Synthesis and Intramolecular [4+2] Cycloaddition Reactions of 4-Pyridazinecarbonitriles with Alkyne Side Chains

Institute of Pharmaceutical Chemistry, University of Vienna, A-1090 Vienna, Austria
*
Author to whom correspondence should be addressed.
Molecules 1998, 3(1), 10-15; https://doi.org/10.3390/30100010
Submission received: 15 October 1997
/
Accepted: 19 November 1997
/
Published: 25 January 1998
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The preparation of a series of new 3-(alkynyl-X)-substituted 4-pyridazinecarbonitriles 2-5 (X = O, NH) is described. The compounds are shown to undergo thermally induced intramolecular Diels-Alder reactions with inverse electron demand, affording the fused benzonitriles 6-8. Incorporation of a 1,2-phenylene unit into the side chain, as in the case of compounds 10 and 13, results in a more favorable conformation of the dienophilic substructure and thus to a pronounced acceleration of the [4+2] cycloaddition reaction.
Introduction
The intramolecular variant of the inverse-electrondemand Diels-Alder reaction of π-deficient N-heteroaromatics such as tetrazines, triazines, diazines and even pyridines has been shown to be of particular value for the construction of a wide variety of fused systems, because - as a consequence of the “entropic assistance” which is associated with the linkage of diene and dienophile - it proceeds with remarkable ease, in many cases even with “unactivated” dienophiles like alkenes or alkynes [1]. In the pyridazine series, for instance, this concept has been applied to the preparation of various indolines [2], indoles [3], and xanthenes [4,5,6], using 1,2-diazines with appropriate olefinic or acetylenic side chains tethered to position 3 of the heterocycle as the starting material. Here, we wish to report on further studies of this reaction type, using 4-pyridazinecarbonitriles as electronically activated diazadienes with either flexible or rigid acetylenic substructures as the side-chain dienophiles.
Results and Discussion
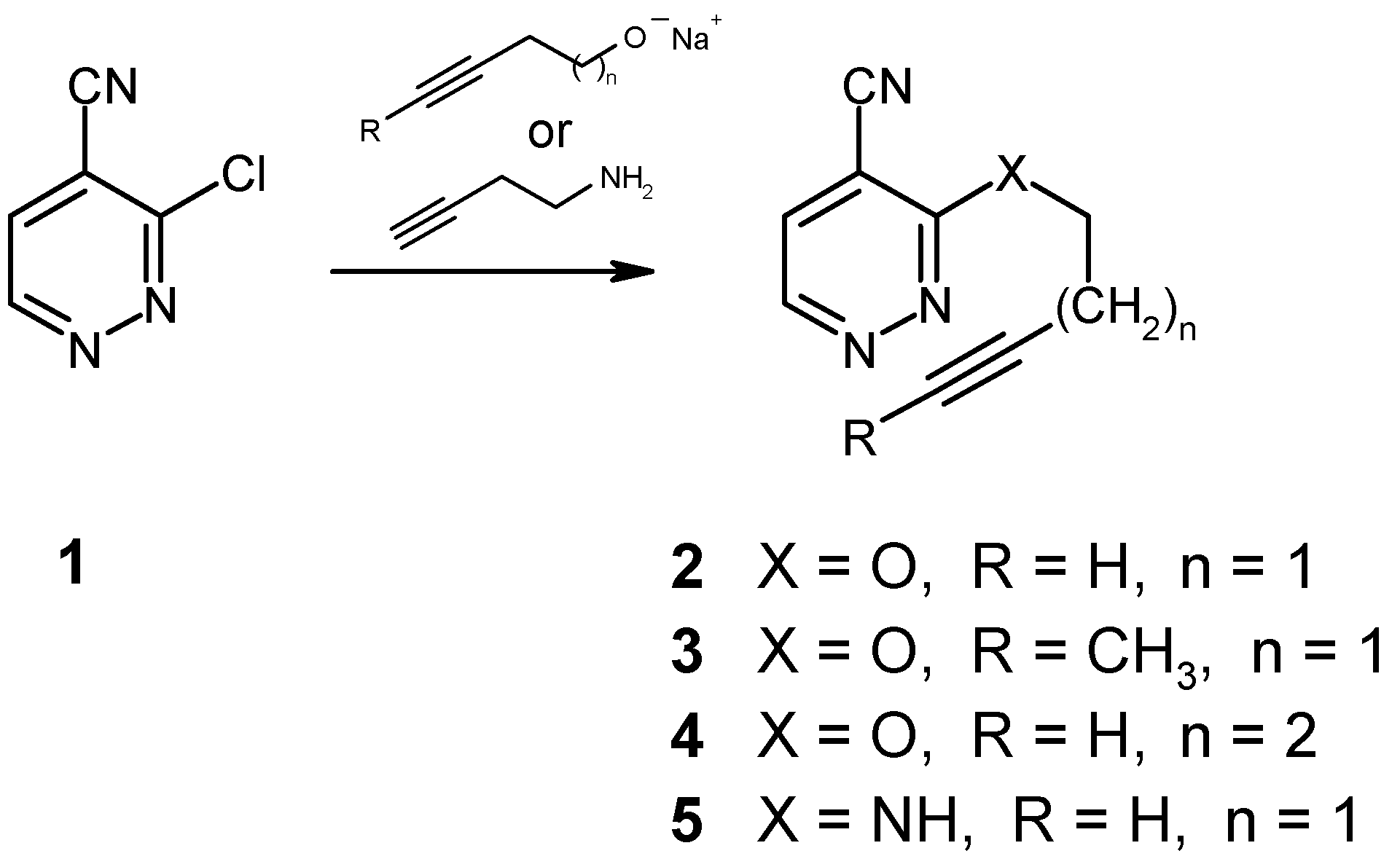
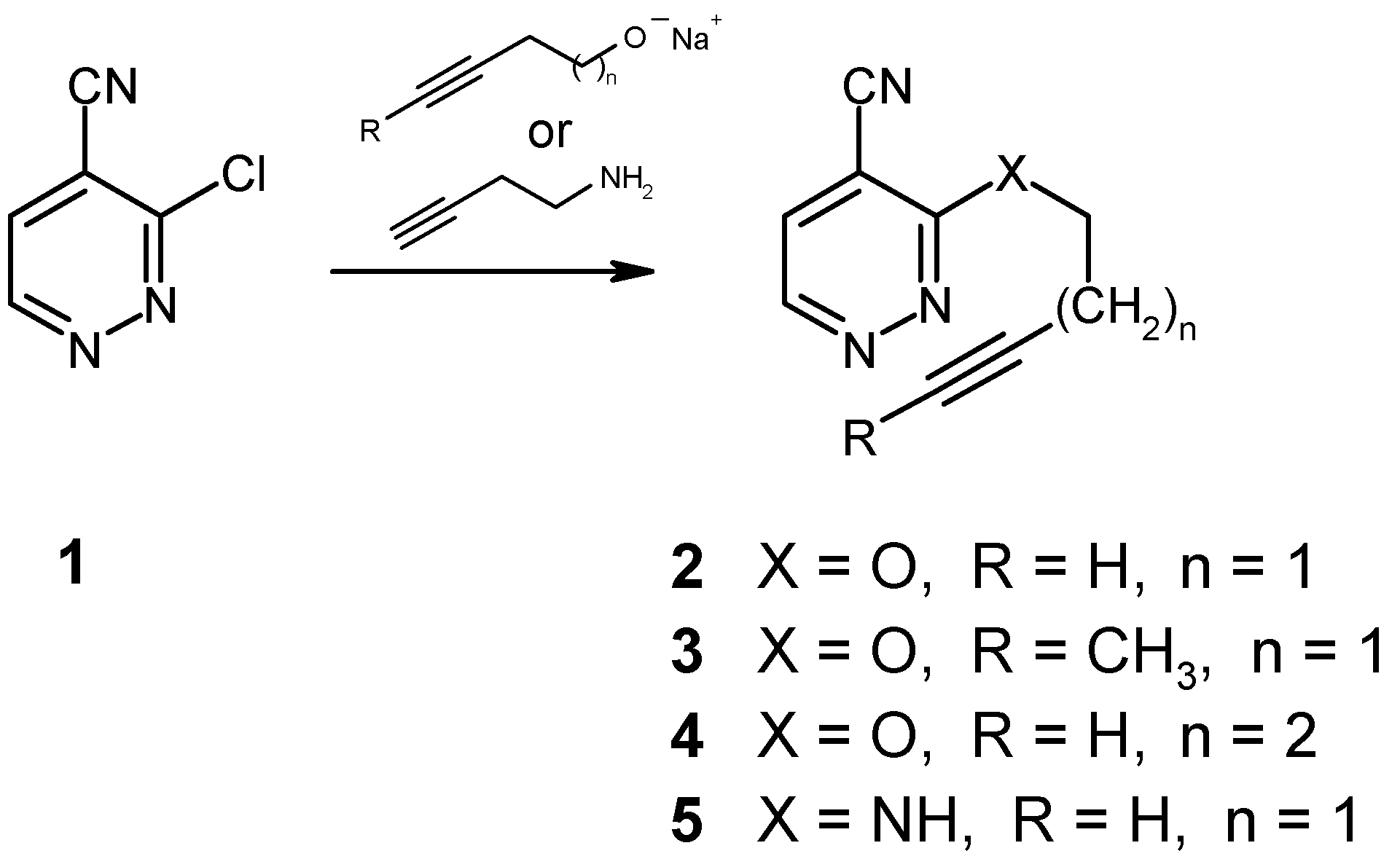
The preparation of the required 3-alkynyloxysubstituted 4-pyridazinecarbonitriles 2-4 as well as that of the amine 5 from readily available 3-chloro-4-pyridazinecarbonitrile (1) [7,8] and the corresponding alkynolates or butynylamine, respectively, is similar to the procedures reported for the synthesis of structurally related pyrimidine and pyrazine derivatives [9,10], although in our case these nucleophilic substitution reactions had to be carried out at much lower temperatures in order to minimize the formation of by-products.
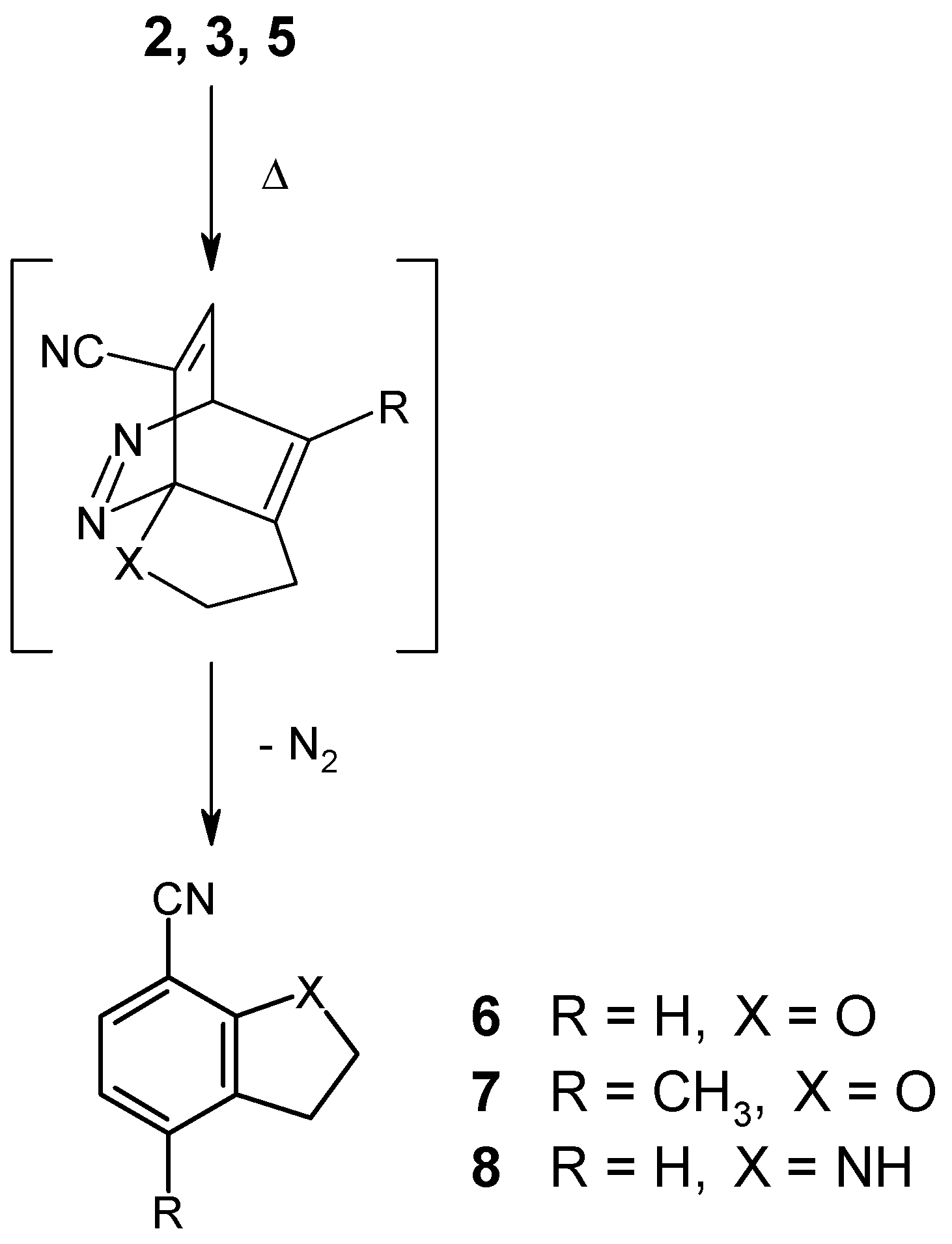
Heating of the ether 2 in an inert solvent (bromobenzene) to 150°C was found to affect the expected cycloaddition reaction, followed by loss of nitrogen in a spontaneous cycloreversion step. After 96 hours, the transformation was complete (NMR monitoring), giving the known dihydrobenzofuran 6 [11] in 79% yield. It should be noted in this context that earlier attempts to cyclize the analogous 6-(alkynyloxy)-3-chloropyridazines had failed [2]. Thus, the presence of an electronwithdrawing group like the cyano function in compound 2 must be regarded as essential.
Scheme 1.
Scheme 2.
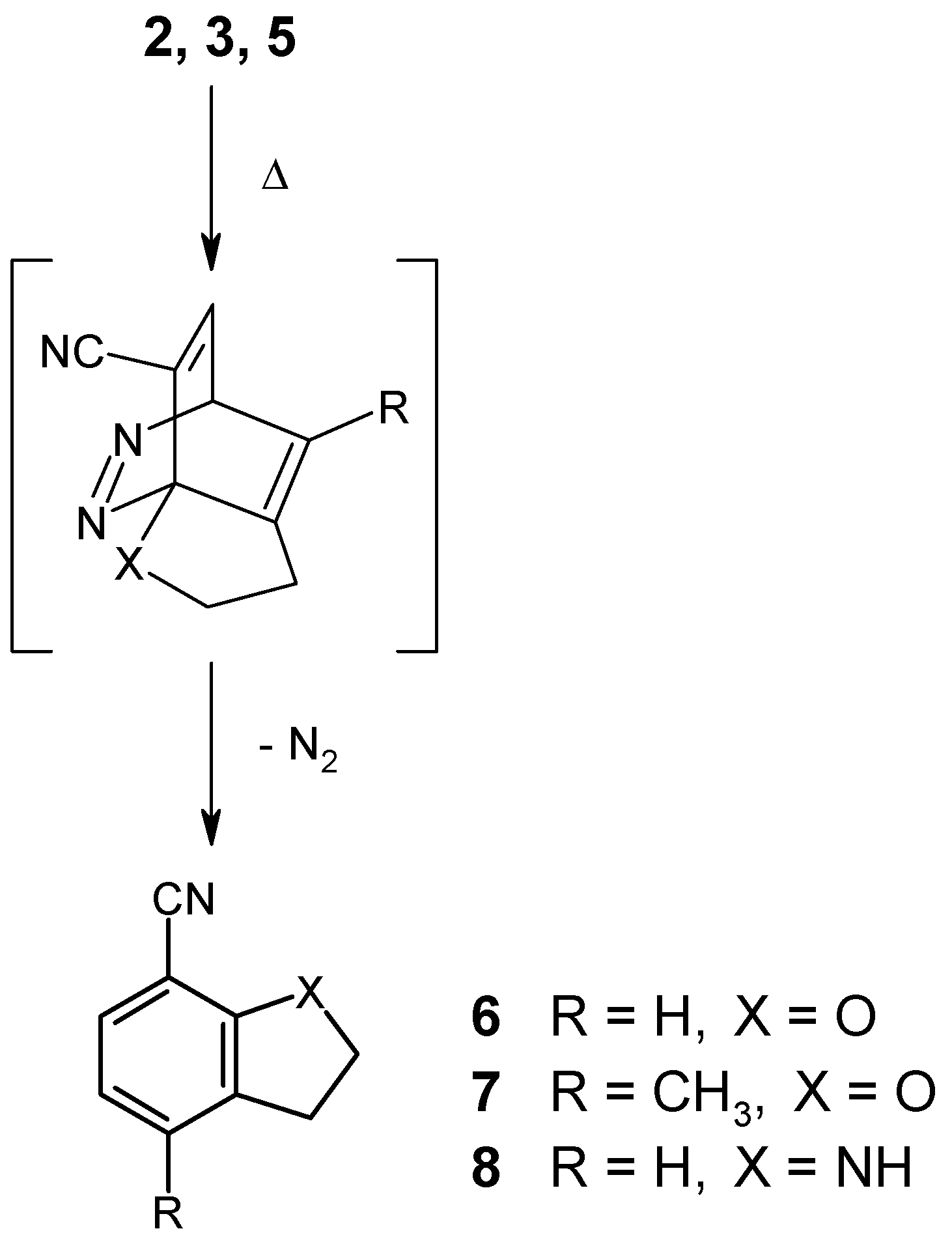
Not surprisingly, the 3-pentynyl ether 3 was found to react significantly slower under identical conditions, as a result of the steric hindrance which is introduced by the terminal methyl group at the alkyne moiety. Here, completion of the reaction takes 36 days at reflux temperature in bromobenzene solution, affording the new disubstituted dihydrobenzofuran 7 in 67% yield. Elongation of the spacer chain by one methylene group, as in the case of the 4-pentyn-1-yloxy compound 4, however, was found to result in a complete loss of reactivity, even under very drastic conditions. Obviously, too much of the required “entropic assistance” is lost with the longer spacer. On the other hand, we were surprised to find that the butynylamino compound 5 does undergo a thermally induced intramolecular cycloaddition reaction, despite the unfavorable effect of the electron-donating amino function. Employment of a higher-boiling solvent (1,2,4-trichlorobenzene), together with a reaction temperature of 180°C for a period of 6 days, permitted the preparation of the indolinecarbonitrile 8 [12] in 49% yield. This successful transformation again shows the beneficial electronic effect of the nitrile function, particularly in view of the fact that comparable cycloadditions with butynylamino-substituted pyrimidines and pyrazines, lacking a cyano group, had failed unless the NH function had been converted into an N-acetyl derivative [9,10].
As an extension of the studies described so far, we investigated the cycloaddition behavior of 4-pyridazinecarbonitriles bearing an acetylenic side chain with a 1,2-phenylene unit incorporated. By this structural feature, the dienophile is fixed in a favorable geometry, hence the [4+2] cycloaddition step should be significantly facilitated, as was observed previously with other ring systems [13,14].
Scheme 3.
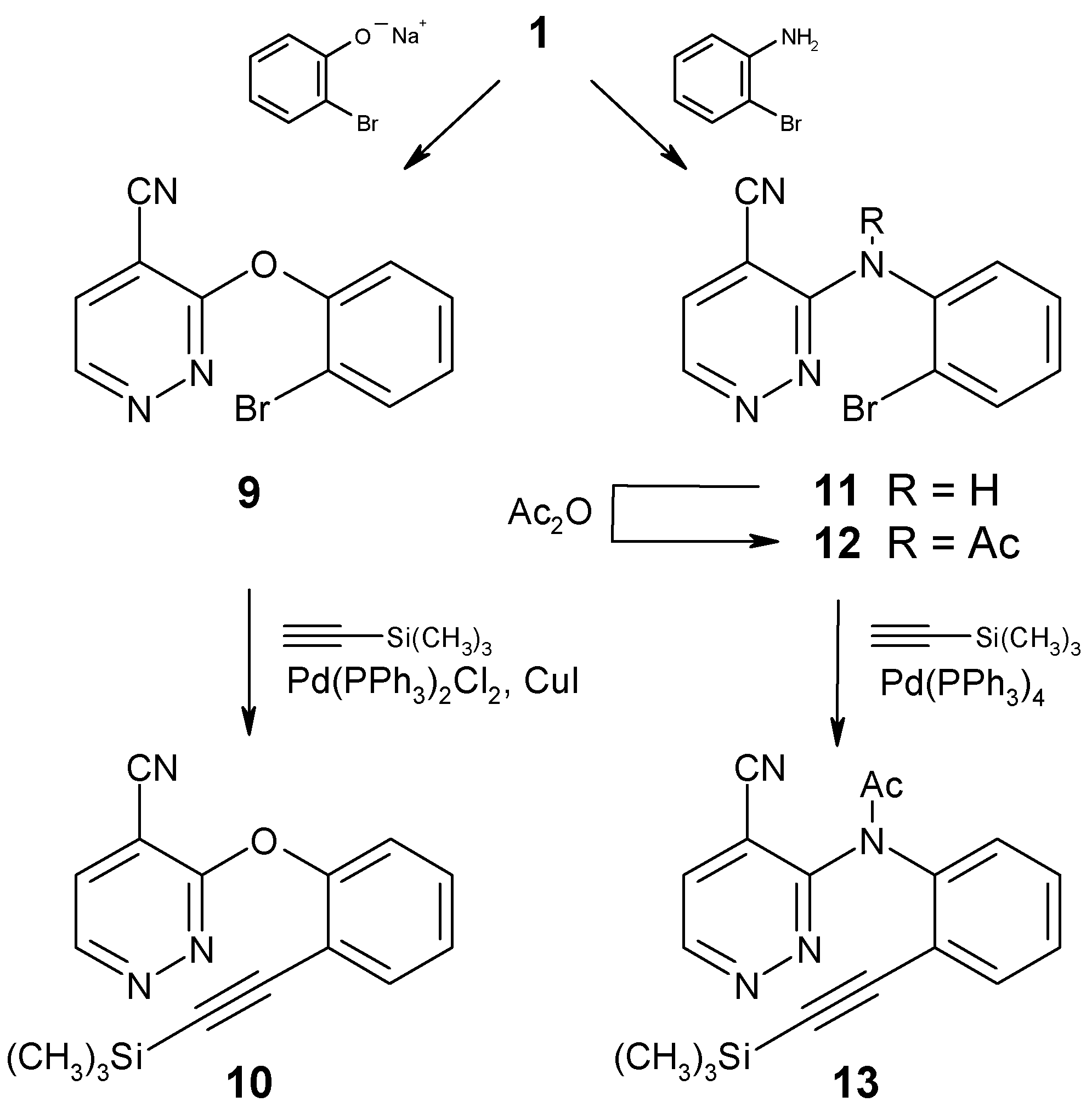
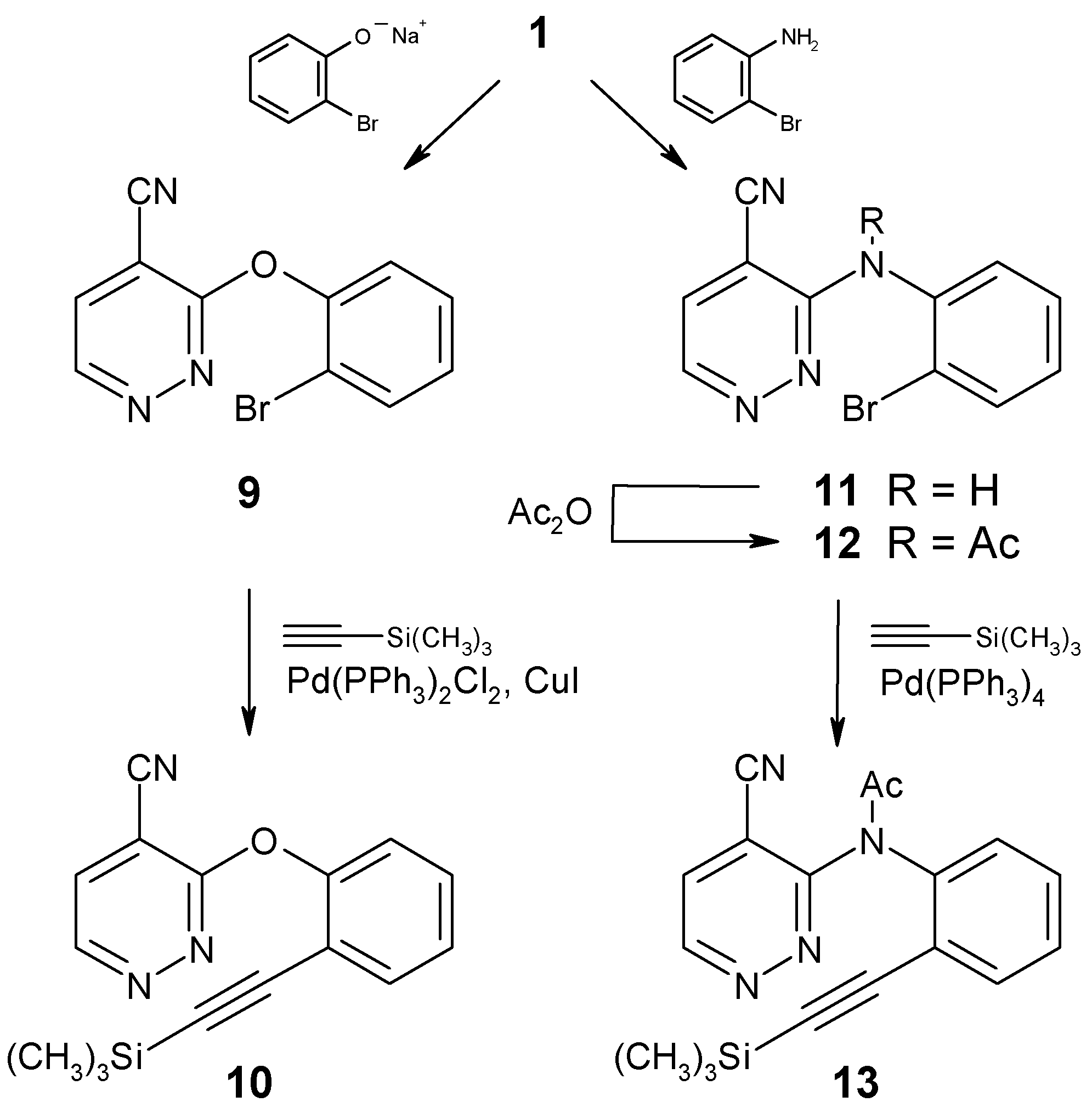
The requisite cycloaddition educts were prepared from the chloro nitrile 1, following the pathways displayed in Scheme 3. Nucleophilic substitution of the chloro function by sodium 2-bromophenolate gave the diaryl ether 9, which was subjected to Pd0-catalyzed cross-coupling with trimethylsilylacetylene to afford the alkyne 10 in moderate yield. In a similar fashion, 1 was reacted with 2-bromoaniline to give the diarylamine 11, which - after N-acetylation - was cross-coupled with the acetylene unit to yield compound 13.
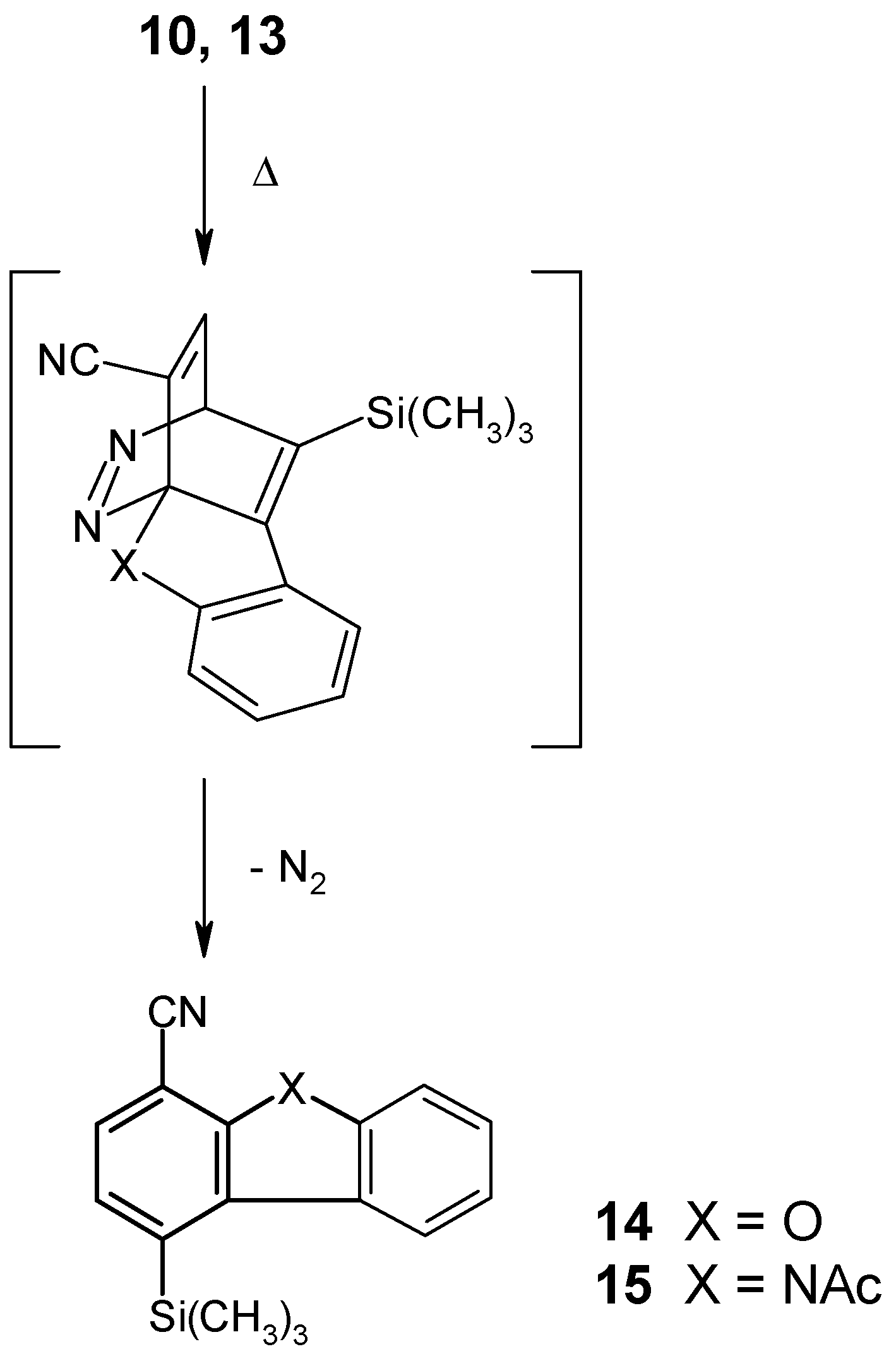
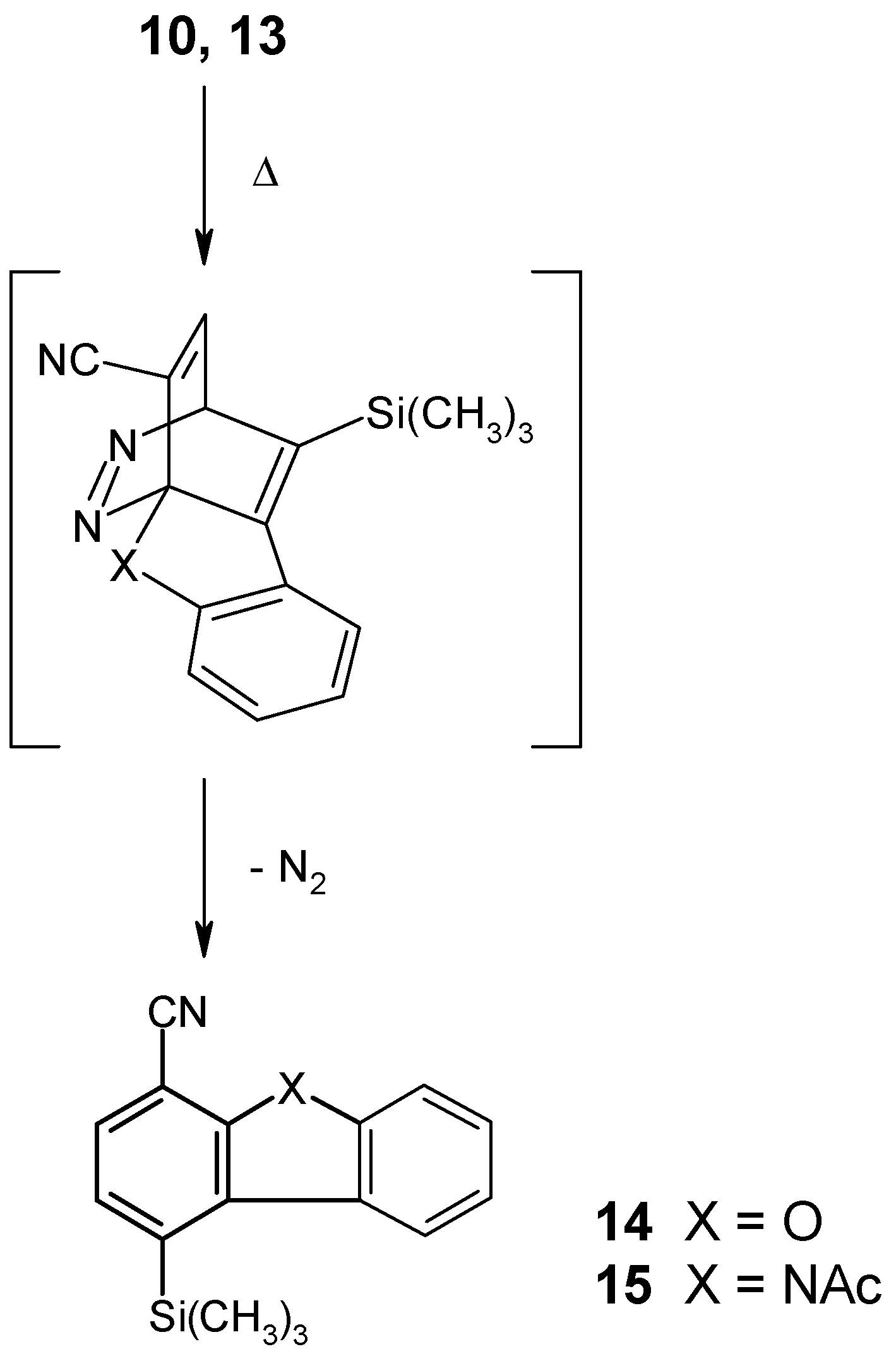
When the ether 10 as well as the amide 13 were heated in bromobenzene solution to 150°C, an intramolecular cycloaddition reaction was found to take place very smoothly within a few hours, despite the considerable steric hindrance of the dienophilic substructure which is caused by the bulky trimethylsilyl group. This clearly shows the important role of the fixed geometry of the dienophilic side chain in 10 and 13, compared with their much more flexible counterparts in compounds 2-5, with respect to the reaction rate in this type of cycloaddition. The new tricyclic nitriles 14 and 15 thus could be obtained in 62% and 65% yield, respectively.
Scheme 4.
Conclusion
The sequence described provides a convenient access to several bicyclic and tricyclic aromatic carbonitriles and demonstrates the usefulness of the intramolecular Diels-Alder reaction of appropriately substituted pyridazines for the construction of fused heterocyclic systems.
Experimental
General
Melting points (uncorrected) were determined on a Kofler hot-stage microscope. 1H NMR spectra were recorded on a Bruker AC 80 (80 MHz) spectrometer, using TMS as internal reference. IR spectra were taken on a Jasco IRA-1 instrument. High-resolution mass spectra were obtained on a Finnigan MAT 8230 spectrometer. For medium pressure liquid chromatography (MPLC), Merck Lobar® columns, packed with LiChroprep Si 60, 40-63 μm, were used. Column chromatography was carried out on Merck Kieselgel 60, 63-200 μm, thin layer chromatography was done on Merck aluminium sheets pre-coated with Kieselgel F254. Microanalyses were performed at the Institute of Physical Chemistry (Microanalytical Laboratory), University of Vienna..
3-(3-Butyn-1-yloxy)-4-pyridazinecarbonitrile (2)
To a solution of 3-butyn-1-ol (280 mg, 4 mmol) in dry THF (12 ml) sodium hydride (105 mg of a 80% suspension in paraffin, 3.5 mmol) was added in small portions. The mixture was stirred at room temperature until the effervescence had ceased, then it was cooled to -60°C. A solution of 1 [7,8] (280 mg, 2 mmol) in dry THF (3 ml) was added, and stirring was continued for 7 hours, allowing the temperature to rise to -30°C. The mixture was taken up in diethyl ether (100 ml), washed with water, and dried (Na2SO4). Removal of the solvents, followed by recrystallization from ethyl acetate/light petroleum afforded compound 2 (300 mg, 87%) as colorless crystals, mp 85-86°C. IR (KBr): 3280, 3060, 2240, 1580, 1550, 1470, 1450, 1365, 1290, 1110, 1000, 870, 770 cm-1. 1H NMR (CDCl3): δ 9.05 (d, J = 4.7 Hz, 1H, H-6), 7.66 (d, J = 4.7 Hz, 1H, H-5), 4.77 (t, J = 6.9 Hz, 2H, O-CH2-C), 2.80 (dt, J = 6.9, 2.7 Hz, 2H, C-CH2-C), 2.03 (t, J = 2.7 Hz, 1H, ≡CH). Anal. Calcd. for C9H7N3O: C, 62.42; H, 4.07; N, 24.26. Found: C, 62.60; H, 4.23; N, 24.37.
3-(3-Pentyn-1-yloxy)-4-pyridazinecarbonitrile (3)
Preparation as described for compound 2, starting from 3-pentyn-1-ol (336 mg, 4 mmol) and 1 (280 mg, 2 mmol). Purification by MPLC (eluent: light petroleum/ethyl acetate, 3+1), followed by recrystallization from n-pentane gave the title compound 3 (270 mg, 72%) as colorless crystals, mp 57-60°C. IR (KBr): 3070, 3050, 2930, 2860, 2240, 1590, 1550, 1480, 1440, 1400, 1370, 1310, 1280, 1000, 870, 770 cm-1. 1H NMR (CDCl3): δ 9.01 (d, J = 4.7 Hz, 1H, H-6), 7.63 (d, J = 4.7 Hz, 1H, H-5), 4.69 (t, J = 7.0 Hz, 2H, O-CH2-C), 2.80-2.50 (m, 2H, C-CH2-C), 1.74 (t, J = 2.5 Hz, 3H, ≡C-CH). Anal. Calcd. for C10H9N3O: C, 64.16; H, 4.85; N, 22.45. Found: C, 64.02; H, 5.11; N, 22.24.
3-(4-Pentyn-1-yloxy)-4-pyridazinecarbonitrile (4)
Preparation as described for compound 2, starting from 4-pentyn-1-ol (336 mg, 4 mmol) and 1 (280 mg, 2 mmol). Purification by MPLC (eluent: light petroleum/ ethyl acetate, 3+1) gave the title compound 4 (210 mg, 56%) as an almost colorless oil. IR (KBr): 3300, 3080, 2970, 2940, 2250, 2130, 1590, 1550, 1450, 1370, 1350, 1290, 1030, 950, 865, 775 cm-1. 1H NMR (CDCl3): δ 9.01 (d, J = 4.8 Hz, 1H, H-6), 7.63 (d, J = 4.8 Hz, 1H, H-5), 4.74 (t, J = 5.9 Hz, 2H, O-CH2-C), 2.50-1.90 (m, 5H, C-CH2CH2-C≡CH). HR-MS Calcd. for C10H9N3O: 187.0746. Found: 187.0735.
3-(3-Butyn-1-ylamino)-4-pyridazinecarbonitrile (5)
To a solution of the chloro nitrile 1 [7,8] (280 mg, 2 mmol) in dry THF (12 ml) 3-butyn-1-ylamine [15] (552 mg, 8 mmol) was added, and the mixture was stirred at room temperature for 26 hours. It was then taken up in diethyl ether (100 ml), washed with water, and dried (Na2SO4).The residue left on evaporation was purified by column chromatography (eluent: light petroleum/ethyl acetate, 3+1), followed by recrystallization from light petroleum to afford the title compound 5 (203 mg, 59%) as colorless crystals, mp 98-99°C. IR (KBr): 3380, 3280, 3200, 3130, 3080, 2960, 2240, 2130, 1580, 1560, 1510, 1440, 1420, 1360, 1160, 1070, 860, 840, 770 cm-1. 1H NMR (CDCl3): δ 8.72 (d, J = 4.8 Hz, 1H, H-6), 7.37 (d, J = 4.8 Hz, 1H, H-5), 5.60 (br t, 1H, NH), 3.86 (q, J = 6.3 Hz, 2H, N-CH2-C), 2.63 (dt, J = 6.3, 2.6 Hz, 2H, C-CH2-C), 2.06 (t, J = 2.6 Hz, 1H, ≡CH). Anal. Calcd. for C9H8N4: C, 62.78; H, 4.68; N, 32.54. Found: C, 62.99; H, 4.64; N, 32.66.
[4+2] Cycloaddition Reaction of Compound 2
A solution of the ether 2 (130 mg, 0.75 mmol) in bromobenzene (1 ml) was heated to 150°C under an Ar atmosphere for 96 hours. The solvent was removed under reduced pressure and the residue was purified by column chromatography (eluent: light petroleum/ethyl acetate, 3+1) and subsequent recrystallization from n-pentane gave 2,3-dihydro-7-benzofurancarbonitrile (6) [11] (86 mg, 79%) as colorless needles, mp 53-54°C (ref.[11]: 54.5-56°C, from ethanol).
[4+2] Cycloaddition Reaction of Compound 3
A solution of the ether 3 (160 mg, 0.85 mmol) in bromobenzene (1 ml) was heated to 150°C under an Ar atmosphere for 36 days. The solvent was removed under reduced pressure and the residue was purified by column chromatography (eluent: light petroleum/ethyl acetate, 3+1) and subsequent recrystallization from light petroleum gave 2,3-dihydro-4-methyl-7-benzofurancarbonitrile (7) (82 mg, 67%) as colorless crystals, mp 79-80°C. IR (KBr): 2980, 2930, 2860, 2220, 1620, 1590, 1470, 1450, 1420, 1225, 1190, 920, 810, 720 cm-1. 1H NMR (CDCl3): δ 7.19 (d, J = 7.9 Hz, 1H, H-6), 6.68 (d, J = 7.9 Hz, 1H, H-5), 4.72 (t, J = 8.8 Hz, 2H, O-CH2-C), 3.16 (t, J = 8.8 Hz, 2H, C-CH2-C), 2.27 (s, 3H, CH3). Anal. Calcd. for C10H9NO: C, 75.45; H, 5.70; N, 8.80. Found: C, 75.24; H, 5.61; N, 8.70.
[4+2] Cycloaddition Reaction of Compound 5
A solution of the amine 5 (86 mg, 0.5 mmol) in 1,2,4-trichlorobenzene (1 ml) was heated to 180°C under an Ar atmosphere for 6 days. The solvent was removed under reduced pressure and the residue was purified by column chromatography (eluent: light petroleum/ethyl acetate, 3+1) and subsequent recrystallization from n-pentane gave 7-indolinecarbonitrile (8) [12] (35 mg, 49%) as colorless crystals, mp 60.5-61°C (ref.[12]: 66-67°C, from diethyl ether/hexane).
3-(2-Bromophenoxy)-4-pyridazinecarbonitrile (9)
Sodium (138 mg, 6 mmol) was cut into small pieces and dissolved in 2-bromophenol (4 ml). Compound 1 [7,8] (837 mg, 6 mmol) was added, and the mixture was stirred at 40°C for 91 hours. It was then taken up in CH2Cl2, washed with water, and dried (Na2SO4). The volatile components were removed under reduced pressure and the residue was dissolved in diethyl ether. The solution was washed with 1M NaOH and water, then it was dried (Na2SO4) and evaporated. Recrystallization from ethanol gave compound 9 (1119 mg, 68%) as colorless crystals, mp 133-134°C. IR (KBr): 3040, 2240, 1540, 1450, 1410, 1360, 1270, 1200, 1020, 890, 770, 730 cm-1. 1H NMR (CDCl3): δ 9.14 (d, J = 4.8 Hz, 1H, H-6), 7.78 (d, J = 4.8 Hz, 1H, H-5), 7.75-7.00 (m, 4H, aryl-H). Anal. Calcd. for C11H6BrN3O: C, 47.85; H, 2.19; N, 15.22. Found: C, 48.05; H, 2.29; N, 15.16.
3-[2-(Trimethylsilylethynyl)phenoxy]-4-pyridazinecarbonitrile (10)
To a suspension of the bromo compound 9 (552 mg, 2 mmol) in dry triethylamine (7 ml), dimethylformamide was added dropwise until a clear solution was obtained. After addition of Pd(PPh3)2Cl2 (25 mg), CuI (5 mg) and trimethylsilylacetylene (392 mg, 4 mmol), the mixture was stirred at 50°C in a sealed tube under an Ar atmosphere for 7 days. The volatile components were removed under reduced pressure and the residue was purified by column chromatography (eluent: light petroleum/ethyl acetate, 1+1), followed by MPLC (eluent: light petroleum/ethyl acetate, 3+1) to afford compound 10 (200 mg, 34%) as a yellow oil which slowly decomposed. IR (KBr): 2960, 2930, 2860, 2170, 1470, 1430, 1370, 1280, 870, 850 cm-1. 1H NMR (CDCl3): δ 9.11 (d, J = 4.8 Hz, 1H, H-6), 7.73 (d, J = 4.8 Hz, 1H, H-5), 7.70-7.20 (m, 4H, aryl-H), 0.04 (s, 9H, Si(CH3)3).
3-(2-Bromophenylamino)-4-pyridazinecarbonitrile (11)
A mixture of compound 1 [7,8] (280 mg, 2 mmol) and 2-bromoaniline (1032 mg, 6 mmol) was heated to 70°C for 27 hours. The excess reagent was distilled off under reduced pressure and the residue was subjected to column chromatography (eluent: light petroleum/ethyl acetate, 1+1). Subsequent recrystallization from ethanol gave compound 11 (170 mg, 31%) as almost colorless needles, mp 137.5-139°C. IR (KBr): 3360, 3080, 2240, 1600, 1580, 1550, 1500, 1440, 1410, 1160, 1040, 900, 860, 750 cm-1. 1H NMR (CDCl3): δ 8.93 (d, J = 4.9 Hz, 1H, H-6), 7.54 (d, J = 4.9 Hz, 1H, H-5), 8.62 (dd, J = 8.2, 1.6 Hz, 1H, aryl-H), 7.80-6.85 (m, 4H, NH, aryl-H). Anal. Calcd. for C11H7BrN4: C, 48.02; H, 2.56; 20.36. Found: C, 48.31; H, 2.41; N, 20.42.
N-(2-Bromophenyl)-N-(4-cyanopyridazin-3-yl)acetamide (12)
A mixture of compound 11 (275 mg, 1 mmol) and anhydrous sodium acetate (82 mg, 1 mmol) in acetic anhydride (10 ml) was stirred at 50°C for 24 hours. The volatile components were removed under reduced pressure and the residue was subjected to column chromatography (eluent: light petroleum/ethyl acetate, 1+1). Subsequent recrystallization from ethanol gave compound 12 (228 mg, 72%) as colorless crystals, mp 148-149.5°C. IR (KBr): 3080, 1680, 1580, 1540, 1470, 1420, 1360, 1270, 1050, 1000, 860, 750 cm-1. 1H NMR (CDCl3): δ 9.21 (d, J = 5.0 Hz, 1H, H-6), 7.78 (d, J = 5.0 Hz, 1H, H-5), 7.77-7.20 (m, 4H, aryl-H), 2.17 (s, 3H, COCH3). Anal. Calcd. for C13H9BrN4O: C, 49.23; H, 2.86; N, 17.67. Found: C, 49.47; H, 3.08; N, 17.81.
N-(4-Cyanopyridazin-3-yl)-N-[2-(trimethylsilylethynyl)-phenyl]acetamide (13)
To a suspension of compound 12 (476 mg, 1.5 mmol) and Pd(PPh3)4 (20 mg) in dry triethylamine (5 ml), trimethylsilylacetylene (295 mg, 3 mmol) and a few drops of dimethylformamide were added. The mixture was stirred at 50°C in a sealed tube under an Ar atmosphere for 14 days. The volatile components were removed under reduced pressure and the residue was purified by column chromatography (eluent: light petroleum/ethyl acetate, 3+1), followed by MPLC (using the same eluent) and subsequent recrystallization from n-pentane to afford compound 13 (195 mg, 39%) as colorless plates, mp 149-152°C (dec.). IR (KBr): 3100, 2960, 2240, 2160, 1700, 1540, 1490, 1425, 1350, 1270, 1250, 850, 760 cm-1. 1H NMR (CDCl3): δ 9.18 (d, J = 5.0 Hz, 1H, H-6), 7.72 (d, J = 5.0 Hz, 1H, H-5), 7.65-7.30 (m, 4H, aryl-H), 2.23 (s, 3H, COCH3), 0.16 (s, 9H, Si(CH3)3). Anal. Calcd. for C18H18N4OSi: C, 64.64; H, 5.42; N, 16.75. Found: C, 64.46; H, 5.34; N, 16.91.
[4+2] Cycloaddition Reaction of Compound 10
A solution of compound 10 (50 mg, 0.17 mmol) in bromobenzene (1 ml) was heated to 150°C under an Ar atmosphere for 2 hours. The solvent was removed under reduced pressure and the residue was purified by column chromatography (eluent: light petroleum) and subsequent recrystallization from light petroleum to give 1-trimethylsilyl-4-dibenzofurancarbonitrile (14) (28 mg, 62%) as colorless needles, mp 163-164°C. IR (KBr): 2970, 2950, 2900, 2230, 1450, 1370, 1260, 1250, 1200, 950, 850, 820, 740, 720 cm-1. 1H NMR (CDCl3): δ 8.15-8.00 (m, 1H, aryl-H), 7.80-7.30 (m, 5H, aryl-H), 0.54 (s, 9H, SiCH3)3). Anal. Calcd. for C16H15NOSi: C, 72.41; H, 5.70; N, 5.28. Found: C, 72.16; H, 5.49; N, 5.09.
[4+2] Cycloaddition Reaction of Compound 13
A solution of compound 13 (50 mg, 0.15 mmol) in bromobenzene (1 ml) was heated to 150°C under an Ar atmosphere for 3.5 hours. The solvent was removed under reduced pressure and the residue was purified by column chromatography (eluent: light petroleum/ethyl acetate, 3+1), followed by recrystallization from n-pentane to afford N-acetyl-4-trimethylsilyl-1-carbazolecarbonitrile (15) (30 mg, 65%) as colorless crystals, mp 131-132°C. IR (KBr): 3090, 2960, 2230, 1740, 1610, 1580, 1460, 1430, 1360, 1280, 1260, 1200, 850, 830, 760 cm-1. 1H NMR (CDCl3): δ 8.30-7.90 (m, 2H, aryl-H), 7.80-7.30 (m, 4H, aryl-H), 2.89 (s, 3H, COCH3), 0.56 (s, 9H, Si(CH3)3). Anal. Calcd. for C18H18N2OSi: C, 70.55; H, 5.92; N, 9.14. Found: C, 70.31; H, 6.01; N, 8.89.
References
- For some excellent reviews cf.: Boger, D.L. Tetrahedron 1983, 39, 2869. ; Boger, D.L. Chem. Rev. 1986, 86, 781. ; Taylor, E.C. Bull. Soc. Chim. Belg. 1988, 97, 599. ; Marcelis, A.T.M.; van der Plas, H.C. Trends in Heterocyclic Chem. 1991, 1, 111. ; Sauer, J. Bull. Soc. Chim. Belg. 1992, 101, 521.
- Boger, D.L.; Coleman, R.S. J. Org. Chem. 1984, 49, 2240.
- Boger, D.L.; Zhang, M. J. Am. Chem. Soc. 1991, 113, 4230.
- Jojima, T.; Takeshiba, H.; Konotsune, T. Chem. Pharm. Bull. 1972, 20, 2191.
- Jojima, T.; Takeshiba, H.; Kinoto, T. Chem. Pharm. Bull. 1976, 24, 1581.
- Jojima, T.; Takeshiba, H.; Kinoto, T. Chem. Pharm. Bull. 1976, 24, 1588.
- Dornow, A.; Abele, W. Chem. Ber. 1964, 97, 3349.
- Yanai, M.; Takeda, S.; Mitsuoka, T. Chem. Pharm. Bull. 1977, 25, 1708. [CrossRef]
- Frissen, A.E.; Marcelis, A.T.M.; van der Plas, H.C. Tetrahedron 1989, 45, 803.
- de Bie, D.A.; Ostrowicz, A.; Geurtsen, G.; van der Plas, H.C. Tetrahedron 1988, 44, 2977.
- Borrevang, P.; Andersen, H.S. PCT Int. Appl. 1991. WO 9100,862. Chem. Abstr. 1988, 115, 8798e.
- Adachi, M.; Matsumura, H.; Sugasawa, T. Eur. Pat. Appl. 1988. EP 257,583. Chem. Abstr. 1988, 109, 73156j.
- Stolle, W.A.W.; Marcelis, A.T.M.; van der Plas, H.C. Tetrahedron 1989, 45, 6511.
- Haider, N.; van der Plas, H.C. Tetrahedron 1990, 46, 3641.
- Taylor, E.C.; Pont, J.L. Tetrahedron Lett. 1987, 379.
- Sample Availability: Available from the authors.
© 1998 MDPI. All rights reserved
Share and Cite
MDPI and ACS Style
Fülep, G.; Haider, N. Synthesis and Intramolecular [4+2] Cycloaddition Reactions of 4-Pyridazinecarbonitriles with Alkyne Side Chains. Molecules 1998, 3, 10-15. https://doi.org/10.3390/30100010
AMA Style
Fülep G, Haider N. Synthesis and Intramolecular [4+2] Cycloaddition Reactions of 4-Pyridazinecarbonitriles with Alkyne Side Chains. Molecules. 1998; 3(1):10-15. https://doi.org/10.3390/30100010
Chicago/Turabian StyleFülep, Günther, and Norbert Haider. 1998. "Synthesis and Intramolecular [4+2] Cycloaddition Reactions of 4-Pyridazinecarbonitriles with Alkyne Side Chains" Molecules 3, no. 1: 10-15. https://doi.org/10.3390/30100010