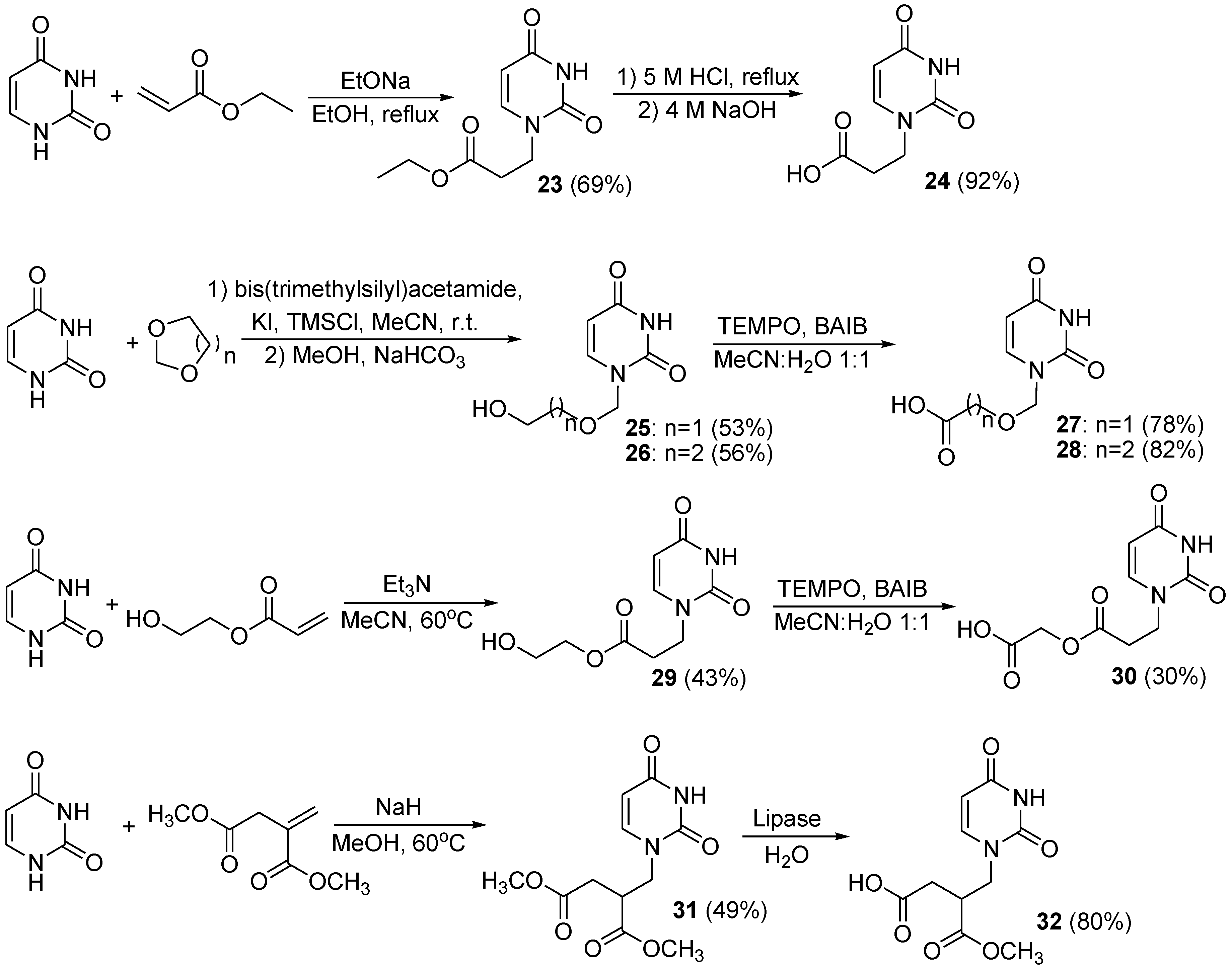
3.2.4. Synthesis of Glycoconjugates 33–54
General procedure. The corresponding amine derivative 17–22 (0.25 mmol) and acyclic uracil derivative 24, 27, 28, 30, or 32 (0.40 mmol) were dissolved in dry THF (6 mL) and MeOH (1 mL). The CDMT (70 mg, 0.40 mmol) and N-methylmorpholine (55 mg, 0.55 mmol) were added. The resulting mixture was microwaved in a reactor set at 50 °C for 1.5–4 h. The progress of the reaction was monitored on TLC plate in toluene:AcOEt (1:1) solvent system. After completion, the reaction mixtures were concentrated, dissolved in CH2Cl2 (50 mL), washed with water (20 mL), saturated NaHCO3 (20 mL), and with brine (20 mL). The organic layer was dried over MgSO4, the adsorbent was filtered off and the filtrate was concentrated to give crude products 33–54 which were purified directly by column chromatography with an appropriate solvent system as indicated.
Glycoconjugate (33) Starting from amine derivative 17 and uracil derivative 24, purified by column chromatography in CHCl3:MeOH solvent system (100:1 do 25:1 (v/v)) to give thick syrup (75 mg, 37%): 134.5 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.78 (m, 2H, CH2CO), 3.57 (dd, 1H, J = 1.7 Hz, J = 10.7 Hz, H-6a), 3.67 (dd, 1H, J = 8.8 Hz, J = 10.0 Hz, H-4), 3.72 (dd, 1H, J = 3.8 Hz, J = 10.9 Hz, H-6b), 3.81 (dd~t, 1H, J = 9.2 Hz, H-3), 3.92–4.01 (m, 3H, H-2, CH2N), 4.13 (ddd, 1H, J = 2.0 Hz, J = 3.5 Hz, J = 10.1 Hz, H-5), 4.35, 4.51 (qAB, 2H, J = 12.1 Hz, CH2Ph), 4.60, 4.78 (qAB, 2H, J = 11.3 Hz, CH2Ph), 4.48, 4.83 (qAB, 2H, J = 11.0 Hz, CH2Ph), 4.78, 4.97 (qAB, 2H, J = 10.9 Hz, CH2Ph), 5.42 (d, 1H, J = 7.9 Hz, H-5ur), 6.47 (d, 1H, J = 5.4 Hz, H-1), 7.12–7.34 (m, 22H, H-Ph, H-6ur, H-3pyr), 7.95 (dd, 1H, J = 2.5 Hz, J = 8.7 Hz, H-4pyr), 8.54 (d, 1H, J = 2.5 Hz, H-6pyr), 9.14 (s, 1H, NH), 10.68 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 35.48 (CH2CO), 46.10 (CH2N), 68.53 (C-6), 72.38 (C-5), 72.17, 73.21, 75.07, 75.64 (CH2Ph), 77.18 (C-4); 79.10 (C-2), 82.83 (C-3), 83.96 (C-1), 101.67 (C-5ur), 124.14 (C-3pyr), 127.60, 127.67, 127.77, 128.80, 127.82, 127.96, 127.98, 128.29, 128.33, 128.34 (C-Ph, C-4pyr), 132.70 (C-5pyr), 137.59, 137.82, 138.14, 138.59 (C-Ph), 141.47 (C-6pyr), 146.42 (C-6ur), 151.19 (C-2pyr), 151.43 (C-2ur), 164.88 (C-4ur), 169.06 (NHCO). HRMS (ESI) (m/z): [M + Na]+ calcd for C46H46N4NaO8S, 837.2934; found, 837.3016.
Glycoconjugate (34) Starting from amine derivative 18 and uracil derivative 24, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 (v/v)) to give thick syrup (136 mg, 67%): 70.3 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.71 (m, 2H, CH2CO), 3.47 (dd, 1H, J = 6.1 Hz, J = 9.5 Hz, H-6a), 3.54 (dd, 1H, J = 6.7 Hz, J = 9.6 Hz, H-6b), 3.75 (dd, 1H, J = 2.8 Hz, J = 9.9 Hz, H-3), 3.89 (m, 2H, CH2N), 3.96 (m, 1H, H-4), 4.28 (m, 1H, H-5), 4.29, 4.34 (qAB, 2H, J = 11.7 Hz, CH2Ph), 4.40 (dd, 1H, J = 5.4 Hz, J = 9.9 Hz, H-2), 4.64, 4.75 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.72, 4.84 (qAB, 2H, J = 11.9 Hz, CH2Ph), 4.55, 4.93 (qAB, 2H, J = 11.4 Hz, CH2Ph), 5.34 (d, 1H, J = 7.9 Hz, H-5ur), 6.44 (d, 1H, J = 5.4 Hz, H-1), 7.12–7.39 (m, 22H, H-Ph, H-6ur, H-3pyr), 7.92 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.50 (d, 1H, J = 2.6 Hz, H-6pyr), 9.10 (s, 1H, NH), 10.75 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 46.04 (CH2N), 35.44 (CH2CO), 68.64 (C-6), 71.38 (C-5), 72.34, 73.16, 73.31, 74.79 (CH2Ph), 74.88 (C-4), 75.87 (C-2); 79.66 (C-3); 84.67 (C-1), 101.63 (C-5ur), 124.11 (C-3pyr), 127.47, 127.54, 127.57, 127.64, 127.80, 128.09, 128.19, 128.22, 128.29, 128.31 (C-Ph, C-4pyr), 132.61 (C-5pyr), 137.85, 137.97, 138.50, 138.57 (C-Ph), 141.43 (C-6pyr), 143.46 (C-6ur), 151.42 (C-2pyr), 151.56 (C-2ur), 164.89 (C-4ur), 169.03 (NHCO). HRMS (ESI) (m/z): [M + Na]+ calcd for C46H46N4NaO8S, 837.2934; found, 837.2966.
Glycoconjugate (35) Starting from amine derivative 17 and uracil derivative 27, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 (v/v)) to give thick syrup (81 mg, 39%): 122.0 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 3.57 (dd, 1H, J = 1.9 Hz, J = 10.8 Hz, H-6a), 3.65–3.75 (m, 2H, H-6b, H-4), 3.82 (dd~t, 1H, J = 9.2 Hz, H-3), 3.97 (dd, 1H, J = 5.4 Hz, J = 9.6 Hz, H-2), 4.13 (ddd, 1H, J = 1.9 Hz, J = 3.5 Hz, J = 10.1 Hz, H-5), 4.21 (s, 2H, CH2N), 4.37, 4.52 (qAB, 2H, J = 12.0 Hz, CH2Ph), 4.64, 4.77 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.49, 4.83 (qAB, 2H, J = 10.8 Hz, CH2Ph), 4.79, 4.98 (qAB, 2H, J = 10.9 Hz, CH2Ph), 5.77 (d, 1H, J = 7.9 Hz, H-5ur), 6.51 (d, 1H, J = 5.4 Hz, H-1), 7.13–7.34 (m, 22H, H-Ph, H-6ur, H-3pyr), 8.05 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.62 (d, 1H, J = 2.6 Hz, H-6pyr), 8.85 (s, 1H, NH), 9.30 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 68.02 (CH2N), 68.51 (C-6), 72.43 (C-5), 72.25, 73.27, 75.07, 75.65 (CH2Ph), 77.18 (C-4), 78.02 (CH2O), 79.13 (C-2), 82.87 (C-3), 83.91 (C-1), 103.61 (C-5ur), 124.21 (C-3pyr), 127.57, 127.61, 127.66, 128.78, 127.83, 127.84, 127.97, 128.03, 128.16, 128.29, 128.33, 128.35 (C-Ph, C-4pyr), 131.78 (C-5pyr), 137.61, 137.85, 138.19, 138.62 (C-Ph), 141.33 (C-6pyr), 143.41 (C-6ur), 151.18 (C-2pyr), 151.84 (C-2ur), 162.94 (C-4ur), 166.86 (NHCO). HRMS (ESI) (m/z): [M + Na]+ calcd for C46H46N4NaO9S, 853.2883; found, 853.2696.
Glycoconjugate (36) Starting from amine derivative 18 and uracil derivative 27, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 (v/v)) to give thick syrup (145 mg, 70%): 96.5 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3) δ 3.46 (dd, 1H, J = 6.0 Hz, J = 9.6 Hz, H-6a), 3.55 (dd, 1H, J = 6.8 Hz, J = 9.6 Hz, H-6b), 3.77 (dd, 1H, J = 2.8 Hz, J = 10.0 Hz, H-3), 3.98 (dd, 1H, J = 1.0 Hz, J = 2.8 Hz, H-4), 4.18 (s, 2H, CH2N); 4.29 (dd~t, 1H, J = 6.7 Hz, H-5), 4.30, 4.36 (qAB, 2H, J = 11.7 Hz, CH2Ph), 4.42 (dd, 1H, J = 5.4 Hz, J = 10.0 Hz, H-2), 4.67, 4.78 (qAB, 2H, J = 11.6 Hz, CH2Ph); 4.73, 4.86 (qAB, 2H, J = 11.9 Hz, CH2Ph), 4.56, 4.95 (qAB, 2H, J = 11.4 Hz, CH2Ph), 5.11 (s, 2H, CH2O), 5.75 (d, 1H, J = 7.9 Hz, H-5ur), 6.48 (d, 1H, J = 5.4 Hz, H-1), 7.15–7.38 (m, 22H, H-Ph, H-6ur, H-3pyr), 8.02 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.58 (d, 1H, J = 2.6 Hz, H-6pyr), 8.82 (s, 1H, NH), 9.40 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3) δ 68.00 (CH2N), 68.59 (C-6), 71.40 (C-5), 72.43, 73.22, 73.34, 74.83 (CH2Ph), 74.94 (C-4), 75.91 (C-2), 78.00 (CH2O), 79.70 (C-3), 84.59 (C-1), 103.54 (C-5ur), 124.18 (C-3pyr), 127.49, 127.55, 127.57, 127.60, 127.72, 127.85, 128.10, 128.20, 128.24, 128.30, 128.33 (C-Ph, C-4pyr), 131.69 (C-5pyr), 137.88, 138.00, 138.54, 138.60 (C-Ph), 141.24 (C-6pyr), 143.46 (C-6ur), 151.14 (C-2pyr), 152.26 (C-2ur); 163.07 (C-4ur), 166.86 (NHCO). HRMS (ESI) (m/z): [M + Na]+ calcd for C46H46N4NaO9S, 853.2883; found, 853.2769.
Glycoconjugate (37) Starting from amine derivative 17 and uracil derivative 28, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 (v/v)) to give thick syrup (103 mg, 49%): 133.4 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.66 (t, 2H, J = 5.7 Hz, CH2CO), 3.56 (dd, 1H, J = 1.8 Hz, J = 10.8 Hz, H-6a), 3.64–3.75 (m, 2H, H-6b, H-4), 3.82 (dd~t, 1H, J = 9.2 Hz, H-3), 3.88 (t, 2H, J = 5.8 Hz, CH2O), 3.95 (dd, 1H, J = 5.4 Hz, J = 9.5 Hz, H-2), 4.14 (ddd, 1H, J = 1.9 Hz, J = 3.5 Hz, J = 10.0 Hz, H-5), 4.35, 4.51 (qAB, 2H, J = 12.0 Hz, CH2Ph), 4.61, 4.75 (qAB, 2H, J = 11.5 Hz, CH2Ph), 4.48, 4.83 (qAB, 2H, J = 10.8 Hz, CH2Ph), 4.78, 4.97 (qAB, 2H, J = 10.8 Hz, CH2Ph), 5.13 (s, 2H, CH2N), 5.77 (d, 1H, J = 7.9 Hz, H-5ur), 6.45 (d, 1H, J = 5.4 Hz, H-1), 7.11–7.34 (m, 22H, H-Ph, H-6ur, H-3pyr), 8.04 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.47 (d, 1H, J = 2.6 Hz, H-6pyr), 8.85 (s, 1H, NH), 9.54 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 37.29 (CH2CO), 65.30 (CH2O), 68.50 (C-6), 72.32 (C-5), 72.20, 73.27, 75.07, 75.65 (CH2Ph), 77.18 (C-4); 77.23 (CH2N), 79.13 (C-2), 82.82 (C-3), 84.04 (C-1), 103.25 (C-5ur), 124.36 (C-3pyr), 127.58, 127.62, 127.67, 128.78, 127.83, 127.86, 127.97, 128.00, 128.20, 128.29, 128.33, 128.35 (C-Ph, C-4pyr), 132.80 (C-5pyr), 137.59, 137.79, 138.17, 138.60 (C-Ph), 141.07 (C-6pyr), 143.53 (C-6ur), 151.07 (C-2pyr), 151.33 (C-2ur), 163.33 (C-4ur), 166.29 (NHCO). HRMS (ESI) (m/z) [M + Na]+ calc for C47H48N4NaO9S, 867.3040; found, 867.3086.
Glycoconjugate (38) Starting from amine derivative 18 and uracil derivative 28, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 (v/v)) to give thick syrup (126 mg, 60%): 134.8 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.64 (t, 2H, J = 5.7 Hz, CH2CO), 3.45 (dd, 1H, J = 6.0 Hz, J = 9.5 Hz, H-6a), 3.54 (dd, 1H, J = 6.9 Hz, J = 9.5 Hz, H-6b), 3.77 (dd, 1H, J = 2.8 Hz, J = 10.0 Hz, H-3), 3.88 (t, 2H, J = 5.7 Hz, CH2O), 3.97 (dd, 1H, J = 0.9 Hz, J = 2.7 Hz, H-4), 4.26–4.31 (m, 2H, H-5, CHHPh), 4.35 (d, 1H, J = 11.8 Hz, CHHPh), 4.41 (dd, 1H, J = 5.4 Hz, J = 10.0 Hz, H-2), 4.65, 4.76 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.72, 4.84 (qAB, 2H, J = 11.8 Hz, CH2Ph), 4.55, 4.94 (qAB, 2H, J = 11.4 Hz, CH2Ph), 5.09 (s, 2H, CH2N), 5.68 (d, 1H, J = 7.9 Hz, H-5ur), 6.43 (d, 1H, J = 5.4 Hz, H-1), 7.14–7.38 (m, 22H, H-Ph, H-6ur, H-3pyr), 8.01 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.58 (d, 1H, J = 2.6 Hz, H-6pyr), 8.82 (s, 1H, NH), 9.44 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 37.30 (CH2CO), 65.28 (CH2O), 68.59 (C-6), 71.30 (C-5), 72.38, 73.22, 73.35, 74.83 (CH2Ph), 74.93 (C-4), 75.91 (C-2), 77.20 (CH2N), 79.67 (C-3), 84.74 (C-1), 103.20 (C-5ur), 124.39 (C-3pyr), 127.49, 127.55, 127.56, 127.61, 127.73, 127.83, 128.10, 128.20, 128.24, 128.30, 128.32 (C-Ph, C-4pyr), 132.70 (C-5pyr), 137.87, 137.99, 138.55, 138.60 (C-Ph), 141.00 (C-6pyr), 143.49 (C-6ur), 152.25 (C-2ur), 151.48 (C-2pyr), 163.28 (C-4ur), 169.25 (NHCO). HRMS (ESI) (m/z): [M + Na]+ calcd for C47H48N4NaO9S, 867.3040; found, 867.3057.
Glycoconjugate (39) Starting from amine derivative 17 and uracil derivative 30, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 (v/v)) to give thick syrup (85 mg, 39%): 3.1 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.90 (dd~t, 2H, J = 5.8 Hz, J = 0.7 Hz, CH2CO), 3.57 (dd, 1H, J = 1.9 Hz, J = 10.8 Hz, H-6a), 3.68 (dd, 1H, J = 9.1 Hz, J = 9.9 Hz, H-4), 3.73 (dd, 1H, J = 3.9 Hz, J = 10.9 Hz, H-6b), 3.82 (dd~t, 1H, J = 9.2 Hz, H-3), 3.96 (dd, 1H, J = 5.4 Hz, J = 9.5 Hz, H-2), 4.06 (dd~t, 2H, J = 6.0 Hz, CH2N), 4.13 (ddd, 1H, J = 1.9 Hz, J = 3.7 Hz, J = 10.1 Hz, H-5), 4.37, 4.52 (qAB, 2H, J = 11.9 Hz, CH2Ph), 4.64, 4.77 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.48, 4.84 (qAB, 2H, J = 10.5 Hz, CH2Ph), 4.79, 4.98 (qAB, 2H, J = 10.9 Hz, CH2Ph), 4.74 (s, 2H, CH2O), 5.69 (d, 1H, J = 7.9 Hz, H-5ur), 6.50 (d, 1H, J = 5.4 Hz, H-1), 7.11–7.35 (m, 22H, H-Ph, H-6ur, H-3pyr), 8.02 (dd, 1H, J = 2.5 Hz, J = 8.7 Hz, H-4pyr), 8.48 (s, 1H, NH), 8.57 (d, 1H, J = 2.5 Hz, H-6pyr), 9.07 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 33.56 (CH2CO), 45.09 (CH2N), 63.60 (CH2O), 68.55 (C-6), 72.40 (C-5), 72.28, 73.29, 75.10, 75.68 (CH2Ph), 77.20 (C-4), 79.13 (C-2), 82.87 (C-3), 83.88 (C-1), 102.68 (C-5ur), 124.18 (C-3pyr), 127.60, 127.63, 127.69, 127.81, 127.85, 127.86, 128.04, 128.31, 128.35, 128.38, 128.69 (C-Ph, C-4pyr), 131.62 (C-5pyr), 137.61, 137.83, 138.18, 138.62 (C-Ph), 141.62 (C-6pyr), 144.90 (C-6ur), 151.03 (C-2pyr), 152.13 (C-2ur), 163.20 (C-4ur), 165.28 (NHCO), 169.83 (COO). HRMS (ESI) (m/z): [M + Na]+ calcd for C48H48N4NaO10S, 895.2989; found, 895.2917.
Glycoconjugate (40) Starting from amine derivative 18 and uracil derivative 30, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 (v/v)) to give thick syrup (105 mg, 48%): 103.2 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.82 (dd~t, 2H, J = 5.8 Hz, CH2CO), 3.47 (dd, 1H, J = 6.2 Hz, J = 9.6 Hz, H-6a), 3.53 (dd, 1H, J = 6.6 Hz, J = 9.6 Hz, H-6b), 3.76 (dd, 1H, J = 2.9 Hz, J = 10.0 Hz, H-3), 3.91–4.01 (m, 3H, H-4, CH2N), 4.28 (dd~t, 1H, J = 6.7 Hz, H-5), 4.30, 4.36 (qAB, 2H, J = 11.8 Hz, CH2Ph), 4.41 (dd, 1H, J = 5.4 Hz, J = 10.0 Hz, H-2), 4.66, 4.77 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.73, 4.85 (qAB, 2H, J = 11.8 Hz, CH2Ph), 4.55, 4.94 (qAB, 2H, J = 11.4 Hz, CH2Ph), 4.69 (s, 2H, CH2O), 5.61 (d, 1H, J = 7.9 Hz, H-5ur), 6.46 (d, 1H, J = 5.4 Hz, H-1), 7.14–7.37 (m, 22H, H-Ph, H-6ur, H-3pyr), 7.96 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.53 (d, 1H, J = 2.6 Hz, H-6pyr), 8.76 (s, 1H, NH), 9.81 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3) δ 33.28 (CH2CO), 45.06 (CH2N), 63.47 (CH2O), 68.66 (C-6), 71.39 (C-5), 72.40, 73.22, 73.34, 74.83 (CH2Ph), 74.92 (C-4), 75.86 (C-2), 79.67 (C-3), 84.56 (C-1), 102.33 (C-5ur), 124.09 (C-3pyr), 127.48, 127.50, 127.57, 127.63, 127.73, 127.82, 128.12, 128.21, 128.24, 128.30, 128.32 (C-Ph, C-4pyr), 131.78 (C-5pyr), 137.76, 137.97, 138.49, 138.57 (C-Ph), 141.50 (C-6pyr), 145.26 (C-6ur), 151.16 (C-2pyr), 152.18 (C-2ur), 163.85 (C-4ur), 165.49 (NHCO), 170.09 (COO). HRMS (ESI) (m/z): [M + Na]+ calcd for C48H48N4NaO10S, 895.2989; found, 895.2966.
Glycoconjugate (41) Starting from amine derivative 17 and uracil derivative 32, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 (v/v)) to give thick syrup (69 mg, 31%): 123.5 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.77–2.92 (m, 2H, CH2CO), 3.31–3.40 (m, 1H, CH), 3.56 (dd, 1H, J = 1.7 Hz, J = 10.8 Hz, H-6a), 3.64–3.79 (m, 5H, H-6b, H-4, CH3), 3.82 (dd~t, 1H, J = 9.2 Hz, J = 0.8 Hz, H-3), 3.95 (dd, 1H, J = 5.4 Hz, J = 9.6 Hz, H-2), 3.98–4.08 (m, 1H, CHHN), 4.10–4.19 (m, 2H, H-5, CHHN), 4.36 4.51 (qAB, 2H, J = 12.0 Hz, CH2Ph), 4.62, 4.76 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.50, 4.83 (qAB, 2H, J = 11.4 Hz, CH2Ph), 4.78, 4.98 (qAB, 2H, J = 10.9 Hz, CH2Ph), 5.70 (d, 1H, J = 7.9 Hz, H-5ur), 6.47 (d, 1H, J = 5.4 Hz, H-1), 7.12–7.33 (m, 22H, H-Ph, H-6ur, H-3pyr), 7.99 (dd, 1H, J = 2.8 Hz, J = 8.7 Hz, H-4pyr), 8.49 (d, 1H, J = 2.5 Hz, H-6pyr), 8.81 (s, 1H, NH), 9.96 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 35.90 (CH2COO), 41.11 (CH), 49.47 (CH2N), 52.62 (CH3), 68.50 (C-6), 72.32 (C-5), 72.18, 73.27, 75.06, 75.65 (CH2Ph), 77.18 (C-4), 79.12 (C-2), 82.84 (C-3), 83.99 (C-1), 102.67 (C-5ur), 124.27 (C-3pyr), 127.57, 127.61, 127.65, 127.77, 127.82, 127.86, 127.97, 128.02, 128.12, 128.28, 128.32, 128.35 (C-Ph, C-4pyr), 132.73 (C-5pyr), 137.60, 137.80, 138.18, 138.62 (C-Ph), 141.13 (C-6pyr), 145.51 (C-6ur), 151.09 (C-2pyr), 152.16 (C-2ur), 163.67 (C-4ur), 168.92 (NHCO), 172.62 (COOCH3). HRMS (ESI) (m/z): [M + Na]+ calcd for C49H50N4NaO10S, 909.3145; found, 909.3187.
Glycoconjugate (42): Starting from amine derivative 18 and uracil derivative 32, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 [v/v]) to give thick syrup (125 mg, 56%): 99.0 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 3.29–3.38 (m, 1H, CH), 3.46 (dd, 1H, J = 6.0 Hz, J = 9.6 Hz, H-6a), 3.55 (dd, 1H, J = 6.9 Hz, J = 9.6 Hz, H-6b), 3.72 (s, 3H, CH3), 3.77 (dd, 1H, J = 2.8 Hz, J = 10.0 Hz, H-3), 3.98 (dd, 1H, J = 1.1 Hz, J = 2.7 Hz, H-4), 4.04 (m, 1H, CHHN), 4.14 (m, 1H, CHHN), 4.28 (m, 1H, H-5), 4.29, 4.35 (qAB, 2H, J = 11.7 Hz, CH2Ph), 4.42 (dd, 1H, J = 5.4 Hz, J = 10.0 Hz, H-2), 4.66, 4.77 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.72, 4.85 (qAB, 2H, J = 11.8 Hz, CH2Ph), 4.56, 4.94 (qAB, 2H, J = 11.4 Hz, CH2Ph), 5.70 (d, 1H, J = 7.9 Hz, H-5ur), 6.44 (d, 1H, J = 5.4 Hz, H-1), 7.12–7.39 (m, 22H, H-Ph, H-6ur, H-3pyr), 7.96 (dd, 1H, J = 2.8 Hz, J = 8.7 Hz, H-4pyr), 8.45 (d, 1H, J = 2.8 Hz, H-6pyr), 8.53 (s, 1H, NH), 9.57 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 35.96 (CH2COO), 41.09 (CH), 49.49 (CH2N), 52.64 (CH3), 68.60 (C-6), 71.31 (C-5), 72.41, 73.22, 73.37, 74.82 (CH2Ph), 74.93 (C-4), 75.94 (C-2), 79.68 (C-3), 84.67 (C-1), 102.66 (C-5ur), 124.26 (C-3pyr), 127.50, 127.54, 127.57, 127.60, 127.73, 128.87, 128.10, 128.15, 128.20, 128.25, 128.30, 128.33 (C-Ph, C-4pyr), 132.45 (C-5pyr), 137.90, 138.00, 138.56, 138.63 (C-Ph), 141.08 (C-6pyr), 145.39 (C-6ur), 151.73 (C-2pyr), 151.96 (C-2ur), 163.46 (C-4ur), 168.75 (NHCO), 172.58 (COOCH3). HRMS (ESI) (m/z): [M + Na]+ calcd for C49H50N4NaO10S, 909.3145; found, 909.3117.
Glycoconjugate (43) Starting from amine derivative 19 and uracil derivative 24, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 20:1 (v/v)) to give thick syrup (58 mg, 32%): −23.1 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.65–2.85 (m, 2H, CH2CO), 3.57 (dd~t, 1H, J = 9.2 Hz, H-4), 3.69 (m, 2H, H-6a, H-6b), 3.81 (dd~t, 1H, J = 9.0 Hz, H-3), 3.89 (dd, 1H, J = 5.3 Hz, J = 9.4 Hz, H-2), 3.95–4.07 (m, 3H, CH2N, H-5), 4.61, 4.73 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.63, 4.86 (qAB, 2H, J = 10.9 Hz, CH2Ph), 4.79, 4.96 (qAB, 2H, J = 11.0 Hz, CH2Ph), 5.45 (d, 1H, J = 7.7 Hz, H-5ur), 6.44 (d, 1H, J = 5.2 Hz, H-1), 7.18–7.34 (m, 17H, H-Ph, H-6ur, H-3pyr), 8.00 (dd, 1H, J = 2.4 Hz, J = 8.6 Hz, H-4pyr), 8.52 (d, 1H, J = 2.4 Hz, H-6pyr), 9.42 (s, 1H, NH), 11.03 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 35.65 (CH2CO), 46.29 (CH2N), 61.47 (C-6), 73.35 (C-5), 72.27, 75.08, 75.56 (CH2Ph), 77.05 (C-4), 79.12 (C-2), 82.64 (C-3), 83.48 (C-1), 101.62 (C-5ur), 124.50 (C-3pyr), 127.60, 127.80, 127.83, 127.91, 127.98, 128.01, 128.34, 128.36, 128.44, 128.53 (C-Ph, C-4pyr), 132.68 (C-5pyr), 137.55, 138.06, 138.56 (C-Ph), 141.63 (C-6pyr), 146.56 (C-6ur), 150.99 (C-2pyr), 151.45 (C-2ur), 165.31 (C-4ur), 169.58 (NHCO). HRMS (ESI) (m/z): [M + Na]+ calcd for C39H40N4NaO8S, 747.2465; found, 747.2447.
Glycoconjugate (44) Starting from amine derivative 20 and uracil derivative 24, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 20:1 (v/v)) to give thick syrup (45 mg, 25%): 48.0 (c 0.5, CHCl3). 1H-NMR (400 MHz, CDCl3) δ 2.64–2.78 (m, 2H, CH2CO), 3.50 (dd, 1H, J = 4.0 Hz, J = 11.5 Hz, H-6a), 3.78 (dd, 1H, J = 2.8 Hz, J = 8.6 Hz, H-3), 3.84–3.96 (m, 4H, H-4, H-6b, CH2N), 4.18 (m, 1H, H-5), 4.26 (dd, 1H, J = 4.5 Hz, J = 8.4 Hz, H-2), 4.63, 4.73 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.70, 4.79 (qAB, 2H, J = 11.8 Hz, CH2Ph), 4.57, 4.83 (qAB, 2H, J = 11.6 Hz, CH2Ph), 5.41 (d, 1H, J = 7.8 Hz, H-5ur), 6.27 (d, 1H, J = 4.5 Hz, H-1), 7.22–7.37 (m, 16H, H-Ph, H-6ur), 7.19 (d, 1H, J = 8.7 Hz, H-3pyr), 7.94 (dd, 1H, J = 2.3 Hz, J = 8.7 Hz, H-4pyr), 8.46 (d, 1H, J = 2.3 Hz, H-6pyr), 9.25 (s, 1H, NH), 10.60 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3) δ 35.41 (CH2CO), 46.96 (CH2N), 61.23 (C-6), 73.55 (C-5); 72.62, 73.61, 74.03 (CH2Ph), 74.60 (C-4), 76.54 (C-2), 78.53 (C-3), 82.61 (C-1), 101.71 (C-5ur), 124.57 (C-3pyr), 127.61, 127.64, 127.76, 127.83, 127.96, 128.20, 128.28, 128.33, 128.38, 128.39 (C-Ph, C-4pyr), 132.948 (C-5pyr), 137.81, 138.13, 138.41 (C-Ph), 141.41 (C-6pyr), 146.30 (C-6ur), 151.10 (C-2pyr), 151.45 (C-2ur), 164. 80 (C-4ur), 169.31 (NHCO). HRMS (ESI) (m/z): [M + Na]+ calcd for C39H40N4NaO8S, 747.2465; found, 747.2487.
Glycoconjugate (45) Starting from amine derivative 19 and uracil derivative 27, purified by column chromatography in CHCl3: MeOH solvent system (100:1 to 20:1 (v/v)) to give thick syrup (68 mg, 37%): 133.0 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 3.56 (dd, 1H, J = 8.7 Hz, J = 9.8 Hz, H-4), 3.67 (dd, 1H, J = 4.6 Hz, J = 12.0 Hz, H-6a), 3.73 (dd, 1H, J = 2.5 Hz, J = 12 Hz, H-6b), 3.85 (dd~t, 1H, J = 9.0 Hz, H-3), 3.92 (dd, 1H, J = 5.3 Hz, J = 9.4 Hz, H-2), 4.05 (ddd, 1H, J = 2.6 Hz, J = 4.5 Hz, J = 9.8 Hz, H-5), 4.22 (s, 2H, CH2N), 4.63, 4.77 (qAB, 2H, J = 11.4 Hz, CH2Ph), 4.64, 4.87 (qAB, 2H, J = 11.4 Hz, CH2Ph), 4.80, 4.98 (qAB, 2H, J = 10.9 Hz, CH2Ph), 5.17 (s, 2H, CH2O), 5.79 (d, 1H, J = 7.9 Hz, H-5ur), 6.48 (d, 1H, J = 5.3 Hz, H-1), 7.24–7.34 (m, 17H, H-Ph, H-6ur, H-3pyr), 8.05 (dd, 1H, J = 2.6 Hz, J = 8.6 Hz, H-4pyr), 8.65 (d, 1H, J = 2.6 Hz, H-6pyr), 8.86 (s, 1H, NH), 9.08 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 61.76 (C-6), 68.03 (CH2N), 73.21 (C-5), 72.34, 75.07, 75.62 (CH2Ph), 77.07 (C-4), 78.16 (CH2O), 79.25 (C-2), 82.69 (C-3), 83.48 (C-1), 103.71 (C-5ur), 124.60 (C-3pyr), 127.62, 127.85, 127.96, 128.02, 128.05, 128.17, 128.36, 128.40, 128.45 (C-Ph, C-4pyr), 131.89 (C-5pyr), 137.56, 138.06, 138.58 (C-Ph), 141.42 (C-6pyr), 143.30 (C-6ur), 151.14 (C-2pyr), 151.51 (C-2ur), 162.70 (C-4ur), 166.89 (NHCO). HRMS (ESI) (m/z): [M + Na] + calcd for C39H40N4NaO9S, 763.2414; found, 763.2427.
Glycoconjugate (46) Starting from amine derivative 20 and uracil derivative 27, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 20:1 (v/v)) to give thick syrup (43 mg, 23%): 51.6 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 3.52 (dd, 1H, J = 3.9 Hz, J = 11.7 Hz, H-6a), 3.79 (dd, 1H, J = 2.8 Hz, J = 8.6 Hz, H-3), 3.89–4.00 (m, 2H, H-4, H-6b), 4.14 (s, 2H, CH2N), 4.20 (m, 1H, H-5), 4.28 (dd, 1H, J = 4.6 Hz, J = 8.4 Hz, H-2), 4.64, 4.74 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.70, 4.79 (qAB, 2H, J = 11.8 Hz, CH2Ph), 4.58, 4.83 (qAB, 2H, J = 11.5 Hz, CH2Ph), 5.05 (s, 2H, CH2O), 5.66 (d, 1H, J = 7.9 Hz, H-5ur), 6.29 (d, 1H, J = 4.5 Hz, H-1), 7.17–7.41 (m, 17H, H-Ph, H-6ur, H-3pyr), 8.05 (dd, 1H, J = 2.4 Hz, J = 8.7 Hz, H-4pyr), 8.53 (d, 1H, J = 2.4 Hz, H-6pyr), 9.14 (s, 1H, NH), 10.02 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 68.04 (CH2N), 61.23 (C-6), 73.52 (C-5), 72.62, 73.64, 74.05 (CH2Ph), 74.67 (C-4), 76.53 (C-2), 78.07 (CH2O), 78.53 (C-3), 82.67 (C-1), 103.33 (C-5ur), 124.63 (C-3pyr), 127.59, 127.64, 127.75, 127.84, 127.94, 128.19, 128.28, 128.32, 128.38 (C-Ph, C-4pyr), 132.23 (C-5pyr), 137.79, 138.08, 138.38 (C-Ph), 141.31 (C-6pyr), 143.71 (C-6ur), 151.44 (C-2pyr), 151.54 (C-2ur), 163.49 (C-4ur), 167.54 (NHCO). HRMS (ESI) (m/z): [M + Na] + calcd for C39H40N4NaO9S, 763.2414; found, 763.2471.
Glycoconjugate (47) Starting from amine derivative 19 and uracil derivative 28, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 20:1 (v/v)) to give thick syrup (81 mg, 43%): 126.6 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.62 (t, 2H, J = 5.3 Hz, CH2CO), 3.49 (dd, 1H, J = 8.7 Hz, J = 9.4 Hz, H-4), 3.64 (dd, 1H, J = 5.3 Hz, J = 12.0 Hz, H-6a), 3.73 (dd, 1H, J = 2.1 Hz, J = 12 Hz, H-6b), 3.80–3.94 (m, 4H, H-2, H-3, CH2O), 4.08 (ddd, 1H, J = 2.3 Hz, J = 4.9 Hz, J = 9.5 Hz, H-5), 4.59, 4.74 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.60, 4.85 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.78, 4.95 (qAB, 2H, J = 11.0 Hz, CH2Ph), 5.07 (s, 2H, CH2N), 5.65 (d, 1H, J = 7.9 Hz, H-5ur), 6.39 (d, 1H, J = 5.2 Hz, H-1), 7.17–7.36 (m, 17H, H-Ph, H-6ur, H-3pyr), 8.00 (dd, 1H, J = 2.3 Hz, J = 8.7 Hz, H-4pyr), 8.49 (d, 1H, J = 2.3 Hz, H-6pyr), 8.86 (s, 1H, NH), 9.96 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 37.29 (CH2CO), 61.68 (C-6), 65.34 (CH2O), 73.23 (C-5), 72.23, 75.01, 75.57 (CH2Ph), 77.00 (C-4), 77.22 (CH2N), 79.24 (C-2), 82.54 (C-3), 83.63 (C-1), 103.04 (C-5ur), 124.96 (C-3pyr), 127.61, 127.81, 127.83, 127.93, 127.97, 128.33, 128.37, 128.42 (C-Ph, C-4pyr), 131.82 (C-5pyr), 137.54, 138.00, 138.50 (C-Ph), 141.15 (C-6pyr), 143.80 (C-6ur), 150.56 (C-2pyr), 151.42 (C-2ur), 163.78 (C-4ur), 169.68 (NHCO). HRMS (ESI) (m/z): [M + Na] + calcd for C40H42N4NaO9S, 777.2570; found, 777.2588.
Glycoconjugate (48) Starting from amine derivative 20 and uracil derivative 28, purified by column chromatography in CHCl3: MeOH solvent system (100:1 to 20:1 (v/v)) to give thick syrup (77 mg, 41%): 84.2 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.57 (t, 2H, J = 5.2 Hz, CH2CO), 3.51 (dd, 1H, J = 3.8 Hz, J = 11.6 Hz, H-6a), 3.75–3.85 (m, 3H, H-3, CH2O), 3.86–3.97 (m, 2H, H-6b, H-4), 4.20 (m, 1H, H-5), 4.29 (dd, 1H, J = 4.7 Hz, J = 8.7 Hz, H-2), 4.64, 4.74 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.70, 4.79 (qAB, 2H, J = 12.0 Hz, CH2Ph), 4.57, 4.84 (qAB, 2H, J = 11.6 Hz, CH2Ph), 5.02 (s, 2H, CH2N), 5.59 (d, 1H, J = 7.9 Hz, H-5ur), 6.29 (d, 1H, J = 4.6 Hz, H-1), 7.19–7.36 (m, 17H, H-Ph, H-6ur, H-3pyr), 7.99 (dd, 1H, J = 2.4 Hz, J = 8.7 Hz, H-4pyr), 8.42 (d, 1H, J = 2.4 Hz, H-6pyr), 9.03 (s, 1H, NH), 10.09 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 37.23 (CH2CO), 55.28 (CH2O), 61.35 (C-6), 73.55 (C-5), 72.53, 73.46, 74.14 (CH2Ph), 74.76 (C-4), 76.43 (C-2), 77.17 (CH2N), 78.70 (C-3), 83.00 (C-1), 102.89 (C-5ur), 124.84 (C-3pyr), 127.55, 127.60, 127.71, 127.80, 127.90, 128.19, 128.29, 128.34, 128.36 (C-Ph, C-4pyr), 132.20 (C-5pyr), 137.81, 138.11, 138.39 (C-Ph); 141.04 (C-6pyr), 143.85 (C-6ur), 150.70 (C-2pyr), 151.38 (C-2ur), 163.89 (C-4ur), 169.80 (NHCO). HRMS (ESI) (m/z): [M + Na]+ calcd for C40H42N4NaO9S, 777.2570; found, 777.2578.
Glycoconjugate (49) Starting from amine derivative 19 and uracil derivative 30, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 20:1 (v/v)) to give thick syrup (96 mg, 49%): 127.4 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.62 (dd~t, 2H, J = 5.3 Hz, CH2CO), 3.48 (dd, 1H, J = 8.6 Hz, J = 9.8 Hz, H-4), 3.59 (dd, 1H, J = 5.5 Hz, J = 12.0 Hz, H-6a), 3.73 (dd, 1H, J = 2.1 Hz, J = 11.9 Hz, H-6b), 3.80–3.94 (m, 4H, H-2, H-3, CH2N), 4.08 (ddd, 1H, J = 2.3 Hz, J = 4.9 Hz, J = 9.5 Hz, H-5), 4.59, 4.74 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.60, 4.85 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.78, 4.96 (qAB, 2H, J = 11.0 Hz, CH2Ph), 5.07 (s, 2H, CH2O), 5.67 (d, 1H, J = 8.0 Hz, H-5ur), 6.51 (d, 1H, J = 5.4 Hz, H-1), 7.17–7.36 (m, 17H, H-Ph, H-6ur, H-3pyr), 7.93 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.43 (d, 1H, J = 2.3 Hz, H-6pyr), 8.33 (s, 1H, NH), 10.06 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 37.29 (CH2CO), 45.16 (CH2N), 61.60 (C-6), 65.34 (CH2O), 73.04 (C-5), 72.14, 74.95, 75.54 (CH2Ph), 77.20 (C-4), 79.23 (C-2), 82.51 (C-3), 83.66 (C-1), 102.88 (C-5ur), 124.90 (C-3pyr), 127.77, 127.89, 127.83, 127.91, 127.94, 128.28, 128.33, 128.37 (C-Ph, C-4pyr), 132.13 (C-5pyr), 137.52, 137.98, 138.49 (C-Ph), 141.71 (C-6pyr), 144.08 (C-6ur), 150.71 (C-2pyr), 151.06 (C-2ur), 163.48 (C-4ur), 168.10 (COO), 172.40 (NHCO). HRMS (ESI) (m/z): [M + Na]+ calcd for C41H42N4NaO10S, 805.2519; found, 805.2631.
Glycoconjugate (50) Starting from amine derivative 20 and uracil derivative 30, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 20:1 (v/v)) to give thick syrup (92 mg, 47%): 87.5 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.83 (m, 2H, CH2CO), 3.45 (dd, 1H, J = 3.7 Hz, J = 11.9 Hz, H-6a), 3.84–4.05 (m, 5H, H-3, H-4, H-6b, CH2N), 4.16 (m, 1H, H-5), 4.27 (dd, 1H, J = 4.5 Hz, J = 8.4 Hz, H-2), 4.65, 4.75 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.70, 4.79 (qAB, 2H, J = 11.9 Hz, CH2Ph), 4.57, 4.83 (qAB, 2H, J = 11.6 Hz, CH2Ph), 4.66 (s, 2H, CH2O), 5.66 (d, 1H, J = 7.8 Hz, H-5ur), 6.24 (d, 1H, J = 4.4 Hz, H-1), 7.20–7.36 (m, 17H, H-Ph, H-6ur, H-3pyr), 7.95 (dd, 1H, J = 2.7 Hz, J = 8.7 Hz, H-4pyr), 8.40 (d, 1H, J = 2.6 Hz, H-6pyr), 8.75 (s, 1H, NH), 10.16 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 33.26 (CH2CO), 45.56 (CH2N), 61.46 (C-6), 63.45 (CH2O), 73.35 (C-5), 72.62, 73.55, 74.00 (CH2Ph), 74.57 (C-4), 76.63 (C-2), 78.40 (C-3), 82.99 (C-1), 103.19 (C-5ur), 125.11 (C-3pyr), 127.63, 127.82, 127.93, 128.18, 128.34, 128.37, 128.40, 128.32 (C-Ph, C-4pyr), 132.17 (C-5pyr), 137.81, 138.11, 138.41 (C-Ph), 141.36 (C-6pyr), 143.77 (C-6ur), 150.81 (C-2pyr), 151.30 (C-2ur), 162.97 (C-4ur), 168.09 (NHCO), 171.33 (COO). HRMS (ESI) (m/z): [M + Na]+ calcd for C41H42N4NaO10S, 805.2519; found, 805.2601.
Glycoconjugate (51) Starting from amine derivative 19 and uracil derivative 32, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 20:1 (v/v)) to give thick syrup (60 mg, 30%): 120.8 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.78 (dd, 1H, J = 6.5 Hz, J = 16.4 Hz, CH2CO), 2.85 (dd, 1H, J = 6.3 Hz, J = 16.3 Hz, CH2CO), 3.34 (m, 1H, CH), 3.57 (dd~t, 1H, J = 9.2 Hz, H-4), 3.65 (dd, 1H, J = 5.0 Hz, J = 12.0 Hz, H-6a), 3.68–3.76 (m, 4H, H-6b, CH3), 3.81 (dd~t, 1H, J = 8.8 Hz, H-3), 3.90 (dd, 1H, J = 5.2 Hz, J = 9.4 Hz, H-2), 4.02–4.13 (m, 3H, CH2N, H-5), 4.61, 4.75 (qAB, 2H, J = 11.8 Hz, CH2Ph), 4.61, 4.86 (qAB, 2H, J = 10.8 Hz, CH2Ph), 4.79, 4.96 (qAB, 2H, J = 10.9 Hz, CH2Ph), 5.67 (d, 1H, J = 7.9 Hz, H-5ur), 6.40 (d, 1H, J = 5.2 Hz, H-1), 7.19–7.34 (m, 17H, H-Ph, H-6ur, H-3pyr), 7.98 (dd, 1H, J = 2.5 Hz, J = 8.6 Hz, H-4pyr), 8.49 (d, 1H, J = 2.5 Hz, H-6pyr); 9.06 (d, 1H, NH), 10.11 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 35.80 (CH2COO), 40.97 (CH), 49.25 (CH2N), 52.63 (CH3), 61.71 (C-6), 73.21 (C-5), 72.25, 75.03, 75.59 (CH2Ph), 77.15 (C-4), 79.23 (C-2), 82.58 (C-3), 83.62 (C-1), 102.57 (C-5ur), 124.90 (C-3pyr), 127.61, 127.82, 127.84, 127.94, 127.98, 128.00, 128.34, 128.37, 128.43 (C-Ph, C-4pyr), 133.00 (C-5pyr), 137.53, 138.02, 138.53 (C-Ph), 141.15 (C-6pyr), 145.53 (C-6ur), 150.58 (C-2pyr), 152.02 (C-2ur), 163.90 (C-4ur), 169.09 (NHCO), 172.75 (COOCH3). HRMS (ESI) (m/z): [M + Na]+ calcd for C42H44N4NaO10S, 819.2676; found, 819.2681.
Glycoconjugate (52) Starting from amine derivative 20 and uracil derivative 32, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 20:1 (v/v)) to give thick syrup (46 mg, 23%): 72.4 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.77 (dd, 1H, J = 6.5 Hz, J = 16.4 Hz, CH2CO), 2.84 (dd, J = 6.0 Hz, J = 16.2 Hz, CH2CO), 3.21 (m, 1H, CH), 3.50 (dd, 1H, J = 4.2 Hz, J = 11.7 Hz, H-6a), 3.70 (s, 3H, CH3), 3.80 (dd, 1H, J = 2.9 Hz, J = 8.6 Hz, H-3), 3.88–3.97 (m, 2H, H-6b, H-4), 4.02–4.07 (m, 2H, CH2N), 4.20 (m, 1H, H-5), 4.27 (dd, 1H, J = 4.5 Hz, J = 8.6 Hz, H-2), 4.64, 4.74 (qAB, 2H, J = 11.5 Hz, CH2Ph), 4.71, 4.80 (qAB, 2H, J = 11.8 Hz, CH2Ph), 4.58, 4.84 (qAB, 2H, J = 11.6 Hz, CH2Ph), 5.64 (d, 1H, J = 7.9 Hz, H-5ur), 6.28 (d, 1H, J = 4.4 Hz, H-1), 7.21 (d, 1H, J = 8.7 Hz, H-3pyr), 7.24–7.36 (m, 16H, H-Ph, H-6ur), 7.99 (dd, 1H, J = 2.5 Hz, J = 8.7 Hz, H-4pyr), 8.46 (d, 1H, J = 2.5 Hz, H-6pyr), 9.02 (s, 1H, NH), 9.88 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 35.85 (CH2COO), 40.97 (CH), 49.24 (CH2N), 52.59 (CH3), 61.28 (C-6), 73.54 (C-5), 72.63, 73.59, 74.01 (CH2Ph), 74.64 (C-4), 76.59 (C-2), 78.49 (C-3), 82.64 (C-1), 102.57 (C-5ur), 124.68 (C-3pyr), 127.63, 127.66, 127.76, 127.85, 127.97, 128.21, 128.33, 128.40 (C-Ph, C-4pyr), 132.98 (C-5pyr), 137.82, 138.12, 138.42 (C-Ph), 141.06 (C-6pyr), 145.46 (C-6ur), 150.99 (C-2pyr), 151.86 (C-2ur), 163.72 (C-4ur), 169.07 (NHCO), 172.76 (COOCH3). HRMS (ESI) (m/z): [M + Na]+ calcd for C42H44N4NaO10S, 819.2676; found, 819.2684.
Glycoconjugate (53) Starting from amine derivative 21 and uracil derivative 28, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 (v/v)) to give thick syrup (114 mg, 70%): −0.8 (c 0.5, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.01, 2.02, 2.03, 2.04 (4s, 12H, CH3CO), 2.70 (t, 2H, J = 5.7 Hz, CH2O), 3.87 (ddd, 1H, J = =2.4 Hz, J = 4.6 Hz, J = 10.0 Hz, H-5), 3.95 (t, 2H, J = 5.8 Hz, CH2O), 4.11 (dd, 1H, J = 2.3 Hz, J = 12.4 Hz, H-6a), 4.25 (dd, 2H, J = 4.7 Hz, J = 12.3 Hz, H-6b), 5.11–5.24 (m, 4H, CH2N, H-2, H-4), 5.34 (dd~t, 1H, J = 9.3 Hz, H-3), 5.60 (d, 1H, J = 10.4 Hz, H-1), 5.75 (d, 1H, J = 7.9 Hz, H-5ur), 7.23 (dd, 1H, J = 0.4 Hz, J = 8.7 Hz, H-3pyr), 7.34 (d, 1H, J = 7.9 Hz, H-6ur), 8.05 (dd, 1H, J = 2.5 Hz, J = 8.7 Hz, H-4pyr), 8.54 (dd, 1H, J = 2.5 Hz, H-6pyr), 8.58 (s, 1H, NH), 9.80 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 20.56, 20.57, 20.64, 20.71 (CH3CO), 37.30 (CH2CO), 61.99 (C-6); 65.20 (CH2O), 68.27 (C-4), 69.58 (C-2), 73.98 (C-3), 75.78 (C-5), 77.24 (CH2N), 82.54 (C-1), 103.23 (C-5ur), 123.90 (C-3pyr), 128.23 (C-4pyr), 132.22 (C-5pyr), 141.13 (C-6pyr), 143.63 (C-6ur), 149.40 (C-2pyr), 151.39 (C-2ur), 163.54 (C-4ur), 169.38 (NHCO), 169.42, 169.51, 170.13, 170.73 (CH3CO). HRMS (ESI) (m/z): [M + Na]+ calcd for C27H32N4NaO13S, 675.1584; found, 675.1590.
Glycoconjugate (54) Starting from amine derivative 22 and uracil derivative 28, purified by column chromatography in CHCl3:MeOH solvent system (100:1 to 25:1 (v/v)) to give thick syrup (106 mg, 65%): 0.1 (c 0.5, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 2.00, 2.01, 2.02, 2.16 (4s, 12H, CH3CO), 2.70 (t, 2H, J = 5.7 Hz, CH2CO), 3.96 (t, 2H, J = 5.7 Hz, CH2O), 4.04–4.15 (m, 3H, H-6a, H-6b, H-5), 5.15–5.23 (s, 3H, H-3, CH2N), 5.38 (dd~t, 1H, J = 10.1 Hz, H-2), 5.48 (d, 1H, J = 3.3 Hz, H-4), 5.60 (d, 1H, J = 10.3 Hz, H-1), 5.76 (d, 1H, J = 7.9 Hz, H-5ur), 7.27 (d, 1H, J = 8.7 Hz, H-3pyr), 7.31 (d, 1H, J = 7.9 Hz, H-6ur), 8.08 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.40 (s, 1H, NH), 8.51 (d, 1H, J = 2.6 Hz, H-6pyr), 9.47 (bs, 1H, NH). 13C-NMR (100 MHz, CDCl3): δ 20.58, 20.67, 20.75 (CH3CO), 37.35 (CH2CO), 61.26 (C-6), 65.19 (CH2O), 66.92 (C-2), 67.29 (C-4), 72.02 (C-3), 74.44 (C-5), 77.27 (CH2N), 83.06 (C-1), 103.33 (C-5ur), 123.86 (C-3pyr), 128.28 (C-4pyr), 133.09 (C-5pyr), 141.12 (C-6pyr), 143.48 (C-6ur), 149.76 (C-2pyr), 151.30 (C-2ur), 163.25 (C-4ur), 169.29 (NHCO), 169.69, 170.03, 170.24, 170.49 (CH3CO). HRMS (ESI) (m/z): [M + Na]+ calcd for C27H32N4NaO13S, 675.1584; found, 675.1592.
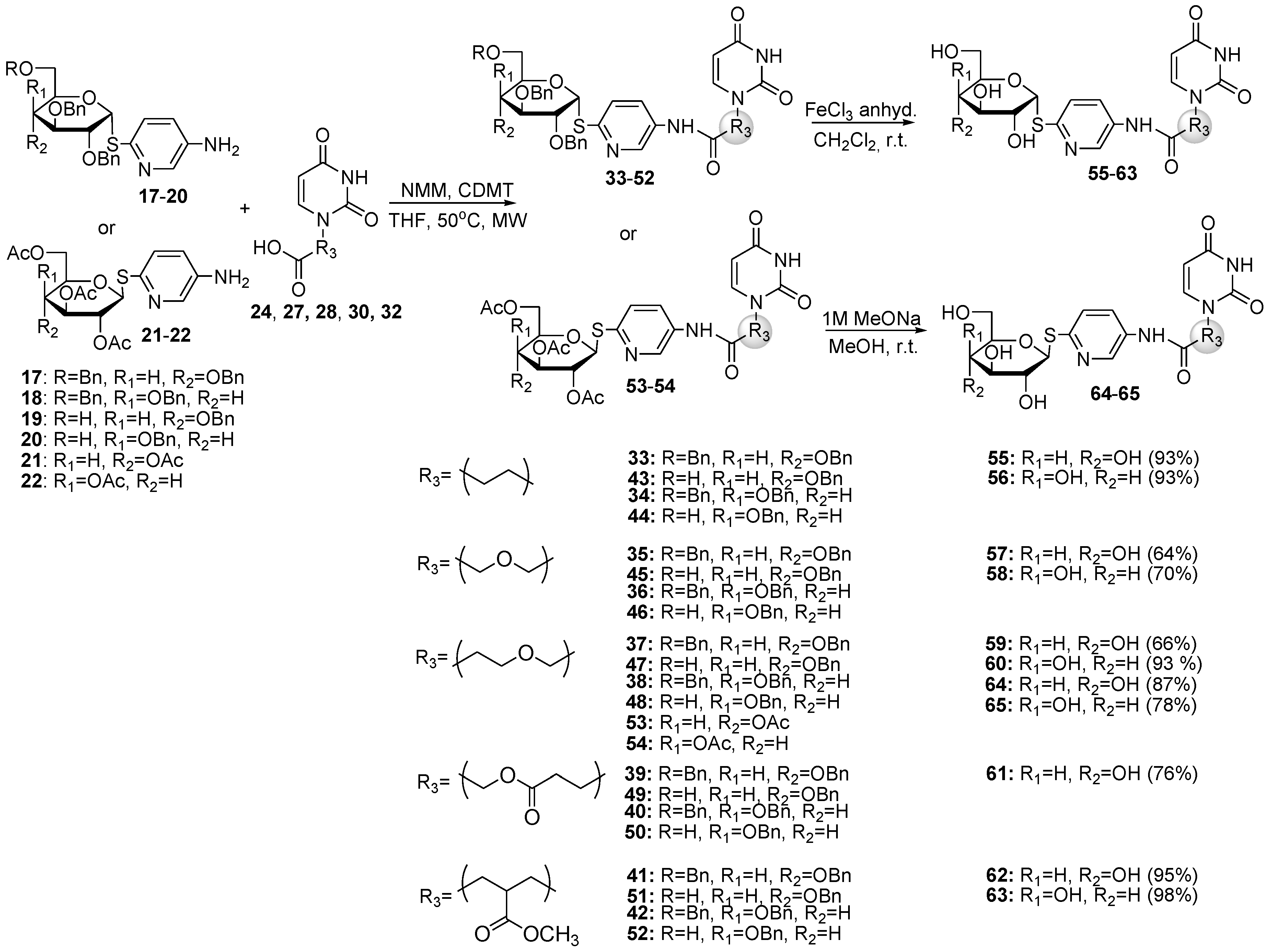
3.2.5. Protecting Groups Removal
Debenzylation: Corresponding glycoconjugate 33–52 (0.06 mmol) was dissolved in dry CH2Cl2 (2 mL) and anhydrous FeCl3 (97 mg, 0.60 mmol) was added. The resulting mixture was stirred under argon. After 30 minutes the reaction mixture was diluted with CH2Cl2 (10 mL) and washed with water. Resulting emulsion was centrifuged (6000 rpm) and the supernatant was collected and evaporated. The residue was dissolved in MeOH (5 mL), the silica gel was added, and solvent was evaporated and purified by column chromatography with CHCl3:MeOH solvent system (10:1 to 3:1 [v/v]).
Glycoconjugate (55) White solid (24 mg, 93%): 149.2 (c 0.3, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 2.83 (t, 2H, J = 6.3 Hz, CH2CO), 3.39 (dd, 1H, J = 8.9 Hz, J = 9.8 Hz, H-4), 3.55 (m, 1H, H-3), 3.66–3.75 (m, 2H, H-6a, H-6b), 3.84 (dd, 1H, J = 5.4 Hz, J = 9.8 Hz, H-2), 3.93 (ddd, J = 3.0 Hz, J = 4.5 Hz, J = 9.8 Hz, 1H, H-5), 4.09 (t, 2H, J = 6.3 Hz, CH2N), 5.62 (d, 1H, J = 7.9 Hz, H-5ur), 6.10 (d, 1H, J = 5.4 Hz, H-1), 7.48 (d, 1H, J = 8.7 Hz, H-3pyr), 7.64 (d, 1H, J = 7.9 Hz, H-6ur), 7.92 (dd, 1H, J = 2.7 Hz, J = 8.7 Hz, H-4pyr), 8.62 (d, 1H, J = 2.7 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD): δ 36.18 (CH2CO), 46.36 (CH2N), 62.35 (C-6), 71.43 (C-4), 72.93 (C-2), 75.49 (C-5), 76.01 (C-3), 88.12 (C-1), 101.94 (C-5ur), 125.96 (C-3pyr), 129.77 (C-4pyr), 134.75 (C-5pyr), 142.11 (C-6pyr), 147.99 (C-6ur), 152.69 (C-2ur), 153.07 (C-2pyr), 166.75 (C-4ur), 171.33 (NHCO). HRMS (ESI) (m/z): [M + H]+ calcd for C18H23N4O8S, 455.1237; found, 455.1241.
Glycoconjugate (56) White solid (24 mg, 93%): 83.6 (c 0.5, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 2.84 (t, 2H, J = 6.4 Hz, CH2CO), 3.62–3.72 (m, 3H, H-6a, H-6b, H-3), 3.98 (dd, 1H, J = 1.5 Hz, J = 3.4 Hz, H-4), 4.10 (t, 2H, J = 6.4 Hz, CH2N), 4.16–4.20 (m, 1H, H-5), 4.23 (dd, 1H, J = 5.5 Hz, J = 10.2 Hz, H-2), 5.62 (d, 1H, J = 7.9 Hz, H-5ur), 6.13 (d, 1H, J = 5.5 Hz, H-1), 7.51 (d, 1H, J = 8.6 Hz, H-3pyr), 7.65 (d, 1H, J = 7.9 Hz, H-6ur), 7.92 (dd, 1H, J = 2.5 Hz, J = 8.6 Hz, H-4pyr), 8.63 (d, 1H, J = 2.5 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD) δ 36.21 (CH2CO), 46.35 (CH2N), 62.39 (C-6), 69.57 (C-2), 70.68 (C-4), 72.52 (C-3), 74.07 (C-5), 88.65 (C-1), 101.96 (C-5ur), 126.25 (C-3pyr), 129.79 (C-4pyr), 134.75 (C-5pyr), 142.12 (C-6pyr), 147.98 (C-6ur), 152.70 (C-2ur), 153.21 (C-2pyr), 166.76 (C-4ur), 171.36 (NHCO). HRMS (ESI) (m/z): [M + H]+ calcd for C18H22N4O8S, 455.1237; found, 455.1237.
Glycoconjugate (57) White solid (17 mg, 64%): 145.9 (c 0.3, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 3.40 (dd, 1H, J = 8.9 Hz, J = 9.9 Hz, H-4), 3.55 (m, 1H, H-3), 3.67–3.76 (m, 2H, H-6a, H-6b), 3.84 (dd, 1H, J = 5.4 Hz, J = 9.8 Hz, H-2), 3.93 (ddd, J = 3.1 Hz, J = 4.4 Hz, J = 9.8 Hz, 1H, H-5), 4.30 (s, 2H, CH2N), 5.30 (s, 2H, CH2O), 5.71 (d, 1H, J = 7.9 Hz, H-5ur), 6.13 (d, 1H, J = 5.4 Hz, H-1), 7.50 (dd, 1H, J = 8.7 Hz, H-3pyr), 7.73 (d, 1H, J = 7.9 Hz, H-6ur), 7.99 (dd, 1H, J = 2.4 Hz, J = 8.7 Hz, H-4pyr), 8.70 (d, 1H, J = 2.4 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD): δ 62.34 (C-6), 69.57 (CH2N), 71.42 (C-4), 72.92 (C-2), 75.52 (C-5), 76.01 (C-3), 79.07 (CH2O), 88.04 (C-1), 103.23 (C-5ur), 125.79 (C-3pyr), 130.25 (C-4pyr), 134.04 (C-5pyr), 142.61 (C-6pyr), 146.42 (C-6ur), 153.19 (C-2ur), 153.69 (C-2pyr), 166.34 (C-4ur), 170.39 (NHCO). HRMS (ESI) (m/z): [M + H]+ calcd for C18H23N4O9S, 471.1186; found, 471.1184.
Glycoconjugate (58) White solid (19 mg, 70%): 107.6 (c 0.5, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 3.63–3.73 (m, 3H, H-6a, H-6b, H-3), 3.98 (dd, 1H, J = 1.2 Hz, J = 3.3 Hz, H-4), 4.18 (ddd, 1H, J = 1.5 Hz, J = 5.3 Hz, J = 6.7 Hz, H-5), 4.24 (dd, 1H, J = 5.5 Hz, J = 10.2 Hz, H-2), 4.30 (s, 2H, CH2N), 5.30 (s, 2H, CH2O), 5.72 (d, 1H, J = 7.9 Hz, H-5ur), 6.16 (d, 1H, J = 5.5 Hz, H-1), 7.52 (dd, 1H, J = 0.6 Hz, J = 8.7 Hz, H-3pyr), 7.69 (d, 1H, J = 7.9 Hz, H-6ur), 7.99 (dd, 1H, J = 2.5 Hz, J = 8.7 Hz, H-4pyr), 8.70 (d, 1H, J = 2.5 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD): δ 62.40 (C-6), 69.56 (CH2N, C-2), 70.67 (C-4), 72.53 (C-3), 74.12 (C-5), 79.08 (CH2O), 88.56 (C-1), 103.23 (C-5ur), 126.04 (C-3pyr), 130.23 (C-4pyr), 134.02 (C-5pyr), 142.58 (C-6pyr), 146.44 (C-6ur), 153.17 (C-2ur), 153.81 (C-2pyr), 166.32 (C-4ur), 170.39 (NHCO). HRMS (ESI) (m/z): [M + H]+ calcd for C18H23N4O9S, 471.1186; found, 471.1187.
Glycoconjugate (59) White solid (18 mg, 66%): 67.5 (c 0.5, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 2.65 (t, 2H, J = 5.9 Hz, CH2CO), 3.55 (m, 1H, H-3), 3.64–3.76 (m, 2H, H-6a, H-6b), 3.84 (dd, 1H, J = 5.4 Hz, J = 9.8 Hz, H-2), 3.93 (m, 1H, H-5), 5.17 (s, 2H, CH2N), 5.65 (d, 1H, J = 7.9 Hz, H-5ur), 6.10 (d, 1H, J = 5.4 Hz, H-1), 7.49 (dd, 1H, J = 0.7 Hz, J = 8.7 Hz, H-3pyr), 7.61 (d, 1H, J = 7.9 Hz, H-6ur), 7.95 (dd, 1H, J = 2.6 Hz, J = 8.9 Hz, H-4pyr), 8.62 (d, 1H, J = 2.6 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD): δ 38.08 (CH2CO), 62.38 (C-6), 66.33 (CH2O), 71.43 (C-4), 72.95 (C-2), 75.49 (C-5), 76.01 (C-3), 78.19 (CH2N), 88.15 (C-1), 103.00 (C-5ur), 125.99 (C-3pyr), 129.76 (C-4pyr), 134.87 (C-5pyr), 142.07 (C-6pyr), 146.21 (C-6ur), 152.70 (C-2ur), 153.22 (C-2pyr), 166.45 (C-4ur), 172.12 (NHCO). HRMS (ESI) (m/z): [M + H]+ calcd for C19H25N4O9S, 485.1342; found, 485.1339.
Glycoconjugate (60) White solid (25 mg, 93%): 130.2 (c 1.0, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 2.64 (t, 2H, J = 5.9 Hz, CH2CO), 3.64–3.73 (m, 3H, H-6a, H-6b, H-3), 3.91 (t, 2H, J = 5.9 Hz, CH2O), 3.98 (dd, 1H, J = 1.6 Hz, J = 3.3 Hz, H-4), 4.19 (ddd, 1H, J = 1.5 Hz, J = 5.3 Hz, J = 6.7 Hz, H-5), 4.24 (dd, 1H, J = 5.5 Hz, J = 10.2 Hz, H-2), 5.18 (s, 2H, CH2N), 5.65 (d, 1H, J = 7.9 Hz, H-5ur), 6.14 (d, 1H, J = 5.5 Hz, H-1), 7.51 (dd, 1H, J = 0.6 Hz, J = 8.7 Hz, H-3pyr), 7.62 (d, 1H, J = 7.9 Hz, H-6ur), 7.94 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.64 (d, 1H, J = 2.6 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD): δ 38.07 (CH2CO), 62.42 (C-6), 66.33 (CH2O), 69.57 (C-2), 70.70 (C-4), 72.51 (C-3), 74.07 (C-5), 78.20 (CH2N), 88.66 (C-1), 102.99 (C-5ur), 126.24 (C-3pyr), 129.70 (C-4pyr), 134.88 (C-5pyr), 142.06 (C-6pyr), 146.26 (C-6ur), 152.96 (C-2ur), 153.06 (C-2pyr), 166.44 (C-4ur), 172.13 (NHCO). HRMS (ESI) (m/z): [M + H]+ calcd for C19H25N4O9S, 485.1342, found, 485.1342.
Glycoconjugate (61) White solid (18 mg, 76%): 90.2 (c 0.2, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 2.93 (t, 2H, J = 6.3 Hz, CH2CO), 3.55 (dd, 1H, J = 8.8 Hz, J = 10.0 Hz, H-3), 3.67–3.75 (m, 2H, H-6a, H-6b), 3.84 (dd, 1H, J = 5.4 Hz, J = 9.8 Hz, H-2), 3.93 (ddd, 1H, J = 2.9 Hz, J = 4.6 Hz, J = 9.8 Hz, H-5), 4.07 (t, 2H, J = 6.3 Hz, CH2N), 4.74 (s, 2H, CH2O), 5.61 (d, 1H, J = 7.9 Hz, H-5ur), 6.13 (d, 1H, J = 5.4 Hz, H-1), 7.50 (dd, 1H, J = 0.6 Hz, J = 8.7 Hz, H-3pyr), 7.65 (d, 1H, J = 7.9 Hz, H-6ur), 7.95 (dd, 1H, J = 2.7 Hz, J = 8.7 Hz, H-4pyr), 8.60 (dd, 1H, J = 0.6 Hz, J = 2.7 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD): δ 33.61 (CH2CO), 46.04 (CH2N), 62.38 (C-6), 63.91 (CH2O), 71.44 (C-4), 72.94 (C-2), 75.54 (C-5), 76.04 (C-3), 88.03 (C-1), 101.97 (C-5ur), 125.84 (C-3pyr), 130.06 (C-4pyr), 134.14 (C-5pyr), 142.34 (C-6pyr), 148.01 (C-6ur), 152.75 (C-2ur), 153.65 (C-2pyr), 166.79 (C-4ur), 168.29 (COO), 172.25 (NHCO). HRMS (ESI) (m/z): [M + H]+ calcd for C20H25N4O10S, 513.1291, found, 513.1294.
Glycoconjugate (62) White solid (29 mg, 95%): 105.4 (c 1.0, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 2.73 (dd, 1H, J = 5.8 Hz, J = 16.2 Hz, CHHCO), 2.84 (dd, 1H, J = 7.7 Hz, J = 16.2 Hz, CHHCO), 3.39 (dd, 1H, J = 8.9 Hz, J = 9.9 Hz, H-4), 3.55 (m, 1H, H-3), 3.68 (s, 3H, CH3), 3.65–3.75 (m, 2H, H-6a, H-6b); 3.83 (dd, 1H, J = 5.4 Hz, J = 9.8 Hz, H-2), 3.93 (ddd, J = 3.0 Hz, J = 4.5 Hz, J = 9.8 Hz, 1H, H-5), 4.05 (ddd, 2H, J = 7.0 Hz, J = 14.0 Hz, J = 24.0 Hz, CH2N), 5.64 (d, 1H, J = 7.9 Hz, H-5ur), 6.10 (d, 1H, J = 5.4 Hz, H-1), 7.48 (dd, 1H, J = 0.5 Hz, J = 8.7 Hz, H-3pyr), 7.57 (d, 1H, J = 7.9 Hz, H-6ur), 7.91 (dd, 1H, J = 2.5 Hz, J = 8.7 Hz, H-4pyr), 8.61 (d, 1H, J = 0.5 Hz, J = 2.5 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD): δ 36.57 (CH2CO), 41.89 (CH), 50.62 (CH2N), 52.81 (CH3), 62.35 (C-6), 71.43 (C-4), 72.94 (C-2), 75.48 (C-5), 76.00 (C-3), 88.15 (C-1), 102.31 (C-5ur), 125.99 (C-3pyr), 129.66 (C-4pyr), 134.83 (C-5pyr), 142.02 (C-6pyr), 147.51 (C-6ur), 151.77 (C-2ur), 152.94 (C-2pyr), 166.71 (C-4ur), 171.31 (NHCO), 174.57 (COOCH3). HRMS (ESI) (m/z): [M + H] + calcd for C21H27N4O10S, 527.1448; found, 527.1450.
Glycoconjugate (63) White solid (31 mg, 98%): 43.5 (c 0.25, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 2.74 (dd, 1H, J = 5.8 Hz, J = 16.2 Hz, CHHCO), 2.85 (dd, 1H, J = 7.7 Hz, J = 16.2 Hz, CHHCO), 3.37 (m, 1H, CH), 3.64–3.72 (m, 6H, H-6a, H-6b, H-3, CH3), 3.98 (dd, 1H, J = 1.2 Hz, J = 3.3 Hz, H-4), 4.06 (ddd, 2H, J = 7.0 Hz, J = 14.0 Hz, J = 20.1 Hz, CH2N), 4.18 (ddd, 1H, J = 1.3 Hz, J = 5.4 Hz, J = 6.7 Hz, 1H, H-5), 4.23 (dd, 1H, J = 5.5 Hz, J = 10.1 Hz, H-2), 5.64 (d, 1H, J = 7.9 Hz, H-5ur), 6.12 (d, 1H, J = 5.5 Hz, H-1), 7.51 (dd, 1H, J = 0.5 Hz, J = 8.7 Hz, H-3pyr), 7.59 (d, 1H, J = 7.9 Hz, H-6ur), 7.91 (dd, 1H, J = 2.6 Hz, J = 8.7Hz, H-4pyr), 8.61 (dd, 1H, J = 0.5 Hz, J = 2.6 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD): δ 36.56 (CH2CO), 41.88 (CH), 49.64 (CH2N), 52.83 (CH3), 62.39 (C-6), 69.58 (C-2), 70.68 (C-4), 72.52 (C-3), 74.07 (C-5), 88.68 (C-1), 101.28 (C-5ur), 126.28 (C-3pyr), 129.66 (C-4pyr), 134.84 (C-5pyr), 142.01 (C-6pyr), 147.59 (C-6ur), 152.86 (C-2ur), 153.07 (C-2pyr), 166.60 (C-4ur), 171.32 (NHCO), 174.55 (COOCH3). HRMS (ESI) (m/z): [M + H] + calcd for C21H26N4O10NaS, 549.1267; found, 549.1262.
Deacetylation: The corresponding glycoconjugate 53 or 54 (0.12 mmol) was dissolved in MeOH (10 mL) and 1 M MeONa in MeOH (0.2 mmol, 0.2 mL) was added. The resulting mixture was stirred at room temperature. The progress of the reaction was monitored on TLC plate in MeOH:CHCl3 (2:1) solvent system. After completion (30 min.), the reaction mixture was neutralised with silica gel, the solvent was evaporated purified by column chromatography with CHCl3: MeOH solvent system (10:1 to 2:1 [v/v]).
Glycoconjugate (64) Purified by column chromatography in CHCl3:MeOH solvent system (10:1 to 2:1 (v/v)) to give white solid (50 mg, 87%): −43.4 (c 1.0, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 2.64 (t, 2H, J = 5.9 Hz, CH2O), 3.32–3.47 (m, 4H, H-2, H-3, H4, H-5), 3.66 (dd, 1H, J = 5.6 Hz, J = 12.1 Hz, H-6a), 3.85 (dd, 1H, J = 2.2 Hz, J = 12.1 Hz, H-6b), 3.90 (t, 2H, J = 5.9 Hz, CH2O), 5.08 (d, 1H, J = 9.9 Hz, H-1), 5.17 (s, 2H, CH2N), 5.64 (d, 1H, J = 7.9 Hz, H-5ur), 7.46 (dd, 1H, J = 0.6 Hz, J = 8.7 Hz, H-3pyr), 7.60 (d, 1H, J = 7.9 Hz, H-6ur), 7.97 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.60 (dd, 1H, J = 0.6 Hz, J = 2.6 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD) δ 38.09 (CH2CO), 62.78 (C-6), 66.33 (CH2O), 71.31 (C-4), 73.89 (C-2), 78.18 (CH2N), 79.71 (C-3), 82.16 (C-5), 86.91 (C-1), 103.01 (C-5ur), 125.38 (C-3pyr), 129.77 (C-4pyr), 134.92 (C-5pyr), 141.88 (C-6pyr), 146.18 (C-6ur), 152.85 (C-2pyr), 152.95 (C-2ur), 166.44 (C-4ur), 172.09 (NHCO). HRMS (ESI) (m/z): [M + H]+ calcd for C19H25N4O9S, 485.1342; found 485.1341.
Glycoconjugate (65) Purified by column chromatography in CHCl3:MeOH solvent system (10:1 to 2:1 (v/v)) to give white solid ( 45 mg, 78%): −34. 7 (c 1.0, CH3OH). 1H-NMR (400 MHz, CD3OD): δ 2.64 (t, 2H, J = 5.9 Hz, CH2CO), 3.56 (dd, 1H, J = 3.3 Hz, J = 9.2 Hz, H-3), 3.64–3.78 (m, 4H, H-2, H-5, H-6a, H-6b), 3.90 (t, 2H, J = 5.9 Hz, CH2O), 3.93 (d, 1H, J = 3.1 Hz, H-4), 5.04 (d, 1H, J = 9.9 Hz, H-1), 5.17 (s, 2H, CH2N), 5.65 (d, 1H, J = 7.9 Hz, H-5ur), 7.49 (dd, 1H, J = 0.7 Hz, J = 8.7 Hz, H-3pyr), 7.60 (d, 1H, J = 7.9 Hz, H-6ur), 7.95 (dd, 1H, J = 2.6 Hz, J = 8.7 Hz, H-4pyr), 8.59 (dd, 1H, J = 0.5 Hz, J = 2.6 Hz, H-6pyr). 13C-NMR (100 MHz, CD3OD): δ 38.09 (CH2CO), 62.69 (C-6), 66.34 (CH2O), 70.52 (C-4), 70.90 (C-2), 76.35 (C-3), 78.18 (CH2N), 80.84 (C-5), 87.41 (C-1), 103.01 (C-5ur), 125.06 (C-3pyr), 129.78 (C-4pyr), 134.74 (C-5pyr), 141.78 (C-6pyr), 146.19 (C-6ur), 152.97 (C-2pyr), 153.34 (C-2ur), 166.47 (C-4ur), 172.08 (NHCO). HRMS (ESI) (m/z): [M + H]+ calcd for C19H25N4O9S, 485.1342; found 485.1337.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}