
Beyond Typical Electrolytes for Energy Dense Batteries

Materials Research Department, Toyota Research Institute of North America, Ann Arbor, MI 48105, USA
Molecules 2020, 25(8), 1791; https://doi.org/10.3390/molecules25081791
Submission received: 17 December 2019
/
Revised: 27 March 2020
/
Accepted: 31 March 2020
/
Published: 14 April 2020
(This article belongs to the Special Issue Advances in Hydrogen Storage Materials for Energy Utilization)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The ever-rising demands for energy dense electrochemical storage systems have been driving interests in beyond Li-ion batteries such as those based on lithium and magnesium metals. These high energy density batteries suffer from several challenges, several of which stem from the flammability/volatility of the electrolytes and/or instability of the electrolytes with either the negative, positive electrode or both. Recently, hydride-based electrolytes have been paving the way towards overcoming these issues. Namely, highly performing solid-state electrolytes have been reported and several key challenges in multivalent batteries were overcome. In this review, the classes of hydride-based electrolytes reported for energy dense batteries are discussed. Future perspectives are presented to guide research directions in this field.
1. Introduction
Over the past few decades, nickel-metal hydride and later lithium-ion batteries, have been thrusted to the forefront of societal electrification owing to their energy density and reliable performances. For instance, the relatively high specific energy densities of Li-ion batteries (ca at least 2 times that in nickel-metal hydride) and higher specific power have positioned them to play an increasingly important role in driving technologies encompassing miniature and portable devices (i.e., cell phones, laptops), medium scale (plug-in hybrid PHEV and electric vehicles EVs) up to large scale stationary and grid applications [1].However, recently there have been increasing demands for highly performing batteries beyond those based on typical Li-ion [2]. In particular, the energy and power densities that can be offered by Li-ion batteries are insufficient to meet those needs. This has been fueling R&D efforts towards battery chemistries capable of meeting these requirements that include solid-state batteries and those based on high capacity metals such as Li and Mg. For example, in contrast to batteries utilizing liquid electrolytes, the use of solid-state electrolytes allows for efficient bipolar stacking design of batteries that decreases the dead space between single cells thereby increasing the overall energy density whilst eliminating the use of volatile liquid electrolytes [3,4]. In addition, the high hardness of solid electrolytes was suggested to mitigate the occurrence of Li dendrites that form upon Li metal battery charge/discharge. On the other hand, batteries that use multivalent metals have been promising a high energy density option that uses benign and earth abundant metals such as Mg (volumetric storage capacity is 3832 mAh/cm3 vs. 2061 mAh/cm3 for Li metal) and eliminates safety hazards encountered with Li metal.
Enabling batteries beyond Li-ion mandates the presence of highly performing battery core components (anode, cathode, electrolyte) capable of supporting competitive battery performances. Electrolytes, which are the media that connect the positive (cathode) and negative (anode) electrodes, are positioned to play a critical role in enabling these technologies. However, whilst numerous families of materials have been proposed for these batteries, they fall short of simultaneously meeting the challenging technological needs. For example, highly conducting Li+ solid-state electrolytes such as sulfides suffer from limitations in their electrochemical stability window [3,5]. In addition, electrolytes used in multivalent batteries had severe limitations caused by their reliance on complex chloro-reagents or were altogether incapable of supporting highly efficient performances [2,6,7] Therefore, there have been dedicated efforts that target the development of new materials positioned to overcome these challenges.
The past decade has witnessed unexpected and rapid developments of hydrides-type materials as competent solid and liquid electrolytes poised to offer potential solutions [8]. This review explains how these advancements came to fruition, summarizes the classes of hydride-based electrolytes and highlights key advancements made thus far.
2. Explanation of the Review’s Structure
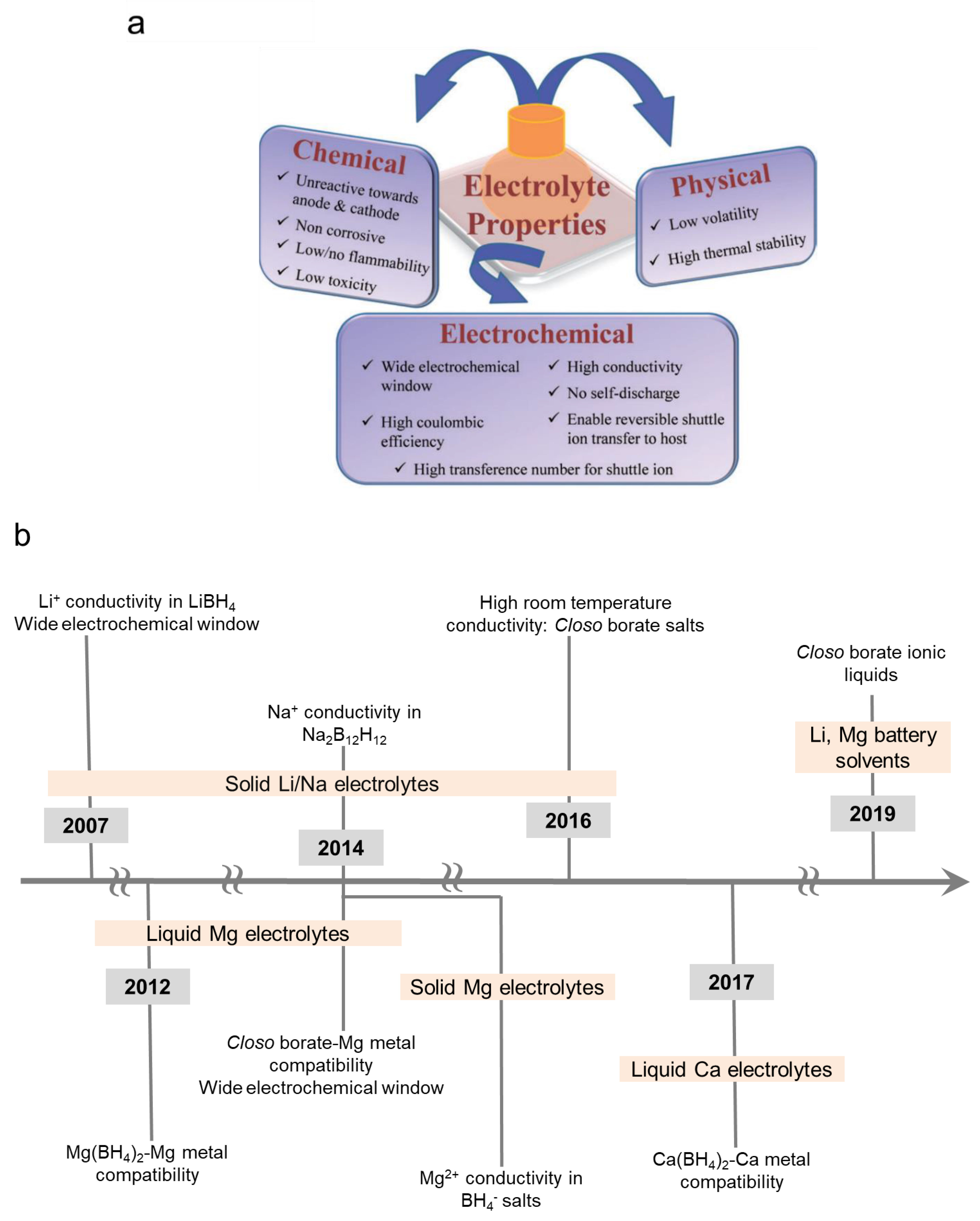
The development of practical electrolytes for energy storage devices is a very challenging endeavor as they are required to meet a myriad of requirements (Figure 1) [9]. All of these demands are based on a battery cell design that allows for optimized battery performances and offer a commercially viable energy storage system. For example, high conductivity is required to enable acceptable battery discharge/charge rate capabilities; wide electrochemical window is needed to prevent electrolyte decomposition that can result in battery failure (i.e., electrolyte is compatible with battery electrodes); absence of no-self discharge or electronically insulating electrolyte is required to prevent battery capacity loss whilst low/non-volatility is highly desired to minimize safety risks and lower manufacturing costs. This review explains how hydride electrolytes for mono and multivalent batteries have been shown to meet several of these requirements which included the demonstration of high conductivity, wide electrochemical window and non/low volatility.
Hydrides are materials characterized by the presence of a hydrogen atom bound with many elements forming ionic (i.e., negatively charged H− or protonic H+ form), covalent, or interstitial systems [8]. In particular, those constituting of anions formed by H− covalently bonding to a metal such as boron have been attracting tremendous attention over the past few years as battery electrolytes. The high potential of hydrides as electrolytes was first reported in the borohydride salts and was manifested by two key accomplishments [8]: (1) The discovery of high Li+ ion conductivity (ca 10−3 S/cm at 423K) and wide electrochemical window (5V) in the solid state (discussed in Section 3) and (2) The design of boron-hydrogen Mg battery liquid electrolytes that are highly compatible with the passivation prone Mg metal (>90% Mg deposition/stripping efficiency) (discussed in Section 4). A timeline depicting key developments is captured in Figure 1b.
The research of hydrides as solid-state electrolytes for monovalent batteries has been motivated by improving the room temperature conductivities of Li+ and Na+ ions (Section 3). High room temperature conductivities in the order of 10−3 S/cm, which placed hydride solid-state electrolytes amongst the best-known electrolytes, was the culmination of these efforts. These studies are discussed in Section 3.1, which is dedicated to strongly hydridic salts, and Section 3.2, which discusses electrolyte salts with a weak hydridic nature. Section 4 explains the advancements made in the field of hydrides as electrolytes for multivalent batteries wherein higher than 99% Mg deposition/stripping coulombic efficiency (>90% for Ca deposition/stripping), coupled with a wide electrochemical stability window exceeding 5 V for Mg salts, resulted in major breakthroughs in this field. Section 4.1 discusses the research of hydride as liquid electrolytes for Mg and Ca batteries that was driven by enabling a high compatibility with these metals. These studies unveiled the limitations of liquid electrolytes employing salts with a highly hydritic nature as was manifested by the low anodic stability of the borohydride salts (i.e., <2 V vs. Mg/Mg2+). Section 4.2 discusses the design concepts that improved the anodic stability of hydrides whilst maintaining the compatibility with Mg metal accomplished using closo-borate salts. The findings reported in Section 4.1 and Section 4.2 most recently paved the way for important Mg metal-hydride fundamental interfacial studies (Section 4.3) and the design of solid-state Mg electrolytes (Section 4.4). Section 5 discusses a very recent new application of hydrides as non-volatile and highly stable battery solvents (ionic liquids), which are amongst the best known ionic liquid electrolytes due to their wide electrochemical window (>4 V) and compatibility with both monovalent and multivalent metals. Finally, Section 6 showcases key battery demonstrations of hydrides as electrolytes in mono and multivalent batteries.
3. Solid-State Hydride Electrolytes for Monovalent Batteries
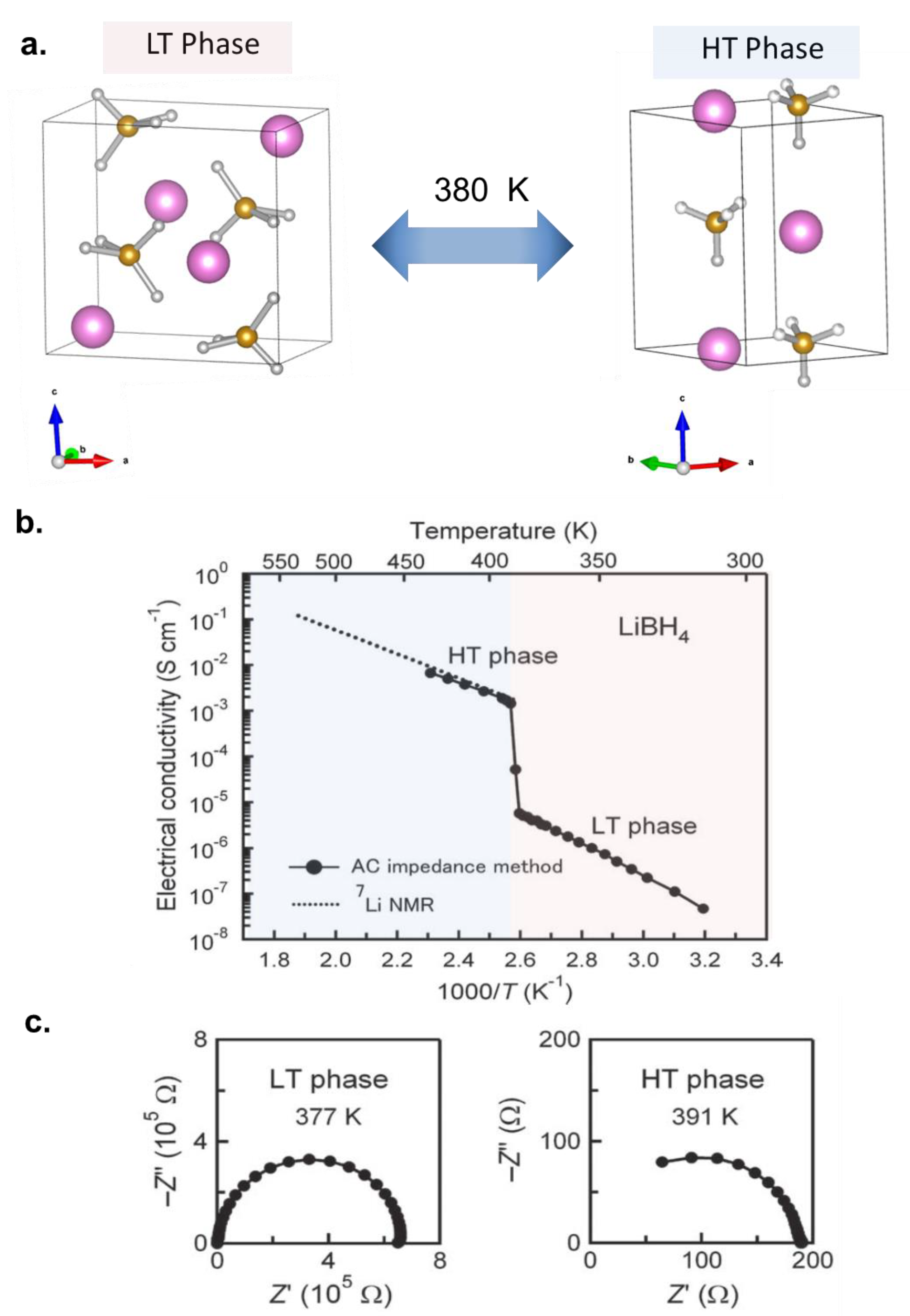
The observation of appreciable conductivity at room temperature in a hydride was first reported close to four decades ago in Li2NH (ca 10−4 S/cm, ca 10−2 S/cm at 400 K) [10]. The ionic conduction was mediated by Frenkel pair defects and/or charged vacancies and the cation diffusion occurred via pathways, which involved octahedral to tetrahedral jumps along the (001) crystallographic direction. However, its poor electrochemical stability (0.7 V vs. Li+/Li) and high reactivity made it less attractive amongst other solid-state electrolytes researched at that time. This interest in the solid-state cationic conductivity in hydrides was revived from 2007 with the unexpected discovery of high Li+ mobility after microwave hearting of LiBH4 [11]. This salt has been known to undergo phase transition from orthohombic to hexagonal phases at 380 K [12], however, the mobility of Li+ was revealed from permittivity measurements that demonstrated the presence of conductive heat loss in the hexagonal phase which suggested Li+ ion mobility (hydrogen is immobile as it is covalently bonded in BH4−). Electrochemical impedance measurements (Figure 2) further confirmed the presence of high Li+ conductivity in the hexagonal high temperature phase (in the order of 10−3 S/cm). In this phase, the high mobility of Li+ ions was explained by the presence of rotational disorder in the BH4−, the formation of metastable interstitial sites and the alignment of both the Li+ and BH4− along the a and b axes thereby permitting unobstructed migration of Li+ in both directions [13,14,15]. The salt was shown to have high oxidative stability beyond 5 V vs. Li+/Li, however this stability was later shown to be apparent and not representative of the thermodynamic stability of this salt. For example, the anodic stability was measured to be about 2 V vs. Li/Li+, when large surface area carbon was infused within the electrolyte [16]. The seminal discovery of Li+ high mobility in the solid state triggered research interests to investigate hydrides as solid-state electrolytes for Li+ and Na+ ion batteries. These are described in the next Sub-Sections.
3.1. Development of Highly Conductive Solid Electrolytes
In an effort to lower the phase transition temperature of LiBH4 to a superconductor, i.e., enable high conductivity at room temperature, the concept of stabilizing the hexagonal phase through the use of halide dopants (Br−, Cl− and I−), used to partially substitute the BH4− anion through forming solid solutions, was conceived and investigated [17,18,19]. This approach was proven most successful for (1 − x)Li(BH4) + xLiI solid solution, where a hexagonal crystal at room temperature (x > 13%) could be formed using mechanochemical synthesis and demonstrated high Li+ conductivities, i.e., 0.1 × 10−3 S/cm at 303 K for the solid solution with 40% LiI.
Beyond the aforementioned concept, other studies increased the room temperature ionic conductivities through the formation of mixed borohydride-amide salts as was shown for Li+ in trigonal Li2(BH4)(NH2) and cubic Li4(BH4)(NH2)3 where high conductivities in the order of 10−4 S/cm were achieved at room temperature [20]. The key for enabling these high conductivities was attributed to the presence of multiple Li+ crystallographic sites [20]. Interestingly, the activation energy for Li4(BH4)(NH2)3 (0.26 eV) was lower than the hexagonal superconductor LiBH4 phase(0.53 eV) [13,20]. It is worth noting that Li2(BH4)(NH2) became a molten salt at a relatively low temperature (onset ca 350 K). Further increase of the LiBH4/LiNH2 to 1:2 resulted in high conductivities (6.4 × 10−3 S/cm at 313 K) through the formation of cubic Li3(BH4)(NH2)2 phase (isostructural to Li4(BH4)(NH2)3) [21]. This salt underwent partial melting at 313 K, so it would be insightful to determine the contribution of the liquid phase to the overall bulk conductivity.
Other borohydride salts studied that increased the conductivity of Li+ included mixed-cation mixed-anion borohydrides [22,23]. In particular, in LiM(BH4)3Cl (M = cation), the disordered distribution of Li+ increased the ionic conductivity at 293 K to 1.02 × 10−4, 3.5 × 10−4, 2.3 × 10−4 S/cm for Ce, Gd and La-containing compounds, respectively. In addition, bimetallic borohydride oxide [24] i.e., LiCa3(BH4)(BO3)2 had a Li+ conductivity of 2.5 × 10−6 S/cm at room temperature which increased by about an order of magnitude when prepared with excess lithium and doped with either heterovalent Na+ or homovalent Sr2+(conductivities in the order of 10−5 S/cm at room temperature) [25]. A structural investigation of LiCa3(BH4)(BO3)2 showed that the borohydride was not directly involved in the conduction mechanism as (calcium borohydride substructure served to stabilize the percolating pathway) the conduction paths were composed exclusively of BO33− anions, which formed faced connected tetra- and octahedra accessible sites for Li+ jumps and suggested the vacancy-dependent mobility of Li+.
Alternate approaches that sought to increase the conductivity of LiBH4 utilized composites which included those with nano oxides or sulfides. In the former case, high room temperature conductivity (10−4 S/cm) was observed for the LiBH4–SiO2 nanocomposite prepared by melt infiltration of the borohydride into a mesoporous inorganic silica scaffold (Figure 3) [26,27]. The study suggested that the high mobility of Li+ was not related to stabilization of the hexagonal phase but was presumably caused by the high density of defects and low diffusion barriers at the interface between the two solids, which resulted from disorder, strain and space-charge regions [27,28]. Follow up studies demonstrated a similar effect using Al2O3 [29] and C60 [30] additives (note that the electronic conductivity in the C60 [30] composites could be higher than that desired from a solid electrolyte). The formation of composites with sulfides was first reported for LiBH4:P2S5, where the preparation process resulted in a material that went beyond a composite formation evident from a new crystalline 90LiBH4:10P2S5 phase that exhibited a high conductivity in the order of 10−3 S cm−1 around 300 K [31]. The combination of LiBH4 with glassy sulfide glass electrolytes xLiBH4–(100 − x):0.75Li2S:0.25P2S5 [32] using ball milling resulted in improved conductivity (i.e., at x = 33 it showed 1.6 × 10−3 S/cm) and low activation energy (0.30 eV). Interestingly, Raman spectroscopy suggested that BH4− was in the same state as that occurring in the hexagonal superconducting LiBH4 phase, where high rotational freedom and delocalized negative charge weakened the electrostatic interactions between Li+ and BH4−. The extension of composite studies to Li(BH4)3I resulted in similar improvements [33]. Other composites that yielded varying degrees of conductivity improvements, albeit less than those achieved with the sulfides, included composites with other borohydrides as Ca(BH4)2 [34], NaBH4 [35], with hydrides such as MgH2 [36], or borohydride-hydride ternary mixtures [37], and mixtures with halides such as NaCl [38].
Neutral molecules that are adducted to LiBH4 were also investigated to increase the room temperature conductivity, as was demonstrated for solid-state lithium borohydride ammoniates Li(NH3)xBH4, x = 0.5 and 1.0 [39]. A dramatic change in the ionic conductivity was attributed to the formation of defects induced by the partial desorption of NH3. For example, for Li(NH3)BH4, the conductivity increased from 1.5 ×10−6 S/cm at 303 K to 2.21 x 10−3 S/cm at temperatures > 313 K. Note that a closed system is required for these electrolytes so that the equilibrium pressure of ammonia is achieved, otherwise, reformation of LiBH4 will occur. Detailed structural analyses are desired to establish the conduction mechanism in these salts.
Unlike the case for the lithium salts, the formation of borohydrides that are highly Na+ conductive was less successful [40,41,42]. For example at room temperature, the conductivity of Na+ in NaBH4, NaNH2, and Na3(BH4)2I were in the range 10−10–10−9 S/cm. Disordered distribution of Na+ resulted in increased mobility, as was reported for Na2(BH4)(NH2) (ca 10−6 S/cm at 300 K), however the conductivity remained inferior to that reported in the lithium salts [40,41].
Inspired by the promising results in borohydrides, other complex hydrides such as alanates and amides were investigated [41,43]. In these cases, no transition to a super conducting phase was observed in the alanates leading to relatively low cationic mobilities [43]. Likewise, with the exception of Li2NH, imides and amides were less successful in demonstrating a good potential as solid-state electrolytes [44,45,46].
3.2. Towards Achieving High Li+, Na+ Mobilities: Closo-Borates
Investigations of ionic conduction mechanisms in borohydrides showed that it is not only vacancy-dependent but also based on the “paddle-wheel” mechanism [47,48,49] wherein the high rotational mobility of the (MyXn)δ− units promotes the ionic conductivity and lowers the activation energy [50,51]. This concept inspired investigating boron clusters (closo-borates)-based salts as electrolytes. These are found as undesirable byproducts that form during the thermal dehydrogenation of borohydrides [52]. Namely, the structural evolution of Li2B12H12 with temperature indicated possible Li+ conduction following transformation at 638 K to a disordered phase (β phase) with a frustrated Li+ lattice [53]. In addition, a solid-state nuclear magnetic resonance (NMR) study of the spin-lattice relaxation in Na2B12H12 salts revealed a substantial increase in the reorientational jump rates of B12H122− accompanied by the fast translational diffusion of Na+ following a first-order transition near 520 K [54]. Interestingly, investigating closo-borates as solid electrolytes for Li and Na batteries was reported concurrent with the report of Mg closo-borate salts as competent liquid electrolytes for Mg batteries- owing to their compatibility with Mg metal anode and high anodic stability- as discussed in Section 4 [9].
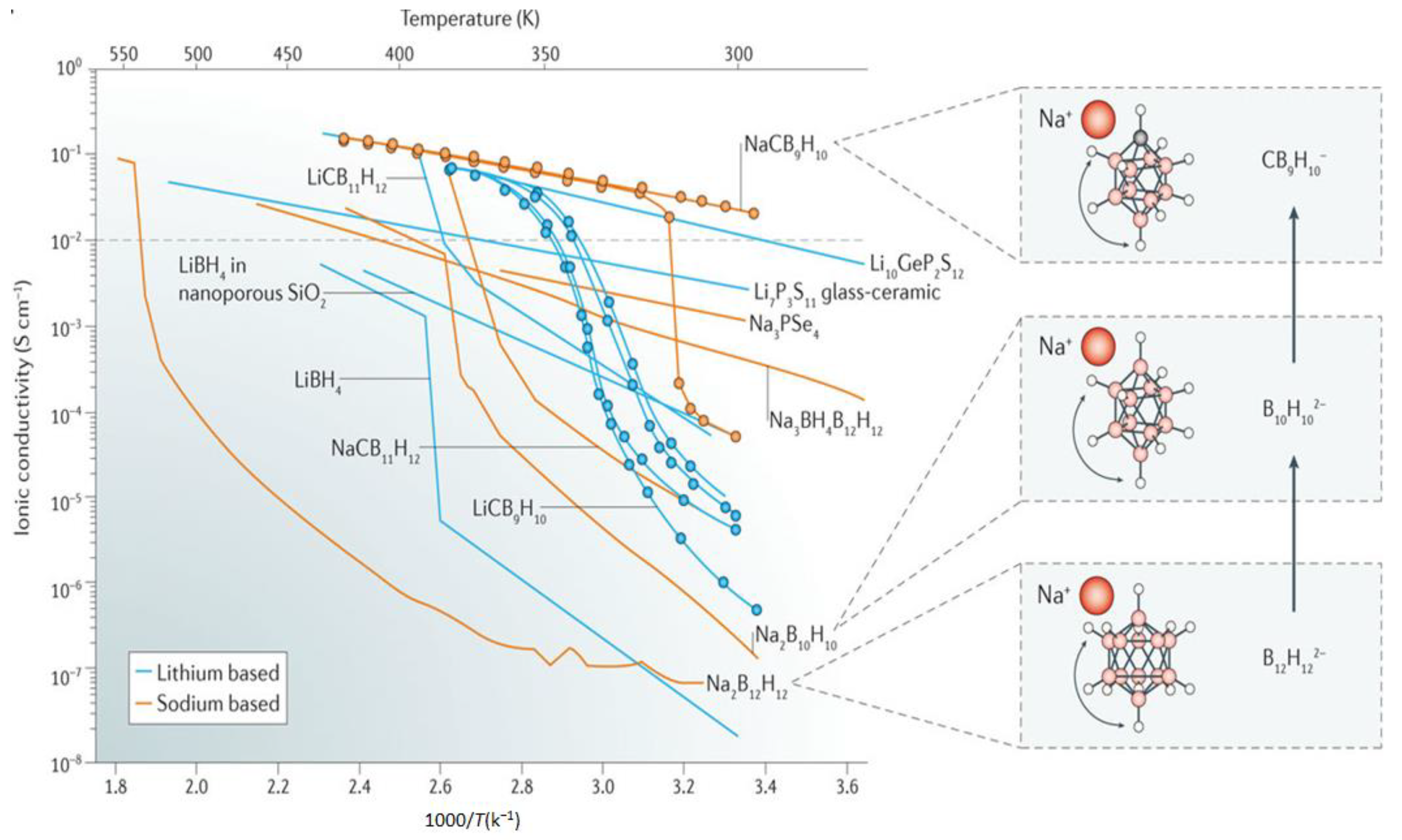
High cationic translational mobility in boron clusters was first reported for hydroborates in Na2B12H12 where high Na+ conductivity in the order of 10−1 S/cm, near 540 K was obtained (Figure 3) [55]. These conductivities were observed following transition from a low-temperature (ordered monoclinic) to a high-temperature (disordered, body-centered cubic) phase and were consistent with 23Na NMR measurements that showed enhancements in Na+ cation jump rate (>2 × 108 jumps/s) in the high temperature polymorph [54]. Lowering the transition temperature to a super conductor was reported for Na2B10H10, where a disordered face-centered cubic phase (>360 K), allowed for high Na conductivities in the order of 0.01 S/cm at 383 K [56]. One source of this improvement was speculated to result from the less spherical B10H102− anion which offered improved cationic diffusion. Results suggested that the anion dynamics was one major contributors to the conduction of the cation. For example, ab initio molecular dynamics (AIMD) simulations showed that the Li+ ion diffusivity in β-Li2B12H12 would be reduced by three orders of magnitude if the anion was constrained and immobile [57]. Computational studies however also showed that anionic dynamics (rotation/vibrations), is not the only factor that dictates the cationic mobility, rather, it is a complex interplay between the density of accessible diffusion sites, anionic dynamics and the nature of local bonding [58].
The relatively high mobility of Na+ in closo-borates triggered further studies to further improve the room temperature ionic conductivity. Similar approaches to those proven successful in enhancing the conductivity of Li+ in LiBH4 were implemented such as partial substitution with another anion including BH4− and a halide (Br, Cl, I) or with a cation like Li+ [59,60]. The absence of order–disorder transition and high room temperature conductivity (0.5 × 10−3 S/cm) for Na3BH4B12H12 suggested that the anion dynamics were a less important contributor to the superconductivities and that structural factors were more at play in this case. Cationic substitution was reported to enhance the ionic conductivity as in LiNaB12H12 (i.e., at 550K, the conductivity was eight times higher than Na2B12H12). Both Li+ and Na+ cations were mobile; Na+ became more mobile between 393 K to 433 K as suggested by the decreased Li+ transference number, respectively, from 0.91 to 0.71 [60]. However, these improvements in conductivity were later suggested to result from the inadvertent presence of other cluster fragments, i.e., B10H102−, produced from the synthesis of the salts using B10H14. For example, intentional inclusion of B10H102− in the structure of Na2B12H12 or simply preparing a binary complex of these anions dramatically enhanced the cationic conductivity [61,62]. It is noteworthy to mention that similar effects of these fragments on improving the cationic conductivity of Li2B12H12 (10−4 S/cm), prepared from B10H14 [63]; believed to result from a brief ball milling, were also suggested [64]. Systematic studies of possible extended ball milling effects on conductivity improvement in several boron cluster salts free from other undetermined cluster fragments were later reported, aided by powder x-ray diffraction (PXRD) and Quasielastic Neutron Scattering (QENS). The main finding was that ball milling resulted in the presence of the high temperature disordered phase at room temperature in the processed materials, which suggested a stabilized room temperature disordered form [65]. Conductivity enhancements were drastic, i.e., at room temperature the conductivity for Na2B12H12 was three orders of magnitude higher than that of the pristine sample. It is interesting that these ball-milled electrolytes consisted of both the low conducting and superconducting phases, where the latter phase was present in a form of interconnected nanocrystallites that were distributed in larger crystallites, which exhibited typical bulk-like conductivities [65]. Revisiting the ball milling effects on the conductivity of fragment-free Li2B12H12 (measured in the order of 10−5 S/cm at room temperature) was conducted in a later study. This work revealed that the improvements were due to Li+ and H deficiencies and proposed that these enhancements were unlikely to result from a stabilization effect of the high temperature phase as was evident from structural analysis. [66] What stands out in this study is the finding that deficiencies of H in boron clusters, that are stabilized by the cation, could be utilized to improve cationic mobilities.
To further improve the cationic conductivity, the utilization of carborate boron clusters, which carry a mononvalent negative charge, was investigated for LiCB11H12 and NaCB11H12 [67]. Interestingly, LiCB11H12 and NaCB11H12 underwent transition to a superconducting (>0.1 S/cm) disordered phase at much lower temperatures, 400 K and 380 K, respectively (Figure 3), accompanied by a high rate of anion reorientational jumps (1010–1011 jumps/s). The cluster’s monovalent charge, presence of less neighbors (cation:anion molar ratio = 1:1), and increased lattice constant were hypothesized to cause these enhanced conductivities. These were confirmed from ab initio molecular dynamics (AIMD) which also demonstrated that formation of a dipole (carbon atoms), creates a frustrated lattice and counteracts the ability of the phase to order, thereby reducing the transition temperature to superconducting phases [68].
Inspired from improvements in conductivity observed using the smaller B10H102−, studies examined Li and NaCB9H10 salts and demonstrated impressive room temperature conductivities of about 0.03 S/cm in the Na salt (Figure 3). Achieving this conductivity required preheating (i.e., sample conditioning to access the superconducting phase) to about 425 K [69]. Note that systematic studies on the effect of thermal cycling on the room temperature stability of the disordered superconducting phase are needed to discern the origin of these drastic improvements, especially given the hysteresis observed. Substantial improvements were also found for the Li-based salt (Figure 3), i.e., 0.03 S/cm at 354 K. As the case in NaCB11H12 and LiCB11H12 salts, the dipole produced by the carbon atom could influence the anion orientations and create a frustrated landscape that results in high cationic mobility at lower temperatures.
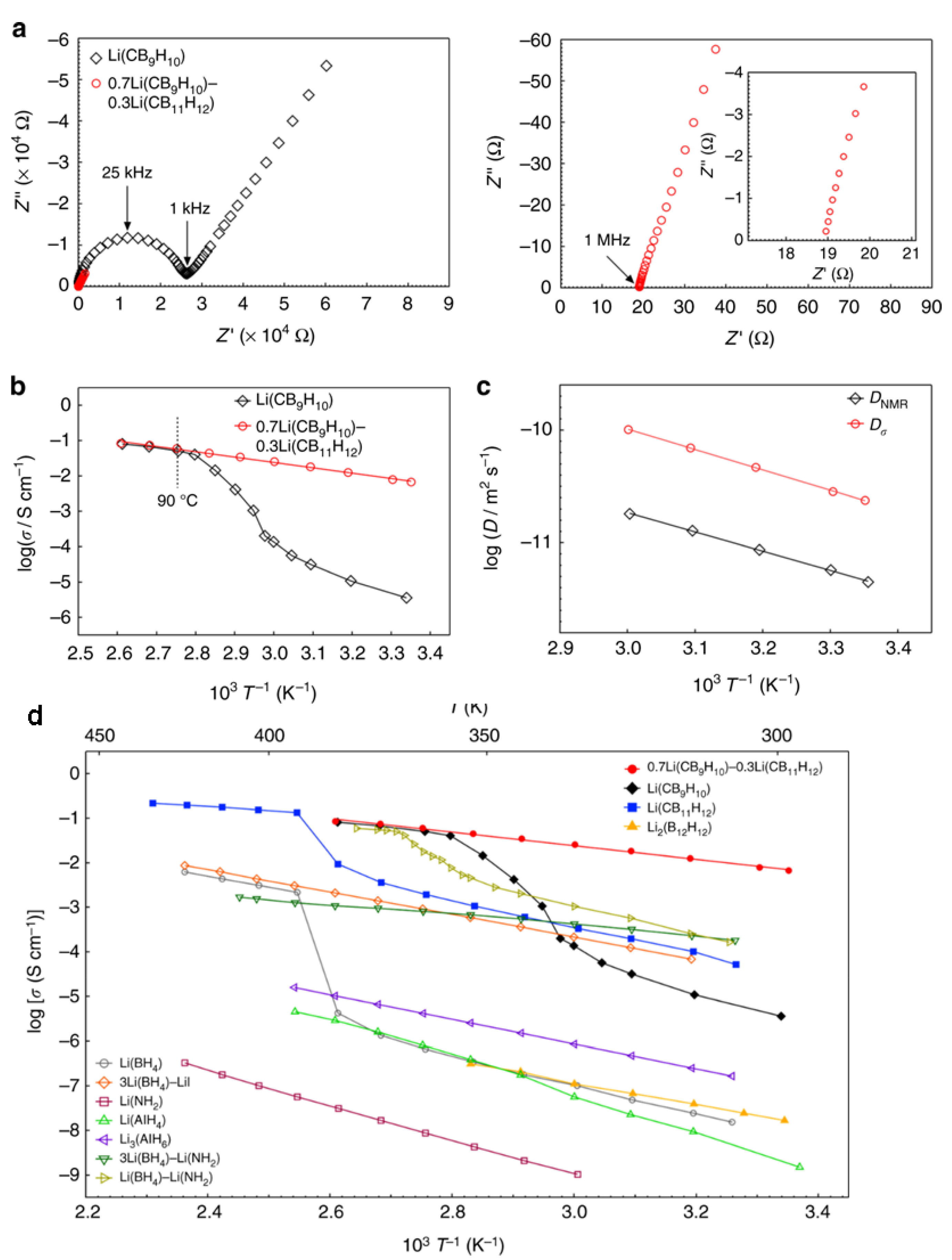
Further studies with Na and Li carborate salts targeted stabilization studies of the high temperature superconducting phase and were inspired by those implemented in borohydrides and hydroborates. Stabilization of the disordered phase was studied through formation of what was suggested as mixed (CB9H10:CB11H12)2− Li and Na salts [64,65]. These salts were produced from mechanochemical treatment or simple mixing of (CB9H10:CB11H12)2− Li and Na salts in the 1:1 molar ratio in aqueous solutions. The absence of phase transitions suggested some sort of stabilization effect on the disordered phases. High ionic conductivity of Li+ was found (10−3 S/cm at 300 K), and impressive Na+ conductivity was reported (6x10−2 S/cm at 300 K). A recent report examined the Li salt conductivity in mixed 7:3 molar CB9H10−:CB11H12− prepared by ball milling also demonstrated very high conductivity of 6.7 × 10−3 S/cm at room temperature without thermal activation (Figure 4) [70]. Other studies examined mixing Na carborate CB11H12− with hydroborate B12H12− salts and also showed high conductivities (2 × 10−3 S/cm at room temperature), albeit lower than those observed for closo-carborates [71]. Studies in carborate-type derivatives based on several nido-carborates demonstrated inferior conductivities to those reported in closo-carborates [72] (note: surprisingly a high value in the order of 10−3 S/cm at 300 K was reported for what was described as α-NaCB11H14, it remains unclear why the conductivity was much higher than that in typical NaCB11H14, 10−6 S/cm at 300 K). Although, the electrochemical stabilities of these specific salts were not reported, they are expected to exhibit a much narrower window compared to the closo-borates owing to the known limited stability of the nido-anions [72].
One approach sought to increase the conductivity in B12H122− by confinement in a nanoporous silica SBA scaffold, which was inspired from LiBH4 studies discussed above [73]. However, nanoconfinement in the case of Li2B12H12 was not effective as was evident from a very low conductivity compared to that of the bulk material (i.e., 10−7 S/cm at room temperature).
It is worth noting there have been studies that investigated the cationic conductivities in boron clusters that were halogenated. Replacing the hydrogens in the B12H122− anion with halogens in the Na salt was counterproductive and showed dramatically low conductivities at room temperature, where high conductivities in the order of 10−1 S/cm, could only be achieved at temperatures exceeding 673 K. This behavior was suggested to result from restriction in cluster’s reorientational mobility, caused by increased anionic mass wherein strong bonds to Na+ as a result of directional charge distribution on the halogen atoms, contributed to this behavior [74]. A recent computational study supported this finding and suggested that partial halogenation may somewhat reduce conductivity losses in halogenated B12H122−, however the conductivity will be lower than the hydrogenated form [75].
4. Electrolytes for Multivalent Batteries
Recently, there have been increased interests in post lithium batteries that include multivalent storage cations such as Al3+, Ca2+ or Mg2+ [2]. These elements are abundant and offer possibilities for battery cost reductions. In addition, their use in the form of metallic anodes offer a safer alternative to reactive Li metal. In particular, magnesium batteries attracted lots of interest due to their high energy density potential. The Mg metal redox potential is low (−2.4 V vs. SHE) and it has high volumetric capacity (3832 mAh/cm vs. 2062 mAh/cm and 1136 mAh/cm, for Li and Na, respectively) [6]. One key hurdle in these batteries is the passivation of Mg metal, due to electrolyte decomposition, in most common salts/solvents in addition to the sluggish diffusion kinetics of Mg2+ in solid-state host structures likely caused by its divalent charge [76,77].
Until recently, the Mg passivation challenge has limited the choice of electrolytes to a handful of chloride base salts/reagents as they were not shown to passivate Mg metal [2,6]. These electrolytes, while highly performing, are corrosive, have limited anodic stability (i.e., limited by Cl− redox potential) and negatively interfere with cation insertion in the cathode [78]. Although designing other alternatives that overcome these challenges seemed farfetched, boron hydrogen compounds were discovered to offer highly competent and practical Mg electrolytes. As similar passivation issues that occur on Mg metal are encountered for Ca metal, strategies that were successful in Mg batteries were shown as applicable to Ca batteries.
4.1. Liquid Borohydride Electrolytes: Achieving Compatibility with Mg Metal
The notion that simple ionic Mg salts could not function in Mg batteries was altered in 2012, where Mg(BH4)2 was demonstrated as a first example of halogen-free, simple-type ionic electrolyte in Mg-based batteries [79]. The heart of this concept was that the high reductive stability of Mg(BH4)2 (i.e., reducing agent) would allow it to withstand the low potential environment of Mg metal anode thereby preventing Mg metal passivation. Ethereal solutions of Mg(BH4)2 in tetrahydrofuran (THF) solvent enabled reversible Mg plating/stripping. This proof of concept sparked research efforts towards understanding the scarcely explored aprotic solution chemistry of hydrides in order to improve the performance of these electrolytes by determining key factors that govern their electrochemical behavior. Spectroscopic analyses (FTIR, NMR) revealed that the electrochemical performance was dictated by poor dissociation in low dielectric solvents such as THF (owing to the relatively ionic nature of the salt). This was overcome using ethereal solvents with more electron donating oxygen sites such as 1,2 dimethoxyethane (DME) and the addition of additives that further dissociate Mg(BH4)2 such as LiBH4, through potential complexation of the acidic cation with the BH4−. The combination of the aforementioned strategies resulted in a competent performance (Figure 5) with high current densities (25 mA/cm2 stripping peak current), low deposition (−0.3 V)/stripping (0 V) overpotentials and excellent Mg deposition/stripping coulombic efficiency (94%). These findings demonstrated that simple ionic salts could be made compatible with the magnesium metal if the anion in the salt has sufficient reductive stability, thereby creating a new design space of highly performing electrolytes for Mg batteries. The electrochemical performance was found to be governed by the extent of salt dissociation per spectroscopic analyses which revealed the presence of strong association in these electrolytes. The amount of the complex cation MgBH4+ (i.e., charge carrier) tracked with improved electrochemical performance. Follow up studies examined other chelating solvents such diglyme (DGM) and tetraglyme, which further demonstrated the direct relationship between the number of electrodonating oxygen sites and the coulombic efficiency of Mg plating/stripping [80,81]. In addition, computations were used to understand the role of coordination and the effects of the solvent on the solubility and dissociation in these electrolytes [82,83]. Significant and irreversible salt agglomeration in all glymes ranging from THF to tetraglyme was found in all non-dilute borohydride salt solutions [82]. The agglomeration rate and diffusivity of Mg2+ in longer chain ethers such as tetraglyme were at their lowest and tracked with the solvent’s self-diffusivity.
The report of Mg(BH4)2 as a competent electrolyte sparked interests in revisiting other ionic salts such as magnesium bis(trifluoromethanesulfonyl)imide Mg(TFSI)2, known to passivate Mg metal [84,85]. Similar solution chemistry dominated by Mg association with the anion was found in Mg(TFSI)2 electrolytes [86], which underscored the importance of the high reductive stability of the BH4− anion in preventing passivation of Mg metal.
Mg(BH4)2 was applied to mitigate the passivation effects that take place in the presence of Mg passivating anions, i.e., TFSI, present in inorganic salts (Mg(TFSI)2) [87], in organic salts (ionic liquids solvents) [88,89] and as a reagent in complex electrolyte solutions such as alkoxyborates (i.e., mixing the acidic tris(2H-hexafluoroisopropyl) borate (THFPB) with Mg(BH4)2 to study Mg-S batteries) [90]. Note that in the latter case, reaction of the reductive borohydride with the acidic additive was indicated thereby partially transforming the borohydride (further investigation is needed to identify the new species). Another approach that was applied to modify Mg(TFSI)2 solutions with BH4− was using a BH4− anion with hydride groups that were partially substituted with phenol [91]. Both 0.5 M Mg(TFSI)2 and 0.15 M Mg(B(OPh)3H)2 were combined in diglyme and exhibited low deposition overpotentials (ca −0.64 V); however, the coulombic efficiency remained at low as 64% even after extended cycling [91].
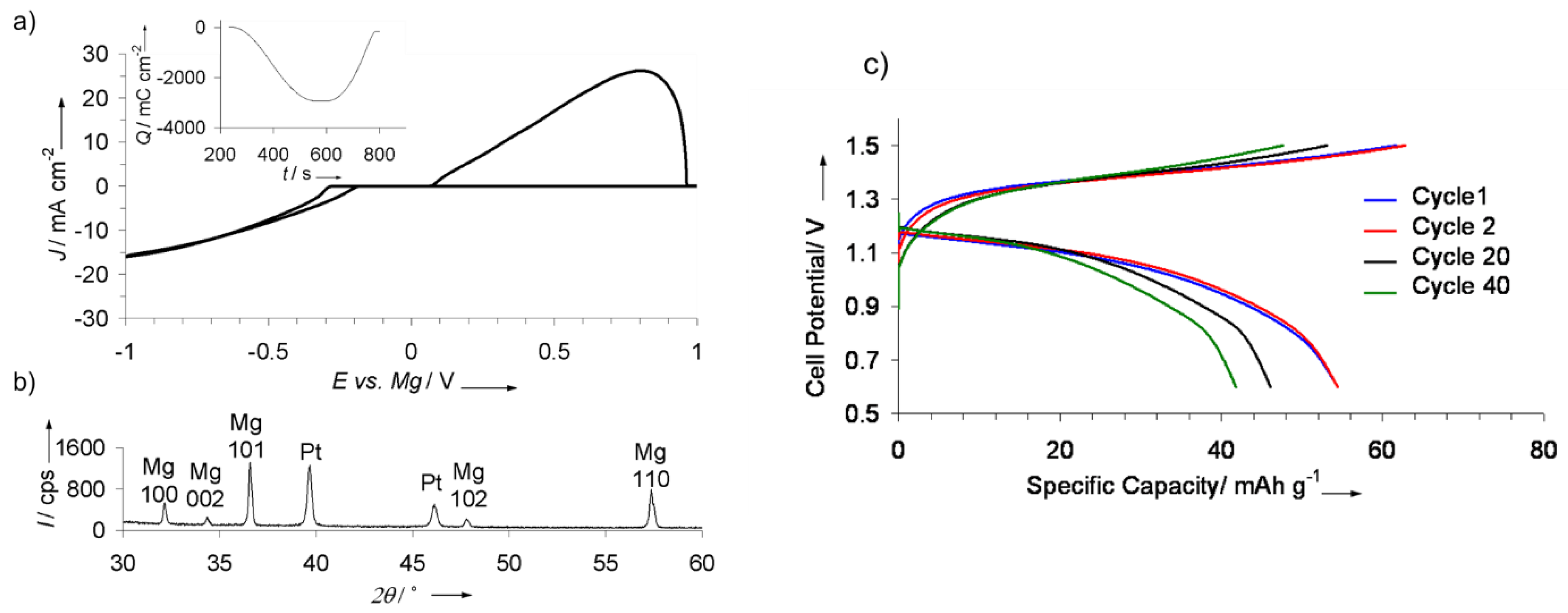
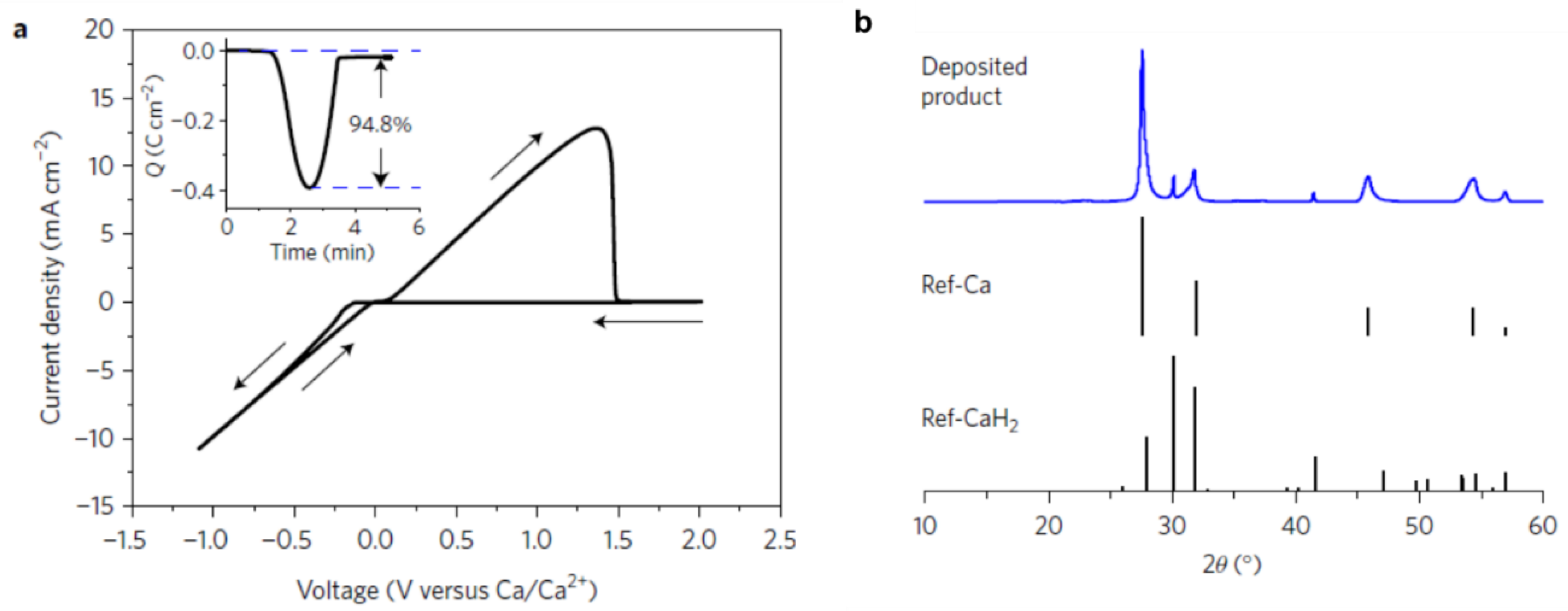
The successes with utilizing magnesium borohydride in Mg batteries later inspired examining borohydrides electrolytes for Ca batteries, wherein Ca metal anode is notorious for being easily passivated by common electrolytes. Ca(BH4)2 in tetrahydrofuran (THF) electrolyte was remarkably capable of plating/stripping Ca metal with high coulombic efficiencies, high purity and current densities (Figure 6) [92].
4.2. Electrolytes with Wide Electrochemical Window: Closo-Borate Salts
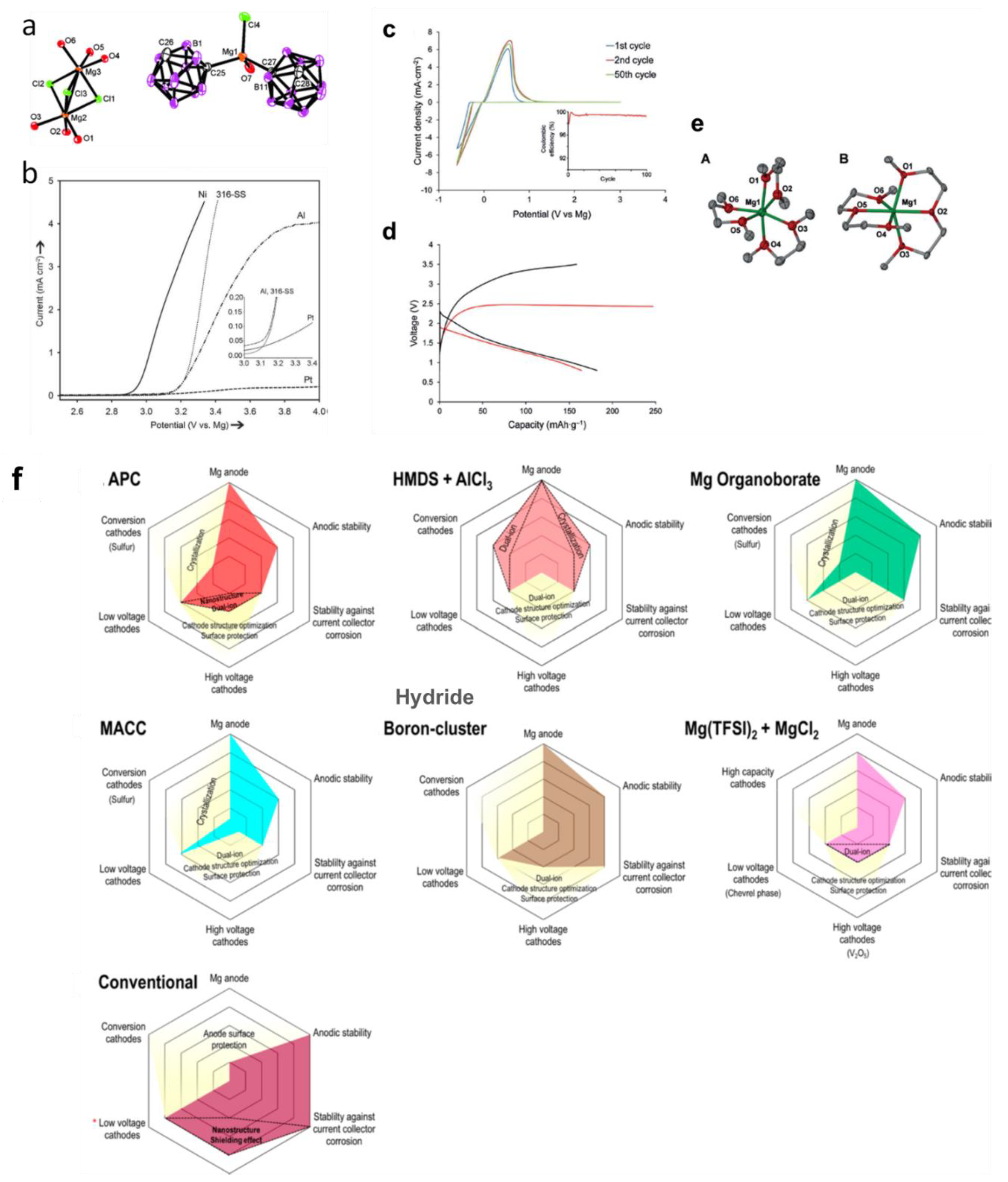
The stability against electrochemical oxidation of borohydrides in ethereal solutions was found low in all magnesium borohydride based electrolytes (i.e., 1.7, 2.2 and 2.3 V vs. Mg/Mg2+) on platinum, stainless steel and glassy carbon electrodes, respectively) [79]. This, coupled with the highly nucleophilic nature, limit the use of borohydrides electrolytes under a high voltage environment and with cathodes susceptible to nucleophilic attacks such as S or some oxides. To increase the oxidative stability of the Mg(BH4)2 electrolytes, and minimize their hydridic reactive character, strengthening of the B-H bond through forming 3-dimensional B-B bonds as in icosahedral boron clusters (hydroborates and carborates) was proposed and demonstrated using closo-carborates [9,93,94]. The concept was that these clusters would retain the high compatibility of the B-H based salt with Mg metal, whilst expanding the electrochemical stability window. Computational and experimental support of this concept was first presented using the dicarborate anion, where carboranyl magnesium chloride electrolyte (1-(1,7-C2B10H11)MgCl) was reported with high oxidative stability (3.3 V vs. Mg) (Figure 7). The electrolyte also demonstrated the high compatibility of the carborate anion with Mg metal (i.e., columbic efficiency was ca 100%). Based on this proof of concept, a new simple salt based on the more weakly coordinating monocarborate anion CB11H12− was synthesized and reported with high solubility in triglyme and tetraglyme (>1M) [94]. The electrolyte efficiently cycled Mg metal (>98%) with very low overpotentials, had high chemical stability and was non corrosive. In addition, unlike the case in the borohydride electrolytes, the Mg2+ in this electrolyte was found be unassociated with the anion as revealed in the crystal structure of the isolated salt (Figure 7). It is worth mentioning that the oxidative stability of Mg(CB11H12)2 (measured in acetonitrile solvent), was found to be very high (4.9 V vs. Mg/Mg2+) which exceeds that of all ether solvents. These remarkable properties qualified the report of this electrolyte as a breakthrough in the field of Mg electrolytes [2]. A follow up study reported the retention of Mg metal compatibility after fluorination of the C-H bond in the CB11H12− anion, as was apparent from high coulombic efficiencies (96%) and low overpotentials (measured in 5 mM salt concentrations in triglyme) [95].
4.3. Hydride Interfaces in Multivalent Batteries: Significance and Nature
It has been generally accepted that the formation of interfaces on the surface of multivalent metal anodes such as Mg would lead to passivation phenomena, thereby prohibiting the function of the battery. This is interestingly in stark contrast with Li or Na based batteries, where these interfaces or solid electrolyte interfaces (SEIs) (resulting from decomposition of the electrolyte), are permeable to Na+, Li+ and are instrumental in enabling a stable performance of the anode [97,98]. The root causes of these passivating phenomena are not well understood and could likely result from the divalent nature of these ions which results in low diffusivity in the solid state.
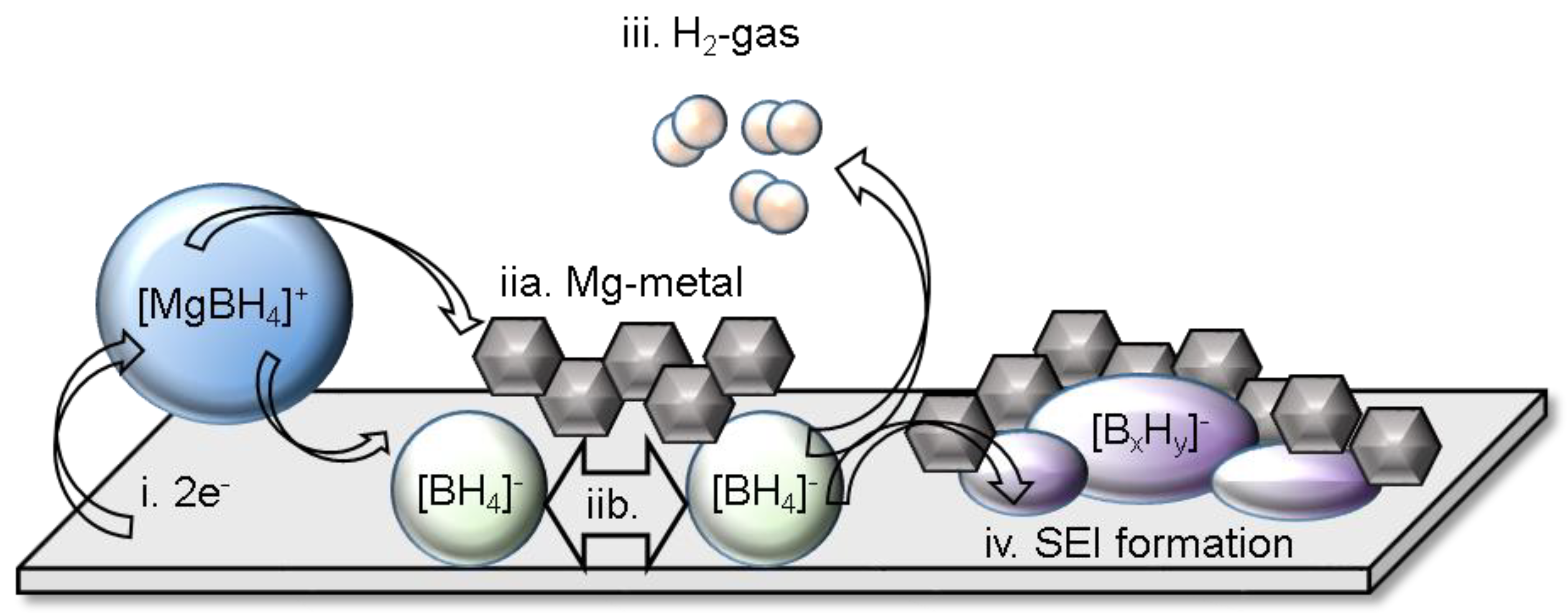
Recently, research into the function of hydrides electrolytes in Mg and Ca batteries revealed the presence of Mg and Ca interfaces that could support highly efficient deposition/stripping of these multivalent metals. This indeed alters the notion of what has been widely understood about factors required to support the function of these anodes [2]. The first evidence of Mg2+ permeable SEI in Mg batteries was shown in the borohydride solutions [99]. The properties of the interface were demonstrated to play an important role in the performance of the electrolyte. For example, the presence of the SEI was observed in Mg/Mg symmetric cells and was found to be dependent on the type of solvent and borohydride additive used, i.e., the use of monoglyme solvent and LiBH4 additive (in the 1:3 molar Mg(BH4)2:LiBH4/DME electrolyte) resulted in the lowest cell overpotentials [99]. Therefore detailed investigations of the nature of this interface and morphology were conducted using operando electrochemical-synchrotron soft X-ray absorption (sXAS) and transmission electron microscopy (TEM) [99]. Chemical transformation of the BH4−, accompanied by H2 gas release, into B12H122−, was observed to occur during the Mg deposition process (Figure 8). It is worth noting that the formation of Mg rich, Li poor alloy was observed in the 1:6 molar Mg(BH4)2:LiBH4/diglyme electrolyte and was suggested to play a role in the Mg deposition process [100]. However, the formation of this alloy seems to be limited thus far to this LiBH4 rich mixture as Li presence was not found in the 1:3 molar Mg(BH4)2:LiBH4/DME electrolyte [99]. SEI presence was also revealed in Mg(CB11H12)2 electrolytes and constituted of Mg, B and C species, in addition to a crystalline phase indexed to be close to a MgB2O5 like species [101,102]. The deposited magnesium was found to be in the form of Mg nanoparticles (ca 10 nm) embedded in an amorphous matrix of SEI material whose growth was captured. This morphology and resultant SEI enabled cycling Mg/Mg symmetric cells under unprecedented high current rate conditions (10 mA cm−2) [102].
In Ca batteries, the presence of CaH2 on the surface of Ca metal, formed from the reaction between the THF solvent and Ca metal was revealed [92]. The non-reactivity of Ca(BH4)2 with the hydride could be the source of the high compatibility in this case. No detailed investigation of possible SEI formation under high currents were reported and further understanding of factors that govern these performances is desired.
4.4. Solid-State Electrolytes
The divalent positive charge carried by cations such as Mg2+ leads to sluggish diffusion kinetics in solid-state structures. As such, designing electrolytes with acceptable Mg2+ conductivity has been very challenging. However, the report of highly Mg compatible Mg(BH4)2-based materials as liquid electrolytes, stimulated research that investigated Mg borohydride-based salts as solid electrolytes. This was first demonstrated for Mg(BH2)(NH2), selected due to the presence of cavities large enough to enable magnesium ion conduction through the hopping mechanism. Conductivity of about 10−3 mS/cm was measured at 423 K for Mg(BH4)(NH2), which is three orders of magnitude higher than that in Mg(BH4)2, presumably due to the shorter distance between the two nearest Mg atoms (3.59 Å in Mg(BH4)(NH2) vs. 4.32 Å in Mg(BH4)2) (Figure 9) [103].
Higher conductivities at lower temperatures were reported for cis-Mg(en)(BH4)2, en = NH2(CH2)2NH2 [104]. The material was obtained by the mechanochemical processing of the ethylene diamine and Mg(BH4)2 powder mixture. The mobility of Mg2+ ions in cis-Mg(en)(BH4)2 increased from 5 × 10−8 S/cm to 6 x 10−5 S/cm in the temperature range 303–343 K (Figure 9).
Polymer-electrolyte systems typically based on polyethylene oxide (PEO) were also reported for Mg batteries. The nanocomposite polymer electrolyte based on PEO, Mg(BH4)2 and MgO, allowed for reversible Mg deposition/dissolution with 98% coulombic efficiency, however the conductivity was not reported [105].
5. Hydrides’ Potential as Stable Solvents: Ionic Liquid Electrolytes
To eliminate hazards associated with the use of volatile and flammable battery solvents, ionic liquids, which are salts in the liquid state at room temperature, represent an alternative option [106]. Ionic liquids can have high thermal stabilities and appreciable conductivities which make them attractive for use as solvents in batteries. The most successfully used ionic liquids are those for Li and Na batteries and couple an organic cation (ammonium, phosphonium or pyrrolidinium cations) with a fluorinated anion such as bis(fluorosulfonyl)imide ((FSO2)2N−, FSI) or bis(trifluoromethanosulfonyl) imide TFSI− ((CF3SO2)2N−) [106]. However, one of the key challenges in designing ionic liquids lies in the stability of the electrolyte itself or both the electrolyte/anode and the electrolyte/cathode interphases. For instance, an anion with insufficient oxidative stability can limit the choice of cathode material. Likewise, anions susceptible to decompose under the highly reductive anode environment can partially or fully passive the anode. This has been particularly true in magnesium batteries, where fluorinated anions such as TFSI− passivate the Mg metal surface thereby prohibiting the use of ionic liquid solvents (unless highly reducing salts/reagents are added) [6]. Recently, as discussed in Section 4, Mg deposition/stripping was shown possible in electrolytes based on Mg(BH4)2 salt and TFSI− based ionic liquids [88,89]. Reported systems consisted of a pyrrolidinium cation with dangling PEGylated chains [89] or with ether-functionalized ammonium cation N2(20201)3+ [88]. The incorporation of chelating ethereal moieties was inspired by previous studies of enhanced Mg(BH4)2 dissociation and performance in ethers with increased number of oxygen electron donors (Section 4) [79,80]. For example, ionic liquids based on methyl polyethylene glycol MPEG3PyrTFSI (three ether oxygens) and MPEG7PyrTFSI were superior to N-butyl-N-methylpyrrolidinium BMPyrTFSI [89]. Further studies showed that ionic liquid solutions with small concentrations of Mg(BH4)2 (i.e.,18 mM) in 0.3 M Mg(TFSI)2/tetraglyme, N-methyl-N-butyl pyrrolidinium TFSI (1:2 molar) allowed for some reversible Mg deposition/plating (initial coulombic efficiency ca 87% that degraded with cycling). Interestingly, despite the presence of Mg(BH4)2, the anodic stability was reported to be up to 3 V, however it dropped to ca 2.1 V in cycled solutions [107].
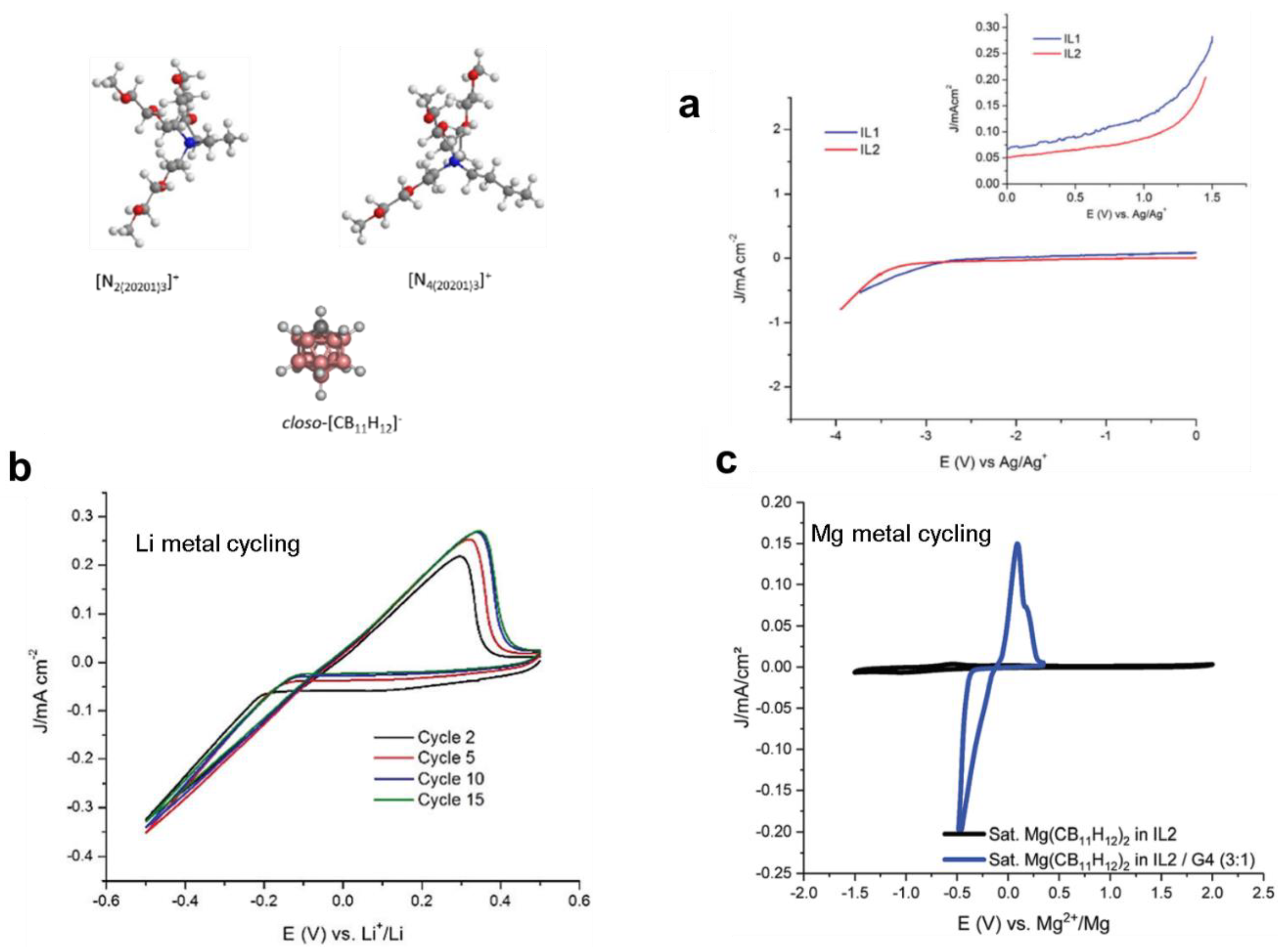
In the aforementioned studies, given the use of borohydrides, the true oxidative stabilities of these solutions were low (<2.5 V). Therefore, to eliminate challenges associated with the use of strongly reducing agents, a new recent alternate approach designed, for the first time, ionic liquids based on closo-borates for Mg batteries [108]. This was inspired from the high compatibility of the carborate anion as was revealed from the Mg salts analogues (Section 4). In addition, the high oxidative stability and weakly coordinating nature of these anion makes them highly desirable for Li and Na batteries. Consideration of ionic liquids based on closo-borates in energy storage devices in general has been ignored given the very high melting temperature of these salts attributed to the rigid structure of the anion. However, recently, this challenge was overcome by creating ionic liquids which incorporate the highly stable closo-monocarborate anion with ammonium cations decorated with flexible alkoxy ligands (N2(20201)3+ and N4(20201)3+), which compensated for the rigidity of the anion. These ionic liquids were reported to remain in the liquid phase down to −52 °C, had high conductivity (in the order of 10−4 S/cm) and were highly dissociated (Figure 10) [108]. This placed them amongst best performing ionic liquids being considered for batteries. Importantly, they were used to demonstrate reversible Li and Mg deposition/stripping thereby marking the introduction of new competent ionic liquids family to energy storage devices [108].
6. Demonstrations of Hydride Electrolytes in Batteries
Successful demonstrations of batteries require electrolytes that are not only highly conductive, but that are also highly compatible with both the anode and the cathode. Compatibility means: (1) the electrolyte is stable chemically and electrochemically upon contact with the electrode’s components (this includes active materials, other components such as conductive additives, etc.). The consequence of this is the absence of any decomposition products from the electrolyte on the surface of the electrode and (2) the electrolyte is unstable in the environment of the electrode and decomposes generating a surface film on the electrode. These decomposition products however, allow for the passage of the charge carriers (i.e., cations) and importantly serve as a barrier to prevent further decomposition of the electrolyte. The surface layers formed on the anode side are referred to as SEI (Solid Electrolyte Interface) as discussed in Section 4 and those formed on the cathode are called CEIs (Cathode Electrolyte Interfaces). The electrolyte–electrode interactions are complex in nature and highly dependent on the electrode and its components.
The first example of the successful application of hydrides electrolytes in a stably cycling battery cell was reported in a Mg battery for Mg(BH4)2–LiBH4/DME electrolyte with a magnesium anode and a Chevrel phase Mo6S8 cathode (Figure 5). The cell was operated at room temperature and delivered the expected performance from the Mo6S8 cathode [79]. A follow-up study using a similar battery (rate of C/10), delivered a capacity of 99 mAh/g during the initial discharge (vs. 129 mAh/g of the theoretical value) and then retained 90% of this capacity for over 300 cycles [80].
LiBH4 application as a solid-state electrolyte in all solid-state batteries was first reported in an electrochemical cell with Li metal anode and LiCoO2 cathode [109]. The battery performance was evaluated at 393 K, which ensured the stability of the highly conductive LiBH4 phase. Due to the nucleophilic nature of borohydrides, parasitic reactions between LiBH4 and LiCoO2 occurred leading to high interfacial resistance and significant capacity loss. To mitigate this interaction, amorphous Li2PO4 was introduced between electrolyte and the cathode and somewhat improved the battery cycling performance; i.e., 97% of the initial discharge capacity (89 mAh/g) after 30 cycles. The hydritic nature of the borohydride called for investigating other cathodes wherein stable interface formation between LiBH4 and TiS2 cathode was reported [41,110]. The solid-state battery consisted of TiS2 cathode, LiBH4 electrolyte and Li anode and was successfully cycled 300 times at 393 K and a rate of 0.2 C. Although, the initial discharge capacity was only 80 mAh/g (vs. 239 mAh/g of the theoretical value), at the second cycle the parameter reached 205 mAh/g with close to 100% coulombic efficiency. The cell’s durability was explained by the chemical/electrochemical oxidation of the borohydride just below the TiS2 surface. This was accompanied by hydrogen release and formation of a new phase, identified to most likely be Li2B12H12. Later studies examined this cathode with other LiBH4 based electrolytes such as Li4(BH4)3I, combined with either TiS2/LiBH4 or TiS2/Li4(BH4)3I composite electrodes [111]. The higher durability of the cell consisting of TiS2/Li4(BH4)3I composite electrodes underscored the importance of the high mechanical plasticity in the I−-substituted LiBH4.
The feasibility of lithium-sulfur (Li-S) conversion batteries utilizing a borohydride electrolyte was demonstrated at 393 K [112]. The Li-S cell delivered 1140 mAh/g at 0.05 C rate, during the first discharge, which corresponded to 70% of sulfur utilization ratio. Over the next 45 cycles, the discharge capacity remained as high as 730 mAh/g with nearly 100% coulombic efficiency. Further studies using the more Li+ conductive electrolytes such as LiBH4–nanoconfined LiBH4 [113] LiCl [114] or allowed for lowering the operating temperature of the Li-S cell. The surprising performances of these batteries, despite the highly reactive nature of borohydrides towards S, suggest formation of a CEI (Cathode Electrolyte Interface) that is analogous to that observed on TiS2; i.e., closo-borates. In fact, just recently, excellent Li/S battery performance was reported in a 7:3 molar [CB9H10−:CB11H12]2− Li salt [70].
The low oxidative stability of the borohydride electrolytes limited the demonstration of high voltage batteries. This was particularly apparent in liquid borohydride electrolytes used in Mg batteries, which unlike solid-state electrolytes, lacked the presence of the kinetic barriers that could extend the oxidative decomposition onset. However, the high stability of Mg(CB11H11)2 in tetraglyme allowed for cycling the high voltage cathode α-MnO2 (initial discharge capacity of 170 mAh/g, Figure 7). This was the first successful example of a Mg coin cell operated at voltages close to 3 V vs. Mg2+/Mg (i.e.,4.2 V vs. Li+/Li) (Figure 7) [94]. On the other hand, one of the best demonstrations of a high voltage solid-state battery (3V), was for the Na metal battery employing Na2(B12H12)0.5(B10H10)0.5 electrolyte which was operated for 250 cycles [115]. One key to this successful demonstration was the high oxidative stability of the electrolyte and accomplishing a good contact between the cathode (NaCrO2, theoretical capacity of 120 mAh/g) and the electrolyte (this was achieved by dissolving the latter in anhydrous methanol and dispersing NaCrO2 into the solution, followed by drying in vacuum and heat treatment at 543 K in order to recrystallize the electrolyte). A follow-up study utilized isopropanol in order to recrystallize the electrolyte in the cathode at a lower temperature (453 K) [116].
Hydride electrolytes were also examined in batteries employing insertion and alloy anodes in liquid Mg batteries (i.e., good cycling was reported for Bi anode in a borohydride electrolyte [117]), solid-state lithium ion (i.e., nanostructured Bi2Te3/LiBH4 composite [118] or Si with Li2B12H12 [18]) and sodium ion batteries (Na3Sn anode in Na(B12H12)0.5(B10H10)0.5 electrolyte) [116]. It is noted that performance losses due to challenges associated with anodes such as Sn and Si occurred and made it difficult to establish the potential advantage of hydrides in these batteries.
7. Conclusions
Several decades ago, hydrides were widely investigated as hydrogen storage materials and their potential to electrochemical energy storage devices was limited to the Ni-MH batteries. However, just over a decade ago, this has been altered owing to two key discoveries: First is the discovery of high solid-state ionic conductivity in LiBH4, which opened the doors for designing hydrides as highly conducting solid-state electrolytes for Li and Na batteries. The second was owing to the creation of borohydrides electrolytes that are highly compatible with Mg metal. This opened a new design space in Mg electrolytes as it resulted in highly competent boron hydrogen-based electrolytes which overcome key challenges and changed several preconceptions about what an ideal electrolyte needs to be like. These advancements have also served to inspire investigating several new families of electrolytes for Mg batteries.
The high Li+ and Na+ conductivities in closo-borate solid-state electrolytes allowed for battery demonstrations and have placed them amongst the top performing solid-state electrolytes being investigated. On the other hand, closo-carborate liquid electrolytes have enabled the demonstrations of high voltage Mg batteries for the first time in battery coin cells.
The journey of hydrides in the field of energy dense batteries is just beginning. Studies of key factors that govern the performances of these materials would be advantageous for future developments. In addition, the high costs associated with closo-borates could represent a barrier towards their practical use and therefore overcoming this challenge would drive the consideration of these materials as future battery electrolytes.
Funding
Funding for meeting costs associated with the preparation of this review was provided by Toyota Motor Corporation.
Conflicts of Interest
The author declares no conflict of interest.
References
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Aurbach, D. Promise and reality of post-lithium-ion batteries with high energy densities. Nat. Rev. Mater. 2016, 1, 16013. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, Y.; Lotsch, B.; Hu, Y.-S.; Li, H.; Janek, J.; Nazar, L.F.; Nan, C.-W.; Maier, J.; Armand, M.; et al. New horizons for inorganic solid state ion conductors. Energy Environ. Sci. 2018, 11, 1945–1976. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Jiang, L.; Yu, Y.; Sun, J. Progress of enhancing the safety of lithium ion battery from the electrolyte aspect. Nano Energy 2019, 55, 93–114. [Google Scholar] [CrossRef]
- Kato, Y.; Hori, S.; Saito, T.; Suzuki, K.; Hirayama, M.; Mitsui, A.; Yonemura, M.; Iba, H.; Kanno, R. High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 2016, 1, 16030. [Google Scholar] [CrossRef]
- Mohtadi, R.; Mizuno, F. Magnesium batteries: Current state of the art, issues and future perspectives. Beilstein J. Nanotechnol. 2014, 5, 1291–1311. [Google Scholar] [CrossRef]
- Ponrouch, A.; Palacin, M.R. On the road toward calcium-based batteries. Curr. Opin. Electrochem. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Mohtadi, R.; Orimo, S.-I. The renaissance of hydrides as energy materials. Nat. Rev. Mater. 2016, 2, 16091. [Google Scholar] [CrossRef]
- Tutusaus, O.; Mohtadi, R. Paving the Way towards Highly Stable and Practical Electrolytes for Rechargeable Magnesium Batteries. ChemElectroChem 2015, 2, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Boukamp, B.A.; Huggins, R.A. Ionic-Conductivity in Lithium Imide. Phys. Lett. A 1979, 72, 464–466. [Google Scholar] [CrossRef]
- Matsuo, M.; Nakamori, Y.; Orimo, S.; Maekawa, H.; Takamura, H. Lithium superionic conduction in lithium borohydride accompanied by structural transition. Appl. Phys. Lett. 2007, 91, 224103. [Google Scholar] [CrossRef]
- Nakamori, Y.; Orimo, S.; Tsutaoka, T. Dehydriding reaction of metal hydrides and alkali borohydrides enhanced by microwave irradiation. Appl. Phys. Lett. 2006, 88, 112104. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, M.; Orimo, S. Lithium Fast-Ionic Conduction in Complex Hydrides: Review and Prospects. Adv. Energy Mater. 2011, 1, 161–172. [Google Scholar] [CrossRef]
- Buchter, F.; Lodziana, Z.; Mauron, P.; Remhof, A.; Friedrichs, O.; Borgschulte, A.; Zuttel, A.; Sheptyakov, D.; Strassle, T.; Ramirez-Cuesta, A.J. Dynamical properties and temperature induced molecular disordering of LiBH4 and LiBD4. Phys. Rev. B 2008, 78, 094302. [Google Scholar] [CrossRef] [Green Version]
- Remhof, A.; Lodziana, Z.; Martelli, P.; Friedrichs, O.; Zuttel, A.; Skripov, A.V.; Embs, J.P.; Strassle, T. Rotational motion of BH4 units in MBH4 (M=Li,Na,K) from quasielastic neutron scattering and density functional calculations. Phys. Rev. B 2010, 81, 214304. [Google Scholar] [CrossRef] [Green Version]
- Asakura, R.; Duchêne, L.; Kühnel, R.-S.; Remhof, A.; Hagemann, H.; Battaglia, C. Electrochemical Oxidative Stability of Hydroborate-Based Solid-State Electrolytes. ACS Appl. Energy Mater. 2019, 2, 6924–6930. [Google Scholar] [CrossRef]
- Maekawa, H.; Matsuo, M.; Takamura, H.; Ando, M.; Noda, Y.; Karahashi, T.; Orimo, S.I. Halide-Stabilized LiBH4, a Room-Temperature Lithium Fast-Ion Conductor. J. Am. Chem. Soc. 2009, 131, 894–895. [Google Scholar] [CrossRef]
- Oguchi, H.; Matsuo, M.; Hummelshoj, J.S.; Vegge, T.; Norskov, J.K.; Sato, T.; Miura, Y.; Takamura, H.; Maekawa, H.; Orimo, S. Experimental and computational studies on structural transitions in the LiBH4-LiI pseudobinary system. Appl. Phys. Lett. 2009, 94, 141912. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, R.; Karahashi, T.; Kumatani, N.; Noda, Y.; Ando, M.; Takamura, H.; Matsuo, M.; Orimo, S.; Maekawa, H. Room temperature lithium fast-ion conduction and phase relationship of LiI stabilized LiBH4. Solid State Ion. 2011, 192, 143–147. [Google Scholar] [CrossRef]
- Matsuo, M.; Remhof, A.; Martelli, P.; Caputo, R.; Ernst, M.; Miura, Y.; Sato, T.; Oguchi, H.; Maekawa, H.; Takamura, H.; et al. Complex Hydrides with (BH4)(-) and (NH2)(-) Anions as New Lithium Fast-Ion Conductors. J. Am. Chem. Soc. 2009, 131, 16389–16391. [Google Scholar] [CrossRef]
- Yan, Y.; Kühnel, R.-S.; Remhof, A.; Duchêne, L.; Reyes, E.C.; Rentsch, D.; Łodziana, Z.; Battaglia, C. A Lithium Amide-Borohydride Solid-State Electrolyte with Lithium-Ion Conductivities Comparable to Liquid Electrolytes. Adv. Energy Mater. 2017, 7, 1700294. [Google Scholar] [CrossRef]
- Ley, M.B.; Ravnsbæk, D.B.; Filinchuk, Y.; Lee, Y.-S.; Janot, R.; Cho, Y.W.; Skibsted, J.; Jensen, T.R. LiCe(BH4)3Cl, a New Lithium-Ion Conductor and Hydrogen Storage Material with Isolated Tetranuclear Anionic Clusters. Chem. Mater. 2012, 24, 1654–1663. [Google Scholar] [CrossRef]
- GharibDoust, S.P.; Brighi, M.; Sadikin, Y.; Ravnsbaek, D.B.; Cerny, R.; Skibsted, J.; Jensen, T.R. Synthesis, Structure, and Li-Ion Conductivity of LiLa(BH4)(3)X, X = Cl, Br, I.J. Phys. Chem. C 2017, 121, 19010–19021. [Google Scholar] [CrossRef]
- Lee, Y.S.; Filinchuk, Y.; Lee, H.S.; Suh, J.Y.; Kim, J.W.; Yu, J.S.; Cho, Y.W. On the Formation and the Structure of the First Bimetallic Borohydride Borate, LiCa3(BH4)(BO3)(2). J. Phys. Chem. C 2011, 115, 10298–10304. [Google Scholar] [CrossRef]
- Didelot, E.; Cerny, R. Ionic conduction in bimetallic borohydride borate, LiCa3(BH4)(BO3)(2). Solid State Ion. 2017, 305, 16–22. [Google Scholar] [CrossRef]
- Ngene, P.; Adelhelm, P.; Beale, A.M.; de Jong, K.P.; de Jongh, P.E. LiBH4/SBA-15 Nanocomposites Prepared by Melt Infiltration under Hydrogen Pressure: Synthesis and Hydrogen Sorption Properties. J. Phys. Chem. C 2010, 114, 6163–6168. [Google Scholar] [CrossRef]
- Blanchard, D.; Nale, A.; Sveinbjornsson, D.; Eggenhuisen, T.M.; Verkuijlen, M.H.; Vegge, T.; Kentgens, A.P.M.; de Jongh, P.E. Nanoconfined LiBH4 as a Fast Lithium Ion Conductor. Adv. Funct. Mater. 2015, 25, 184–192. [Google Scholar] [CrossRef]
- Verdal, N.; Udovic, T.J.; Rush, J.J.; Liu, X.F.; Majzoub, E.H.; Vajo, J.J.; Gross, A.F. Dynamical Perturbations of Tetrahydroborate Anions in LiBH4 due to Nanoconfinement in Controlled-Pore Carbon Scaffolds. J. Phys. Chem. C 2013, 117, 17983–17995. [Google Scholar] [CrossRef]
- Choi, Y.S.; Lee, Y.S.; Choi, D.J.; Chae, K.H.; Oh, K.H.; Cho, Y.W. Enhanced Li Ion Conductivity in LiBH4-Al2O3 Mixture via Interface Engineering. J. Phys. Chem. C 2017, 121, 26209–26215. [Google Scholar] [CrossRef]
- Teprovich, J.A.; Colon-Mercado, H.R.; Ward, P.A.; Peters, B.; Giri, S.; Zhou, J.; Greenway, S.; Compton, R.N.; Jena, P.; Zidan, R. Experimental and Theoretical Analysis of Fast Lithium Ionic Conduction in a LiBH4-C-60 Nanocomposite. J. Phys. Chem. C 2014, 118, 21755–21761. [Google Scholar] [CrossRef]
- Unemoto, A.; Wu, H.; Udovic, T.J.; Matsuo, M.; Ikeshoji, T.; Orimo, S. Fast lithium-ionic conduction in a new complex hydride-sulphide crystalline phase. Chem. Commun. 2016, 52, 564–566. [Google Scholar] [CrossRef]
- Yamauchi, A.; Sakuda, A.; Hayashi, A.; Tatsumisago, M. Preparation and ionic conductivities of (100-x)(0.75Li(2)S center dot 0.25P(2)S(5))center dot xLiBH(4) glass electrolytes. J. Power Sources 2013, 244, 707–710. [Google Scholar] [CrossRef]
- El Kharbachi, A.; Hu, Y.; Yoshida, K.; Vajeeston, P.; Kim, S.; Sorby, M.H.; Orimo, S.; Fjellvag, H.; Hauback, B.C. Lithium ionic conduction in composites of Li(BH4)(0.75)I-0.25 and amorphous 0.75Li(2)S center dot 0.25P(2)S(5) for battery applications. Electrochim. Acta 2018, 278, 332–339. [Google Scholar] [CrossRef]
- Sveinbjörnsson, D.; Blanchard, D.; Myrdal, J.S.G.; Younesi, R.; Viskinde, R.; Riktor, M.D.; Norby, P.; Vegge, T. Ionic conductivity and the formation of cubic CaH2 in the LiBH4–Ca(BH4)2 composite. J. Solid State Chem. 2014, 211, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Xiang, M.; Zhang, Y.; Zhan, L.; Zhu, Y.; Guo, X.; Chen, J.; Wang, Z.; Li, L. Study on xLiBH4-NaBH4 (x = 1.6, 2.3, and 4) composites with enhanced lithium ionic conductivity. J. Alloy. Compd. 2017, 729, 936–941. [Google Scholar] [CrossRef]
- López-Aranguren, P.; Berti, N.; Dao, A.H.; Zhang, J.; Cuevas, F.; Latroche, M.; Jordy, C. An all-solid-state metal hydride–Sulfur lithium-ion battery. J. Power Sources 2017, 357, 56–60. [Google Scholar] [CrossRef]
- Xiang, M.; Zhang, Y.; Zhu, Y.; Guo, X.; Chen, J.; Li, L. Ternary LiBH4-NaBH4-MgH2 composite as fast ionic conductor. Solid State Ion. 2018, 324, 109–113. [Google Scholar] [CrossRef]
- Xiang, M.; Zhang, Y.; Lin, H.; Zhu, Y.; Guo, X.; Chen, J.; Li, L. LiBH4-NaX (X = Cl, I) composites with enhanced lithium ionic conductivity. J. Alloy. Compd. 2018, 764, 307–313. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, Y.; Song, T.; Miyaoka, H.; Shinzato, K.; Miyaoka, H.; Ichikawa, T.; Shi, S.; Zhang, X.; Isobe, S.; et al. Ammonia, a Switch for Controlling High Ionic Conductivity in Lithium Borohydride Ammoniates. Joule 2018, 2, 1522–1533. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, M.; Kuromoto, S.; Sato, T.; Oguchi, H.; Takamura, H.; Orimo, S. Sodium ionic conduction in complex hydrides with [BH4](-) and [NH2](-) anions. Appl. Phys. Lett. 2012, 100, 203904. [Google Scholar] [CrossRef] [Green Version]
- Unemoto, A.; Matsuo, M.; Orimo, S. Complex Hydrides for Electrochemical Energy Storage. Adv. Funct. Mater. 2014, 24, 2267–2279. [Google Scholar] [CrossRef]
- Matsuo, M.; Oguchi, H.; Sato, T.; Takamura, H.; Tsuchida, E.; Ikeshoji, T.; Orimo, S. Sodium and magnesium ionic conduction in complex hydrides. J. Alloy. Compd. 2013, 580, S98–S101. [Google Scholar] [CrossRef]
- Oguchi, H.; Matsuo, M.; Sato, T.; Takamura, H.; Maekawa, H.; Kuwano, H.; Orimo, S. Lithium-ion conduction in complex hydrides LiAlH(4) and Li(3)AlH(6). J. Appl. Phys. 2010, 107, 096104. [Google Scholar] [CrossRef]
- Li, W.; Wu, G.T.; Xiong, Z.T.; Feng, Y.P.; Chen, P. Li+ ionic conductivities and diffusion mechanisms in Li-based imides and lithium amide. Phys. Chem. Chem. Phys. 2012, 14, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Rijssenbeek, J.; Gao, Y.; Hanson, J.; Huang, Q.; Jones, C.; Toby, B. Crystal structure determination and reaction pathway of amide-hydride mixtures. J. Alloy. Compd. 2008, 454, 233–244. [Google Scholar] [CrossRef]
- Wu, G.T.; Xiong, Z.T.; Liu, T.; Liu, Y.F.; Hu, J.J.; Chen, P.; Feng, Y.P.; Wee, A.T.S. Synthesis and characterization of a new ternary imide-Li2Ca(NH)(2). Inorg. Chem. 2007, 46, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Martelli, P.; Remhof, A.; Borgschulte, A.; Ackermann, R.; Strassle, T.; Embs, J.P.; Ernst, M.; Matsuo, M.; Orimo, S.I.; Zuttel, A. Rotational Motion in LiBH4/LiI Solid Solutions. J. Phys. Chem. A 2011, 115, 5329–5334. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Ley, M.B.; Jensen, T.R.; Filinchuk, Y. Nuclear Magnetic Resonance Studies of BH4 Reorientations and Li Diffusion in LiLa(BH4)(3)Cl. J. Phys. Chem. C 2013, 117, 14965–14972. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Ley, M.B.; Jensen, T.R.; Cho, Y.W. Lithium Ion Disorder and Conduction Mechanism in LiCe(BH4)3Cl. J. Phys. Chem. C 2016, 120, 19035–19042. [Google Scholar] [CrossRef]
- Jansen, M. Volume Effect or Paddle-Wheel Mechanism—Fast Alkali-Metal Ionic-Conduction in Solids with Rotationally Disordered Complex Anions. Angew. Chem. Int. Ed. Engl. 1991, 30, 1547–1558. [Google Scholar] [CrossRef]
- Verdal, N.; Udovic, T.J.; Rush, J.J.; Wu, H.; Skripov, A.V. Evolution of the Reorientational Motions of the Tetrahydroborate Anions in Hexagonal LiBH4-Lil Solid Solution by High-Q Quasielastic Neutron Scattering. J. Phys. Chem. C 2013, 117, 12010–12018. [Google Scholar] [CrossRef]
- Orimo, S.-I.; Nakamori, Y.; Eliseo, J.R.; Züttel, A.; Jensen, C.M. Complex Hydrides for Hydrogen Storage. Chem. Rev. 2007, 107, 4111–4132. [Google Scholar] [CrossRef] [PubMed]
- Paskevicius, M.; Pitt, M.P.; Brown, D.H.; Sheppard, D.A.; Chumphongphan, S.; Buckley, C.E. First-order phase transition in the Li2B12H12 system. Phys. Chem. Chem. Phys. 2013, 15, 15825–15828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skripov, A.V.; Babanova, O.A.; Soloninin, A.V.; Stavila, V.; Verdal, N.; Udovic, T.J.; Rush, J.J. Nuclear Magnetic Resonance Study of Atomic Motion in A2B12H12 (A = Na, K, Rb, Cs): Anion Reorientations and Na+ Mobility. J. Phys. Chem. C 2013, 117, 25961–25968. [Google Scholar] [CrossRef]
- Udovic, T.J.; Matsuo, M.; Unemoto, A.; Verdal, N.; Stavila, V.; Skripov, A.V.; Rush, J.J.; Takamura, H.; Orimo, S. Sodium superionic conduction in Na2B12H12. Chem. Commun. 2014, 50, 3750–3752. [Google Scholar] [CrossRef]
- Udovic, T.J.; Matsuo, M.; Tang, W.S.; Wu, H.; Stavila, V.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; Rush, J.J.; et al. Exceptional Superionic Conductivity in Disordered Sodium Decahydro-closo-decaborate. Adv. Mater. 2014, 26, 7622–7626. [Google Scholar] [CrossRef]
- Varley, J.B.; Kweon, K.; Mehta, P.; Shea, P.; Heo, T.W.; Udovic, T.J.; Stavila, V.; Wood, B.C. Understanding Ionic Conductivity Trends in Polyborane Solid Electrolytes from Ab Initio Molecular Dynamics. ACS Energy Lett. 2017, 2, 250–255. [Google Scholar] [CrossRef]
- Kweon, K.E.; Varley, J.B.; Shea, P.; Adelstein, N.; Mehta, P.; Heo, T.W.; Udovic, T.J.; Stavila, V.; Wood, B.C. Structural, Chemical, and Dynamical Frustration: Origins of Superionic Conductivity in closo-Borate Solid Electrolytes. Chem. Mater. 2017, 29, 9142–9153. [Google Scholar] [CrossRef]
- Sadikin, Y.; Brighi, M.; Schouwink, P.; Cerny, R. Superionic Conduction of Sodium and Lithium in Anion-Mixed Hydroborates Na3BH4B12H12 and (Li0.7Na0.3)(3)BH4B12H12. Adv. Energy Mater. 2015, 5, 1501016. [Google Scholar] [CrossRef]
- He, L.; Li, H.-W.; Nakajima, H.; Tumanov, N.; Filinchuk, Y.; Hwang, S.-J.; Sharma, M.; Hagemann, H.; Akiba, E. Synthesis of a Bimetallic Dodecaborate LiNaB12H12 with Outstanding Superionic Conductivity. Chem. Mater. 2015, 27, 5483–5486. [Google Scholar] [CrossRef] [Green Version]
- Duchene, L.; Kuhnel, R.S.; Rentsch, D.; Remhof, A.; Hagemann, H.; Battaglia, C. A highly stable sodium solid-state electrolyte based on a dodeca/deca-borate equimolar mixture. Chem. Commun. 2017, 53, 4195–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Sato, T.; Unemoto, A.; Matsuo, M.; Ikeshoji, T.; Udovic, T.J.; Orimo, S. Fast sodium ionic conduction in Na2B10H10-Na2B12H12 pseudo-binary complex hydride and application to a bulk-type all-solid-state battery. Appl. Phys. Lett. 2017, 110, 103901. [Google Scholar] [CrossRef] [Green Version]
- Teprovich, J.A.; Colon-Mercado, H.; Washington Ii, A.L.; Ward, P.A.; Greenway, S.; Missimer, D.M.; Hartman, H.; Velten, J.; Christian, J.H.; Zidan, R. Bi-functional Li2B12H12 for energy storage and conversion applications: Solid-state electrolyte and luminescent down-conversion dye. J. Mater. Chem. A 2015, 3, 22853–22859. [Google Scholar] [CrossRef]
- Tang, W.S.; Yoshida, K.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; Dimitrievska, M.; Stavila, V.; Orimo, S.-I.; Udovic, T.J. Stabilizing Superionic-Conducting Structures via Mixed-Anion Solid Solutions of Monocarba-closo-borate Salts. ACS Energy Lett. 2016, 1, 659–664. [Google Scholar] [CrossRef]
- Tang, W.S.; Matsuo, M.; Wu, H.; Stavila, V.; Unemoto, A.; Orimo, S.-I.; Udovic, T.J. Stabilizing lithium and sodium fast-ion conduction in solid polyhedral-borate salts at device-relevant temperatures. Energy Storage Mater. 2016, 4, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Toyama, N.; Oguchi, H.; Sato, T.; Takagi, S.; Ikeshoji, T.; Orimo, S.-I. Fast Lithium-Ion Conduction in Atom-Deficient closo-Type Complex Hydride Solid Electrolytes. Chem. Mater. 2018, 30, 386–391. [Google Scholar] [CrossRef]
- Tang, W.S.; Unemoto, A.; Zhou, W.; Stavila, V.; Matsuo, M.; Wu, H.; Orimo, S.-I.; Udovic, T.J. Unparalleled lithium and sodium superionic conduction in solid electrolytes with large monovalent cage-like anions. Energy Environ. Sci. 2015, 8, 3637–3645. [Google Scholar] [CrossRef] [Green Version]
- Dimitrievska, M.; Shea, P.; Kweon, K.E.; Bercx, M.; Varley, J.B.; Tang, W.S.; Skripov, A.V.; Stavila, V.; Udovic, T.J.; Wood, B.C. Carbon Incorporation and Anion Dynamics as Synergistic Drivers for Ultrafast Diffusion in Superionic LiCB11H12 and NaCB11H12. Adv. Energy Mater. 2018, 8, 1703422. [Google Scholar] [CrossRef]
- Tang, W.S.; Matsuo, M.; Wu, H.; Stavila, V.; Zhou, W.; Talin, A.A.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; et al. Liquid-like Ionic Conduction in Solid Lithium and Sodium Monocarba-closo-decaborates near or at Room Temperature. Adv. Energy Mater. 2016, 6, 1502237. [Google Scholar] [CrossRef]
- Kim, S.; Oguchi, H.; Toyama, N.; Sato, T.; Takagi, S.; Otomo, T.; Arunkumar, D.; Kuwata, N.; Kawamura, J.; Orimo, S.-I. A complex hydride lithium superionic conductor for high-energy-density all-solid-state lithium metal batteries. Nat. Commun. 2019, 10, 1081. [Google Scholar] [CrossRef] [Green Version]
- Brighi, M.; Murgia, F.; Łodziana, Z.; Schouwink, P.; Wołczyk, A.; Cerny, R. A mixed anion hydroborate/carba-hydroborate as a room temperature Na-ion solid electrolyte. J. Power Sources 2018, 404, 7–12. [Google Scholar] [CrossRef]
- Tang, W.S.; Dimitrievska, M.; Stavila, V.; Zhou, W.; Wu, H.; Talin, A.A.; Udovic, T.J. Order–Disorder Transitions and Superionic Conductivity in the Sodium nido-Undeca(carba)borates. Chem. Mater. 2017, 29, 10496–10509. [Google Scholar] [CrossRef]
- Yan, Y.; Rentsch, D.; Battaglia, C.; Remhof, A. Synthesis, stability and Li-ion mobility of nanoconfined Li2B12H12. Dalton Trans. 2017, 46, 12434–12437. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.R.S.; Paskevicius, M.; Jørgensen, M.; Jensen, T.R. Halogenated Sodium-closo-Dodecaboranes as Solid-State Ion Conductors. Chem. Mater. 2017, 29, 3423–3430. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Sethio, D.; Lawson Daku, L.M.; Hagemann, H. Theoretical Study of Halogenated B12HnX(12–n)2– (X = F, Cl, Br). J. Phys. Chem. A 2019, 123, 1807–1813. [Google Scholar] [CrossRef]
- Yoo, H.D.; Shterenberg, I.; Gofer, Y.; Gershinsky, G.; Pour, N.; Aurbach, D. Mg rechargeable batteries: An on-going challenge. Energy Environ. Sci. 2013, 6, 2265–2279. [Google Scholar] [CrossRef]
- Aurbach, D.; Cohen, Y.; Moshkovich, M. The Study of Reversible Magnesium Deposition by In Situ Scanning Tunneling Microscopy. Electrochem. Solid-State Lett. 2001, 4, A113–A116. [Google Scholar] [CrossRef]
- Dong, H.; Tutusaus, Y.L.O.; Mohtadi, R.; Zhang, Y.; Hao, F.; Yao, Y. Directing Mg-storage chemistry in organic polymers towards high-energy Mg batteries. Joule 2018, 3, 782–793. [Google Scholar] [CrossRef] [Green Version]
- Mohtadi, R.; Matsui, M.; Arthur, T.S.; Hwang, S.J. Magnesium borohydride: From hydrogen storage to magnesium battery. Angew. Chem. Int. Ed. 2012, 51, 9780–9783. [Google Scholar] [CrossRef]
- Shao, Y.; Liu, T.; Li, G.; Gu, M.; Nie, Z.; Engelhard, M.; Xiao, J.; Lv, D.; Wang, C.; Zhang, J.-G.; et al. Coordination Chemistry in magnesium battery electrolytes: How ligands affect their performance. Sci. Reports 2013, 3, 3130. [Google Scholar] [CrossRef]
- Tuerxun, F.; Abulizi, Y.; NuLi, Y.N.; Su, S.J.; Yang, J.; Wang, J.L. High concentration magnesium borohydride/tetraglyme electrolyte for rechargeable magnesium batteries. J. Power Sources 2015, 276, 255–261. [Google Scholar] [CrossRef]
- Samuel, D.; Steinhauser, C.; Smith, J.G.; Kaufman, A.; Radin, M.D.; Naruse, J.; Hiramatsu, H.; Siegel, D.J. Ion Pairing and Diffusion in Magnesium Electrolytes Based on Magnesium Borohydride. ACS Appl. Mater. Interfaces 2017, 9, 43755–43766. [Google Scholar] [CrossRef] [PubMed]
- Deetz, J.D.; Cao, F.; Wang, Q.; Sun, H. Exploring the Liquid Structure and Ion Formation in Magnesium Borohydride Electrolyte Using Density Functional Theory. J. Electrochem. Soc. 2018, 165, A61–A70. [Google Scholar] [CrossRef]
- Orikasa, Y.; Masese, T.; Koyama, Y.; Mori, T.; Hattori, M.; Yamamoto, K.; Okado, T.; Huang, Z.-D.; Minato, T.; Tassel, C.; et al. High energy density rechargeable magnesium battery using earth-abundant and non-toxic elements. Sci. Rep. 2014, 4, 5622. [Google Scholar] [CrossRef] [Green Version]
- Sa, N.; Rajput, N.N.; Wang, H.; Key, B.; Ferrandon, M.; Srinivasan, V.; Persson, K.A.; Burrell, A.K.; Vaughey, J.T. Concentration dependent electrochemical properties and structural analysis of a simple magnesium electrolyte: Magnesium bis(trifluoromethane sulfonyl)imide in diglyme. RSC Adv. 2016, 6, 113663–113670. [Google Scholar] [CrossRef]
- Rajput, N.N.; Qu, X.; Sa, N.; Burrell, A.K.; Persson, K.A. The Coupling between Stability and Ion Pair Formation in Magnesium Electrolytes from First-Principles Quantum Mechanics and Classical Molecular Dynamics. J. Am. Chem. Soc. 2015, 137, 3411–3420. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.Z.; Rajput, N.N.; Wan, C.; Shao, Y.; Deng, X.; Jaegers, N.R.; Hu, M.; Chen, Y.; Shin, Y.; Monk, J.; et al. 25Mg NMR and computational modeling studies of the solvation structures and molecular dynamics in magnesium based liquid electrolytes. Nano Energy 2018, 46, 436–446. [Google Scholar] [CrossRef]
- Kar, M.; Ma, Z.; Azofra, L.M.; Chen, K.; Forsyth, M.; MacFarlane, D.R. Ionic liquid electrolytes for reversible magnesium electrochemistry. Chem. Commun. 2016, 52, 4033–4036. [Google Scholar] [CrossRef] [Green Version]
- Watkins, T.; Kumar, A.; Buttry, D.A. Designer Ionic Liquids for Reversible Electrochemical Deposition/Dissolution of Magnesium. J. Am. Chem. Soc. 2016, 138, 641–650. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, Z.; Li, J.; Qiao, L.; Lu, C.; Tang, K.; Dong, S.; Ma, J.; Liu, Y.; Zhou, X.; et al. Multifunctional Additives Improve the Electrolyte Properties of Magnesium Borohydride Toward Magnesium–Sulfur Batteries. ACS Appl. Mater. Interfaces 2018, 10, 23757–23765. [Google Scholar] [CrossRef]
- Hebié, S.; Ngo, H.P.K.; Leprêtre, J.-C.; Iojoiu, C.; Cointeaux, L.; Berthelot, R.; Alloin, F. Electrolyte Based on Easily Synthesized, Low Cost Triphenolate–Borohydride Salt for High Performance Mg(TFSI)2-Glyme Rechargeable Magnesium Batteries. ACS Appl. Mater. Interfaces 2017, 9, 28377–28385. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, X.; Chen, Y.; Jin, L.; Kuss, C.; Bruce, P.G. Plating and stripping calcium in an organic electrolyte. Nat. Mater. 2017, 17, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, T.J.; Mohtadi, R.; Arthur, T.S.; Mizuno, F.; Zhang, R.; Shirai, S.; Kampf, J.W. Boron Clusters as Highly Stable Magnesium-Battery Electrolytes. Angew. Chem. Int. Ed. 2014, 53, 3173–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutusaus, O.; Mohtadi, R.; Arthur, T.S.; Mizuno, F.; Nelson, E.G.; Sevryugina, Y.V. An Efficient Halogen-Free Electrolyte for Use in Rechargeable Magnesium Batteries. Angew. Chem. Int. Ed. 2015, 54, 7900–7904. [Google Scholar] [CrossRef] [PubMed]
- Hahn, N.T.; Seguin, T.J.; Lau, K.-C.; Liao, C.; Ingram, B.J.; Persson, K.A.; Zavadil, K.R. Enhanced Stability of the Carba-closo-dodecaborate Anion for High-Voltage Battery Electrolytes through Rational Design. J. Am. Chem. Soc. 2018, 140, 11076–11084. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Sahadeo, E.; Noked, M.; Lee, S.B. Mapping the Challenges of Magnesium Battery. J. Phys. Chem. Lett. 2016, 7, 1736–1749. [Google Scholar] [CrossRef]
- Xu, K. Electrolytes and Interphases in Li-Ion Batteries and Beyond. Chem. Rev. 2014, 114, 11503–11618. [Google Scholar] [CrossRef]
- Xu, K. Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries. Chem. Rev. 2004, 104, 4303–4418. [Google Scholar] [CrossRef]
- Arthur, T.S.; Glans, P.-A.; Singh, N.; Tutusaus, O.; Nie, K.; Liu, Y.-S.; Mizuno, F.; Guo, J.; Alsem, D.H.; Salmon, N.J.; et al. Interfacial Insight from Operando XAS/TEM for Magnesium Metal Deposition with Borohydride Electrolytes. Chem. Mater. 2017, 29, 7183–7188. [Google Scholar] [CrossRef]
- Chang, J.; Haasch, R.T.; Kim, J.; Spila, T.; Braun, P.V.; Gewirth, A.A.; Nuzzo, R.G. Synergetic Role of Li+ during Mg Electrodeposition/Dissolution in Borohydride Diglyme Electrolyte Solution: Voltammetric Stripping Behaviors on a Pt Microelectrode Indicative of Mg–Li Alloying and Facilitated Dissolution. ACS Appl. Mater. Interfaces 2015, 7, 2494–2502. [Google Scholar] [CrossRef]
- Tutusaus, O.; Mohtadi, R.; Singh, N.; Arthur, T.S.; Mizuno, F. Study of Electrochemical Phenomena Observed at the Mg Metal/Electrolyte Interface. ACS Energy Lett. 2017, 2, 224–229. [Google Scholar] [CrossRef]
- Singh, N.; Arthur, T.S.; Tutusaus, O.; Li, J.; Kisslinger, K.; Xin, H.L.; Stach, E.A.; Fan, X.; Mohtadi, R. Achieving High Cycling Rates via In Situ Generation of Active Nanocomposite Metal Anodes. ACS Appl. Energy Mater. 2018, 1, 4651–4661. [Google Scholar] [CrossRef]
- Higashi, S.; Miwa, K.; Aoki, M.; Takechi, K. A novel inorganic solid state ion conductor for rechargeable Mg batteries. Chem. Commun. 2014, 50, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Roedern, E.; Kuhnel, R.S.; Remhof, A.; Battaglia, C. Magnesium Ethylenediamine Borohydride as Solid-State Electrolyte for Magnesium Batteries. Sci. Rep. 2017, 7, 46189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.Y.; Rajput, N.N.; Hu, J.Z.; Hu, M.; Liu, T.B.; Wei, Z.H.; Gu, M.; Deng, X.C.; Xu, S.C.; Han, K.S.; et al. Nanocomposite polymer electrolyte for rechargeable magnesium batteries. Nano Energy 2015, 12, 750–759. [Google Scholar] [CrossRef] [Green Version]
- MacFarlane, D.R.; Forsyth, M.; Howlett, P.C.; Kar, M.; Passerini, S.; Pringle, J.M.; Ohno, H.; Watanabe, M.; Yan, F.; Zheng, W.; et al. Ionic liquids and their solid-state analogues as materials for energy generation and storage. Nat. Rev. Mater. 2016, 1, 15005. [Google Scholar] [CrossRef]
- Ma, Z.; Forsyth, M.; MacFarlane, D.R.; Kar, M. Ionic liquid/tetraglyme hybrid Mg[TFSI]2 electrolytes for rechargeable Mg batteries. Green Energy Environ. 2018, 4, 146–153. [Google Scholar] [CrossRef]
- Kar, M.; Tutusaus, O.; MacFarlane, D.R.; Mohtadi, R. Novel and versatile room temperature ionic liquids for energy storage. Energy Environ. Sci. 2018, 12, 566–571. [Google Scholar] [CrossRef]
- Takahashi, K.; Hattori, K.; Yamazaki, T.; Takada, K.; Matsuo, M.; Orimo, S.; Maekawa, H.; Takamura, H. All-solid-state lithium battery with LiBH4 solid electrolyte. J. Power Sources 2013, 226, 61–64. [Google Scholar] [CrossRef]
- Unemoto, A.; Ikeshoji, T.; Yasaku, S.; Matsuo, M.; Stavila, V.; Udovic, T.J.; Orimo, S.-I. Stable Interface Formation between TiS2 and LiBH4 in Bulk-Type All-Solid-State Lithium Batteries. Chem. Mater. 2015, 27, 5407–5416. [Google Scholar] [CrossRef]
- Unemoto, A.; Nogami, G.; Tazawa, M.; Taniguchi, M.; Orimo, S. Development of 4V-Class Bulk-Type All-Solid-State Lithium Rechargeable Batteries by a Combined Use of Complex Hydride and Sulfide Electrolytes for Room Temperature Operation. Mater. Trans. 2017, 58, 1063–1068. [Google Scholar] [CrossRef] [Green Version]
- Unemoto, A.; Yasaku, S.; Nogami, G.; Tazawa, M.; Taniguchi, M.; Matsuo, M.; Ikeshoji, T.; Orimo, S.-I. Development of bulk-type all-solid-state lithium-sulfur battery using LiBH4 electrolyte. Appl. Phys. Lett. 2014, 105, 083901. [Google Scholar] [CrossRef]
- Das, S.; Ngene, P.; Norby, P.; Vegge, T.; de Jongh, P.E.; Blanchard, D. All-Solid-State Lithium-Sulfur Battery Based on a Nanoconfined LiBH4 Electrolyte. J. Electrochem. Soc. 2016, 163, A2029–A2034. [Google Scholar] [CrossRef] [Green Version]
- Atsushi, U.; ChunLin, C.; Zhongchang, W.; Motoaki, M.; Tamio, I.; Shin-ichi, O. Pseudo-binary electrolyte, LiBH 4 –LiCl, for bulk-type all-solid-state lithium-sulfur battery. Nanotechnology 2015, 26, 254001. [Google Scholar]
- Duchene, L.; Kuhnel, R.S.; Stilp, E.; Cuervo Reyes, E.; Remhof, A.; Hagemann, H.; Battaglia, C. A sTable 3 V all-solid-state sodium-ion battery based on a closo-borate electrolyte. Energy Environ. Sci. 2017, 10, 2609–2615. [Google Scholar] [CrossRef]
- Duchêne, L.; Kim, D.H.; Song, Y.B.; Jun, S.; Moury, R.; Remhof, A.; Hagemann, H.; Jung, Y.S.; Battaglia, C. Crystallization of closo-borate electrolytes from solution enabling infiltration into slurry-casted porous electrodes for all-solid-state batteries. Energy Storage Mater. 2019, 26, 543–549. [Google Scholar] [CrossRef]
- Shao, Y.; Gu, M.; Li, X.; Nie, Z.; Zuo, P.; Li, G.; Liu, T.; Xiao, J.; Cheng, Y.; Wang, C.; et al. Highly Reversible Mg Insertion in Nanostructured Bi for Mg Ion Batteries. Nano Lett. 2014, 14, 255–260. [Google Scholar] [CrossRef]
- Singh, R.; Kumari, P.; Rathore, R.K.; Shinzato, K.; Ichikawa, T.; Verma, A.S.; Saraswat, V.K.; Awasthi, K.; Jain, A.; Kumar, M. LiBH4 as solid electrolyte for Li-ion batteries with Bi2Te3 nanostructured anode. Int. J. Hydrog. Energy 2018, 43, 21709–21714. [Google Scholar] [CrossRef]
Figure 1.
(a) Key properties required for competent battery electrolytes. Reprinted with permission from reference [9], copyright 2014 WILEY-VCH. (b) Timeline depicting key advancements made in the development of hydrides as battery electrolytes.
Figure 1.
(a) Key properties required for competent battery electrolytes. Reprinted with permission from reference [9], copyright 2014 WILEY-VCH. (b) Timeline depicting key advancements made in the development of hydrides as battery electrolytes.

Figure 2.
Properties of solid-state electrolytes for Li batteries: (a) Phase transition in LiBH4, (b) Arrhenius plot depicting the ionic conductivity of LiBH4, (c) Impedance spectra of the low and high temperature LiBH4 phases. Panels b and c are reproduced with permission from reference [11], American Institute of Physics.
Figure 2.
Properties of solid-state electrolytes for Li batteries: (a) Phase transition in LiBH4, (b) Arrhenius plot depicting the ionic conductivity of LiBH4, (c) Impedance spectra of the low and high temperature LiBH4 phases. Panels b and c are reproduced with permission from reference [11], American Institute of Physics.

Figure 3.
Arrhenius plots depicting the cationic conductivity in a variety of hydrides. Reprinted with permission from reference. [8], copyright 2016 Macmillan Publishers Limited, part of Springer Nature.
Figure 3.
Arrhenius plots depicting the cationic conductivity in a variety of hydrides. Reprinted with permission from reference. [8], copyright 2016 Macmillan Publishers Limited, part of Springer Nature.

Figure 4.
Lithium ion conductivity of 7:3 molar (CB9H10:CB11H12)2− Li salt. (a) Nyquist plots at room temperature and magnified Nyquist plots, (b) Arrhenius plots of the lithium ion conductivity, (c) Arrhenius plots of the diffusion coefficients calculated from the impedance and nuclear magnetic resonance (NMR) measurements, (d) Arrhenius plots comparing the conductivity for variety of Li salts. Reprinted with permission from reference [70], copyright 2019 Macmillan Publishers Limited, part of Springer Nature.
Figure 4.
Lithium ion conductivity of 7:3 molar (CB9H10:CB11H12)2− Li salt. (a) Nyquist plots at room temperature and magnified Nyquist plots, (b) Arrhenius plots of the lithium ion conductivity, (c) Arrhenius plots of the diffusion coefficients calculated from the impedance and nuclear magnetic resonance (NMR) measurements, (d) Arrhenius plots comparing the conductivity for variety of Li salts. Reprinted with permission from reference [70], copyright 2019 Macmillan Publishers Limited, part of Springer Nature.

Figure 5.
Performance of Mg borohydride electrolyte: (a) Cyclic voltammogram (inset shows deposition/stripping charge balance) in 1:3 molar Mg(BH4)2:LiBH4 in 1,2 dimethoxyethane (DME), (b) XRD results following galvanostatic deposition of Mg metal, (c) Battery cycling with this electrolyte. Reprinted from reference [79] with permission. Copyright © 2012, Wiley-VCH Verlag GmbH and Co. KGaA, Weinheim.
Figure 5.
Performance of Mg borohydride electrolyte: (a) Cyclic voltammogram (inset shows deposition/stripping charge balance) in 1:3 molar Mg(BH4)2:LiBH4 in 1,2 dimethoxyethane (DME), (b) XRD results following galvanostatic deposition of Mg metal, (c) Battery cycling with this electrolyte. Reprinted from reference [79] with permission. Copyright © 2012, Wiley-VCH Verlag GmbH and Co. KGaA, Weinheim.

Figure 6.
Ca(BH4)2/THF electrolyte for Ca battery: (a) Electrochemical results of calcium plating/stripping in 1.5 M Ca(BH4)2/THF, (b) Powder X-ray diffraction demonstrating Ca plating from the electrolyte. Reprinted with permission from reference [92], copyright 2017 Macmillan Publishers Limited, part of Springer Nature.
Figure 6.
Ca(BH4)2/THF electrolyte for Ca battery: (a) Electrochemical results of calcium plating/stripping in 1.5 M Ca(BH4)2/THF, (b) Powder X-ray diffraction demonstrating Ca plating from the electrolyte. Reprinted with permission from reference [92], copyright 2017 Macmillan Publishers Limited, part of Springer Nature.

Figure 7.
For 1-(1,7-C2B10H11)MgCl electrolyte: (a) Molecular structure and (b) Oxidative stability. Reprinted from reference [93] with permission from Wiley-VCH Verlag GmbH and Co. KGaA, Weinheim, Copyright 2014. For Mg(CB11H12)2/tetraglyme electrolyte: (c) Cyclic voltammograms on Pt, (d) Initial discharge–charge profiles of a rechargeable Mg battery with Mg(CB11H12)2/tetraglyme (black line) and chlorophenyl aluminate electrolyte APC (red line) as the electrolyte, a Mg anode, and α-MnO2 cathode, (e) Mg cation coordination environment in monoglyme (A) and diglyme (B). Reprinted from reference [94] with permission from Wiley-VCH Verlag GmbH and Co. KGaA, Weinheim, Copyright 2015. (f) Position of hydride liquid electrolytes (boron clusters) amongst other Mg battery electrolytes. Reprinted from reference [96] with permission from the American Chemical Society, Copyright 2016.
Figure 7.
For 1-(1,7-C2B10H11)MgCl electrolyte: (a) Molecular structure and (b) Oxidative stability. Reprinted from reference [93] with permission from Wiley-VCH Verlag GmbH and Co. KGaA, Weinheim, Copyright 2014. For Mg(CB11H12)2/tetraglyme electrolyte: (c) Cyclic voltammograms on Pt, (d) Initial discharge–charge profiles of a rechargeable Mg battery with Mg(CB11H12)2/tetraglyme (black line) and chlorophenyl aluminate electrolyte APC (red line) as the electrolyte, a Mg anode, and α-MnO2 cathode, (e) Mg cation coordination environment in monoglyme (A) and diglyme (B). Reprinted from reference [94] with permission from Wiley-VCH Verlag GmbH and Co. KGaA, Weinheim, Copyright 2015. (f) Position of hydride liquid electrolytes (boron clusters) amongst other Mg battery electrolytes. Reprinted from reference [96] with permission from the American Chemical Society, Copyright 2016.

Figure 8.
Formation of interface in Mg interface in Mg(BH4)2:LiBH4/DME electrolyte. Reprinted from reference [99], permission of the American Chemical Society, copyright 2017.
Figure 8.
Formation of interface in Mg interface in Mg(BH4)2:LiBH4/DME electrolyte. Reprinted from reference [99], permission of the American Chemical Society, copyright 2017.

Figure 9.
Temperature dependence of ionic conductivity for selected magnesium solid-state electrolytes. The structure of Mg(BH4)(NH2) is shown. Reproduced from reference [104] permission from Springer Nature, Copyright 2017.
Figure 9.
Temperature dependence of ionic conductivity for selected magnesium solid-state electrolytes. The structure of Mg(BH4)(NH2) is shown. Reproduced from reference [104] permission from Springer Nature, Copyright 2017.

Figure 10.
For the closo-carborate ionic liquids (structure shown): (a) Oxidative and reductive stability, (b) and (c) are Li and Mg electrochemical deposition/stripping, respectively. Reproduced from reference [108] with permission from the Royal Society of Chemistry, Copyright 2019.
Figure 10.
For the closo-carborate ionic liquids (structure shown): (a) Oxidative and reductive stability, (b) and (c) are Li and Mg electrochemical deposition/stripping, respectively. Reproduced from reference [108] with permission from the Royal Society of Chemistry, Copyright 2019.

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mohtadi, R. Beyond Typical Electrolytes for Energy Dense Batteries. Molecules 2020, 25, 1791. https://doi.org/10.3390/molecules25081791
AMA Style
Mohtadi R. Beyond Typical Electrolytes for Energy Dense Batteries. Molecules. 2020; 25(8):1791. https://doi.org/10.3390/molecules25081791
Chicago/Turabian StyleMohtadi, Rana. 2020. "Beyond Typical Electrolytes for Energy Dense Batteries" Molecules 25, no. 8: 1791. https://doi.org/10.3390/molecules25081791