
Synthesis and Coordination Behavior of a Flexible Bis(phosphinoferrocene) Ligand


Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 40 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(8), 2054; https://doi.org/10.3390/molecules23082054
Submission received: 25 July 2018
/
Revised: 11 August 2018
/
Accepted: 15 August 2018
/
Published: 16 August 2018
(This article belongs to the Special Issue Ferrocene and Ferrocene-Containing Compounds)
Abstract
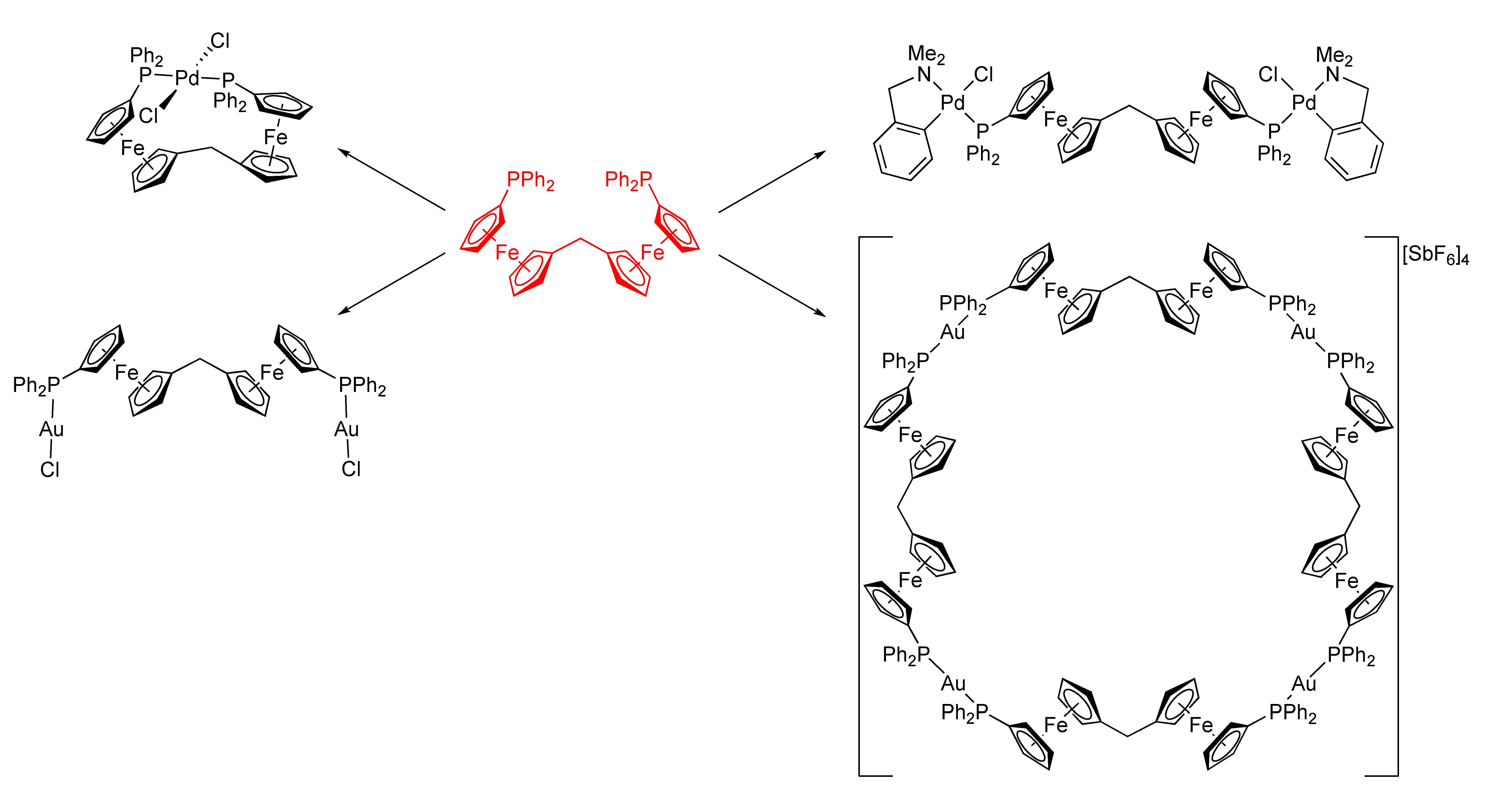
:A symmetrical flexible bis(phosphinoferrocene) derivative, viz. bis[1′-(diphenylphosphino)ferrocenyl]methane (1), was prepared and studied as a ligand in Pd(II) and Au(I) complexes. The reactions of 1 with [PdCl2(cod)] (cod = cycloocta-1,5-diene) and [Pd(μ-Cl)(LNC)]2 (LNC = [2-(dimethylamino-κN)methyl]phenyl-κC1) produced bis(phosphine) complex trans-[PdCl2(1-κ2P,P′)] (4), wherein the ligand spans trans positions in the square-planar coordination sphere of Pd(II) and the tetranuclear, P,P-bridged complex [(μ(P,P′)-1){PdCl(LNC)}2] (5), respectively. In reactions with the Au(I) precursors [AuCl(tht)] and [Au(tht)2][SbF6] (tht = tetrahydrothiophene), ligand 1 gave rise to tetranuclear Au2Fe2 complex [(μ(P,P′)-1)(AuCl)2] (6) and to symmetrical macrocyclic tetramer [Au4(μ(P,P′)-1)4][SbF6]4 (7). All compounds were characterized by spectroscopic methods. In addition, the structures of compound 1, its synthetic precursor bis[1′-(diphenylphosphino)ferrocenyl]methanone (3), and all aforementioned Pd(II) and Au(I) complexes were determined by single-crystal X-ray diffraction analysis (some in solvated form).

1. Introduction
Phosphinoferrocene ligands have been extensively studied in recent years, particularly for their numerous applications as versatile supporting ligands in coordination chemistry and catalysis [1,2,3,4]. Although numerous simple and chiral ferrocene phosphines are available, compounds with two or more connected ferrocene units as the molecular scaffold remain less common. Representative examples of chiral bis(phosphinoferrocene) ligands include BIFEP (I in Scheme 1) and its analogs with various phosphine substituents [5,6], diphosphines from the TRAP family (II) [7,8], and phosphinoferrocenyl analogs of the Trost’s chiral pocket ligand [9] (compound III and its stereoisomers) [10,11]. Non-chiral ferrocene phosphines of this type are mainly represented by biferrocene phosphines isomeric to the aforementioned BIFEP ligands, compounds IV [12,13]. However, their potentially less structurally confined counterparts, in which two equivalent 1′-(phosphino)ferrocenyl units are connected by one- or two-carbon bridges, are so far represented only by compound V. This diphosphine was obtained as a side-product from the reaction of 1′-(diphenylphosphino)-1-acetylferrocene with 1′-(diphenylphosphino)-1-lithioferrocene, and neither its properties nor its coordination behavior have been studied yet [14].
The lack of bis(phosphinoferrocene) ligands with flexible connecting groups led us to design and to prepare a new diphosphine comprising two identical phosphinoferrocene units connected by a methylene linker, viz. bis [1′-(diphenylphosphino)ferrocenyl]methane (1 in Scheme 1). In this contribution, we describe the synthesis of this compound and its coordination behavior towards Pd(II) and Au(I) ions with various auxiliary ligands in addition to providing a detailed structural characterization (including structure determination) of all compounds prepared.
2. Results and Discussion
2.1. Synthesis of Ligand 1
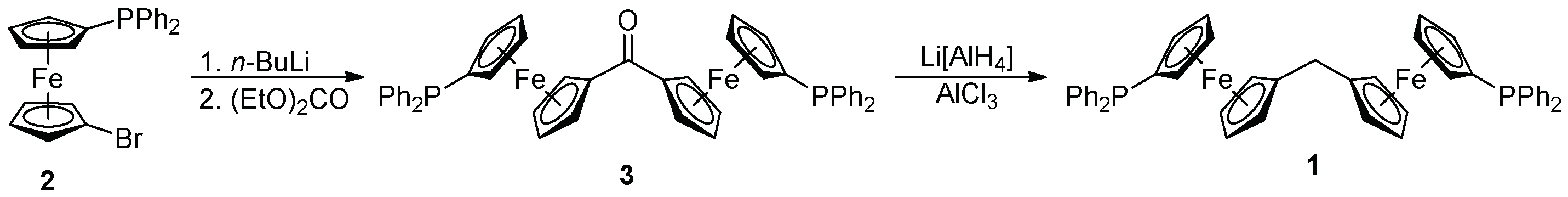
Diphosphine 1 was prepared in two steps from 1′-(diphenylphosphino)-1-bromoferrocene (compound 2 in Scheme 2). This bromide was lithiated with n-butyllithium and the resulting lithio intermediate immediately reacted with diethyl carbonate to give symmetrical ketone 3 in an 88% isolated yield [15,16]. Upon treating with Li[AlH4]/AlCl3 in tetrahydrofuran [17], this ketone smoothly deoxygenated, thereby producing the target compound 1 as an air-stable, orange solid in a 71% yield, following chromatographic purification and crystallization from hot heptane.
The conversion of 3 into 1 was clearly identified in the NMR spectra, mainly through the replacement of the C=O resonance (δC 198.61) by signals due to the methylene linker at δH 2.91 and δC 28.98. The spectra also indicated the symmetrical nature of 1 and 3, by showing a single set of signals due to the two chemically equivalent phosphinoferrocene moieties in both cases, and further confirmed that the phosphine moieties remained intact during the synthesis (δP −16.1 and −17.5 for 1 and 3, respectively; cf. δP −18.1 for 2 [18]). Notably, the ν(C=O) band in the IR spectrum of 3 was observed at 1599 cm−1, that is, at a lower energy than that of the corresponding band of benzophenone (1666 cm−1, Nujol mull [19]) and even of diferrocenyl methanone (1609 cm−1 [20]). Both the relatively low δC(C=O) chemical shift and the energy of the ν(C=O) band suggest an extensive conjugation in the molecule of 3. These findings correspond with the structural data.
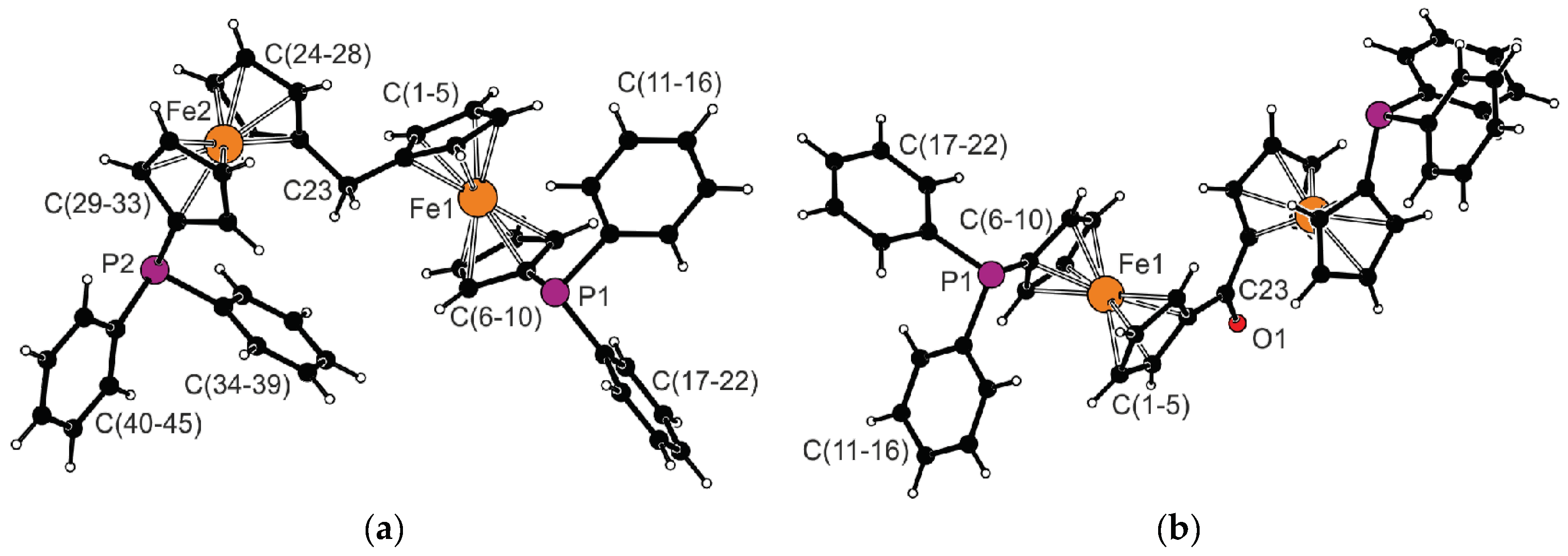
The molecular structures of 1 and 3 determined by single-crystal X-ray diffraction analysis are shown in Figure 1. Pertinent structural parameters are outlined in Table 1. The ferrocene moieties in the structure of diphosphine 1 show regular geometries with similar Fe–C distances (the difference in individual Fe–C distances is smaller than 0.03 Å for an individual Fe atom) and tilt angles well below 5°. However, they differ by the mutual positioning of the substituents bonded at positions 1 and 1′ of the ferrocene core. The substituents in the ferrocene unit comprising atom Fe1 are moved to a farther position than in the other ferrocene fragment (Fe2; compare τ angles in Table 1). The two ferrocene units are mutually rotated, as shown by the dihedral angle of the cyclopentadienyl planes C(1–5) and C(24–28), 80.6(1)°. A similar tilting has been noted for differocenylmethane (82.9(2)°) [20]. The angle at the connecting atom C23, as well as the C1–C23 and C24–C23 distances determined for 1, fall within the common ranges [21].
The C=O bond in the crystal structure of ketone 3 coincides with the crystallographic two-fold axis (space group C2/c). Therefore, only the half the molecule is structurally independent. Even in this structure, the ferrocene moieties adopt the usual geometry (Δ(Fe–C) ≈ 0.02 Å) and are negligibly tilted (≈2°). The length of the C=O bond (1.231(3) Å) is identical (within the margins of error) to the C=O bond length determined for diferrocenyl methanone (1.237(3) Å) [22]. The cyclopentadienyl planes connected by the central C=O unit in the molecule of 3 subtend a dihedral angle of 28.7(1)°.
2.2. Preparation and Characterization of Pd(II) and Au(I) Complexes
Two soft transition metal ions were chosen to evaluate the coordination preferences of diphosphine 1, viz. Pd(II) with a d8 configuration and a strong preference for the formation of square planar complexes [23] and Au(I) with a more diverse coordination behavior reflecting the lack of crystal field stabilization for a particular coordination arrangement [24]. The palladium precursors used were [PdCl2(cod)] (cod = cycloocta-1,5-diene) serving as the source of the PdCl2 fragment with two vacant coordination sites, and the dipalladium complex [(LNC)Pd(μ-Cl)]2 (LNC = 2-[(dimethylamino-κN)mehyl]phenyl-κC1) as the source of the (LNC)PdCl fragment with only one accessible coordination site and with two adjacent coordination sites blocked by the ortho-metallated ligand. The reaction of the former precursor with diphosphine 1 in equimolar amounts led exclusively to red trans-chelate complex 1, whereas the similar reaction of 1 with [(LNC)Pd(μ-Cl)]2 produced orange tetranuclear Pd2Fe2 complex 5 (Scheme 3).
The coordination of the phosphine moieties in Pd(II) complexes 4 and 5 was indicated by shifts of their 31P-NMR resonances to a lower field. For instance, complex 4 displayed a singlet at δP 16.0, which is similar to that of the non-chelate analog trans-[PdCl2(FcPPh2-κP)2] (Fc = ferrocenyl; δP 15.8) [25]. In addition, the 13C-NMR signals of the 31P-coupled carbons (i.e., carbons of the phosphinylated cyclopentadienyl and phenyl rings) were observed as characteristic triplets resulting from virtual coupling in the AXX′ spin system 13C-31P-M-31P-12C (M = Pd) [26]. Such a feature is typical of transition metal bis-phosphine complexes. Conversely, compound 5 gave rise to a singlet 31P-NMR resonance at δP 33.0, which corresponds to the position of the signals of [(LNC)PdCl(Ph2PfcY-κP)2] (fc = ferrocene-1,1′-diyl, Y = CH2(2-pyridyl): δP 33.1 in CD2Cl2 [27], Y = CH2OMe: δP 33.0 in CDCl3 [28]) and demonstrates the equivalence of the chemically identical parts of this complex.
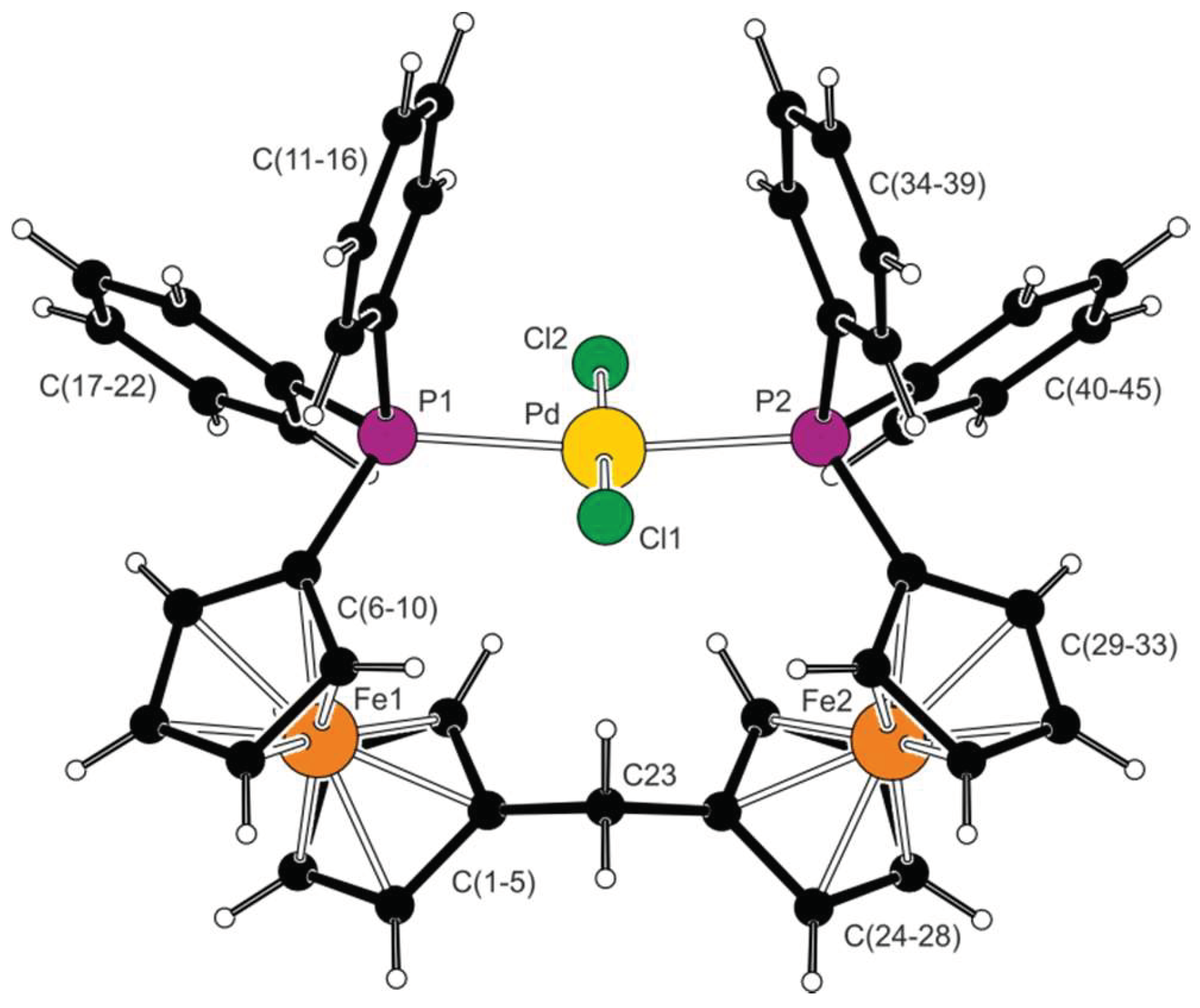
Recrystallization of complex 4 from chloroform-hexane provided crystals of the stoichiometric solvate 4·CHCl3, which was structurally characterized by X-ray diffraction analysis. A view of the complex molecule is shown in Figure 2, and the selected geometric parameters are listed in Table 2. The molecule has near-mirror symmetry; however, this is not reflected in the symmetry of the crystal assembly (i.e., the halves of the complex molecule are crystallographically independent but structurally virtually identical) [29]. In such an arrangement, the diphenylphosphino substituents are located on the same side of the diferrocenylmethane scaffold, forming a pocket that accommodates the central atom. The ferrocene units contribute to the lateral placement of the phosphine substituent, adopting a 1,2 or synclinal eclipsed configuration (ideal value: τ = 72° [30]).
The Pd–Cl and Pd–P distances in 4·CHCl3 are similar to those reported for the aforementioned compound trans-[PdCl2(FcPPh2-κP)2] [25]. Notably, the two independent Pd–P distances in 4·CHCl3 are almost identical, whereas the Pd–Cl bond lengths differ by nearly 0.03 Å, with the Pd–Cl2 bond directed from the ligand pocket shortened. When viewed along the Cl1⋅⋅⋅Cl2 direction, the palladium atom appears displaced toward the ferrocene units. An alternative view from above the coordination plane reveals an outward displacement for the Pd atom. Consequently, the P1-Pd-P2 angle of 167.49(2)° substantially differs from the ideal 180°, whereas the Cl1-Pd-Cl2 angle is considerably less affected (174.79(2)°). The distortion of the coordination sphere is already indicated by the τ(4) parameter of the tetracoordinate species, being 0.14. The ideal square planar and tetrahedral arrangements would be τ(4) = 0.00 and 1.00, respectively [31].
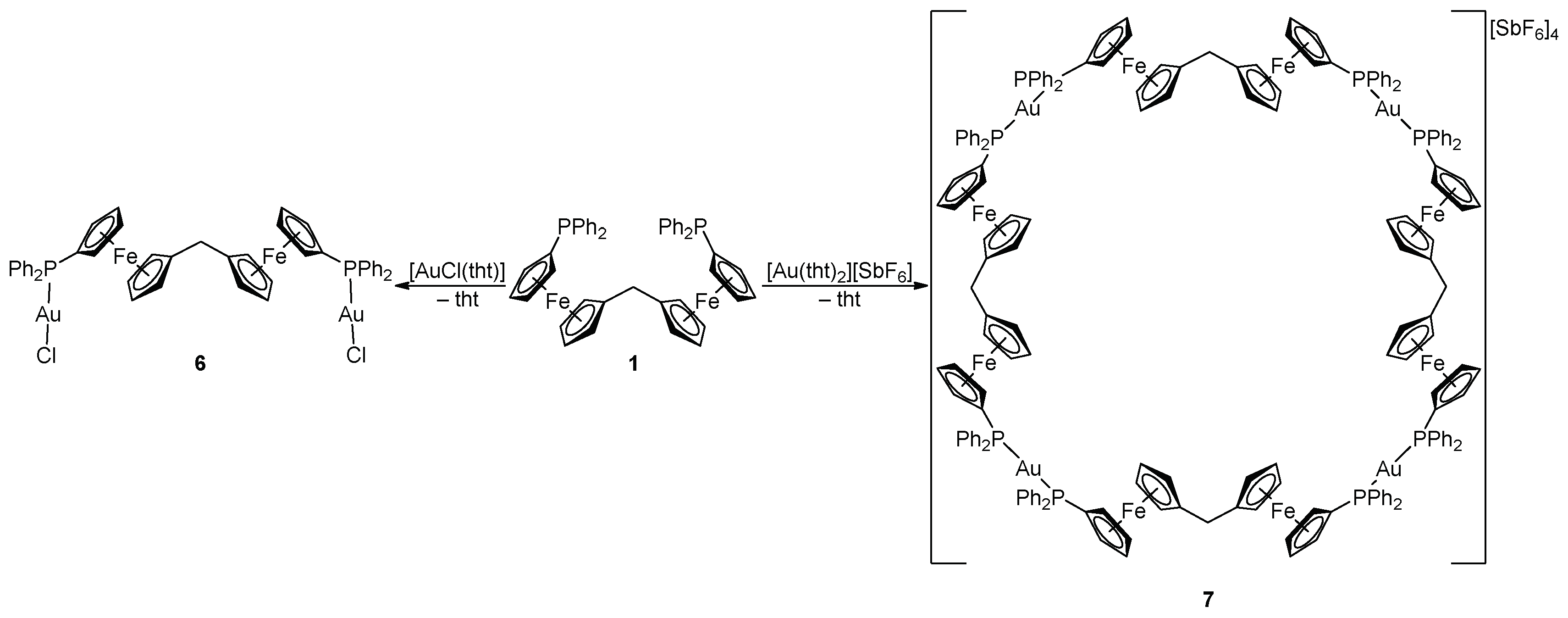
The reaction of ligand 1 with [AuCl(tht)] (tht = tetrahydrothiophene) at the 1:Au molar ratio of 1:2 produced the anticipated neutral digold complex 6 as a yellow solid (Scheme 4). When the amount of gold precursor was decreased to 1 equiv., another product was formed. Unfortunately, this product could not be unequivocally characterized due to its poor solubility and reluctance to crystallize. Changing the gold precursor to [Au(tht)2][SbF6], which lacks a firmly bound halide ligand, the reaction at a 1:1 1:Au molar ratio led to the unexpected, cationic macrocyclic tetragold complex 7 (Scheme 4). Compound 7 is cleanly and reproducibly formed and can be isolated in solvated form as a yellow solid in a high yield (93%) by precipitation. The preferential formation of tetrameric 7 over other possible oligomers may be explained by the tetrameric assembly leading to a favorable linear geometry at the gold centers, and possibly by the template effect of the hexafluoroantimonate(V) counter ions also (see the crystal structure below).
The 1H- and 13C-NMR spectra of 6 show the signals of the diphosphine ligand at the expected position, whereas the 31P resonance is characteristically shifted to a lower field (δP 29.3 in CDCl3, cf. δP 27.6 for [AuCl(FcPPh2-κP)] in the same solvent [32]). The formulation of 6 is supported by ESI mass spectra showing monocationic fragments attributable to [M – Cl]+ at m/z 1181. In the case of 7, the 1H- and 13C-NMR spectra also display all expected signals of the equivalent ferrocene ligands. The 13C-NMR resonances of phosphorus-coupled carbons appear as characteristic virtual triplets (similar to 4), which further corroborates the formulation of 7 as a symmetrical bis(phosphine) complex. The 31P-NMR signal of 7 appears further shifted to a lower field (δP 40.5 in CD2Cl2), that is, to the position reported for [Au(Ph2PfcSPh-κP)2]ClO4 (δP 40.1) [33]. The ESI mass spectrum of 7 showed fragment ions at m/z 4501 (monoisotopic mass) attributable to the cations [Au4(1)4(SbF6)3]+, thus suggesting that the tetrameric structure of 7 is retained even in solution (at least partly).
The solid-state structures of both Au(I) complexes were determined by single-crystal X-ray diffraction analysis. The molecular structure of complex 6 is shown in Figure 3, and the relevant structural parameters are outlined in Table 3. In the crystal state, the molecules of the complex reside on the crystallographic two-fold axes passing through the connecting carbon atom C23. The imposed symmetry results in an antiparallel arrangement of the equivalent halves of the complex molecule and the P–Au–Cl subunits (as opposed to the complex molecule in 4·CHCl3, which adopts a near-mirror symmetry).
The Au–Cl and Au–P distances in 6 do not differ from those previously reported for the monogold complex [AuCl(FcPPh2-κP)] (2.280(2) and 2.228(2) Å, respectively [32]) and the Au-donor bonds are arranged to form a virtually linear P−Au−Cl fragment (≈176°). The two phosphine moieties and the P−Au−Cl fragments are directed toward opposite sides of the central differocenylmethane unit, and their P−C bonds subtend an angle of 110.53(2)°. The ferrocene cyclopentadienyls assume a near-synclinal eclipsed conformation and are tilted by 3.5(2)°. The P–C bonds in 6 are slightly shorter than in free 1. These bonds are apparently strengthened by the donation of the phosphorus lone pair and by the associated electron density shift from the conjugated substituents toward the phosphorus atom.
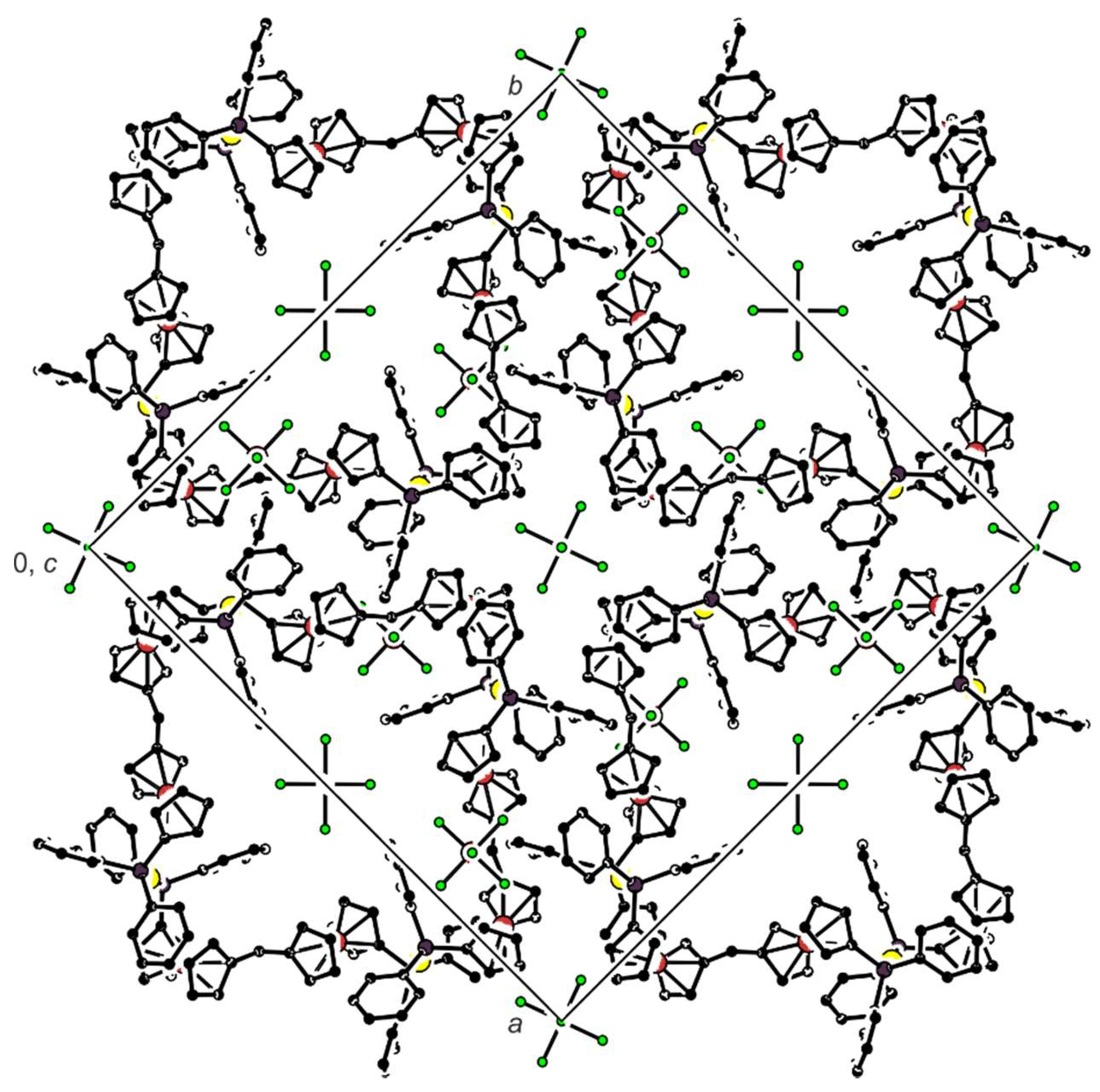
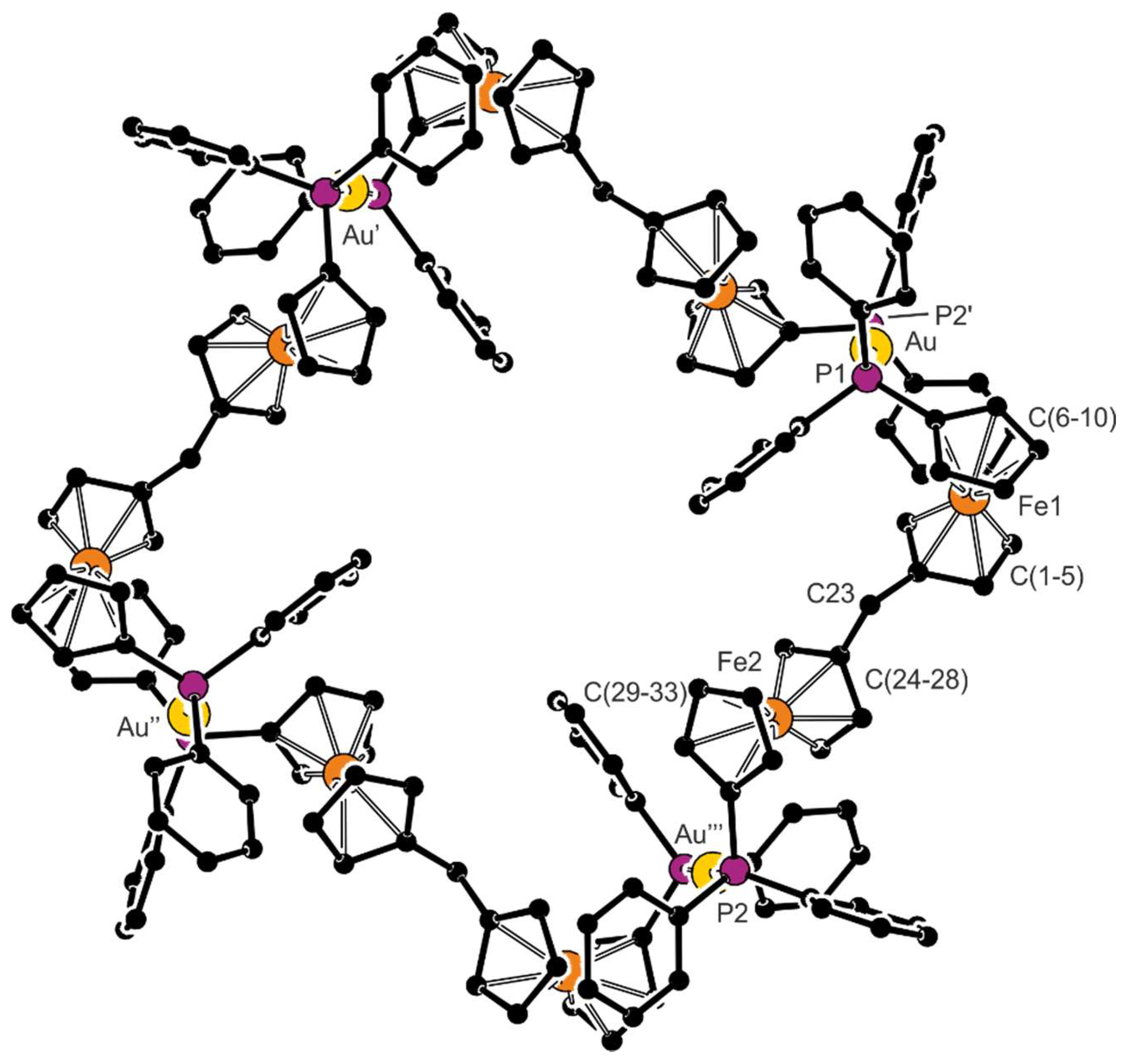
Compound 7 crystallizes in solvated form, but the crystallization solvents are severely disordered within very large, columnar structural voids. Nonetheless, the overall symmetry of the crystal assembly is rather high (tetragonal, space group I4/m). The complex cations lie over a crystallographic four-fold inversion axis; hence, only one Au(1)+ subunit is symmetrically independent. The stacking of the complex cations results in the formation of channels oriented along the crystallographic axis c, which accommodate the counter anions and the solvent molecules (see the packing diagram in Figure 4). A view of the tetragold cation in the structure of 7 is shown in Figure 5. Selected geometric parameters are presented in Table 4.
The Au–P bonds in 7 are significantly longer than in 6 (by approximately 0.08 Å) but similar to those determined for the discrete bis(phoshine) complex [Au2(Ph2PfcCN-κP)2][SbF6]2 (Au–P: 2.3104(8) and 2.3140(8) Å) [34]. Qualitatively similar differences can also be detected in the pair of triphenylphosphine complexes, [AuCl(PPh3)] (2.228(1) Å) [35] and [Au(PPh3)2][SbF6] (2.319 and 2.324 Å) [36], and are consistent with a larger trans influence [37] of the phosphine ligands that destabilize each other. Similarly to 6, the P−C bonds in 7 are shorter than in the free ligand 1.
Although the ferrocene cyclopentadienyls in the two structurally independent ferrocene moieties (comprising Fe1 and Fe2) are both almost perfectly eclipsed, they differ in conformation. In the first ferrocene moiety (Fe1), the substituents in positions 1 and 1′ adopt a synclinal eclipsed (or 1,2) conformation. Conversely, in the other (Fe2), they are rotated to a more distant position assuming an anticlinal eclipsed (or 1,3) conformation.
3. Experimental
3.1. Materials and Methods
All syntheses were performed under an argon atmosphere using standard glassware. Compound 2 [18], [AuCl(tht)] [38], and [Au(tht)2][SbF6] [34] were prepared according to the literature methods. All other chemicals were purchased from commercial suppliers (Sigma-Aldrich, Alfa-Aesar, UK) and were used as received. Tetrahydrofuran (THF) and dichloromethane were dried with a PureSolv MD5 solvent purification system (Innovative Technology, Inc., Amesbury, MA, USA). Solvents were used without any additional purification during workup, chromatography, and crystallizations (Lach-Ner, Neratovice, Czech Republic).
The NMR spectra were recorded at 25 °C on a Varian UNITY Inova 400 spectrometer (Palo Alto, CA, USA) operating at 399.95 MHz, 100.58 MHz, and 161.90 MHz for 1H, 13C, and 31P, respectively. Chemical shifts (δ in ppm) are given relative to internal tetramethylsilane (1H and 13C) or to external 85% H3PO4 (31P). In addition to the standard notation of signal multiplicity (s = singlet, d = doublet, t = triplet, etc.), vt and vq are used to denote virtual multiplets arising from the AA′BB′ and AA′BB′X spin systems (A, B = 1H, X = 31P) constituted by the protons in the C- and Ph2P-substituted cyclopentadienyl rings, respectively. FTIR spectra were measured on a Thermo Fisher Nicolet 6700 spectrometer (Waltham, MA, USA) in the range of 400–4000 cm−1. Electrospray ionization (ESI) mass spectra were recorded using a Compact QTOF-MS spectrometer (Bruker Daltonics, Billerica, MA, USA). Elemental analyses were performed using a Perkin-Elmer PE 2400 CHN analyzer (Waltham, MA, USA). The amount of residual clathrated solvent (if any) was verified by NMR spectroscopy.
3.2. Syntheses
3.2.1. Synthesis of 3
1′-(Diphenylphosphino)-1-bromoferrocene (2; 2.25 g, 5.0 mmol) was dissolved in anhydrous THF (50 mL) in a reaction flask equipped with a stirring bar. The solution was cooled to −78 °C in a dry ice/ethanol bath and treated with n-butyllithium (2.0 mL of 2.5 M in hexanes, 5.0 mmol). The resulting mixture was stirred at −78 °C for 20 min before adding neat diethyl carbonate (0.30 mL, 2.5 mmol). The cooling bath was removed, continuously stirring at room temperature overnight. Next, the reaction was terminated by adding saturated aqueous NaHCO3 (40 mL), and the mixture was extracted with methyl tert-butyl ketone (40 mL). The organic layer was separated and washed with saturated aqueous NaHCO3 (40 mL) and brine (2 × 40 mL). The partly separated product was redissolved by adding dichloromethane (50 mL). The organic phase was dried over MgSO4 and evaporated, leaving a crude product, which was purified by chromatography over a silica gel column using dichloromethane-methanol (100:1, v/v) as the eluent. The methanol content in the mobile phase was gradually increased up to 75:1 (v/v), which formed three bands. The two first, minor orange bands, containing non-polar side-products and impurities, were discarded; the third, a major red band, was collected and evaporated to isolate ketone 3 as a red solid. Yield: 1.68 g (88%). Single crystals used for structure determination were grown from warm ethyl acetate.
1H-NMR (CDCl3): δ 4.09 (vq, J′ = 1.8 Hz, 4H), 4.34 (vt, J′ = 1.7 Hz, 4H), 4.36 (vt, J′ = 2.0 Hz, 4H), 4.80 (vt, J′ = 2.0 Hz, 4H) (4 × CH of fc); 7.31–7.39 (m, 20 H, CH of PPh2). 13C{1H}-NMR (CDCl3): δ 71.39 (s, 4C, CH of fc), 73.16 (s, 4C, CH of fc), 73.40 (d, JPC = 3 Hz, 4C, CH of fc), 74.31 (d, JPC = 14 Hz, 4C, CH of fc), 77.91 (d, 1JPC = 9 Hz, 2C, Cipso–P of fc), 80.58 (s, 2C, Cipso–CO of fc), 128.25 (d, JPC = 7 Hz, 8C, CH of PPh2), 128.71 (s, 4C, CH of PPh2), 133.47 (d, JPC = 20 Hz, 8 C, CH of PPh2), 138.47 (d, 1JPC = 10 Hz, 4 C, Cipso–P of PPh2), 198.61 (s, 1 C, C=O). 31P{1H}-NMR (CDCl3): δ −17.5 (s). IR (Nujol): νmax 1599 s, 1569 w, 1543 w, 1479 m, 1432 s, 1375 s, 1327 w, 1309 m, 1291 s, 1197 m, 1094 w, 1067 m, 1059 m, 1051 m, 1027 m, 998 w, 890 w, 832 s, 810 m, 772 s, 743 s, 699 s, 637 w, 599 w, 499 s, 480 s, 463 m, 447 m, 406 w cm−1. ESI + MS: m/z 767.1 ([M + H]+). Anal. Calc. for C45H36Fe2OP2 (766.4) C 70.52, H 4.73%. Found: C 70.17, H 4.55%.
3.2.2. Synthesis of Diphosphine 1
Ketone 3 (1.68 g, 2.2 mmol) was dissolved in anhydrous THF (120 mL) in a reaction flask equipped with a gas inlet, a septum, and a stirring bar. Solid Li[AlH4] (0.33 g, 8.8 mmol) was added, and the mixture was stirred at room temperature for 30 min. Then, anhydrous AlCl3 was introduced (1.17 g, 8.8 mmol), and the resultant mixture was stirred overnight. The reaction mixture was quenched by carefully adding saturated aqueous NaHCO3 (50 mL) and diluting with ethyl acetate (50 mL). The organic layer was separated and consecutively washed with saturated aqueous NaHCO3 (2 × 50 mL) and brine (50 mL). The aqueous layers were combined and back-extracted with ethyl acetate (50 mL). The combined organic layers were dried over MgSO4 and evaporated. The crude product was taken up with a little dichloromethane and transferred to the top of a silica gel column. Elution with hexane-ethyl acetate (5:1, v/v) provided a single orange band (side-products remained adsorbed at the top of the column), which was collected and evaporated. The residue was dissolved in hot ethyl acetate (50 mL), and the solution was poured into hot heptane (30 mL). The crystalline product, which separated upon cooling to 4 °C, was isolated by suction to give pure 1 as an orange crystalline solid. Yield: 1.17 g (71%). Single crystals were grown btained from hot heptane.
1H-NMR (CDCl3): δ 2.91 (s, 2H, CH2), 3.89–3.94 (m, 8H), 4.01 (vq, J′ = 1.9 Hz, 4H, CH of fc), 4.27 (vt, J′ = 1.8 Hz, 4H, CH of fc); 7.30–7.41 (m, 20H, CH of PPh2). 13C{1H}-NMR (CDCl3): δ 28.98 (s, 1C, CH2), 68.53 (s, 4C, CH of fc), 69.36 (s, 4C, CH of fc), 75.13 (d, JPC = 4 Hz, 4C, CH of fc), 73.47 (d, JPC = 15 Hz, 4C, CH of fc), 75.61 (d, 1JPC = 5 Hz, 2C, Cipso–P of fc), 88.91 (s, 2C, Cipso–CH2 of fc), 128.09 (d, JPC = 7 Hz, 8C, CH of PPh2), 128.42 (s, 4C, CH of Ph), 133.51 (d, JPC = 19 Hz, 8C, CH of PPh2), 139.29 (d, 1JPC = 10 Hz, 4C, Cipso–P of PPh2). 31P{1H}-NMR (CDCl3): δ −16.1 (s). IR (Nujol): νmax 1583 w, 1568 w, 1433 s, 1324 w, 1310 m, 1292 w, 1276 w, 1221 w, 1193 m, 1095 m, 1068 m, 1035 m, 1025 s, 1000 w, 970 vw, 928 m, 917 w, 889 w, 835 s, 819 s, 756 s, 742 s, 698 vs, 630 m, 584 vw, 529 m, 494 vs, 464 s, 456 m, 440 m cm−1. ESI + MS: m/z 753.1 ([M + H]+). Anal. Calc. for C45H38Fe2P2 (752.4): C 71.83, H 5.09%. Found C 71.71, 5.14%.
3.2.3. Synthesis of Complex 4
Ligand 1 (75.2 mg, 0.10 mmol) and [PdCl2(cod)] (28.5 mg, 0.10 mmol) were dissolved in dichloromethane (5 mL) in a small reaction flask equipped with a stirring bar. The resulting solution was stirred for 3 h and then evaporated. The residue was dissolved in chloroform and crystallized by adding hexane as a top layer. The crystals formed over several days were filtered off and dried under vacuum. Yield of 4: 91.7 mg (98%), red needles. Crystals of 4·CHCl3 used for X-ray diffraction analysis were obtained by liquid-phase diffusion of hexane into a chloroform solution.
1H-NMR (CDCl3, 50 °C): δ 4.14 (vt, J′ = 1.8 Hz, 4H), 4.48 (vt, J′= 1.8 H, 4H), 4.57 (br s, 4H) (3 × CH of fc); 4.80 (br s, 2H, CH2), 4.90 (br s, 4H, CH of fc), 7.29–7.40 (m, 12H, CH of PPh2), 7.56–7.64 (m, 8H, CH of PPh2). 13C{1H}-NMR (CDCl3): δ 31.60 (s, 1C, CH2), 68.39 (s, 4 C, CH of fc), 70.21 (t, J′ = 27 Hz, 2C, Cipso–P of fc), 71.38 (br s, 4C, CH of fc), 71.75 (br s, 4C, CH of fc), 90.1 (s, 2C, Cipso–CH2 of fc), 127.42 (t, J′ = 5 Hz, 8C, CH of PPh2), 130.03 (s, 4C, CH of PPh2), 131.10 (t, J′ = 26 Hz, 4 C, Cipso–P of PPh2), 134.01 (t, J′ = 6 Hz, 8 C, CH of PPh2). 31P{1H}-NMR (CDCl3): δ 16.0 (s). ESI+ MS: m/z 893.0 ([M − Cl]+). Anal. Calcd. for C45H38Cl2Fe2P2Pd (929.7): C 58.13, H 4.12%. Found: C 57.80, H 4.29%.
3.2.4. Synthesis of Complex 5
Ligand 1 (75.2 mg, 0.10 mmol) and [(LNC)Pd(μ-Cl)]2 (55.2 mg, 0.10 mmol) were dissolved in dichloromethane (5 mL), and the resulting mixture was stirred for 3 h. Following evaporation under vacuum, the crude product was dissolved in a minimum amount of dichloromethane and precipitated by adding pentane. The formed solid was filtered off and dried under vacuum. Yield of 5: 95.0 mg (77%), orange amorphous solid.
1H-NMR (CDCl3): δ 2.87 (d, 4JPH = 2.7 Hz, 12H, N(CH3)2), 3.38 (s, 2H, CH2), 4.07 (d, 4JPH = 2.2 Hz, 4H, CH2N), 4.24 (d of vt, J ≈ 1.0 Hz and 2.0 Hz, 4H, CH of fc), 4.26 (vt, J′ = 1.8 Hz, 4H, CH of fc), 4.28 (vq, J′ = 1.9 Hz, 4H, CH of fc), 4.45 (vt, J′ = 1.9 Hz, 4H, CH of fc), 6.31 (ddd, J = 7.6, 6.3, 1.2 Hz, 2H, C6H4), 6.42 (td, J = 7.6, 1.4 Hz, 2H, C6H4), 6.84 (td, J = 7.3, 1.2 Hz, 2H, C6H4), 7.02 (dd, J = 7.4, 1.6 Hz, 2H, C6H4), 7.29–7.36 (m, 8H, CH of PPh2), 7.38–7.43 (m, 4H, CH of PPh2), 7.52–7.59 (m, 8H, CH of PPh2). 13C{1H}-NMR (CDCl3): δ 29.25 (s, 1C, CH2), 50.15 (d, 3JPC = 3 Hz, 4C, N(CH3)2), 70.00 (s, 4C, CH of fc), 70.64 (s, 4C, CH of fc), 72.67 (d, JPC = 7 Hz, 4C, CH of fc), 72.89 (d, 1JPC = 60 Hz, 2C, Cipso–P of fc), 73.60 (d, 3JPC = 3 Hz, 2C, CH2N), 75.78 (d, JPC = 10 Hz, 4C, CH of fc), 89.35 (s, Cipso–CH2 of fc), 122.51 (s, 2C, CH of C6H4), 123.74 (s, 2C, CH of C6H4), 124.80 (d, JPC = 6 Hz, 2C, CH of C6H4), 127.79 (d, JPC = 11 Hz, 8C, CH of PPh2), 131.02 (d, JPC = 2 Hz, 4C, CH of PPh2), 131.87 (d, 1JPC = 50 Hz, 4 C, Cipso of PPh2), 134.37 (d, JPC = 12 Hz, 8 C, CH of PPh2), 138.44 (d, JPC = 10 Hz, 2C, CH of C6H4), 148.29 (d, JPC = 2 Hz, 2C, Cipso of C6H4), 151.99 (s, 2C, Cipso of C6H4). 31P{1H}-NMR (CDCl3): δ 33.0 (s). Anal. Calcd. for C63H62Cl2Fe2N2P2Pd2 (1304.5): C 58.00, H 4.79, N 2.15%. Found C 57.67, H 4.76, N 1.92%.
3.2.5. Synthesis of Complex 6
Ligand 1 (75.2 mg, 0.10 mmol) and [AuCl(tht)] (64.0 mg, 0.20 mmol) were dissolved in dichloromethane (5 mL), and the resulting mixture was stirred for 3 h. Subsequent evaporation afforded crude product, which was triturated successively with diethyl ether and pentane until the characteristic odor of tetrahydrothiophene was no longer perceptible and then dried under vacuum. Yield of 6·0.25CH2Cl2: 103 mg (71%), orange amorphous solid. Single crystals were obtained upon recrystallization of the compound from chloroform-hexane.
1H-NMR (CDCl3): δ 3.25 (s, 2H, CH2), 3.97 (vt, J′ = 1.8 Hz, 4 H, CH of fc), 4.17 (vt, J′ = 1.9 Hz, 4H, CH of fc), 4.23–4.25 (m, 4H, CH of fc), 4.57–4.58 (m, 4H, CH of fc), 7.41–7.52 (m, 12H, CH of PPh2), 7.54–7.62 (m, 8H, CH of Ph). 13C{1H}-NMR (CDCl3): δ 29.05 (s, 1C, CH2), 68.72 (d, 1JPC = 74 Hz, 2 C, Cipso–P of fc), 69.37 (s, 4C, CH of fc), 70.74 (s, 4C, CH of fc), 73.44 (d, JPC = 9 Hz, 4C, CH of fc), 74.34 (d, JPC = 14 Hz, 4H, CH of fc), 90.12 (s, 2C, Cipso–CH2 of fc), 128.87 (d, JPC = 12 Hz, 8C, CH of Ph), 130.92 (d, 1JPC = 64 Hz, 4C, Cipso–P of PPh2), 131.58 (d, JPC = 2 Hz, 4H, CH of PPh2), 133.53 (d, JPC = 14 Hz, 8H, CH of PPh2). 31P{1H}-NMR (CDCl3): δ 29.2 (s). ESI + MS: m/z 1181.0 ([M − Cl]+). Anal. Calc. for C45H38Au2Cl2Fe2P2·0.25CH2Cl2 (1238.5): C 43.88, H 3.13%. Found: C 43.81, H 3.01%.
3.2.6. Synthesis of Complex 7
Ligand 1 (152 mg, 0.20 mmol) and [Au(tht)2][SbF6] (123 mg, 0.20 mmol) were dissolved in dry dichloromethane (5 mL) and the resulting orange solution was stirred at room temperature for 3 h. Then, the volatiles were removed under vacuum, and the solid residue was repeatedly triturated with diethyl ether and pentane, consecutively, to remove residual tetrahydrothiophene. The powdery residue was dissolved in a minimum amount of dichloromethane and precipitated by adding an excess of pentane. The separated solid was filtered off and dried under vacuum. Yield of 7: 221 mg (93%), yellow flakes. Crystals used for structure determination were obtained from 1,2-dichloroethane-methyl tert-butyl ketone by liquid-phase diffusion.
1H-NMR (CD2Cl2): δ 4.06 (vt, J′ = 1.7 Hz, 16H, CH of fc), 4.31 (s, 16H, CH of fc), 4.36 (s, 24H, CH of fc and CH2), 4.78 (s, 16H, CH of fc), 7.54–7.66 (m, 80, CH of PPh2).13C{1H}-NMR (CD2Cl2): δ 32.80 (s, 4C, CH2), 68.31 (t, J′ = 35 Hz, 8 C, Cipso–PPh2), 69.53 (s, 16C, CH of fc), 72.08 (s, 16C, CH of fc), 73.81 (t, JPC = 4 Hz, 16C, CH of fc), 75.43 (t, JPC = 7 Hz, 16C, CH of fc), 88.96 (s, 8C, Cipso–CH2), 130.00 (t, JPC = 6 Hz, 32H, CH of PPh2), 130.04 (t, JPC = 28 Hz, 16C, Cipso–P of PPh2; partly obscured by the signal due to CH PPh2), 132.86 (s, 16C, CH of PPh2), 133.90 (t, JPC = 7 Hz, 32C, CH of PPh2). 31P{1H}-NMR (CD2Cl2): δ 40.5 (s). ESI + MS: m/z 4501.0 ([M − SbF6]+). Anal. Calc. for C180H152Au4F24Fe8P8Sb4 (4740.5): C 45.60, H 3.23%. Found: C 45.34, H 2.97%.
3.3. X-ray Crystallography
The diffraction data θmax = 26.0° or 27.5° (data completeness ≥ 99.5%) were collected at 120(2) K or 150(2) K using either a Nonius KappaCCD diffractometer with a Bruker Apex-II image plate detector (compounds 3 and 1) or a Bruker D8 VENTURE Kappa Duo PHOTON100 diffractometer with a IµS micro-focus X-ray tube (other compounds) equipped with a Cryostream Cooler (Oxford Cryosystems, Oxford, United Kindgom). Mo Kα radiation was used in all cases.
The structures were solved by direct methods (SHELXT) [39] and refined using a full-matrix, least-squares routine based on F2 with SHELXL-97 [40]. The non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were included in their calculated positions and refined using the “riding model” with Uiso(H) set to a multiple of Ueq of their bonding carbon atom. The structure of compound 7 contained disordered solvent molecules within the columnar cavities. The contribution of the solvent, which could not be resolved in terms of atomic positions, was modeled as diffuse electron density using PLATON SQUEEZE [41].
Relevant crystallographic data, data collection, and structure refinement parameters are presented in Table 5. All geometric calculations were performed using a recent version of the program PLATON [42], which was also used to prepare the structural diagrams. Complete structural data were deposited with the Cambridge Structural Database (CCDC deposition numbers: 1858205–1858209). These data are available free of charge at http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44-1223-336-033; E-mail: [email protected]).
4. Conclusions
In summary, we have reported a facile synthesis of an achiral bis(phosphinoferrocene) ligand 1. This compound was fully characterized and further demonstrated to coordinate as a normal terminal ligand coordinating two metal centers (P-monodentate donor) in 5 and in 6, as a trans-chelating ligand in 4, or as a P,P′-bridging ligand forming an unusual macrocyclic tetragold complex 7. The crystal structures determined that all reported complexes (except for 5) demonstrate the flexible nature of ligand 1, which can easily change conformationally (via tilting of the ferrocene moieties and rotation of their cyclopentadienyl rings) to meet the steric demands of the particular central atom to which it is coordinated.
Author Contributions
P.Š. conceived the study and, in collaboration with the other co-authors, interpreted the collected data and wrote this article; Z.L. performed all syntheses; J.S. and Z.L. collected and analyzed the spectroscopic data; I.C. recorded the X-ray diffraction data and solved the crystal structures.
Funding
This research was supported by the Charles University Research Centre program (project No. UNCE/SCI/014).
Conflicts of Interest
The authors declare no conflict of interest. The funding agency had no role in the experimental design, data interpretation, and manuscript preparation or in the decision to publish the results.
References and Notes
- Togni, A.; Hayashi, T. Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Chemistry; VCH: Weinheim, Germany, 1995; ISBN 978-3-527-61559-9. [Google Scholar]
- Štěpnička, P. Ferrocenes: Ligands, Materials and Biomolecules; Wiley: Chichester, UK, 2008; ISBN 978-0-470-03585-6. [Google Scholar]
- Atkinson, R.C.J.; Gibson, V.C.; Long, N.J. The syntheses and catalytic applications of unsymmetrical ferrocene ligands. Chem. Soc. Rev. 2004, 33, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Gómez Arrayás, R.; Adrio, J.; Carretero, J.C. Recent applications of chiral ferrocene ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, M.; Yamauchi, A.; Takegawa, T.; Ito, Y. Synthesis of 2,2′′-bis(diphenylphosphino)-1,1′′-biferrocene, a planar chiral bisphosphine, and its palladium(II) complex. J. Chem. Soc. Chem. Commun. 1991, 874–875. [Google Scholar] [CrossRef]
- Espino, G.; Xiao, L.; Puchberger, M.; Mereiter, K.; Spindler, F.; Manzano, B.R.; Jalón, F.A.; Weissensteiner, W. Synthesis, coordination behaviour, structural features and use in asymmetric hydrogenations of bifep-type biferrocenes. Dalton Trans. 2009, 2751–2763. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, M.; Hamashima, H.; Ito, Y. Catalytic asymmetric synthesis with trans-chelating chiral diphosphine ligand TRAP: rhodium-catalyzed asymmetric Michael addition of. alpha-cyano carboxylates. J. Am. Chem. Soc. 1992, 114, 8295–8296. [Google Scholar] [CrossRef]
- Sawamura, M.; Hamashima, H.; Sugawara, M.; Kuwano, R.; Ito, Y. Synthesis and structures of trans-chelating chiral diphosphine ligands bearing aromatic p-substituents, (S,S)-(R,R)- and (R,R)-(S,S)-2,2′′-bis[1-(diarylphosphino)ethyl]-1,1′′-biferrocene (ArylTRAPs) and their transition metal complexes. Organometallics 1995, 14, 4549–4558. [Google Scholar] [CrossRef]
- Trost, B.M.; van Vranken, D.L.; Bingel, C. A modular approach for ligand design for asymmetric allylic alkylations via enantioselective palladium-catalyzed alkylations. J. Am. Chem. Soc. 1992, 114, 9327–9343. [Google Scholar] [CrossRef]
- You, S.-L.; Hou, X.-L.; Dai, L.-X.; Cao, B.-X.; Sun, J. Novel bis-N-[2-(diphenylphosphino)ferrocenyl- carbonyl]diaminocyclohexane ligands: Application in asymmetric allylic alkylation of imino esters with simple allyl carbonate. Chem. Commun. 2000, 1933–1934. [Google Scholar] [CrossRef]
- Longmire, J.M.; Wang, B.; Zhang, X. Highly efficient kinetic resolution of 2-cyclohexenyl acetate in Pd-catalyzed allylic alkylation. Tetrahedron Lett. 2000, 41, 5435–5439. [Google Scholar] [CrossRef]
- Horikoshi, R.; Mochida, T.; Torigoe, R.; Yamamoto, Y. Preparation and electrochemical properties of polynuclear organometallic complexes derived from ferrocene-containing bidentate ligands. Eur. J. Inorg. Chem. 2002, 2002, 3197–3203. [Google Scholar] [CrossRef]
- Lohan, M.; Milde, B.; Heider, S.; Speck, J.M.; Krausse, S.; Schaarschmidt, D.; Rueffer, T.; Lang, H. Synthesis, electrochemistry, spectroelectrochemistry, and solid-state structures of palladium biferrocenylphosphines and their use in C, C cross-coupling reactions. Organometallics 2012, 31, 2310–2326. [Google Scholar] [CrossRef]
- Butler, I.R.; Cullen, W.R. The synthesis of α-N,N-dimethyl-1′-diphenylphosphinoferrocenylethylamine and related ligands. Can. J. Chem. 1983, 61, 147–153. [Google Scholar] [CrossRef]
- Štěpnička, P. 1’-Functionalised ferrocene phosphines: Synthesis, coordination chemistry and catalytic applications. In Ferrocenes: Ligands, Materials and Biomolecules; Štěpnička, P., Ed.; Wiley: Chichester, UK, 2008; Chapter 5; pp. 177–204. ISBN 978-0-470-03585-6. [Google Scholar]
- Barreiro, E.M.; Broggini, D.F.D.; Adrio, L.A.; White, A.J.P.; Schwenk, R.; Togni, A.; Hii, K.K. Gold(I) complexes of conformationally constricted chiral ferrocenyl phosphines. Organometallics 2012, 31, 3745–3754. [Google Scholar] [CrossRef]
- Podlaha, J.; Štěpnička, P.; Gyepes, R.; Mareček, V.; Lhotský, A.; Polášek, M.; Kubišta, J.; Nejezchleba, M. Hydrophobic ferrocene derivatives as potential standards in electrochemistry. Collect. Czech. Chem. Commun. 1997, 62, 185–198. [Google Scholar] [CrossRef]
- Butler, I.R.; Davies, R.L. A rapid convenient synthesis of ferrocene-based triphos analogue ligands. Synthesis 1996, 1350–1354. [Google Scholar] [CrossRef]
- Spectral Database for Organic Compounds SDBS. Benzophenone, SDBS No. 1294. Available online: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/cre_index.cgi (accessed on 23 July 2018).
- Ferguson, G.; Glidewell, C.; Opromolla, G.; Zakaria, C.M.; Zanello, P. The redox behaviour of some bis-ferrocenyl compounds: crystal and molecular structures of diferrocenylmethane and diferrocenylmethanol. J. Organomet. Chem. 1996, 517, 183–190. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, S1–S19. [Google Scholar] [CrossRef]
- Bratych, N.; Hassall, K.; White, J. Redetermination of the structure of diferrocenyl ketone at low temperature. Acta Crystallogr. Sect. E Struct. Rep. Online 2003, 59, m33–m35. [Google Scholar] [CrossRef]
- Barnard, C.J.; Russell, M.J.H. Palladium. In Comprehesive Coordination Chemistry; Wilkinson, G., Gillard, R.D., McCleverty, J.A., Eds.; Pergamon Press: Oxford, UK, 1997; Volume 5, Chapter 51; pp. 1099–1170. ISBN 0-08-035948-5. [Google Scholar]
- Puddephatt, R.J. Gold. In Comprehesive Coordination Chemistry; Wilkinson, G., Gillard, R.D., McCleverty, J.A., Eds.; Pergamon Press: Oxford, UK, 1997; Volume 5, Chapter 55; pp. 862–923. ISBN 0-08-035948-5. [Google Scholar]
- Zábranský, M.; Císařová, I.; Štěpnička, P. Synthesis, coordination, and catalytic use of 1′-(diphenylphosphino) ferrocene-1-sulfonate anion. Organometallics 2018, 37, 1615–1626. [Google Scholar] [CrossRef]
- Hersh, W.H. False AA’X spin-spin coupling systems in 13C-NMR: Examples involving phosphorus and a 20-year-old mystery in off-resonance decoupling. J. Chem. Educ. 1997, 74, 1485–1488. [Google Scholar] [CrossRef]
- Štěpnička, P.; Schulz, J.; Klemann, T.; Siemeling, U.; Císařová, I. Synthesis, structural characterization, and catalytic evaluation of palladium complexes with homologous ferrocene-based pyridylphosphine ligands. Organometallics 2010, 29, 3187–3200. [Google Scholar] [CrossRef]
- Zábranský, M.; Machara, A.; Císařová, I.; Štěpnička, P. Palladium(II) complexes of homologated ferrocene phosphanylether and thioether ligands. Eur. J. Inorg. Chem. 2017, 2017, 4850–4860. [Google Scholar] [CrossRef]
- Notably, the apparent “molecular” symmetry is not diminished by the solvent molecule, which is located rather symmetrically above the phosphinylated cyclopentadienyl rings.
- Gan, K.-S.; Hor, T.S.A. 1,1′-Bis(diphenylphosphino)ferrocene. Coordination chemistry, organic syntheses, and catalysis. In Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Chemistry; Togni, A., Hayashi, T., Eds.; VCH: Weinheim, Germany, 1995; Chapter 1; pp. 3–104. ISBN 978-3-527-61559-9. [Google Scholar]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ 4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Roessler, K.; Rueffer, T.; Walfort, B.; Packheiser, R.; Holze, R.; Zharnikov, M.; Lang, H. Synthesis, characterization and electrochemical behavior of unsymmetric transition metal-terminated biphenyl ethynyl thiols. J. Organomet. Chem. 2007, 692, 1530–1545. [Google Scholar] [CrossRef]
- Aguado, J.E.; Canales, S.; Gimeno, M.C.; Jones, P.G.; Laguna, A.; Villacampa, M.D. Group 11 complexes with unsymmetrical P, S and P, Se disubstituted ferrocene ligands. Dalton Trans. 2005, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Škoch, K.; Císařová, I.; Štěpnička, P. Synthesis and catalytic use of gold(I) complexes containing a hemilabile phosphanylferrocene nitrile donor. Chem. Eur. J. 2015, 21, 15998–16004. [Google Scholar] [CrossRef] [PubMed]
- Dunstan, S.P.C.; Healy, P.C.; Sobolev, A.N.; Tiekink, E.R.T.; White, A.H.; Williams, M.L. Isomorphism in the structural chemistry of two-coordinate adducts of diphenyl(2-formylphenyl)phosphine and triphenylphosphine with gold(I) halides. J. Mol. Struct. 2014, 1072, 253–259. [Google Scholar] [CrossRef]
- Inagaki, F.; Matsumoto, C.; Okada, Y.; Maruyama, N.; Mukai, C. Air-stable cationic gold(I) catalyst featuring a z-type ligand: Promoting enyne cyclizations. Angew. Chem. Int. Ed. 2014, 54, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Appleton, T.G.; Clark, H.C.; Manzer, L.E. The trans-influence: Its measurement and significance. Coord. Chem. Rev. 1973, 10, 335–422. [Google Scholar] [CrossRef]
- Usón, R.; Laguna, A.; Laguna, M. (Tetrahydrothiophene) gold(I) or gold(III) complexes. Inorg. Synth. 1989, 26, 85–91. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Platon squeeze: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are available from the authors within one year from publication of this article. |
Scheme 1.
Examples of bis(phosphinoferrocene ligands) and the structure of the newly prepared compound 1. For clarity, chiral donors are presented without indicating chirality.
Scheme 1.
Examples of bis(phosphinoferrocene ligands) and the structure of the newly prepared compound 1. For clarity, chiral donors are presented without indicating chirality.

Scheme 2.
Synthesis of diphosphine 1.

Figure 1.
Views of the molecular structures of (a) diphosphine 1 and (b) ketone 3; note that the unlabeled half of the molecule of ketone 3 is generated by the crystallographic two-fold axis passing through the C23=O1 bond.
Figure 1.
Views of the molecular structures of (a) diphosphine 1 and (b) ketone 3; note that the unlabeled half of the molecule of ketone 3 is generated by the crystallographic two-fold axis passing through the C23=O1 bond.

Scheme 3.
Synthesis of Pd(II) complexes 4 and 5 (cod = cycloocta-1,5-diene).

Figure 2.
View of the complex molecule in the structure of 4·CHCl3.

Scheme 4.
Synthesis of Au(I) complexes 6 and 7 (tht = tetrahydrothiophene).

Figure 3.
View of the molecular structure of complex 6; prime-labeled atoms are generated by crystallographic symmetry.
Figure 3.
View of the molecular structure of complex 6; prime-labeled atoms are generated by crystallographic symmetry.

Figure 4.
Simplified packing diagram of 7 as viewed along the crystallographic c axis; Hydrogen atoms are omitted for clarity. Note that the disordered solvent is not shown.
Figure 4.
Simplified packing diagram of 7 as viewed along the crystallographic c axis; Hydrogen atoms are omitted for clarity. Note that the disordered solvent is not shown.

Figure 5.
View of the complex cation in the structure of complex 7; prime-labeled atoms are generated by crystallographic symmetry.
Figure 5.
View of the complex cation in the structure of complex 7; prime-labeled atoms are generated by crystallographic symmetry.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected distances and angles for compounds 1 and 3 (in Å and deg) a.
| Parameter | 1 (Fe1) | Parameter | 1 (Fe2) | Parameter | 3 (Fe1) b |
|---|---|---|---|---|---|
| Fe–C | 2.030(2)–2.057(2) | Fe–C | 2.032 (2)–2.051 (2) | Fe–C | 2.028 (2)–2.055 (2) |
| tilt | 4.1 (1) | tilt | 1.6 (1) | tilt | 2.4 (1) |
| τ | −88.7 (2) | τ | −67.4 (2) | τ | −162.7 (1) |
| P1-C6 | 1.805 (2) | P2-C29 | 1.805 (2) | P1-C6 | 1.817 (2) |
| P1-C11 | 1.838 (2) | P2-C34 | 1.824 (2) | P1-C11 | 1.835 (2) |
| P1-C17 | 1.839 (2) | P2-C40 | 1.838 (2) | P1-C17 | 1.838 (2) |
| C1-C23 | 1.500 (3) | C24-C23 | 1.511 (3) | C1-C23 | 1.472 (2) |
| C1-C23-C24 | 110.8 (2) | – | – | C1-C23-C1′ | 122.4 (2) |
a Definitions: Fe–C the range of Fe–C distances for the particular ferrocene moiety; τ is the torsion angle C1–CgC–CgP–C6 (or similar), where the CgC and CgP denote the centroids of the C- and P-substituted cyclopentadienyl ring, respectively; tilt is the dihedral angle of the least-squares cyclopentadienyl planes surrounding the particular Fe atom. b Further data: C23–O1 = 1.231(3) Å, C1–C23–O1 = 118.8(1)°.
Table 2.
Selected geometric data for 4·CHCl3 a.
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| Pd-P1 | 2.3390 (5) | P1-Pd-Cl1 | 86.14 (2) |
| Pd-P2 | 2.3401 (5) | P1-Pd-Cl2 | 92.95 (2) |
| Pd-Cl1 | 2.3167 (5) | P2-Pd-Cl1 | 86.09 (2) |
| Pd-Cl2 | 2.2906 (5) | P2-Pd-Cl2 | 93.90 (2) |
| Fe1-C | 2.039 (2)–2.061 (2) | Fe2-C | 2.034 (2)–2.058 (2) |
| tilt (Fe1) | 0.71 (9) | tilt (Fe2) | 1.68 (9) |
| τ(Fe1) | 71.2 (1) | τ(Fe2) | −71.2 (1) |
| P1-C6 | 1.805 (2) | P2-C29 | 1.806 (2) |
| P1-C11 | 1.822 (2) | P2-C34 | 1.821 (2) |
| P1-C17 | 1.827 (2) | P2-C40 | 1.824 (2) |
| C1-C23 | 1.509 (2) | C24-C23 | 1.507 (2) |
| Fe1, …, Fe2 | 6.1653 (6) | C1-C23-C24 | 113.0 (1) |
a The parameters are defined as for the free ligand; see footnote to Table 1.
Table 3.
Selected geometric parameters for complex 6 a.
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| Au–P | 2.2277 (7) | P–Au–Cl | 176.15 (3) |
| Au–Cl | 2.2881 (7) | Au···Au′ | 12.2322 (7) |
| Fe–C | 2.029 (3)–2.057 (3) | P-C6 | 1.781 (3) |
| tilt (Fe) | 3.5 (2) | P-C11 | 1.819 (3) |
| τ(Fe) | −77.2 (2) | P-C17 | 1.815 (2) |
| Fe···Au | 4.4147 (5) | C1-C23 | 1.502 (3) |
| Fe⋅⋅⋅Fe′ | 6.3509 (8) | C1-C23-C1′ | 109.3 (3) |
a The parameters are defined as for the free ligand; see footnote to Table 1.
Table 4.
Selected geometric parameters for the complex cation in the structure of 7 a.
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| Au-P1 | 2.311 (2) | P1-Au-P2′ | 174.87 (5) |
| Au-P2′ | 2.309 (2) | Au···Au’ b | 11.6898 (9) |
| P1⋅⋅⋅P2 | 10.890 (2) | Au···Au″ c | 16.531 (1) |
| Fe1-C | 2.018 (5)–2.063 (6) | Fe2-C | 2.022 (6)–2.056 (5) |
| tilt (Fe1) | 2.2 (3) | tilt (Fe2) | 3.7 (4) |
| τ(Fe1) | −77.3 (4) | τ(Fe2) | −144.4 (4) |
| P1-C6 | 1.786 (5) | P2-C29 | 1.792 (6) |
| P1-C11 | 1.822 (5) | P2-C34 | 1.813 (5) |
| P1-C17 | 1.817 (5) | P2-C40 | 1.805 (5) |
| C1-C23 | 1.491 (7) | C23-C24 | 1.503 (8) |
Table 5.
Summary of relevant crystallographic data and structure refinement parameters.
| Compound | 1 | 3 | 4·CHCl3 | 6 | 7 |
|---|---|---|---|---|---|
| Formula | C45H38Fe2P2 | C45H36Fe2OP2 | C46H39Cl5Fe2P2Pd | C45H38Au2Cl2Fe2P2 | C180H152Au4F24Fe8P8Sb4 |
| M | 752.39 | 766.38 | 1049.06 | 1217.22 | 4740.43 |
| Crystal system | monoclinic | monoclinic | triclinic | monoclinic | tetragonal |
| Space group | P21/c (No. 14) | C2/c (No. 15) | P–1 (No. 2) | C2/c (No. 15) | I4/m (No. 87) |
| T/K | 150 (2) | 150 (2) | 150 (2) | 150 (2) | 120(2) |
| a/Å | 15.6501 (4) | 18.7605 (5) | 8.9665 (4) | 16.8308 (5) | 27.6537 (10) |
| b/Å | 13.2675 (3) | 6.5050 (1) | 13.2550 (6) | 8.9762 (3) | 27.6537 (10) |
| c/Å | 17.2689 (4) | 29.1239 (7) | 18.5420 (8) | 26.5919 (9) | 28.6005 (13) |
| α/deg | 90 | 90 | 82.774 (2) | 90 | 90 |
| β/deg | 91.126 (1) | 97.5130 (10) | 81.222 (1) | 94.7570 (10) | 90 |
| γ/deg | 90 | 90 | 77.846 (1) | 90 | 90 |
| V/Å3 | 3585.0 (2) | 3523.7 (1) | 2119.1 (2) | 4003.6 (2) | 21,872 (2) |
| Z | 4 | 4 | 2 | 4 | 4 |
| F(000) | 1560 | 1584 | 1056 | 2328 | 9184 |
| μ (Mo Kα)/mm−1 | 0.931 | 0.950 | 1.520 | 8.262 | 3.787 |
| Diffrns collected | 28,736 | 25,843 | 80,051 | 31,375 | 101,295 |
| Independent diffrns | 8239 | 4054 | 9744 | 4596 | 10,972 |
| Observed diffrns a | 6050 | 3492 | 8989 | 4430 | 8548 |
| Rintb/% | 3.23 | 3.30 | 2.53 | 2.15 | 4.47 |
| No. of param. | 442 | 227 | 505 | 241 | 524 |
| Rc obsd diffrns/% | 3.62 | 3.27 | 2.12 | 1.67 | 3.61 |
| R, wRc all data/% | 5.82, 9.43 | 3.99, 8.72 | 2.43, 5.15 | 1.79, 3.59 | 5.51, 10.1 |
| Δρ/e Å−3 | 0.48, −0.35 | 0.95, −0.25 | 0.65, −0.79 | 1.16, −0.56 | 2.29, −3.45 |
a Diffractions with I > 2σ(I). b Definitions: Rint = Σ|Fo2 − Fo2(mean)|/ΣFo2, where Fo2(mean) denotes the average intensity of symmetry-equivalent diffractions. c R = Σ||Fo| − |Fc||/Σ|Fo|, wR = [Σ{w(Fo2 − Fc2)2}/Σw(Fo2)2]½.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schulz, J.; Leitner, Z.; Císařová, I.; Štěpnička, P. Synthesis and Coordination Behavior of a Flexible Bis(phosphinoferrocene) Ligand. Molecules 2018, 23, 2054. https://doi.org/10.3390/molecules23082054
AMA Style
Schulz J, Leitner Z, Císařová I, Štěpnička P. Synthesis and Coordination Behavior of a Flexible Bis(phosphinoferrocene) Ligand. Molecules. 2018; 23(8):2054. https://doi.org/10.3390/molecules23082054
Chicago/Turabian StyleSchulz, Jiří, Zdeněk Leitner, Ivana Císařová, and Petr Štěpnička. 2018. "Synthesis and Coordination Behavior of a Flexible Bis(phosphinoferrocene) Ligand" Molecules 23, no. 8: 2054. https://doi.org/10.3390/molecules23082054