
Theoretical Calculations for Highly Selective Direct Heteroarylation Polymerization: New Nitrile-Substituted Dithienyl-Diketopyrrolopyrrole-Based Polymers

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
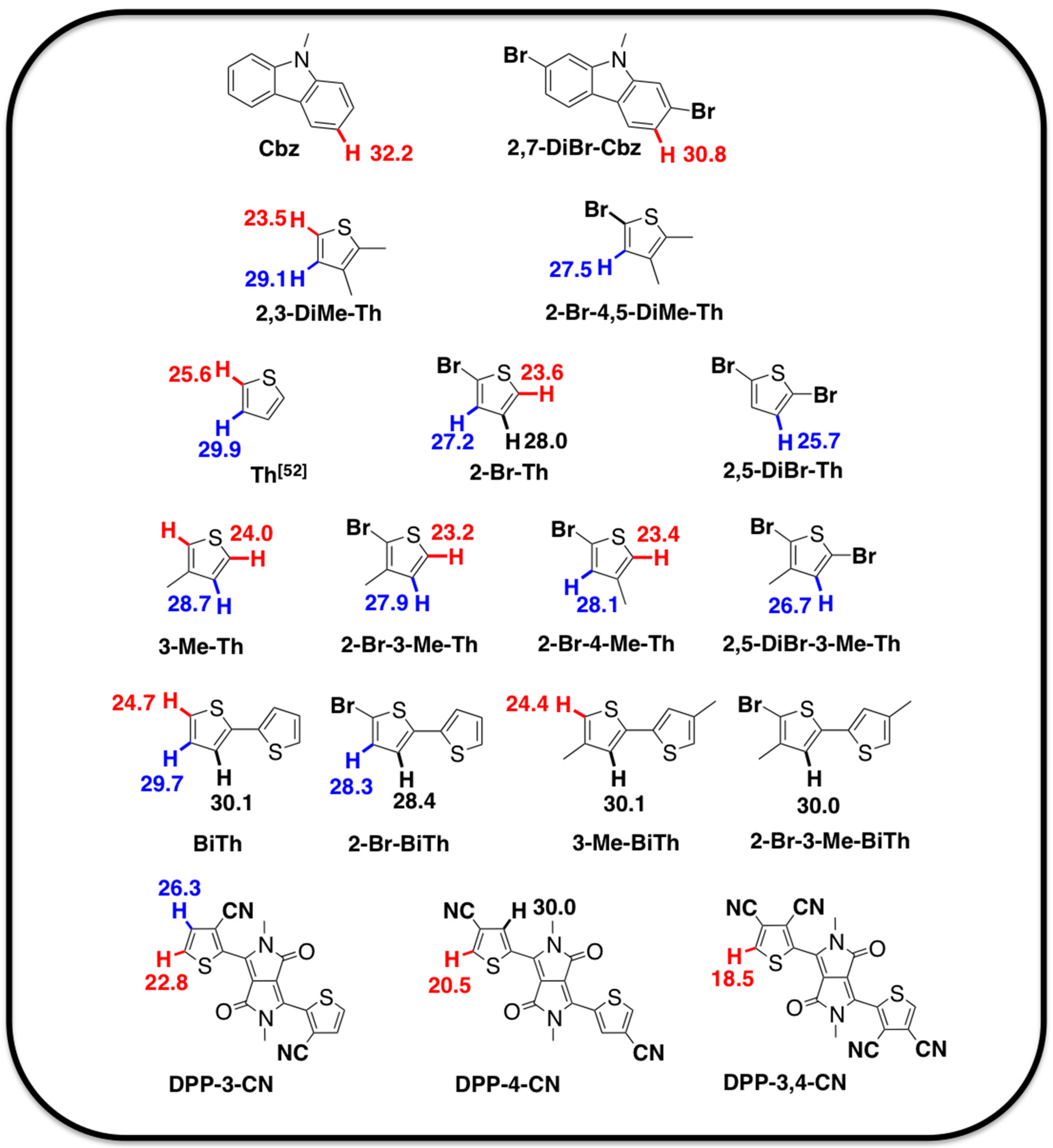
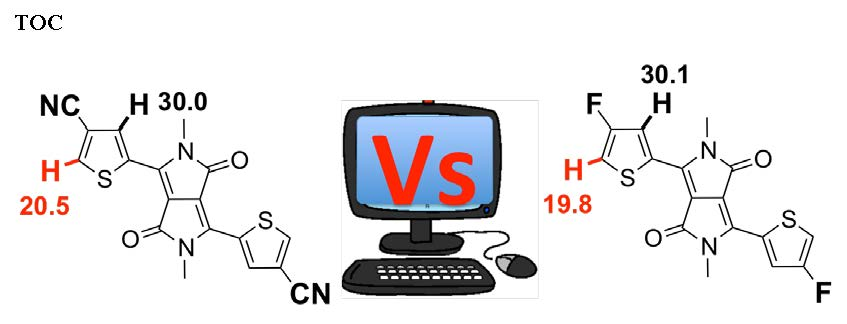
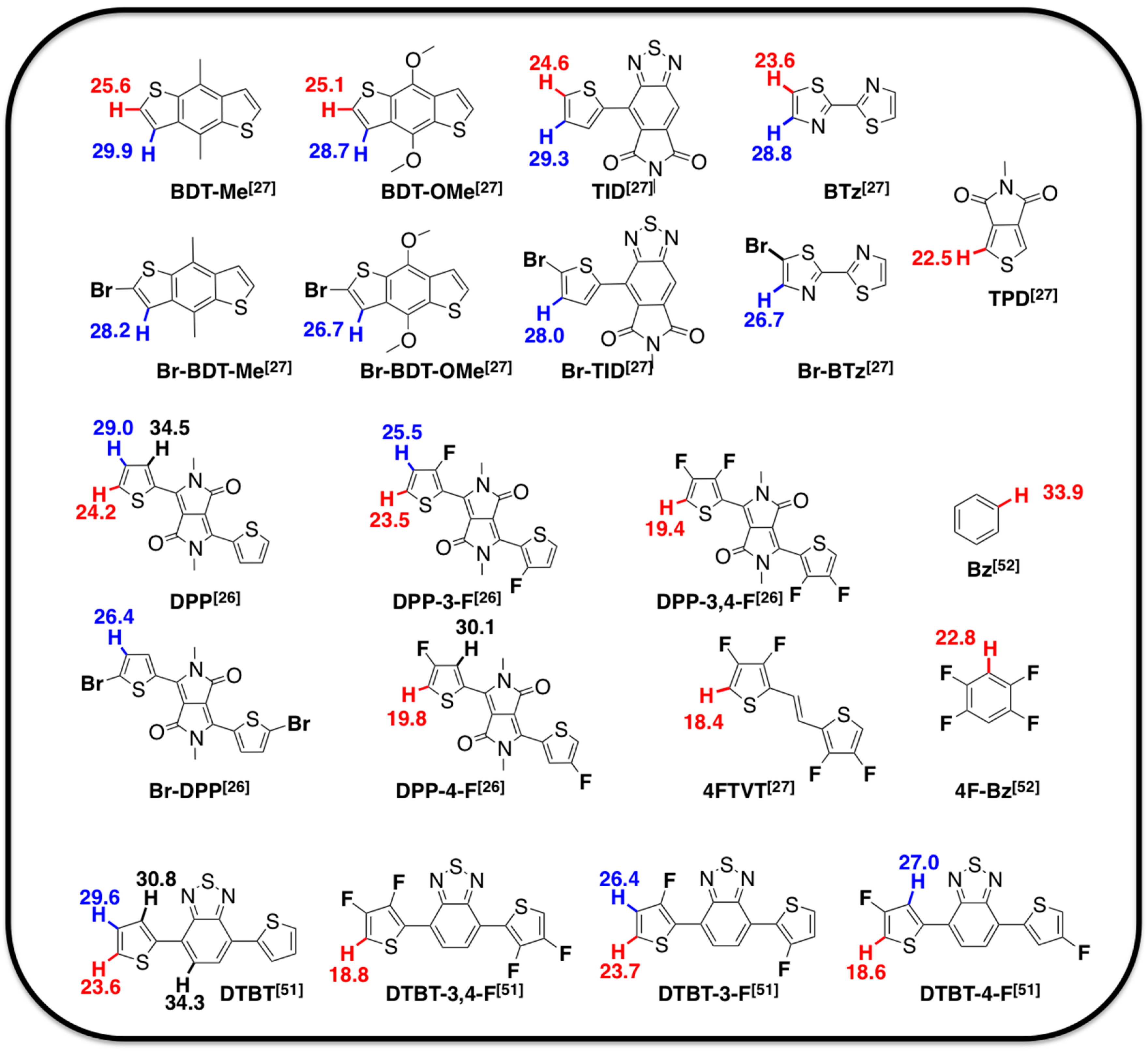
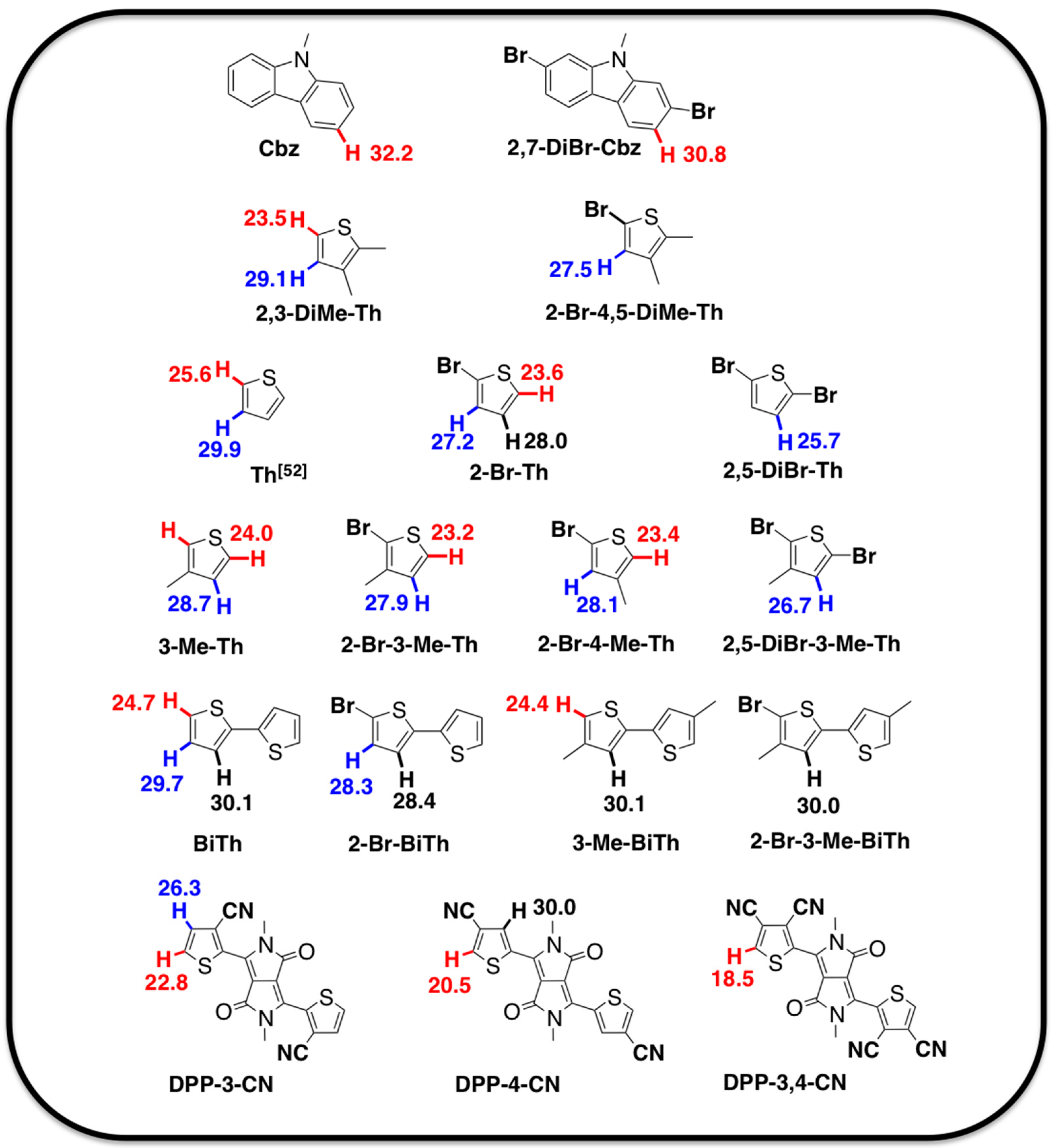
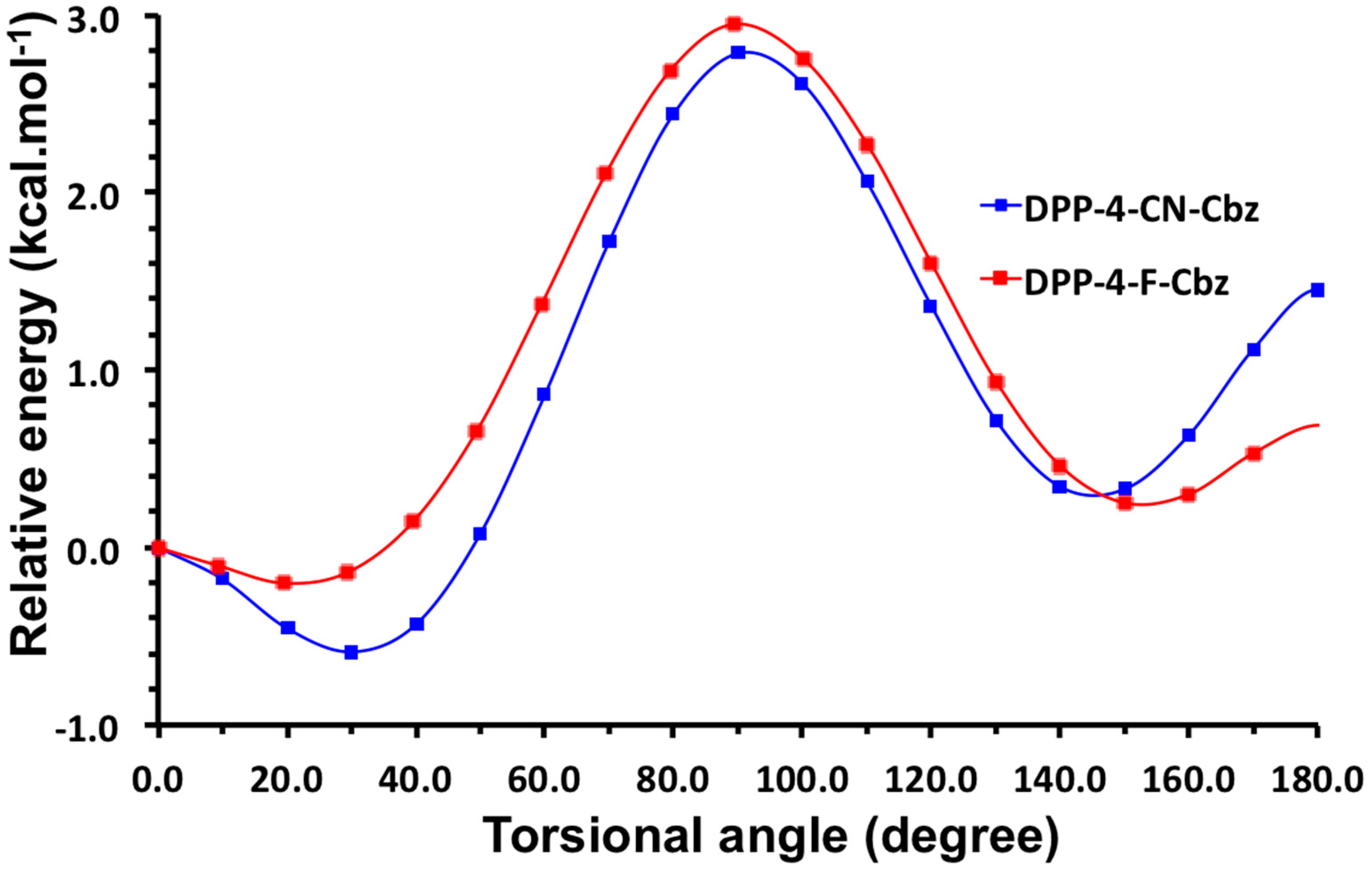
2.1. Theoretical Calculations
2.2. Justification for Cyano-Dithienyl-DPP (CNDT-DPP)
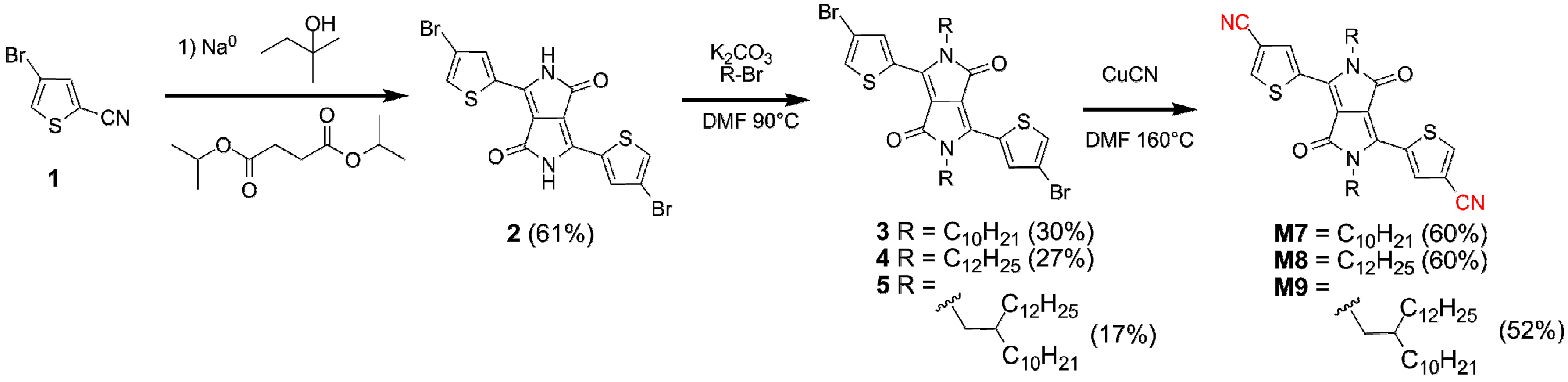
2.3. Synthesis of Monomers
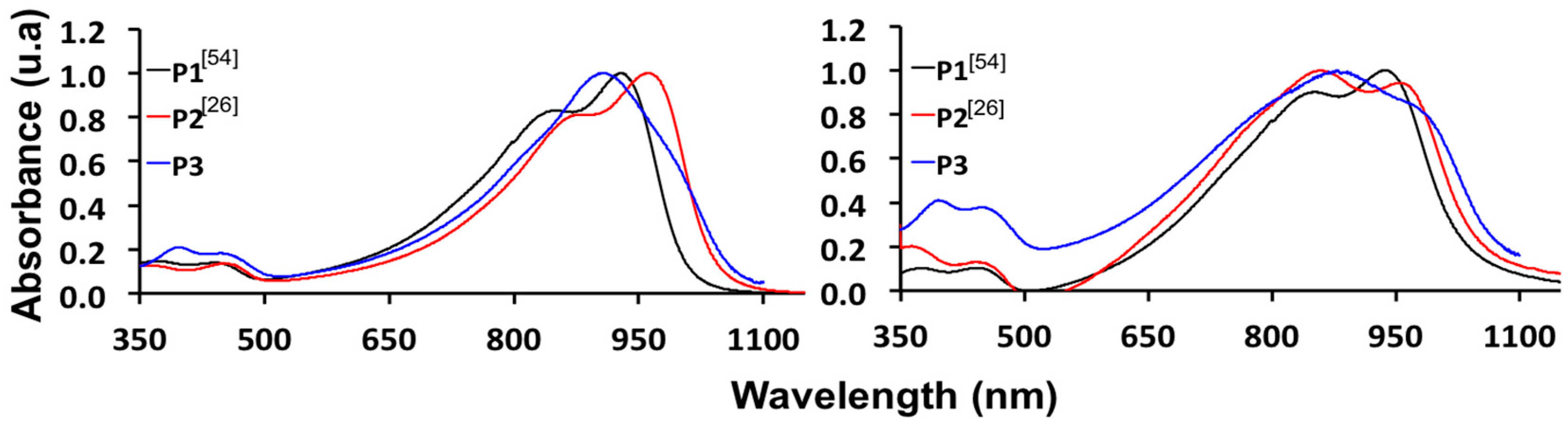
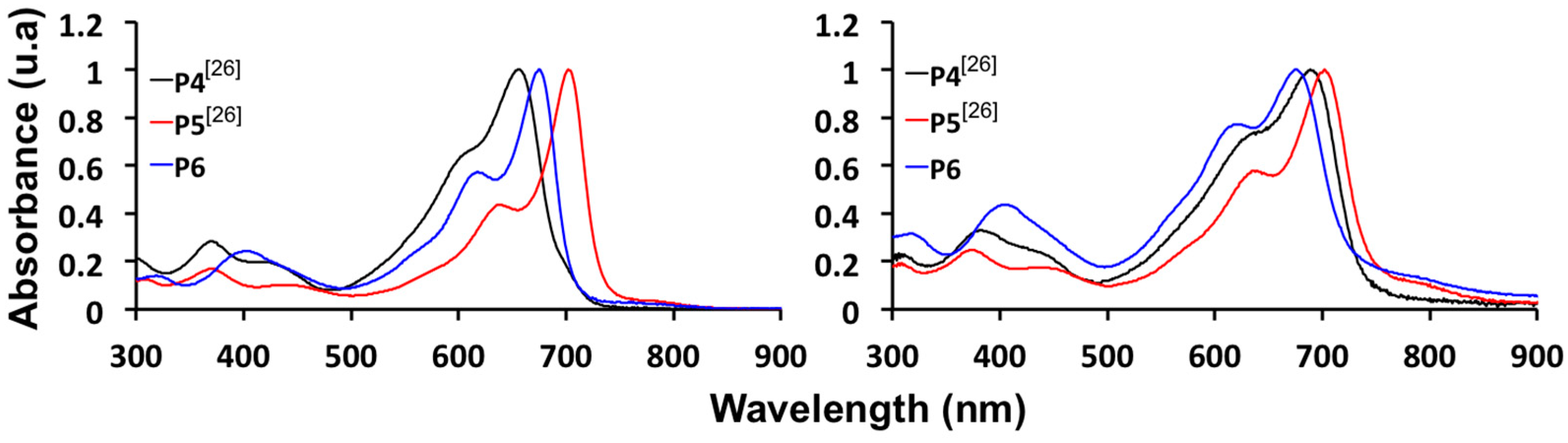
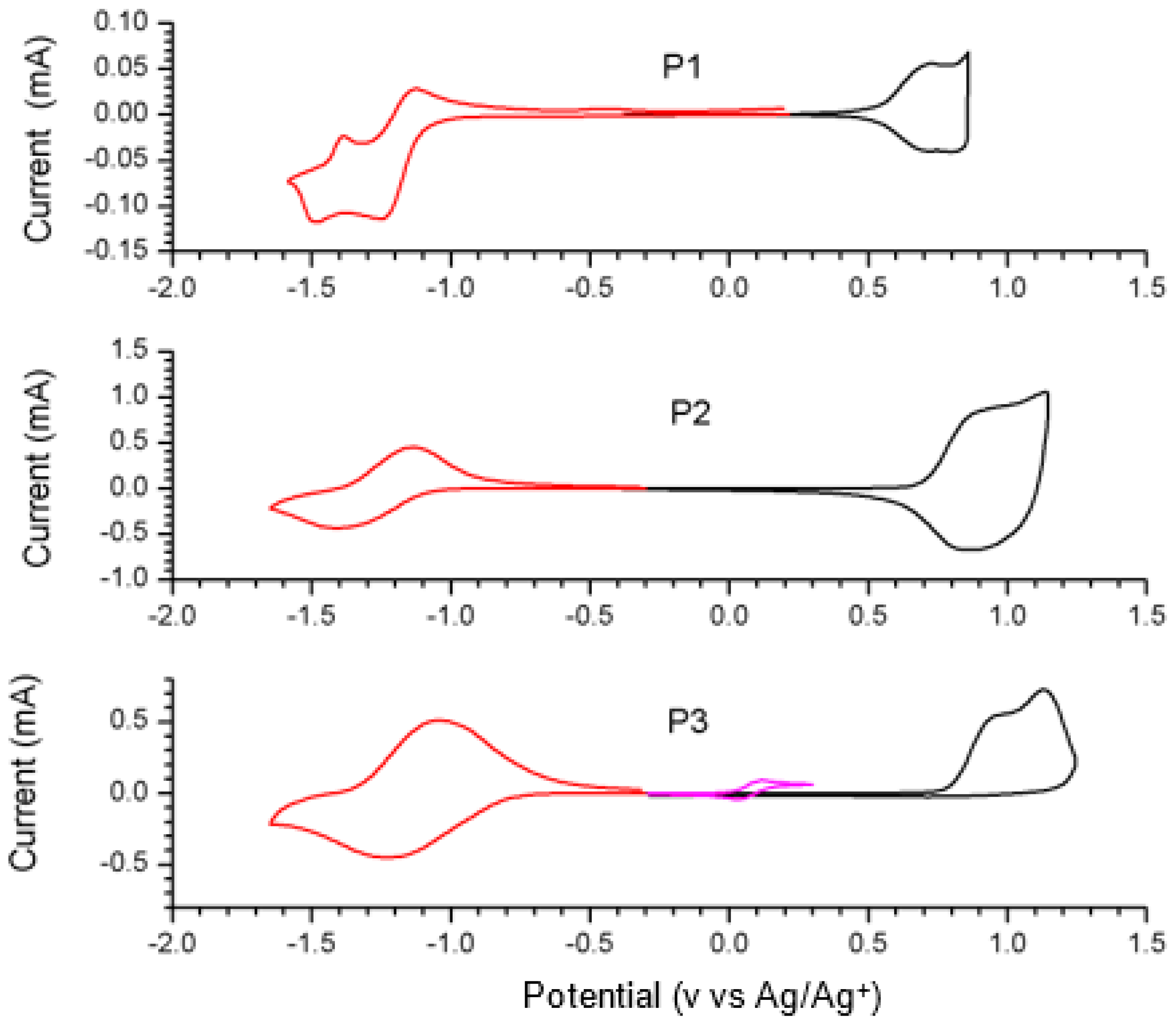
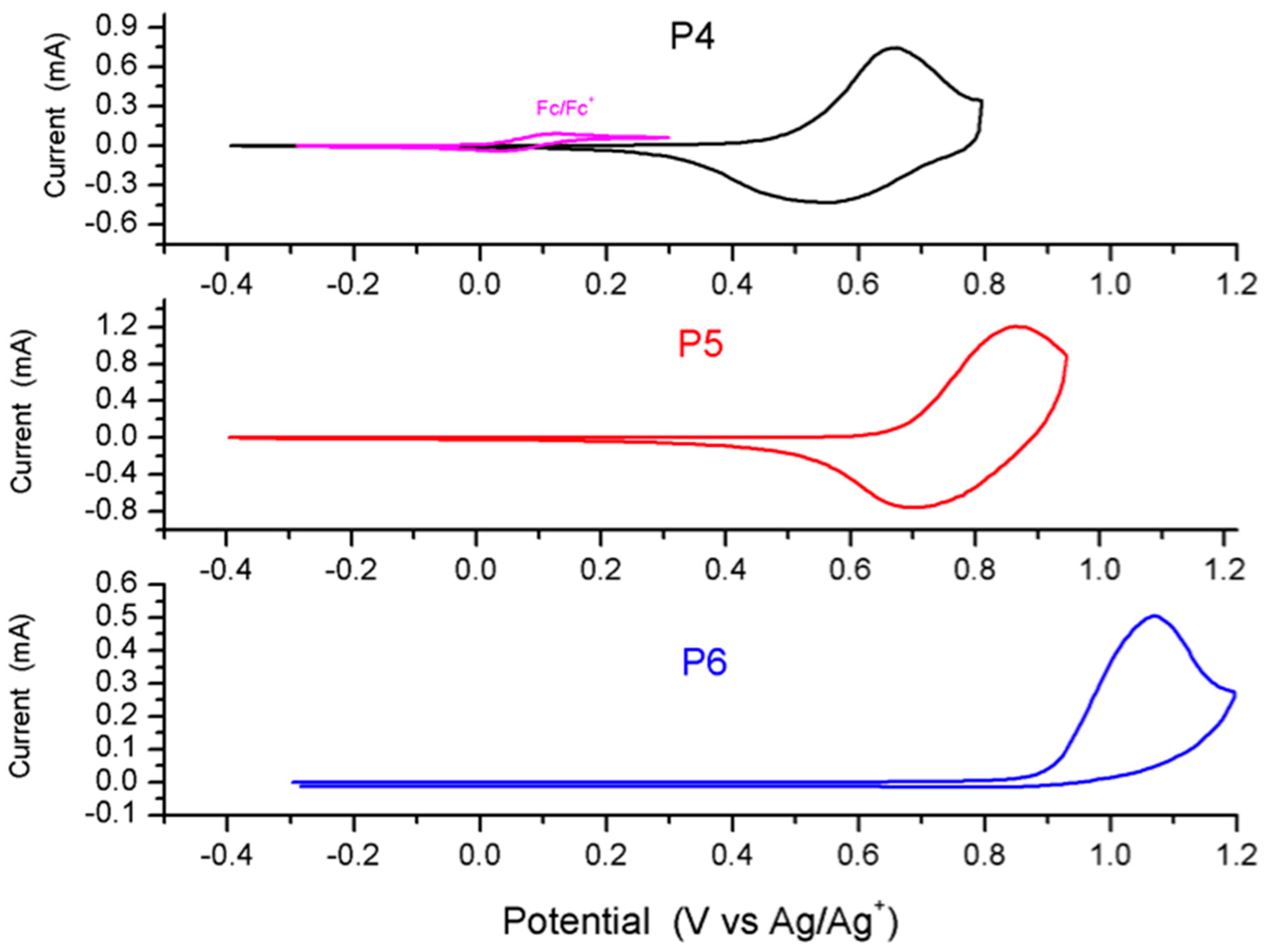
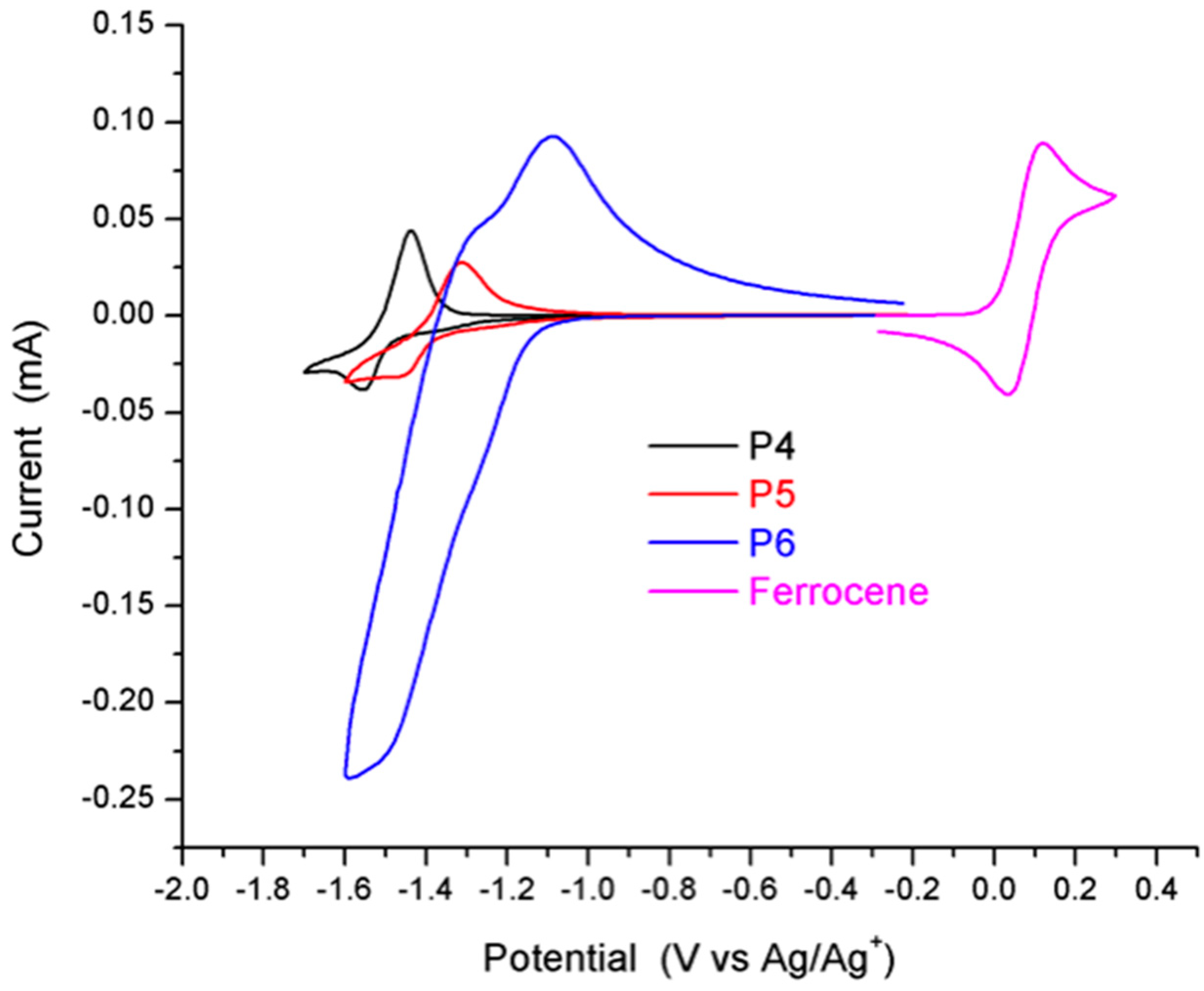
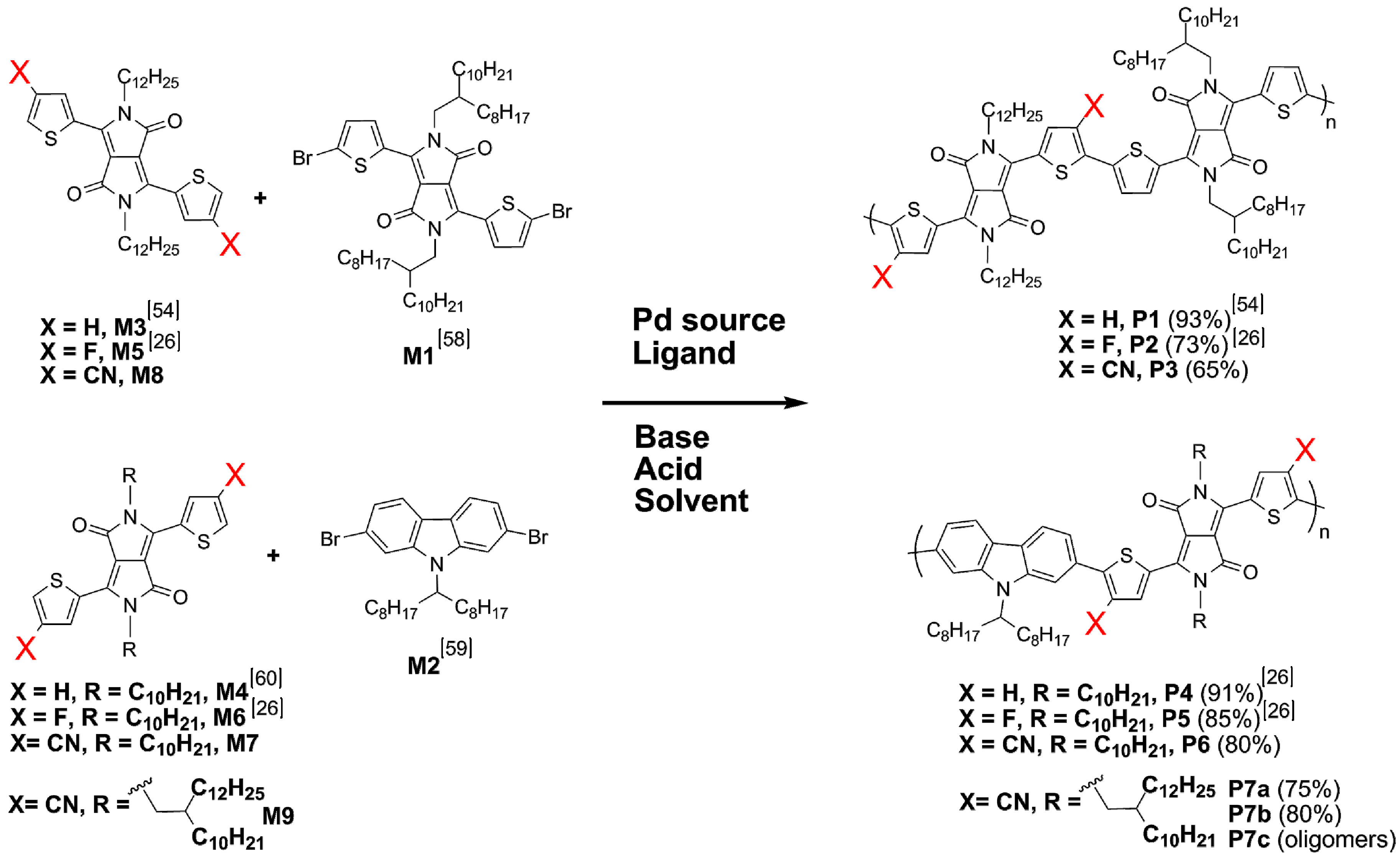
2.4. Synthesis and Characterization of Polymers
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Monomers and Polymers
3.2.1. Synthesis of 3,6-(4-Bromothiophen-2-yl)pyrrolo[3,4-c]-pyrrole-1,4-dione (2)
3.2.2. Synthesis of 3,6-(4-Bromothiophen-2-yl)-2,5-bis(decyl)-pyrrolo[3,4-c]pyrrole-1,4-dione (3)
3.2.3. Synthesis of 3,6-(4-Bromo-thiophen-2-yl)-2,5-bis(dodecyl)-pyrrolo[3,4-c]pyrrole-1,4-dione (4)
3.2.4. Synthesis of 3,6-(4-Bromo-thiophen-2-yl)-2,5-di(2′-decyltetradecyl)-pyrrolo[3,4-c]pyrrole-1,4-dione (5)
3.2.5. Synthesis of 3,6-(4-Cyano-thiophen-2-yl)-2,5-bis(decyl)-pyrrolo[3,4-c]pyrrole-1,4-dione (M7)
3.2.6. Synthesis of 3,6-(4-Cyano-thiophen-2-yl)-2,5-bis(dodecyl)-pyrrolo[3,4-c]pyrrole-1,4-dione (M8)
3.2.7. Synthesis of 3,6-(4-Cyano-thiophen-2-yl)-2,5-bis(2-decyltetradecyl)-pyrrolo[3,4-c]pyrrole-1,4-dione (M9)
3.2.8. Synthesis of P3
3.2.9. Synthesis of P6
3.2.10. Synthesis of P7a
3.2.11. Synthesis of P7b
3.2.12. Synthesis of P7c
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, L.; Zheng, T.; Wu, Q.; Schneider, A.M.; Zhao, D.; Yu, L. Recent advances in bulk heterojunction polymer solar cells. Chem. Rev. 2015, 115, 12666–12731. [Google Scholar] [CrossRef] [PubMed]
- Etxebarria, I.; Ajuria, J.; Pacios, R. Solution-processable polymeric solar cells: A review on materials, strategies and cell architectures to overcome 10%. Org. Electron. 2015, 19, 34–60. [Google Scholar] [CrossRef]
- Sondergaard, R.; Hösel, M.; Angmo, D.; Larsen-Olsen, T.T.; Krebs, F.C. Roll-to-roll fabrication of polymer solar cells. Mater. Today 2012, 15, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Hösel, M.; Dam, H.F.; Krebs, F.C. Development of lab-to-fab production equipment across several length scales for printed energy technologies, including solar cells. Energy Tech. 2015, 3, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.T.; Tsai, C.Y.; Wang, C.M.; Chang, Y.F.; Meng, H.F.; Chen, Z.K.; Lin, H.W.; Zan, H.W.; Horng, S.F.; Lai, Y.C.; et al. High-efficiency polymer solar cells by blade coating in chlorine-free solvents. Org. Electron. 2014, 15, 893–903. [Google Scholar] [CrossRef]
- Berny, S.; Blouin, N.; Distler, A.; Egelhaaf, H.J.; Krompiec, M.; Lohr, A.; Lozman, O.R.; Morse, G.E.; Nanson, L.; Pron, A.; et al. Solar trees: First large-scale demonstration of fully solution coated, semitransparent, flexible organic photovoltaic modules. Adv. Sci. 2016, 3, 1500342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ye, L.; Hou, J. Breaking the 10% Efficiency Barrier in Organic Photovoltaics: Morphology and device optimization of well-known PBDTTT polymers. Adv. Energy Mater. 2016, 6, 1502529. [Google Scholar] [CrossRef]
- Ouyang, X.; Peng, R.; Ai, L.; Zhang, X.; Ge, Z. Efficient polymer solar cells employing a non-conjugated small electrolyte. Nat. Photonics 2015, 9, 520–524. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y.; Mai, C.K.; Collins, S.D.; Bazan, G.C.; Nguyen, T.Q.; Heeger, A.J. Polymer homo-tandem solar cells with best efficiency of 11.3%. Adv. Mater. 2015, 27, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Li, S.; Yao, H.; Zhang, S.; Zhang, Y.; Yang, B.; Hou, J. Molecular optimization enables over 13% in organic solar cells. J. Am. Chem. Soc. 2017, 139, 7148–7151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qin, Y.; Zhu, J.; Hou, J. Over 14% Efficiency in polymer solar cells enable by a chlorinated polymer donor. Adv. Mater. 2018, 30, 1800868. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Yun, H.J.; Chung, D.S.; Kwon, S.K.; Kim, Y.H. Record high hole mobility in polymer semiconductors via side-chain engineering. J. Am. Chem. Soc. 2013, 135, 14896–14899. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.J.; Kang, S.J.; Xu, Y.; Kim, S.O.; Kim, Y.H.; Noh, Y.Y.; Kwon, S.K. Dramatic inversion of charge polarity in Diketopyrrolopyrrole-based organic field-effect transistors via simple nitrile group substitution. Adv. Mater. 2014, 26, 7300–7307. [Google Scholar] [CrossRef] [PubMed]
- Mercier, L.G.; Leclerc, M. Direct (Hetero)Arylation: A new tool for polymer chemists. Acc. Chem. Res. 2013, 46, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Bura, T.; Blaskovits, T.; Leclerc, M. Direct(hetero)arylation polymerization (DHAP): Trends and perspectives. J. Am. Chem. Soc. 2016, 138, 10056–10071. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, J.-R.; Grenier, F.; Blaskovits, T.; Beaupré, S.; Leclerc, M. Direct (Hetero)arylation polymerization: Simplicity for conjugated polymers synthesis. Chem. Rev. 2016, 116, 14225–14274. [Google Scholar] [CrossRef] [PubMed]
- Nitti, A.; Po, R.; Bianchi, G.; Pasini, D. Direct Arylation strategies in the synthesis of n-extended monomers for organic polymeric solar cells. Molecules 2017, 22, 21. [Google Scholar] [CrossRef] [PubMed]
- Hendsbee, A.; Li, Y. Performance Comparisons of polymer semiconductors synthesized by Direct (Hetero)Arylation Polymerization (DHAP) and conventional methods for organic thin film transistors and organic photovoltaics. Molecules 2018, 23, 1255. [Google Scholar] [CrossRef] [PubMed]
- Gobalasingham, N.S.; Thompson, B.C. Direct Arylation Polymerization: A guide to optimal conditions for effective conjugated polymer. Prog. Polym. Sci. 2018, 83, 135–201. [Google Scholar] [CrossRef]
- Nakabayashi, K. Direct Arylation Polycondensation as conjugated polymer synthesis methodology. Polym. J. 2018, 50, 475–483. [Google Scholar] [CrossRef]
- Mercier, L.G.; Pron, A.; Leclerc, M. Direct Arylation/Heteroarylation Polycondensation Reaction. In Conjugated Polymers: A Practical Guide to Synthesis; RSC Publishing: Cambridge, UK, 2014; pp. 422–442. [Google Scholar]
- Leclerc, M.; Beaupré, S. Direct (Hetero)arylation Polymerization, Synthetic Methods for Conjugated Polymers and Carbon Materials; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2017; pp. 131–158. [Google Scholar]
- Iizuka, E.; Wakioka, M.; Ozawa, F. Mixed-ligand approach to palladium-catalyzed direct arylation polymerization: Synthesis of donor-acceptor polymers with Dithienosilole (DTS) and Thienopyrroledione (TPD) units. Macromolecules 2015, 48, 2989–2993. [Google Scholar] [CrossRef]
- Iizuka, E.; Wakioka, M.; Ozawa, F. Mixed-ligand approach to palladium-catalyzed Direct Arylation Polymerization: Effective prevention of structural defects using diamines. Macromolecules 2016, 49, 3310–3317. [Google Scholar] [CrossRef]
- Wakioka, M.; Takahashi, R.; Ichihara, N.; Ozawa, F. Mixed-ligand approach to palladium-catalyzed Direct Arylation Polymerization: Highly selective synthesis of n-Conjugated polymers with Diketopyrrolopyrrole units. Macromolecules 2017, 50, 927–934. [Google Scholar] [CrossRef]
- Bura, T.; Beaupré, S.; Ibraikulov, O.A.; Légaré, M.-A.; Quinn, J.; Lévêque, P.; Heiser, T.; Li, Y.; Leclerc, N.; Leclerc, M. New fluorinated dithienyl-diketopyrrolo-pyrrole monomers and polymers for organic electronics. Macromolecules 2017, 50, 7080–7090. [Google Scholar] [CrossRef]
- Bura, T.; Beaupré, S.; Légaré, M.-A.; Quinn, J.; Rochette, E.; Blaskovits, J.T.; Fontaine, F.-G.; Pron, A.; Li, Y.; Leclerc, M. Direct Heteroarylation Polymerization: Guidelines for defect-free conjugated polymers. Chem. Sci. 2017, 8, 3913–3925. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Ko, S.J.; Kim, T.; Morin, P.O.; Walker, B.; Lee, B.H.; Leclerc, M.; Kim, J.Y.; Heeger, A.J. Small-bandgap polymer solar cells with unprecedented short-circuit current density and high fill factor. Adv. Mater. 2015, 27, 3318–3324. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, Y.; Tan, H.S.; Guo, Y.; Di, C.A.; Yu, G.; Liu, Y.; Lin, M.; Lim, S.H.; Zhou, Y.; et al. A stable solution-processed polymer semiconductor with record high-mobility for printed transitors. Sci. Rep. 2012, 2, 754. [Google Scholar] [CrossRef] [PubMed]
- Farnum, D.G.; Mehta, G.; Moore, G.G.I.; Siegel, F.P. Attempted reformatskii reaction of benzonitrile, 1,4-diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole. A lactam analogue of pentalene. Tetrahedron Lett. 1974, 15, 2549–2552. [Google Scholar] [CrossRef]
- Wallquist, O.; Lenz, R. 20 years of DPP pigments—Future perspectives. Macromol. Symp. 2002, 187, 617–629. [Google Scholar] [CrossRef]
- Grzybowski, M.; Gryko, D.T. Diketopyrrolopyrrole: Synthesis, Reactivity, and Optical Properties. Adv. Opt. Mater. 2015, 3, 280–320. [Google Scholar] [CrossRef]
- Yi, Z.; Wang, S.; Liu, Y. Design of high-mobility diketopyrrolopyrrole-based π-conjugated copolymers for organic thin-film transistors. Adv. Mater. 2015, 27, 3589–3606. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hendriks, K.H.; Wienk, M.M.; Janssen, R.A.J. Diketopyrrolopyrrole polymers for organic solar cells. Acc. Chem. Res. 2016, 49, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Dubnik, A.S.; Aldrich, T.J.; Eastham, N.D.; Chang, R.P.H.; Facchetti, A.; Marks, T.J. Tin-free direct C–H arylation polymerization for high photovoltaic efficiency conjugated copolymers. J. Am. Chem. Soc. 2016, 48, 15699–15709. [Google Scholar]
- Nitti, A.; Signorile, M.; Boiocchi, M.; Bianchi, G.; Po, R. PBDTTPD for plastic solar cells via Pd(PPh3)4-catalyzed direct(hetero)arylation polymerization. J. Mater. Chem. A 2016, 4, 17163–17170. [Google Scholar]
- Greve, D.R.; Apperloo, J.J.; Janssen, R.A.J. Synthesis and characterisation of novel regioregular polythiophenes 2-tuning the redox properties. Eur. J. Org. Chem. 2001, 3437–3443. [Google Scholar] [CrossRef]
- Liu, M.S.; Jiang, X.; Liu, S.; Herguth, P.; Jen, A.K.-Y. Effect of cyano substituents on electron affinity and electron-transporting properties of conjugated polymers. Macromolecules 2002, 35, 3532–3538. [Google Scholar] [CrossRef]
- Seri, M.; Bolognesi, M.; Chen, Z.; Lu, S.; Koopman, W.; Facchetti, A.; Muccini, M. Fine structural tuning of cyanated dithieno[3,2-b:2′,3′-d]silole-oligothiophene copolymers: Synthesis, characterization, and photovoltaic response. Macromolecules 2013, 46, 6419–6430. [Google Scholar] [CrossRef]
- Qiu, M.; Brandt, R.G.; Niu, Y.; Bao, X.; Yu, D.; Wang, N.; Han, L.; Yu, L.; Xia, S.; Yang, R. Theoretical study on the rational design of cyano-substituted P3HT materials for OSCs: Substitution effect on the improvement of photovoltaic performance. J. Phys. Chem. C 2015, 119, 8501–8511. [Google Scholar] [CrossRef]
- Xiong, Y.; Qiao, X.; Li, H. Nitrile-substituted thienyl and phenyl units as building blocks for high performance n-type polymer semiconductors. Polym. Chem. 2015, 6, 6579–6584. [Google Scholar] [CrossRef]
- Nakabayashi, K.; Otani, H.; Mori, H. Benzodithiophene-based low band-gap polymers with deep HOMO levels: synthesis, characterization, and photovoltaic performance. Polym. J. 2015, 47, 617–623. [Google Scholar] [CrossRef]
- Kim, H.G.; Kim, M.; Clement, J.A.; Lee, J.; Shin, J.; Hwang, H.; Sin, D.H.; Cho, K. Energy level engineering of donor polymers via inductive and resonance effects for polymer solar cells: effects of cyano and alkoxy substituents. Chem. Mater. 2015, 27, 6858–6868. [Google Scholar] [CrossRef]
- Casey, A.; Han, Y.; Fei, Z.; White, A.J.P.; Anthopoulos, T.D.; Heeney, M. Cyano substituted benzothiadiazole: a novel acceptor inducing n-type behaviour in conjugated polymers. J. Mater. Chem. C 2015, 3, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Casey, A.; Dimitrov, S.D.; Shakya-Tuladhar, P.; Fei, Z.; Nguyen, M.; Han, Y.; Anthopoulos, T.D.; Durrant, J.R.; Heeney, M. Effect of systematically tuning conjugated donor polymer lowest unoccupied molecular orbital levels via cyano substitution on organic photovoltaic device performance. Chem. Mater. 2016, 28, 5110–5120. [Google Scholar] [CrossRef]
- Casey, A.; Green, J.P.; Shakya Tuladhar, P.; Kirkus, M.; Han, Y.; Anthopoulos, T.D.; Heeney, M. Cyano substituted benzotriazole based polymers for use in organic solar cells. J. Mater. Chem. A 2017, 5, 6465–6470. [Google Scholar] [CrossRef] [Green Version]
- Heuvel, R.; van Franeker, J.J.; Janssen, R.A.J. Energy level tuning of poly(phenylene-alt-dithienobenzothiadiazole)s for low photon energy loss solar cells. Macromol. Chem. Phys. 2017, 218, 1600502. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.; He, R.; Shen, W. Theoretical investigations on fluorinated and cyano copolymers for improvements of photovoltaic performances. Phys. Chem. Chem. Phys. 2014, 16, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Suraru, S.-L.; Zschieschang, U.; Klauk, H.; Würthner, F. Diketopyrrolopyrrole as a p-channel organic semiconductor for high performance OTFTs. Chem. Commun. 2011, 47, 1767–1769. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-W.; Pan, Q.-Q.; Li, S.-B.; Duan, Y.-A.; Geng, Y.; Zhang, M.; Su, Z.-M. A theoretical exploration of the effect of fluorine and cyano substitutions in diketopyrrolopyrrole-based polymer donor for organic solar cells. J. Mol. Graph. 2017, 77, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.; Bura, T.; Beaupré, S.; Légaré, M.-A.; Sun, J.-P.; Hill, I.G.; Leclerc, M. Fluorinated Thiophene-Based Synthons: Polymerization of 1,4-dialkoxybenzene and Fluoro-Dithieno-2,1,3-benzothiadiazole by Direct Heteroarylation. Macromolecules 2017, 50, 4658–4667. [Google Scholar] [CrossRef]
- Gorelski, S.I. Origins of regioselectivity if the palladium-catalyzed (aromatic) C–H bond metalation-deprotonation. Coord. Chem. Rev. 2013, 257, 153–164. [Google Scholar] [CrossRef]
- Guo, C.; Quinn, J.; Sun, B.; Li, Y. Dramatically different charge transport properties of bisthienyl diketopyrrolopyrrole-bithiazole copolymers synthesized via two direct (hetero)arylation polymerization routes. Polym. Chem. 2016, 7, 4515–4524. [Google Scholar] [CrossRef]
- Pouliot, J.R.; Sun, B.; Leduc, M.; Najari, A.; Li, Y.; Leclerc, M. A high mobility dpp-based polymer obtained via direct(hetero)arylation polymerization. Polym. Chem. 2015, 6, 278–282. [Google Scholar] [CrossRef]
- Ha, J.S.; Kim, K.H.; Choi, D.H. 2,5-Bis(2-octyldodecyl)pyrrolo[3,4-c]pyrrole-1,4-(2H,5H)-dione-Based Donor–Acceptor Alternating Copolymer Bearing 5,5′-Di(thiophen-2-yl)-2,2′-biselenophene Exhibiting 1.5 cm2·V−1·s−1 Hole Mobility in Thin-Film Transistors. J. Am. Chem. Soc. 2011, 133, 10364–10367. [Google Scholar] [CrossRef] [PubMed]
- Blouin, N.; Michaud, A.; Gendron, D.; Wakim, S.; Plesu, R.; Belletête, M.; Durocher, G.; Tao, Y.; Leclerc, M. Towards a Rational Design of Poly(2,7-carbazole) Derivatives for Solar Cells. J. Am. Chem. Soc. 2008, 130, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Gendron, D.; Aïch, B.R.; Najari, A.; Tao, Y.; Leclerc, M. A High-Mobility Low-Bandgap Poly(2,7-carbazole) Derivative for Photovoltaic Applications. Macromolecules 2009, 42, 2891–2894. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Singh, S.P.; Sonar, P. A High Mobility P-Type DPP-Thieno[3,2-b]thiophene Copolymer for Organic Thin-Film Transistors. Adv. Mater. 2010, 22, 4862–4866. [Google Scholar] [CrossRef] [PubMed]
- Conwell, E. Primary Photoexcitations. In Conjugated Polymers: Molecular Excitons Versus Semiconductor Band Model; Sariciftci, N.S., Ed.; World Scientific: Singapore, 1997. [Google Scholar]
- Zhu, Y.; Champion, R.D.; Jenekhe, S.A. conjugated donor−acceptor copolymer semiconductors with large intramolecular charge transfer: Synthesis, optical properties, electrochemistry, and field effect carrier mobility of thienopyrazine-based copolymers. Macromolecules 2006, 39, 8712–8719. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ᴆ | Yield | Reaction Time | Td | ||
|---|---|---|---|---|---|
| kg mol−1 | % | min | °C | ||
| P1 [54] | 16 | 2.4 | 93 | 1200 | n.a |
| P2 [26] | 22 | 3.0 | 73 | 1200 | 430 |
| P3 | n.a. | n.a. | 65 | 10 | 420 |
| P4 [26] | 44 | 3.0 | 91 | 960 | 420 |
| P5 [26] | 125 | 3.0 | 85 | 30 | 420 |
| P6 | 25 | 3.5 | 80 | 40 | 405 |
| P7a | 20 | 1.7 | 75 | 240 | 405 |
| P7b | 35 | 2.4 | 80 | 40 | 410 |
| P7c | oligomers | - | - | ||
| HOMO a | LUMO a | LUMO b | λmax | ||||
|---|---|---|---|---|---|---|---|
| Solution | Film | ||||||
| eV | eV | eV | eV | nm | nm | eV | |
| P1 [54] | −5.13 | −3.82 | 1.31 | −3.96 | 820/920 | 846/927 | 1.17 |
| P2 [26] | −5.35 | −3.69 | 1.66 | −4.20 | 887/962 | 859/967 | 1.15 |
| P3 | −5.44 | −3.96 | 1.48 | −4.28 | 907 | 881 | 1.16 |
| P4 [26] | −5.11 | −3.46 | 1.65 | −3.43 | 622/655 | 630/689 | 1.68 |
| P5 [26] | −5.33 | −3.61 | 1.72 | −3.68 | 639/702 | 639/702 | 1.65 |
| P6 | −5.54 | −3.79 | 1.75 | −3.86 | 621/674 | 620/676 | 1.68 |
| P7a | -5.54 | -3.79 | 1.75 | −3.86 | 620/673 | 621/674 | 1.73 |
| P7b | −5.54 | −3.79 | 1.75 | −3.86 | 621/674 | 621/676 | 1.72 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bura, T.; Beaupré, S.; Légaré, M.-A.; Ibraikulov, O.A.; Leclerc, N.; Leclerc, M. Theoretical Calculations for Highly Selective Direct Heteroarylation Polymerization: New Nitrile-Substituted Dithienyl-Diketopyrrolopyrrole-Based Polymers. Molecules 2018, 23, 2324. https://doi.org/10.3390/molecules23092324
Bura T, Beaupré S, Légaré M-A, Ibraikulov OA, Leclerc N, Leclerc M. Theoretical Calculations for Highly Selective Direct Heteroarylation Polymerization: New Nitrile-Substituted Dithienyl-Diketopyrrolopyrrole-Based Polymers. Molecules. 2018; 23(9):2324. https://doi.org/10.3390/molecules23092324
Chicago/Turabian StyleBura, Thomas, Serge Beaupré, Marc-André Légaré, Olzhas A. Ibraikulov, Nicolas Leclerc, and Mario Leclerc. 2018. "Theoretical Calculations for Highly Selective Direct Heteroarylation Polymerization: New Nitrile-Substituted Dithienyl-Diketopyrrolopyrrole-Based Polymers" Molecules 23, no. 9: 2324. https://doi.org/10.3390/molecules23092324