Orientation of Laurdan in Phospholipid Bilayers Influences Its Fluorescence: Quantum Mechanics and Classical Molecular Dynamics Study

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
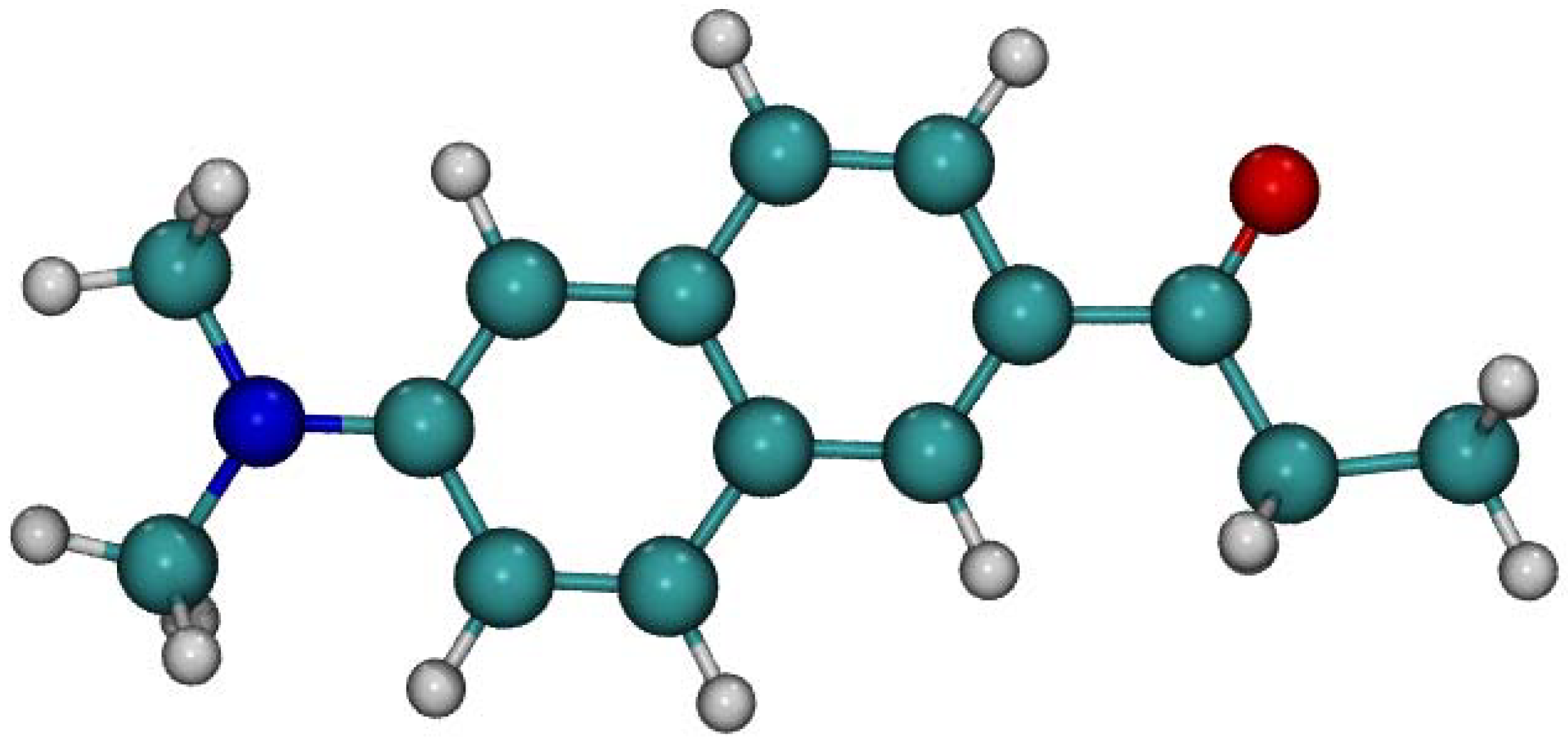
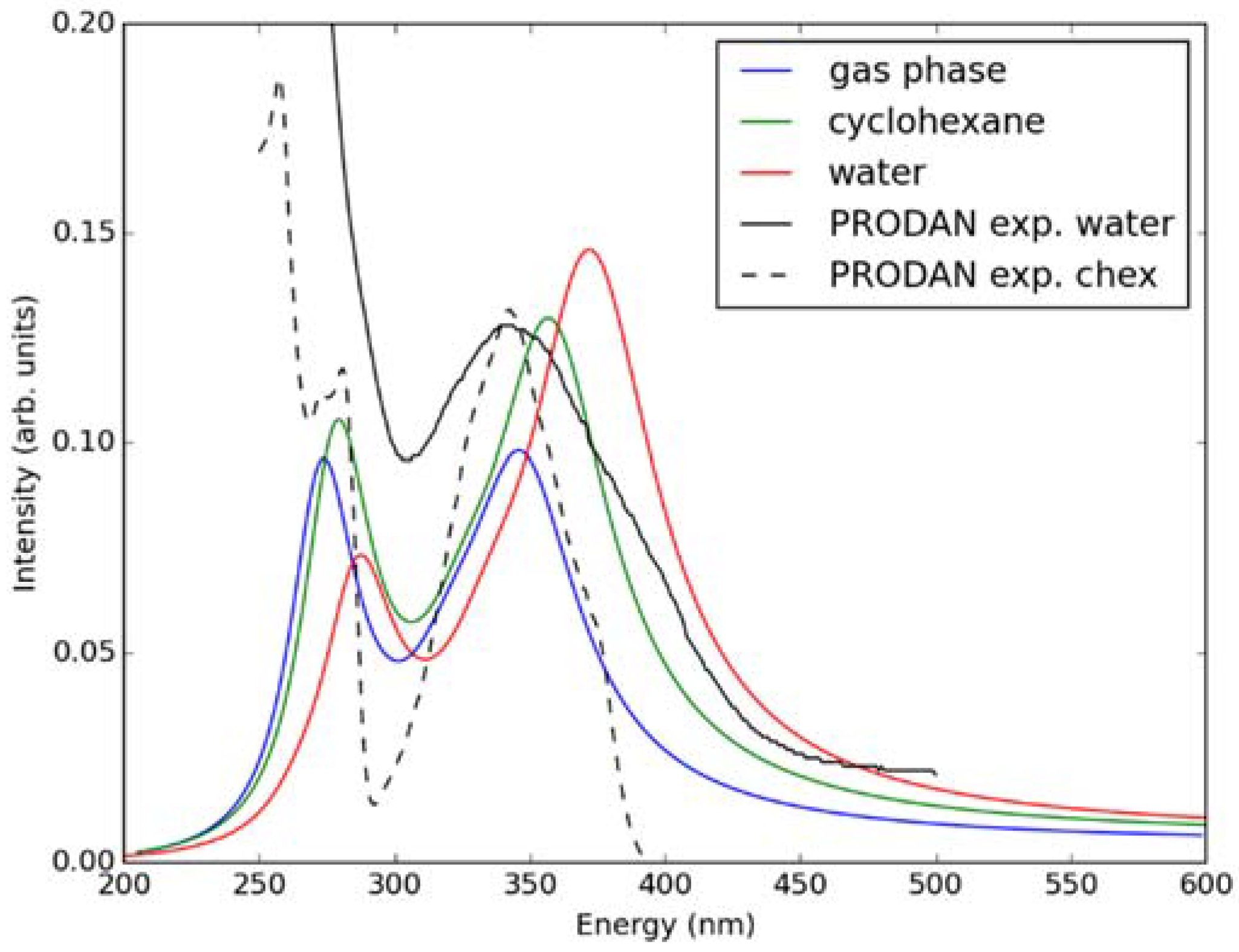
2.1. Ground State Absorption Spectra–Benchmarking the TD-DFT Method
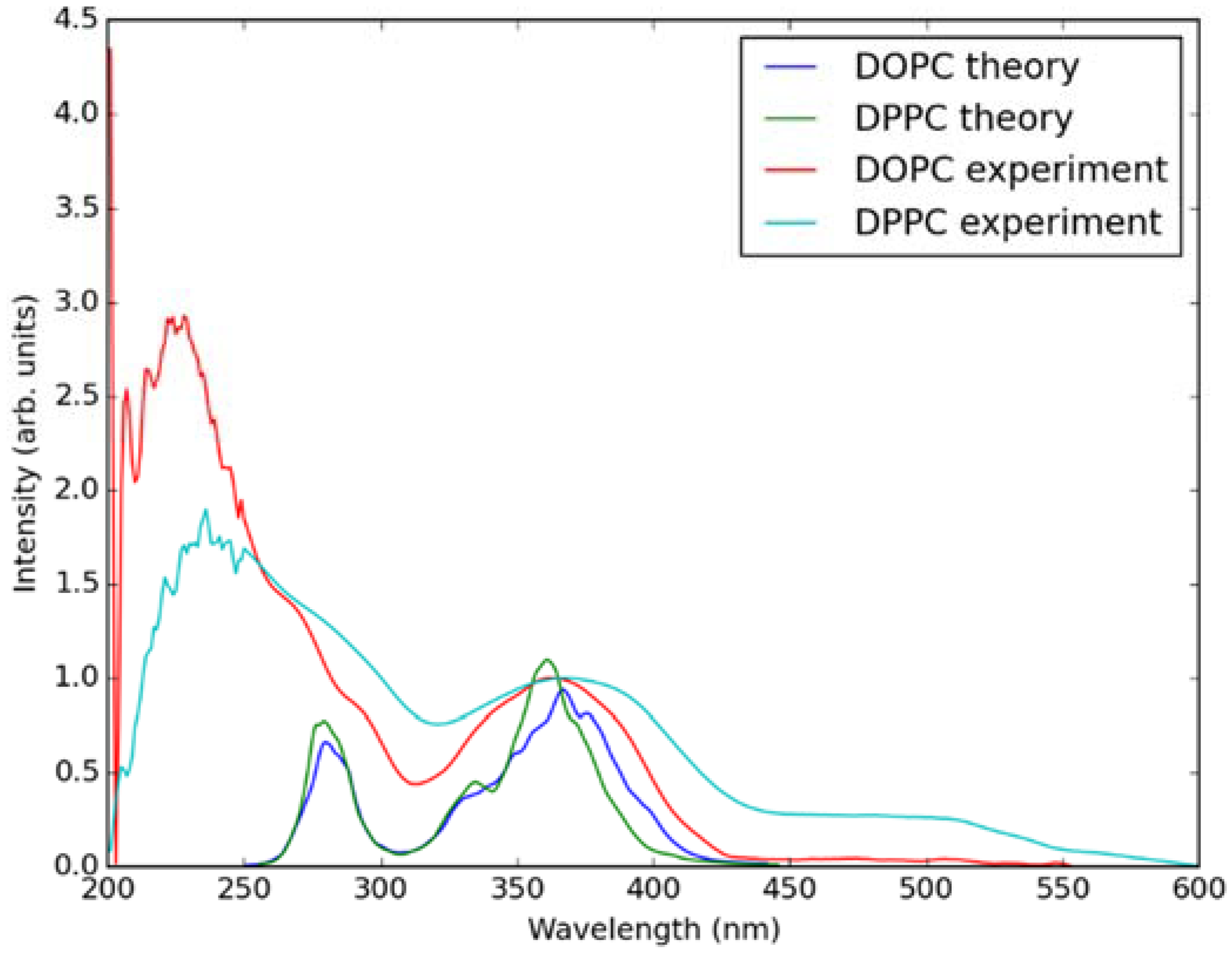
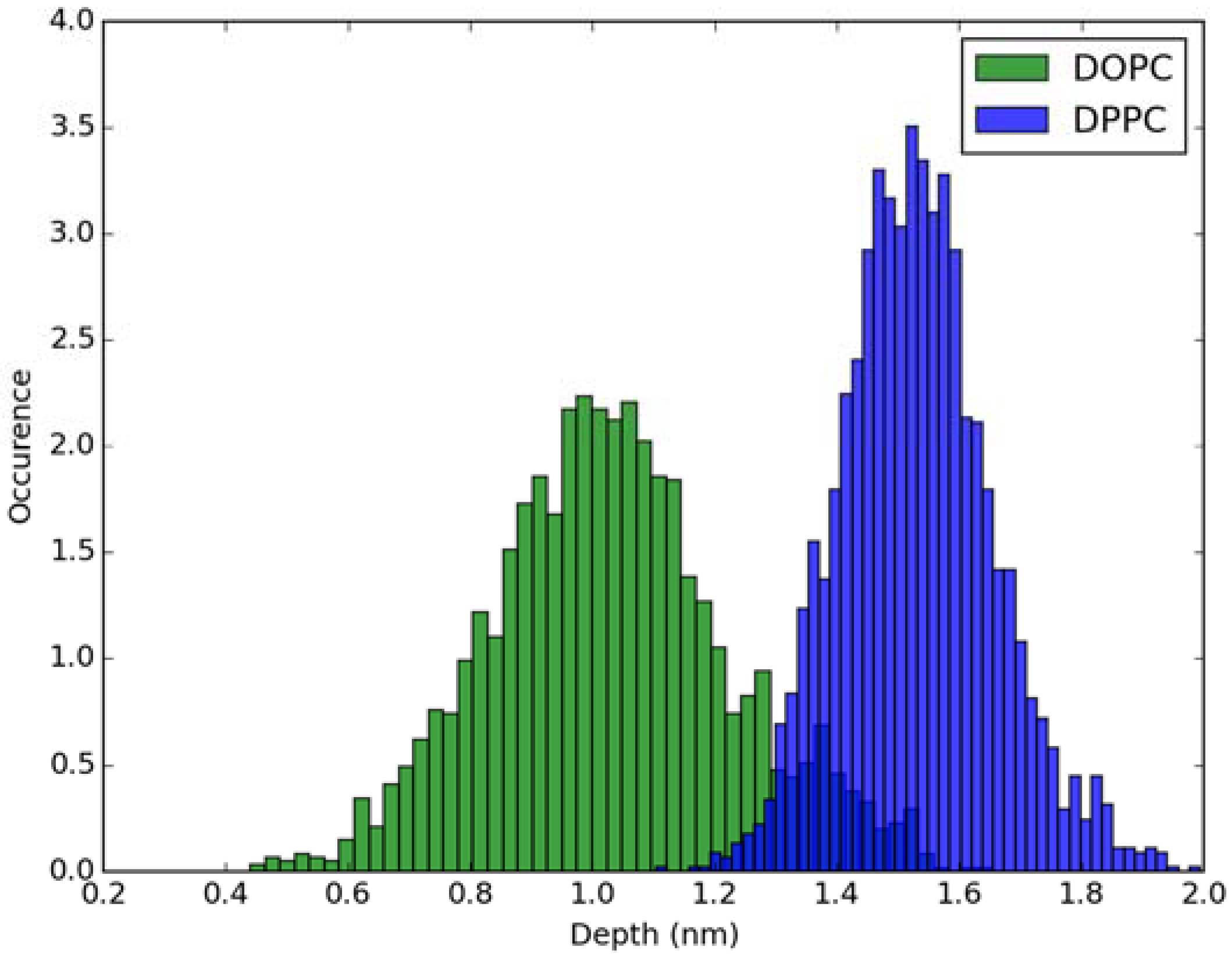
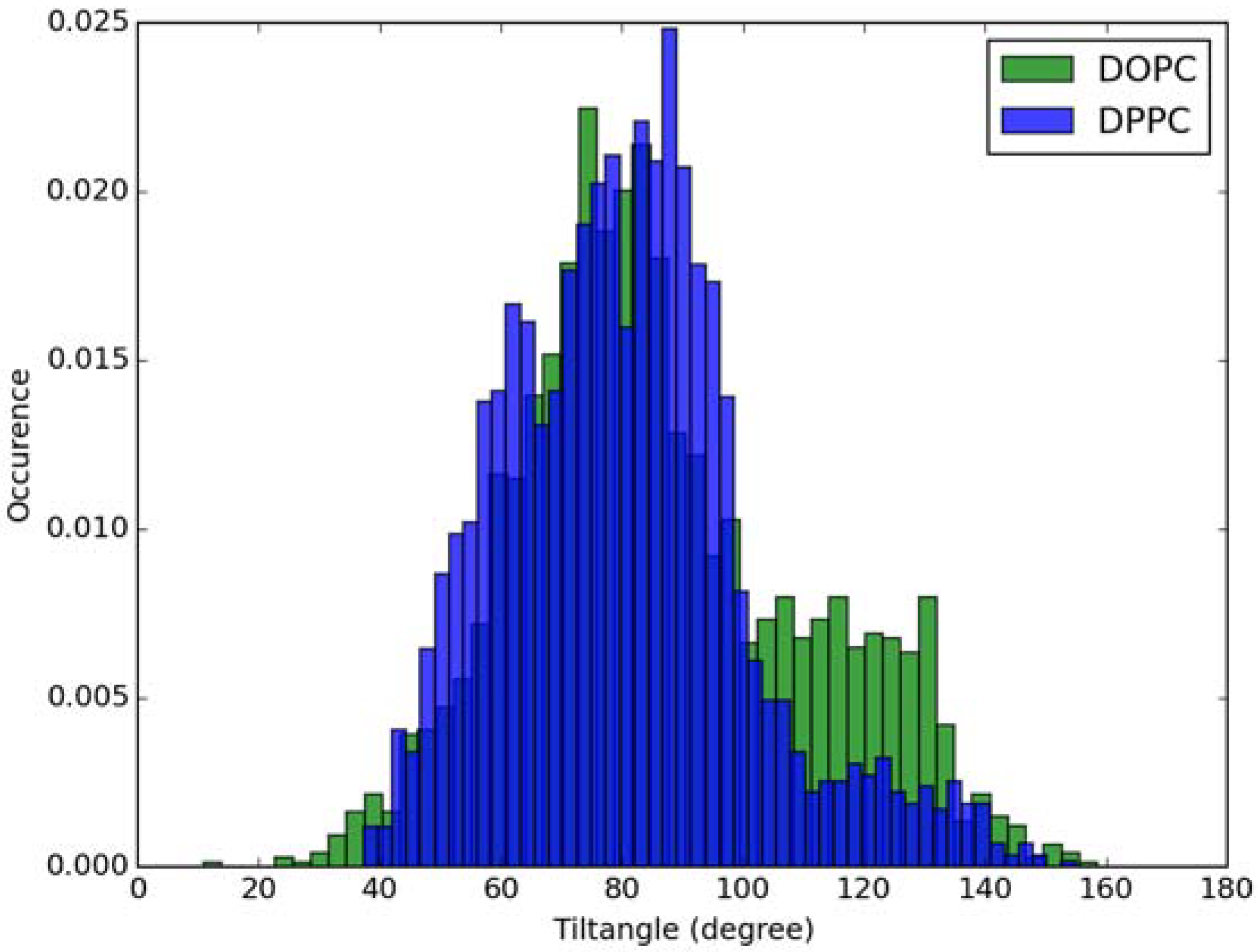
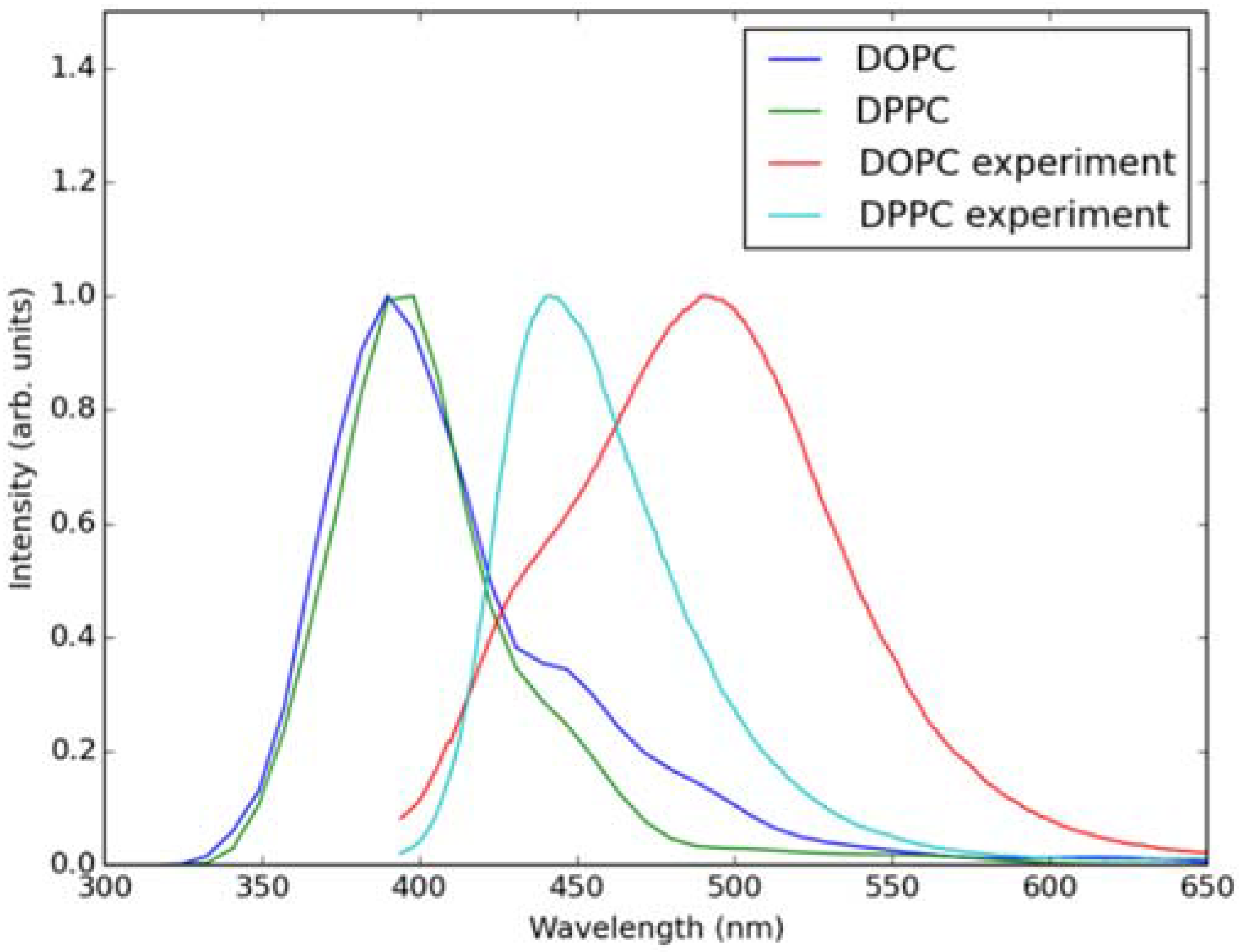
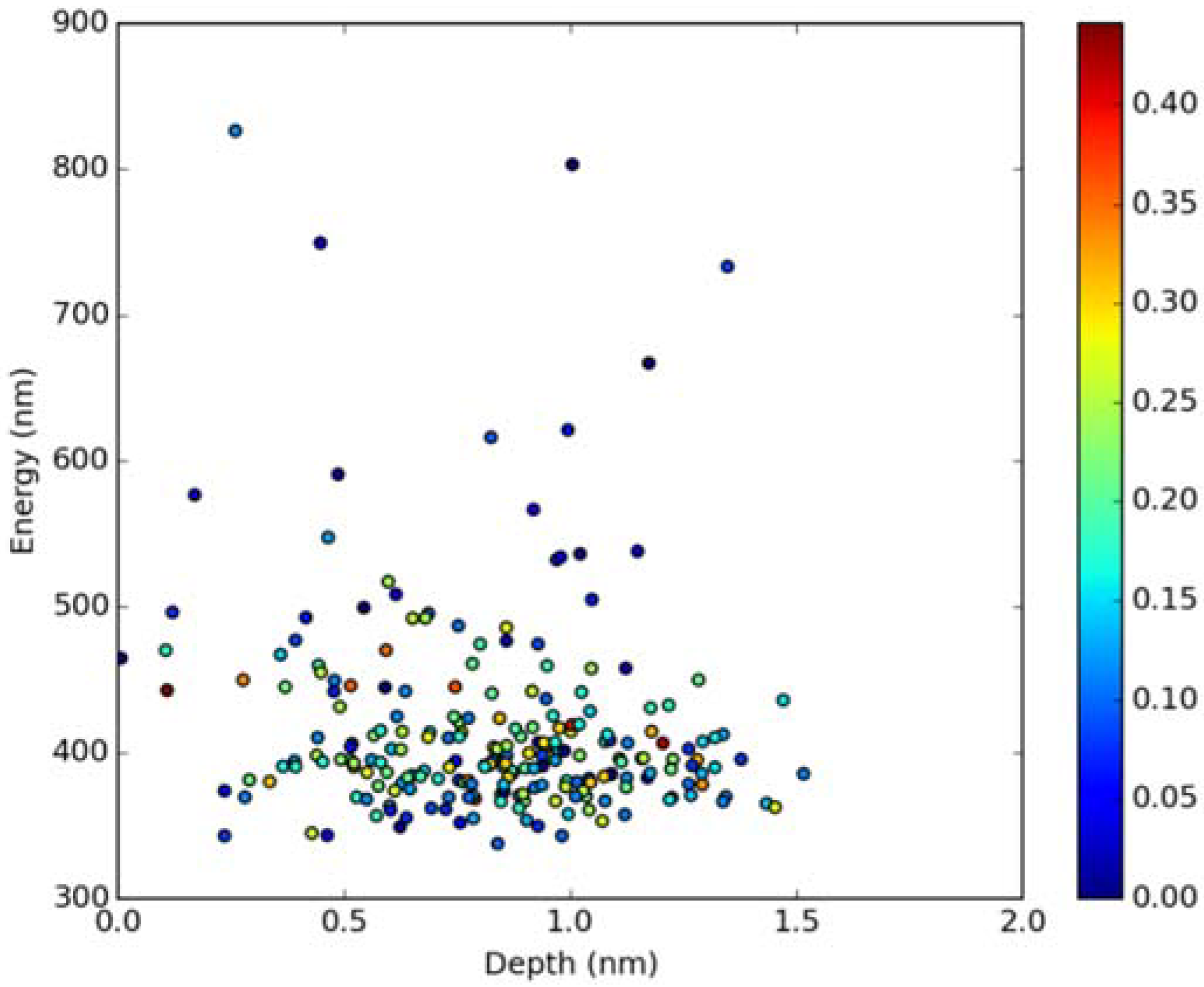
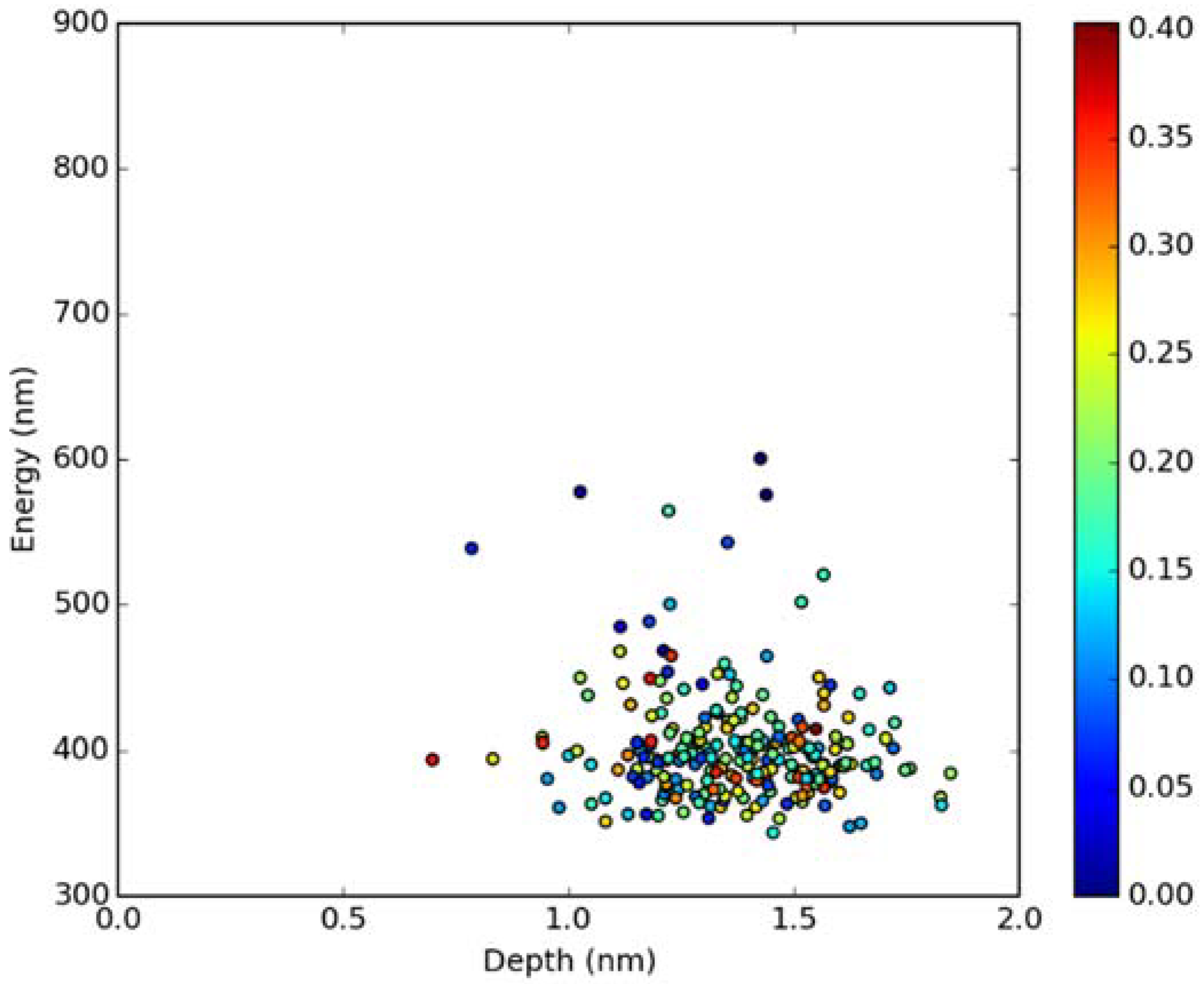
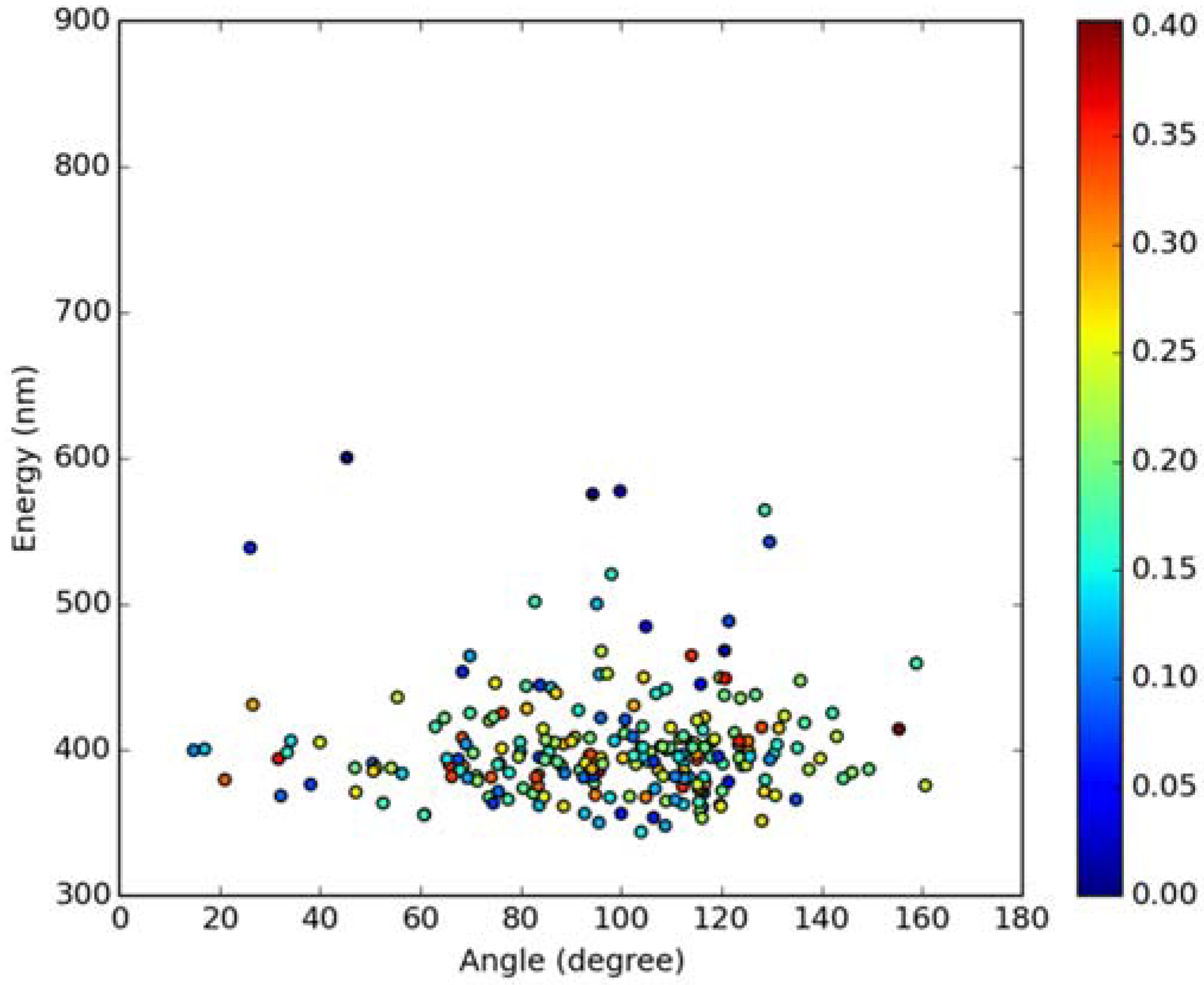
2.2. Absorption Spectra in Lipid Bilayers
2.3. Emission Spectra in Lipid Bilayers
3. Materials and Methods
3.1. Quantum Chemical Calculations
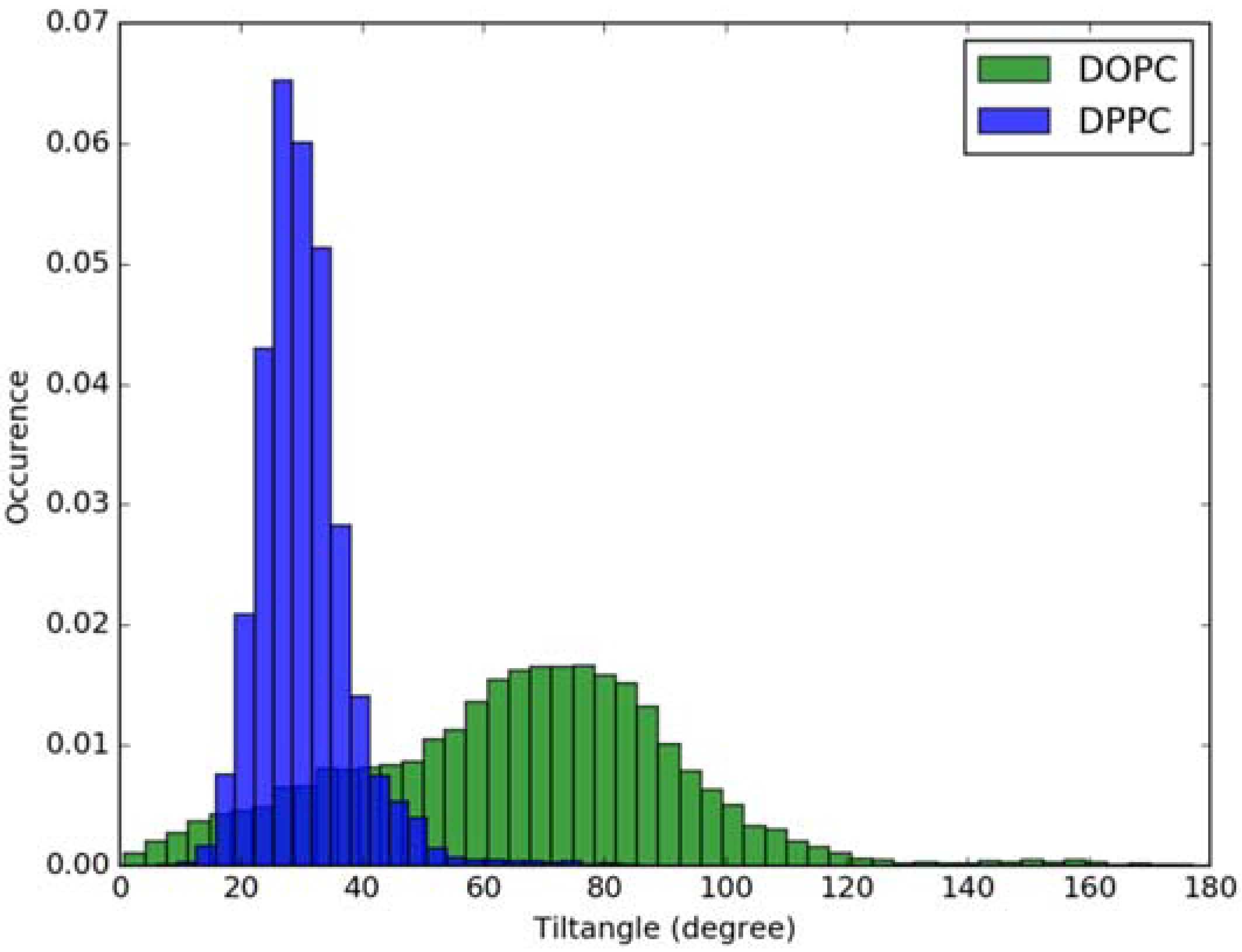
3.2. Classical MD Simulations
3.3. Simulations of Absorption and Emission Spectra in Lipid Bilayers
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lakowicz, J. Instrumentation for fluorescence spectroscopy.Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 1999. [Google Scholar]
- Bagatolli, L.; Gratton, E. Two-photon fluorescence microscopy observation of shape changes at the phase transition in phospholipid giant unilamellar vesicles. Biophy. J. 1999, 77, 2090–2101. [Google Scholar] [CrossRef]
- Simons, K.; Gerl, M.J. Revitalizing membrane rafts: New tools and insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Parasassi, T.; De Stasio, G.; Ravagnan, G.; Rusch, R.; Gratton, E. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of laurdan fluorescence. Biophy. J. 1991, 60, 179–189. [Google Scholar] [CrossRef]
- Barucha-Kraszewska, J.; Kraszewski, S.; Ramseyer, C. Will c-laurdan dethrone laurdan in fluorescent solvent relaxation techniques for lipid membrane studies? Langmuir 2013, 29, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Osella, S.; Murugan, N.A.; Jena, N.K.; Knippenberg, S. Investigation into biological environments through (non) linear optics: A multiscale study of laurdan derivatives. J. Chem. Theory Comput. 2016, 12, 6169–6181. [Google Scholar] [CrossRef] [PubMed]
- Cwiklik, L.; Aquino, A.J.A.; Vazdar, M.; Jurkiewicz, P.; Pittner, J.; Hof, M.; Lischka, H. Absorption and fluorescence of prodan in phospholipid bilayers: A combined quantum mechanics and classical molecular dynamics study. J. Phys. Chem. A 2011, 115, 11428–11437. [Google Scholar] [CrossRef] [PubMed]
- Runge, E.; Gross, E.K. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 1984, 52, 997. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The pbe0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Jambeck, J.P.; Lyubartsev, A.P. Derivation and systematic validation of a refined all-atom force field for phosphatidylcholine lipids. J. Phys. Chem. B 2012, 116, 3164–3179. [Google Scholar] [CrossRef] [PubMed]
- Vega, C.; de Miguel, E. Surface tension of the most popular models of water by using the test-area simulation method. J. Chem. Phys. 2007, 126, 154707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field (vol 25, pg 1157, 2004). J. Comput. Chem. 2005, 26, 1157–1174. [Google Scholar]
- Frisch, M.; Trucks, G.; Schlegel, H.B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision A. 02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Barucha-Kraszewska, J.; Kraszewski, S.; Jurkiewicz, P.; Ramseyer, C.; Hof, M. Numerical studies of the membrane fluorescent dyes dynamics in ground and excited states. Biochim. Biophys. Acta-Biomembr. 2010, 1798, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, G.; Harder, E.; Vorobyov, I.V.; Roux, B.; MacKerell, A.D. A polarizable model of water for molecular dynamics simulations of biomolecules. Chem. Phys. Lett. 2006, 418, 245–249. [Google Scholar] [CrossRef]
- Pederzoli, M.; Sobek, L.; Brabec, J.; Kowalski, K.; Cwiklik, L.; Pittner, J. Fluorescence of prodan in water: A computational qm/mm md study. Chem. Phys. Lett. 2014, 597, 57–62. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Barbatti, M.; Ruckenbauer, M.; Plasser, F.; Pittner, J.; Granucci, G.; Persico, M.; Lischka, H. Newton-x: A surface-hopping program for nonadiabatic molecular dynamics. Rev. Comput. Mol. Sci. 2014, 4, 26–33. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Excited State | PBE0/aug-cc-pVDZ | PBE0/aug-cc-pVTZ | B3LYP/aug-cc-pVDZ | B3LYP/aug-cc-pVTZ |
| S1 | 3.57 (0.28) | 3.57 (0.28) | 3.43 (0.28) | 3.44 (0.28) |
| S2 | 3.81 (0.48 × 10−4) | 3.83 (0.58 × 10−4) | 3.71 (0.33 × 10−3) | 3.73 (0.52 × 10−1) |
| S3 | 3.85 (0.77 × 10−1) | 3.86 (0.72 × 10−1) | 3.73 (0.55 × 10−3) | 3.74 (0.56 × 10−3) |
| S4 | 4.47 (0.15 × 10−1) | 4.49 (0.15 × 10−1) | 4.22 (0.13 × 10−1) | 4.23 (0.13 × 10−1) |
| S5 | 4.54 (0.3) | 4.54 (0.31) | 4.36 (0.25) | 4.36 (0.26) |
| Excited State | PBE0/cc-pVDZ | PBE0/cc-pVTZ | B3LYP/cc-pVDZ | B3LYP/cc-pVTZ |
| S1 | 3.64 (0.26) | 3.65 (0.27) | 3.52 (0.26) | 3.52 (0.27) |
| S2 | 3.83(0.21 × 10−4) | 3.86 (0.41 × 10−4) | 3.74 (0.15 × 10−4) | 3.78 (0.35 × 10−4) |
| S3 | 3.95 (0.62 × 10−1) | 3.94 (0.73 × 10−1) | 3.82 (0.45 × 10−1) | 3.81 (0.52 × 10−1) |
| S4 | 4.65 (0.36) | 4.64 (0.34) | 4.48 (0.31) | 4.46 (0.29) |
| S5 | 5.13 (0.14 × 10−3) | 5.15 (0.21 × 10−3) | 4.88 (0.38 × 10−4) | 4.91 (0.26 × 10−4) |
| Excited State | PBE0aug-/cc-pVDZ | PBE0/aug-cc-pVTZ | B3LYP/aug-cc-pVDZ | B3LYP/aug-cc-pVTZ |
| S1 | 3.33 (0.46) | 3.34 (0.45) | 3.19 (0.45) | 3.21 (0.44) |
| S2 | 3.69 (0.89 × 10−1) | 3.70 (0.89 × 10−1) | 3.57 (0.62 × 10−1) | 3.58 (0.65 × 10−1) |
| S3 | 3.96 (0.54 × 10−4) | 3.98 (0.67 × 10−4) | 3.88 (0.61 × 10-4) | 3.92 (0.74 × 10−4) |
| S4 | 4.32 (0.21) | 4.33 (0.23) | 4.12 (0.18) | 4.15 (0.20) |
| S5 | 4.67 (0.16 × 10−1) | 4.67 (0.15 × 10−1) | 4.43 (0.14 × 10−1) | 4.44 (0.14 × 10−1) |
| Excited State | PBE0/cc-pVDZ | PBE0/cc-pVTZ | B3LYP/cc-pVDZ | B3LYP/cc-pVTZ |
| S1 | 3.44 (0.37) | 3.44 (0.41) | 3.32 (0.37) | 3.31 (0.41) |
| S2 | 3.82 (0.10) | 3.80 (0.10) | 3.69 (0.73 × 10−1) | 3.68 (0.74 × 10−1) |
| S3 | 3.98 (0.39 × 10−4) | 4.04 (0.64 × 10−4) | 3.90 (0.34 × 10−4) | 3.97 (0.58 × 10−4) |
| S4 | 4.47 (0.29) | 4.44 (0.27) | 4.28 (0.24) | 4.26 (0.22) |
| S5 | 5.11 (0.16 × 10−1) | 5.09 (0.45) | 4.95 (0.27) | 4.95 (0.33) |
| Excited State | PBE0/aug-cc-pVDZ | PBE0aug-/cc-pVTZ | B3LYP/aug-cc-pVDZ | B3LYP/aug-cc-pVTZ |
| S1 | 3.46 (0.39) | 3.47 (0.39) | 3.33 (0.39) | 3.34 (0.39) |
| S2 | 3.79 (0.92 × 10−1) | 3.80 (0.88 × 10−1) | 3.67 (0.66 × 10−1) | 3.67 (0.63 × 10−1) |
| S3 | 3.86 (0.58 × 10−4) | 3.88 (0.63 × 10−4) | 3.77 (0.52 × 10−4) | 3.79 (0.59 × 10-4) |
| S4 | 4.44 (0.32) | 4.44 (0.34) | 4.26 (0.27) | 4.26 (0.29) |
| S5 | 4.55 (0.19 × 10−1) | 4.56 (0.18 × 10−1) | 4.30 (0.16 × 10−1) | 4.32 (0.16 × 10−1) |
| Excited State | PBE0/cc-pVDZ | PBE0/cc-pVTZ | B3LYP/cc-pVDZ | B3LYP/cc-pVTZ |
| S1 | 3.55 (0.34) | 3.55 (0.36) | 3.43 (0.34) | 3.43 (0.37) |
| S2 | 3.89 (0.33 × 10−4) | 3.89 (0.91 × 10−1) | 3.77 (0.60 × 10−1) | 3.77 (0.66 × 10−1) |
| S3 | 3.90 (0.82 × 10−1) | 3.902 (0.70 × 10−4) | 3.79 (0.52 × 10−) | 3.84 (0.52 × 10−4) |
| S4 | 4.56 (0.40) | 4.54 (0.38) | 4.38 (0.34) | 4.36 (0.33) |
| S5 | 5.15 (0.37) | 5.13 (0.44) | 4.95 (0.29 × 10−3) | 4.98 (0.33) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wasif Baig, M.; Pederzoli, M.; Jurkiewicz, P.; Cwiklik, L.; Pittner, J. Orientation of Laurdan in Phospholipid Bilayers Influences Its Fluorescence: Quantum Mechanics and Classical Molecular Dynamics Study. Molecules 2018, 23, 1707. https://doi.org/10.3390/molecules23071707
Wasif Baig M, Pederzoli M, Jurkiewicz P, Cwiklik L, Pittner J. Orientation of Laurdan in Phospholipid Bilayers Influences Its Fluorescence: Quantum Mechanics and Classical Molecular Dynamics Study. Molecules. 2018; 23(7):1707. https://doi.org/10.3390/molecules23071707
Chicago/Turabian StyleWasif Baig, Mirza, Marek Pederzoli, Piotr Jurkiewicz, Lukasz Cwiklik, and Jiri Pittner. 2018. "Orientation of Laurdan in Phospholipid Bilayers Influences Its Fluorescence: Quantum Mechanics and Classical Molecular Dynamics Study" Molecules 23, no. 7: 1707. https://doi.org/10.3390/molecules23071707