
Design, Synthesis and Cancer Cell Growth Inhibition Evaluation of New Aminoquinone Hybrid Molecules


Laboratory of Bioorganic Chemistry, Department of Physics, University of Trento, Via Sommarive 14, 38123 Trento, Italy
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(12), 2224; https://doi.org/10.3390/molecules24122224
Submission received: 4 June 2019
/
Revised: 11 June 2019
/
Accepted: 12 June 2019
/
Published: 14 June 2019
(This article belongs to the Special Issue Design and Synthesis of Organic Molecules as Antineoplastic Agents)
Abstract
:Molecular hybridization has proven to be a successful multi-target strategy in the design and development of new antitumor agents. Based on this rational approach, we have planned hybrid molecules containing covalently linked pharmacophoric units, present individually in compounds acting as inhibitors of the cancer protein targets tubulin, human topoisomerase II and ROCK1. Seven new molecules, selected by docking calculation of the complexes with each of the proteins taken into consideration, have been efficiently synthesized starting from 2,3-dichloro-1,4-naphtoquinone or 6,7-dichloro-5,8-quinolinquinone. By screening the full National Cancer Institute (NCI) panel, including 60 human cancer cell lines, four molecules displayed good and sometimes better growth inhibition GI50 than the ROCK inhibitor Y-27632, the Topo II inhibitor podophyllotoxin and the tubulin inhibitor combretastatin A-4. The relative position of N,N heteroatoms in the structures of the tested compounds was crucial in affecting bioactivity and selectivity. Furthermore, compound 3 (2-(4-(2-hydroxyethyl)piperazin-1-yl)-3-(3,4,5-trimethoxyphenoxy)naphthalene-1,4-dione) emerged as the most active in the series, showing a potent and selective inhibition of breast cancer BT-549 cells (GI50 < 10 nM).
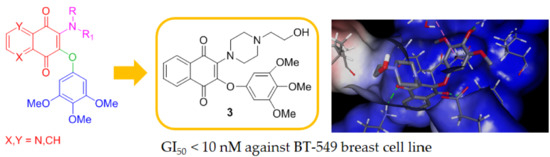
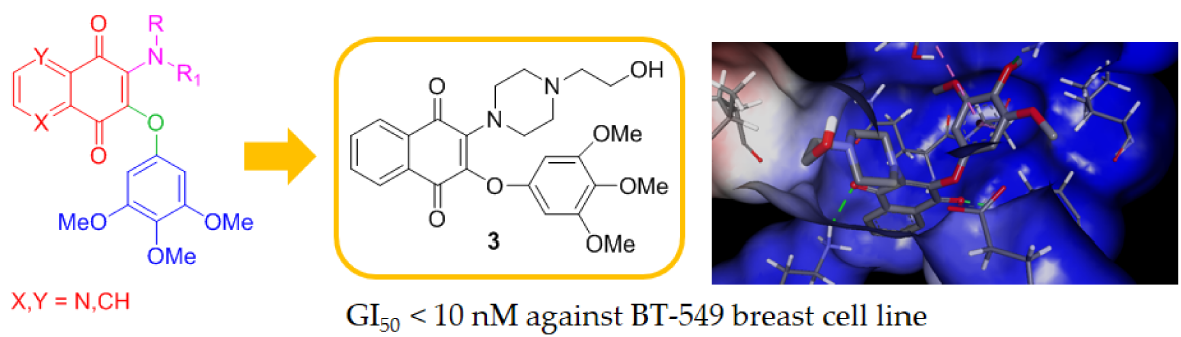
1. Introduction
Cancer is one of the prominent causes of death worldwide, and there is an urgent need to introduce new therapeutic agents due to resistance and severe side effects shown by the currently available drugs. Using molecules to act on different tumor targets simultaneously has shown a higher therapeutic potential than single-target chemotherapy. In the recent rational design of anticancer drugs, molecular hybridization is a promising approach. A hybrid molecule usually contains two or more pharmacophore scaffolds present in single therapeutically active agents, connected together in a new single structure by covalent bonds. The selection of these moieties is based on the strategy of combining structures and pharmacological activities of known drugs and bioactive natural or synthetic compounds [1]. The aim of this strategy is to obtain new therapeutic agents that are able not only to reduce undesirable side effects of the parent drugs, but also to display a modified selectivity profile, a higher affinity and a better therapeutic effect than the administration of a combination of the single-target drugs [2]. Hybrid molecules have demonstrated more favorable pharmacokinetic and pharmacodynamic parameters, in addition to dual or multiple modes of action, due to their ability to inhibit more than one biological target [1]. It is therefore understandable that the investigation of new hybrid anticancer drugs has recently become of great therapeutic interest.
Tubulin is a protein composed of microtubules which are the main components of the cellular cytoskeleton. They play a pivotal role in proliferation, migration and mitosis. Molecules able to bind this protein interfere with microtubule polymerization and depolymerization, inducing cell cycle arrest and leading to apoptosis in cancer cells. A variety of molecules have been proven to act efficiently as tubulin inhibitors, but their therapeutic use is limited by toxicity and development of resistance. Molecular hybridization has been used with good results to target this protein [3].
Topoisomerases (Topo I and Topo II) are enzymes that change the topological state of DNA through the breaking and rejoining of DNA strands. They have a relevant role in replication, recombination, transcription and preservation of genome stability. A series of currently used anticancer drugs and molecules in clinical trials act as inhibitors of these enzymes, stabilizing the DNA-Topo complex by intercalation between DNA base pairs. Therefore, inhibition of human topoisomerases is a promising target in the development of new antitumor agents [4].
Rho-associated kinases, known as two isoforms, ROCK1 and ROCK2, have been shown to induce stress fiber formation, cancer cell migration and metastasis. ROCK protein expression is elevated in several types of cancer. In a series of both in vitro and in vivo studies, advantages have been demonstrated by blocking these proteins, obtaining results especially in reducing tumor growth and in preventing metastases. The data support the potential of ROCKs as targets against tumors [5].
We report here on the efficient synthesis of seven new amino-quinone derivatives, designed as hybrid molecules that combine structural units present in inhibitors of tubulin, topoisomerase II and ROCK as tumor targets, and selected by docking calculations as ligands of these proteins. The evaluation on the growth inhibition of cancer cells by the in vitro NCI screening has been related to the structural features of these hybrid molecules.
2. Results and Discussion
2.1. In Silico Molecular Modeling
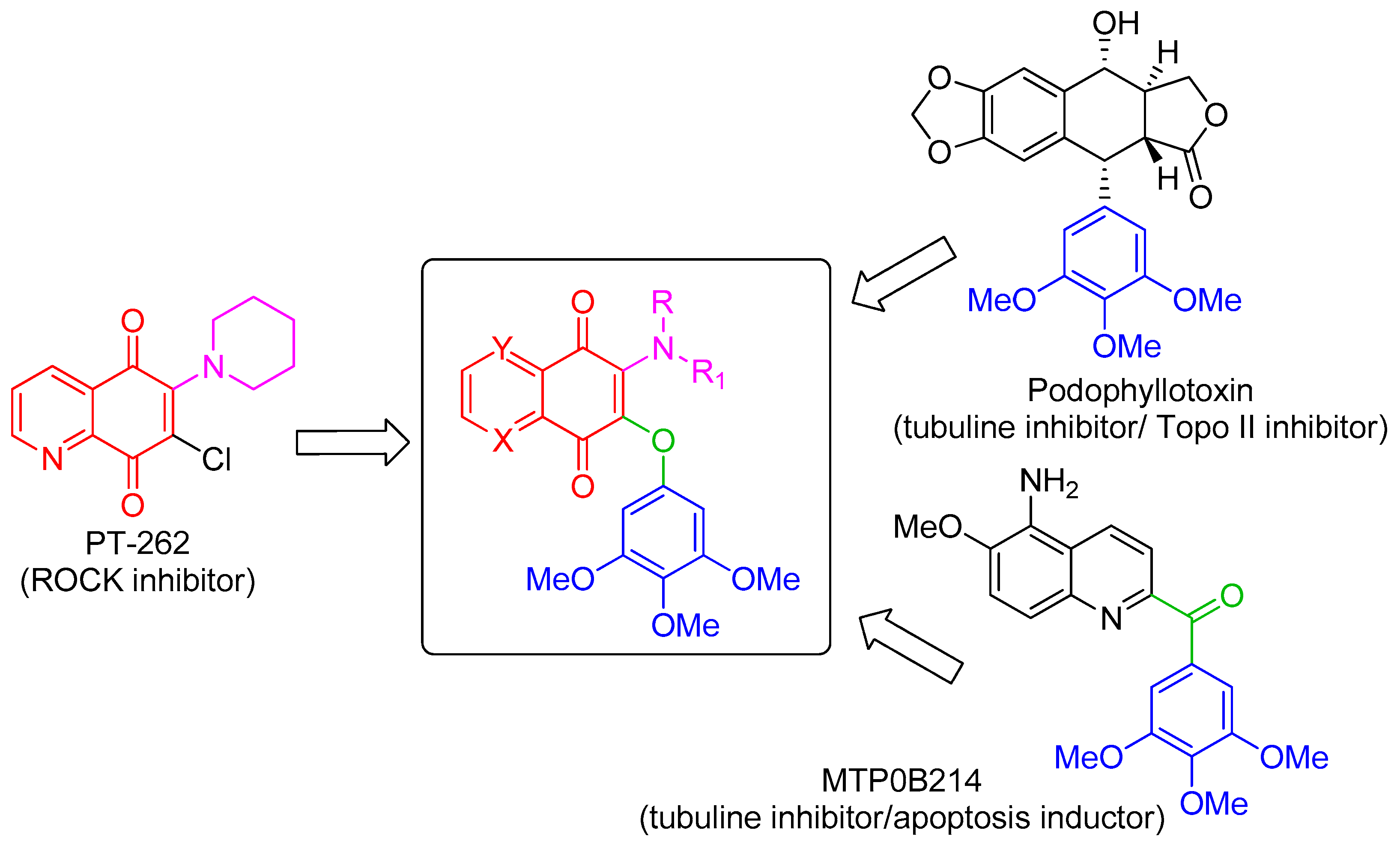
The 3,4,5-trimethoxyphenyl (TMP) unit is a structural feature of anti-neoplastic molecules. It is the case of the natural phenols combretastatins, whose biological activities have been attributed to the presence of this moiety’s ability to target tubulin by the inhibition of microtubule formation. Other representative examples are given by colchicine and podophyllotoxin (Figure 1) [6], which make this pharmacophore a peculiar structural motif among the effective tubulin inhibitors studied in the last decades, with a special value in the design of new antitumor drugs. In addition, podophyllotoxin derivatives acting as topoisomerase II inhibitors are drugs commonly used in clinical oncology [7]. MPT0B214, also showing the TMP unit (Figure 1), inhibits tubulin polymerization and induces apoptosis through mitochondria-mediated pathways [8]. Naphthoquinone and quinolinequinones cores are other representative chemical scaffolds for the development of antitumor agents, which are present in structures of natural and synthetic bioactive products [9,10]. Their molecular mechanism includes reactive oxygen species (ROS) generation mediated by naphtoquinone oxidoreductase 1 (NQO1) bioreduction [11]. Quinolinequinone is a structural unit also present in 7-chloro-6-piperidinyl-quinoline-5,8-dione (PT-262, Figure 1), a synthetic molecule showing an effective inhibition of ROCK kinase activities [12].
Based on these evidences, the structures considered in this molecular hybridization design show a quinone unit with X, Y as CH or N, substituted by both a linear or cyclic amine and a 3,4,5 trimethoxyphenyl ether (Figure 1). A very good overlap has been observed for the energy-minimized structures of PT-262 and podophyllotoxin in cases where quinolinequinone is substituted by piperidine as a cyclic amine (corresponding to 1b in Scheme 1) in the planned molecule, showing an N,N-anti configuration (as in PT-262 (Figure S1)).
In silico screening of the interactions with the target proteins tubulin, human topoisomerase II β and human ROCK1 (Table S1) via docking calculation permitted the selection of the new molecules 1a–c, 2a–c and 3 (Scheme 1). Their drug-likeness was computationally predicted regarding the physico-chemical properties relevant for the development of a drug. By using the free web tool SwissADME, a series of descriptors were calculated, taking into account lipophilicity, size, polarity, solubility, flexibility and unsaturation [13]. All the designed molecules showed a behavior that respected the parameters necessary for good bioavailability (Table S2).
2.2. Synthesis of Compounds 1a–c, 2a–c and 3
The desired products were easily accessible using the common precursors 4 and 5, by the reaction carried out at room temperature in DMSO in the presence of potassium carbonate of 3,4,5-trimethoxyphenol with 2,3-dicholoro-1,4-naphtoquinone or 6,7-dichloro-5,8-quinolinequinone respectively (Scheme 1), via a proposed mechanism involving a Michael addition followed by chloride elimination [14]. The following treatment of compound 4 with piperidine, N,Ndimethylethane-1,2 diamine or 2-(piperazin-1-yl)ethan-1-ol in dichloromethane at room temperature provided the products 1a, 2a and 3, respectively, with global yields in the range 77% ÷ 79%, as evaluated after chromatographic purification of the products. It has been reported that similar quinones with one chlorine and one alkoxyl unit preferentially react by replacing the latter group [15,16], and therefore the use of the symmetric disubstituted 3,4,5-trimethoxyphenoxyl reagent was essential. Through a similar procedure, the products 1b, 1c, 2b and 2c were obtained starting from the quinolinequinone 5. Pure regioisomers 1b and 1c with a 40:60 ratio resulted from chromatographic separation on silica gel by elution with dichloromethane/methanol/triethylamine 95:5:0.1. Similarly, 2b and 2c were isolated in a 42:58 ratio. Structural assignments of the regioisomer pairs 1b/2b and 1c/2c were based on long-range hetero-correlations observed by HMBC experiments. In detail, taking as references the data obtained for similar isomers [17]: (i) 3J(1H, 13C) couplings of the signal at 8.35 ppm for H-8 with 183.2 ppm for C(1)=O in 1b, and 8.36 ppm with 182.0 ppm for 2b, supported the N,N-anti configuration; (ii) 3J(1H, 13C) couplings of the signal at 8.34 ppm for H-5 with 177.1 ppm for C(1)=O in 1c, and 8.38 ppm with 176.3 ppm for 2c, supported the N,N-syn configuration (Scheme 1).
2.3. Biological Evaluation
Compounds 1a–c, 2a–c and 3 were subjected to in vitro growth percentage activity on the NCI full panel containing 60 human cancer cell lines at a single dose at 10 μM. From this evaluation compounds 1a, 1c and 2a were considered inactive (Table 1), whereas 1b, 2b, 2c and 3 were selected for further investigation at five concentration levels. Table 2 reports GI50 data (defined as the concentration values of the molecules inhibiting the growth of cancer cells by 50%) in comparison with the values reported in the NCI database for the ROCK inhibitor Y-27632, the Topo II inhibitor podophyllotoxin and the tubulin inhibitor combretastatin A-4 [18]. The GI50 values of the new tested compounds resulted in 10 nM ÷ 10 µM range.
Submicromolar activities were observed for compound 1b (Table 2), in particular against central nervous system (CNS) cancer for most of the tested cell lines (Figure 2a). Compound 2b showed the highest inhibitions on MDA-MB-468 breast cancer cells (GI50 = 0.133 µM) and on both HCT-116 and SW-620 colon cell lines (GI50 0.277 and 0.318 µM, respectively, Table 2). Notably, the relative structural position of N,N heteroatoms is crucial in affecting bioactivity and selectivity. In fact, compared with 2b, its N,N-syn regioisomer 2c displayed a lower activity corresponding to a growth inhibition in the micromolar range on the same cancer cell lines. A more pronounced disparity was observed for the pair 1b/1c, where the N,N-syn isomer 1c showed no activity. This is evident taking into account the values observed for all the synthesized compounds reported in Table 1 when expressed as the mean dose percentage of single high doses (10−5 M) from the full NCI 60 cell panel. Moreover, the structures lacking of the N atom on quinone unit as 1a and 2a provided inactive compounds (Table 1), whereas product 3 having additional heteroatoms on the C-2 substituent (specifically an NCH2CH2OH moiety) when compared with the inactive 1a, emerged as the most active molecule. Product 3 displayed submicromolar inhibition and was about 160 and 100 times more active than podophyllotoxin and combretastatin A-4, respectively, on T-47D breast cancer lines (Table 2). Furthermore, 3 emerged as the most promising in the series, with a selective GI50 value lower than 10 nM on BT-549 breast cancer cell line (Figure 2b).
3. Materials and Methods
3.1. Chemistry
3.1.1. General
All reagents were purchased from Sigma Aldrich and used without further purification. Preparation of 6,7-Dichloroquinoline-5,8-dione followed a reported procedure [14,19]. The reaction yields were calculated for the products after chromatographic purification. Thin layer chromatography (TLC): Merck silica gel F254 or reversed phase Merck RP-18 F254, with visualization using UV light. Flash chromatography (FC): Merck Si 15–25 μm. Preparative thin layer chromatography (PLC): 20 × 20 cm Merck Kieselgel 60 F254 0.5-mm plates. NMR spectra were recorded on a Bruker-Avance 400 spectrometer using a 5-mm BBI probe 1H at 400 MHz and 13C at 100 MHz in CDCl3 (relative to δH 7.25 and δC 77.00 ppm), with δ values in ppm and J values in Hz; assignments were supported by heteronuclear single quantum correlation (HSQC) and heteronuclear multiple bond correlation (HMBC) experiments. Electrospray ionization (ESI)-MS mass spectra were recorded using a Bruker Esquire-LC spectrometer by direct infusion of a methanol solution (source temperature 300 °C, drying gas N2, 4 L min−1, scan range m/z 100 ÷ 1000). Electron ionization (EI) mass spectra (m/z; rel%) and high resolution (HR)-EI data were recorded with a Kratos-MS80 mass spectrometer, heating at 213 °C for 2a–c, at 276 °C for 1a–c and at 417 °C for compound 3, using a home-built computerized acquisition software.
3.1.2. Typical Reaction Procedure for Precursors 4 and 5
Anhydrous potassium carbonate (414 mg, 3.00 mmol, 1.5 equiv) was added to a solution of 2,3-dichloro-1,4-naphtoquinone (227 mg, 1.00 mmol, 1.0 equiv) or 6,7-dichloro-5,8-quinolinquinone (228 mg, 1.00 mmol, 1.0 equiv) and 3,4,5-trimethoxyphenol (384 mg, 2.00 mmol, 2.00 equiv) in 2.5 mL of dry DMSO, and the reaction mixture was stirred at room temperature for 48 h. The mixture was decanted to remove inorganic salt, and partitioned between dichloromethane/water (X3). The combined organic extracts were washed with water, dried over anhydrous Na2SO4 and concentrated in vacuo to give a solid that was purified by silica gel FC eluting with hexane/EtOAc (from 9:1 to 6:4 v/v) for 4, and with dichloromethane/methanol/triethylamine (96:4:0.1 v/v) for 5.
2,3-Bis(3,4,5-trimethoxyphenoxy)naphthalene-1,4-dione (4). TLC (hexane: EtOAc = 1:1 v/v): Rf = 0.50. Light orange solid. Yield: 87%. 1H-NMR (400 MHz, CDCl3) δ 8.15 and 7.79 (two m, 2H each, H-5/H-8 and H-6/H-7), 6.11 (s, 4H, H-2′, H-6′, H-2′and H-6′), 3.75 (s, 6H, -OCH3), 3.72 (s, 12H, -OCH3). 13C-NMR (100 MHz, CDCl3) δ 180.4 (C=O), 153.7, 134.7, 134.5, 130.7, 126.5, 94.4, 60.7 (-OCH3), 56.0(-OCH3). Significant HMBC correlations: 8.15 ppm with 180.4 ppm; 7.79 ppm with 130.7 ppm. ESI(+)-MS: m/z 545 [M + Na]+. HRMS(EI) calcd. for C28H26O10, 522.15260, found 522.15248.
6,7-Bis(3,4,5-trimethoxyphenoxy)quinoline-5,8-dione (5). TLC (CH2Cl2/MeOH = 95:5 v/v, with two drops of Et3N): Rf = 0.90. Brown solid. Yield: 93%. 1H-NMR (400 MHz, CDCl3) δ 9.11 (br d, J = 4.9 Hz, 1H, H-6), 8.49 (d, J = 7.5 Hz, 1H, H-8), 7.75 (dd, J = 7.5, 4.9 Hz 1H, H-7), 6.12 and 6.08 (two s, 2H each, H-2′, H-6′, H-2′and H-6′), 3.76 (s, 6H, -OCH3), 3.73 (s, 12H, -OCH3). 13C-NMR (100 MHz, CDCl3) δ 179.7 (C=O), 178.8 (C=O), 153.6, 153.5, 152.4, 146.7, 144.7, 135.5, 134.6, 127.8, 127.7, 94.0, 60.2 (-OCH3), 55.5 (-OCH3). Significant HMBC correlations: 9.11 ppm with 146.7 ppm; 8.49 ppm with 179.7 ppm; 7.75 ppm with 127.7 ppm; 6.12 and 6.10 ppm with 152.4, 146.7 and 134.6 ppm; 3.76 ppm with 134.6 ppm; 3.73 ppm with 152.4 ppm ESI(+)-MS: m/z 524 [M + H]+, 545 [M + Na]+, 562 [M + K]+. HRMS(EI) calcd. for C27H25NO10, 523.14785, found 523.14778.
3.1.3. Typical Reaction Procedure for the Synthesis of Compounds 1a–c, 2a–c and 3
A mixture of compound 4 or 5 (52.2 mg, 0.10 mmol, 1.0 equiv) and the suitable amine (0.20 mmol, 2.0 equiv) in 2 mL of anhydrous dichloromethane was stirred at room temperature for 24 h. The solvent was removed in vacuo and the residue was purified by PLC eluting with hexane/EtOAc 1:1 (v/v) for 1a and CH2Cl2/MeOH/Et3N 95:5:0.1 (v/v) for the other compounds.
2-(Piperidin-1-yl)-3-(3,4,5-trimethoxyphenoxy)naphthalene-1,4-dione (1a). TLC (hexane: EtOAc = 1:1 v/v): Rf = 0.74. Red solid. Yield: 91%. 1H-NMR (400 MHz, CDCl3) δ 8.02 and 7.65 (two m, 2H each, H-5/H-8 and H-6/H-7), 6.18 (s, 2H, H-2′ and H-6′), 3.77 (s, 9H, three –OCH3), 3.43 (br s, 4H, H-2′and H-6′), 1.65 (br s, 6H, H-3′, H-4′and H-5′). 13C-NMR (100 MHz, CDCl3) δ 184.1 and 178.4 (two C=O), 154.3, 142.2, 134.2, 133.3, 132.3, 126.2, 96.2, 56.2 (-OCH3), 51.4, 28.2. Significant HMBC correlations: 8.02 ppm with 184.1 and 178.4 ppm; 7.65 ppm with 132.3 ppm. ESI(+)-MS: m/z 462 [M + K]+, 446[M + Na]+, 424 [M + H]+. HRMS(EI) calcd. for C24H25NO6, 423.16819, found 423.16810.
6-(Piperidin-1-yl)-7-(3,4,5-trimethoxyphenoxy)quinoline-5,8-dione (1b). TLC (CH2Cl2/MeOH = 95:5 v/v, with two drops of Et3N): Rf = 0.93. Violet solid. Yield: 40% (as single regioisomer). 1H-NMR (400 MHz, CDCl3) δ 8.97 (br d, J = 4.9 Hz, 1H, H-6), 8.35 (br d, J = 7.9 Hz, 1H, H-8), 7.59 (m, 1H, H-7), 6.20 (s, 2H, H-2′ and H-6′), 3.79 and 3.77 (two s, 9H, OCH3), 3.45 (m, 4H, H-2′ and H-6′), 1.65 (m, 6H, H-3′, H-4′and H-5′). 13C-NMR (100 MHz, CDCl3) δ 183.2 (C=O), 154.9, 154.0, 147.3, 145.4, 139.7, 135.7, 133.7, 128.5, 126.1, 91.8, 56.7, 51.4, 29.2, 24.0. Significant HMBC correlations: 8.35 ppm with 183.2 and 128.5 ppm; 7.59 ppm with 128.5 ppm; 6.20 ppm with 139.8 ppm; 3.45 ppm with 145.4 and 29.2 ppm. ESI(+)-MS: m/z 447 [M + Na]+. HRMS(EI) calcd. for C23H24N2O6, 424.16344, found 424.16329.
7-(Piperidin-1-yl)-6-(3,4,5-trimethoxyphenoxy)quinoline-5,8-dione (1c). TLC (CH2Cl2/MeOH = 95:5 v/v, with two drops of Et3N): Rf = 0.90. Violet solid. Yield: 60% (as single regioisomer). 1H-NMR (400 MHz, CDCl3) δ 8.93 (br d, J = 4.8 Hz, 1H, H-7), 8.34 (br d, J = 7.9 Hz, 1H, H-5), 7.61 (m, 1H, H-6), 6.19 (s, 2H, H-2′ and H-6′), 3.78 (s, 9H, OCH3), 3.50 (br s, 4H, H-2′ and H-6′), 1.69 (m, 6H, H-3′, H-4′ and H-5′). 13C-NMR (100 MHz, CDCl3) δ 182.4 and 177.1 (two C=O), 154.4 154.1, 151.6, 151.1, 147.7, 134.2, 133.5, 127.9, 92.9, 56.2, 52.6, 26.9, 25.0. Significant HMBC correlations: 8.93 ppm with 151.1 and 127.9 ppm; 8.34 ppm with 177.1 and 151.1 ppm; 7.61 ppm with 154.4 ppm; 6.19 ppm with 154.1 and 133.5 ppm. ESI(+)-MS: m/z 463 [M + K]+, 447 [M + Na]+, 425 [M + H]+. HRMS(EI) calcd. for C23H24N2O6, 424.16344, found 424.16328.
2-((2-(Dimethylamino)ethyl)amino)-3-(3,4,5-trimethoxyphenoxy)naphthalene-1,4-dione (2a). TLC (CH2Cl2/MeOH = 95:5 v/v, with two drops of Et3N): Rf = 0.30. Orange solid. Yield: 90%. 1H-NMR (400 MHz, CDCl3) δ 8.05 (d, J = 7.5 Hz, 2H, H-5 and H-8), 7.70 and 7.61 (two t, J = 7.7 Hz, 2H, H-6 and H-7), 6.40 (br s, 1H, NH), 6.22 (s, 2H, H-2′ and H-6′), 3.77 (s, 6H, two –OCH3), 3.75 (s, 3H, –OCH3), 3.56 (q, J = 5.5 Hz, 2H, H-1′), 2.49 (t, J = 5.5 Hz, 2H, H-2′), 2.22 (s, 6H, N(CH3)2). 13C-NMR (100 MHz, CDCl3) δ 182.4 and 177.8 (C=O), 155.1, 134.2, 133.3, 132.1, 129.8, 126.4, 92.6, 58.9 (C-2′), 55.9 (-OCH3), 44.6 (N(CH3)2), 41.7 (C-1′). Significant HMBC correlations: 8.05 ppm with 182.4 and 177.8 ppm; 7.65 ppm with 132.3 ppm; 7.70 ppm with 134.2 ppm; 7.61 ppm with 132.1 ppm; 6.22 ppm with 155.1 and 133.3 ppm. ESI(+)-MS: m/z 449 [M + Na]+, 427 [M + H]+. HRMS(EI) calcd. for C23H26N2O6, 426.17909, found 426.17896.
6-((2-(Dimethylamino)ethyl)amino)-7-(3,4,5-trimethoxyphenoxy)quinoline-5,8-dione (2b). TLC (CH2Cl2/MeOH = 95:5 v/v, with two drops of Et3N): Rf = 0.28. Orange solid. Yield: 42% (as single regioisomer). 1H-NMR (400 MHz, CDCl3) δ 8.99 (br d, J = 4.8 Hz, 1H, H-6), 8.36 (br d, J = 7.9 Hz, 1H, H-8), 7.55 (dd, J = 7.9, 4.8 Hz, 1H, H-7), 6.46 (br s, 1H, NH), 6.23 (s, 2H, H-2′and H-6′), 3.78 and 3.77 (two s, 9H, three –OCH3), 3.57 (m, 2H, H-1′), 2.49 (br t, J = 5.3 Hz, 2H, H-2′,), 2.22 (s, 6H, N(CH3)2). 13C-NMR (100 MHz,CDCl3) δ 182.0 (C=O), 176.1 (C=O), 154.6, 154.4, 148.6, 136.9, 133.7, 130.1, 123.8, 93.2, 57.8 (-OCH3), 57.7 (C-2′), 44.5 (-N(CH3)2), 41.1(C-1′). Significant HMBC correlations: 8.99 ppm with 123.8 ppm; 8.36 ppm with 182.0, 154.4 and 148.6 ppm; 7.55 ppm with 154.4 and 130.1 ppm; 6.24 ppm with 154.6 and 133.7 ppm; 3.78 and 3.77 ppm with 154.6 and 133.7 ppm; 2.49 ppm with 41.1 ppm; 2.23 ppm with 57.7 ppm. ESI(+)-MS: m/z 450 [M + Na]+, 428 [M + H]+. HRMS(EI) calcd. for C22H25N3O6, 427.17434, found 427.17446.
7-((2-(Dimethylamino)ethyl)amino)-6-(3,4,5-trimethoxyphenoxy)quinoline-5,8-dione (2c). TLC (CH2Cl2/MeOH = 95:5 v/v, with two drops of Et3N): Rf = 0.31. Orange solid. Yield: 58% (as single regioisomer). 1H-NMR (400 MHz, CDCl3) δ 8.92 (br d, J = 4.6 Hz, 1H, H-7), 8.38 (br d, J = 7.9 Hz, 1H, H-5), 7.62 (dd, J = 7.8, 4.5 Hz, 1H, H-6), 6.53 (br s, 1H, NH), 6.22 (s, 2H, H-2′ and H-6′), 3.78 (s, 9H, three –OCH3), 3.61 (m, 2H, H-1′), 2.50 (br t, J = 5.7 Hz, 2H, H-2′), 2.22 (s, 6H, N(CH3)2). 13C-NMR (100 MHz, CDCl3) δ 180.9 (C=O), 176.3 (C=O), 155.0, 153.2, 151.4, 147.2, 135.1, 133.6, 132.2, 127.0, 93.2, 58.5 (C-2′), 56.5 (-OCH3), 44.7 (-N(CH3)2), 41.2 (C-1′). Significant HMBC correlations: 8.92 ppm with 147.2, 135.1 and 127.0 ppm; 8.38 ppm with 180.9 (small), 176.3, 153.2 and 147.2 ppm; 7.62 ppm with 153.2, 135.1 and 132.2 ppm; 6.22 ppm with 155.0 and 133.6 ppm; 3.78 ppm with 155.0 and 133.6 ppm. ESI(+)-MS: m/z 450 [M + Na]+. HRMS(EI) calcd. for C22H25N3O6, 427.17434, found 427.17427.
2-(4-(2-Hydroxyethyl)piperazin-1-yl)-3-(3,4,5-trimethoxyphenoxy)naphthalene-1,4-dione (3). TLC (CH2Cl2/MeOH = 95:5 v/v, with two drops of Et3N): Rf = 0.30. Red solid. Yield: 88%. 1H-NMR (400 MHz, CDCl3) δ 8.01 and 7.66 (two m, 2H each, H-5/H-8 and H-6/H-7), 6.17 (s, 2H, H-2′ and H-6′), 3.77 (s, 9H, OCH3), 3.60 (m, 2H, CH2OH), 3.52 (m, 4H, H-2′), 2.59 (m, 4H, H-2′), 2.54 (m, 2H, NCH2). 13C-NMR (100 MHz, CDCl3) δ 183.7 and 178.6 (C=O), 154.0, 133.6, 133.5, 132.3, 131.7, 126.2, 92.6, 56.6, 55.9 (OCH3), 54.2, 49.8. Significant HMBC correlations: 8.01 ppm with 183.7 and 178.6 ppm; 7.66 ppm with 132.3 and 131.7 ppm; 6.17 ppm with 154.0 and 133.5 ppm. ESI(+)-MS: m/z 491 [M + Na]+, 469 [M + H]+. HRMS(EI) calcd. for C25H28N2O7, 468.18965, found 468.18951.
3.2. Computational Analysis
Calculations were carried out using Autodock Vina 1.1.2 [20], adopting a reported procedure [21]. The structures of human ROCK 1 (PDB ID: 2ETK), human topoisomerase II β (PDB ID: 3QX3) and tubulin (PDB ID: 5JCB) were determined by X-ray crystallography with a resolution of 2.9, 2.2 and 2.3 Å respectively. For the docking calculation, a grid box of 16 × 16 × 24 Å in x, y, z directions was created with a spacing of 1.00 Å, and centered at x = 51.964, y = 101.296, z = 29.213 for 2ETK; a grid box of 22 × 18 × 18 Å in x, y, z directions was created with a spacing of 1.00 Å, and centered at x = 32.884, y = 95.413, z = 50.785 for 3QX3; and a grid box of 18 × 28 × 26 Å in x, y, z directions was created with a spacing of 1.00 Å, centered at x = −13.614, y = 9.720, z = 20.911 for 5JCB. Results were expressed as energy associated to each ligand–enzyme complex in terms of Gibbs free energy values (Table S1). The visual ligand–enzyme interactions were displayed using Discovery Studio Visualizer v.19.1.0.18287 [22]. ADME predictions were performed using the online server Swiss-ADME [23].
3.3. Biological Evaluation
The synthesized compounds were evaluated for their in vitro activity against cancer cell lines by the National Cancer Institute (NCI-USA) following its anticancer drug development program based on automated sulforhodamine blue (SRB) cytotoxicity assay. The screening was a two-stage process, where after a first evaluation was carried out against the full panel of cell lines at a single dose of 10 μM, with the compounds exhibiting significant growth inhibition being tested at five concentration levels [24].
4. Conclusions
Based on the known benefits of considering a molecular hybridization approach in the design and development of new antitumor agents, we have planned new hybrid molecules containing pharmacophoric units, which present individually in compounds acting as inhibitors of the cancer protein targets tubulin, human topoisomerase II and ROCK1. Docking calculation of the complexes with each protein allowed us to select seven molecules, structurally characterized by a naphtoquinone or quinolinequinone moiety, and substituted by both a cyclic or functionalized amine and a 3,4,5-trimethoxyphenyl group.
The evaluation of human cancer cell inhibition by the seven synthetic compounds provided a qualitative structure–activity relationship study. What is more, compound 3 emerged as the most active in the series, displaying a selective nanomolar inhibition of breast cancer BT-549 cells. According to these promising findings and their easily accessible synthesis, the molecules herein reported are worthy of further biological investigation.
Supplementary Materials
The following are available online. Table S1: Energy data from docking calculation by Autodock Vina for 1a–c, 2a–c and 3, in comparison with original and reference ligands; Table S2: ADME prediction of 1a–c, 2a–c, 3 and reference compounds evaluated by on-line Server Swiss-ADME; Figure S1: Overlapping of the energy minimized structures 1b, PT-262 and podophyllotoxin; Figures S2–S23: NMR spectra of compounds 1a–c, 2a–c, 3, 4 and 5.
Author Contributions
Conceptualization, A.D.; software, A.D.; investigation, A.D.; writing—original draft preparation, A.D and I.M.; writing—review and editing, I.M.; supervision, I.M.; funding acquisition, I.M.
Funding
This research received no external funding.
Acknowledgments
The authors thank the National Cancer Institute (NCI) at Bethesda, USA for the antitumor screening tests of the compounds. We are grateful to Adriano Sterni, university of Trento for mass spectra recording.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Abbot, V.; Sharma, P.; Dhiman, S.; Noolvi, M.N.; Patel, H.M.; Bhardwaj, V. Small hybrid heteroaromatics: Resourceful biological tools in cancer research. RSC Adv. 2017, 7, 28313–28349. [Google Scholar] [CrossRef]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Morgan-Fisher, M.; Wewer, U.M.; Yoneda, A. Regulation of ROCK activity in cancer. J. Histochem. Cytochem. 2013, 61, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, S.; Li, X.; Liu, Y.; Su, J.; Chen, J. Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2018, 151, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Li, H.P. Camptothecin and podophyllotoxin derivatives inhibitors of topoisomerase I and II—Mechanisms of Action, Pharmacokinetics and Toxicity Profile. Drug Saf. 2006, 29, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.J.; Lin, C.I.; Liou, J.P.; Kuo, C.C.; Chang, C.Y.; Chen, L.T.; Chang, J.Y. A novel synthetic microtubule inhibitor, mpt0b214 exhibits antitumor activity in human tumor cells through mitochondria-dependent intrinsic pathway. PLoS ONE 2013, 8, e58953. [Google Scholar] [CrossRef] [PubMed]
- Defant, A.; Guella, G.; Mancini, I. Synthesis and in vitro cytotoxicity evaluation of novel naphthindolizinedione derivatives. Arch. Pharm. Chem. Life Sci. 2007, 340, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Defant, A.; Guella, G.; Mancini, I. Synthesis and in-vitro cytotoxicity evaluation of novel naphtindolizinedione derivatives, part II: Improved activity for aza-analogues. Arch. Pharm. Chem. Life Sci. 2009, 342, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.Y.; Wang, P.F.; Lin, H.Y.; Tang, C.Y.; Zhu, H.L.; Yang, Y.H. Naphthoquinones: A continuing source for discovery of therapeutic antineoplastic agents. Chem. Biol. Drug Des. 2018, 91, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Liu, H.F.; Hsu, K.C.; Yang, J.M.; Chen, C.; Liu, K.K.; Hsu, T.S.; Chao, J.I. 7-Chloro-6-piperidin-1-yl-quinoline-5,8-dione (PT-262), a novel ROCK inhibitor blocks cytoskeleton function and cell migration. Biochem. Pharmacol. 2011, 81, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Defant, A.; Guella, G.; Mancini, I. Regioselectivity in the multi-component synthesis of indolizinoquinoline-5,12-dione derivatives. Eur. J. Org. Chem. 2006, 4201–4210. [Google Scholar] [CrossRef]
- Silver, R.F.; Holmes, H.L. Synthesis of some 1,6-naphthoquinones and reactions relating to their use in the study of bacterial growth inhibition. Can. J. Chem. 1968, 46, 1859–1864. [Google Scholar] [CrossRef]
- Egleton, J.E.; Thinnes, C.C.; Seden, P.T.; Laurieri, N.; Lee, S.P.; Hadavizadeh, K.S.; Measures, A.R.; Jones, A.M.; Thompson, S.; Varney, A.; et al. Structure–activity relationships and colorimetric properties of specific probes for the putative cancer biomarker human arylamine N-acetyltransferase 1. Bioorg. Med. Chem. 2014, 22, 3030–3054. [Google Scholar] [CrossRef] [PubMed]
- Defant, A.; Rossi, B.; Viliani, G.; Guella, G.; Mancini, I. Metal-assisted regioselectivity in nucleophilic substitutions: A study by Raman spectroscopy and density functional theory calculations. J. Raman Spectrosc. 2010, 41, 1398–1403. [Google Scholar] [CrossRef]
- NCI. Available online: https://dtp.cancer.gov/dtpstandard/cancerscreeningdata/index.jsp (accessed on 30 May 2019).
- Shaikh, I.A.; Johnson, F.; Grollman, A.P. Streptonigrin.1. Structure-activity relationship among simple bicyclic analogues. Rate dependence of DNA on quinolone reduction potential. J. Med. Chem. 1986, 29, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Bosco, B.; Defant, A.; Messina, A.; Incitti, T.; Sighel, D.; Bozza, A.; Ciribilli, Y.; Inga, A.; Casarosa, S.; Mancini, I. Synthesis of 2,6-diamino-substituted purine derivatives and evaluation of cell cycle arrest in breast and colorectal cancer cells. Molecules 2018, 23, 1996. [Google Scholar] [CrossRef] [PubMed]
- Systèmes, D. BIOVIA, Discovery Studio Modeling Environment, Release 2019; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Swiss ADME. Available online: http://www.swissadme.ch/ (accessed on 28 April 2019).
- NCI (NIH). Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 30 May 2019).
Sample Availability: Samples of the compounds 1a–c, 2a–c and 3 are available from the authors. |
Figure 1.
Design strategy of the target hybrid molecules.

Scheme 1.
Synthesis of molecules 1a–c, 3a–c and 3. Reagents and conditions: (a) K2CO3, DMSO, r.t. 48 h; (b) piperidine, CH2Cl2, r.t. 24 h; (c) N1,N1-dimethylethane-1,2-diamine, CH2Cl2, r.t. 24 h; (d) 2-(piperazin-1-yl)ethan-1-ol, CH2Cl2, r.t. 24 h. Arbitrary numbering is for convenience.
Scheme 1.
Synthesis of molecules 1a–c, 3a–c and 3. Reagents and conditions: (a) K2CO3, DMSO, r.t. 48 h; (b) piperidine, CH2Cl2, r.t. 24 h; (c) N1,N1-dimethylethane-1,2-diamine, CH2Cl2, r.t. 24 h; (d) 2-(piperazin-1-yl)ethan-1-ol, CH2Cl2, r.t. 24 h. Arbitrary numbering is for convenience.

Figure 2.
Dose response curves at NCI fixed protocol: (a) for compound 1b on the indicated central nervous system (CNS) cancer cell lines, and (b) for compound 3 on the indicated breast cancer cell lines. The curves were obtained at five concentrations (log dilutions from 10–4 to 10–8 M). The concentrations of each compound, which inhibited 50% of cancer cell growth (GI50), were deduced and are the values reported in Table 1.
Figure 2.
Dose response curves at NCI fixed protocol: (a) for compound 1b on the indicated central nervous system (CNS) cancer cell lines, and (b) for compound 3 on the indicated breast cancer cell lines. The curves were obtained at five concentrations (log dilutions from 10–4 to 10–8 M). The concentrations of each compound, which inhibited 50% of cancer cell growth (GI50), were deduced and are the values reported in Table 1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Mean dose percent values by one-dose assay (10−5 M) of all the tested compounds in the full NCI 60 cell panel.
Table 1.
Mean dose percent values by one-dose assay (10−5 M) of all the tested compounds in the full NCI 60 cell panel.
| Compound | Mean Growth Percent | Activity |
|---|---|---|
| 1a | 93.74 | Inactive |
| 1b | 20.92 | Active |
| 1c | 67.49 | Inactive |
| 2a | 78.44 | Inactive |
| 2b | −4.33 | Active |
| 2c | 12.79 | Active |
| 3 | 17.88 | Active |
Table 2.
Inhibition of in vitro human cancer cell lines by compounds 1a, 2b, 2c and 3, in comparison with combretastatin A-4, podophyllotoxin and Y-27632 taken as reference compounds.
Table 2.
Inhibition of in vitro human cancer cell lines by compounds 1a, 2b, 2c and 3, in comparison with combretastatin A-4, podophyllotoxin and Y-27632 taken as reference compounds.
| Cytotoxicity GI50 (μM) | |||||||
|---|---|---|---|---|---|---|---|
| 1b | 2b | 2c | 3 | Y-27632 | Podophyllotoxin | Combretastatin A-4 | |
| Cell lines | |||||||
| Leukemia | |||||||
| CCRF-CEM | 2.98 | 2.21 | 2.24 | 2.48 | 31.6 | 0.01 | 0.251 |
| HL-60(TB) | 1.76 | 0.811 | 1.43 | 1.66 | 100 | 0.01 | 0.01 |
| K-562 | 0.354 | 0.659 | 2.90 | 2.19 | 100 | - | 0.316 |
| MOLT-4 | 3.58 | 3.07 | 2.48 | 2.35 | 100 | 0.01 | 0.501 |
| RPMI-8226 | 0.486 | 1.92 | 2.23 | 2.03 | 100 | 0.01 | 0.063 |
| SR | 1.89 | 3.17 | 2.13 | 2.87 | 25.1 | 0.01 | 1.99 |
| Non-Small Cell Lung Cancer | |||||||
| A549/ATCC | 0.482 | 3.43 | 1.87 | 13.8 | 100 | 0.0126 | 0.020 |
| HOP-92 | 10.4 | 1.88 | 2.01 | 1.46 | 1.26 | 0.0316 | 0.100 |
| NCI-H226 | 0.506 | 16.67 | 2.08 | 2.92 | 100 | 0.01 | 0.251 |
| NCI-H23 | 2.11 | 2.25 | 1.76 | 2.22 | 100 | 0.01 | 0.040 |
| NCI-H322M | 6.27 | 2.65 | 7.04 | 1.23 | 100 | 0.01 | 0.063 |
| NCI-H460 | 2.77 | 3.11 | 1.65 | 3.13 | 100 | 0.01 | 0.050 |
| NCI-H522 | 1.47 | 1.46 | 1.39 | 0.571 | 100 | 0.01 | 0.032 |
| Colon Cancer | |||||||
| COLO 205 | 2.79 | 1.10 | 2.44 | 1.80 | 100 | 0.01 | 6.31 |
| HCC-2998 | 5.57 | 1.62 | 4.71 | 12.4 | 100 | 0.0126 | 0.158 |
| HCT-116 | 0.513 | 0.277 | 1.85 | 1.44 | 100 | 0.01 | 0.079 |
| HCT-15 | 0.442 | 2.07 | 2.78 | 1.56 | 100 | 0.0126 | 0.040 |
| HT29 | 3.74 | 2.36 | 3.77 | 2.27 | 100 | 0.01 | 6.31 |
| KM12 | 4.59 | 2.91 | 3.34 | 4.39 | 100 | 0.01 | 0.063 |
| SW-620 | 1.37 | 0.318 | 1.55 | 1.79 | 100 | 0.01 | 0.063 |
| CNS Cancer | |||||||
| SF-268 | 1.94 | 2.39 | 2.87 | 1.73 | 63.1 | 0.01 | 0.063 |
| SF-295 | 0.465 | 9.88 | 3.76 | 5.36 | 100 | 0.01 | 0.032 |
| SF-539 | 0.434 | 1.78 | 1.85 | 2.48 | 100 | 0.01 | 0.025 |
| SNB-19 | 0.450 | 2.00 | 2.86 | 3.41 | 100 | 0.01 | 0.025 |
| SNB-75 | 0.334 | 1.89 | 1.39 | 1.68 | 10 | 0.01 | 1.259 |
| U251 | 0.440 | 2.00 | 2.12 | 3.10 | 100 | 0.01 | 0.079 |
| Melanoma | |||||||
| LOX IMVI | 1.01 | 0.508 | 1.76 | 1.81 | 100 | 0.01 | 0.050 |
| MALME-3M | 2.73 | 0.967 | 2.23 | 3.99 | 100 | 50.1 | 0.631 |
| M14 | 0.552 | 1.84 | 3.07 | 1.96 | 100 | 0.0126 | 0.100 |
| MDA-MB-435 | 1.11 | 1.72 | 1.53 | 1.76 | 100 | 0.01 | 0.010 |
| SK-MEL-2 | 4.16 | 2.03 | 2.01 | 2.01 | 100 | 0.016 | 0.050 |
| SK-MEL-28 | 1.35 | - | 2.00 | 2.34 | 100 | 0.01 | 5.012 |
| SK-MEL-5 | 1.95 | 1.70 | 1.55 | 1.81 | 100 | 0.01 | 0.013 |
| UACC-257 | 0.846 | 1.78 | 1.56 | 3.56 | 100 | 0.01 | 0.063 |
| UACC-62 | 2.53 | 3.17 | 1.60 | 1.79 | 100 | 0.01 | 0.040 |
| Ovarian Cancer | |||||||
| OVCAR-3 | 1.77 | 0.364 | 2.02 | 1.02 | 79.4 | 0.0126 | 0.051 |
| OVCAR-4 | 1.31 | 0.937 | 1.53 | 1.63 | 100 | 0.016 | 1.995 |
| OVCAR-5 | 0.439 | 2.35 | 2.19 | 2.60 | 100 | 0.251 | 3.981 |
| OVCAR-8 | 0.463 | 0.386 | 2.13 | 2.91 | 100 | 0.01 | 0.079 |
| NCI/ADR-RES | 1.06 | 7.40 | 5.95 | 3.19 | 100 | 0.01 | 0.063 |
| SK-OV-3 | 3.28 | 11.6 | 7.49 | 5.31 | 100 | 0.01 | 0.251 |
| Renal Cancer | |||||||
| 786-0 | 2.03 | 2.46 | 2.04 | 1.93 | 100 | 0.016 | 0.631 |
| A498 | 0.640 | 2.17 | 2.09 | 2.04 | 100 | 0.01 | 0.100 |
| ACHN | 1.25 | 1.81 | 2.36 | 1.87 | 100 | 0.01 | 0.199 |
| CAKI-1 | 0.595 | 2.10 | 2.51 | 3.59 | 100 | 0.1 | 0.251 |
| RXF 393 | 0.694 | 2.00 | 2.83 | 1.55 | 100 | 0.01 | 0.398 |
| SN12C | 0.829 | 2.81 | 2.02 | 3.11 | 100 | 0.016 | 0.251 |
| TK-10 | 3.27 | 3.63 | 3.91 | 4.97 | 10 | 0.0316 | 3.162 |
| UO-31 | 1.18 | 1.80 | 1.29 | 1.61 | 100 | 0.016 | 1.000 |
| Prostate Cancer | |||||||
| PC-3 | 0.914 | 2.43 | 2.72 | 3.19 | 100 | 0.01 | 0.010 |
| DU-145 | 2.60 | 3.43 | 4.19 | - | 100 | 0.01 | 0.013 |
| Breast Cancer | |||||||
| MCF7 | 0.509 | 0.304 | 1.20 | 1.15 | 100 | 0.01 | 0.010 |
| MDA-MB-231/ATCC | 4.14 | 2.55 | 1.81 | 2.00 | 100 | 0.01 | 0.016 |
| HS 578T | 0.415 | 3.06 | 2.84 | 2.69 | 100 | 0.01 | 0.010 |
| BT-549 | 0.819 | 2.68 | 4.96 | <0.01 | 100 | 0.01 | 0.020 |
| T-47D | 2.53 | 1.67 | 1.98 | 0.510 | 100 | 79.4 | 50.12 |
| MDA-MB-468 | 1.77 | 0.133 | 0.285 | 1.37 | 100 | 0.01 | 0.079 |
| MGMa) | 1.29 | 1.82 | 2.24 | 2.24 | 91.6 | 1.52 | 1.52 |
a) MGM (mean graph medium) value as average GI50 (µM) over all cell lines investigated.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Defant, A.; Mancini, I. Design, Synthesis and Cancer Cell Growth Inhibition Evaluation of New Aminoquinone Hybrid Molecules. Molecules 2019, 24, 2224. https://doi.org/10.3390/molecules24122224
AMA Style
Defant A, Mancini I. Design, Synthesis and Cancer Cell Growth Inhibition Evaluation of New Aminoquinone Hybrid Molecules. Molecules. 2019; 24(12):2224. https://doi.org/10.3390/molecules24122224
Chicago/Turabian StyleDefant, Andrea, and Ines Mancini. 2019. "Design, Synthesis and Cancer Cell Growth Inhibition Evaluation of New Aminoquinone Hybrid Molecules" Molecules 24, no. 12: 2224. https://doi.org/10.3390/molecules24122224