolidine derivatives could be obtained by a similar procedure starting from 4-O-mesyl-2,3-O-isopropylidene-l-rhamnopyranose, cyclization occurring at C-4 of this hexopyranose.
Nitromethyl anions [72]: In a very simple, two-step one-pot procedure, piperidine iminosugars carrying a 1-C-nitromethyl group were obtained from d-ribof tosylate 25 by reaction with a primary amine in the presence of Et3N, followed by the addition of the nitromethyl anion to the in situ-generated iminium cation. This led to nitromethyl derivatives, such as 30 (Scheme 12), with 1,2-trans diastereoselectivity exclusively. By similar reactions from the d-lyxo isomer of 25, the epimers 31 were obtained, still with dominance of the 1,2-trans isomer (d.r. = 3.1)
Pyrrolidine analogs (e.g., 32) were prepared from 4-O-mesyl-2,3-O-isopropylidene-l-rhamno-pyranose by the same sequence of reactions. Owing to the rich chemistry of the nitromethyl group, 1-C-nitromethyl iminosugar derivatives constitute precursors of a wide variety of further glycoside mimics, as well as to novel polycyclic compounds. Several examples of further functionalization/cyclization by reactions of the nitromethyl group with the nitrogen substituent (allyl, propargyl) were reported by Baskaran and coworkers [71].
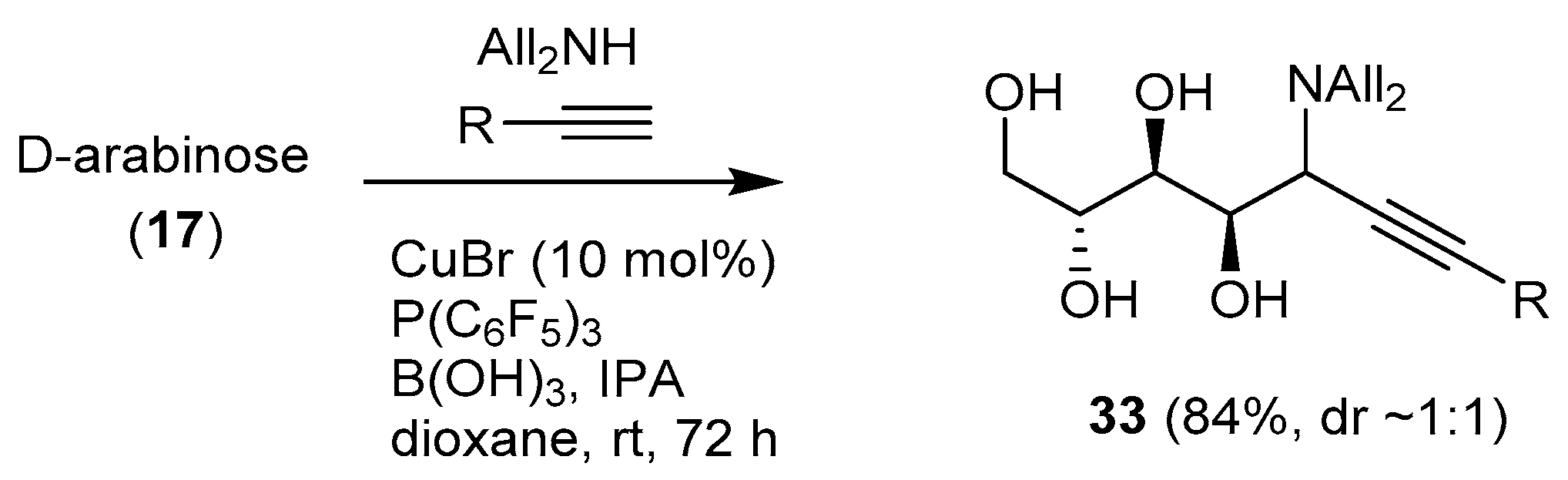
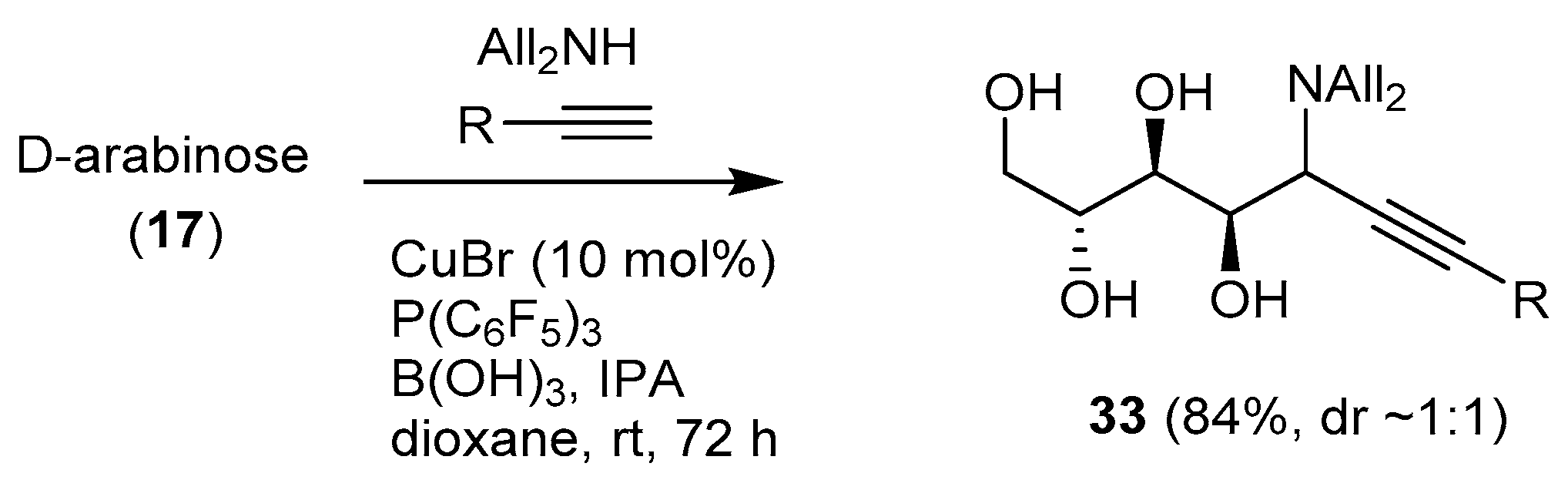
A Cu(I)-catalyzed aminoalkynylation of unprotected aldoses was reported by Kanai et al. [73] (Scheme 13). The one-pot reaction of free sugars (pentoses, d-galactose, l-fucose) with diallylamine, a terminal alkyne, catalytic CuBr, a boron reagent (boric acid), and a ligand (P(C6F5)3) afforded the corresponding chain-extended aminoalditols 33 in good to very good yields and with rather low diastereoselectivity (with some exceptions, the anti product being predominant).
The reaction was applied to substrates of biological significance, including a biotinylated alkyne derivative. The addition products however were not cyclized owing to the unprotected nature of the substrate.
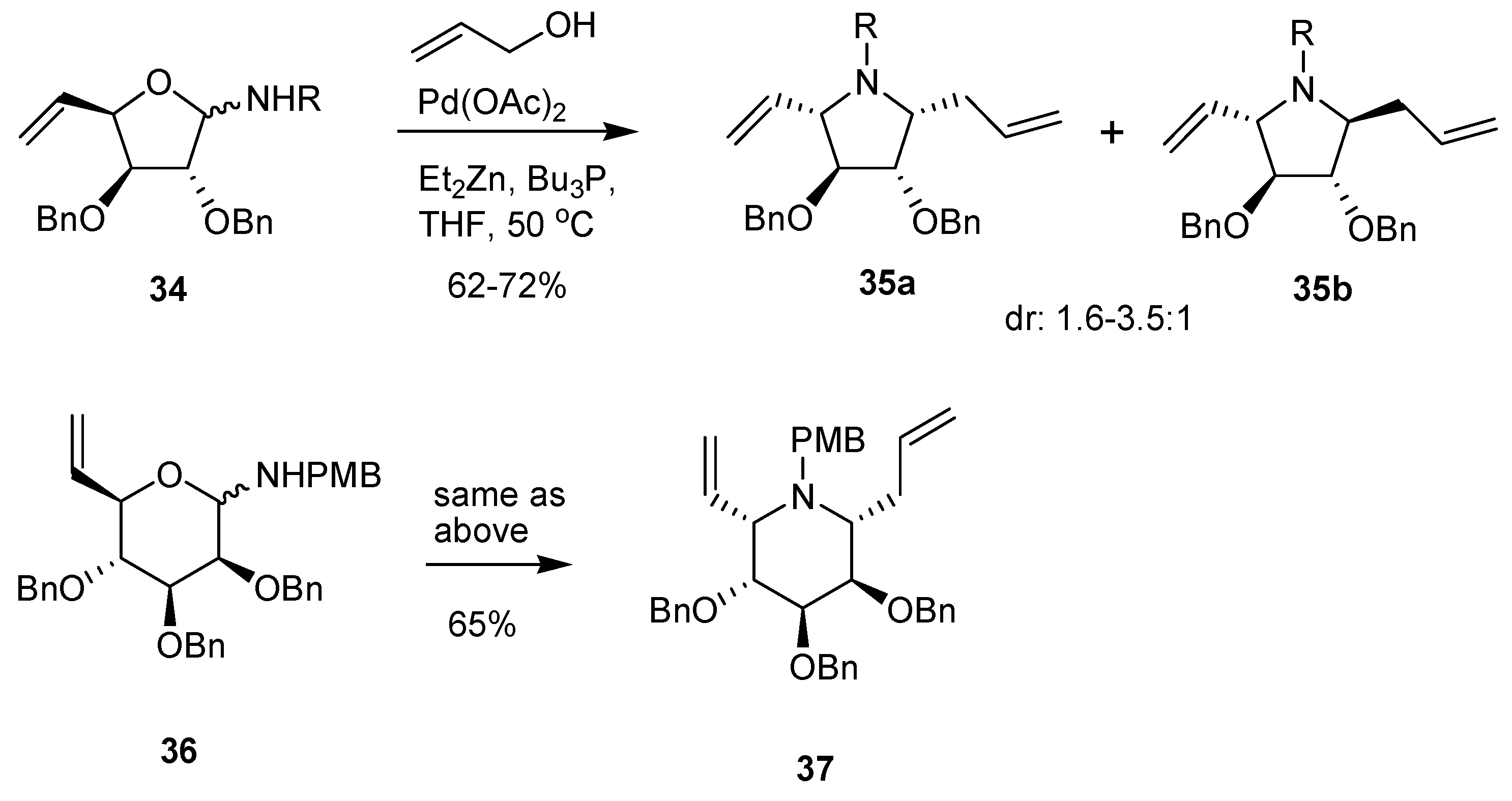
In very recent work, Rao et al. reported an interesting Pd-mediated double allylation process in which the C-N bond is created in the same step as the C-C bond [74]. This procedure requires a substrate (furanosyl- or pyranosylamine) carrying a vinyl group at C-4 or C-5, respectively. Thus, for example, glycosylamine 34 was submitted to reaction with allyl alcohol in the presence of diethylzinc, tributylphosphine, and catalytic Pd(II) acetate, in THF at 50 °C for 24 h, to give directly the cyclized 1-C-allyl, 4-C-vinyl pyrrolidine 35, with low stereoselectivity, however (Scheme 14).
The reaction has also been applied successfully to pyranosylamines. For example, the 6,7-unsaturated mannopyranosylamine 36 gave the 1-C-allylated L-gulo iminoalditol 37 as a single stereoisomer (Scheme 14). The final products obtained (e.g., 37) contain two vinyl groups which were combined by ring-closing metathesis to form several analogs of the calystegins. The mechanism involves a Pd-mediated formation of a nucleophilic allylzinc species which react with the open-chain form of the glycosylamine to give an intermediate containing an allylic alcohol function presumed to be in the form of a ZnEt salt. This undergoes an intramolecular Tsuji-Trost electrophilic allylation of the amine function, thus leading to the formation of the C-N bond and ring-closure.
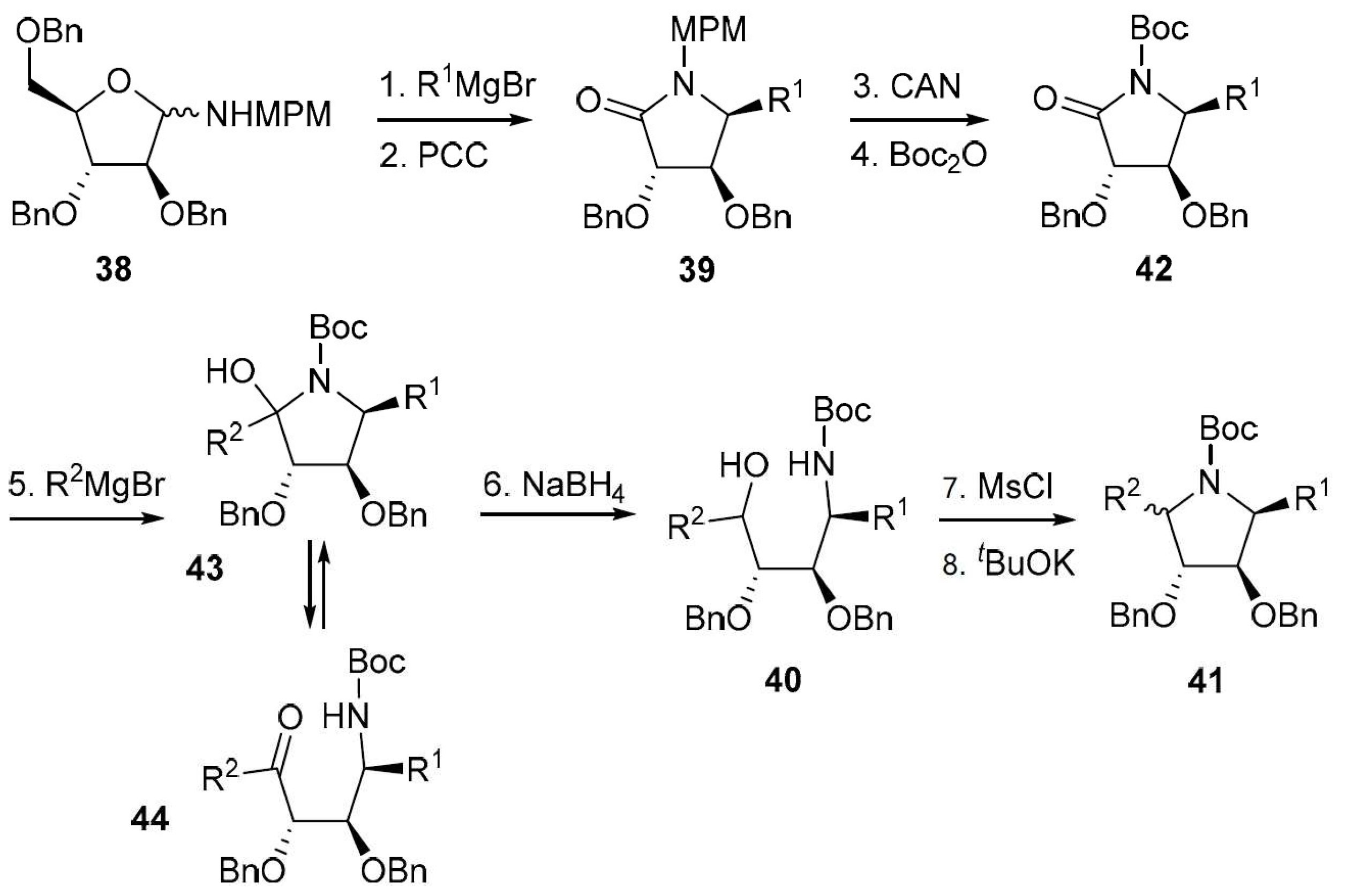
Finally, a modified version of the Nicotra’s procedure was reported in the 1990s by Yoda et al. [75,76]. In this work, the product resulting from the addition of a Grignard reagent (R1 = C4H9, C9H19, Bn) to a glycosylamine such as 38 was submitted to an oxidative chain shortening leading to a carboxylic acid derivative which underwent spontaneous cyclization to a lactam (e.g., 39) (Scheme 15). The interest of this procedure is the possibility to use the lactam for a second alkylation step, by way of the addition of an organometallic reagent (e.g., R2 = C4H9, C9H19, Bn), reduction of the resulting hemiaminal to give 40, and cyclization by an SN2 process.
This sequence leads to pyrrolidine derivatives carrying two different R groups at C-1 and C-4, such as 41. The authors used this procedure to prepare the natural product (+)-preussin [76].
3. N-Benzyl-N-Glycosylhydroxylamines
3.1. Synthesis
In general, 1,2-anti aminoalditol derivatives could be synthesized in a more diverse and effective manner via the addition of lithium and magnesium reagents to N-benzyl-N-glycosyl-hydroxylamines. This approach is indeed more general than the ones described for N-(benzyl)-N-glycosides which require the use of 2,3-O-isopropylidene-protected ribo or lyxo glycosylamines or the isolation of the minor diastereomer of the open-chain adducts.
N-benzyl-N-glycosylhydroxylamines were frequently prepared by heating mixtures of N-benzylhydroxylamine and sugar hemiacetals at 110 °C for 30–60 min under-solvent free conditions [77,78,79]. Alternatively, they were prepared by stirring a suspension of N-benzylhydroxylamine hydrochloride, 3 Å molecular sieves and the hemiacetal derivatives in dry pyridine at room temperature [80].
Various N-benzyl-N-hydroxy-glycosylamines 45a–45h (see Table 1), derived from furanoses and pyranoses, have been prepared and isolated (up to multigram scale) in moderate to good yields (61–88%).
3.2. Addition Reactions
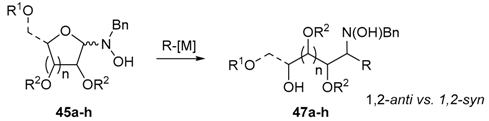
The two anomers of these sugar-hydroxylamines exist in equilibrium with the masked open-chain nitrones (46). Although the equilibrium is largely or completely shifted to cyclic hydroxylamines, these forms undergo reactions with various lithium and magnesium reagents at low temperature to give the corresponding adducts (47), albeit often in inseparable mixtures of diastereomers (route a, Scheme 16) [77,78,80,81].
The results of the addition reactions are summarized in Table 1. In general, an excess of organometallic species in Et2O or THF was added to cooled solution (−75 or −30 °C) of 45a–45h in THF to provide 47a–47h in good yields (52–95%) and moderate to high levels of diastereoselectivity in favor of the 1,2-trans adducts (see entries 1–9, 11, 14) [77,78,81].
In contrast, hexopyranosylamines 45f, 45g, and 45h did not react with 2-lithiothiazole under these conditions [79]. Treatment of these hydroxylamines with 5 equiv. of 2-thiazolylmagnesium bromide in THF at 0 °C was mandatory to afford the diastereomers of thiazolylalkyl-hydroxylamines 47f, 47g, and 47h in good overall yields, but modest selectivities (see entries 10, 13 and 16). Of note, the major diastereomers of 47f and 47h were both syn-adducts, whereas the main product of 47g was an anti-adduct [79]. Obviously, the carbon stereocenter adjacent to the nitrone group affects the selectivity of these addition reactions. Moreover, a 1:1 mixture of diastereomeric adducts were obtained with allylmagnesium bromide (entries 12 and 15). The stereoselectivity was not improved by lowering the reaction temperature to −50 °C, while, at this temperature, the yields dramatically decreased [78].
As a rule, the anti selectivities may be rationalized by a preferential conformation (B) adopted by the open-chain nitrone form 46 due to the metal coordination, involving the nitrone oxygen and the free hydroxyl group. As a consequence, the addition occurs to the less hindered side of this complex to give the anti-product (Scheme 16).
3.3. Cyclizations
Due to the usual difficulty in separating the two diastereomers of compounds 47, the open-chain products were often subjected to numerous synthetic sequences. In general, reductive N-dehydroxylation using a Zn–Cu couple [82] was achieved in good yield (ca. 90%). Then, the resulting benzylamino-1-deoxyalditol derivatives 47 were transformed into pyrrolidine and piperidine iminosugars following standard activation of the free hydroxyl group and cyclization. MsCl in the presence of Et3N was used for the synthesis of N-benzyl-N-glycosides. For the cyclization of compounds 47f–h which do not cyclize under the present conditions, a catalytic amount of tetramethylethylenediamine (TMEDA) was added as a promoter [83], followed by heating the crude product in MeCN at 85 °C.
Overall, collections of 1,2-trans iminosugar-C-glycosides were successfully prepared. Through the stereoselective addition of 2-lithiothiazole and 2-thiazolylmagnesium bromide, access to dideoxyiminoheptitols (e.g., piperidine homoiminosugars) from pyranoses was conveniently achieved via a formal one-carbon chain elongation [79]. This thiazole-to-formyl unmasking protocol was further utilized to generate aza-C-disaccharides as methylene isosteres of O-disaccharides [77].
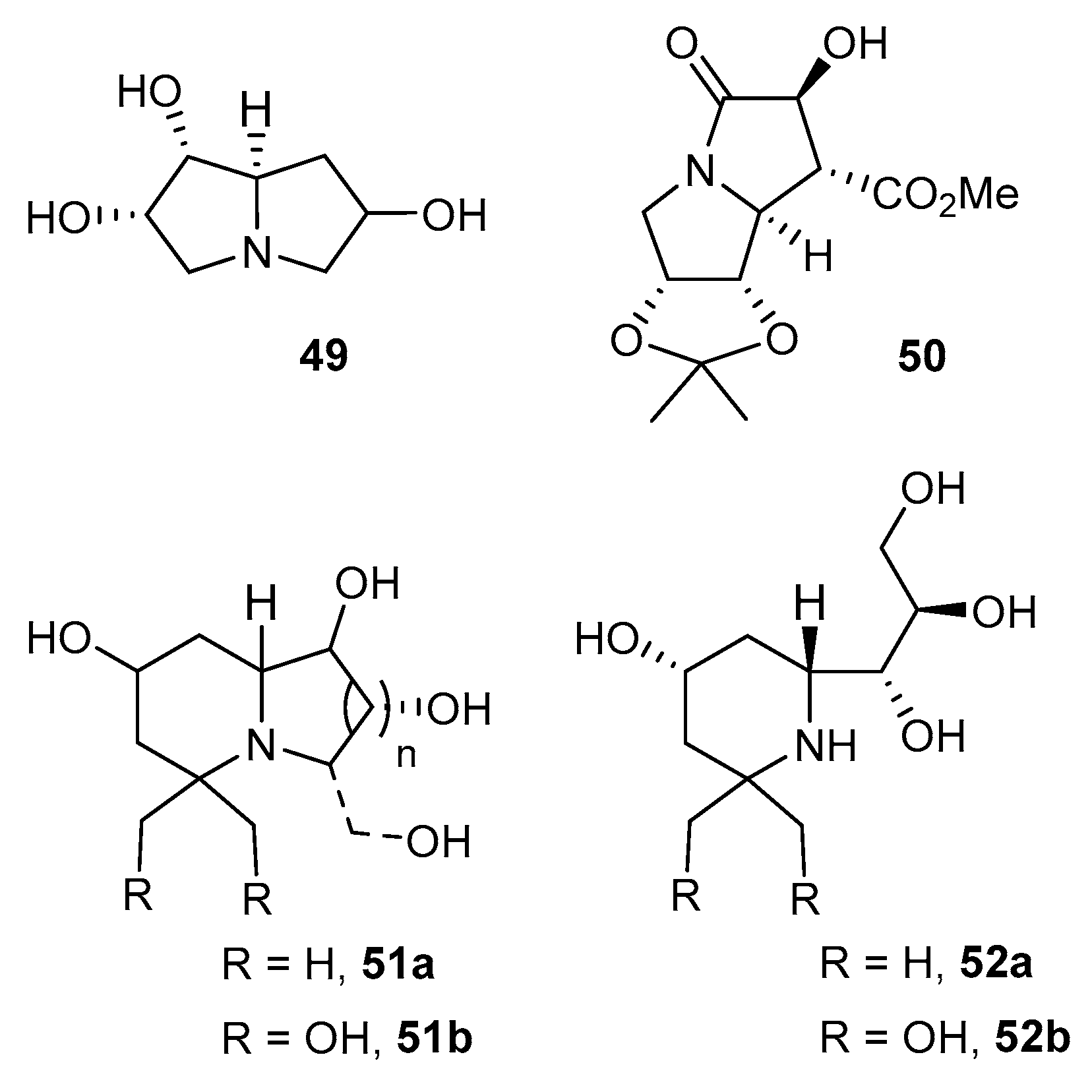
As outlined in Scheme 16 (route b), N-glycosylhydroxylamines may also react as masked nitrones in 1,3-dipolar cycloaddition reactions (see 48). This synthetic approach was undertaken by the group of Argyropoulos to synthesize enantiomerically-pure trihydroxypyrrolizidines of type 49 [84], and by Goti and co-workers to prepare highly-functionalized pyrrolizidine 50 [85]. The nitrones could serve further to react via [1,4]-sigmatropic rearrangement to construct iminosugars of various heterocyclic cores (e.g., 51a, 51b, 52a, 52b, Figure 1) [86].
4. N-(Alkoxycarbonyl)-N-Glycosides
1,2-Syn aminoalditols may also be efficiently synthesized through the addition of silylated nucleophiles to N-(benzyloxycarbonyl)-glycosylamines under Lewis acid catalysis, opening an approach to iminosugar-C-glycosides carrying a greater diversity of aglycon moieties (e.g., allenyl, oxoalkyl, etc.).
4.1. Synthesis
Studies on the addition of silicon-based nucleophiles to semicyclic N,O-acetals possessing an exocyclic nitrogen atom protected by an alkoxycarbonyl group were pioneered by Kobayashi on simple substrates [87,88], and subsequently applied to the sugar series [89].
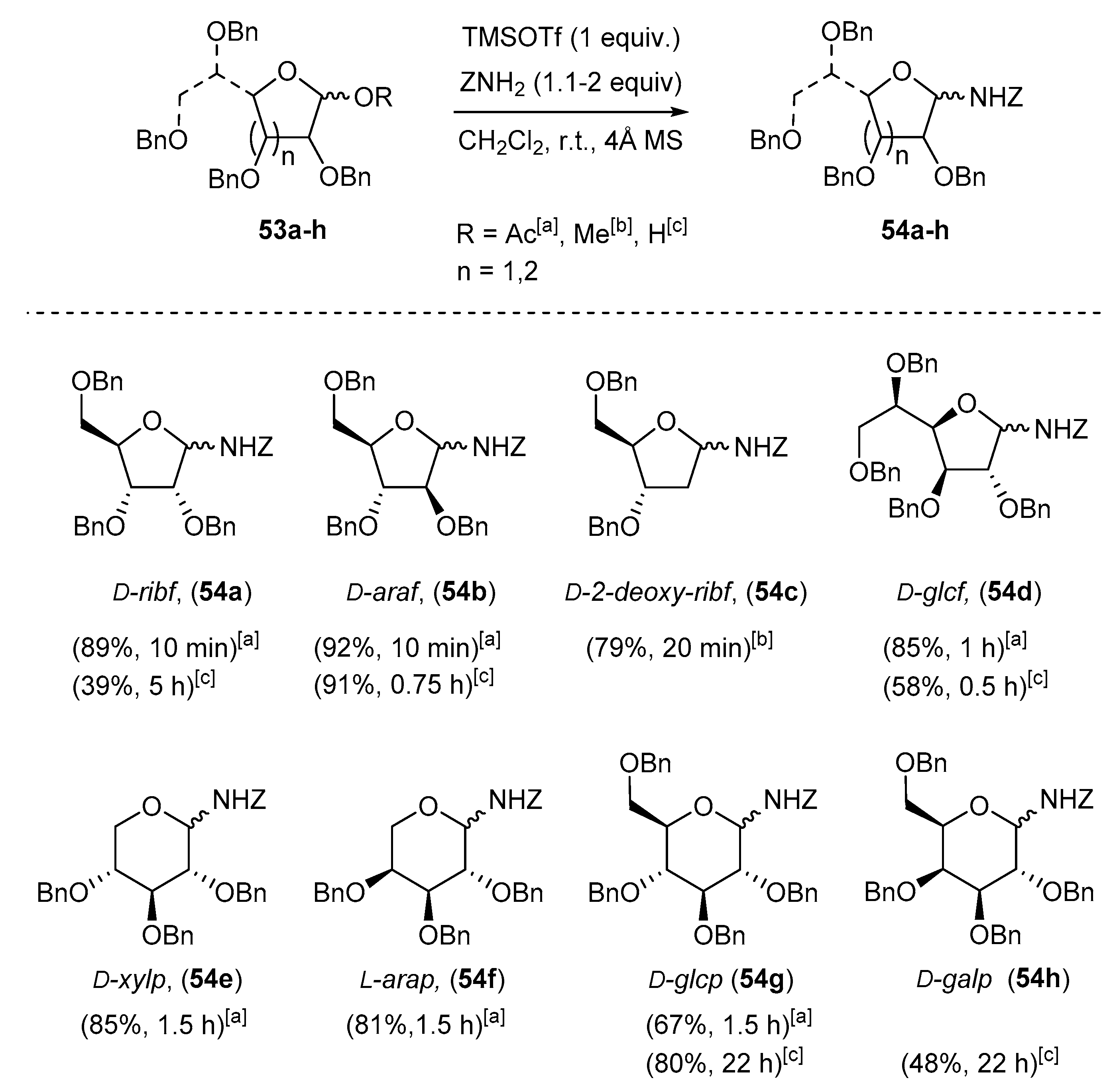
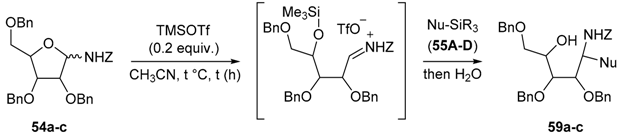
Carbohydrates derived N-benzyloxycarbonyl-N-glycosides are typically protected by O-benzyl substituents (Scheme 17). Their preparation proceeds through dropwise addition of trimethylsilyl trifluoromethanesulfonate (TMSOTf) to a suspension of benzyl carbamate (1.1–2 equiv.) and related glycosyl acetates 53 in CH2Cl2 at room temperature [89,90,91,92]. Since benzyl carbamate is a weak nucleophile and glycosylamines are unstable under aqueous conditions, addition of 4 Å molecular-sieves (MS) is essential to prevent the formation of hydrolyzed products [89].
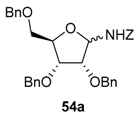
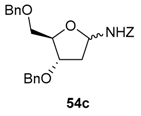
A mixture of α- and β-O-benzyl-N-benzyloxycarbonyl glycosylamines (d-ribof, d-araf, d-glcf, d-glcp, d-xylp, l-arap, etc.) were isolated in good yields (>67%). 2-Deoxy-d-glycofuranosyl-amines were obtained following the same conditions from the O-methyl glycoside derivatives (e.g., 54c) [89]. The choice of the protective groups may be further extended to acetates, but it was shown afterwards that a 2-O-acetyl group is detrimental to the chain extension reaction. An electron withdrawing group at the α-position will, of course, retard the formation of the assumed N-acyliminium intermediate [89].
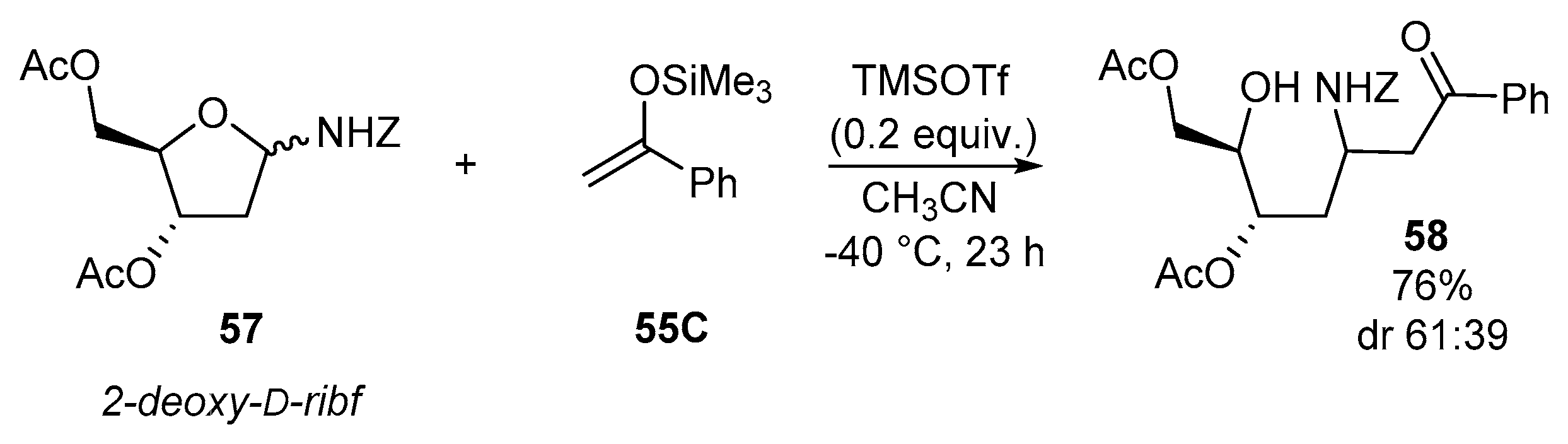
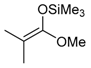
Indeed, no reaction was observed by the addition of silyl enolate 55A to benzyl-(2,3,5-tri-O-acetyl-d-ribofuranosyl)carbamate 56 (Scheme 18), whereas the analogous deoxyribose derivative 57 underwent the addition reaction in good yield (76%) albeit with low stereoselectivity (dr 61:39) [89] (Scheme 19). O-silylated groups could also possibly be employed, but no example has been reported in the sugar series [88].
Using these conditions, hexopyranose-based N-(benzyloxycarbonyl) N,O-acetals were also obtained, but in yields lower than for the more entropically-favored five-membered ring sugars and pentopyranose derivatives. O-Benzyl-N-(2,3,4,6-tetra-O-benzyl-d-glucopyranosyl)carbamate 54g was, thus, obtained in moderate yield (67%) from 1-O-acetyl-2,3,4,6-tetra-O-benzyl-d-glucopyranose 53g (R = H, Scheme 17) [89].
Alternatively, conversion into O-acetyl glycosides is not mandatory. The amination protocol could be performed directly from free sugar hemiacetals using similar conditions [93], although the expected glycosylamines were obtained after longer reaction time (0.75–22 h) (see Scheme 17, above).
It is worth noting that carbonylated glycosylamines have low stability under acidic conditions, limiting their isolation by SiO2-column chromatography, mainly for hexopyranosides. They may also act as activated glycosyl donors, leading to C-glycosyl compounds. As a rule, the stability order is furanosyl > pentopyranosyl > hexopyranosyl derivatives.
4.2. Addition Reactions
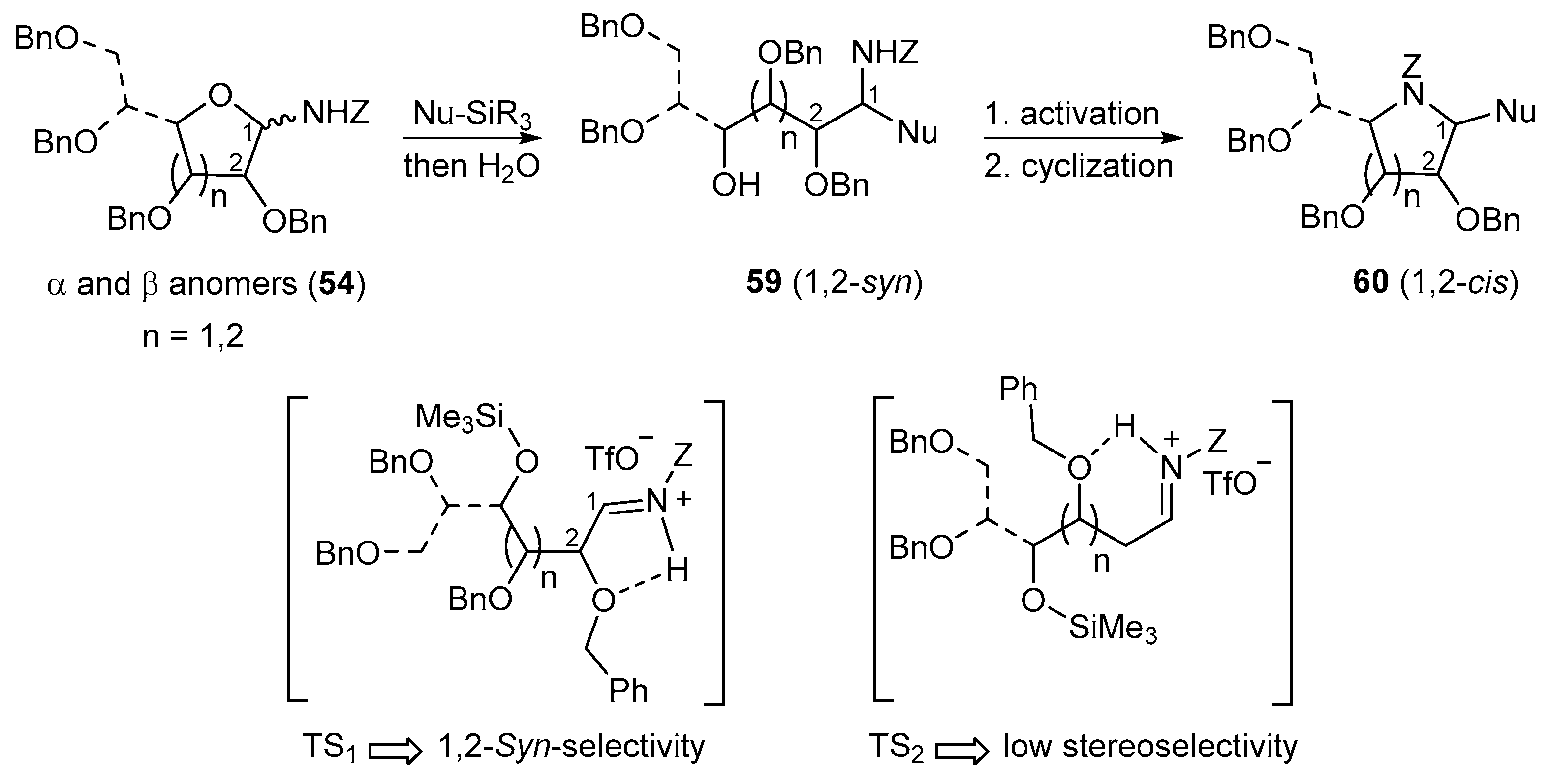
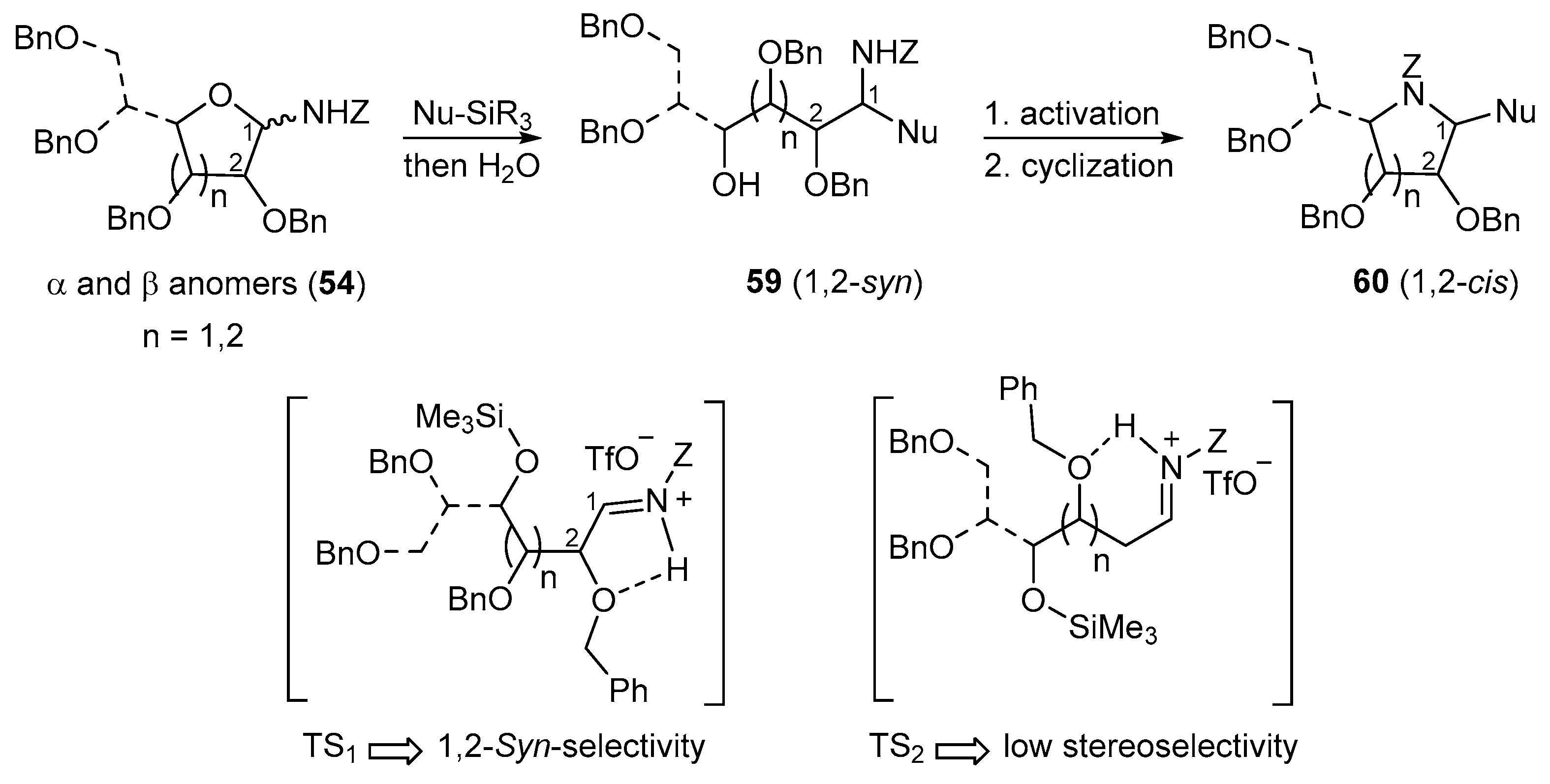
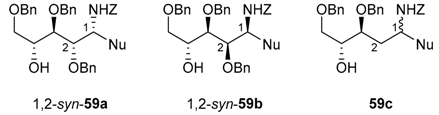
N-Benzyloxycarbonyl-N-glycosides (54) undergo Lewis acid-catalyzed ring-opening reactions with silylated nucleophiles (Nu-SiR3) to give related 1,2-syn-aminoalditols (59) with good to high diastereoselectivity through Cram chelate transition states (TS1, Figure 2). As for N-alkyl- and N-benzyl-N-glycosides, they then provide 1,2-cis-iminosugar-C-glycosyl compounds (e.g., 60) in good yields after activation and a cyclization reaction (Figure 2).
Addition reactions performed in the furanose series were carried out with a sub-stoichiometric amount of TMSOTf and an excess of the silylated nucleophile in CH3CN at low temperature (−40 °C). The main examples are shown below (Table 2, and Scheme 20) [89,90,91]. Importantly, no reaction of 54a and allyltrimethylsilane 55D was observed, while 2-deoxy-d-ribo derivative 54c showed no diastereoselectivity when reacted with acetophenone-derived silyl enol ether 55C. This further highlights the rationale of assuming Cram chelated transition states during the nucleophilic addition process involving the O-alkyl substituent at C-2 (see Figure 2, TS1 vs. TS2) [89].
The synthetic value of this reaction was extended to several functionalities as various important motifs were introduced on an N-glucofuranosylamine derivative 54d (e.g., TMSCN (55E), the silyl enol ether of cyclohexanone (55F), and cyclopentanone (55G), etc., see Scheme 20).
It is noteworthy that a single stereoisomer was formed at the alkylation site α to the carbonyl group in the cycohexanone derivative 59dF, whereas a mixture of two stereoisomers (ratio 2:1) was isolated for the cyclopentanone analogue 59dG. A lower yield (12%) and a low diastereoselectivity were observed for the addition of diethyl trimethylsilyl phosphite, although it was possible to isolate the related α-amino phosphonate product 59dH. Unsaturated aliphatic chains could also be introduced as alkyne or allene moieties in moderate yields using 3-trimethylsilyl-1,2-butadiene 55I or propargyltrimethylsilane 55J as reagents. These compounds are of particular interest as synthetic precursors of disaccharide mimics [90,91].
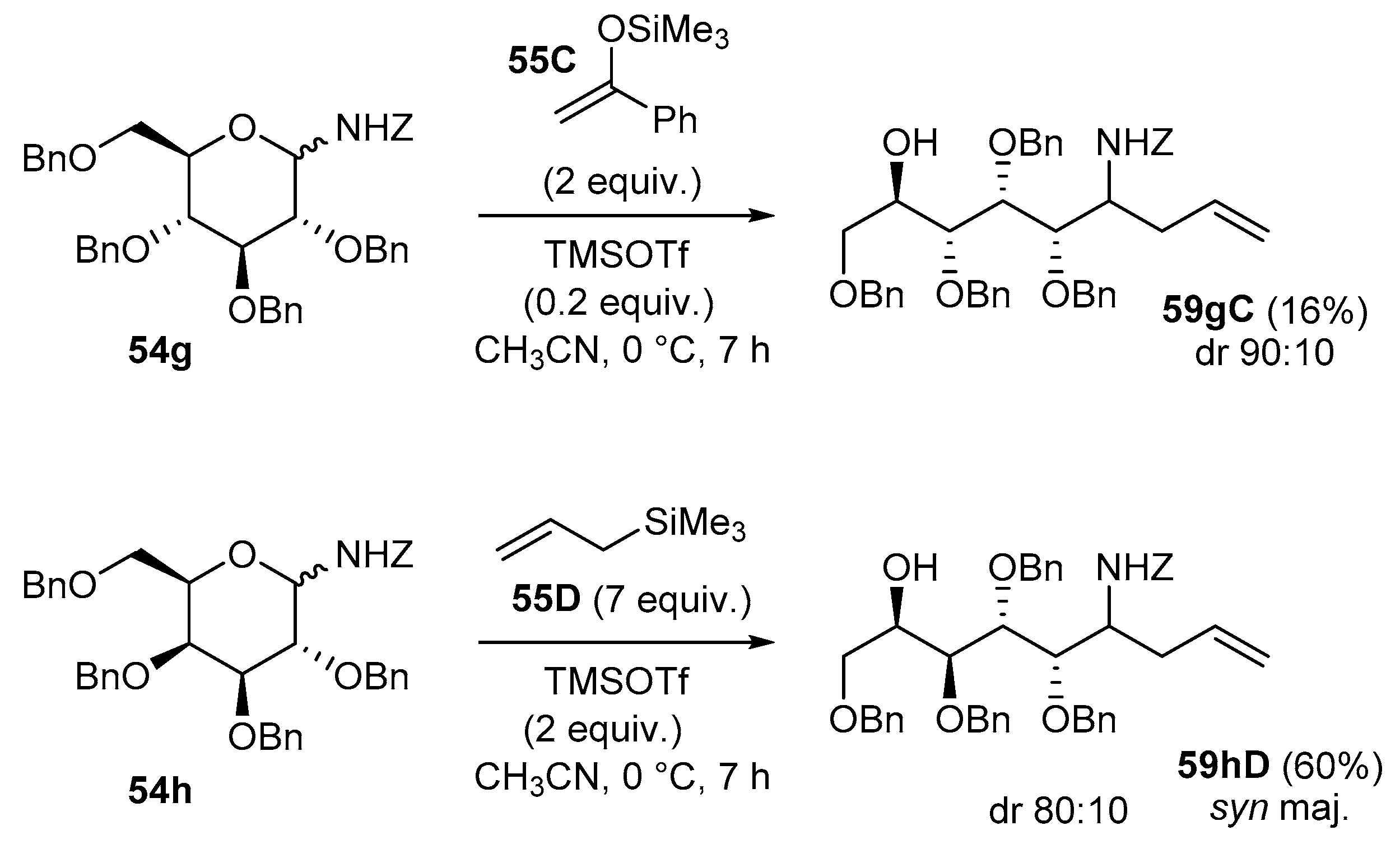
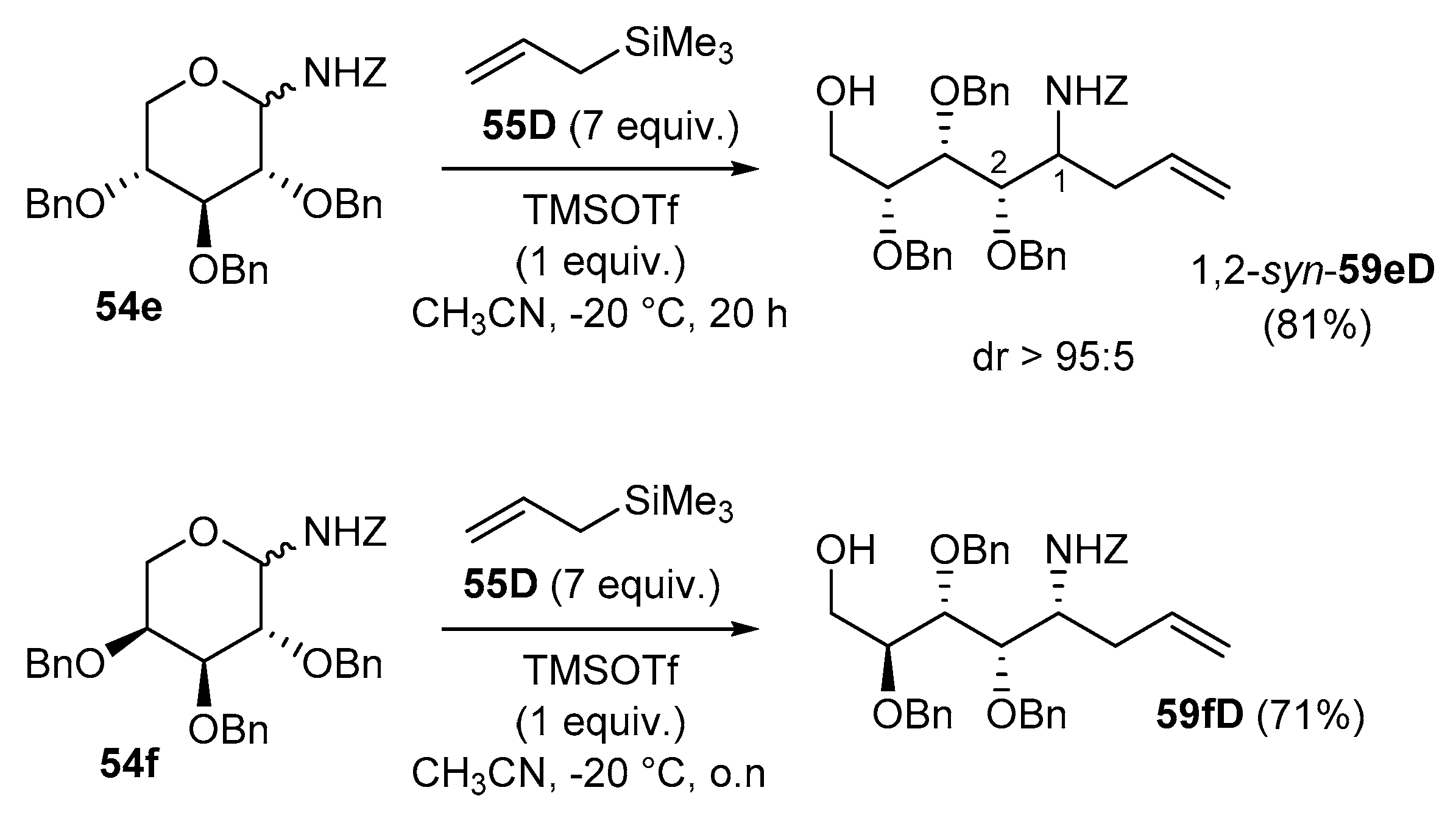
In the pyranose series, however, under the typical reaction conditions, the limited stability of the six-membered glycosylamines commonly preclude the addition reaction even after prolonged reaction time. As depicted in Scheme 21, this was observed for the addition of the silyl enol ether of acetophenone (55C) to the N-Z-protected d-glucopyranosylamine 54g which gave the addition product 59gC in 16% yield [89]. However, the addition of allylTMS 55D onto d-galactosamine 54h could be achieved using 2 equiv. of TMSOTf in a yield of about 60% (59hD) when the isolated reaction mixture was submitted a second time to the same conditions [94].
4.3. Cyclization Reactions
The open-chain silylated iminoalditols were generally cyclized via a mesylation/t-BuOK-treatment sequence. The cyclization step requires a stronger base than in the case of the N-alkylated aminoalditols. Cyclization occurs also through an intramolecular SN2 reaction with inversion at C-4 or C-5. The related protected 1,2-cis iminosugar-C-glycosides are usually obtained in good yields. Of note, retention of the configuration at the carbon atom carrying the free OH group could also be achieved by a sequence of oxidation—intramolecular reductive amination.
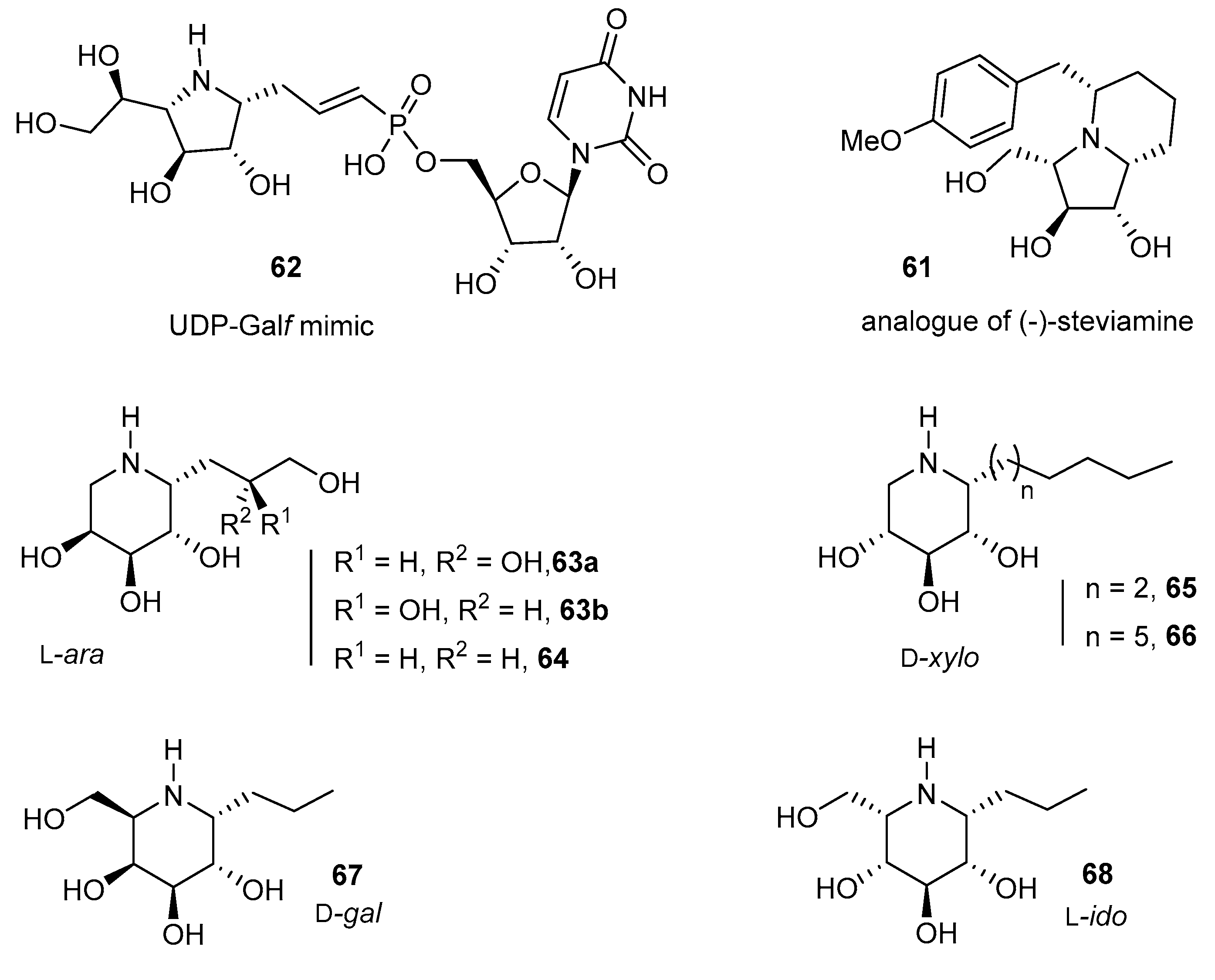
These valuable reactions have been applied to a range of furanosides and a few pyranosides to give a new poly-hydroxylated indolizidine derivative 61 as an analogue of (–)-steviamine [95], UDP-Galf mimics such as 62 [90,91], or potent pharmalogical chaperones (in the l-arabino, d-xylo, d-galacto and l-ido iminosugar-C-glycoside series, see 63–68) for the treatment of glucosidase and galactosidase-linked lysosomal storage disorders (LSDs) (Figure 3) [92,94].
4.4. Miscellaneous
As miscellaneous examples, 2-deoxy glycosylamines of type 69 could be synthesized through vicinal amino chlorination of related glycals 70 [96].
Following a dechlorination protocol, unmasking of the amide functionality and protection of the free amine as its benzyl- or tert-butyl carbamate, glycosylamines 71 and 72 were prepared. Reduction of the N-oxycarbonyl-N-glycosides with LiAlH4 followed by cyclization-deprotection gave fagomines 73a–b and their epimers 74a–b (Scheme 23) [97].
Interestingly, N-Boc-protected glycosylamines 72a and 72b, obtained, respectively, from d-glucal and d-galactal derivatives 70a and 70b could also be treated with excess allylmagnesium bromide to give the ring-opened amino alcohols 75a and 75b in good yields (70% and 75%, respectively) as an inseparable 1:1 mixture of diastereomers (Scheme 24) [98]. The free hydroxyl groups of amino alcohols 75a and 75b were mesylated and cyclized under intramolecular SN2 reactions (after removal of the Boc group and treatment with K2CO3) to give compounds 76 and 77, respectively. After separation of the diastereomers and further elaboration, novel polyhydroxylated quinolizidines of type 78 and 79 were provided in good yields. These molecules could be regarded as analogues of l-1,2-dideoxyhomonojirimycin.
5. N-(tert-Butanesulfinyl) Glycosylamines
Considering the higher stability of the N-tert-butanesulfinyl aldimines and ketimines compared with most N-alkyl, aryl, acyl, and carbonyl Schiff’s bases, as well as the advantages of Ellman’s imines in terms of stereocontrol, N-tert-butanesulfinyl glycosylamines have recently emerged as more versatile synthetic intermediates en route to imino-C-glycosyl compounds.
5.1. Synthesis
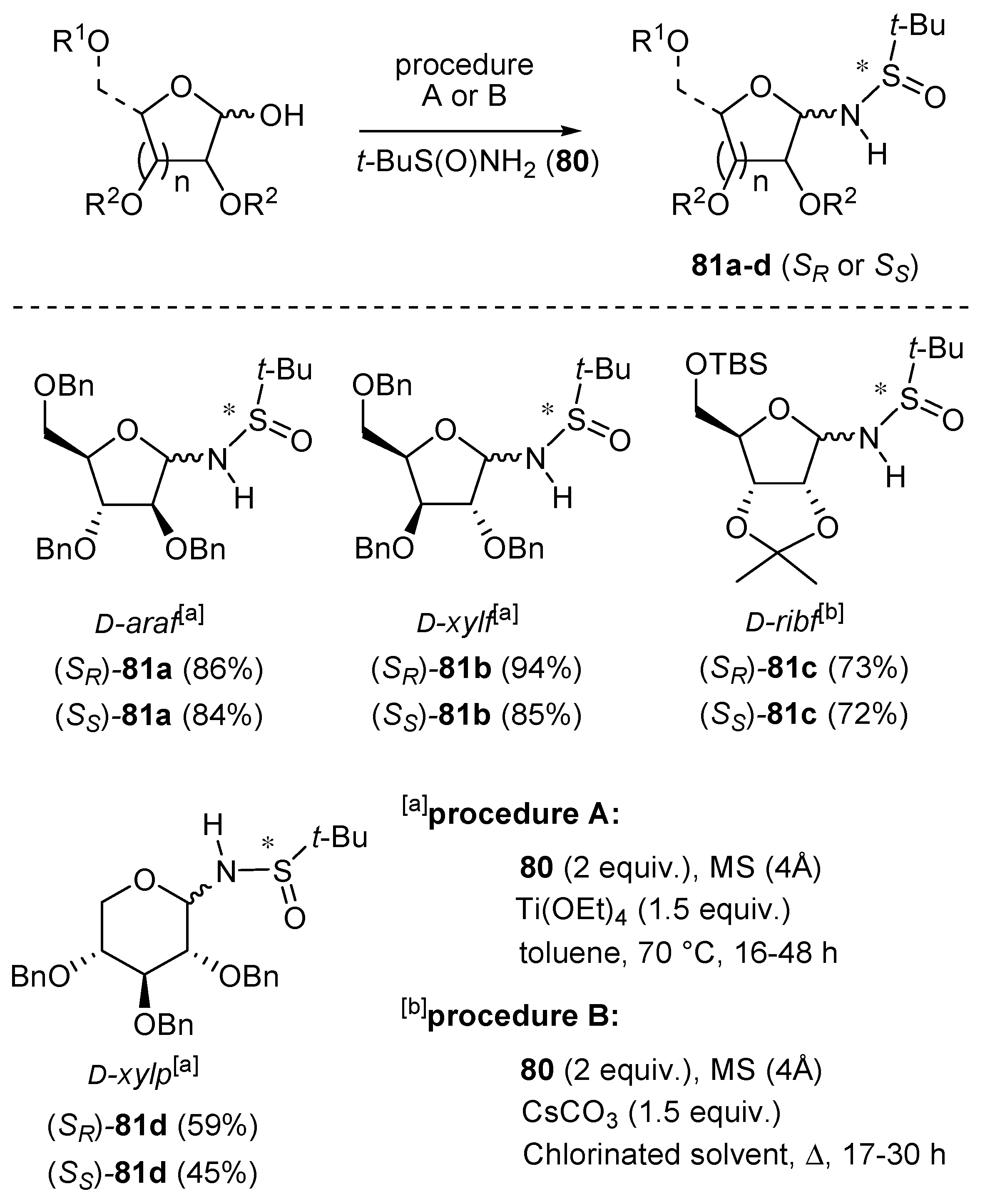
As shown in Scheme 25 (see procedure A), these derivatives are commonly synthesized by the addition of an O-benzyl-protected pentofuranose or pentopyranose to a mixture of (R)- or (S)-2-methyl-2-propanesulfinamide 80 (2 equiv.) and Ti(OEt)4 (1.5 equiv.) in dry toluene. The reaction mixture is heated at 70 °C to give compounds 81a, 81b and 81d in moderate to excellent yields (45–94%) [99,100,101].
Alternatively, N-sulfinyl glycosylamines with different protecting groups may be prepared using a modified protocol (see procedure B). For example, N-tert-butanesulfinyl ribofuranosyl-amines (SR)-81c and (SS)-81c, carrying acid-sensitive groups were prepared in moderate yields by reacting the substrate with (R)- or (S)-Ellman’s sulfinamide in the presence of CsCO3 and molecular sieves for 17–30 h under reflux [99,100].
Interestingly, the N-sulfinylglycosylamines are hydrolytically stable (particularly, in the furanose form), allowing their isolation by normal silica gel (SiO2) chromatography. Furthermore, the two anomers (α/β) of compounds 81 could be separated (SiO2 chromatography), although a slow epimerization was observed when the anomers of (SR)-81a were separately analyzed by NMR spectroscopy as solutions in CDCl3. These glycosylamines can be handled in air at room temperature, but prolonged storage at room temperature results in decomposition over a period of a few days. Stability is greatly increased by storing the N-sulfinyl glycosylamines in closed containers at 5 °C [99].
5.2. Addition Reactions
The N-tert-butanesulfinyl glycosylamines undergo addition reactions with a range of Grignard and lithium reagents to give related 1,2-syn or 1,2-anti-aminoalditols in good yields and moderate to excellent diastereoselectivities [99,101,102,103]. Their reactivity was found to be similar to that of N-alkyl-N-glycosides and of N-benzyl-N-glycosylhydroxylamines with a chemical stability similar to that of the N-Cbz-analogues [99]. In that respect, and as shown by the potential of the chiral sulfinyl auxiliary to direct in some instances the stereoselectivity at C-1, they provide more reliable and versatile synthetic intermediate en route to iminosugar-C-glycosides than previously described N-alkyl and N-alkoxycarbonyl glycosylamines.
5.3. Addition of Grignard Reagents and Cyclizations
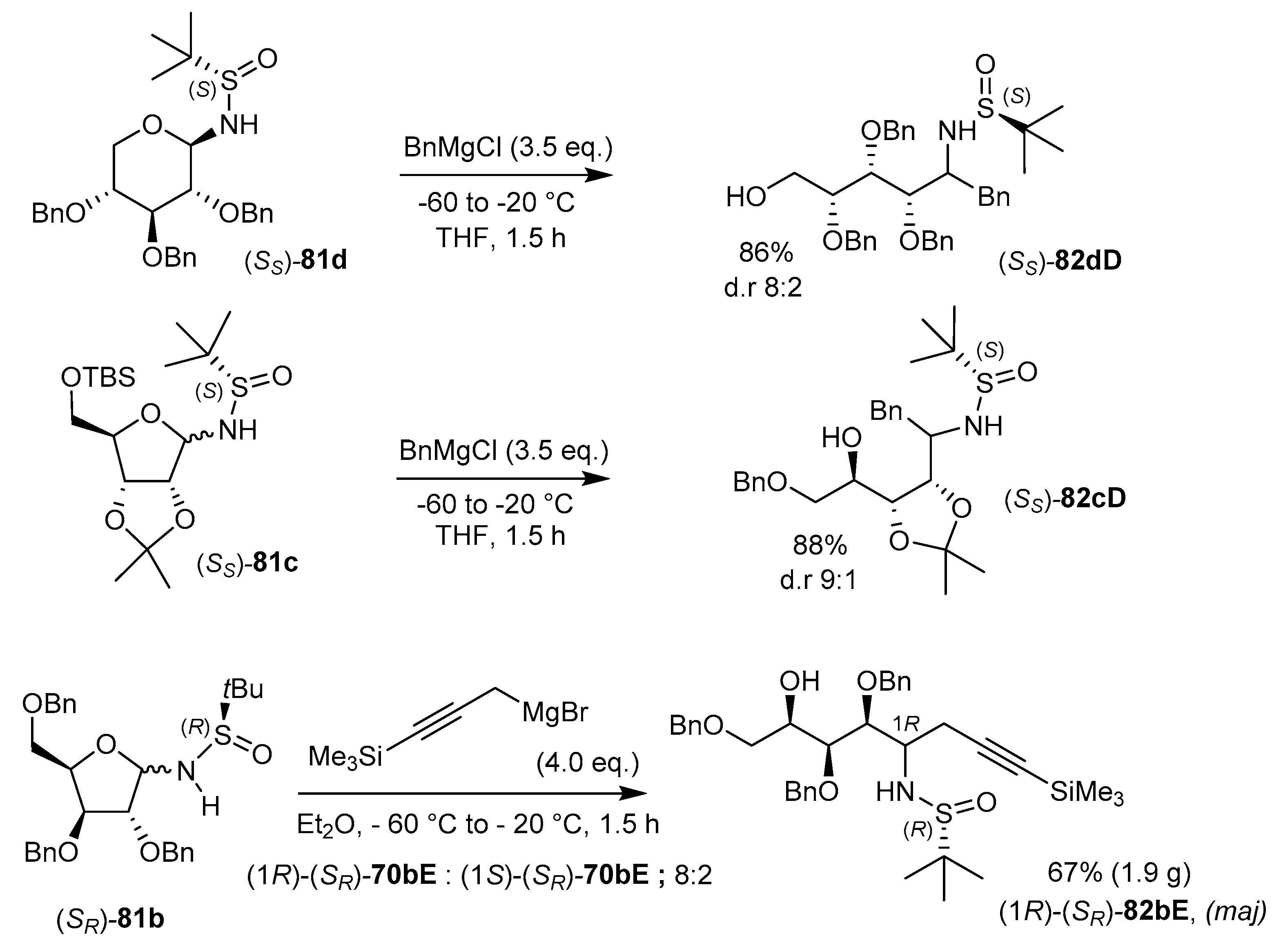
As reported in Table 3, the addition of Grignard reagents (e.g., alk, Ar, all, Bn, propargyl, vinyl, etc.) proceeded usually by adding an excess of the organometallic species to a solution of a N-tert-butanesulfinyl-N-glycoside in THF at −60 °C. The reaction mixture is subsequently allowed to reach −20 °C, over 1.5 h to give the related 1,2-syn aminoalditol derivative 82 in good yield (45–95%) and moderate to excellent diastereoselectivity.
Interestingly, the diastereomeric ratio is often comprised between 7:3 and 10:0 in favor of the 1,2-syn diastereomer, from either epimer at the S-atom ((SS) or (SR)). Thus, in such cases, the chiral N-tert-butanesulfinyl auxiliary does not direct the stereoselectivity at C-1. Importantly, however, the process can be scaled-up (up to 1.9 g of product 82) [102], without erosion of the diastereo-selectivity. The method was implemented to a variety of organomagnesium species (see Table 3) [99], including propargyl Grignard reagents [101]. It may be possible to further enrich the diastereoselectivity significantly by adding LiCl, which resulted in d.r. greater than 90% (entries 3, 7, 12). The addition reactions were extended to different series, namely on glycosylamines derived from 2,3,4-tri-O-benzyl-d-xylopyranose [99] (e.g., 81d), 2,3,5-tri-O-benzyl-d-xylofuranose (e.g., 81b), [101], and the acid-sensitive furanose derivative 81c [99] to afford compounds (SS)-82dD, (SR)-82bE, and (SS)-82cD, respectively, in good yields and good diastereoselectivities (Scheme 26).
Furthermore, although in these series the chiral sulfinyl group does not control the stereochemistry, the diastereoisomers of the 1-C-substituted iminoalditols were all separable by regular SiO2-column chromatography using either (SS)-N- or (SR)-N- or both sulfinyl glycosylamines. This is remarkable since, in the N-benzyl- and other N-alkyl-N-glycoside series, as well as in N-benzylhydroxylamines and N-alkoxycarbonylglycosylamines, difficulties in separating both diastereomers often hampered the synthetic utility of these important scaffolds.
As illustrated in Scheme 27, the N-tert-butanesulfinyl iminoalditols were routinely cyclized under the same conditions as those reported for N-Cbz-N-glycosides (mesylation followed by treatment with t-BuOK). Afterwards, the sulfinyl protecting group was removed with mild acid (HCl in MeOH) to generate the corresponding 1,2-cis imino-C-glycosyl derivatives 83 in good yields (see procedure C) [99,102]. Alternatively, the sulfinyl group may be cleaved first and cyclization promoted from the free amine, which occurs spontaneously (procedure D) [101,103].
The preparation of 1,4-dideoxy-1,4-imino-l-arabinitol scaffolds 84bE tethered to 1,2,3-triazoles carrying (hetero)aromatic systems as simplified uridinyl diphospho-d-galactofuranose (UDP-Galf) mimics were prepared by these methods through the addition of a trimethylsilylpropargyl Grignard reagent to N-sulfinylglycosylamine (SR)-81b, followed by cyclization/deprotection sequences (Scheme 28) [102]. Compound 84bE is a moderate inhibitor of GlFT2, a key galactofuranosyltransferase involved in the assembly of the cell wall of mycobacteria (including the causative agent of tuberculosis, Mycobacterium tuberculosis) [104], and it is essential for mycobacterial viability [105,106].
5.4. Addition of M-CF2P(O)(OEt)2 Metalated Species and Cyclizations
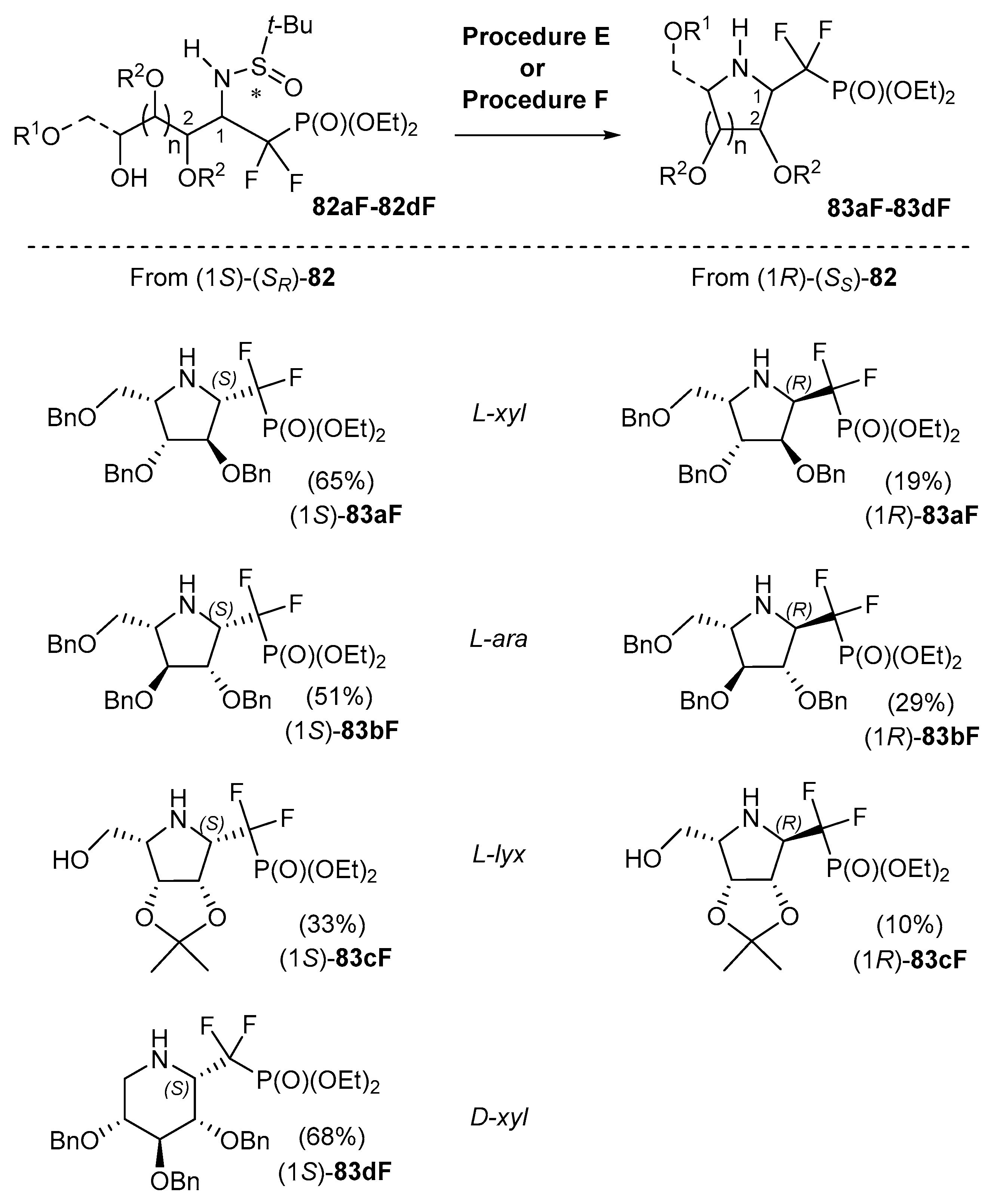
We have recently described an efficient methodology for the introduction of a CF2P(O)(OEt)2 group by the addition of either BrMgCF2P(O)(OEt)2 (prepared by reacting BrCF2P(O)(OEt)2 with i-PrMgCl and LiBr in THF at −75 °C = procedure E) or LiCF2P(O)(OEt)2 (generated from LDA and HCF2P(O)(OEt)2 in THF at −60 °C = procedure F) [101] (Scheme 29).
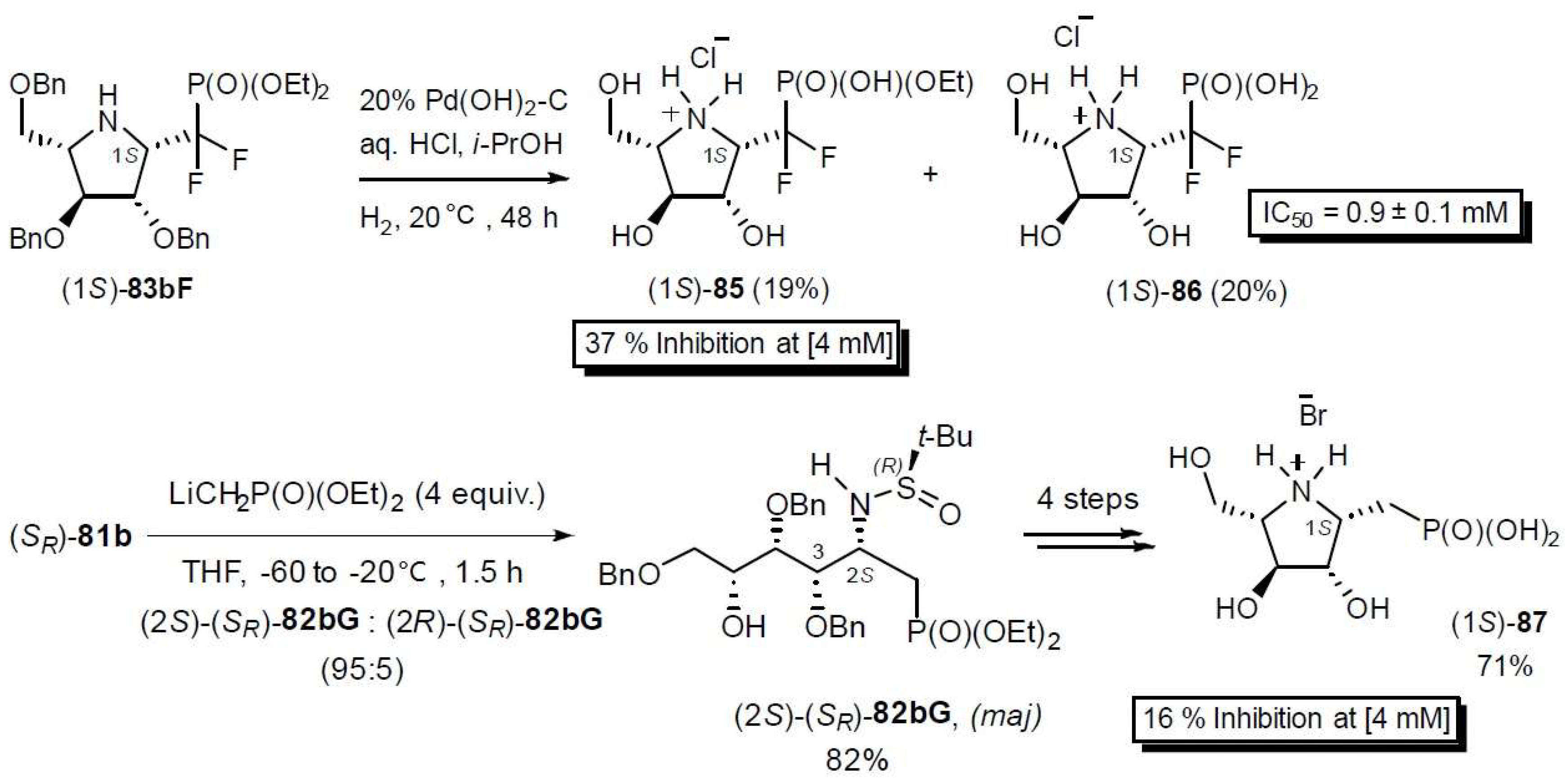
Upon activation/cyclization, the resulting aminoalditols 82aF–82dF lead to 1-C-difluorophosphonomethyl-iminosugar-C-glycosides 83aF–83dF in modest to good overall yields (ca. 3–49% over 3–4 steps). Such compounds are very important mimics of glycosyl phosphates and precursors of sugar nucleotide analogs. Remarkably, the stereoselectivity of the addition of these reagents is tunable, i.e., the pseudoanomeric configuration of the glycosyl phosphate mimics can be chosen by selecting the configuration of the sulfinyl group in the starting N-t-butanesulfinyl glycosylamines. The corresponding N-t-butanesulfinyl iminoalditol derivatives 82aF–82dF were obtained in moderate to good yields (44–88%) and modest to excellent diastereoselectivities (6:4 to 10:0) from compounds 81a–d. Details on the stereochemical effects at play in this process were gained from quantum chemical calculations [101]. These can be exploited to predict the selectivities of future novel substrates. As a rule, glycosylamines (SS)-81 give (1R)-(SS)-83aF–dF (i.e., a pseudo α-anomer) and (SR)-81, (1S)-(SR)-83aF–dF (i.e., a pseudo β-anomer) respectively, as the major products.
Compound (1S)-83bF was deprotected by hydrogenation using Pd(OH)2/C (20%) as the catalyst [103]. Introduction of the −CH2P(O)(OEt)2 moiety was also performed using similar synthetic sequences from (SR)-81b and LiCH2P(O)(OEt)2 [103]. β-Phosphonomethyl- and β-phosphono(difluoromethyl)-1,4-imino-l-arabinitols (1S)-85, (1S)-86, and (1S)-87 were provided in low to moderate overall yields. Compounds 85–87 were found to be moderate inhibitors of the mycobacterial galactofuranosyltransferase GalfT2 (Scheme 30).
6. Conclusions
Since the pioneering studies of Nicotra and co-workers in the early 1990s, and more recent contributions from Kobayashi and Dondoni, N-protected glycosylamines have progressively made their way to become important synthetic scaffolds en route to iminosugar-C-glycosyl compounds. Various types of N-glycosyl derivatives (e.g., in the pentofuranose, pentopyranose, and hexopyranose series) have been reported in the last 20 years and the synthetic reaction sequences giving important iminosugar-C-glycosyl derivatives have frequently been improved. Foremost, they include the direct condensation of a primary amine with a protected sugar hemiacetal, followed by a typical addition, activation, and cyclization reaction sequence. In particular, the valuable N-tert-butanesulfinyl glycosylamines have very recently been developed giving an approach to iminosugar-C-glycosides where the stereoselectivity at C-1 may be tuned. In that respect, this field of research will, hence, surely continue to motivate the scientific community in designing new types of N-glycosidic structures for the synthesis of iminosugars, as well as for their use for therapeutic purposes.
Funding
This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Université d’Orléans, Labex SynOrg (ANR-1-LABX-0029) and the Region Centre-Val de Loire.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Carbohydrates in Chemistry and Biology; Ernst, B.; Hart, G.W.; Sinaÿ, P. (Eds.) Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Bertozzi, C.R.; Kiessling, L.L. Chemical Glycobiology. Science 2001, 291, 2357–2364. [Google Scholar] [CrossRef] [PubMed]
- Hudak, J.E.; Bertozzi, C.R. Glycotherapy: New Advances Inspire a Reemergence of Glycans in Medicine. Chem. Biol. 2014, 21, 16–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Davis, B.G. Realizing the Promise of Chemical Glycobiology. Chem. Sci. 2013, 4, 3381–3394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Sugar Code: Fundamentals of Glycosciences; Gabius, H.-J. (Ed.) Wiley-VCH: Weinheim, Germany, 2009; ISBN 978-3-527-32089-9. [Google Scholar]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Stanley, P.; Hart, G.W.; Aebi, M.; Darvill, A.G.; Kinoshita, T.; Packer, N.H.; Prestegard, J.H.; et al. Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 2017; ISBN 9781621821328. [Google Scholar]
- Sattin, S.; Bernardi, A. Design and synthesis of glycomimetics. Carbohydr. Chem. 2016, 41, 1–25. [Google Scholar] [CrossRef]
- Ernst, B.; Magnani, J.L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 2009, 8, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Biochemistry of Glycoconjugate Glycans. In Comprehensive Glycosciences: From Chemistry to Systems Biology; Kamerling, J.P.; Boons, G.J.; Lee, Y.C.; Suzuki, A.; Taniguchi, N.; Voragen, A.G.J. (Eds.) Elsevier: Oxford, UK, 2007; Volume 3, ISBN 9780444519672. [Google Scholar]
- Spiro, R.G. Protein glycosylation: Nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 2002, 12, 43R–56R. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, D.P.; Scanlan, E.M.; Davis, B.G. Glycoprotein Synthesis: An Update. Chem. Rev. 2009, 109, 131–163. [Google Scholar] [CrossRef] [PubMed]
- Kan, C.; Danishefsky, S.J. Recent Departures in the Synthesis of Peptides and Glycopeptides. Tetrahedron 2009, 65, 9047–9065. [Google Scholar] [CrossRef] [PubMed]
- Larkin, A.; Imperiali, B. The Expanding Horizons of Asparagine-Linked Glycosylation. Biochemistry 2011, 50, 4411–4426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, B.; Depaix, A.; Périgaud, C.; Peyrottes, S. Recent Trends in Nucleotide Synthesis. Chem. Rev. 2016, 116, 7854–7897. [Google Scholar] [CrossRef] [PubMed]
- Takeda, U.; Okada, T.; Takagi, M.; Gomi, S.; Itoh, J.; Sezaki, M.; Ito, M.; Miyadoh, S.; Shomura, T. SF2446, New Benzo[a]naphthacene Quinone Antibiotics. I. Taxonomy and Fermentation of the Producing Strain, Isolation and Characterization of Antibiotics. J. Antibiot. 1988, 41, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Gomi, S.; Sazaki, M.; Itoh, J.; Sezaki, M. SF2446, New Benzo[a]naphthacene Quinone Antibiotics. II. The Structural Elucidation. J. Antibiot. 1988, 41, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Maskey, R.P.; Grün-Wollny, I.; Fiebig, H.H.; Laatsch, H. Akashins A, B, and C: Novel Chlorinated Indigoglycosides from Streptomyces sp. GW 48/1497. Angew. Chem. Int. Ed. 2002, 41, 597–599. [Google Scholar] [CrossRef]
- Link, J.T.; Raghavan, S.; Gallant, M.; Danishefsky, S.J.; Chou, T.C.; Ballas, L.M. Staurosporine and ent-Staurosporine: The First Total Syntheses, Prospects for a Regioselective Approach, and Activity Profiles. J. Am. Chem. Soc. 1996, 118, 2825–2842. [Google Scholar] [CrossRef]
- Belmokhtar, C.A.; Hillion, J.; Ségal-Bendirdjian, E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 2001, 20, 3354–3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonsson, A.; Persson, J.L. Induction of Apoptosis by Staurosporine Involves the Inhibition of Expression of the Major Cell Cycle Proteins at the G2/M Checkpoint Accompanied by Alteration in Erk and Akt Kinase Activities. Anticancer Res. 2009, 29, 2893–2898. [Google Scholar] [PubMed]
- Bush, J.A.; Long, B.H.; Catino, J.J.; Bradner, W.T.; Tomita, K. Production and Biological Activity of Rebeccamycin, a Novel Antitumor Agent. J. Antibiot. 1987, 40, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Snipes, C.E.; Duebelbeis, D.O.; Olson, M.; Hahn, D.R.; Dent, W.H., III; Gilbert, J.R.; Werk, T.L.; Davis, G.E.; Lee-Lu, R.; Graupner, P.R. The Ansacarbamitocins: Polar Ansamitocin Derivatives. J. Nat. Prod. 2007, 70, 1578–1581. [Google Scholar] [CrossRef] [PubMed]
- Eklund, E.A.; Bode, L.; Freeze, H.H. Diseases associated with carbohydrates/glycoconjugates. In Comprehensive Glycoscience. From Chemistry to Systems Biology, 1st ed.; Kamerling, J.P., Boons, G.J., Lee, Y.C., Suzuki, A., Taniguchi, N., Voragen, A.G.J., Eds.; Elsevier: Oxford, UK, 2007; Volume 4, pp. 339–371. ISBN 0-444-52746-X. [Google Scholar]
- Legler, G. Glycoside Hydrolases: Mechanistic Information from Studies with Reversible and Irreversible Inhibitors. Adv. Carbohydr. Chem. Biochem. 1990, 48, 319–384. [Google Scholar] [CrossRef] [PubMed]
- Isbell, H.S.; Frush, H.L. Mutarotation, Hydrolysis, and Rearrangement Reactions of Glycosylamines. J. Org. Chem. 1958, 23, 1309–1319. [Google Scholar] [CrossRef]
- Compain, P.; Chagnault, V.; Martin, O.R. Tactics and strategies for the synthesis of iminosugar C-glycosides: A review. Tetrahedron Asymmetry 2009, 20, 672–711. [Google Scholar] [CrossRef]
- Iminosugars: From Synthesis to Therapeutic Applications; Compain, P.; Martin, O.R. (Eds.) John Wiley & Sons: Chichester, UK, 2007; ISBN 9780470033913. [Google Scholar]
- Horne, G.; Wilson, F.X.; Tinsley, J.; Williams, D.H.; Storer, R. Iminosugars past, present and future: Medicines for tomorrow. Drug Discov. Today 2011, 16, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Nash, R.J.; Kato, A.; Yu, C.-Y.; Fleet, G.W. Iminosugars as therapeutic agents: Recent advances and promising trends. Future Med. Chem. 2011, 3, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Asano, N. Iminosugars: The Potential of Carbohydrate Analogs. In Carbohydrate Chemistry: State of the Art and Challenges for Drug Development; Cipolla, L., Ed.; Imperial College Press: London, UK, 2016; pp. 279–301. ISBN 978-1-78326-720-0. [Google Scholar]
- Germain, D.; Giugliani, R.; Hughes, D.; Mehta, A.; Nicholls, K.; Barisoni, L.; Jennette, C.J.; Bragat, A.; Castelli, J.; Sitaraman, S.; et al. Safety and Pharmacodynamic effects of a pharmacological chaperone on α-galactosidase A activity and globotriaosylceramide clearance in Fabry disease: Report from two phase 2 clinical studies. Orphanet J. Rare Dis. 2012, 7, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J.; Spencer, C.M. Miglitol: A review of its therapeutic potential in type 2 diabetes mellitus. Drugs 2000, 59, 521–549. [Google Scholar] [CrossRef] [PubMed]
- Canda, E.; Kose, M.; Kagnici, M.; Ucar, S.K.; Sozmen, E.Y.; Coker, M. Patients with Gaucher type 1: Switching from imiglucerase to miglustat therapy. Blood Cells Mol. Dis. 2018, 68, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Hernández, D.; Boto, A. Nucleoside Analogues: Synthesis and Biological Properties of Azanucleoside Derivatives. Eur. J. Org. Chem. 2014, 2201–2220, and references cited therein. [Google Scholar] [CrossRef]
- Singh, V.; Evans, G.B.; Lenz, D.H.; Mason, J.M.; Clinch, K.; Mee, S.; Painter, G.F.; Tyler, P.C.; Furneaux, R.H.; Lee, J.E.; et al. Femtomolar Transition State Analogue Inhibitors of 5′-Methylthioadenosine/S-Adenosylhomocysteine Nucleosidase from Escherichia coli. J. Biol. Chem. 2005, 280, 18265–18273. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Singh, V.; Evans, G.B.; Tyler, P.C.; Furneaux, R.H.; Cornell, K.A.; Riscoe, M.K.; Schramm, V.L.; Howell, P.L. Structural Rationale for the Affinity of Pico- and Femtomolar Transition State Analogues of Escherichia coli 5′-Methylthioadenosine/S-Adenosylhomocysteine Nucleosidase. J. Biol. Chem. 2005, 280, 18274–18282. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, E.M.; Garcia Fernández, J.M.; Ortiz Mellet, C. Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: Lessons from Gaucher, GM1-gangliosidosis and Fabry diseases. Chem. Commun. 2016, 52, 5497–5515. [Google Scholar] [CrossRef] [PubMed]
- Bergeron-Brlek, M.; Goodwin-Tindall, J.; Cekic, N.; Roth, C.; Zandberg, W.F.; Shan, X.; Varghese, V.; Chan, S.; Davies, G.J.; Vocadlo, D.J.; et al. A Convenient Approach to Stereoisomeric Iminocyclitols: Generation of Potent Brain-Permeable OGA Inhibitors. Angew. Chem. Int. Ed. 2015, 54, 15429–15433. [Google Scholar] [CrossRef] [PubMed]
- Bergeron-Brlek, M.; Meanwell, M.; Britton, R. Direct synthesis of imino-C-nucleoside analogues and other biologically active iminosugars. Nat. Commun. 2015, 6, 6903–6908. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, M.; Sutherland, M.; Britton, R. Application of sequential proline-catalyzed α-chlorination and aldol reactions in the total synthesis of 1-deoxygalactonojirimycin. Can. J. Chem. 2018, 96, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Behr, J.-B.; Plantier-Royon, R. Addition of organometallics to aldimines, aldoximes, and aldononitriles: A key step towards the synthesis of azasugars. Recent Res. Dev. Org. Chem. 2006, 10, 1–30. [Google Scholar] [CrossRef]
- Carcano, M.; Nicotra, F.; Panza, L.; Russo, G. A New Procedure for the Synthesis of d-Glucosamine α-C-Glycosides. J. Chem. Soc. Chem. Commun. 1989, 287–289. [Google Scholar] [CrossRef]
- Lay, L.; Nicotra, F.; Paganini, A.; Pangrazio, C.; Panza, L. A New Procedure for the Synthesis of Azasugars. Tetrahedron Lett. 1993, 34, 4555–4558. [Google Scholar] [CrossRef]
- Cipolla, L.; Lay, L.; Nicotra, F.; Pangrazio, C.; Panza, L. Synthesis of Azasugars by Grignard Reactions on Glycosylamines. Tetrahedron 1995, 51, 4679–4690. [Google Scholar] [CrossRef]
- Cipolla, L.; La Ferla, B.; Peri, F.; Nicotra, F. A new procedure for the synthesis of C-glycosides of nojirimycin. Chem. Commun. 2000, 1289–1290. [Google Scholar] [CrossRef]
- Cipolla, L.; Palma, A.; La Ferla, B.; Nicotra, F. Synthesis of nojirimycin C-glycosides. J. Chem. Soc. Perkin Trans. 1 2002, 2161–2165. [Google Scholar] [CrossRef]
- Von der Osten, C.H.; Sinskey, A.J.; Barbas, C.F., III; Pederson, R.L.; Wang, Y.-F.; Wong, C.-H. Use of a Recombinant Bacterial Fructose-1,6-diphosphate Aldolase in Aldol Reactions: Preparative Syntheses of 1-Deoxynojirimycin, 1-Deoxymannojirimycin, 1,4-Dideoxy-1,4-imino-D-arabinitol, and Fagomine. J. Am. Chem. Soc. 1989, 111, 3924–3927. [Google Scholar] [CrossRef]
- Wennekes, T.; Overkleeft, H.S. Synthesis and Evaluation of Lipophilic Aza-C-glycosides as Inhibitors of Glucosylceramide Metabolism. Eur. J. Org. Chem. 2010, 1258–1283. [Google Scholar] [CrossRef]
- Lahiri, R.; Suman Reddy, Y.; Kulkarni, S.A.; Vankar, Y.D. Synthesis of unnatural indolizidines, pyrrolizidine and C-alkyl iminosugars from sugar derived hemiaminals. RSC Adv. 2013, 3, 23242–23254. [Google Scholar] [CrossRef]
- Wang, H.Y.; Kato, A.; Kinami, K.; Li, Y.-X.; Fleet, G.W.J.; Yu, C.-Y. Concise synthesis of calystegines B2 and B3 via intramolecular Nozaki–Hiyama–Kishi reaction. Org. Biomol. Chem. 2016, 14, 4885–4896. [Google Scholar] [CrossRef] [PubMed]
- Saha, J.; Peczuh, M.W. Access to Ring-Expanded Analogues of 2-Amino Sugars. Org. Lett. 2009, 11, 4482–4484. [Google Scholar] [CrossRef] [PubMed]
- Saha, J.; Peczuh, M.W. Expanding the Scope of Aminosugars: Synthesis of 2-Amino Septanosyl Glycoconjugates Using Septanosyl Fluoride Donors. Chemistry 2011, 17, 7357–7365. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, M.B.; Chagnault, V.; Postel, D. Synthesis of glycosylamines and glyconamides using molecular iodine. Tetrahedron 2013, 69, 542–550. [Google Scholar] [CrossRef]
- Prasada Rao, J.; Venkataswara Rao, B.; Lakshmi Swarnalatha, J. A new stereoselective approach for N-benzyl amino(hydroxymethyl)-cyclopentitols using RCM. Tetrahedron Lett. 2010, 51, 3083–3087. [Google Scholar] [CrossRef]
- Srinivas Rao, G.; Venkataswara Rao, B. A common and stereoselective strategy for the synthesis of N,O,O,O-tetra-acetyl d-ribo-(2S,3S,4R)-phytosphingosine and 2-epi-jaspine B. Tetrahedron Lett. 2011, 52, 4861–4864. [Google Scholar] [CrossRef]
- Rajender, A.; Prasada Rao, J.; Venkataswara Rao, B. A Divergent and Stereoselective Approach for the Syntheses of Some Polyhydroxylated Indolizidine and Pyrrolizidine Iminosugars. Eur. J. Org. Chem. 2013, 1749–1757. [Google Scholar] [CrossRef]
- Chirke, S.S.; Rajender, A.; Lakshmi, J.K.; Venkataswara Rao, B. A divergent, short and stereoselective approach to pyrrolidine iminosugars: Synthesis of 1,4-dideoxy-1,4-imino-derivatives of d-allitol, d-ribitol, ethyl-erythritol and (–)-2,3-trans-3-4-cis-dihydroxyproline. Tetrahedron Lett. 2015, 56, 1218–1221. [Google Scholar] [CrossRef]
- Kotland, A.; Accadbled, F.; Robeyns, K.; Behr, J.B. Synthesis and Fucosidase Inhibitory Study of Unnatural Pyrrolidine Alkaloid 4-epi-(+)-Codonopsinine. J. Org. Chem. 2011, 76, 4094–4098. [Google Scholar] [CrossRef] [PubMed]
- Hottin, A.; Dubar, F.; Steenackers, A.; Delannoy, P.; Biot, C.; Behr, J.-B. Iminosugar–ferrocene conjugates as potential anticancer agents. Org. Biomol. Chem. 2012, 10, 5592–5597. [Google Scholar] [CrossRef] [PubMed]
- Hottin, A.; Wright, D.W.; Davies, G.J.; Behr, J.-B. Exploiting the Hydrophobic Terrain in Fucosidases with Aryl-Substituted Pyrrolidine Iminosugars. ChemBioChem 2015, 16, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.-J.; Ye, J.-L.; Zhang, H.-K.; Huang, P.-Q. An unexpected high erythro-selection in the Grignard reaction with an N,O-acetal: A concise asymmetric synthesis of indolizidine alkaloid (-)-2-epi-lentiginosine. Tetrahedron 2012, 68, 1750–1755. [Google Scholar] [CrossRef]
- Behr, J.-B.; Mvondo Evina, C.; Phung, N.; Guillerm, G. Synthesis of (difluoromethyl)phosphonate azasugars designed as inhibitors for glycosyl transferases. J. Chem. Soc. Perkin Trans. 1 1997, 1597–1599. [Google Scholar] [CrossRef]
- Gautier-Lefebvre, I.; Behr, J.-B.; Guillerm, G.; Muzard, M. Iminosugars as glycosyltransferase inhibitors: Synthesis of polyhydroxypyrrolidines and their evaluation on chitin synthase activity. Eur. J. Med. Chem. 2005, 40, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.-B.; Guillerm, G. Synthesis of 6-deoxy-homoDMDP and its C(5)-epimer: Absolute stereochemistry of natural products from Hyacinthus orientalis. Tetrahedron Asymmetry 2002, 13, 111–113. [Google Scholar] [CrossRef]
- Behr, J.-B.; Hottin, A.; Ndoye, A. Highly Selective Indium Mediated Allylation of Unprotected Pentosylamines. Org. Lett. 2012, 14, 1536–1539. [Google Scholar] [CrossRef] [PubMed]
- Bosco, M.; Bisseret, P.; Constant, P.; Eustache, J. Synthesis of 2’,3’-dihydrosolanesyl analogues of β-d-arabinofuranosyl-1-monophosphoryldecaprenol with promising antimyco-bacterial activity. Tetrahedron Lett. 2007, 48, 153–157. [Google Scholar] [CrossRef]
- Schönemann, W.; Gallienne, E.; Compain, P.; Ikeda, K.; Asano, N.; Martin, O.R. Synthesis of new β-1-C-alkylated imino-l-iditols: A comparative study of their activity as β-glucocerebrosidase inhibitors. Bioorg. Med. Chem. 2010, 18, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Schönemann, W.; Gallienne, E.; Ikeda-Obatake, K.; Asano, N.; Nakagawa, S.; Kato, A.; Adachi, I.; Gorecki, M.; Frelek, J.; Martin, O.R. Glucosylceramide Mimics: Highly Potent GCase Inhibitors and Selective Pharmacological Chaperones for Mutations Associated with Types 1 and 2 Gaucher Disease. ChemMedChem 2013, 8, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, C.; Engo-Ilanga, F.; Cocaud, C.; Martin, O.R. En route to Novel Furanoside Mimics through Zinc-mediated Propargylation of N-benzyl Glycosylamines using Ultrasound Activation. Synlett 2015, 26, 187–192. [Google Scholar] [CrossRef]
- Senthilkumar, S.; Siva Prasad, S.; Senthil Kumar, P.; Baskaran, S. A diversity oriented one-pot synthesis of novel iminosugar C-glycosides. Chem. Commun. 2014, 50, 1549–1551. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, S.; Siva Prasad, S.; Das, A.; Baskaran, S. One-pot Synthesis of Hydropho-bically Modified Iminosugar C-Alkynylglycosides: Facile Synthesis of Polyhydroxy Tetrahydroindolizidines. Chemistry 2015, 21, 15914–15918. [Google Scholar] [CrossRef] [PubMed]
- Siva Prasad, S.; Senthilkumar, S.; Srivastava, A.; Baskaran, S. Iminosugar C-Nitromethyl Glycosides and Divergent Synthesis of Bicyclic Iminosugars. Org. Lett. 2017, 19, 4403–4406. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Ito, S.; Shimizu, Y.; Kanai, M. Catalytic Anomeric Aminoalkynylation of Unprotected Aldoses. Org. Lett. 2013, 15, 4130–4133. [Google Scholar] [CrossRef] [PubMed]
- Naresh, A.; Marumudi, K.; Kunwar, A.C.; Rao, B.V. Palladium-Catalyzed Double Allylation of Sugar-Imines by Employing Tamaru–Kimura’s Protocol: Access to Unnatural Iminosugars. Org. Lett. 2017, 19, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Yoda, H.; Yamazaki, H.; Kawauchi, M.; Takabe, K. An Efficient Asymmetric Functionalization to Highly Substituted Pyrrolidines. Tetrahedron Asymmetry 1995, 6, 2669–2672. [Google Scholar] [CrossRef]
- Yoda, H.; Yamazaki, H.; Takabe, K. A Novel Synthesis of Enantiomerically Pure Antifungal Agent, (+)-Preussin. Tetrahedron Asymmetry 1996, 7, 373–374. [Google Scholar] [CrossRef]
- Dondoni, A.; Giovannini, P.P.; Perrone, D. New synthesis of Pyrrolidine Homoazasugars via Aminohomologation of Furanoses and Their Use for the Stereoselective Synthesis of Aza-C-disaccharides. J. Org. Chem. 2002, 67, 7203–7214. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Perrone, D. A convenient synthesis of iminosugar-C-glycosides via organometallic addition to N-benzyl-N-glycosylhydroxylamines. Tetrahedron 2003, 59, 4261–4273. [Google Scholar] [CrossRef]
- Dondoni, A.; Nuzzi, A. Access to Piperidine Imino-C-glycosides via Stereoselective Thiazole-Based Aminohomologation of Pyranoses. J. Org. Chem. 2006, 71, 7574–7582. [Google Scholar] [CrossRef] [PubMed]
- Cicchi, S.; Marradi, M.; Corsi, M.; Faggi, C.; Goti, A. Preparation of N-Glycosylhydroxylamines and Their Oxidation to Nitrones for the Enantioselective Synthesis of Isoxazolidines. Eur. J. Org. Chem. 2003, 4152–4160. [Google Scholar] [CrossRef]
- Dondoni, A.; Perrone, D. New entry to pyrrolidine homoazasugars: Conversion of d-arabinose into 2,5-anhydro-2,5-imino-d-glucitol via aminohomologation. Tetrahedron Lett. 1999, 40, 9375–9378. [Google Scholar] [CrossRef]
- Dondoni, A.; Perrone, D.; Rinaldi, M. Grignard Addition to Aldonitrones. Stereochemical Aspects and Application to the Synthesis of C2-Symmetric Diamino Alcohols and Diamino Diols. J. Org. Chem. 1998, 63, 9252–9264. [Google Scholar] [CrossRef]
- Yoshida, Y.; Shimonishi, K.; Sakakura, Y.; Okada, S.; Asano, N.; Tanabe, Y. Facile and Practical Methods for the Sulfonylation of Alcohols Using Ts(Ms)Cl and Me2N(CH2)nNMe2 as a Key Base. Synthesis 1999, 1633–1636. [Google Scholar] [CrossRef]
- Argyropoulos, N.G.; Gkizis, P.; Coutouli-Argyropoulou, E. A convenient synthesis of new enantiomerically pure trihydroxypyrrolizidines using l-erythrose glycosylhydroxylamine as a masked acyclic chiral nitrone. Tetrahedron 2008, 64, 8752–8758. [Google Scholar] [CrossRef]
- Marradi, M.; Corsi, M.; Cicchi, S.; Bonanni, M.; Cardona, F.; Goti, A. N-Glycosylhydroxylamines as Masked Polyhydroxylated Chiral Nitrones in Cycloaddition Reactions: An Access to Pyrrolizidines. Heterocycles 2009, 79, 883–896. [Google Scholar] [CrossRef]
- Malinowski, M.; Rowicki, T.; Guzik, P.; Gryszel, M.; Łapczyński, S.; Wielechowska, M.; Czerwińska, K.; Madura, I.; Sas, W. [1,4]-sigmatropic rearrangement of chiral nitrones and their utilization in the synthesis of new iminosugars. Org. Biomol. Chem. 2016, 14, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Kobayashi, S. Lewis Acid-Catalyzed Ring-Opening Reactions of Semicyclic N,O-Acetals. Org. Lett. 2001, 3, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Hagio, H.; Hirabayashi, R.; Kobayashi, S. Lewis Acid-Catalyzed Ring-Opening Reactions of Semicyclic N,O-Acetals Possessing an Exocyclic Nitrogen Atom: Mechanistic Aspect and Application to Piperidine Alkaloid Synthesis. J. Am. Chem. Soc. 2001, 123, 12510–12517. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Hagio, H.; Kobayashi, S. Lewis Acid Catalyzed Ring-Opening Reactions of Sugar-Derived Semicyclic N,O-Acetals. Helv. Chim. Acta 2002, 85, 3678–3691. [Google Scholar] [CrossRef]
- Liautard, V.; Desvergnes, V.; Martin, O.R. Stereoselective Synthesis of α-C-Substituted 1,4-Dideoxy-1,4-imino-d-galactitols. Toward Original UDP-Galf Mimics via Cross-Metathesis. Org. Lett. 2006, 8, 1299–1302. [Google Scholar] [CrossRef] [PubMed]
- Liautard, V.; Desvergnes, V.; Itoh, K.; Liu, H.-W.; Martin, O.R. Convergent and Stereoselective Synthesis of Iminosugar-Containing Galf and UDP-Galf Mimicks: Evaluation as Inhibitors of UDP-Gal Mutase. J. Org. Chem. 2008, 73, 3103–3115. [Google Scholar] [CrossRef] [PubMed]
- Biela, A.; Oulaïdi, F.; Gallienne, E.; Górecki, M.; Frelek, J.; Martin, O.R. An improved methodology for the synthesis of 1-C-allyl imino-d-xylitol and -l-arabinitol and their rapid functionalization. Tetrahedron 2013, 69, 3348–3354. [Google Scholar] [CrossRef]
- Liautard, V.; Pillard, C.; Desvergnes, V.; Martin, O.R. One-step synthesis of N-protected glycosylamines from sugar hemiacetals. Carbohydr. Res. 2008, 343, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Biela-Banaś, A.; Gallienne, E.; Front, S.; Martin, O.R. Stereoselective synthesis of 1-C-alkyl iminogalactitol derivatives, potential chaperones for galactosidase-linked LSDs: A real challenge. Tetrahedron Lett. 2014, 55, 838–841. [Google Scholar] [CrossRef]
- Chronowska, A.; Gallienne, E.; Nicolas, C.; Kato, A.; Adachi, I.; Martin, O.R. An expeditious synthesis of an analogue of (–)-steviamine by way of the 1,3-dipolar cycloaddition of a nitrile oxide with a 1-C-allyl iminosugar. Tetrahedron Lett. 2011, 52, 6399–6402. [Google Scholar] [CrossRef]
- Rawal, G. K.; Kumar, A.; Tawar, U.; Vankar, Y. D. New Method for Chloroamidation of Olefins. Application in the Synthesis of N-Glycopeptides and Anticancer Agents. Org. Lett. 2007, 9, 5171–5174. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Reddy, B.G.; Vankar, Y.D. Efficient and Stereoselective Syntheses of d- and l-Fagomines and Their Analogues. Eur. J. Org. Chem. 2009, 160–169. [Google Scholar] [CrossRef]
- Kumari, N.; Vankar, Y.D. Synthesis and glycosidase-inhibitory activity of novel polyhydroxylated quinolizidines derived from d-glycals. Org. Biomol. Chem. 2009, 7, 2104–2109. [Google Scholar] [CrossRef] [PubMed]
- Cocaud, C.; Nicolas, C.; Bayle, A.; Poisson, T.; Pannecoucke, X.; Martin, O.R. Synthesis and Reactivity of N-tert-Butanesulfinyl Glycosylamines. Eur. J. Org. Chem. 2015, 4330–4334. [Google Scholar] [CrossRef]
- Cocaud, C.; Nicolas, C.; Désiré, J.; Martin, O.R. One-Step Preparation of Protected N-tert-Butanesulfinyl d-ribo and d-xylo Furanosylamines from Related Sugar Hemiacetals. In Carbohydrate Chemistry: Proven Synthetic Methods; Vogel, C., Murphy, P., Eds.; CRC Press: Boca Raton, FL, USA, 2017; Volume 4, pp. 45–53. ISBN 978-1-4987-2691-7. [Google Scholar]
- Cocaud, C.; Nicolas, C.; Poisson, T.; Pannecoucke, X.; Legault, C.Y.; Martin, O.R. Tunable Approach for the Stereoselective Synthesis of 1-C-Diethylphosphono(difluoromethyl) Iminosugars as Glycosyl Phosphate Mimics. J. Org. Chem. 2017, 82, 2753–2763. [Google Scholar] [CrossRef] [PubMed]
- Cocaud, C.; Maujoin, A.; Zheng, R.B.; Lowary, T.L.; Rodrigues, N.; Percina, N.; Chartier, A.; Buron, F.; Routier, S.; Nicolas, C.; et al. Triazole-Linked Iminosugars and Aromatic Systems as Simplified UDP-Galf Mimics: Synthesis and Preliminary Evaluation as Galf-Transferase Inhibitors. Eur. J. Org. Chem. 2017, 2017, 6192–6201. [Google Scholar] [CrossRef]
- Cocaud, C.; Zheng, R.B.; Lowary, T.L.; Poisson, T.; Pannecoucke, X.; Nicolas, C.; Martin, O.R. 1-C-phosphonomethyl- and 1-C-difluorophosphonomethyl-1,4-imino-l-arabinitols as Galf transferase inhibitors: A comparison. Carbohydr. Res. 2018, 461, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Poulin, M.B.; Lowary, T.L. Chemical Insight into the Mechanism and Specificity of GlfT2, a Bifunctional Galactofuranosyltransferase from Mycobacteria. J. Org. Chem. 2016, 81, 8123–8130. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Jackson, M.; Ma, Y.; McNeil, M. Cell Wall Core Galactofuran Synthesis Is Essential for Growth of Mycobacteria. J. Bacteriol. 2001, 183, 3991–3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Mikusova, K.; Robuck, K.; Scherman, M.; Brennan, P.; McNeil, M. Recognition of Multiple Effects of Ethambutol on Metabolism of Mycobacterial Cell Envelope. Antimicrob. Agents Chemother. 1995, 39, 694–701. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Protected glycosylamines: preparation and use in the synthesis of iminosugar-C-glycosides.
Scheme 1.
Protected glycosylamines: preparation and use in the synthesis of iminosugar-C-glycosides.

Scheme 2.
Addition of Grignard reagents to N-benzylglycosylamines.
Scheme 3.
Addition of Grignard reagents to N-benzylglycosylamines in the ribofuranose series.

Scheme 4.
Addition of Grignard reagents to N-benzylglycosylamines in the erythrofuranose series.

Scheme 5.
Addition of lithium difluoromethylphosphonate to an N-benzyl furanosylamine.
Scheme 6.
Indium-mediated allylation of a free pentose.
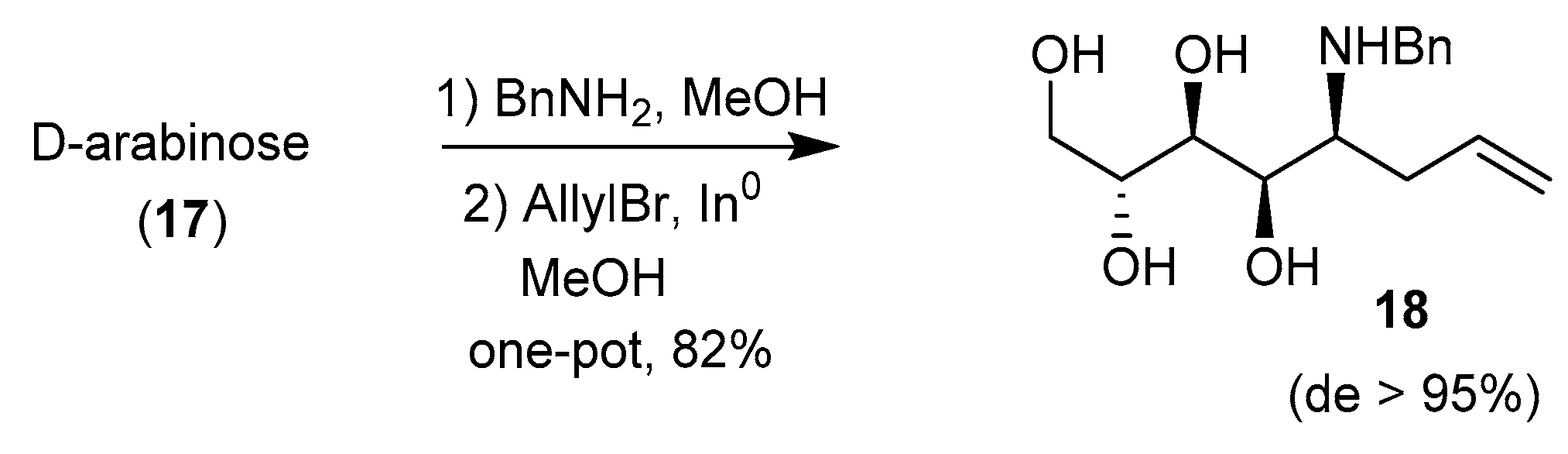
Scheme 7.
Synthesis of the DPA mimic.
Scheme 8.
Synthesis of a Glucosylceramide Mimic.

Scheme 9.
One-pot process leading to C-arylated iminosugars.

Scheme 10.
Synthesis of a tetracyclic iminosugar derivative using tryptophane methyl ester.

Scheme 11.
Synthesis of 1-C-alkynylated iminosugars.
Scheme 12.
Synthesis of 1-C-nitromethyl iminosugars.

Scheme 13.
Cu-mediated alkynylation of an in situ-generated furanosylamine.
Scheme 14.
1-C-allylation of 4- or 5-C-vinylated glycosylamines.

Scheme 15.
Synthesis of 2,5-dialkylated pyrrolidine 2,3-diol.

Scheme 16.
Synthetic approaches to iminosugars using glycosylhydroxylamines.

Figure 1.
Iminosugar-C-glycosyl compounds from N-glycosylhydroxylamines.


Scheme 18.
Attempted ring-opening reactions of O-acetyl-protected furanosylamines.

Scheme 19.
Ring-opening reactions of O-acetyl-protected 2-deoxy furanosylamines.

Figure 2.
Synthesis of iminosugar-C-glycosides from N-Z-glycosylamines and proposed transition states for the acyclic iminium ion intermediate.
Figure 2.
Synthesis of iminosugar-C-glycosides from N-Z-glycosylamines and proposed transition states for the acyclic iminium ion intermediate.


Scheme 22.
Ring-opening reactions of N-Z-pentopyranosylamines 54e and 54f [92].
Scheme 22.
Ring-opening reactions of N-Z-pentopyranosylamines 54e and 54f [92].

Figure 3.
Iminosugar-C-glycosyl compounds.

Scheme 24.
Synthesis of novel polyhydroxylated quinolizidines 78 and 79.



Scheme 27.
Synthesis of iminosugar-C-glycosides from N-t-butanesulfinyl iminoalditols [99,101,102,103].

Scheme 28.
Synthesis of triazole-linked iminosugars and aromatic systems as simplified UDP-Galf mimics [102].
Scheme 28.
Synthesis of triazole-linked iminosugars and aromatic systems as simplified UDP-Galf mimics [102].
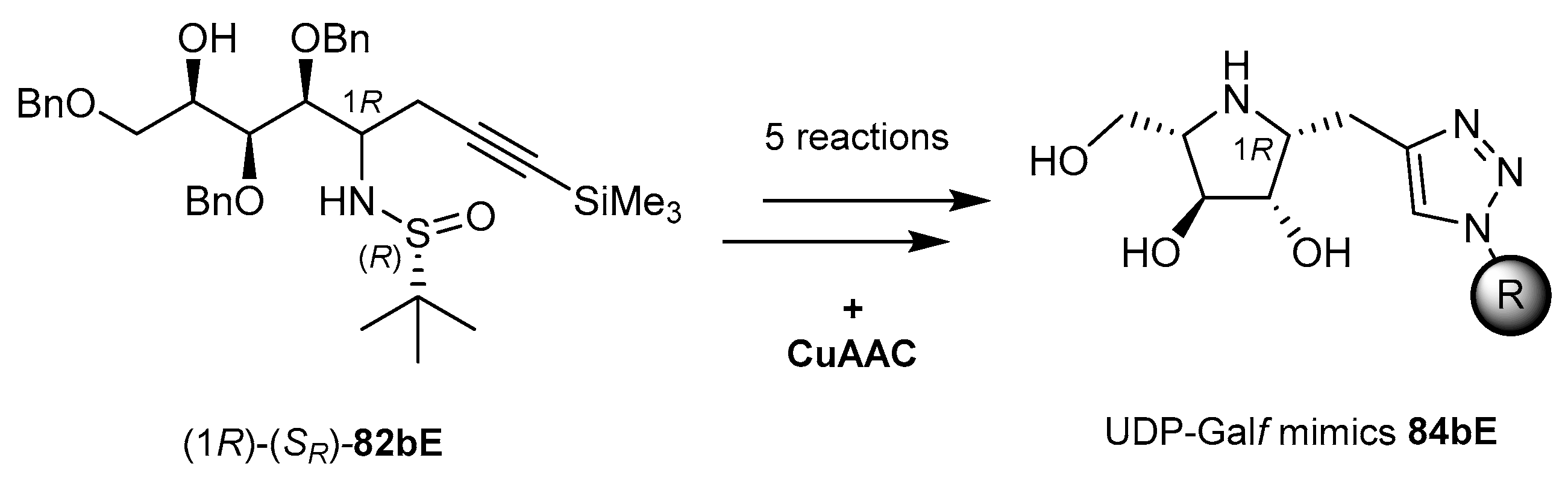
Scheme 29.
Tunable synthesis of 1-C-diethylphosphono(difluoromethyl) iminosugars as glycosyl phosphate mimics [101].
Scheme 29.
Tunable synthesis of 1-C-diethylphosphono(difluoromethyl) iminosugars as glycosyl phosphate mimics [101].

Scheme 30.
Synthesis of 1-C-Difluorophosphonomethyl- and 1-C-phosphonomethyl-1,4-imino-l-arabinitols and their activity as Galf transferase inhibitors [101,103].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
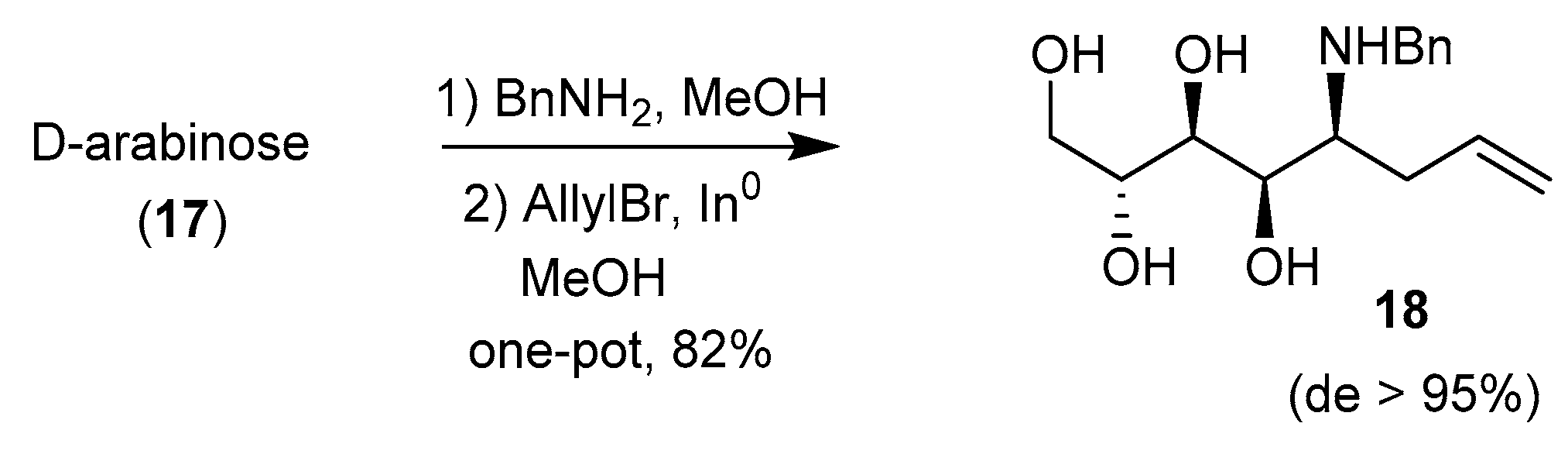{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Addition of organometallic reagents R-[M] to N-Benzyl-N-glycosyl-hydroxylamines [77,78,80,81].
 | ||||
|---|---|---|---|---|
| Entry | Hydroxylamine | R-[M] | Yield (%) [a] | anti/syn [b] |
| 1 | 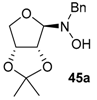 |  | 65 | 98/2 |
| 2 | 45a |  | 72 | 70/30 |
| 3 | 45a |  | 95 | 96/4 |
| 4 | 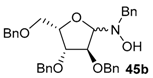 |  | 72 | 80/20 |
| 5 | 45b | 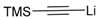 | 82 | 90/10 |
| 6 | 45b |  | 80 | 85/15 |
| 7 |  |  | 72 [c] | ND [d] |
| 8 | 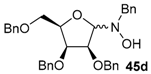 | 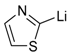 | 52 [c] | 70/30 |
| 9 |  | 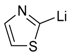 | 75 | 90/10 |
| 10 | 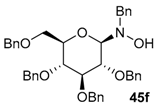 |  | 80 | 25/75 |
| 11 | 45f |  | 85 | 85/15 |
| 12 | 45f | 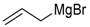 | 85 | 50/50 |
| 13 | 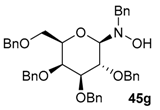 |  | 77 | 33/67 |
| 14 | 45g | 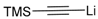 | 85 | 75/25 |
| 15 | 45g | 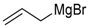 | 90 | 50/50 |
| 16 | 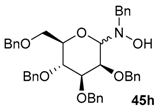 |  | 82 | 60/40 |
 | ||||
[a] Isolated yield of the diastereomeric mixture after flash chromatography; [b] The diastereomeric ratio was determined by 1H-NMR analysis of the crude mixture; [c] Yield of isolated 1,2-anti disatereomer; [d] ND = not determined.
Table 2.
Addition of silicon reagents 55A–D (Nu-SiR3) to N-(benzyloxycarbonyl)-N-glycosides 54a–d [89].
Table 2.
Addition of silicon reagents 55A–D (Nu-SiR3) to N-(benzyloxycarbonyl)-N-glycosides 54a–d [89].
 | |||
 |  |  |  |
 55A | −40 °C, 12 h [a] 59aA (quant.) [b,d] syn:anti 93:7 | −40 °C, 21 h [a] 59bA (62%) [b,d] syn:anti 91:9 | - |
| Me2ClSiH 55B | 0 °C, 6 h [c] 59aB (76%) [b,d] | 0 °C, 9 h [c] 59bB (59%) [b,d] | - |
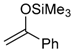 55C | −40 °C, 16 h [a] 59aC (70%) [b,d] syn:anti 93:7 | −40 °C, 37 h [a] 59bC (77%) [b,d] syn:anti 93:7 | −40 °C, 9 h [a] 59cC (80%) [b,d] syn:anti 50:50 |
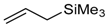 55D | 0 °C, 23 h [a] 59aD (0%) [b,d] | −30 °C, 39 h [a] 59bD (64%) [b,d] syn:anti > 94:6 | - |
 | |||
[a] Reactions were performed using 0.2 equiv. of TMSOTf and 2 equiv. of silylated nucleophile; [b] Isolated yield of the diastereomeric mixture after flash chromatography; [c] Reactions were performed using 1.0 equiv. of TMSOTf and 2 equiv. of Me2ClSiH; [d] Product 59: A Nu = CMe2CO2Me; B Nu = H; C Nu = CH2COPh; D Nu = CH2CH = CH2.
Table 3.
Addition of Grignard reagents onto N-tert-butanesulfinyl glycosylamines (SR)-81a and (SS)-81a [99].
Table 3.
Addition of Grignard reagents onto N-tert-butanesulfinyl glycosylamines (SR)-81a and (SS)-81a [99].
 | |||||
|---|---|---|---|---|---|
| Entry | Reagent (equiv.) | Auxiliary (SR or SS) | Dr [a] | Yield (%) [b,c] | Rf [d] |
| 1 | PhMgBr (3.5) | SR | 7:3 | 69 | 0.3 |
| 2 | PhMgBr (3.5) | SR | 10:0 [e] | 30 | – |
| 3 | PhMgBr (3.5) | SR | 85:15 [e,f] | 60 | – |
| 4 | vinMgBr (6) | SR | 8:2 | 69 | 0.5/0.3 |
| 5 | n-hexMgBr (6) | SR | 6:4 | 45 | 0.5 |
| 6 | BnMgCl (3.5) | SR | 7:3 | 83 | 0.6/0.55 |
| 7 | BnMgCl (3.5) | SR | 9:1 [e] | 95 | |
| 8 | PhMgBr (3.5) | SS | 7:3 | 57 | 0.5/0.2 |
| 9 | vinMgBr (6) | SS | 8:2 | 62 | 0.3/0.1 |
| 10 | n-hexMgBr (6) | SS | 6:4 | 40 | 0.5/0.4 |
| 11 | BnMgCl (3.5) | SS | ~85:15 | 94 | 0.6/0.5 |
| 12 | BnMgCl (3.5) | SS | 97:3 [e] | 83 | – |
| 13 | allMgBr (3.5) | SS | 5:5 | 83 | 0.6/0.4 |
[a] d.r determined on crude mixture using 1H-NMR (250 MHz) spectroscopy; [b] Isolated yield (SiO2 column chromatography); [c] 82aA (R = Ph), 82aB (R = vin), 82aC (R = n-hex), 82aD (R = Bn); [d] (1st eluted fraction/2nd eluted fraction). Rf determined on TLC (SiO2) using PE:EA 5:5 as eluent; [e] Reaction performed with LiCl (3.5 equiv.) as an additive; [f] Reaction mixture allowed to reach 5 °C over a period of 3.5 h.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nicolas, C.; Martin, O.R. Glycoside Mimics from Glycosylamines: Recent Progress. Molecules 2018, 23, 1612. https://doi.org/10.3390/molecules23071612
AMA Style
Nicolas C, Martin OR. Glycoside Mimics from Glycosylamines: Recent Progress. Molecules. 2018; 23(7):1612. https://doi.org/10.3390/molecules23071612
Chicago/Turabian StyleNicolas, Cyril, and Olivier R. Martin. 2018. "Glycoside Mimics from Glycosylamines: Recent Progress" Molecules 23, no. 7: 1612. https://doi.org/10.3390/molecules23071612