Linear Triquinane Sesquiterpenoids: Their Isolation, Structures, Biological Activities, and Chemical Synthesis


1
School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China
2
School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(9), 2095; https://doi.org/10.3390/molecules23092095
Submission received: 22 July 2018
/
Revised: 17 August 2018
/
Accepted: 19 August 2018
/
Published: 21 August 2018
(This article belongs to the Special Issue Natural Product Isolation, Identification and Biological Activity)
Abstract
:Linear triquinane sesquiterpenoids represent an important class of natural products. Most of these compounds were isolated from fungi, sponges, and soft corals, and many of them displayed a wide range of biological activities. On account of their structural diversity and complexity, linear triquinane sesquiterpenoids present new challenges for chemical structure identification and total synthesis. 118 linear triquinane sesquiterpenoids were classified into 8 types, named types I–VIII, based on the carbon skeleton and the position of carbon substituents. Their isolation, structure elucidations, biological activities, and chemical synthesis were reviewed. This paper cited 102 articles from 1947 to 2018.
1. Introduction
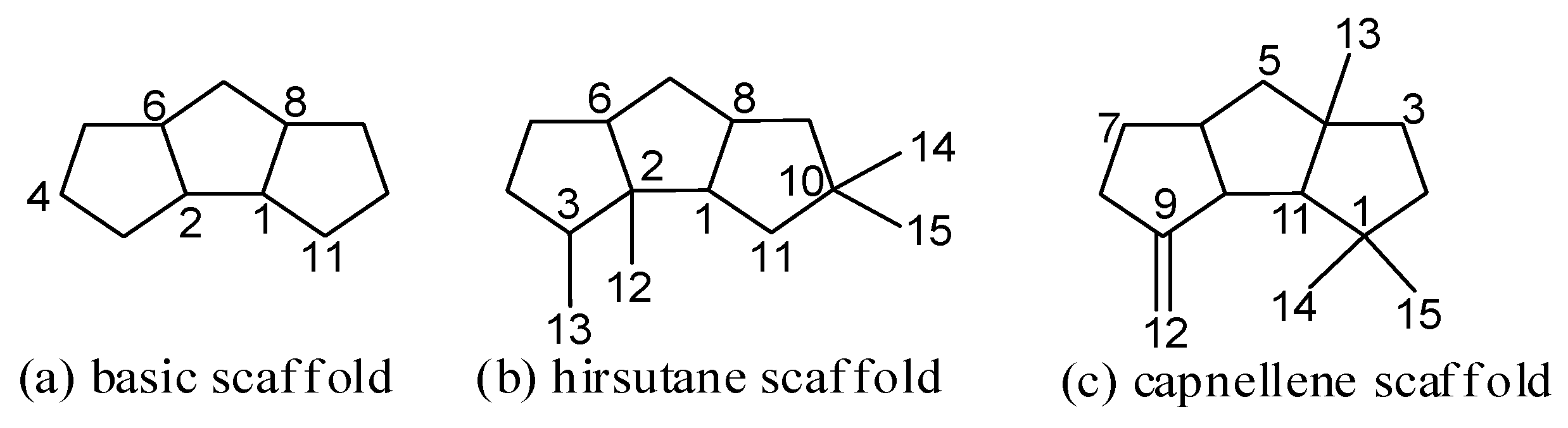
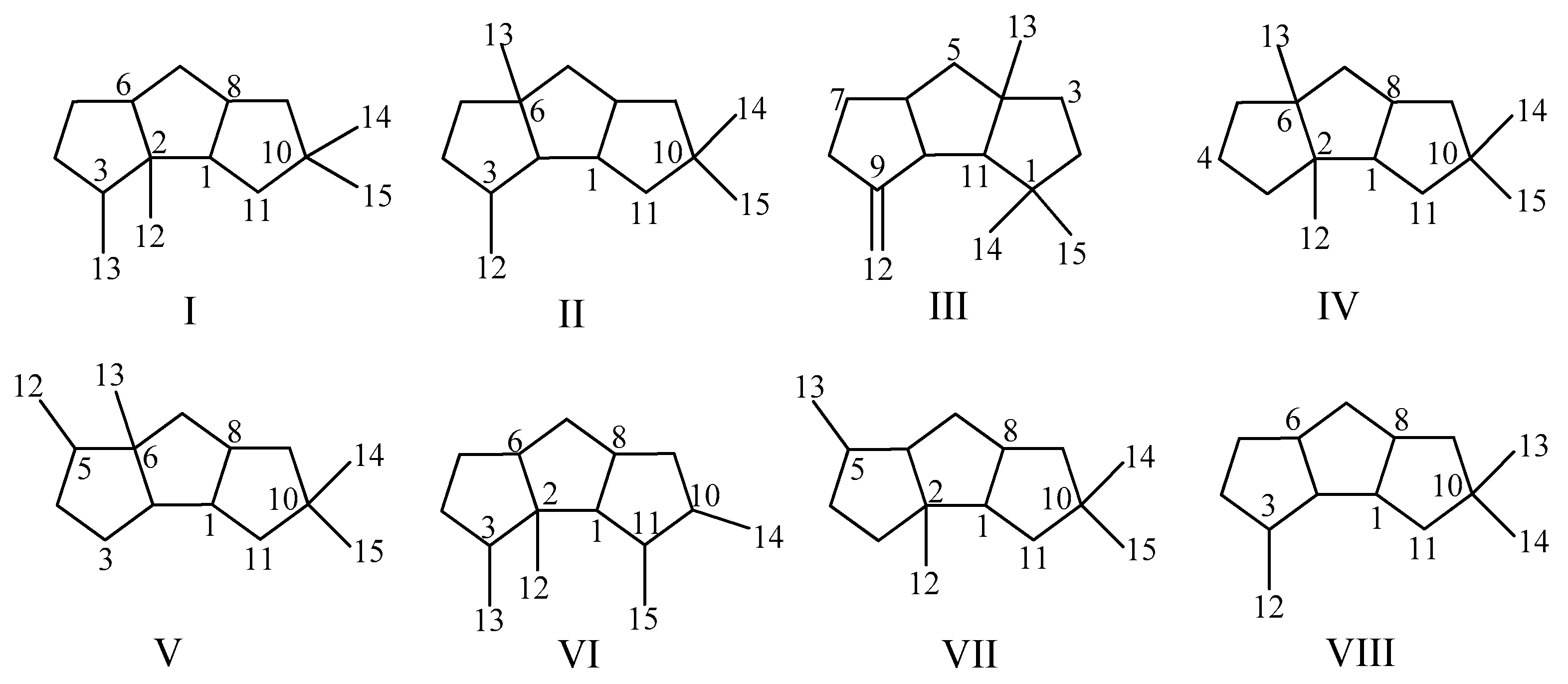
Sesquiterpenoids represent an important class of natural products. Among them, linear triquinane sesquiterpenoids have a basic skeleton 1H-cyclopenta[a]pentalene (Figure 1a) [1]. Since the first linear triquinane sesquiterpenoid, named hirsutic acid C obtained in 1947 [2,3], more than 100 linear triquinane sesquiterpenoids have been isolated, mainly from fungi, sponges, and soft corals. Many compounds displayed a wide range of biological activities, such as cytotoxic, antimicrobial, and anti-inflammatory activities [4,5,6,7]. Hirsutanes and capnellenes are the most common scaffolds of the linear triquinane sesquiterpenoids (Figure 1b,c) [8]. However, many compounds cannot be simply classified into these two types. Here, we categorize the linear triquinane sesquiterpenoids into eight types, types I–VIII, according to the carbon skeleton and the position of carbon substituents (Figure 2). This review gives an overview about the isolation, structure, biological activities, and chemical synthesis of linear triquinane sesquiterpenoids.
2. Isolation and Structure Elucidation
2.1. Type I
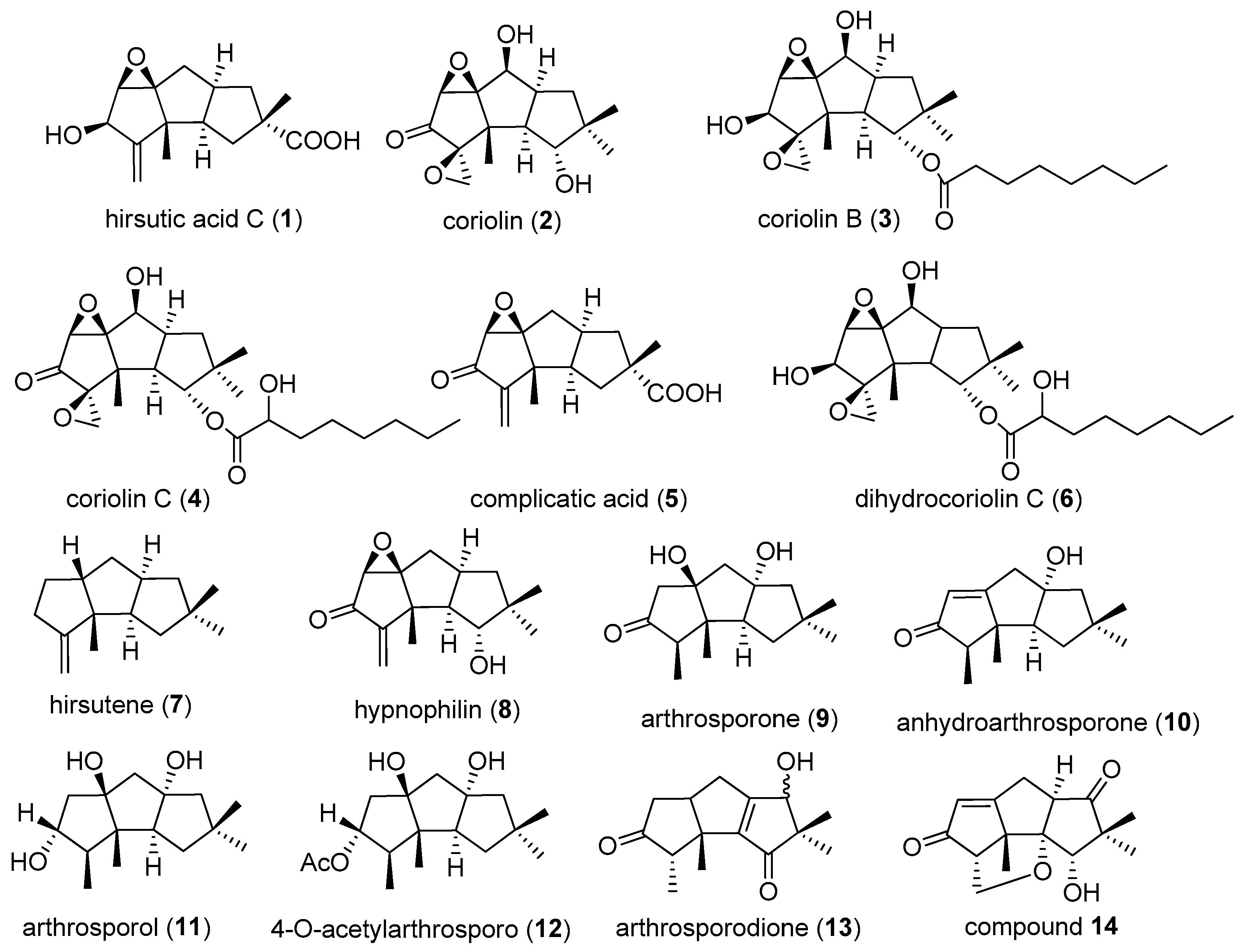
There are four carbon substituents at C-2, 3, 10, 10 in type I compounds (Figure 3). A total of 76 compounds, constitute the largest classification of linear triquinane sesquiterpenoids. Hirsutic acid C (1), was the first linear triquinane sesquiterpenoids isolated from the cultures of Stereum hirsutum in 1947 [2,3]. The antibiotic coriolin (2), coriolin B (3), C (4), and hirsutene (7) were purified from the mycelia of basidiomycetes Coriolus consors [9,10,11]. However, the initial structures of coriolins were not exactly right, afterwards their structures and stereochemistry were revised by the same research group based on spectroscopic analysis, biogenetic consideration and chemical transformations, in 1971 [12]. The revised structures of coriolins showed that they were hirsutane-type sesquiterpenes. (−)-Complicatic acid (5) was first isolated from the fungus Stereum complicatum in 1973 [13]. 5-Dihydrocoriolin C (6) was obtained from Coriolus consors (ATCC 11574) [14].
Hypnophilin (8) was isolated from mycelial cultures of Pleurotellus hypnophilus (Berk.) Sacc. in 1981 [15,16]. Six new hirsutane metabolites, arthrosporone (9), anhydroarthrosporone (10), arthrosporol (11), 4-O-acetylarthrosporo (12), arthrosporodione (13), and compound 14 were obtained from fungus UAMH 4262 by Amouzou in 1989 [4]. Incarnal (15) was purified from the culture fluid of Gloeostereum incarnatum in 1991 [17]. In 1992, gloeosteretriol (16), also named hirsutanol F was obtained from the fluid fermentation products of Gloeostereum incarnatum [18]. From the culture extract of the fungus Lentinus crinitus, 1-desoxy-hypnophilin (17), and hypnophilin (8) were isolated as the most active compounds. In addition to these ketones, their corresponding alcohols 6,7-epoxy-4(15)-hirsutene-5-ol (18) and diol 4 (19) were obtained in 1994 [19].
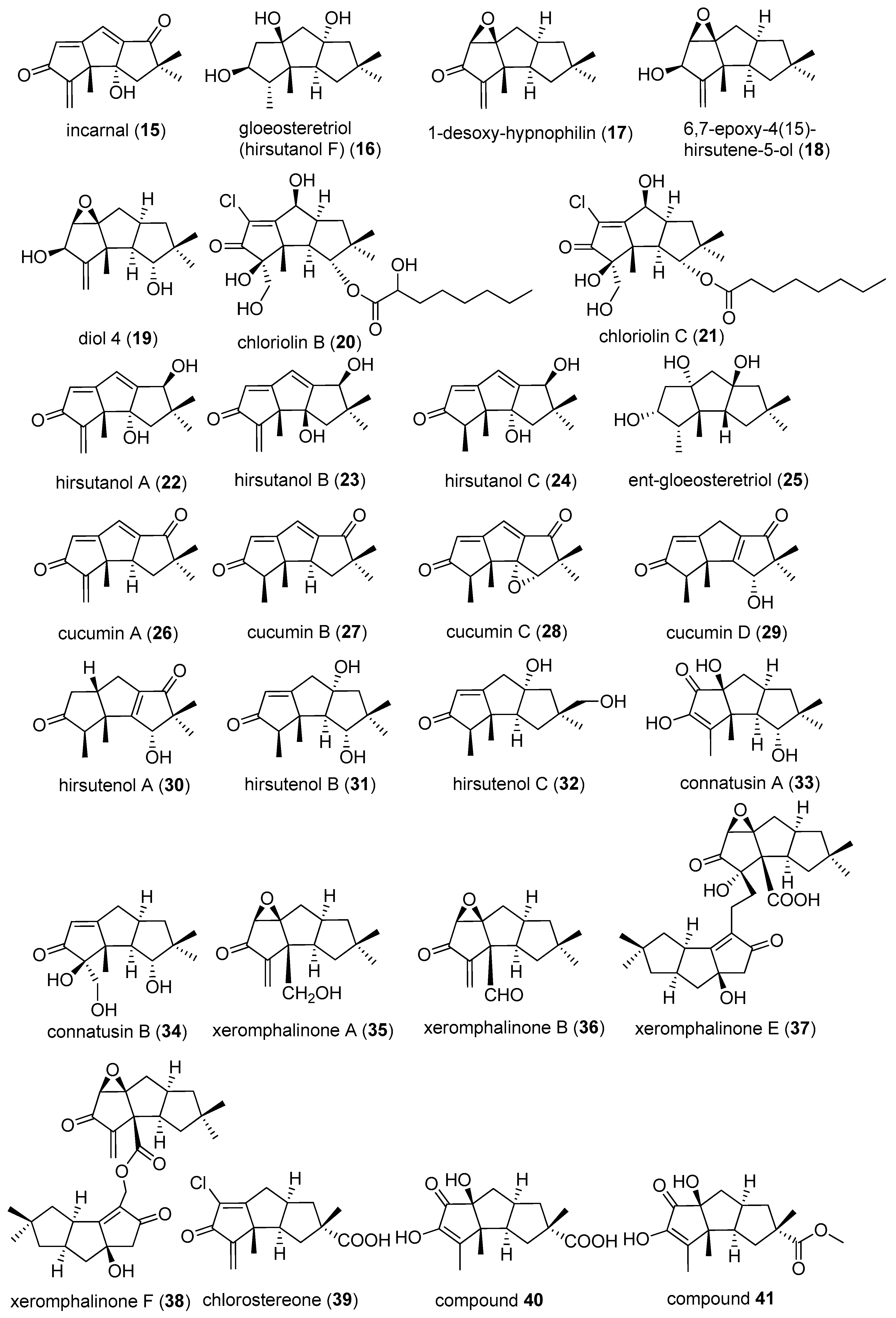
Chloriolin B (20) and C (21) were purified from an unidentified fungus, which was separated from a marine sponge Jaspis aff. johnstoni [20].
Hirsutanols A–C (22–24) and entgloeosteretriol (25) were obtained from an unidentified fungus, separated from an Indo-Pacific sponge Haliclona sp. in 1998, and study showed that hirsutanol B was an epimer of hirsutanol A [5]. In 1998, from mycelial cultures of the agaric Macrocystidia cucumis, cucumis A–D (26–29) were found together with arthrosporone (9) [21]. Hirsutenols A–C (30–32) were isolated in 2002, from the culture broth of the mushroom Stereum hirsutum [22]. Connatusins A (33) and B (34) were isolated from the fungus Lentinus connatus BCC 8996, in 2005 [23].
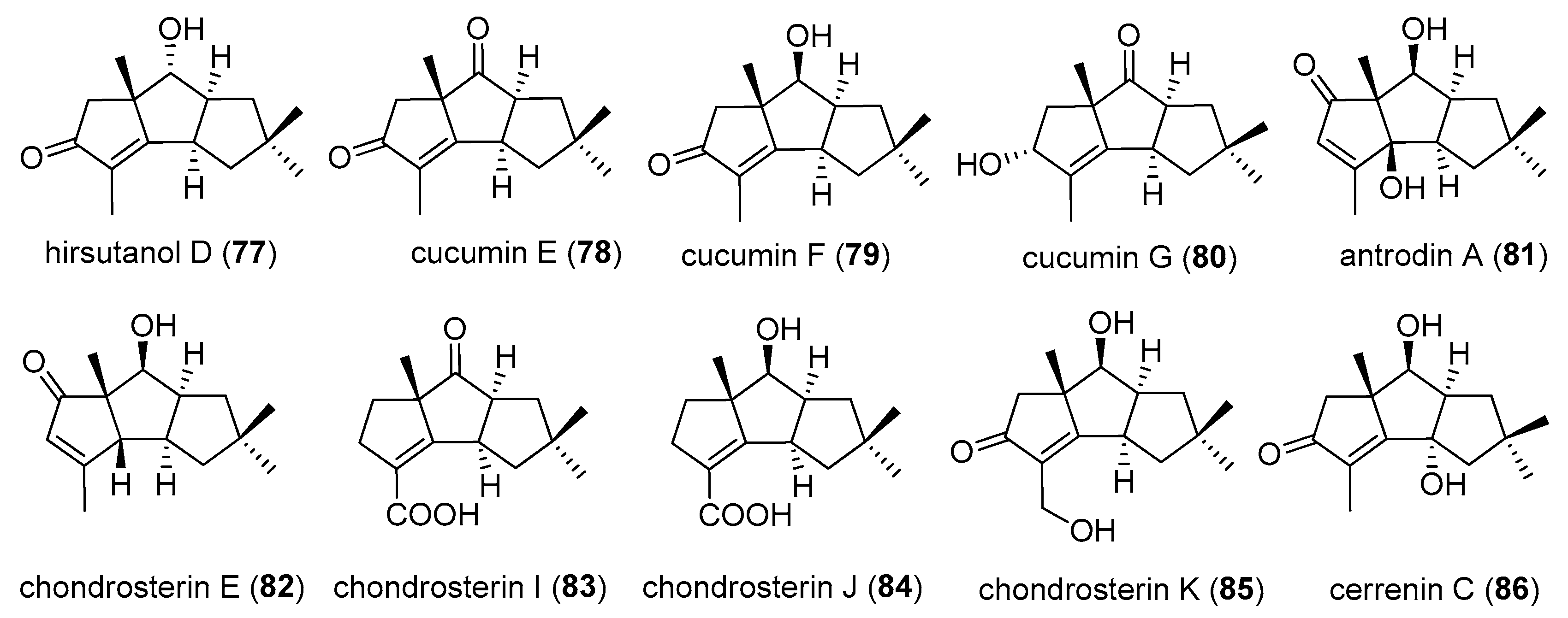
Xeromphalinones A, B, E, F (35–38), were isolated from Xeromphalina sp. by Liermann et al. in 2010. At the same time, they isolated chlorostereone (39) from Stereum sp. [24]. In 2014, Ma and co-workers found three new sesquiterpenoids from the mycelia of Stereum hirsutum. Since they did not name these compounds at that time, we called them compounds 40–42 in proper order, successively [25].
Li and co-workers have studied Chondrostereum sp., which was separated from the soft coral Sarcophyton tortuosum, from the South China Sea. Their investigations found that the metabolites of Chondrostereum sp., were varied depending on the culture media. By altering the fermentation conditions, e.g., carbon and nitrogen source, Chondrostereum sp. can produce highly functionalized hirsutane type compounds with a surprising chemodiversity [26]. From the PDB (potato dextrose broth) medium, chondrosterins A–D (43–46) [26] were isolated, while hirsutanol E (47) [27], chondrosterins L–O (48–51) [28,29] were obtained from GPY (glucose-peptone-yeast) medium.
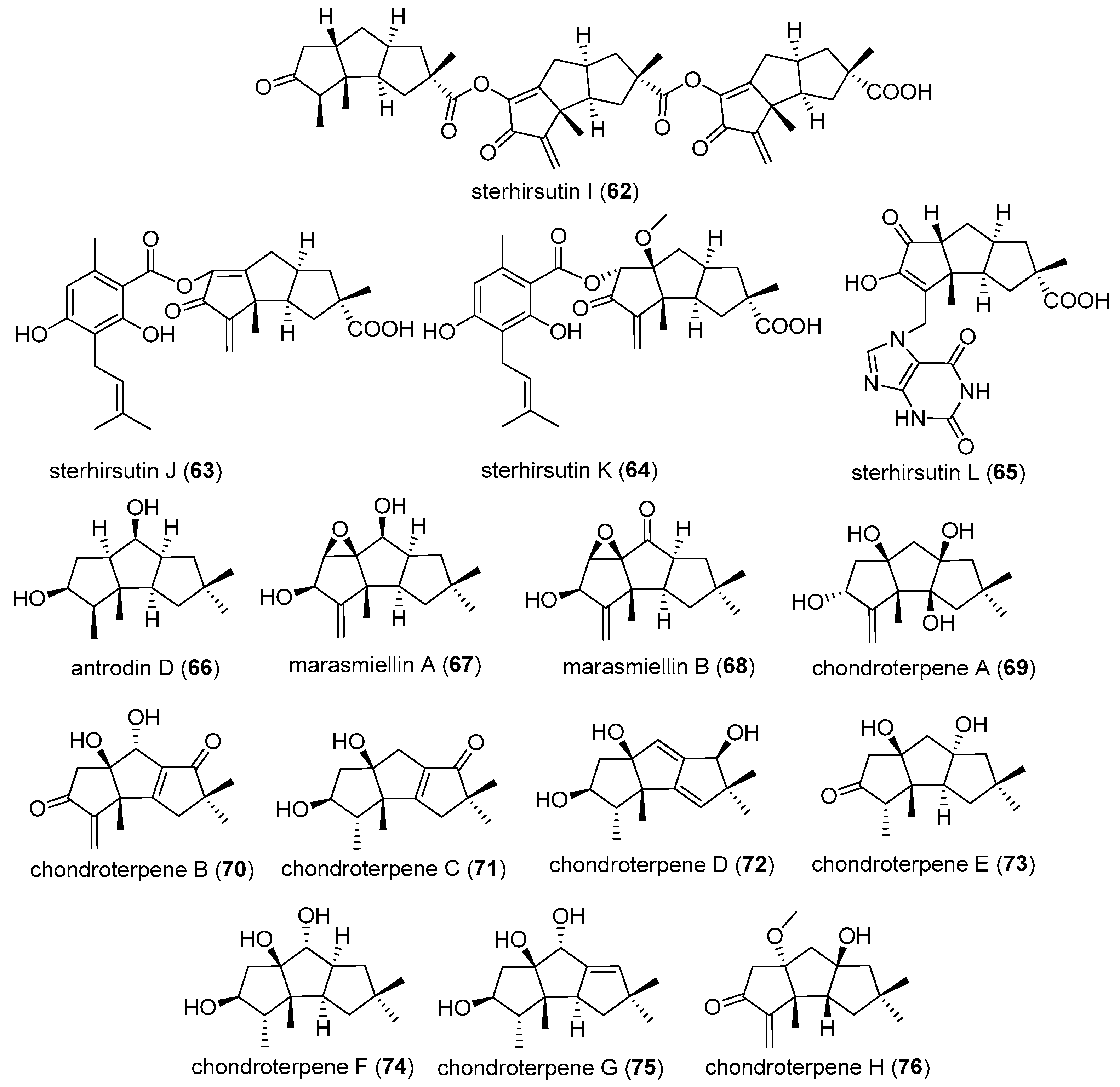
Aiming at finding new secondary metabolites, with bioactivities from fungi collected in the region of Tibet Plateau, a strain of Stereum hirsutum (L515) was isolated from its fruiting body. Sterhirsutins A (52) and B (53), hirsutic acids D (54) and E (55), were isolated from this strain’s solid-substrate fermentation culture in 2014 [30]. Additionally, in 2015, from its ethyl acetate extract, sterhirsutins C–L (56–65) were obtained [31]. Sterhirsutin L was the first sesquiterpene coupled with a xanthine moiety.
Antrodin D (66) together with coriolin (2), dihydrocoriolin C (6), and chloriolin B (20), were isolated from the fermentation of the basidiomycete Antrodiella albocinnamomea in 2015 [32].
Scale-up fermentation and chemical studies of basidiomycetes Marasmiellus sp. BCC 22389 led to the isolation and characterization of marasmiellins A (67) and B (68) in 2016. Marasmiellins are structurally, closely related to coriolins [33].
Separation of the active principles of the fermented broth of a fungus Chondrostereum sp. NTOU4196 which was isolated from the marine red alga Pterocladiella capillacea, resulted in the isolation of chondroterpenes A–H (69–76) in 2017 [34].
2.2. Type II
There were 10 compounds belonging to type II, with four carbon substituents at C-3, 6, 10, 10 (Figure 4). 9 compounds a have carbonyl group.
Hirsutanol D (77) was produced from the terrestrial fungus Corious consors, cultured under both sea water and deionized water media, in 1998 [31]. Cucumins E–G (78–80) were isolated from mycelial extract of the agaric Macrocystidia cucumis, in 1998 [21]. From the fermentation of basidiomycete Antrodiella albocinnamomea, antrodin A (81) was obtained, in 2015 [32]. Chondrosterins E, I–K (82–85) were purified from the marine fungus Chondrostereum sp. [26,28,35]. Cerrenin C (86) was obtained from the broth extract of Cerrena sp. A593 [36].
2.3. Type III
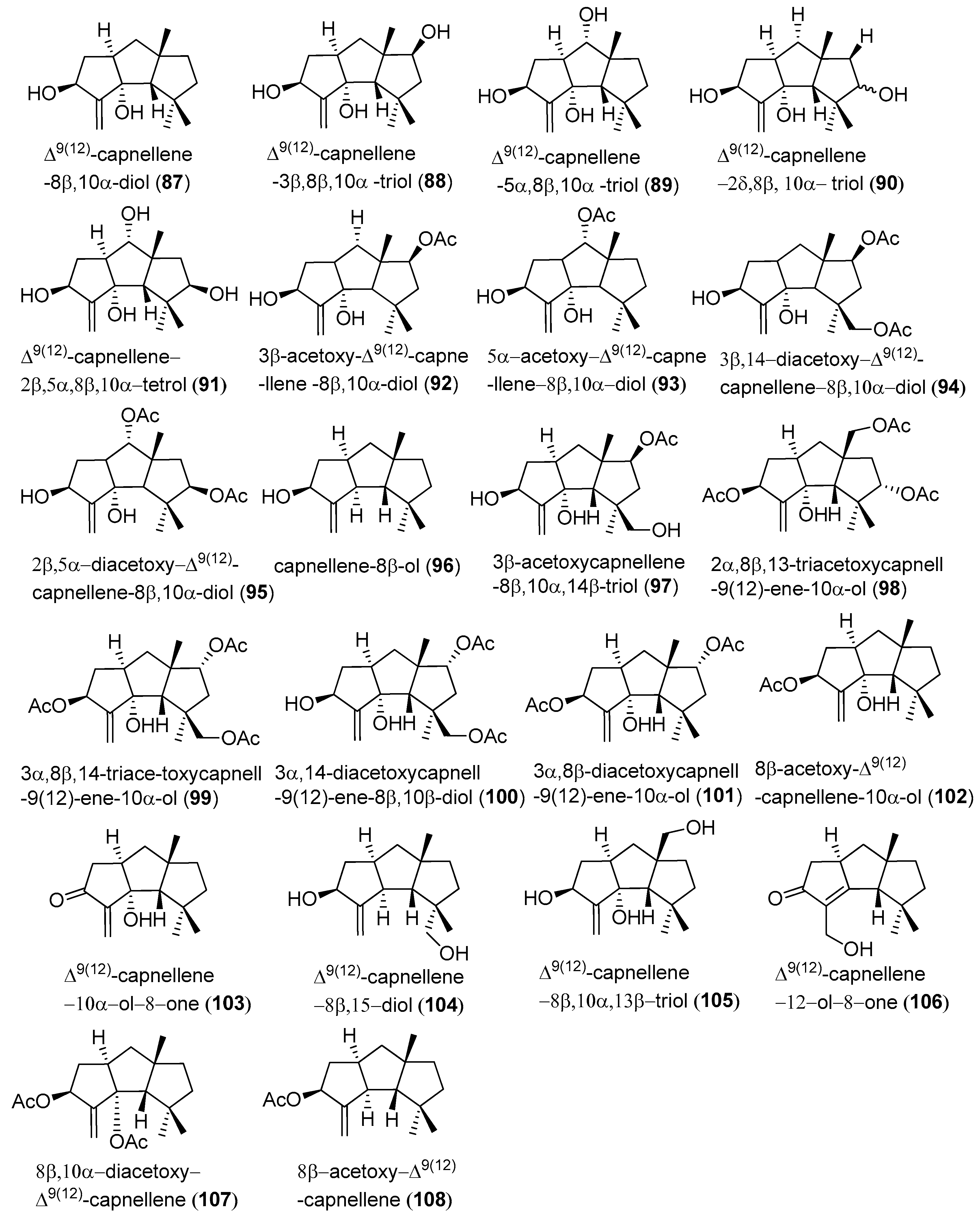
In type III compounds, there are four carbon substituents in C-1, 1, 4, 9 (Figure 5). The most notable feature of these compounds is they contain an off-ring carbon-carbon double bond at C-9.
∆9(12)-capnellene-8β,10α-diol (87), ∆9(12)-capnellene-3β, 8β, 10α-triol (88), ∆9(12)-capnellene-5α,8β,10α-triol (89), and ∆9(12)-capnellene-2ξ,8β,10α-triol (90), were obtained from the soft coral Capnella imbricata, by Sheikh et al. These compounds were the first members of a new sesquiterpene class, which was named capnellane [37]. ∆9(12)-Capnellene-2β,5α,8β,10α-tetrol (91), has been isolated from the alcyonarian Capnella imbricate, by Kaisin et al. Its structure and relative configuration was determined by X-ray diffraction analysis, in 1979 [38]. In 1985, Kaisin and co-workers implemented the isolation of other eight acetylated capnellenes from acetone extraction of fresh colonies of C. imbricate. Four of these compounds were named as 3β-acetoxy-∆9(12)-capnellene-8β,10α-diol (92), 5α-acetoxy-∆9(12)-capnellene-8β,10α-diol (93), 3β,14-diacetoxy-∆9(12)-capnellene-8β,10α-diol (94), and 2β,5α-diacetoxy-∆9(12)-capnellene-8β,10α-diol (95) [39].
In 1998, capnellene-8β-ol (96) and 3β-acetoxycapnellene-8β, 10α,14β-triol (97) were isolated from Capnella imbricata, collected from Mayu Island, Molucca Sea, Indonesia, in 1996 [40]. 2α,8β,13-Triacetoxycapnell-9(12)-ene-10α-ol (98), 3α,8β,14-triace-toxycapnell-9(12)-ene-10α-ol (99), 3α,14-diacetoxycapnell-9(12)-ene-8β,10α-diol (100), and 3α,8β-diacetoxycapnell-9(12)-ene-10α-ol (101), were obtained from the soft coral Dendronephthya rubeola, in 2007 [6].
From the acetone/methylene chloride extracts of the Formosan soft coral Capnella imbricate, 8β-acetoxy-∆9(12)-capnellene-10α-ol (102), ∆9(12)-capnellene-10α-ol-8-one (103), ∆9(12)-capnellene-8β,15-diol (104), ∆9(12)-capnellene-8β,10α, 13β-triol (105), Δ9(10)-capnellene-12-ol-8-one (106), 8β,10α-diacetoxy-∆9(12)-capnellene (107), and 8β-acetoxy-∆9(12)-capnellene (108), were purified in 2008 [7].
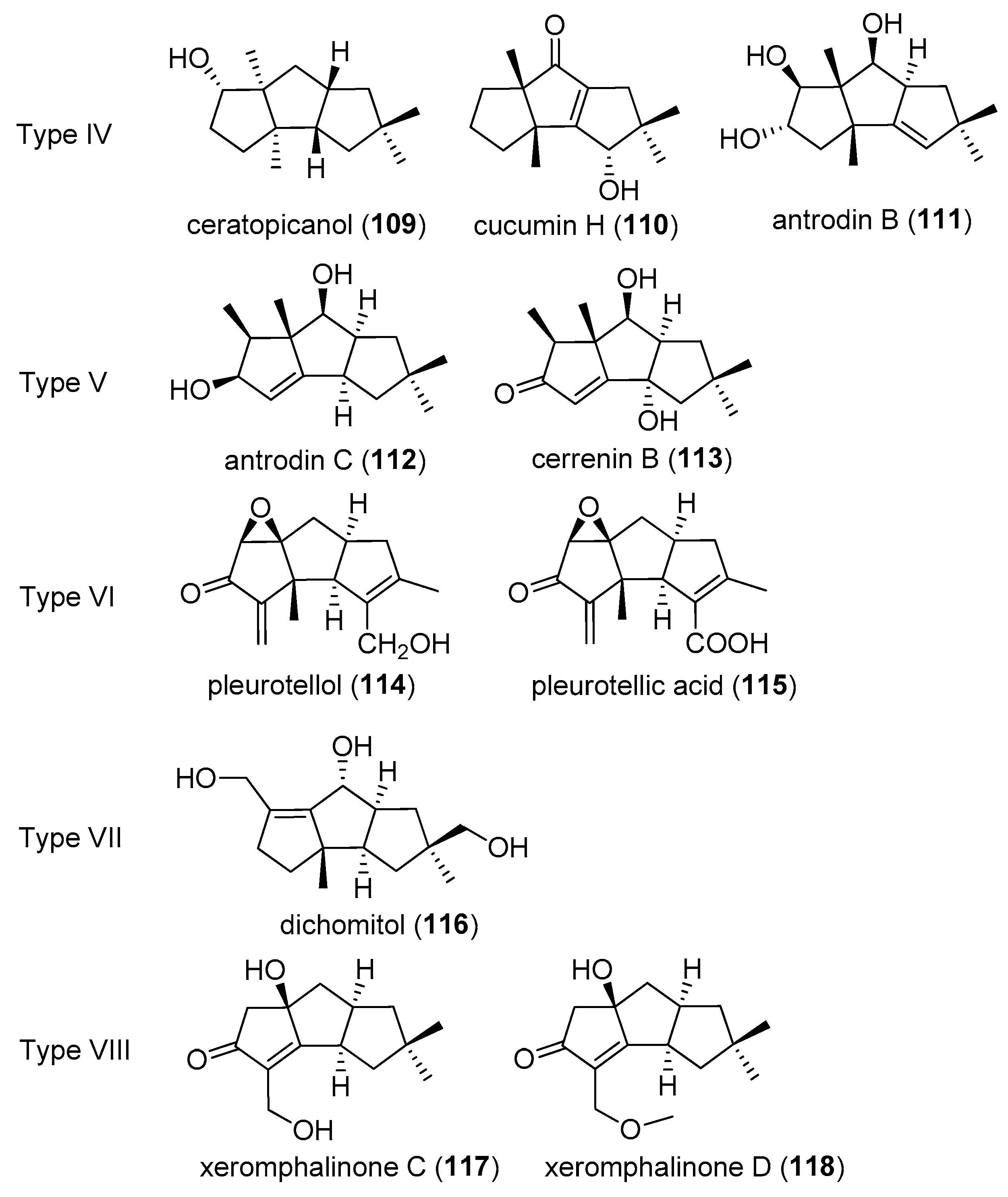
2.4. Type IV, V, VI, VII and VIII
The number of type IV–VIII compounds (Figure 6), is relatively small. It is worth mentioning that type VIII compounds, have one less carbon substituent on the skeleton than other compounds.
Type IV: Ceratopicanol (109) was isolated from the fungus Ceratosystis piceae, in 1988. Ceratosystis piceae (Munch) Bakshi, is a sapwood staining ascomycete, occurring on coniferous loga and lumber [41]. Cucumin H (110) was found in 1998, from mycelial cultures of the agaric Macrocystidia cucumis [21]. From fermentation of Antrodiella albocinnamomea, antrodin B (111) was isolated in 2015 [32].
Type V: Antrodin C (112), was obtained from fermentation of Antrodiella albocinnamomea, in 2015 [32]. Cerrenin B (113) was isolated from the broth extract of Cerrena sp. A593, in 2018 [36].
Type VI: Pleurotellic acid (114) and pleurotellol (115) were isolated from mycelial cultures of Pleurotellus hypnophilus (Berk.), in 1986 [15,16].
Type VII: Dichomitol (116), was purified from mycelial solid cultures of Dichomitus squalens, which was a commonly found white-rot basidiomycete fungus, in 2004 [42].
Type VIII: Xeromphalinones C (117) and D (118), were obtained from Xeromphalina sp. by Liermann et al., in 2010 [24].
From the perspective of source, linear triquinane sesquiterpenoids can be isolated from both marine and terrestrial organisms, however, the proportion of terrestrial species is relatively larger. It is worthwhile to note that, there are several highly productive biological species, such as the mushroom Stereum hirsutum, the soft coral Capnella imbricata, and marine fungus Chondrostereum sp.
Structurally, there are 76 compounds belonging to type I, accounting for the majority of these 118 compounds, and 22 compounds constitute type III. In fact, it was most common to categorize linear triquinanes, according to their ring scaffold with the hirsutanes and capnellenes. Type I compounds, exactly belong to hirsutanes, while type III belong to capnellenes.
As for functional groups, the epoxy bond is easy to form at C-6, 7 positions and the majority of carbonyls are formed at C-4, C-7, and C-9 positions, with a minority of carbonyls formed at C-5. As regards the hydroxyl group, except for C-10, it is possible to form at the other 10 positions.
Additionally, Kutateladze and Kuznetsov used the relatively fast parametric/DFT hybrid computational method DU8+, to analyze the reported NMR data for 90+ natural triquinanes in 2017, which resulted in requiring structure correction of 13 compounds, including 10 linear triquinane sesquiterpenoids, noted in this review [43]. However, this review still used the structures from the original literatures, because only the calculations were not convincing enough.
3. Biological Activity
3.1. Cytotoxicity
Cytotoxicity was a kind of common biological activity reported for linear triquinane sesquiterpenoids, usually expressed as inhibitory concentration (IC50), 50% of the effective dose (ED50), cytotoxic concentration (CC50), lethal concentration (LC50), or growth inhibition (GI50), this value emphasizes the correction for the cell count at time zero) [1]. Among the 118 linear triquinane sesquiterpenoids, many compounds exhibited activities against cancer cell lines (Table 1).
Coriolin B (3) inhibited human breast cancer cell T-47D and CNS cancer cell line SNB-75, with the IC50 values of 0.7 and 0.5 μM, respectively [20].
Toxicity assays were carried out with L929 mouse fibroblasts cells. The IC50 values were 2.4 μg/mL for 1-desoxy-hypnophilin (17) and 0.9 μg/mL for 6,7-epoxy-4(15)-hirsutene-5-ol (18) [19].
Cucumins A–C (26–28) were cytotoxic, with IC100 range from 0.5 to 1 mg/mL for L1210 cells [21]. The cytotoxicities (IC50 value) of xeromphalinones A (35), B (36), F (38) and chlorostereone (39) against Jurkat cells, ranged from 1 to 5 μg/mL (2 to 20 μM). For xeromphalinones C–E (117–118, 37), the IC50 values were above 50 μg/mL [24].
Compound 40 inhibited the growth of HepG2 cells, with IC50 value of 24.41 ± 1.86 μM [25]. Chondrosterin A (43) showed significant cytotoxicity against cancer lines A549, CNE2, and LoVo, with IC50 values of 2.45, 4.95, and 5.47 Μm, respectively, whilst chondrosterins B–E (44–46, 48) were apparently inactive (IC50 > 200 μM), in the assay developed by Li [26]. Additionally, in Hsiao and co-workers’ investigation, chondrosterins A (43) and B (44) and chondroterpene H (76), showed potent cytotoxic activities on BV-2 cells, at a concentration of 20 μM [34]. Chondrosterin J (84) showed potent cytotoxic activities against the cancer cell lines CNE1 and CNE2, with the IC50 values of 1.32 and 0.56 μM, respectively. By comparison, chondrosterin J (84) showed stronger cytotoxic activities than chondrosterin A (43) (CNE2: 4.95 μM), hirsutanol A (22) (CNE1:10.08 μM; CNE2: 12.72 μM), and incarnal (15) (CNE1: 34.13 μM; CNE2: 24.87 μM). In contrast, chondrosterin I (83) was inactive (IC50 values > 200 μM) [35]. Yang and co-workers have investigated the molecular mechanisms of the antitumor activity of hirsutanol A, with the result that hirsutanol A inhibited tumor growth, through triggering ROS production and apoptosis [44]. Chondrosterins K–M (85, 48–49) showed cytotoxicities against seven cancer cell lines CNE1, CNE2, HONE1, SUNE1, A549, GLC82, and HL7702, in vitro [28].
Sterhirsutins A–B (52–53) and hirsutic acid D–E (54–55) showed cytotoxicity against K562 and HCT116 cell lines. They exhibited cytotoxicity against K562 cells, with the IC50 values of 12.97, 16.29, 6.93, and 30.52 μg/mL, respectively. For the HCT116 cell line, they showed cytotoxicity with the IC50 values of 10.74, 16.35, 25.43, and 24.17 μg/mL [30]. Sterhirsutins E–G (58–60) and I (62) also showed antiproliferative activity against K562 and HCT116 cancer cells, with IC50 values ranging from 6 to 20 μM.
Compound 87 was cytotoxic toward human promyelocytic leukemia cell line HL-60, human erythroleukaemia cell line K562, human renal leiomyoblastoma cell line G402, human breast adenocarcinoma cell line MCF-7, human colon-cancer cell line HT 115, and human ovarian cancer cell line A2780, with IC50 values in the range of 0.7–93 μM, and the greatest activity was displayed against K562 at 0.7 μM. Compound 95 showed some selectivity, for the renal leiomyoblastoma and ovarian cancer cell lines. Compound 96 was effective against the K562, as well as HL-60 [40].
Compounds 98–101 showed weak cytotoxic activities against the human cervix carcinoma cell line HeLa, but they also showed weak or moderate antiproliferative effects, against the cell lines K562 and murine fibroblast cell line L-929 [6].
Compounds 87 and 102 exhibited strong activity against HeLa and L-929. Compounds 87, 101, 102 showed similar anti-proliferative activities against K562. The anti-proliferative and cytotoxic activity of compound 84 against L-929 and HeLa cells, is 5.7 and 3.8 times lower than doxorubicin, which was used in the cancer therapy for the treatment of lymphoma, carcinoma, sarcoma, and leukemia [6].
3.2. Antimicrobial Activity
Coriolin (2) showed inhibitory effect on the growth of Gram-positive bacteria at 6.25–12.5 mcg/mL, some of Gram-negative bacteria at 50–100 mcg/mL [9].
(−)-Complicatic acid (5) was against a range of Gram-positive and Gram-negative bacteria [42]. Compounds 9–11 displayed weak antifungal activity [4]. Incarnal (15) inhibited the growth of Gram-positive bacteria at 6.25–12.5 µg/mL, while it had no antibacterial activity against Gram-negative bacteria [17].
3.3. Other Bioactivity
Compounds 22, 43–44, 69–70, and 76, inhibited lipopolysaccharide (LPS)-induced NO production in BV-2 microglial cells, at a concentration of 20 μM. Compounds 22 (5–20 μM) and 69, had less effects on cell viability. At the same concentration, 24 and 70–75 had little effect on viabilities. Only 22, with an α-exomethylene carbonyl moiety, was more potent than 24, on inducing cellular toxicity. Compounds 43 and 76, reduced cellular viability, more than 22. Compounds 43, 44 (20 μM), and 76, all decreased the NO levels to the resting condition. These results suggested that cell death, predominantly caused decreasing NO production. However, the decreased NO levels by 22 (5 μM) and 69, were dependent on inhibition of NO production instead of cell death [34] (Table 3 and Table 4).
Compound 40 exhibited NO inhibitory effect, on the LPS-induced macrophages, with the IC50 value of 15.44 ± 1.07 μM [25].
Sterhirsutin G (60) showed inhibitory activity on the activation of IFNβ promoter at a dose of 10 μM in HEK293 cells, whereas other compounds exhibited no apparent inhibitory effect on the IFNβ promoter activation, under the same experimental conditions. Furthermore, 60 dramatically reduced the SeV-induced mRNA level of IL6, RANTES, and IFNB1. Compared with 60, sterhirsutin E (58) showed no inhibitory activity on the IFNβ promoter activation. These findings implied that sterhirsutin G, has the ability to inhibit the RLRs-mediated antiviral signaling in cells, and was potent to be a leading compound in the treatment of autoimmune diseases [31].
Sterhirsutin K (64) induced autophagy in HeLa cells, and its autophagy-inducing effect was evaluated via quantification of the number of GFP-LC3 dots in cells. Sterhirsutins A–L (52, 53, 56–65) and hirsutic acids D (54) and E (55), were screened using GFP-LC3 stable HeLa cells. As a result, 64 was found to exhibit strong autophagy-inducing activity at a dose of 50 μM. In comparison with hirsutic acid E (55), sterhirsutin J (63) and K (64) showed cytotoxicity rather than autophagy-inducing effect, on GFP-LC3 stable HeLa cells, at the concentration of 50 μM (inhibition rate = 100%). The double bond between C-6 and C-7 in 55 and 63 increased cytotoxicity, but significantly decreased the autophagy-inducing activity [31].
Compound 86 showed a specific inhibition (77%) of the Myc/Max interaction in yeast, and a lower inhibition of the Tax–CREB interaction (27%) and Myc–Miz-1 interaction (34%). This specific inhibition of Myc–Max interaction, is well associated with its antiproliferative effect against the L-929 cell line [6].
Compounds 87, 102, and 103, significantly reduced the levels of the iNOS protein at concentrations of 10 μM, in comparison with control cells stimulated with LPS. 87 and 102 dramatically reduced the levels of the COX-2 protein at a dose of 10 μM, compared with control cells stimulated with LPS [7].
The anti-inflammatory effects of compounds 87, 96, and 103–109 were tested using LPS-stimulated cells. Stimulation of RAW 264.7 cells with LPS led to up-regulation of the pro-inflammatory iNOS and COX-2 proteins [7].
As a summary, among these 118 compounds, there were 56 compounds showing biological activities. Hirsutanes and capnellenes are major active compounds. They have cytotoxic, antimicrobial, anti-inflammatory activities, and so on. Previous reports revealed that the compounds with a α-methylidene oxo group, such as hypnophilin (8) and desoxyhypnophilin (17), possessed stronger antimicrobial or cytotoxic activities [26]. These structures and bioactivities are a source of inspiration, for synthetic chemists and pharmaceutist, to develop innovative methodologies in the field of medicine chemistry.
4. Chemical Synthesis
Linear triquinane sesquiterpenoids, have attracted a lot of interest among synthetic chemists in recent decades, owing to their premium structures and promising biological activities [45]. Their skeletal and functional diversities, also pose a considerable synthetic challenge [46].
4.1. Hirsutane Type
In the early 1970s, chemists began to synthesize hirsutane type compounds, in a variety of ways.
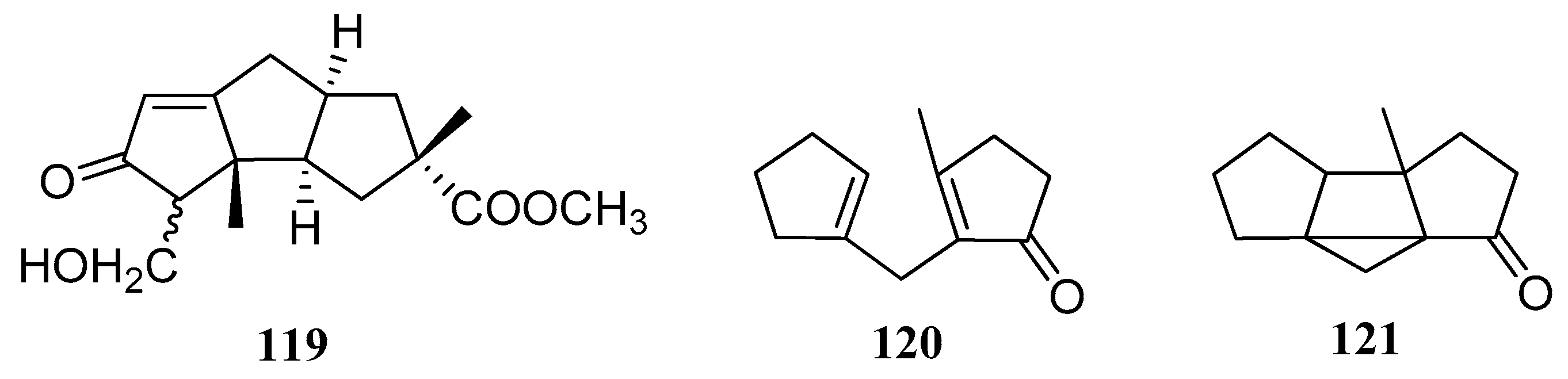
The total synthesis of hirsutic acid C (1), was achieved via the suitably functionalized skeleton compound 119 (Figure 7). In 1974, Hisanobu Hashimoto and co-workers reported a stereospecific conversion of 3α-hydroxymethylene ketone (compound 119) to racemic hirsutic acid [47,48]. In 1978, Kueh and co-workers [49] reported the synthesis of the hirsutane carbon skeleton. They found that the photochemical intramolecular [2 + 2] cycloaddition of dicyclopent-l-enylmethanes (120), followed by in situation of methanol to the strained cyclopropane ring in the presumed intermediate bicycle[2.1.0]pentanes (121), led to an easy synthesis of the ring system.
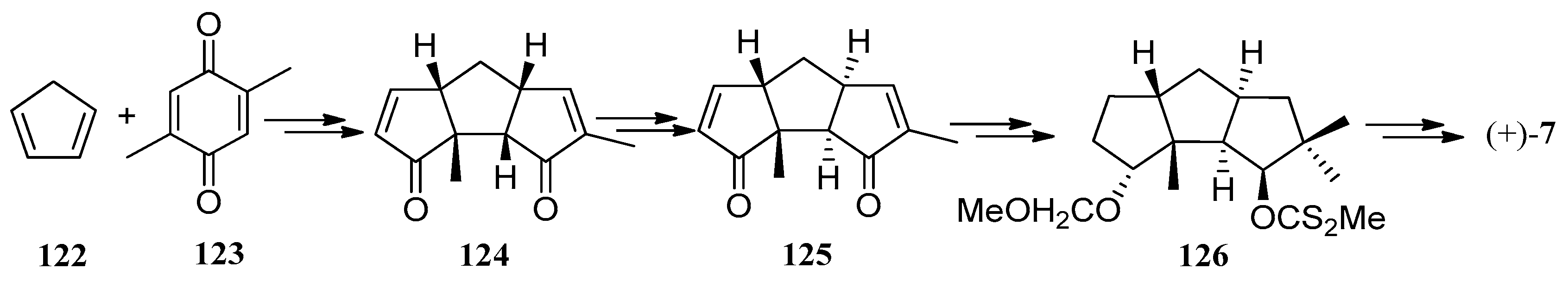
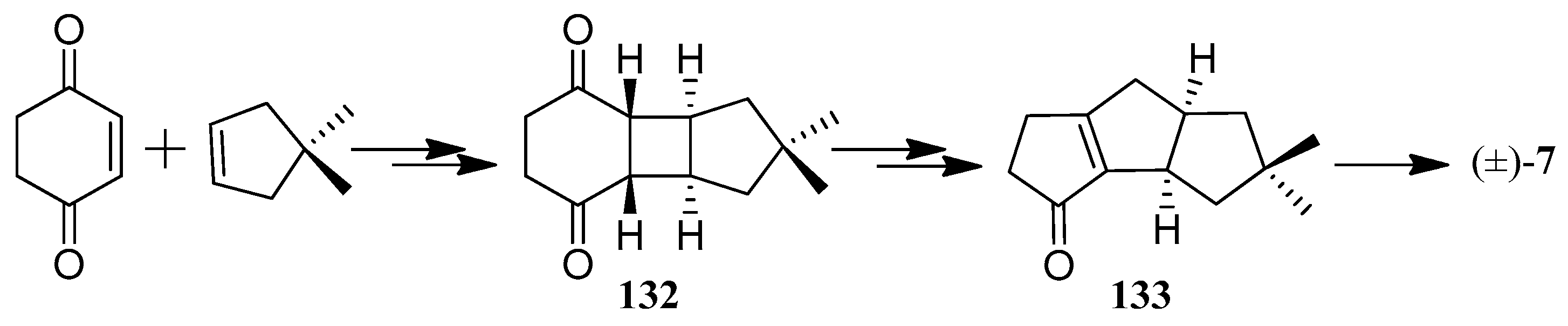
In 1981, Mehta and co-workers developed a simple expedient synthesis of (+)-hirsutene ((+)-7) via a novel photo-thermal metathetic sequence (Scheme 1). There were three distinctive features in this method: (1) the cis-syn-cis-C13-tricyclopentanoid frame (124) was efficiently obtained from cyclopentadiene and 2,5-dimethyl-p-benzoquinone, in three high-yielding steps employing only heat and light as the reagents; (2) compound 124 with the requisite cis-anti-cis system (125), was in ready and remarkable thermal equilibration; and (3) generation of abundant functionality on the tricyclopentanoid frame permits the synthesis of the higher oxygenated members of the hirsutane type compounds [50].
In 1984, Dawson and co-workers reported a simple stereocontrolled sequence to synthesize hirsutene from dicyclopentadiene which contained ring expansion by ketonization of a cyclopropoxide and skeletal rearrangement, through a β-enolate [51]. A primary feature of this synthesis, was that the stereochemistry at three of the four chiral centers was established essentially at the beginning of the sequence, and then the β-enolate rearrangement specifically generated the correct configuration at the fourth center, exclusively.
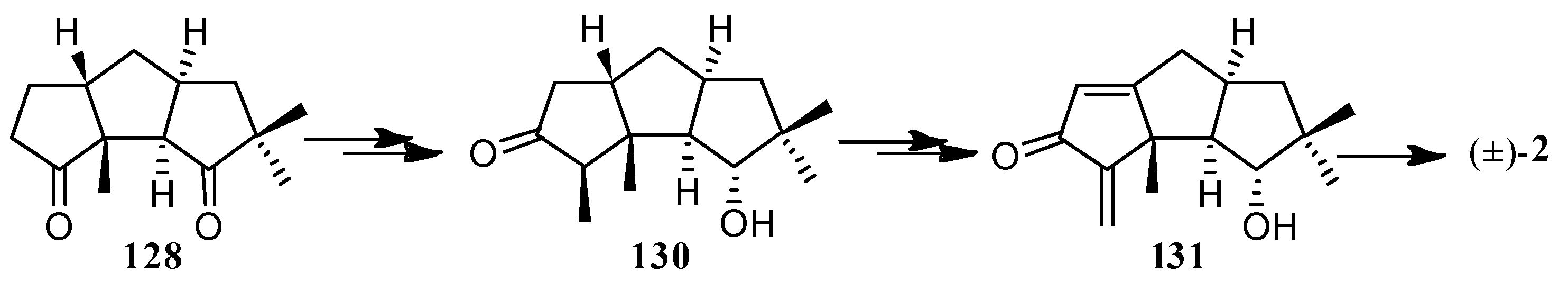
In 1986, Mehta and co-workers [52] developed a simple method for triquinane natural products, based on photo-thermal olefin metathesis of readily available Diels-Alder adducts. They also applied this approach to the total synthesis of (±)-hirsutene ((±)-7) (10 steps, Scheme 2) and (±)-coriolin ((±)-2) (17 steps, Scheme 3).
The trimethylsilyl iodide-mediated rearrangement of a bicyclo[4.2.0]octane-2,5-dione to a bicyclo[3.3.0]-oct-1(6)-en-2-one, was achieved by Oda and co-workers, for a short synthesis of (±)-hirsutene ((±)-7) (Scheme 4) [53].
The photochemical reduction of a δ, ε-unsaturated ketone, was a key step to synthesize (±)-hirsutene ((±)-7), reported by Cossy and co-workers (Scheme 5) [54].
Majetich and co-workers achieved a synthesis of (±)-hirsutene ((±)-7), via the intramolecular addition of two functionalized cyclopentane rings, linked by a one-carbon tether (Scheme 6) [55].
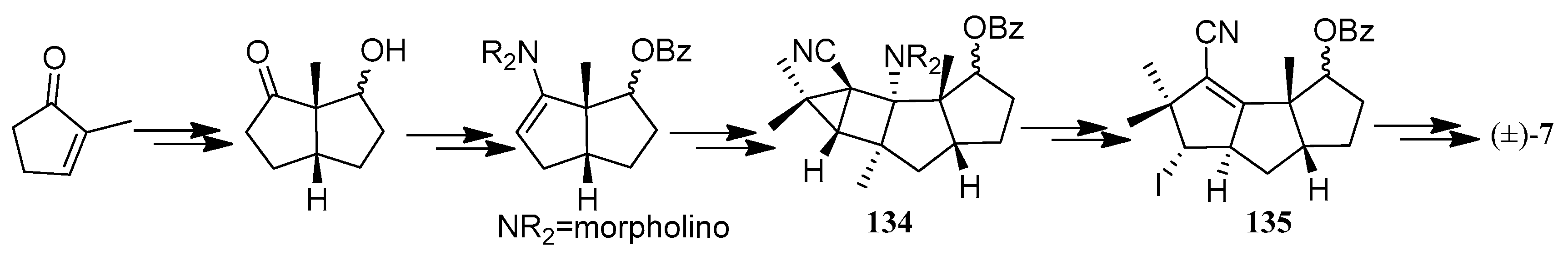
In Franck-Neumann and co-workers’ approach to (±)-hirsutene ((±)-7) (Scheme 7), the key strategy was the region and stereospecific cleavage (134→135) of the central bond in the resulting bicyclo[2.1.0]-pentane moiety [56,57,58].
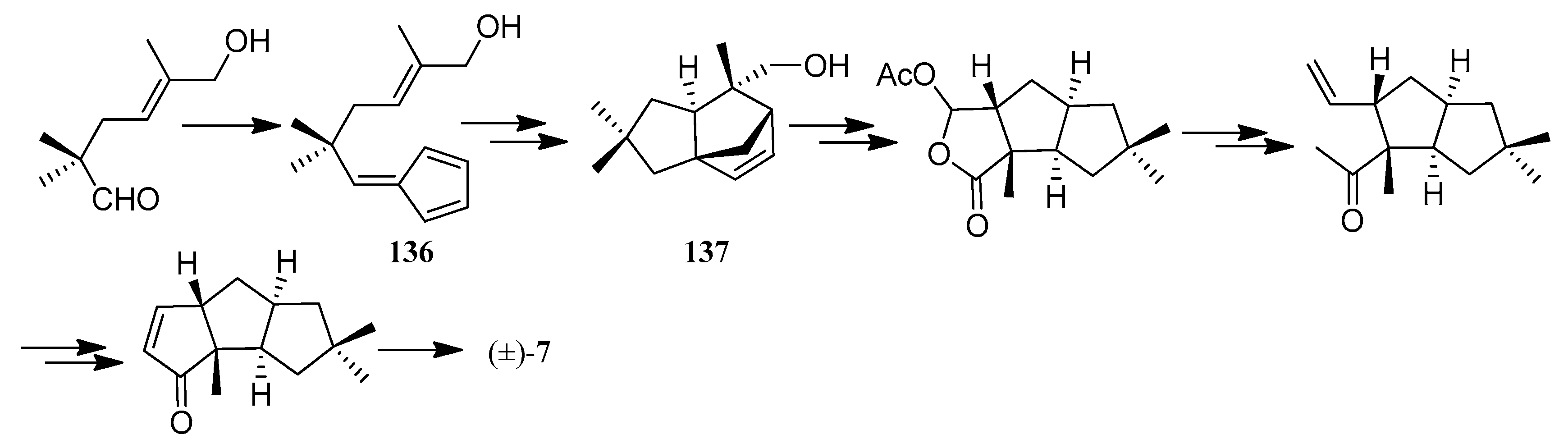
Sternbach and Ensinger achieved a highly efficient 20-step synthesis of (±)-hirsutene ((±)-7) (Scheme 8) [59]. An intramolecular Diels-Alder reaction of the substituted cyclopentadiene (136), which contains all but two of the carbon atoms found in hirsutene, serves as the key step in this sequence.
Castro and co-workers accomplished an enantioselective formal synthesis of hirsutene (7), employing an asymmetric Pauson-Khand bicyclization [60] (Scheme 9).
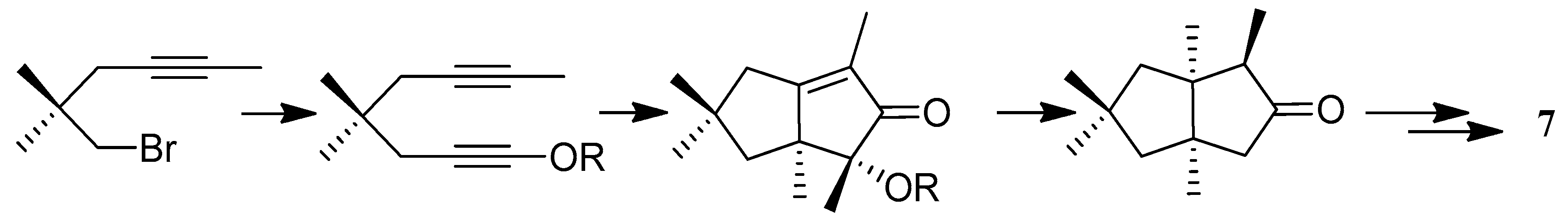
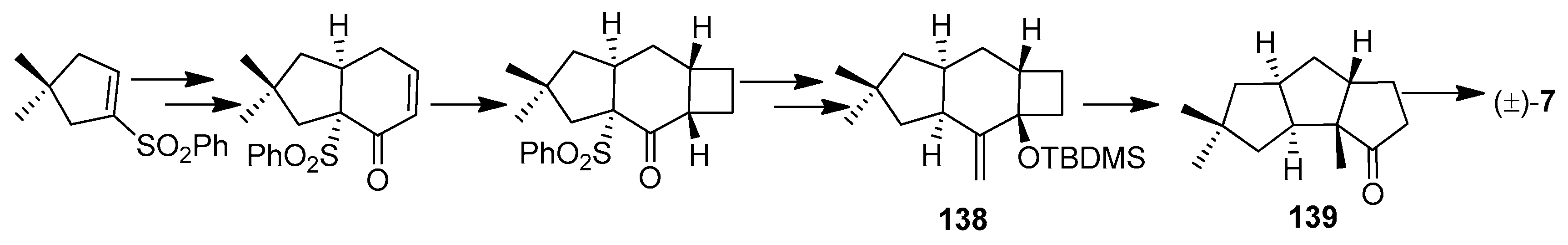
Paquette and co-workers have achieved a total synthesis of (±)-hirsutene ((±)-7) in 9 steps and in 14% overall yield via a twofold cycloadditive scheme, utilizing an iodine catalyzed rearrangement of the methylenetricyclo[6.3.0.03,6]undecane derivative (138) into the norketone (139), a commonly used precursor of hirsutene (7) [61] (Scheme 10).
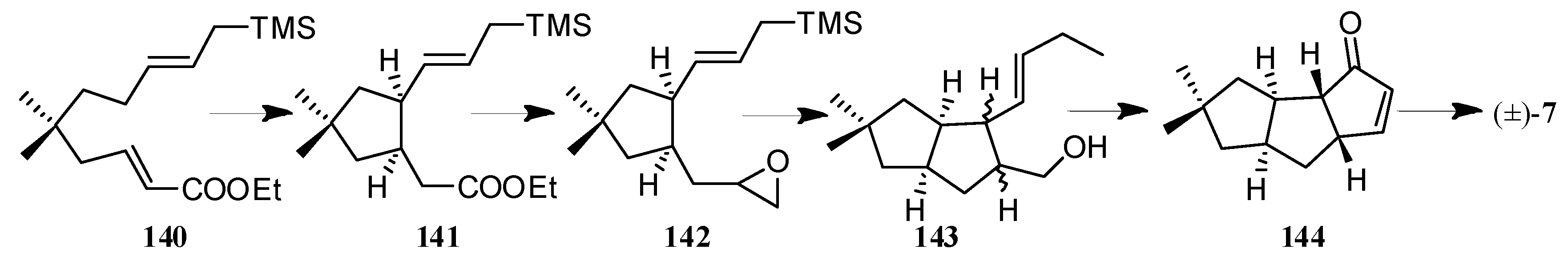
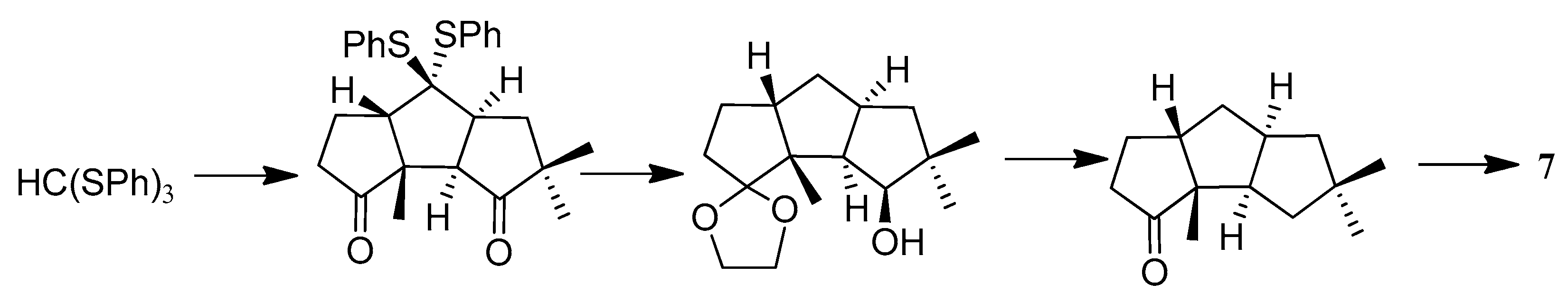
In the formal synthesis of (±)-hirsutene ((±)-7), by Sarkar and co-workers [62,63,64,65] (Scheme 11), two key cyclopentannulation protocols, leading to the diquinane 141, were an efficient intramolecular ene reaction (140→141) and a titanium catalyzed epoxy-allylsilane cyclization (142→143). Then, 143 was further transformed to the 144, via the classical aldol condensation.
Keith and co-workers [66] achieved a highly efficient total synthesis of (±)-hirsutene ((±)-7), by using dilithiomethane equivalent in a novel one-flask [2 + 1 + 2] cyclopentannulation reaction (Scheme 12).
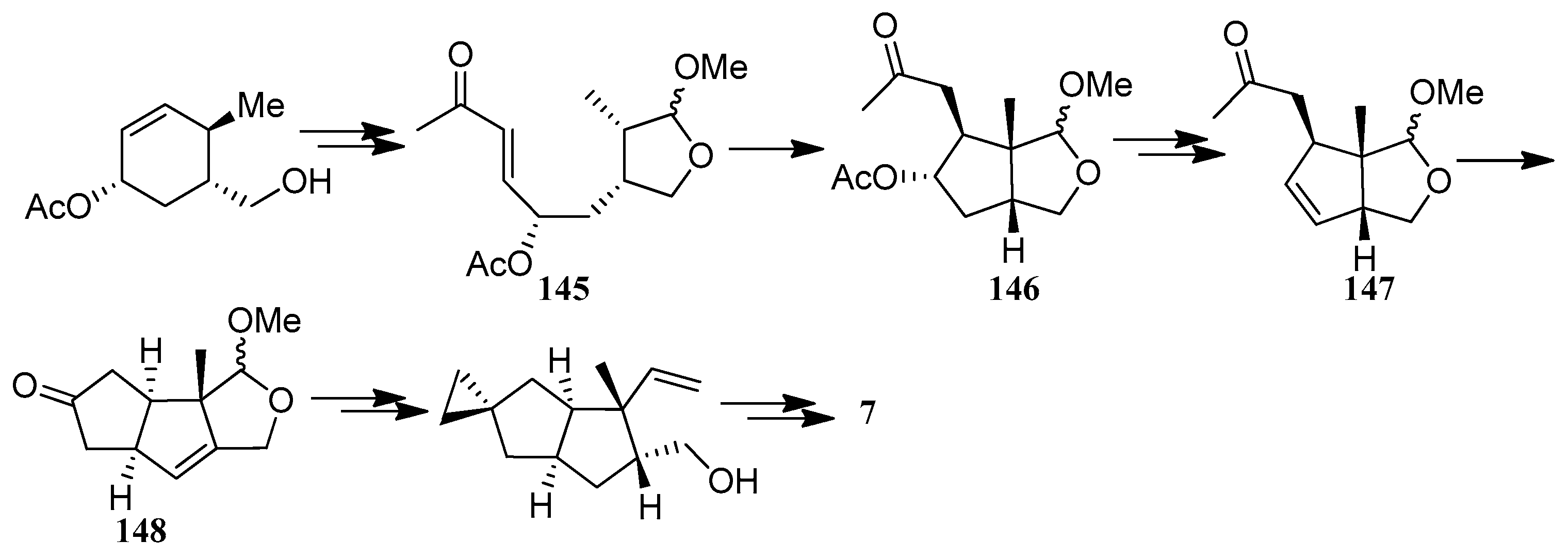
Toyota and co-workers developed a new, highly diastereocontrolled approach, for the synthesis of hirsutene (7), in which the key step utilized an acid catalyzed intramolecular conjugate addition (145→146), and a Pd2+-promoted highly stereocontrolled cyclization (147→148) [67] (Scheme 13).
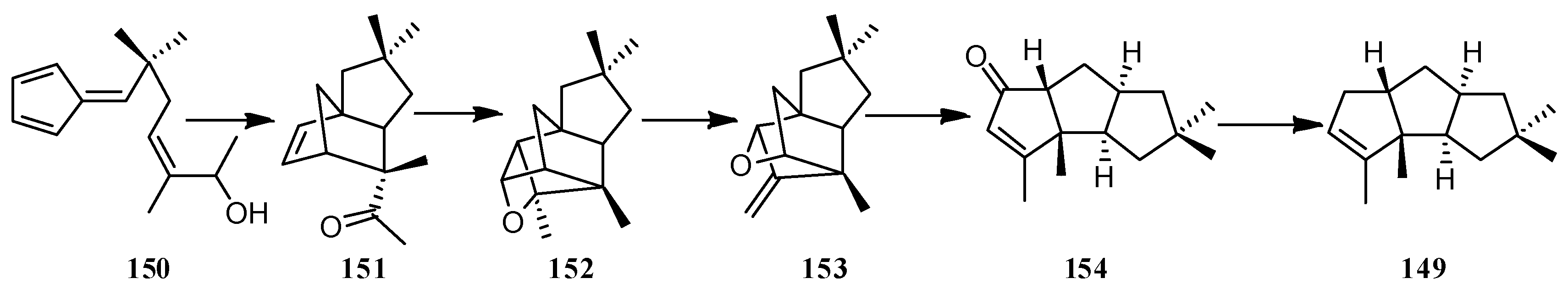
An efficient stereocontrolled synthesis of (±)-endo-hirsutene (149) was achieved, in which the key steps included intramolecular Diels-Alder, Paterno-Buchi reactions, and a reductive fragmentation [68]. (±)-Endo-hirsutene (149), earlier converted to (±)-hirsutene ((±)-7), was furnished from the deoxygenation of triquinane 154 (Scheme 14).
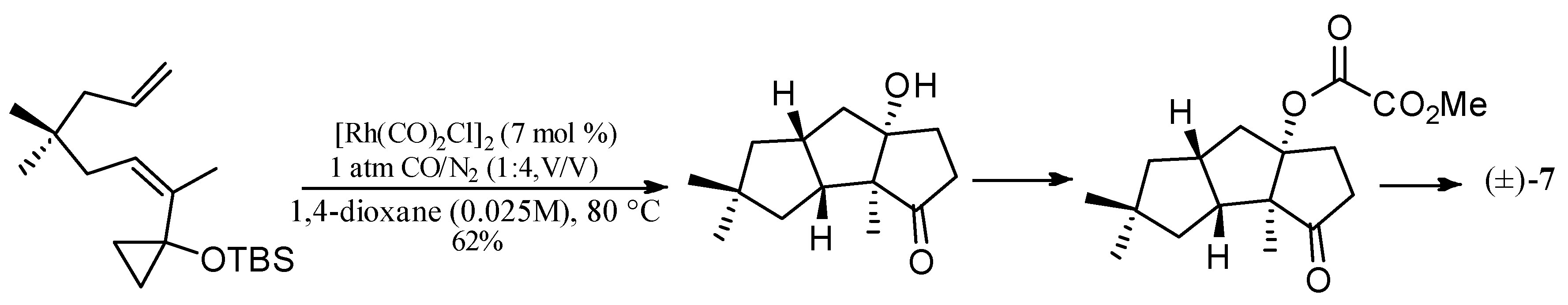
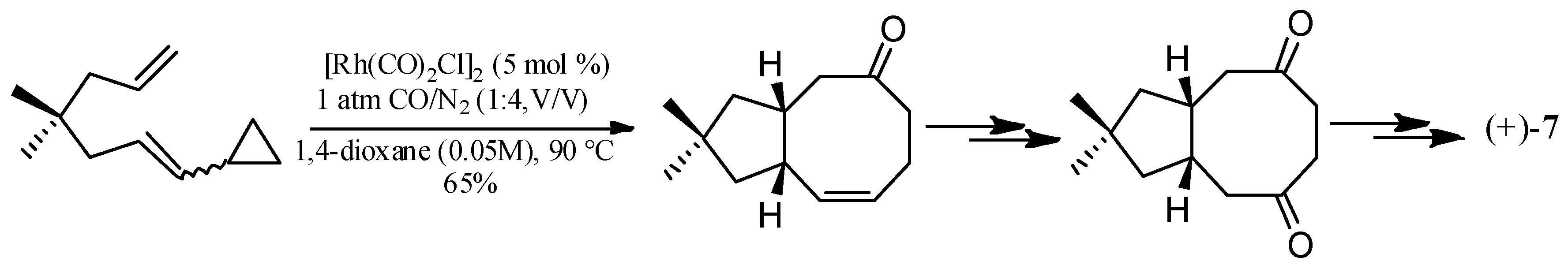
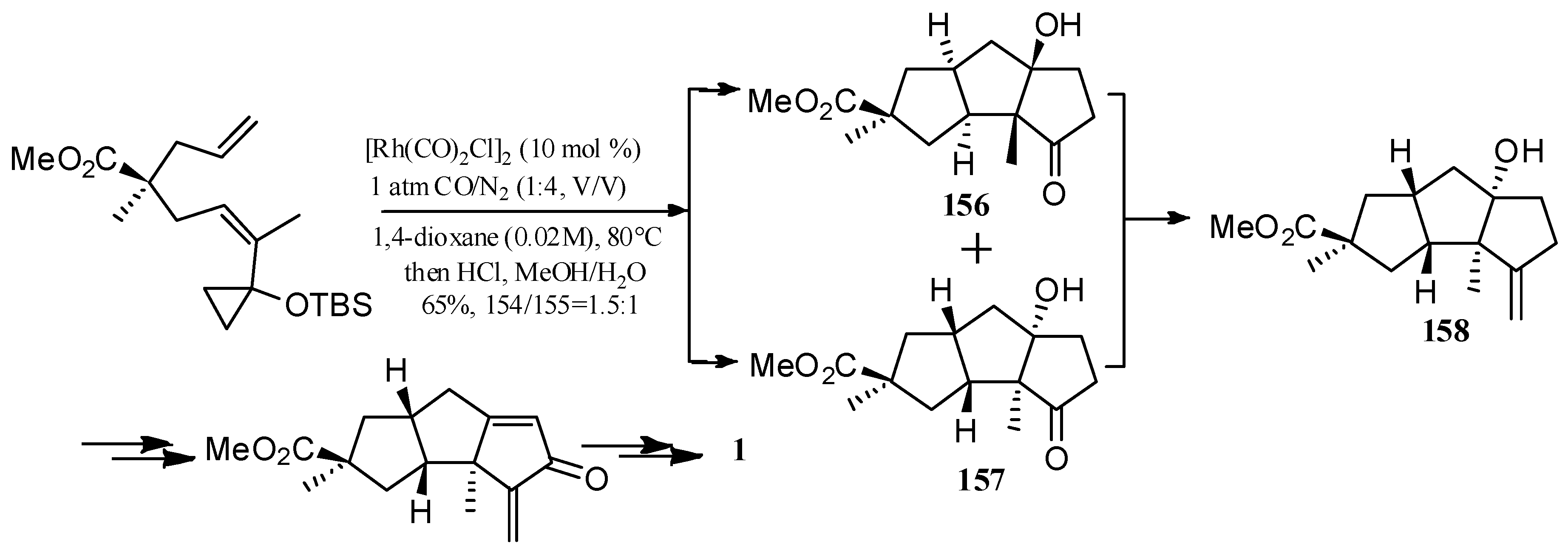
Wang and Yu described a rhodium-catalyzed [5+2+1] cycloaddition of ene-vinylcyclopropanes and CO, and applied this method to synthesize (±)-hirsutene ((±)-7) (Scheme 15), (+)-hirsutene ((+)-7) (Scheme 16), 1-desoxyhypnophilin (155) (Scheme 17), and hirsutic acid C (1) (Scheme 18) [69].
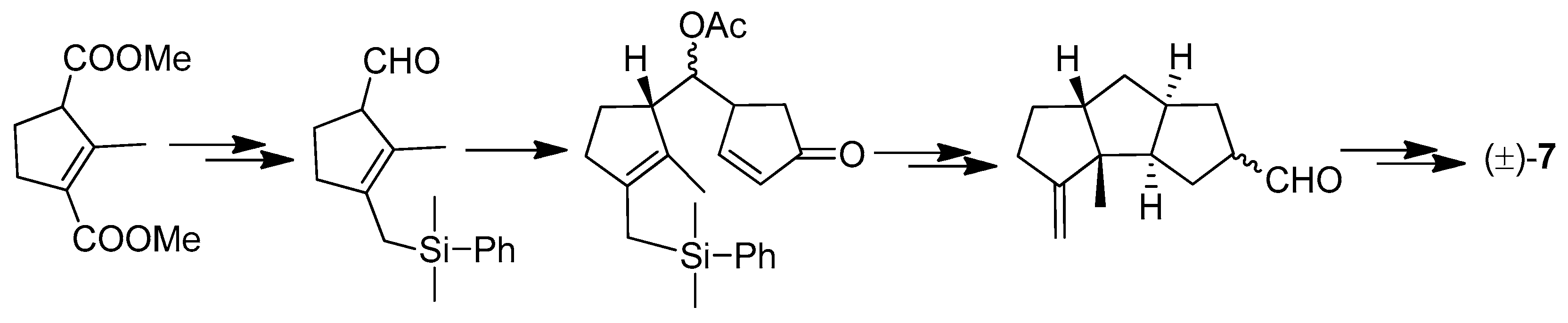
A facile synthesis of the hirsutane framework was reported in 2002 [58]. This method was via a novel [6 + 2] cycloaddition of fulvenes with alkenes, as shown in Scheme 19.
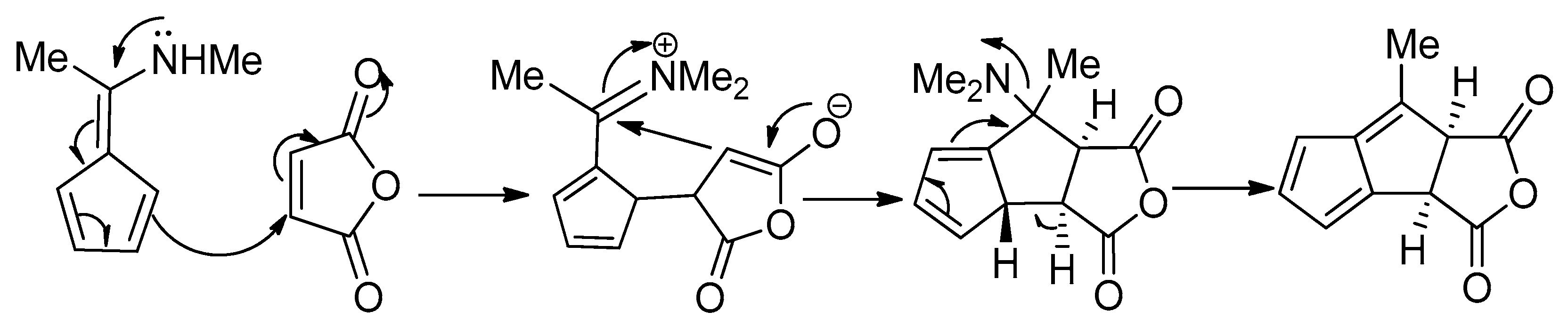
In 2002, Geng and co-workers presented a direct 10-step route from a squarate ester to hypnophilin (8) [70]. The access route involved a combination of chlorination, reduction, dehydration, and oxidation maneuvers in the proper sequence (Scheme 20).
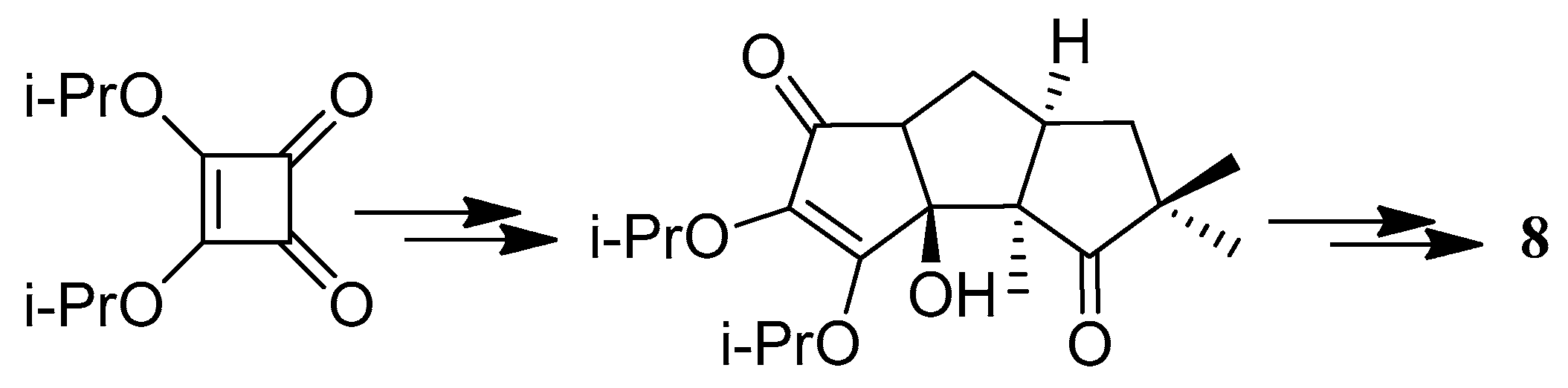
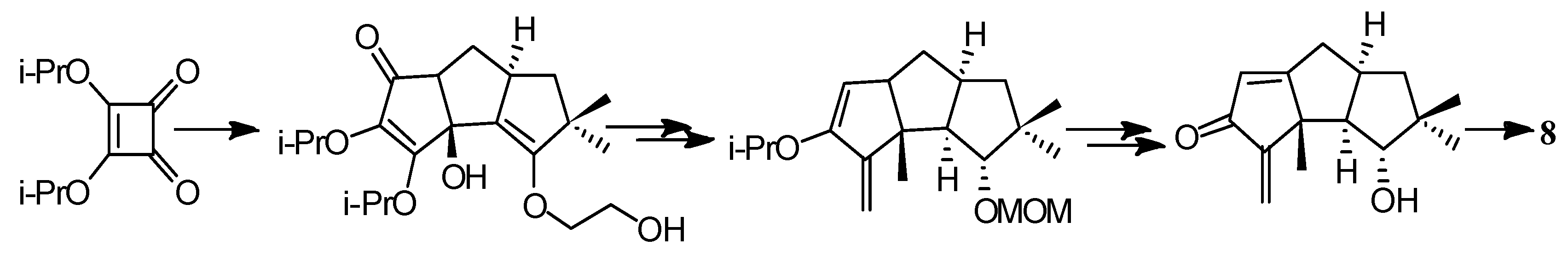
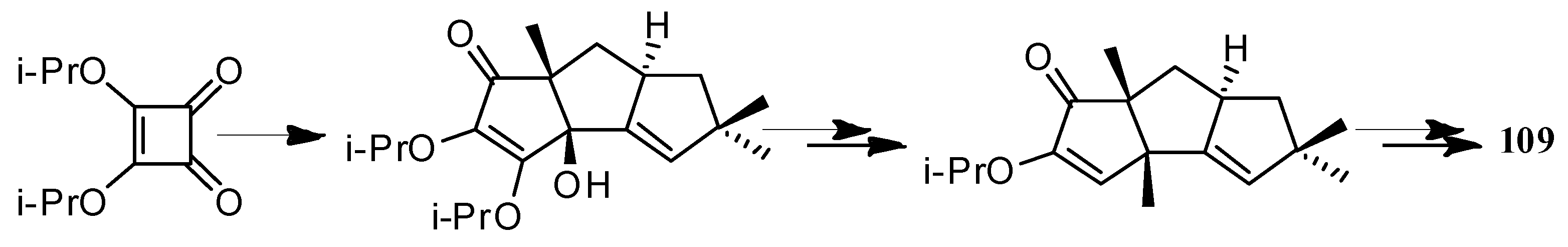
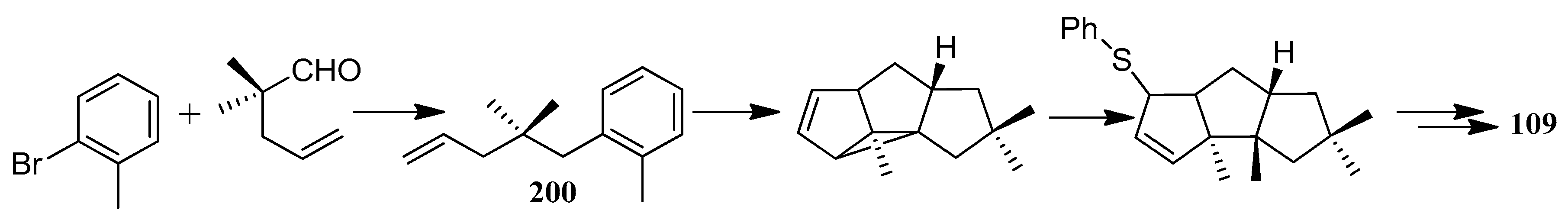
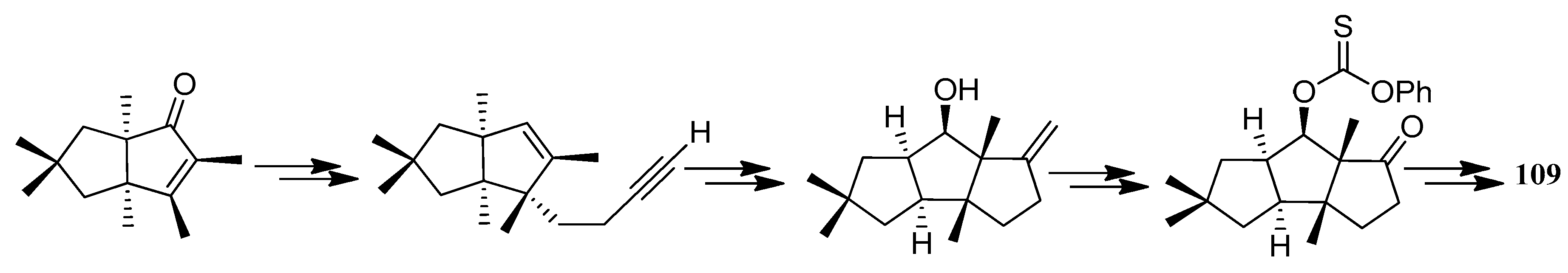
The first application of the squarate ester cascade to natural products synthesis, was realized by Paquette and Geng, in 2002 [71]. Only 10 laboratory steps were needed to achieve the conversion of diisopropyl squarate to hypnophilin (8) (Scheme 21). A penultimate precursor to 8, has previously been transformed to 2 (Scheme 22), thereby achieving a formal synthesis of racemic 2. A rather different strategy was employed to synthesize ceratopicanol (109) (Scheme 23).
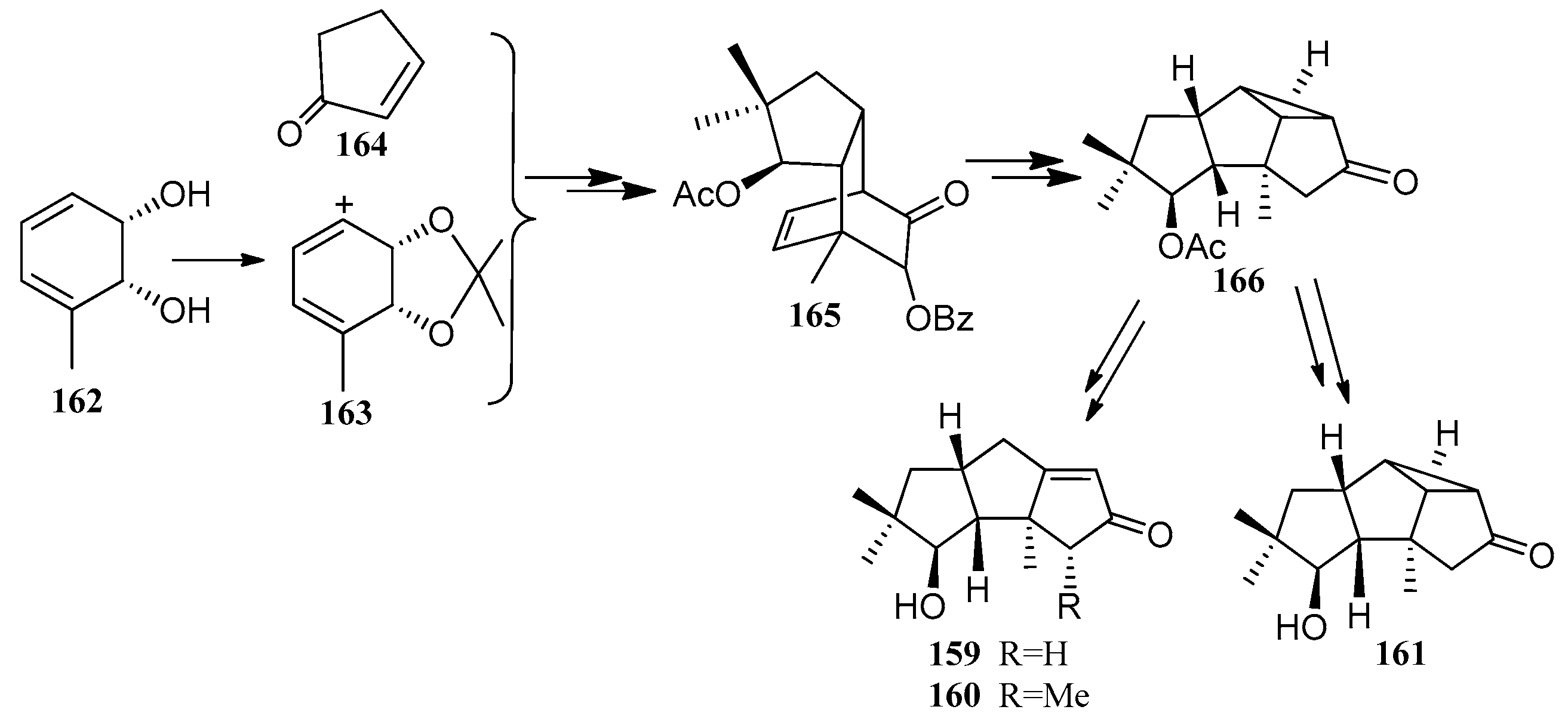
In 2013, Bon and co-workers accomplished the formal total syntheses of (−)-hypnophilin ((−)-8) and (−)-coriolin ((−)-2) [72]. In particular, they reported the synthesis of compound 159, an advanced intermediate related with previously reported syntheses of both compounds (−)-8 and (−)-2. They attempted to prepare its methylated congener 160, and the generation of the isomeric tetracyclic system 161, representing a late-stage precursor to (−)-2 (Scheme 24).
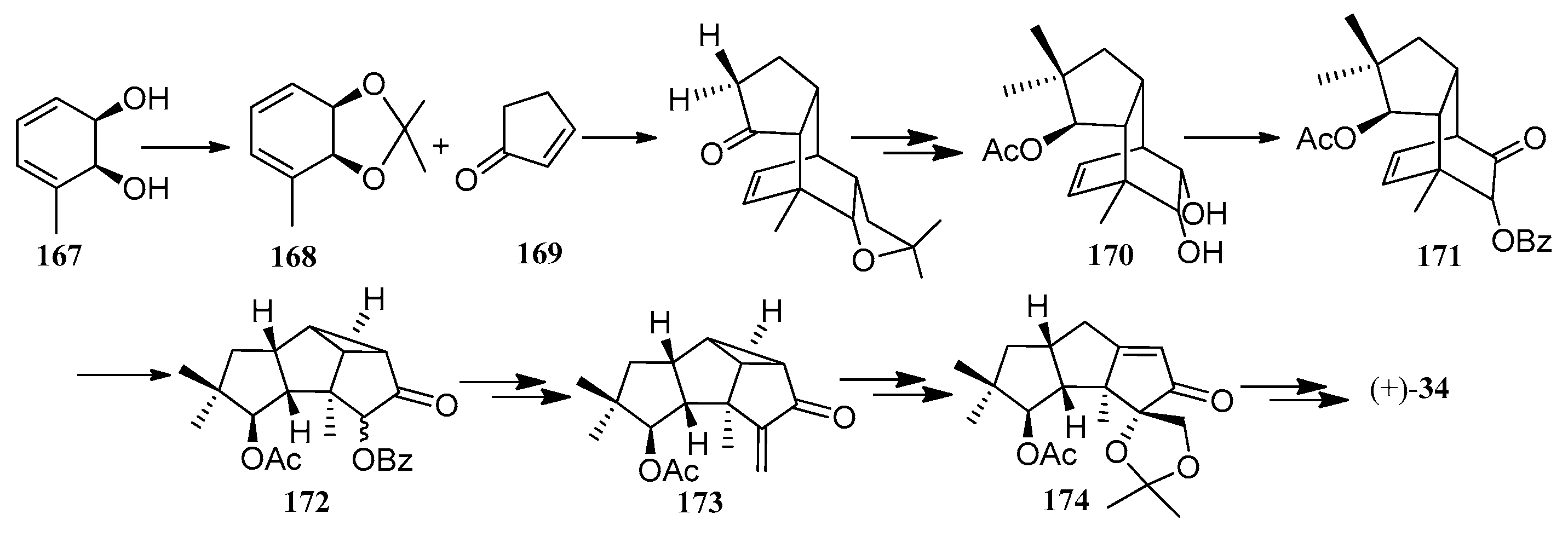
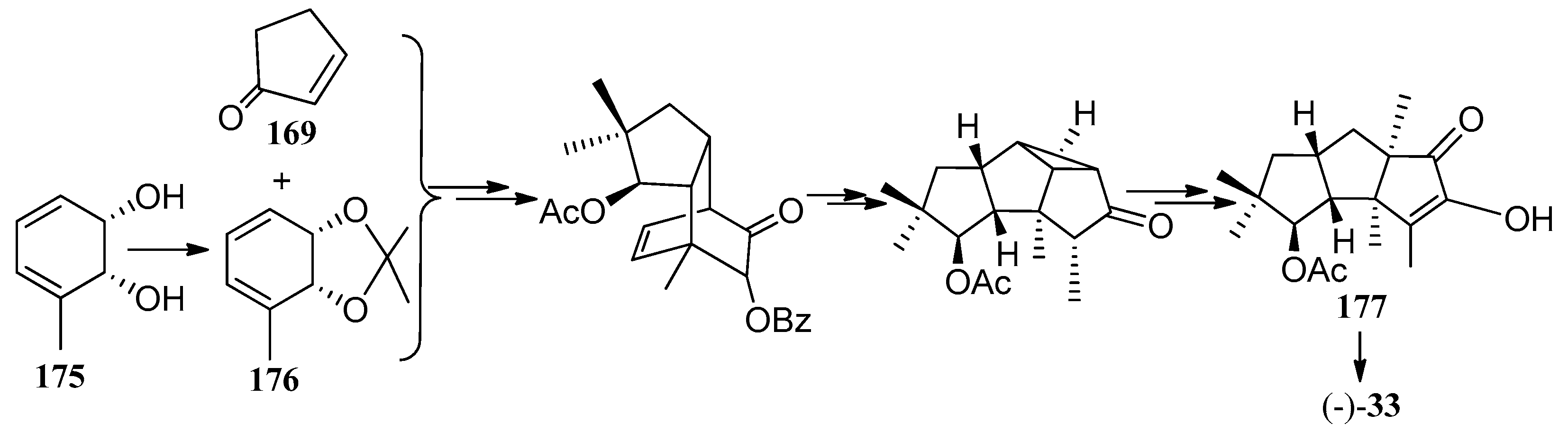
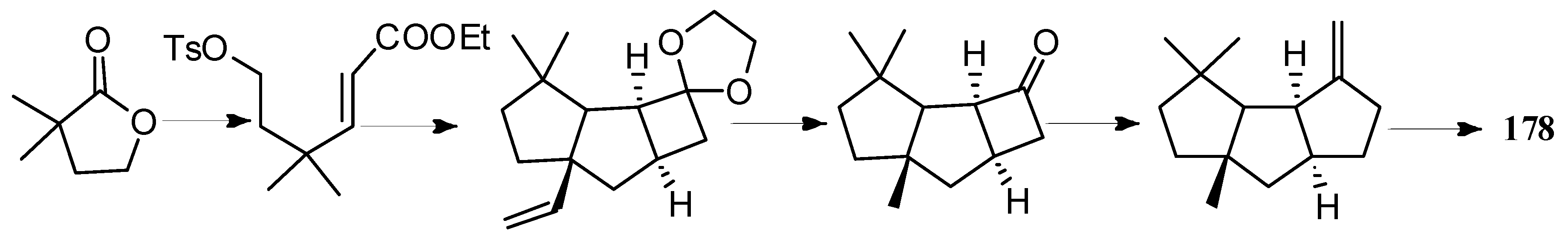
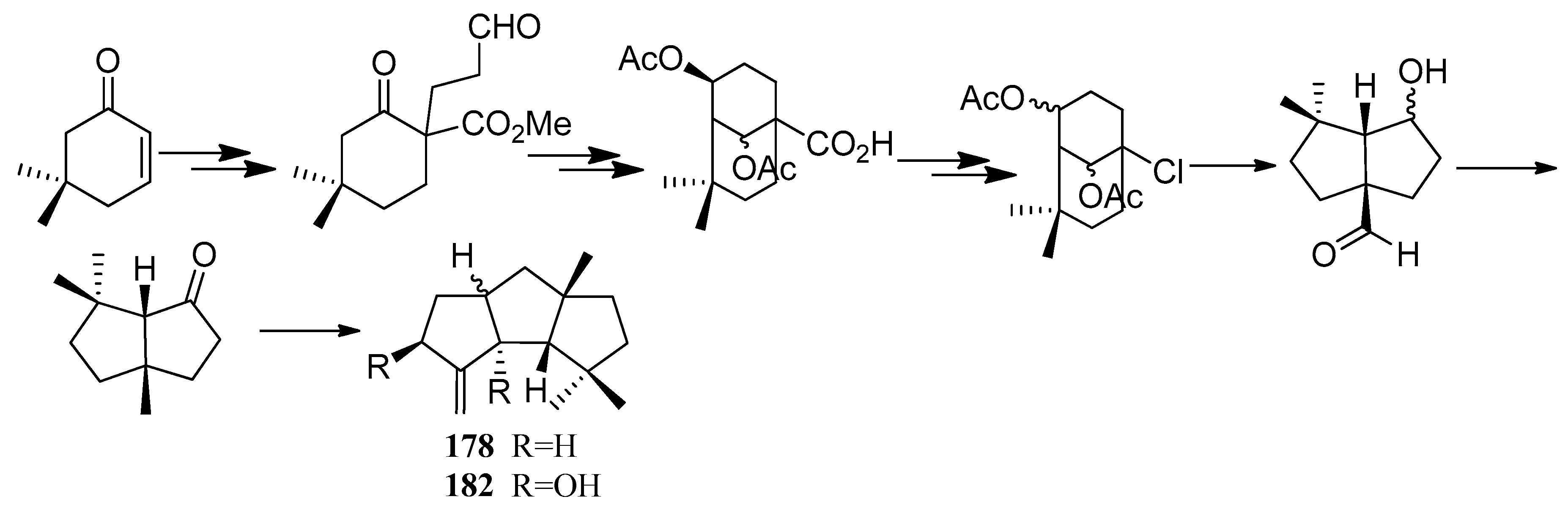
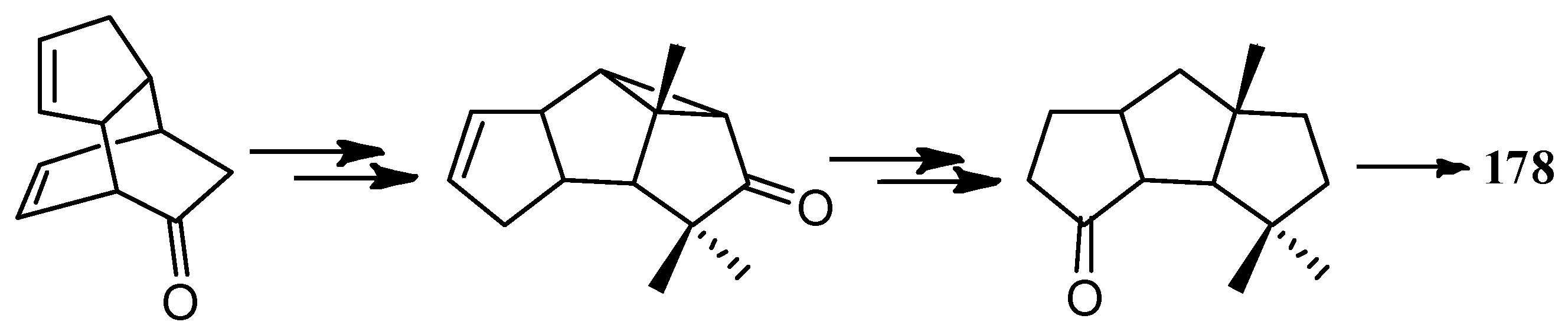
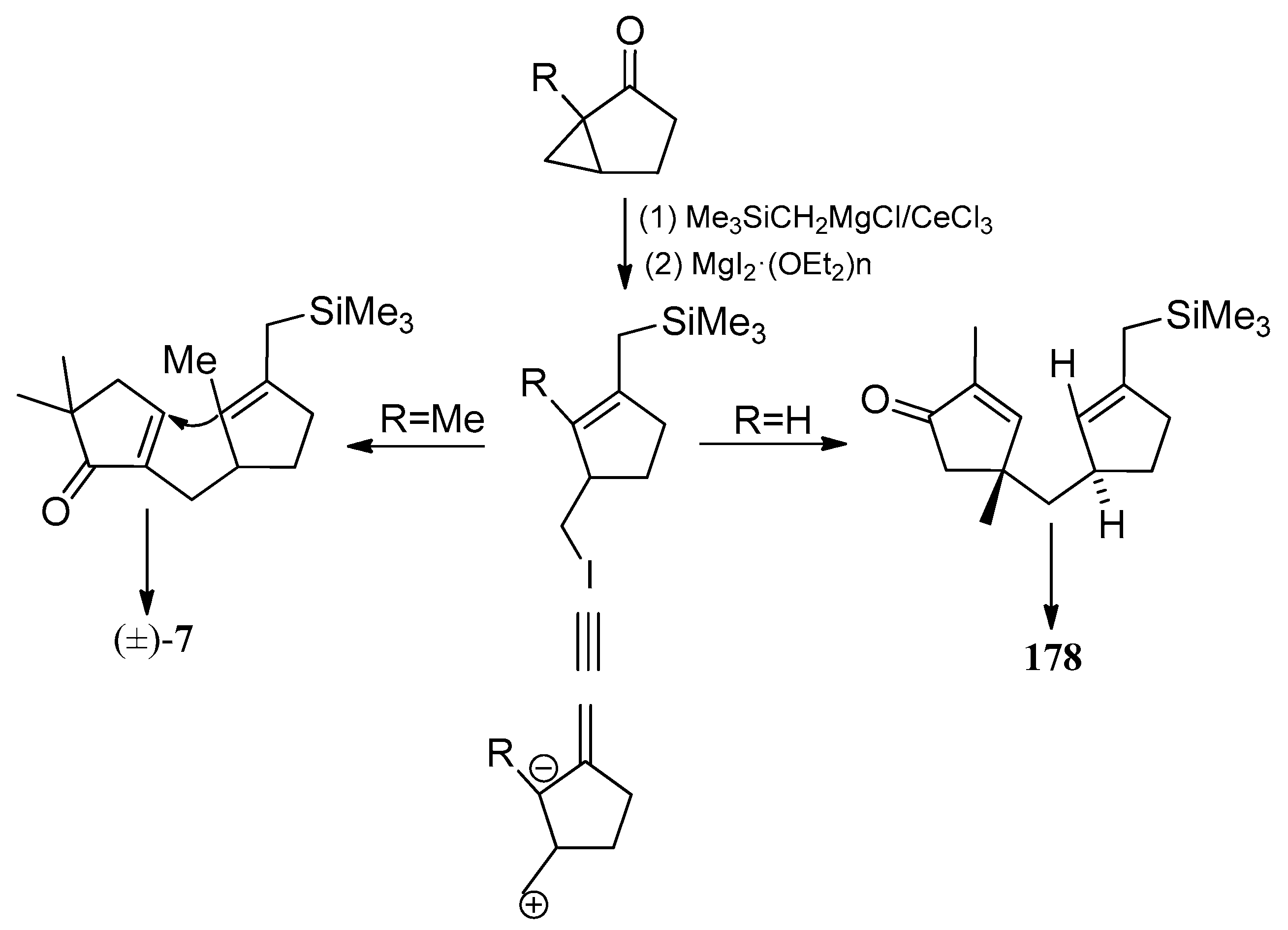
Bon and co-workers showed great interest in the synthesis of the linear triquinane sesquiterpenoids. In 2010, they reported a chemoenzymatic total synthesis of (+)-connatusin B ((+)-34) from toluene [73]. In this synthesis, the starting material employed the cis-1,2-dihydrocatechol (167), which was acquired in enantiomerically pure via the enzymatic dihydroxylation of toluene. Diels-Alder cycloaddition and oxa-di-π-methane rearrangement reactions, represented the key steps in the reaction sequence, leading to the cyclopropane ring-fused linear triquinane (172). Reductive cleavage of the three-membered ring within this framework and types of functional group interconversions, then provided (+)-connatusin B ((+)-34) (Scheme 25). In 2011, the researchers applied this work to the synthesis of (−)-connatusin A ((−)-33). Notably, the final step of this sequence required cleaving the acetate residue within compound 177, to reveal the corresponding alcohol, and they could not secure good yields of the final product. Eventually, after considerable experimentation, they found that the use of acidified DOWEX-50WX-100 resin in 5:1 v/v methanol at 65 °C for 18 h, showed the most effective result and provided a crystalline sample of (−)-connatusin A ((−)-33) in 84% yield (Scheme 26) [74].
4.2. Capnellene Type
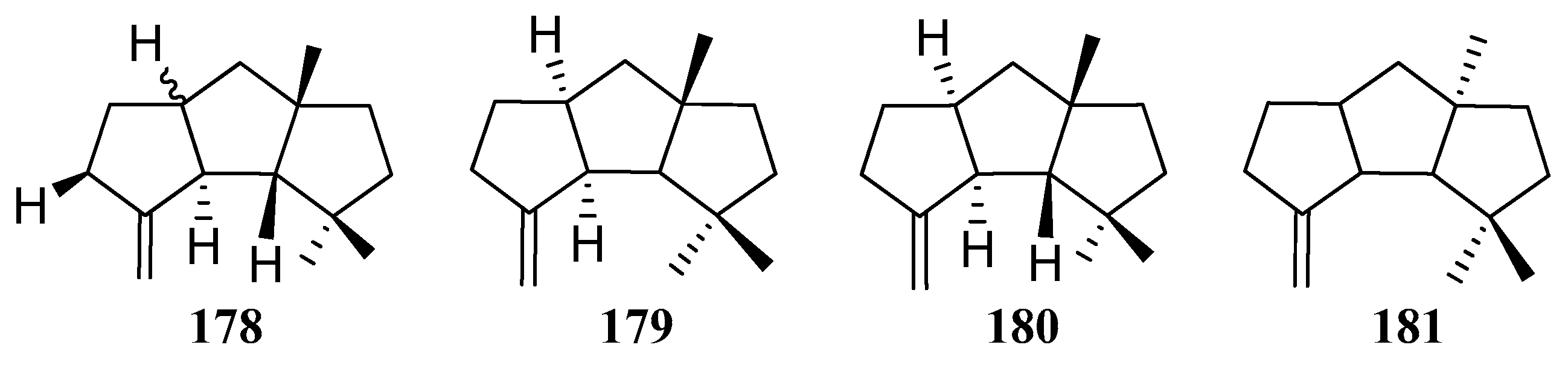
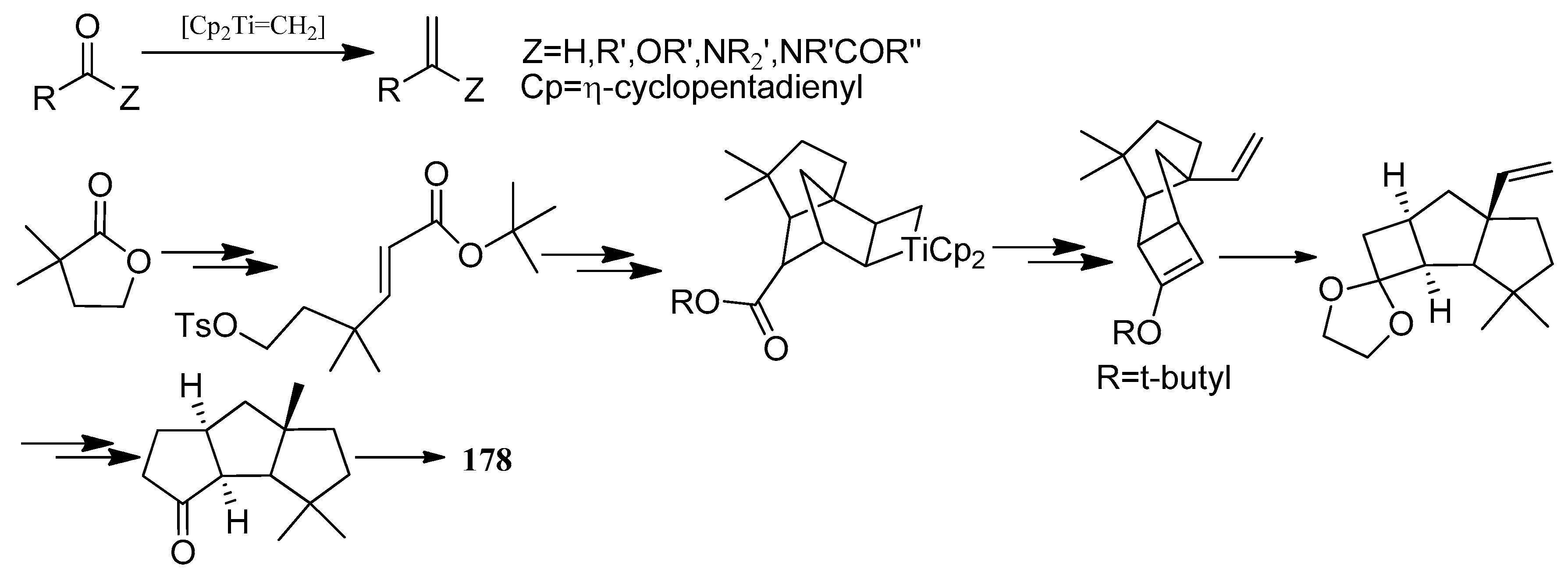
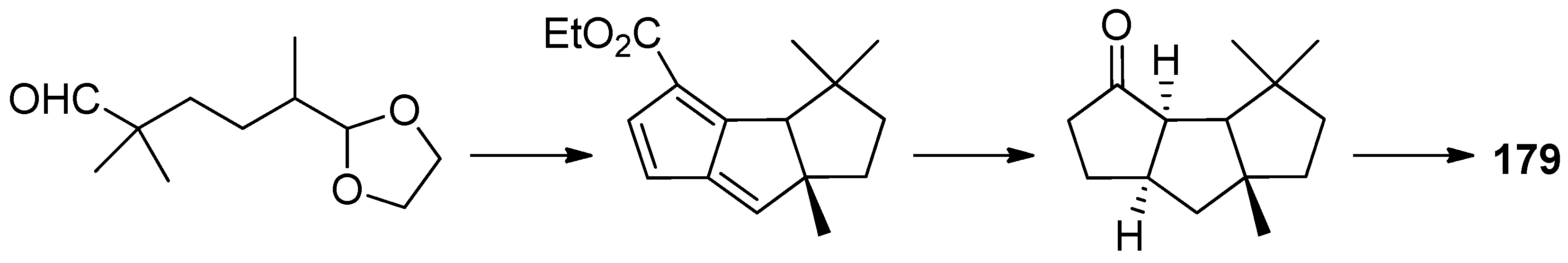
Capnellene is one of the most popular targets for synthesis, mainly owing to its molecular architecture, exocyclic olefinic linkage, disposition of methyl groups, and its role in the defense mechanism and biosynthesis of oxygenated capnellenes [75], especially (±)-∆9(12)-capnellene (178), ∆9(12)-capnellene (179), (−)-∆9(12)-capnellene (180), and (+)-∆9(12)-capnellene (181) (Figure 8). The first total synthesis of (±)-∆9(12)-capnellene (178), was reported in 1981 [76].
Stille and Grubbs [77] realized the synthesis of (±)-∆9(12)-capnellene (178) by using titanium reagents (Scheme 27). Stille and co-workers [78] achieved the total synthesis of 178, through the rearrangement of bicyclo[2.2.1]heptane ring systems by titanocene alkylidene complexes to bicyclo[3.2.0]heptane enol ethers (Scheme 28). A new synthetic strategy for (±)-∆9(12)-capnellene (178) and (±)-∆9(12)-capnellene-8β,10α-diol (182), was presented in 1992 [79]. This strategy was presented as a model study in the synthesis of the pentalanic trimethylketone, which has been used as a key intermediate in the total synthesis of both 178 and 182 (Scheme 29).
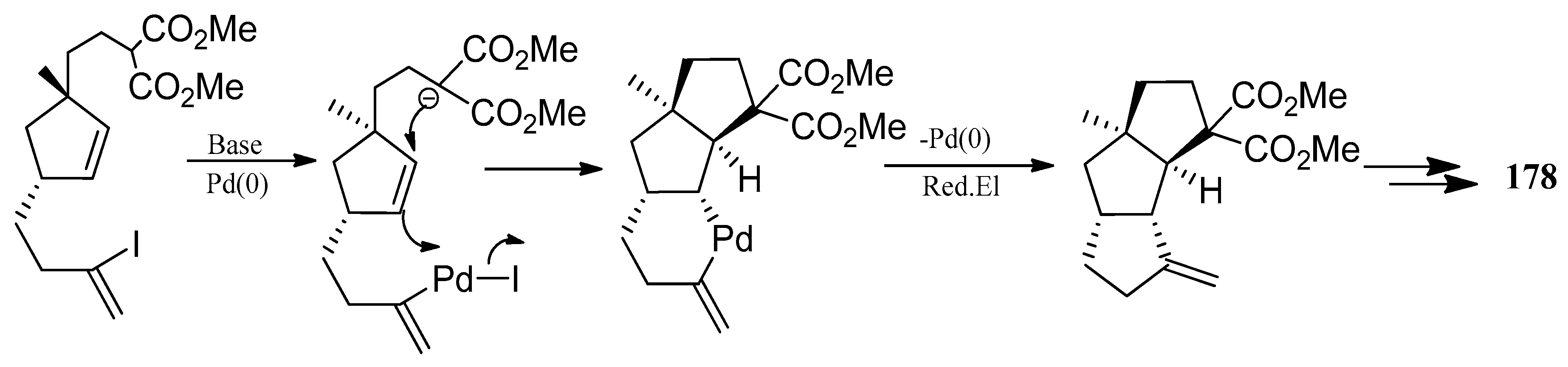
It was worth mentioning that although palladium catalyzed zipper mode cyclization, using Heck reaction conditions had caused widespread concern for the construction of polycyclic system, this methodology was not suitable for the required cis-anti-cis stereochemistry of the capnellane system [61]. However, in 1994 Genevieve Balme achieved the total synthesis of 178, using a palladium-catalyzed bis-cyclization step [80]. Functionalized diquinanes were easily and stereoselectively gained from a new palladium catalyzed cyclisation 183→186 (Scheme 30). Applying the intramolecular version of this strategy, a triquinane should be directly obtained in a single step, by construction of the two outer rings around the central ring (Scheme 31).
In 1997, Singh and co-workers [81] developed a novel synthesis of capnellene from p-cresol and cyclopentadiene, the key steps of the synthesis were inverse demand Diels-Alder reaction and oxa-di-π-methane rearrangement [74]. They also developed a novel and efficient total synthesis of 178 from p-cresol employing π4s + π2s cycloaddition of spiroepoxycyclohexa-2,4-dienone and photochemical 1,2-acyl shift, as key features (Scheme 32).
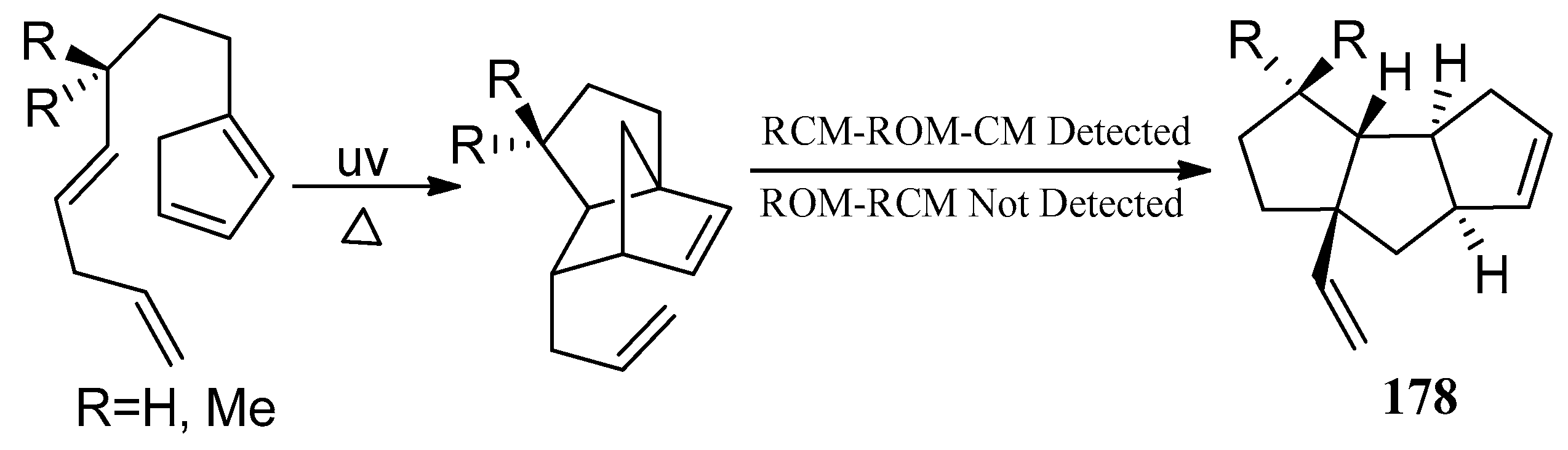
In 2009, Nguyen and co-workers [82] presented a general route to a formal synthesis of 178. This cascade strategy, combined an intramolecular Diels-Alder reaction tandem metathesis protocol to produce the linear cis-anti-cis-tricyclo[6.3.0.02,6]undecane skeleton directly (Scheme 33).
In 2009, Hsu and co-workers [83] developed an efficient and short route from 2-methoxyphenols to polyfunctionalized linear triquinanes, which was characterized by the photochemistry of tricyclo[5.2.2.02,6]undeca-4,10-dien-8-ones. They successfully applied this methodology to the total synthesis of 178, in nine steps, with 20% overall yield by using the strategy of masked o-benzoquinone chemistry (Scheme 34).
In 2013, Shen and Li [84] achieved dual total syntheses of (±)-hirsutene ((±)-7) and (±)-capnellene (178), via a formal [3 + 2] annulation strategy. In this synthesis, the key bifunctional synthons were the cyclic homoiodo allylsilanes, which were readily prepared from the corresponding cyclopropanated cyclopentenones (Scheme 35).
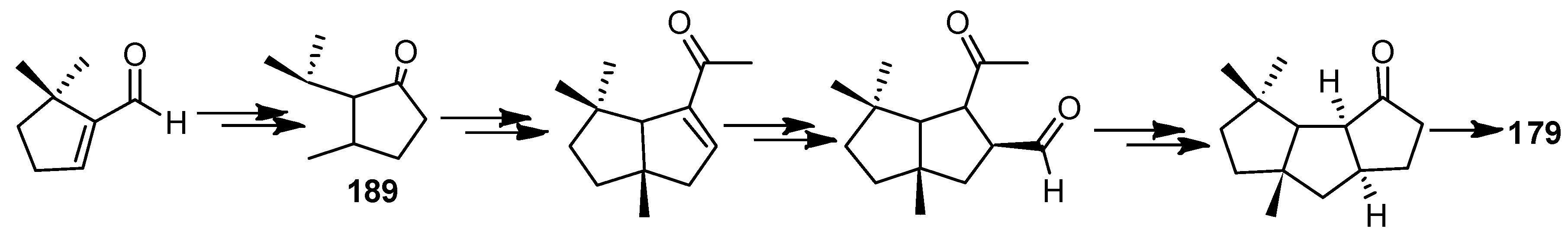
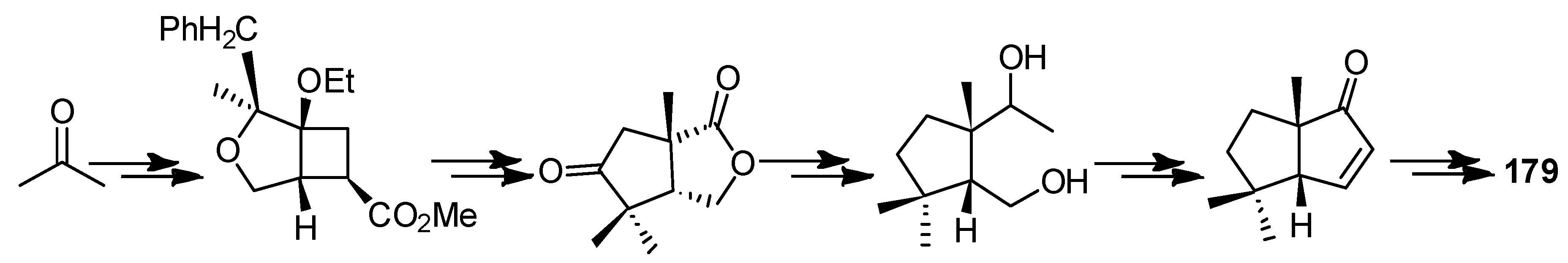
In 1983, a total synthesis of ∆9(12)-capnellene (179) was described by Little and co-workers [85]. They used an intramolecular 1,3-diyl trapping reaction as a primary strategy, to achieve this objective. That ketone 188 corresponded to the desired cis, anti, cis ring-fused product and was confirmed unambiguously by converting it to 179 with a simple Wittig reaction (Scheme 36).
In 1984, a stereocontrolled total synthesis of 179, in 8 steps, from bicyclic enone (189) in 8.3% overall yield was reported (Scheme 37) [86].
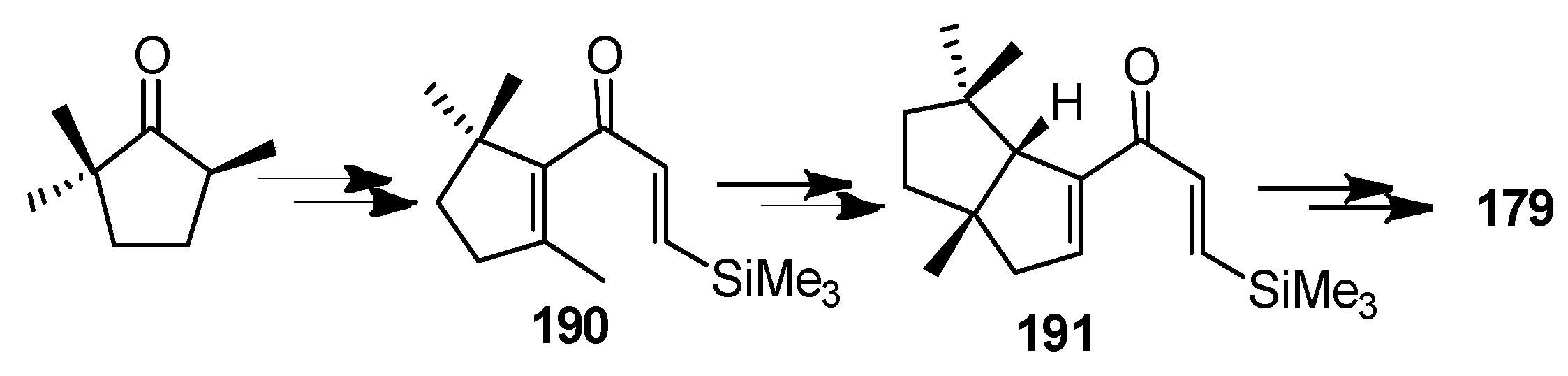
Crisp and co-workers [87] developed a method for palladium-catalyzed carbonylative coupling of vinyl triflates with organostannanes, and this methodology was applied to the total synthesis of the 179. The approach to 179 was based on two intermediates, 190 and 191, which were envisioned as arising from a carbonylative coupling of a vinyl triflate with vinylstannane (Scheme 38).
New type of fulvenes undergo cycloadditions regioselectively to produce synthetically useful tricyclopentanoids, and a formal synthesis of 179 was reported by Ying Wang and co-workwers (Scheme 39) [88].
A direct stereocontrolled route to construct cyclopentanones with substituents up to three contiguous chiral centres, had been achieved in 1998 [89], and this approach was applied in the formal synthesis of 179 (Scheme 40).
In 2009, Lemiere found that cyclopentenylidene gold complexes could be easily formed from vinyl allenes, through a Nazarovlike mechanism. At the same time, polycyclic frameworks may be converted from such carbenes in four different ways [90]: Electrophilic cyclopropanation, C-H insertion, C-C migration, or proton shift. They have studied the selectivity of these different pathways and used their findings to synthesize 179 (Scheme 41).
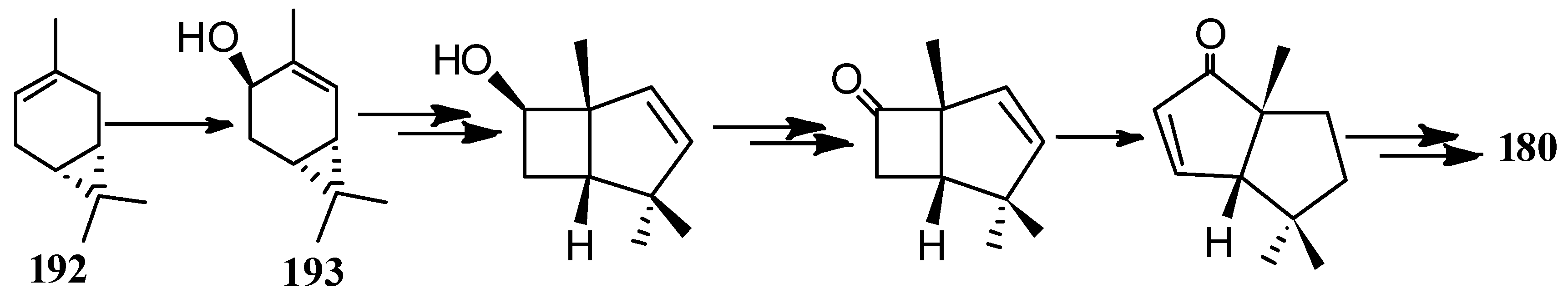
A formal asymmetric synthesis of (−)-capnellene (180) was achieved in 1991, based on the photoinduced vinyl-cyclopropane−cyclopentene rearrangement of the bicyclic alcohol 193, which was obtained from (+)-3-carene (192) (Scheme 42) [91,92].
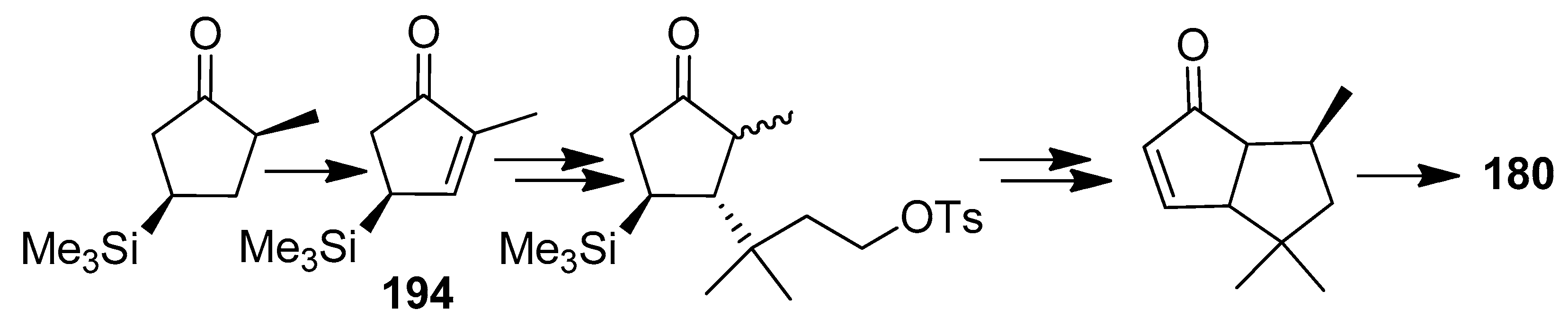
In 1994, Asaoka and co-workers [93] achieved a formal synthesis of 180 by employing silyl group directed stereo control, starting from (−)-2-methyl-4-trimetylsilyl-2-cyclopenten-l-one (194) (Scheme 43). They also noted that compound 194, could be an advanced intermediate in the synthesis of more complex triquinanes.
In 1996, Tanaka and Ogasawara reported [94] the first stereocontrolled synthesis of 180, starting from (−)-oxodicyclopentadiene (195). This synthesis was based on the structure and high functionality of the starting material, which resulted in the stereoselective construction of the requisite stereogenic centres on the triquinane framework easily (Scheme 44).
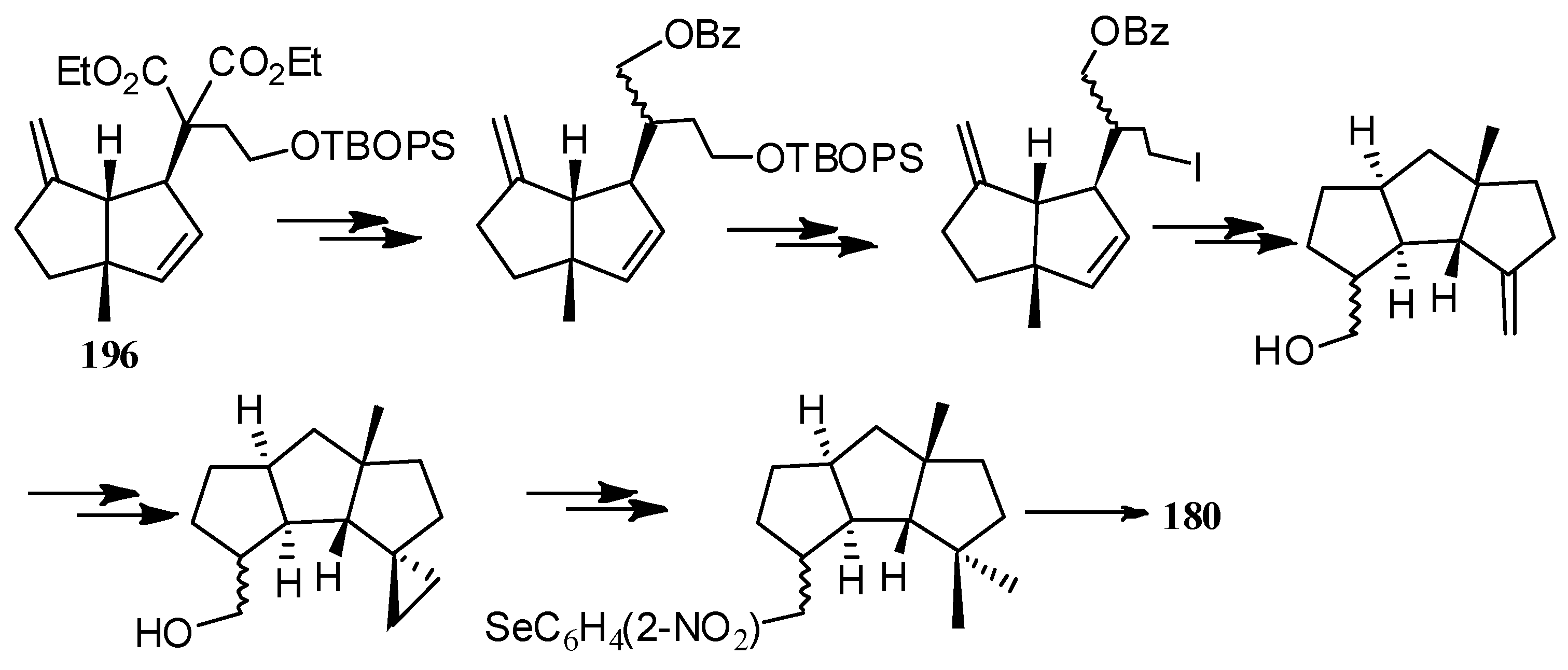
The first asymmetric Heck reaction-carbanion capture process was accomplished in 1996, by Ohshima et al. [95]. They achieved the first catalytic asymmetric total synthesis of 180, in 19 steps, and in 20% overall yield, by using 196 as an asymmetric building block (Scheme 45).
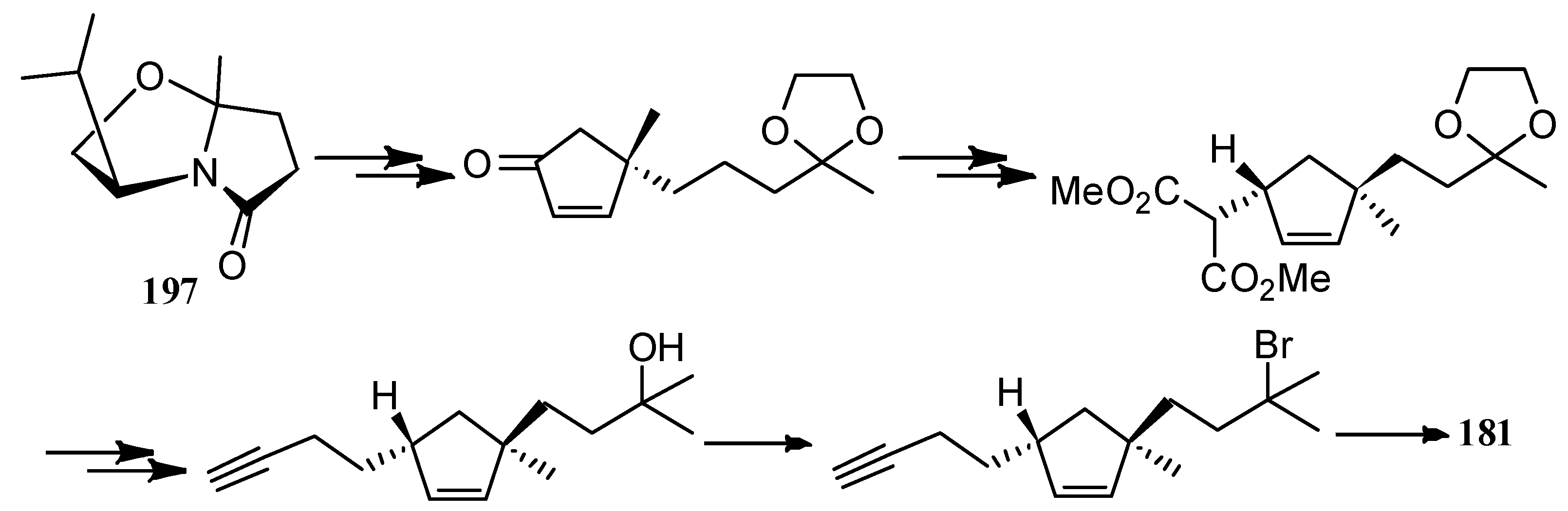
The first asymmetric total synthesis of the unnatural enantiomer of (+)-∆9(12)-capnellene (181) was achieved by Meyers and Bienz, on the basis of the chiral nonracemic bicyclic lactam 197 derived from inexpensive (S)-valinol and levulinic acid (Scheme 46) [96].
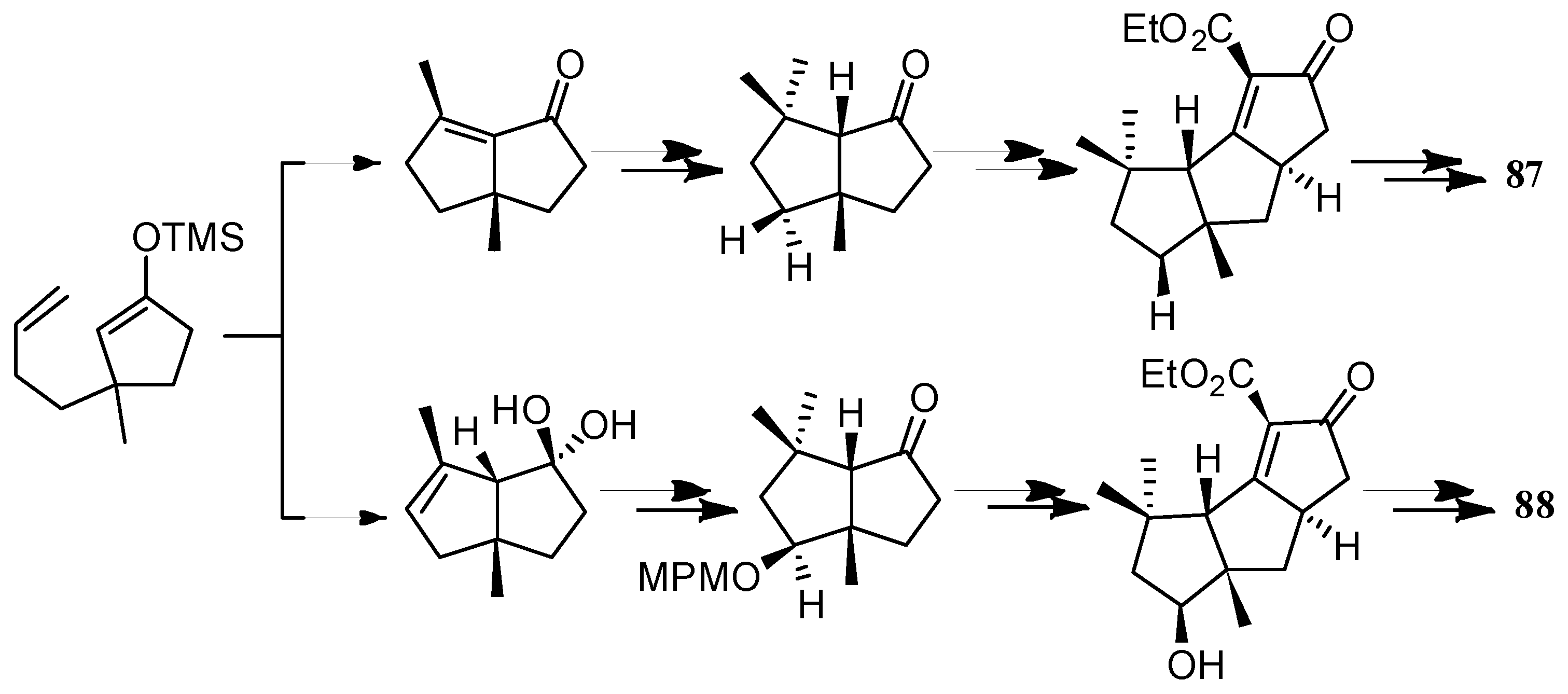
The first total syntheses of ∆9(12)-capnellene-8β,10α-diol (87) and ∆9(12)-capnellene-3β,8β, 10α-troil (88), was achieved by Shibasaki and co-workers [97] (Scheme 47).
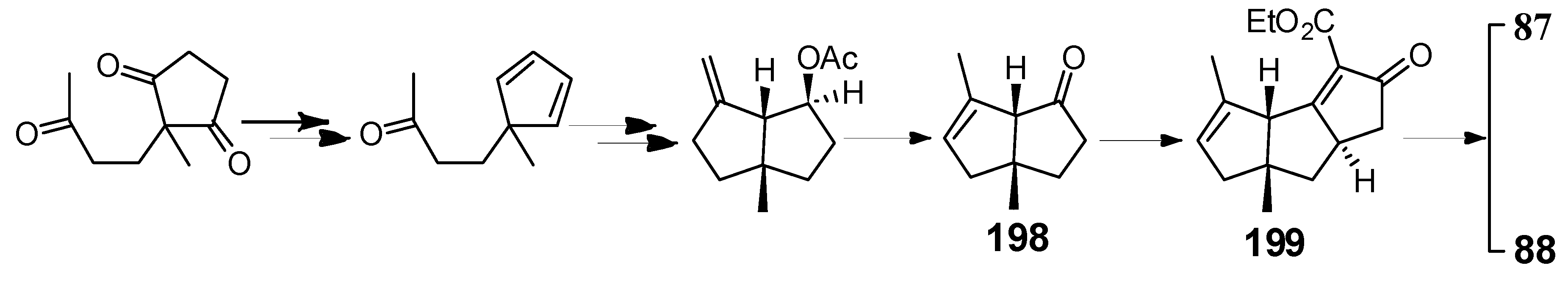
Kagechika and Shibasaki [98] accomplished a catalytic asymmetric synthesis of the key intermediates 198 and 199, for the capnellenols by an asymmetric Heck reaction, followed by an anion capture process. Furthermore, an improved synthetic route to (+)-capnellenols was developed, which led to the synthesis of ∆9(12)-capnellene-8β,10α-diol (87) and ∆9(12)-capnellene-3β,8β,10α-troil (88) (Scheme 48).
4.3. Other Type
In 1996, Baralotto and co-workers [99] proposed a total synthesis of racemic (±)-ceratopicanol ((±)-109), employing seven steps, in which the key one was an intramolecular meta photocycloaddition of the phenylpentene derivative 200, which constituted a typical holosynthon (Scheme 49). In 1996, Clive and co-workers [100] also reported a method to synthesize (+)-ceratopicanol (109) (Scheme 50). This method has been used to form many compounds of the triquinane class.
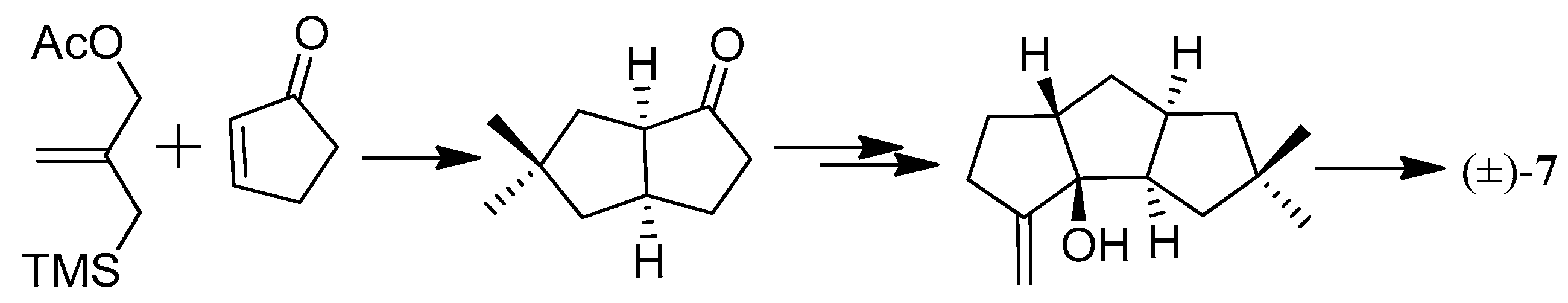
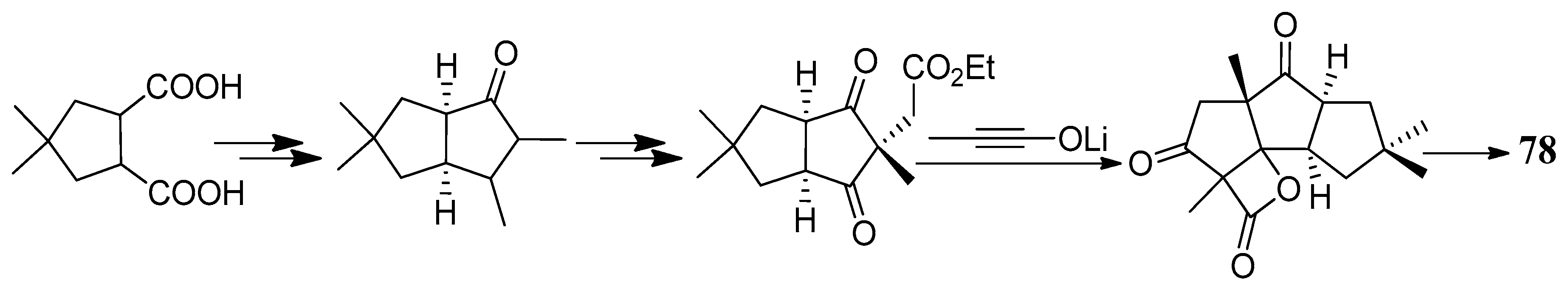
The first total synthesis of cucumin E (78) was achieved in 2002, which used an ynolate-initiated tandem reaction for the construction of the cyclopentenone unit (Scheme 51). This strategy proved the feasibility of a short access to triquinane sesquiterpenoids [45].
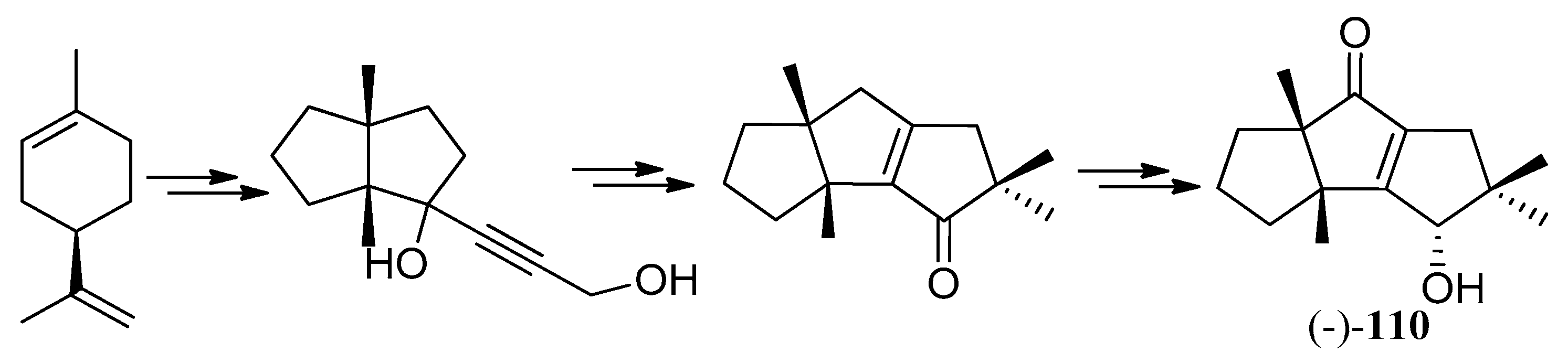
The first total synthesis of the (−)-cucumin H ((−)-110) was reported in 2003 [101] (Scheme 52). This method started from (R)-limonene employing two different cyclopentannulation methodologies, which were to confirm the structure, as well as establish the absolute configuration of the natural product.
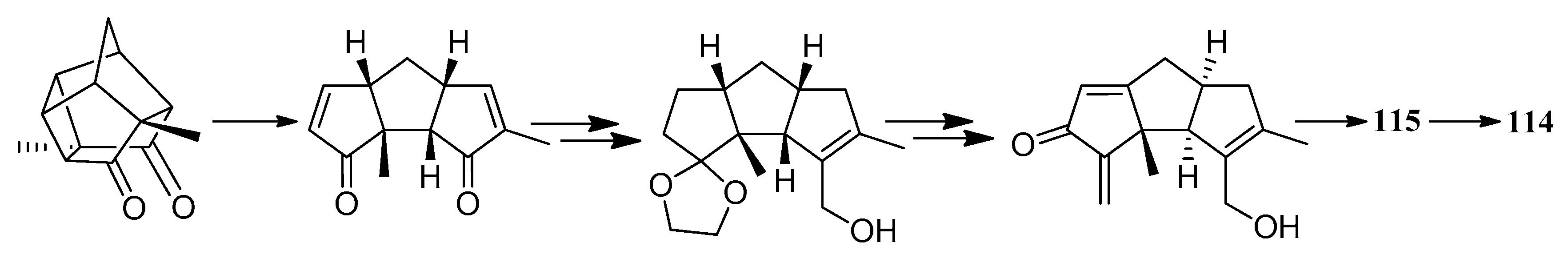
Furthermore in 2003, the first total synthesis of pleurotellic acid (115) and pleurotellol (114) was achieved. One notable feature of this synthesis, was that it followed an adaptation of the versatile photo-thermal metathesis-based approach to triquinane sesquiterpenoids. The other feature was the installation of the sensitive functionalization pattern in the two five-membered rings of the triquinane system, through simple reaction sequences [46] (Scheme 53).
5. Conclusions
From 1947 to July 2018, more than 100 linear triquinane sesquiterpenoids have been isolated, and most of them were isolated from fungi, sponges, and soft corals, among which the mushroom stereum hirsutum, the soft coral capnella imbricata, and marine fungus chondrostereum sp. were typical sources. It is important to note in particular, that Li and co-workers have obtained 19 linear triquinane sesquiterpenoids including chondrosterins A–E, I–O, hirsutanols A, C, E, F, incarnal, arthrosporone, and anhydroarthrosporone, from the marine-derived fungus Chondrostereum sp. Furthermore, the production of secondary metabolites often correlated with a specific stage of morphological differentiation [102]. It indicated that in addition to looking for new sources to produce linear triquinane sesquiterpenoids, we can also find ways to isolate linear triquinane sesquiterpenoids with multiple structural types from one “talented” organism. Many of these compounds displayed a variety of biological activities, such as cytotoxicity, anti-microbial, and anti-inflammatory activities. Given their structural diversity and complexity, and significant biological activities, linear triquinane sesquiterpenoids attracted great interest from synthetic chemists. Among the 8 skeletal types, type I and III are the most popular ones, a number of efficient synthesis methods have been developed to construct the unique skeleton of linear triquinane sesquiterpenoids.
Author Contributions
Y.Q., W.-J.L. and H.-J.L. reviewed the literatures and wrote the paper. L.-P.C. supervised the writing and revised the paper. All authors approved the final version of the manuscript.
Funding
This work was funded by National Natural Science Foundation of China (No. 81872795), Guangdong Natural Science Foundation (No. 2018A030313157), Guangdong Provincial Science and Technology Research Program (Nos. 2015A020216007 and 2016A020222004), National Science and Technology Major Project for New Drug Innovation and Development (No. 2017ZX09305010), and the Fundamental Research Funds for the Central Universities (No. 15ykpy05).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Le, B.F.; Kousara, M.; Chen, L.; Wei, L.; Dumas, F. Tricyclic sesquiterpenes from marine origin. Chem. Rev. 2017, 117, 6110–6159. [Google Scholar] [CrossRef]
- Comer, F.W.; Mocapra, F.; Qureshi, I.H.; Scott, A.I. The structure and chemistry of hirsutic acid. Tetrahedron 1967, 23, 4761–4768. [Google Scholar] [CrossRef]
- Heatley, N.G.; Jennings, M.A.; Florey, H.W. Antibiotics from Stereum hirsutum. Brit. J. Exp. Pathol. 1947, 28, 35–46. [Google Scholar]
- Amouzou, E.; Ayer, W.A.; Browne, L.M. Antifungal sesquiterpenoids from an arthroconidial fungus. J. Nat. Prod. 1989, 52, 1042–1054. [Google Scholar] [CrossRef]
- Wang, G.-Y.-S.; Abrell, L.M.; Avelar, A.; Borgeson, B.M.; Crews, P. New hirsutane based sesquiterpenes from salt water cultures of a marine sponge-derived fungus and the terrestrial fungus Coriolus consors. Tetrahedron 1998, 54, 7335–7342. [Google Scholar]
- Grote, D.; Hanel, F.; Dahse, H.M.; Seifert, K. Capnellenes from the soft coral Dendronephthya rubeola. Chem. Biodivers. 2007, 4, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Wen, Z.H.; Wang, S.K.; Duh, C.Y. Capnellenes from the formosan soft coral Capnella imbricate. J. Nat. Prod. 2008, 71, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Helaly, S.E.; Richter, C.; Thongbai, B.; Hyde, K.D.; Stadler, M. Lentinulactam, a hirsutane sesquiterpene with an unprecedented lactam modification. Tetrahedron Lett. 2016, 57, 5911–5913. [Google Scholar] [CrossRef]
- Takeuchi, T.; Iinuma, H.; Iwanaga, J.; Takahashi, S.; Takita, T.; Umezaw, H. Coriolin, a new basidiomycetes antibiotic. J. Antibiot. 1969, 22, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Iinuma, H.; Takita, T.; Maeda, K.; Umezawa, H. The structures of coriolin B and C. Tetrahedron Lett. 1970, 11, 1637–1639. [Google Scholar] [CrossRef]
- Nozoe, S.; Furukawa, J.; Sankawa, U.; Shibata, S. Isolation, structure and synthesis of hirsutene, a precursor hydrocarbon of coriolin biosynthesis. Tetrahedron Lett. 1976, 17, 195–198. [Google Scholar] [CrossRef]
- Takahashi, S.; Naganawa, H.; Iinuma, H.; Takita, T.; Maeda, K.; Umezawa, H. Revised structure and stereochemistry of coriolins. Tetrahedron Lett. 1971, 12, 1955–1958. [Google Scholar] [CrossRef]
- Mellows, G.; Mantle, P.G.; Feline, T.C.; William, D. Sesquiterpenoid metabolites from Stereum complicatun. J. Phytochemistry 1973, 12, 2717–2720. [Google Scholar] [CrossRef]
- Tanabe, M.; Suzuki, K.T. Biosynthetic studies with carbon-13: The FT-13C NMR spectra of the sesquiterpenoid coriolins. Tetrahedron Lett. 1974, 15, 2271–2274. [Google Scholar] [CrossRef]
- Kupka, J.; Anke, T.; Giannetti, B.M.; Steglich, W. Antibiotics from basidiomycetes XIV. * Isolation and biological characterization of hypnophilin, pleurotellol, and pleurotellic acid from Pleurotellus hypnophilus (Berk.) Sacc. Arch. Microbiol. 1981, 130, 223–227. [Google Scholar] [CrossRef]
- Giannetti, B.M.; Steffan, B.; Steglich, W. Antibiotics from basidiomycetes. Part 24.1: Antibiotics with a rearranged hirsutane skeleton from Pleurotellus Hypnophilus (agaricales). Tetrahedron 1986, 42, 3587–3593. [Google Scholar] [CrossRef]
- Takazawa, H.; Kashino, S. Incarnal, a new antibacterial sesquiterpene from basidiomycetes. Chem. Pham. Bull. 1991, 39, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yue, D.C.; Cheng, K.D.; Wang, S.C.; Yu, K.B.; Zheng, Q.T.; Yang, J.S. Gloeosteretriol, a new sesquiterpene from the fermentation products of Gloeostereum incarnatum S. Ito et Imai. Acta Pharm. Sin. 1992, 27, 33–36. [Google Scholar] [CrossRef]
- Abate, D.; Abraham, W.R. Antimicrobial metabolites from Lentinus crinitus. J. Antibiot. 1994, 47, 1348–1350. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.C.; Varoglu, M.; Abrell, L.; Lobkovsky, E.; Clardy, J. Chloriolins A–C, chlorinated sesquiterpenes produced by fungal cultures separated from a juspis marine sponge. J. Org. Chem. 1994, 59, 6344–6348. [Google Scholar] [CrossRef]
- Hellwig, V.; Dasenbrock, J.; Schumann, S.; Steglich, W.; Leonhardt, K.; Anke, T. New triquinane-type sesquiterpenoids from Macrocystidia cucumis (Basidiomycetes). Eur. J. Org. Chem. 1998, 73–79. [Google Scholar] [CrossRef]
- Yun, B.S.; Lee, I.K.; Cho, Y.; Cho, S.M.; Yoo, I.D. New tricyclic sesquiterpenes from the fermentation broth of Stereum hirsutum. J. Nat. Prod. 2002, 65, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Rukachaisirikul, V.; Tansakul, C.; Saithong, S.; Pakawatchai, C.; Isaka, M.; Suvannakad, R. Hirsutane sesquiterpenes from the fungus Lentinus connatus BCC 8996. J. Nat. Prod. 2005, 68, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Liermann, J.C.; Schuffler, A.; Wollinsky, B.; Birnbacher, J.; Kolshorn, H.; Anke, T.; Opatz, T. Hirsutane-type sesquiterpenes with uncommon modifications from three basidiomycetes. J. Org. Chem. 2010, 75, 2955–2961. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Bao, L.; Han, J.J.; Yang, X.L.; Zhao, F.; Li, S.F.; Song, F.H.; Liu, M.M.; Liu, H.W. New benzoate derivatives and hirsutane type sesquiterpenoids with antimicrobial activity and cytotoxicity from the solid-state fermented rice by the medicinal mushroom Stereum hirsutum. Food Chem. 2014, 143, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Xie, Y.L.; Xie, Z.L.; Chen, Y.; Lam, C.K.; Lan, W.J. Chondrosterins A–E, triquinane-type sesquiterpenoids from soft coral-associated fungus Chondrostereum sp. Mar. Drugs 2012, 10, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Lan, W.J.; Lam, C.K.; Yang, F.; Zhu, X.F. Hirsutane sesquiterpenoids from the marine-derived fungus Chondrostereum sp. Chem. Biodivers. 2011, 8, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Lan, W.J.; Deng, R.; Feng, G.K.; Xu, Q.Y.; Hu, Z.Y.; Zhu, X.F.; Li, H.J. Additional new cytotoxic triquinane-type sesquiterpenoids chondrosterins K–M from the marine fungus Chondrostereum sp. Mar. Drugs 2016, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Lan, W.J.; Li, H.J. Two new hirsutane-type sesquiterpenoids chondrosterins N and O from the marine fungus Chondrostereum sp. Nat. Prod. Res. 2018, 32, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.Y.; Bao, L.; Ren, J.W.; Han, J.J.; Zhang, Z.Y.; Li, Y.; Yao, Y.J.; Cao, R.; Liu, H.W. Sterhirsutins A and B, two new heterodimeric sesquiterpenes with a new skeleton from the culture of Stereum hirsutum collected in Tibet plateau. Org. Lett. 2014, 16, 5092–5095. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.Y.; Ren, J.W.; Sun, L.W.; He, L.W.; Bao, L.; Yue, W.; Sun, Q.M.; Yao, Y.J.; Yin, W.B.; Liu, H.W. Stucturally diverse sesquiterpenes produced by a Chinese Tibet fungus Stereum hirsutum and their cytotoxic and immunosuppressant activities. Org. Lett. 2015, 17, 3098–3101. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.M.; Chen, H.P.; Wang, F.; Li, Z.H.; Feng, T.; Liu, J.K. New triquinane and gymnomitrane sesquiterpenes from fermentation of the basidiomycete Antrodiella albocinnamomea. Fitoterapia 2015, 102, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Palasarn, S.; Sappan, M.; Supothina, S.; Boonpratuang, T. Hirsutane sesquiterpenes from cultures of the basidiomycete Marasmiellus sp. BCC 22389. Nat. Prod. Bioprospect. 2016, 6, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, G.; Chi, W.C.; Pang, K.L.; Chen, J.J.; Kuo, Y.H.; Wang, Y.K.; Cha, H.J.; Chou, S.C.; Lee, T.H. Hirsutane-type sesquiterpenes with inhibitory activity of microglial nitric oxide production from the red alga-derived fungus Chondrostereum sp. NTOU4196. J. Nat. Prod. 2017, 80, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Jiang, W.H.; Liang, W.L.; Huang, J.X.; Mo, Y.F.; Ding, Y.Q.; Lam, C.K.; Qian, X.J.; Zhu, X.F.; Lan, W.J. Induced marine fungus Chondrostereum sp. as a means of producing new sesquiterpenoids chondrosterins I and J by using glycerol as the carbon source. Mar. Drugs 2014, 12, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.X.; Tan, H.B.; Chen, K.; Chen, Y.C.; Li, S.N.; Li, H.H.; Zhang, W.M. Cerrenins A-C, cerapicane and isohirsutane sesquiterpenoids from the endophytic fungus Cerrena sp. Fitoterapia 2018, 129, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, Y.M.; Singy, G.; Kaisin, M.; Eggert, H.; Djerassi, C. Terpenoids—LXXI: Chemical studies of marine invertebrates—XIV. Four representatives of a novel sesquiterpene class—The capnellane skeleton. Tetrahedron 1976, 32, 1171–1178. [Google Scholar] [CrossRef]
- Kaisin, M.; Tursch, B.; Declercq, J.P.; Germain, G.; Meerssche, M.V. Chemical studies of marine invertebrates. xl. Δ9(12)-capnellene-2β,5α,8β, 10α-tetrol, a new sesquiterpene alcohol from the soft coral Capnella imbricate. Bull. Soc. Chim. Belg. 1979, 88, 253–258. [Google Scholar] [CrossRef]
- Kaisin, M.; Braekman, J.C.; Daloze, D.; Tursch, B. Novel acetoxycapnellenes from the alcyonacean capnella imbricate. Tetrahedron 1985, 41, 1067–1072. [Google Scholar] [CrossRef]
- Morris, L.A.; Jaspars, M.; Adamson, K.; Woods, S.; Wallace, H.M. The capnellenes revisited: New structures and new biological activity. Tetrahedron 1998, 54, 12953–12958. [Google Scholar] [CrossRef]
- Hanssen, H.P.; Abranam, W.R. Sesquiterpene alcohols with novel skeletons from the fungus Ceratocystis piceae (ascomycotina). Tetrahedron 1988, 44, 2175–2180. [Google Scholar] [CrossRef]
- Huang, Z.L.; Dan, Y.; Huang, Y.C.; Lin, L.D.; Li, T.H.; Ye, W.H.; Wei, X.Y. Sesquiterpenes from the mycelial cultures of Dichomitus squalens. J. Nat. Prod. 2004, 67, 2121–2123. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, A.G.; Kuznetsov, D.M. Triquinanes and related sesquiterpenes revisited computationally: Structure corrections of hirsutanols B and D, hirsutenol E, cucumin B, antrodins C–E, chondroterpenes A and H, chondrosterins C and E, dichrocephone A, and pethybrene. J. Org. Chem. 2017, 82, 10795–10802. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, W.D.; Deng, R.; Zhang, H.; Tang, J.; Wu, K.W.; Li, D.D.; Feng, G.K.; Lan, W.J.; Li, H.J.; Zhu, X.F. Hirsutanol A, a novel sesquiterpene compound from fungus Chondrostereum sp., induces apoptosis and inhibits tumor growth through mitochondrial-independent ROS production: Hirsutanol A inhibits tumor growth through ROS production. J. Transl. Med. 2013, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Shindo, M.; Sato, Y.; Shishido, K. An ynolate-initiated tandem process giving cyclopentenones: Total synthesis of cucumin E. Tetrahedron Lett. 2002, 43, 5039–5041. [Google Scholar] [CrossRef]
- Mehta, G.; Murthy, A.S.K. The first total synthesis of the novel triquinane natural products pleurotellol and pleurotellic acid. Tetrahedron Lett. 2003, 44, 5243–5246. [Google Scholar] [CrossRef]
- Sakan, F.; Hashimoto, H.; Ichihara, A.; Shirahama, H.; Matsumoto, T. A synthesis of the hirsutane skeleton. Tetrahedron Lett. 1971, 12, 3703–3706. [Google Scholar] [CrossRef]
- Hashimoto, H.; Tsuzuki, K.; Sakan, F.; Shirahama, H.; Matsumoto, T. Total synthesis of dl-hirsutic acid. Tetrahedron Lett. 1974, 15, 3745–3748. [Google Scholar] [CrossRef]
- Kueh, J.S.H.; Mellor, M.; Pattenden, G. Photocyclisations of dicyclopent-1-enyl methanes to tricyclo[6.3.0.02,6]undecanes: A synthesis of the hirsutane carbon skeleton. J. Chem. Soc. Chem. Commun. 1978, 1, 5–6. [Google Scholar] [CrossRef]
- Mehta, G.; Reddy, A.V. Olefin metathesis in polycyclic frames. A total synthesis of hirsutene. J. Chem. Soc. Chem. Commun 1981, 15, 756–757. [Google Scholar] [CrossRef]
- Dawson, B.A.; Ghosh, A.K.; Jurlina, J.L.; Ragauskas, A.J.; Stothers, J.B. A synthesis of hirsutene: A simple route via β-enolization. Can. J. Chem. 1984, 62, 2521–2525. [Google Scholar] [CrossRef]
- Coverdhan, M.; Murthy, A.N.; Reddy, D.S.; Reddy, A.V. A general approach to linearly fused triquinane natural products. Total syntheses of (±)-hirsutene, (±)-coriolin, and (±)-capnellene. J. Am. Chem. Soc. 1986, 108, 3443–3452. [Google Scholar] [CrossRef]
- Iyoda, M.; Kushida, T.; Kitami, S.; Oda, M. An extremely short synthesis of hirsutene. J. Chem. Soc. Chem. Commun. 1986, 1049–1050. [Google Scholar] [CrossRef]
- Cossy, J.; Belotti, D.; Pete, J.P. Photoreductive cyclization: Application to the total synthesis of (±) Hirsutene. Tetrahedron Lett. 1987, 28, 4547–4550. [Google Scholar] [CrossRef]
- Majetich, G.; Defauw, J. Intramolecular additions of allylsilanes in triquinane synthesis. Studies directed toward the total synthesis of (±)-hirsutene. Tetrahedron 1988, 44, 3833–3849. [Google Scholar] [CrossRef]
- Franck-Neumann, M.; Miesch, M.; Lacroix, E.; Metz, B.; Kern, J.M. Access to the Linear Triquinane Skeleton via Bicyclo (2.1. 0) pentane Intermediates. Total Synthesis of the Triquinane Hirsutene. Tetrahedron 1992, 48, 1911–1926. [Google Scholar] [CrossRef]
- Franck-Neumann, M.; Miesch, M.; Lacroix, E. Total synthesis of the natural linear triquinane (±)-hirsutene from a cyclopropene compound. Tetrahedron Lett. 1989, 30, 3529–3532. [Google Scholar] [CrossRef]
- Hong, B.C.; Shr, Y.J.; Wu, J.L.; Gupta, A.K.; Lin, K.J. Novel [6 + 2] cycloaddition of fulvenes with alkenes: A facile synthesis of the anislactone and hirsutane framework. Org. Lett. 2002, 4, 2249–2252. [Google Scholar] [CrossRef] [PubMed]
- Sternbach, D.D.; Ensinger, C.L. Synthesis of polyquinanes. 3. Total synthesis of (±)-hirsutene: The intramolecular Diels-Alder approach. J. Org. Chem. 1990, 55, 2725–2736. [Google Scholar] [CrossRef]
- Castro, J.; Sorensen, H.; Riera, A.; Morin, C.; Moyano, A.; Greene, A.E. Asymmetric approach to Pauson-Khand bicyclization. Enantioselective formal synthesis of hirsutene. J. Am. Chem. Soc. 1990, 112, 9388–9389. [Google Scholar] [CrossRef]
- Paquette, L.A.; Moriarty, K.J.; Chang, S.C. Sequential annulation in molecular construction. A short, stereocontrolled synthesis of (+)-hirsutene. Israel J. Chem. 1991, 31, 195–198. [Google Scholar] [CrossRef]
- Sarkar, T.K.; Ghosh, S.K.; Subba Rao, P.S.V.; Mamdapur, V.R. Cyclopentanoid allylsilanes in synthesis: A stereoselective synthesis of (+)-hirsutene. Tetrahedron Lett. 1990, 31, 3465–3466. [Google Scholar] [CrossRef]
- Sarkar, T.K.; Ghosh, S.K.; Subba Rao, P.S.V.; Satapathi, T.K. Cyclopentanoid allysilanes in synthesis: Generation via intramolecular ene reaction of activated 1,6-dienes and application to the synthesis of functionalized diquinanes. Tetrahedron Lett. 1990, 31, 3461–3464. [Google Scholar] [CrossRef]
- Sarkar, T.K.; Ghosh, S.K.; Subba Rao, P.S.V.; Satapathi, T.K.; Mamdapur, V.R. Cyclopentanoid allylsilanes in synthesis of di- and triquinanes. A stereoselective synthesis of (±)-hirsutene. Tetrahedron 1992, 48, 6897–6908. [Google Scholar] [CrossRef]
- Mehta, G.; Srikrishna, A. Synthesis of polyquinane natural products: An update. Chem. Rev. 1997, 97, 671–719. [Google Scholar] [CrossRef] [PubMed]
- Ramig, K.; Kuzemko, M.A.; McNamara, K.; Cohen, T. Use of a dilithiomethane equivalent in a novel one-flask [2+1+2] cyclopentannulation reaction: A highly efficient total synthesis of (±)-hirsutene. J. Org. Chem. 1992, 57, 1969–1970. [Google Scholar] [CrossRef]
- Toyota, M.; Nishikawa, Y.; Motoki, K.; Yoshida, N.; Fukumoto, K. Pd2+-promoted cyclization in linear triquinane synthesis total synthesis of (±)-hirsutene. Tetrahedron Lett. 1993, 34, 6099–6102. [Google Scholar] [CrossRef]
- Rawal, V.H.; Fabre, A.; Iwasa, S. Photocyclization-fragmentation route to linear triquinanes: Stereocontrolled synthesis of (±)-endo-hirsutene. Tetrahedron Lett. 1995, 36, 6851–6854. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, Z.X. Rhodium-catalyzed [5 + 2 +1] cycloaddition of ene–vinylcyclopropanes and CO: Reaction design, development, application in natural product synthesis, and inspiration for developing new reactions for synthesis of eight-membered carbocycles. Acc. Chem. Res. 2015, 48, 2288–2296. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Liu, J.; Paquette, L.A. Three-component coupling via the squarate ester cascade as a concise route to the bioactive triquinane sesquiterpene hypnophilin. Org. Lett. 2002, 4, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Paquette, L.A.; Geng, F. Applications of the squarate ester cascade to the expeditious synthesis of hypnophilin, coriolin, and ceratopicanol. J. Am. Chem. Soc. 2002, 124, 9199–9203. [Google Scholar] [CrossRef] [PubMed]
- Bon, D.J.Y.D.; Banwell, M.G.; Ward, J.S.; Willis, A.C. From toluene to triquinanes: Formal total syntheses of the sesquiterpenoid natural products (−)-hypnophilin and (−)-coriolin. Tetrahedron 2013, 69, 1363–1368. [Google Scholar] [CrossRef]
- Bon, D.J.Y.D.; Banwell, M.G.; Willis, A.C. A chemoenzymatic total synthesis of the hirsutene-type sesquiterpene (+)-connatusin B from toluene. Tetrahedron 2010, 66, 7807–7814. [Google Scholar] [CrossRef]
- Bon, D.J.Y.D.; Banwell, M.G.; Cade, I.A.; Willis, A.C. The total synthesis of (−)-connatusin A, a hirsutane-type sesquiterpene isolated from the fungus Lentinus connatus BCC8996. Tetrahedron 2011, 67, 8348–8352. [Google Scholar] [CrossRef]
- Singh, V.; Prathap, S.; Porinehu, M. A novel, stereospecific total synthesis of (±)-∆9(12)-capnellene from p-Cresol. Tetrahedron Lett. 1997, 38, 2911–2914. [Google Scholar] [CrossRef]
- Stevens, K.E.; Paquette, L.A. Stereocontrolled total synthesis of (±)-∆9(12)-capnellene. Tetrahedron Lett. 1981, 22, 4393–4396. [Google Scholar] [CrossRef]
- Stille, J.R.; Grubbs, R.H. Synthesis of (±)-∆9(12)-capnellene using titanium reagents. J. Am. Chem. Soc. 1986, 108, 855–856. [Google Scholar] [CrossRef]
- Stille, J.R.; Santarsiero, B.D.; Grubbs, R.H. Rearrangement of bicyclo[2.2.1]heptane ring systems by Titanocene alkylidene complexes to bicyclo[3.2.0]heptane enol ethers. Total synthesis of (±)-∆9(12)-capnellene. J. Org. Chem. 1990, 55, 843–862. [Google Scholar] [CrossRef]
- Augusto, G.; Giovanni, F.; Paolo, B. Bicyclo[3.3.1]nonane approach to triquinanes. Formal synthesis of (±)-∆9(12)-capnellen and (±)-∆9(12)-capnellen-8β-10α-diol. Tetrahedron 1992, 48, 4459–4464. [Google Scholar]
- Balme, G.; Bouyssi, D. Total synthesis of the triquinane marine sesquiterpene (±)-∆9(12)-capnellene using a palladium-catalyzed bis-cyclization step. Tetrahedron 1994, 50, 403–414. [Google Scholar] [CrossRef]
- Singh, V.; Prathap, S.; Porinchu, M. Aromatics to triquinanes: P-cresol to (±)-∆9(12)-capnellene. J. Org. Chem. 1998, 63, 4011–4017. [Google Scholar] [CrossRef]
- Nguyen, N.N.M.; Leclere, M.; Stogaitis, N.; Fallis, A.G. Triquinanes: A “One-Pot” IMDA-Tandem metathesis cascade strategy: Ring-Closing Metathesis (RCM) dominates norbornene ROM! Org. Lett. 2010, 12, 1684–1687. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.S.; Chou, Y.Y.; Tung, Y.S.; Liao, C.C. Photochemistry of tricyclo[5.2.2.02,6]undeca-4,10-dien-8-ones: An efficient general route to substituted linear triquinanes from 2-methoxyphenols. Total synthesis of (±)-∆9(12)-capnellene. Chem.-Eur. J. 2010, 16, 3121–3131. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.J.; Li, W.D.Z. Formal homoiodo allylsilane annulations: Dual total syntheses of (±)-hirsutene and (±)-capnellene. J. Org. Chem. 2013, 78, 7112–7120. [Google Scholar] [CrossRef] [PubMed]
- Little, R.D.; Carroll, G.L.; Petersen, J.L. Total synthesis of the marine natural product ∆9(12)-capnellene. Reversal of regiochemistry in the intramolecular 1,3-diyl trapping reaction. J. Am. Chem. Soc. 1983, 105, 928–932. [Google Scholar] [CrossRef]
- Paquette, L.A.; Stevens, K. Stereocontrolled total synthesis of the triquinane marine sesquiterpene ∆9(12)-capnellene. Can J. Chem. 1984, 62, 2415–2419. [Google Scholar] [CrossRef]
- Crisp, G.T.; Scott, W.J.; Stille, J.K. Palladium-catalyzed carbonylative coupling of vinyl triflates with organostannanes. A total synthesis of (±)-∆9(12)-capnellene. J. Am. Chem. Soc. 1984, 106, 7500–7506. [Google Scholar] [CrossRef]
- Wang, Y.; Mukherjee, D.; Birney, D.; Houk, K.N. Synthesis and reactions of ester-substituted fulvenes. A new route to ∆9(12)-capnellene. J. Org. Chem. 1990, 55, 4504–4506. [Google Scholar] [CrossRef]
- Samajdar, S.; Patra, D.; Ghosh, S. Stereocontrolled approach to highly substituted cyclopentanones. Application in a formal synthesis of ∆9(12)-capnellene. Tetrahedron 1998, 54, 1789–1800. [Google Scholar] [CrossRef]
- Lemiere, G.; Gandon, V.; Cariou, K.; Hours, A.; Fukuyama, T.; Dhimane, A.L.; Fensterbank, L.; Malacria, M. Generation and trapping of cyclopentenylidene gold species: Four pathways to polycyclic compounds. J. Am. Chem. Soc. 2009, 131, 2993–3006. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, H.R.; Nanjundiah, B.S.; Shah, V.G.; Kulkarni, D.G.; Ahuja, J.R. Synthesis of naturally occurring (+)-∆9(12)-capnellene and its antipode: An application of the photo-induced vinylcyclopropane-cyclopentene rearrangement. Tetrahedron Lett. 1991, 1991. 32, 1107–1108. [Google Scholar] [CrossRef]
- Sonawane, H.R.; Naik, V.G.; Bellur, N.S.; Shah, V.G.; Purohit, P.C.; Kumar, M.U.; Kulltarni, D.G.; Ahuja, J.R. Photoinduced vinylcyclopropane-cyclopentene rearrangement: A methodology for chiral bicyclo[3.2.0]heptenes. Formal syntheses of (±)-grandisol and naturally occurring (+)-∆9(12)-capnellene antipode. Tetrahedron 1991, 47, 8259–8276. [Google Scholar] [CrossRef]
- Asaoka, M.; Obuchi, K.; Takei, H. An Enantioselective route to (−)-∆9(12)-capnellene employing silyl group directed stereo control. Tetrahedron 1994, 50, 655–660. [Google Scholar] [CrossRef]
- Tanaka, K.; Ogasawara, K. Stereocontrolled synthesis of natural (−)-∆9(12)-capnellene from a (−)-oxodicyclopentadiene. Chem. Commun. 1996, 15, 1839–1840. [Google Scholar] [CrossRef]
- Ohshima, T.; Kagechika, K.; Adachi, M.; Sodeoka, M.; Shibasaki, M. Asymmetric heck reaction-carbanion capture process. Catalytic asymmetric total synthesis of (−)-∆9(12)-capnellene. J. Am. Chem. Soc. 1996, 118, 7108–7116. [Google Scholar] [CrossRef]
- Meyers, A.I.; Bienz, S. Asymmetric total synthesis of (+)-∆9(12)-capnellene. J. Org. Chem. 1990, 55, 791–798. [Google Scholar] [CrossRef]
- Shibasaki, M.; Mase, T.; Ikegami, S. The first total syntheses of ∆9(12)-capnellene-8β,10α-diol and ∆9(12)-capnellene-3β,8β,10α-triol. J. Am. Chem. Soc. 1986, 108, 2090–2091. [Google Scholar] [CrossRef]
- Kagechika, K.; Shibasaki, M. Asymmetric heck reaction: A catalytic asymmetric synthesis of the key intermediate for ∆9(12)-capnellene-3β,8β,10α-triol and ∆9(12)-capnellene-3β,8β,10α,14-tetrol. J. Org. Chem. 1991, 56, 4093–4094. [Google Scholar] [CrossRef]
- Baralotto, C.; Chanon, M.; Julliard, M. Total synthesis of the tricyclic sesquiterpene (±)-ceratopicanol. An illustration of the holosynthon concept. J. Org. Chem. 1996, 61, 3576–3577. [Google Scholar] [CrossRef] [PubMed]
- Clive, D.L.J.; Magnuson, S.R.; Manning, H.W.; Mayhew, D.L. Cyclopentannulation by an iterative process of sequential claisen rearrangement and enyne radical closure: Routes to triquinane and propellane systems and use in the synthesis of (±)-ceratopicanol. J. Org. Chem. 1996, 61, 2095–2108. [Google Scholar] [CrossRef]
- Srikrishna, A.; Dethe, D.H. Enantiospecific first total synthesis and assignment of absolute configuration of the sesquiterpene (−)-cucumin H. Org. Lett. 2003, 5, 2295–2298. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal secondary metabolism from biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Skeleton of linear triquinane sesquiterpenoids.

Figure 2.
Structure types of linear triquinane sesquiterpenoids.

Figure 3.
Structures of type I compounds.




Figure 4.
Structures of type II compounds.

Figure 5.
Structures of type III compounds.

Figure 6.
Structures of type VI–VIII compounds.

Figure 7.
Structures of compounds 119–121.

Scheme 1.
The simple expedient synthesis of (+)-hirsutene ((+)-7) via a novel photo-thermal metathetic sequence.
Scheme 1.
The simple expedient synthesis of (+)-hirsutene ((+)-7) via a novel photo-thermal metathetic sequence.

Scheme 2.
The total synthesis of (±)-hirsutene ((±)-7).

Scheme 3.
The total synthesis of (±)-coriolin ((±)-2).

Scheme 4.
The short synthesis of (±)-7.

Scheme 5.
The synthesis of (±)-7 utilizing photochemical reduction.

Scheme 6.
The synthesis of (±)-7 via the intramolecular addition.

Scheme 7.
The synthesis of (±)-7 via the region and stereospecific cleavage.

Scheme 8.
The highly efficient synthesis of (±)-7.

Scheme 9.
The enantioselective formal synthesis of hirsutene (7).

Scheme 10.
The total synthesis of (±)-7 via a twofold cycloaddition.

Scheme 11.
The formal synthesis of (±)-7 via an intramolecular ene reaction and a titanium catalyzed epoxy-allylsilane cyclization.
Scheme 11.
The formal synthesis of (±)-7 via an intramolecular ene reaction and a titanium catalyzed epoxy-allylsilane cyclization.

Scheme 12.
The total synthesis of (±)-7 via a novel one-flask [2+1+2] cyclopentannulation reaction.

Scheme 13.
The synthesis of 7 utilizing an acid catalyzed intramolecular conjugate addition and a Pd2+-promoted highly stereocontrolled cyclization.
Scheme 13.
The synthesis of 7 utilizing an acid catalyzed intramolecular conjugate addition and a Pd2+-promoted highly stereocontrolled cyclization.

Scheme 14.
The stereocontrolled synthesis of (±)-endo-hirsutene (149) via intramolecular Diels-Alder, Paterno-Buchi reactions and a reductive fragmentation.
Scheme 14.
The stereocontrolled synthesis of (±)-endo-hirsutene (149) via intramolecular Diels-Alder, Paterno-Buchi reactions and a reductive fragmentation.

Scheme 15.
The synthesis of (±)-7.

Scheme 16.
The synthesis of (+)-hirsutene ((+)-7).

Scheme 17.
The synthesis of 1-desoxyhypnophilin (155).

Scheme 18.
The synthesis of hirsutic acid C (1).

Scheme 19.
The facile synthesis of the hirsutane framework.

Scheme 20.
The synthesis of hypnophilin (8) via a combination of chlorination, reduction, dehydration and oxidation maneuvers.
Scheme 20.
The synthesis of hypnophilin (8) via a combination of chlorination, reduction, dehydration and oxidation maneuvers.

Scheme 21.
The conversion of diisopropyl squarate to 8.

Scheme 22.
The formal synthesis of racemic coriolin (2).

Scheme 23.
The synthesis of ceratopicanol (109).

Scheme 24.
The synthesis of compound 159, an advanced intermediate of both compounds (−)-8 and (−)-2 and the synthesis of the isomeric tetracyclic system 161, a late-stage precursor to (−)-2.
Scheme 24.
The synthesis of compound 159, an advanced intermediate of both compounds (−)-8 and (−)-2 and the synthesis of the isomeric tetracyclic system 161, a late-stage precursor to (−)-2.

Scheme 25.
The synthesis of (+)-connatusin B ((+)-34) via reductive cleavage of the three-membered ring within this framework and kinds of functional group interconversions.
Scheme 25.
The synthesis of (+)-connatusin B ((+)-34) via reductive cleavage of the three-membered ring within this framework and kinds of functional group interconversions.

Scheme 26.
The synthesis of (−)-connatusin A ((−)-33).

Figure 8.
Structures of compounds 178–181.

Scheme 27.
The synthesis of (±)-∆9(12)-capnellene (178) by using titanium reagents.

Scheme 28.
The total synthesis of 178 through the rearrangement of bicyclo[2.2.1]heptane ring systems.
Scheme 28.
The total synthesis of 178 through the rearrangement of bicyclo[2.2.1]heptane ring systems.

Scheme 29.
The total synthesis of both 178 and 182.

Scheme 30.
The synthesis of 186 from a new palladium catalyzed cyclisation.

Scheme 31.
The total synthesis of 178 using a palladium-catalyzed bis-cyclization step.

Scheme 32.
The total synthesis of 178 from p-cresol employing π4s + π2s cycloaddition.

Scheme 33.
The formal synthesis of 178 via an intramolecular Diels-Alder reaction tandem metathesis protocol.
Scheme 33.
The formal synthesis of 178 via an intramolecular Diels-Alder reaction tandem metathesis protocol.

Scheme 34.
The total synthesis of 178 by using the strategy of masked o-benzoquinone chemistry.

Scheme 35.
The total syntheses of (±)-7 and 178 via a formal [3 + 2] annulation strategy.

Scheme 36.
The total synthesis of ∆9(12)-capnellene (179) by using an intramolecular 1, 3-diyl trapping reaction as a primary strategy.
Scheme 36.
The total synthesis of ∆9(12)-capnellene (179) by using an intramolecular 1, 3-diyl trapping reaction as a primary strategy.

Scheme 37.
The stereocontrolled total synthesis of 179.

Scheme 38.
The total synthesis of 179 via palladium-catalyzed carbonylative coupling of vinyl triflates with organostannanes.
Scheme 38.
The total synthesis of 179 via palladium-catalyzed carbonylative coupling of vinyl triflates with organostannanes.

Scheme 39.
The formal synthesis of 179 by using new type of fulvenes undergo cycloadditions regioselectively.
Scheme 39.
The formal synthesis of 179 by using new type of fulvenes undergo cycloadditions regioselectively.

Scheme 40.
The formal synthesis of 179 via the construction of cyclopentanones with substituents up to three contiguous chiral centres.
Scheme 40.
The formal synthesis of 179 via the construction of cyclopentanones with substituents up to three contiguous chiral centres.

Scheme 41.
The synthesis of 179.

Scheme 42.
The formal asymmetric synthesis of (−)-capnellene (180).

Scheme 43.
The formal synthesis of 180 by employing silyl group directed stereo control.

Scheme 44.
The stereocontrolled synthesis of 180 based on the structure and high functionality of the starting material.
Scheme 44.
The stereocontrolled synthesis of 180 based on the structure and high functionality of the starting material.

Scheme 45.
The first catalytic asymmetric total synthesis of 180 by using 196 as an asymmetric building block.
Scheme 45.
The first catalytic asymmetric total synthesis of 180 by using 196 as an asymmetric building block.

Scheme 46.
The first asymmetric total synthesis of the unnatural enantiomer of (+)-∆9(12)-capnellene (181).
Scheme 46.
The first asymmetric total synthesis of the unnatural enantiomer of (+)-∆9(12)-capnellene (181).

Scheme 47.
The first total syntheses of ∆9(12)-capnellene-8β,10α-diol (87) and ∆9(12)-capnellene-3β,8β, 10α-troil (88).
Scheme 47.
The first total syntheses of ∆9(12)-capnellene-8β,10α-diol (87) and ∆9(12)-capnellene-3β,8β, 10α-troil (88).

Scheme 48.
The synthesis of 87 and 88.

Scheme 49.
The total synthesis of racemic (±)-ceratopicanol ((±)-109) via intramolecular meta photocycloaddition.
Scheme 49.
The total synthesis of racemic (±)-ceratopicanol ((±)-109) via intramolecular meta photocycloaddition.

Scheme 50.
The synthesis of 109.

Scheme 51.
The first total synthesis of cucumin E (78) by using an ynolate-initiated tandem reaction for the construction of the cyclopentenone unit.
Scheme 51.
The first total synthesis of cucumin E (78) by using an ynolate-initiated tandem reaction for the construction of the cyclopentenone unit.

Scheme 52.
The first total synthesis of the (−)-cucumin H ((−)-110).

Scheme 53.
The first total synthesis of pleurotellic acid (115) and pleurotellol (114).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
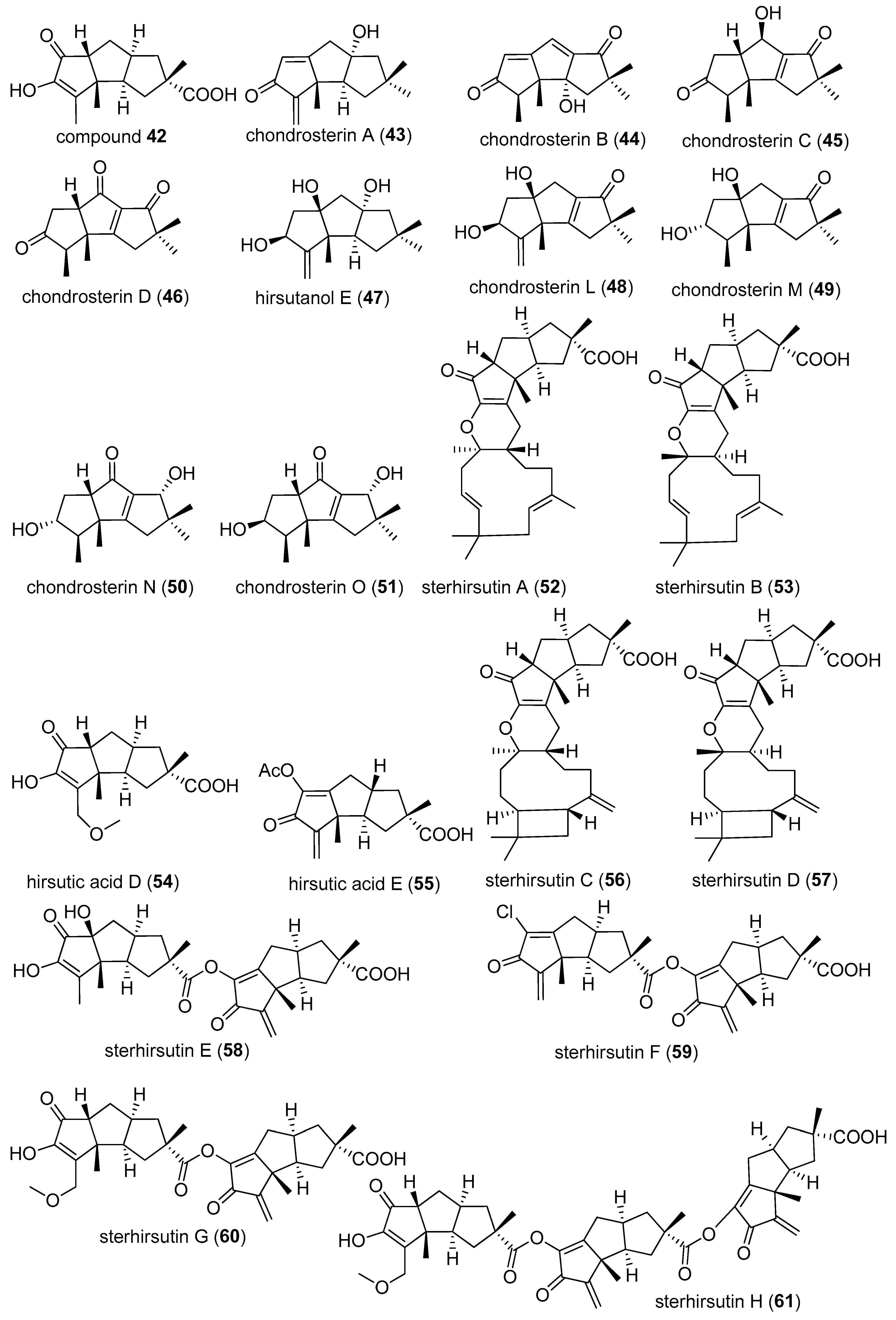{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
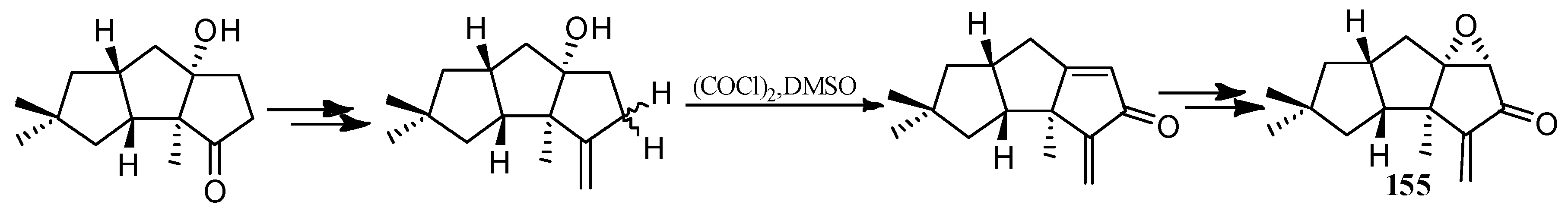{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Cytotoxicity of linear triquinane sesquiterpenoids.
| Cell Lines | Cytotocicity |
|---|---|
| CNE1 | 15 IC50 = 34.13 μM, 22 IC50 = 10.08 μM, 48 IC50 = 33.55 μM, 49 IC50 = 42.00 μM, 84 IC50 = 1.32 μM, 85 IC50 = 17.66 μM |
| CNE2 | 15 IC50 = 24.87 μM, 22 IC50 = 12.72 μM, 43 IC50 = 4.95μM, 48 IC50 = 22.50 μM, 49 IC50 = 44.08 μM, 84 IC50 = 0.56 μM, 85 IC50 = 12.03 μM |
| HONE1 | 22 IC50 = 17.40 μM, 48 IC50 = 34.60 μM, 49 IC50 = 46.11 μM, 85 IC50 = 22.06 μM |
| SUNE1 | 22 IC50 = 3.50 μM, 48 IC50 = 30.40 μM, 49 IC50 = 58.83 μM, 85 IC50 = 16.44 μM |
| A549 | 22 IC50 = 11.96 μM, 43 IC50 = 2.45μM, 48 IC50 = 29.67 μM, 49 IC50 = 49.58 μM, 85 IC50 = 23.51 μM |
| GLC82 | 22 IC50 = 10.11 μM, 48 IC50 = 37.47 μM, 49 IC50 = 55.90 μM, 85 IC50 = 18.08 μM |
| HL7702 | 22 IC50 = 9.76 μM, 48 IC50 = 34.26 μM, 49 IC50 = 56.40 μM, 85 IC50 = 22.14 μM |
| K562 | 52 IC50 = 12.97 μg/mL, 53 IC50 = 16.29 μg/mL, 54 IC50 = 6.93 μg/mL, 55 IC50 = 30.52 μg/mL, 87 IC50 = 0.7 μM, 96 IC50 = 4.6 μM, 97 IC50 = 24 μM, 98 GI50 = 126.9 ± 3.0 μm/L, 99 GI50 = 126.9 ± 2.0 μm/L, 100 GI50 = 142.0 ± 4.7 μm/L, 101 GI50 = 142.0 ± 4.7 μm/L, Doxorubicin GI50 = 1.0 ± 0.6 μm/L |
| HCT116 | 52 IC50 = 10.74 μg/mL, 53 IC50 = 16.35 μg/mL, 54 IC50 = 25.43 μg/mL, 55 IC50 = 24.17 μg/mL |
| HL-60 | 87 IC50 = 51 μM, 96 IC50 = 68 μM, 97 IC50 = 713 μM |
| G402 | 87 IC50 = 42–51 μM, 96 IC50 > 4500 μM, 97 IC50 = 52 μM |
| MCF-7 | 87 IC50 = 93 μM, 96 IC50 > 4500 μM, 97 IC50 = 1029 μM |
| HT115 | 87 IC50 = 63 μM, 96 IC50 > 4500 μM |
| A2780 | 87 IC50 = 9.7 μM, 96 IC50 = 6.6 μM, 97 IC50 = 32 μM, |
| HeLa | 98 CC50 = 126.9 ± 1.8 μm/L, 99 CC50 = 126.9 ± 2.5μm/L, 100 CC50 = 142.0 ± 2.7 μm/L, 101 CC50 = 125.0 ± 2.0 μm/L, Doxorubicin CC50 = 2.0 ± 0.8 μm/L |
| L-929 | 17 IC50 = 2.4 μg/mL, 18 IC50 = 0.9 μg/mL, 98 GI50 = 126.9 ± 2.7 μm/L, 99 GI50 = 126.9 ± 2.9 μm/L, 100 GI50 = 126.4 ± 3.0 μm/L, 101 GI50 = 99.1 ± 1.8 μm/L, Doxorubicin GI50 = 1.2 ± 0.6 μm/L |
| T-47D | 3 IC50 = 0.7 μM |
| SNB-75 | 3 IC50 = 0.5 μM |
| HepG2 | 40 IC50 = 24.41 ± 1.86 μM |
| LoVo | 43 IC50 = 5.47 μM |
Table 2.
Antimicrobial activity of compounds 17 and 18 (MIC50, μg/mL).
| Test Organisms | 17 | 18 |
|---|---|---|
| Bacillus cereus DSM 318 | 2–5 | >100 |
| Staphylococcus aureus ATCC 13709 | 10–25 | >100 |
| Escherichia coli ATCC 9637 | >100 | >100 |
| Salmonella gallinarum ATCC 9184 | 50–100 | 50–100 |
| Mycobacterium smegmatis ATCC 607 | 25–50 | 50–100 |
| Candida albicans ATCC 10231 | 50–100 | >100 |
| Candida tropicalis DSM 1346 | >100 | >100 |
| Rhodotorula glutinis DSM 70398 | >100 | >100 |
| Aspergillus niger (spores) DSM 737 | 1–2 | 25–50 |
| Aspergillus flavus (spores) BD 27 | 2–5 | 25–50 |
| Mucor rouxii (spores) DSM 1691 | 2–5 | 25–50 |
Table 3.
Inhibitory effect of compounds 22, 43–44, 69–70 and 76 in LPS-induced NO production in microglial BV-2 cells.
Table 3.
Inhibitory effect of compounds 22, 43–44, 69–70 and 76 in LPS-induced NO production in microglial BV-2 cells.
| Compounds (20 μM) | NO (μM) |
|---|---|
| 22 | 8 ± 2 |
| 43 | 4 ± 1 |
| 44 | 4 ± 1 |
| 69 | 8 ± 2 |
| 70 | 10 ± 2 |
| 76 | 4 ± 1 |
Table 4.
Effect of compounds 22, 43–44, 69–70, and 76 in LPS-induced NO production in microglial viability.
Table 4.
Effect of compounds 22, 43–44, 69–70, and 76 in LPS-induced NO production in microglial viability.
| Compounds | Viability (%) | |||
|---|---|---|---|---|
| 2 μM | 5 μM | 10 μM | 20 μM | |
| 22 | 94 ± 1 | 87 ± 3 | 77 ± 8 | 39 ± 5 |
| 43 | nt * | nt | 22 ± 3 | 12 ± 1 |
| 44 | nt | nt | 23 ± 7 | 11 ± 1 |
| 69 | 93 ± 3 | 82 ± 5 | 73 ± 4 | 67 ± 3 |
| 70 | 95 ± 5 | 95 ± 4 | 89 ± 9 | 69 ± 5 |
| 76 | 47 ± 6 | 30 ± 5 | 11 ± 2 | 11 ± 2 |
* nt = not tested.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Qiu, Y.; Lan, W.-J.; Li, H.-J.; Chen, L.-P. Linear Triquinane Sesquiterpenoids: Their Isolation, Structures, Biological Activities, and Chemical Synthesis. Molecules 2018, 23, 2095. https://doi.org/10.3390/molecules23092095
AMA Style
Qiu Y, Lan W-J, Li H-J, Chen L-P. Linear Triquinane Sesquiterpenoids: Their Isolation, Structures, Biological Activities, and Chemical Synthesis. Molecules. 2018; 23(9):2095. https://doi.org/10.3390/molecules23092095
Chicago/Turabian StyleQiu, Yi, Wen-Jian Lan, Hou-Jin Li, and Liu-Ping Chen. 2018. "Linear Triquinane Sesquiterpenoids: Their Isolation, Structures, Biological Activities, and Chemical Synthesis" Molecules 23, no. 9: 2095. https://doi.org/10.3390/molecules23092095