A Short and Efficient Synthesis of (R)-Phoracantholide I

Bio-Organic Division, Bhabha Atomic Research Centre, Mumbai - 400 085, India
*
Author to whom correspondence should be addressed.
Molecules 1998, 3(2), 44-47; https://doi.org/10.3390/30200044
Submission received: 16 January 1998
/
Accepted: 26 January 1998
/
Published: 13 February 1998
{kind=link}
Abstract
:A short and efficient chemoenzymatic synthesis of the title compound has been developed. The salient features of the synthesis were i) preparation of the required chiron via a porcine pancreatic lipase (PPL) catalyzed esterification of a ω-functionalized 2-alkanol and ii) use of easily accessible and inexpensive materials/ reagents.
Introduction
Enzymes show great promise as efficient catalysts in asymmetric synthesis and several biocatalytic protocols have been developed [1a,b] for enantioselective organic transformations. In this context, the utility of biocatalysts for the preparation of useful chirons has been appreciated in recent times. In addition, other attributes of enzyme chemistry viz. functional specificity, regio- and chemoselectivity, high catalytic rate even under moderate conditions etc. provide useful advantages for designing suitable chirons [1b]. Thus, a chemoenzymatic protocol ensures good enantiocontrol and higher efficiency of the synthesis by providing operationally simple and shorter protocol. With this aim, we have developed [2a-e] several enzymatic protocols for the preparation of useful chirons and applied [3] them to the syntheses of different bioactive compounds.
Earlier, we formulated a lipase catalyzed trans-esterification strategy for the resolution of alkan-2-ols which however, were not useful synthons due to the lack of adequate functionality. Hence, we tested the applicability of the above method for the resolution of ω-functionalized alkan-2-ols using 1-tert-butyldimethylsilyloxy-8-hydroxynonane (7) as the model compound. Subsequently, this was derivatized to (R)-phoracantholide I ((R)-I, or 10) which is presented in this paper. Compound R-I constitutes [4] one of the components of the highly odoriferous defense secretion from the metasternal gland of the eucarypt longicorn, Phoracantha synonyma. Since natural (R)-I is known [5] to possess (R)-configuration, we have prepared the natural isomer only. Several syntheses of (R)-I in racemic [6] and chiral forms [7a-c] have already been reported. However, most of these involved multiple steps/ and or used rather inaccessible and expensive reagents. In comparison, the present one (Scheme 1) involving a chemoenzymatic route is by far the simplest and most efficient synthesis of (R)-I.
Results and Discussion
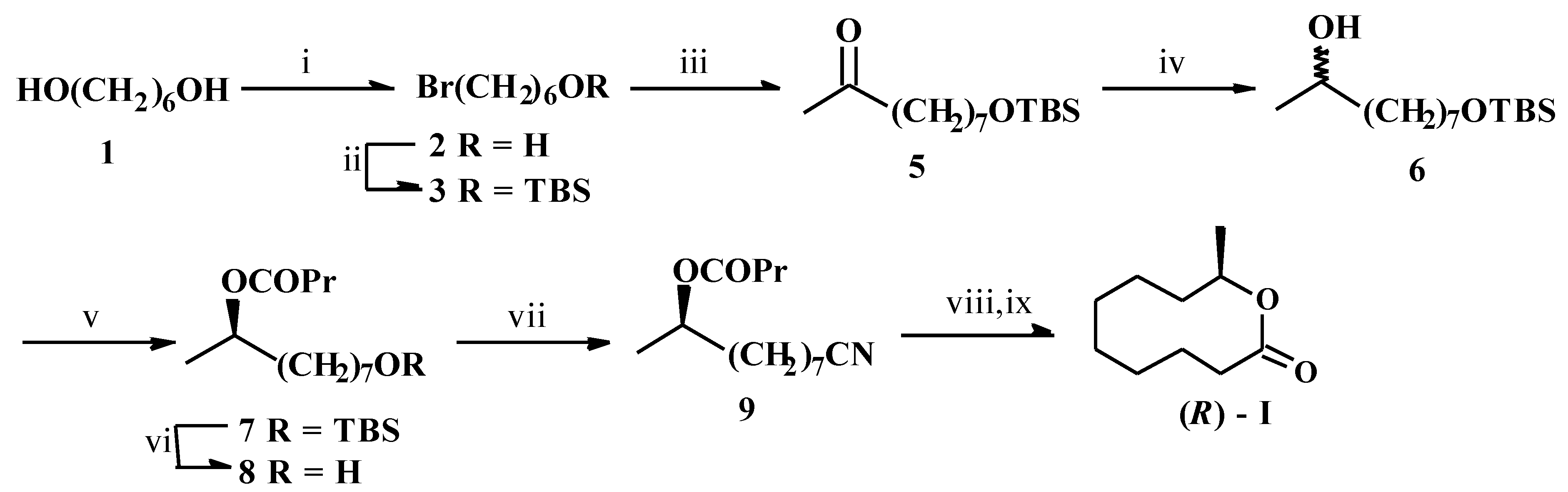
Thus, hexane-1,6-diol (1) was partially brominated [8] to give the known compound 2 [9] which on silylation furnished 3. α-Alkylation of methyl acetoacetate (4) with 3 followed by alkaline hydrolysis gave the ketone 5. Its reduction with NaBH4 afforded the key carbinol synthon 6 which was subjected to porcine pancreatic lipase (PPL) catalyzed trans-acylation under different conditions viz. different acylating agents and solvents, and extent of conversion. The best result was obtained with trifluoroethyl butyrate (TFEB) as the acyl donor in diisopropyl ether when at 30% conversion, the (R)-butyrate 7 and (S)-6 were obtained with 93% and 78% ees respectively. The %ee of the butyrate 7 was assayed by the GLC analysis of its (R)-MTPA ester on 3% OV-17 column GLC, temperature programming 140-240 °C, 4 °C/min, tR = 18.6 min and 19.2 min for (R)-6 and (S)-6 respectively. The configurational assignments were inferred from the general trend [3] of PPL catalyzed acylation of 2-alkanols and their derivatives. For further confirmation, the ester 7 was converted to the known [10] compound 2-nonanol via alkaline hydrolysis, pyranylation, desilylation, mesylation, LAH reduction and depyranylation. Comparison of the sign of its optical rotation with those reported confirmed its (R)- configuration.
Scheme 1.
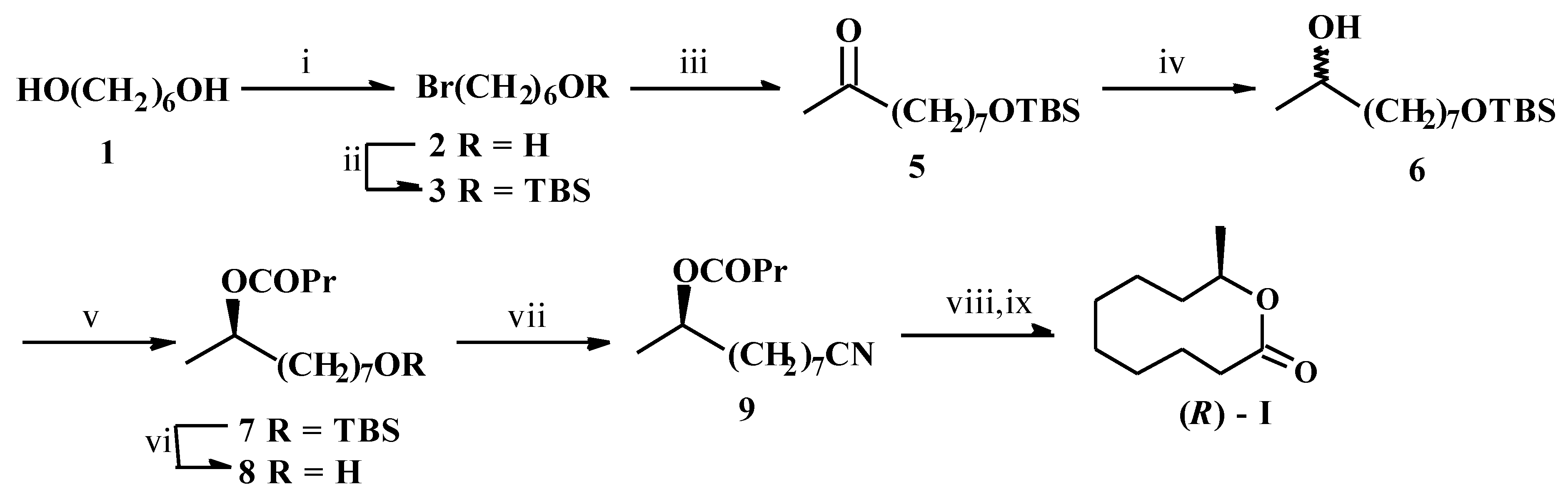
i) 48%HBr/benzene/ Δ, ii) TBSCl/triethylamine/DMAP/CH 2Cl2, iii) CH3COCH2CO2Me (4)/NaOMe/MeOH; Aqueous NaOH/H +, iv) NaBH 4/MeOH, v) PPL/TFEB/diisopropyl ether, vi) Bu 4NF/THF, vii) p-TsCl/pyridine/ CH2Cl2; KCN/DMSO/ Δ, viii) Conc. HCl/Δ, ix) Alcoholic KOH; H +.
For the synthesis, 7 was desilylated to give the alcohol 8. Its tosylation followed by reaction with KCN afforded the cyanohydrin derivative 9. This on acidic hydrolysis, deesterification with alkali and subsequent acidification furnished (R)-I. Its spectral and chiroptical data were similar to those reported [7a]. Based on the optical purity of the starting chiron and the literature data of the [α]D-value of (R)-I, the synthetic compound should be 93% optically pure.
Experimental Section
All the boiling points are uncorrected. The IR spectra were scanned with a Perkin-Elmer spectrophotometer model 837. The 1H-NMR spectra were recorded in CDCl3 with a Bruker AC-200 (200 MHz) instrument. The optical rotations were measured with a Jasco DIP 360 polarimeter. The GLC analyses were carried out using Shimadzu GC-7A chromatograph fitted with stainless steel column and flame ionization detector using 3% OV-17 (2 Mt. x 0.5 mm) column and a N2 flow rate 40 mL/min. Anhydrous reactions were carried out under Ar using freshly dried solvents. The organic extracts were dried over anhydrous Na2SO4. For enzymatic resolution PPL (Sigma, sp. act. 52.4 units/mg) was used as obtained.
1-tert-Butyldimethylsilyloxy-6-bromohexane (3)
A mixture of hexane-1,6-diol (11.8 g, 0.1 mol) and 48% aq. HBr (16.9 mL, 0.1 mol) in benzene (150 mL) was refluxed for 30 h with continuous azeotropic removal of water by a Dean-Stark apparatus. The mixture was washed with water, 10% aqueous NaHCO3, water, brine and dried. Removal of solvent followed by distillation gave pure 2. yield: 10.8 g (60%); bp: 88-89°C/2 mm, (lit. [9] bp: 88-90°C/2 mm); IR: 3350, 1480, 1460 cm-1; 1H-NMR: δ 1.2-2.1 (m, partially D2O exchangeable, 9H), 3.54 (t, J = 7 Hz, 2H), 3.72 (t, J = 7 Hz, 2H).
A mixture of 2 (1.81 g, 0.01 mol), TBSCl (1.66 g, 0.011 mol), triethylamine (1.53 mL, 0.011 mol) and DMAP (0.122 mg, 1 mmol) in CH2Cl2 (50 mL) was stirred at room temperature for 14 h. The mixture was poured into water, the organic layer separated and the aqueous layer extracted with CHCl3. The combined organic extract was washed with water, and brine, dried and concentrated. The crude product was purified by column chromatography (silica gel, 0-10% EtOAc/hexane) to give 3. Yield: 2.6 g (88%); IR: 1455, 1440 cm-1; 1H-NMR: δ 0.12 (s, 6H), 0.88 (s, 9H), 1.3-1.9 (m, 8H), 3.4-3.6 (m, 4H).
1-tert-Butyldimethylsilyloxynonan-8-one (5)
To a stirred solution of NaOMe [prepared from Na (0.343 g, 14.9 mmol) in anhydrous MeOH (20 mL)] was added 4 (1.73 g, 14.9 mmol). After the initial reaction subsided, the mixture was refluxed for 0.5 h, cooled to room temperature and the bromide 3 (4.0 g, 13.6 mmol) in MeOH (20 mL) added to it followed by NaI (0.1 g). The mixture was refluxed for 18 h, cooled to room temperature and concentrated in vacuo. Aqueous 2N NaOH (20 mL) was added to the residue and stirring continued for 12 h at room temperature. It was acidified to pH 6 with aqueous 2N HCl, extracted with ether, the ether layer washed with water and brine and finally dried. Removal of solvent followed by column chromatography (silica gel, 0-10% EtOAc-hexane) of the residue gave pure 5. Yield: 1.77 g (48%); IR: 1720, 1460, 1450 cm-1; 1H-NMR: δ 0.12 (s, 6H), 0.88 (s, 9H), 1.1-1.6 (m, 10H), 2.16 (s, 3H), 2.34 (t, J = 7 Hz, 2H), 3.59 (t, J = 6 Hz, 2H). Anal. Calcd. for C15H32O2Si: %C 66.11, %H 11.84; Found: %C 66.29, %H 12.06.
1-tert-Butyldimethylsilyloxynonan-8-ol (6)
To a cooled (0°C) and stirred solution of 5 (2.1 g, 7.72 mmol) in MeOH (30 mL) was added NaBH4 (0.15 g, 3.95 mmol) in portions. After 1 h, the reaction was quenched with solid NH4Cl, the mixture concentrated and the residue taken up in ether. The ether layer was washed with water and brine and dried. Removal of solvent under reduced pressure followed by column chromatography of the residue over silica gel (0-15% EtOAc/hexane) gave pure 6. Yield: 1.65 g (78%); IR: 3360, 1465, 1100 cm-1; 1H-NMR: δ 0.12 (s, 6H), 0.9 (s, 9H), 1.18 (d, J = 6 Hz, 3H), 1.3-1.7 (m, 12H), 2.36 (s, D2O exchangeable, 1H), 3.63 (t, J = 6 Hz, 2H), 3.7-3.9 (m, 1H). Anal. Calcd. for C15H34O2Si: %C 65.63, %H 12.48; Found: %C 65.48, %H 12.31.
(8R)-1-tert-Butyldimethylsilyloxy-8-butyroxynonane (7)
A mixture of (+)-6 (2.0 g, 7.30 mmol), TFEB (1.86 g, 10.9 mmol) and PPL (0.5 g) in diisopropyl ether (40 ml) was magnetically stirred. After 18 h, at 30% conversion (cf. GLC), the reaction mixture was filtered and the residue chromatographed (silica gel, 0-20% EtOAc/hexane) to obtain pure (R)-7. Yield: 0.652 g (26%); [α]D24 +14.18 (c 1.4, CHCl 3); IR: 1740, 1470, 1230 cm-1; 1H-NMR: δ 0.12 (s, 6H), 0.9 (s, 12H), 1.2 (d, J = 6 Hz, 3H), 1.3-1.6 (m, 14H), 2.3 (t, J = 6 Hz, 2H), 3.68 (t, J = 6 Hz, 2H), 4.0-4.3 (m, 1H). Anal. Calcd. for C19H40O3Si: %C 66.22, %H 11.70; Found: %C 66.08, %H 11.61. (S)-6: yield: 1.26 g (63%); [α]D24 -9.36 (c 0.7, CHCl3).
(8R)-8-Butyroxynonan-1-ol (8)
To a cooled (-78°C) and stirred solution of 7 (0.6 g, 1.74 mmol) in THF (10 mL) was added Bu4NF (1.74 mL, 1.74 mmol). The mixture was stirred at the same temperature for 3 h and then kept at 0°C for 12 h. It was diluted with water, extracted with ether and the ether extract washed with water and brine. Solvent removal and column chromatography of the residue (silica gel, 0-15% EtOAc/hexane) gave 8. Yield: 0.336 g (84%);[α]D24 +8.34 (c 0.86, CHCl3); IR: 3380, 1740, 1460, 1230 cm-1; 1H-NMR: δ 0.9 (dist. t, 3H), 1.16 (d, J = 6 Hz, 3H), 1.2-1.6 (m, 14H), 2.1 (s, D2O exchangeable, 1H), 2.4 (t, J = 6 Hz, 2H), 3.63 (t, J = 6 Hz, 2H), 4.1-4.3 (m, 1H). Anal. Calcd. for C13H26O3: %C 67.78, %H 11.38; Found: %C 66.86, %H 11.61.
(9R)-9-Butyroxynonanonitrile (9)
To a cooled (0°C) and stirred solution of 8 (0.336 g, 1.46 mmol) and pyridine (0.230 mL, 2.89 mmol) in CH2Cl2 (10 mL) was added p-TsCl (0.550 g, 2.89 mmol). After stirring for 2 h at the same temperature, the mixture was kept in the freezer for 12 h. It was poured into ice-water, the organic layer separated and the aqueous part extracted with CHCl3. The combined organic extract was washed with water, and brine and dried. Removal of solvent in vacuo followed by column chromatography (silica gel, 0-5% EtOAc/hexane) of the residue furnished the corresponding tosylate. yield: 0.500 g (89%).
A mixture of the above compound (0.5 g, 1.3 mmol) and KCN (0.254 g, 3.9 mmol) in DMSO (20 mL) was gently heated at 80°C for 18 h. After cooling to room temperature, the mixture was poured into a large excess of cold water and extracted with ether. The ether layer was thoroughly washed with water, and brine. After drying, the extract was concentrated and the crude product was purified by column chromatography (silica gel, 0-5% EtOAc/hexane) to furnish pure 9. Yield: 0.212 g (68%); IR: 2200, 1730, 1240 cm-1; 1H-NMR: δ 0.88 (dist. t, 3H), 1.16 (d, J = 6 Hz, 3H), 1.2-1.6 (m, 14H), 2.1-2.3 (m, 4H), 4.0-4.2 (m, 1H). Anal. Calcd. for C14H25O2N: %C 70.25, %H 10.53, %N 5.85; Found: %C 70.48, %H 10.61, %N 6.10.
(9R)-Phoracantholide I ((R)-I or 10)
A mixture of compound 9 (0.210 g, 0.88 mmol) and conc. HCl (2.0 mL) was stirred for 16 h at room temperature and subsequently at 80°C for 6 h. The mixture was extracted with CHCl3, the organic extract washed with water, brine and dried. After concentration, the resultant crude product was directly taken up in alcoholic KOH (5 mL) and stirred for 16 h. The mixture was concentrated in vacuo, the residue dissolved in water, acidified with conc. HCl to pH 2 and extracted with ether. The ether layer was washed with water, brine and dried. Solvent removal followed by preparative TLC (silica gel, 10% EtOAc/hexane) furnished pure (R)-I (10). Yield: 0.091 g (61%); [α]D24 -32.4 (c 0.7, CHCl3) (lit. [7a] [α]D22 -35.1 (c 1.15, CHCl3)); IR: 1720, 1260, 1240 cm-1; PMR: δ 1.18 (d, J = 6 Hz, 3H), 1.2-1.6 (m, 12H), 2.1-2.4 (m, 2H), 4.4-4.8 (m, 1H). Anal. Calcd. for C10H18O2: %C 70.55, %H 10.66; Found: %C 70.38, %H 10.81.
References
- Davies, H. G.; Green, R. H.; Kelly, D. R.; Roberts, S. M. Biotransformations in Preparative Organic Chemistry. The Use of Isolated Enzymes and Whole Cells Systems in Synthesis; Academic: London, 1989. [Google Scholar] Drauz, K.; Waldmann, H. Enzyme Catalysis In Organic Synthesis; VCH Verlagsgesellschaft mbH, Weinheim and VCH Publishers Inc.: New York NY, USA, 1995. [Google Scholar]
- Sharma, A.; Sankaranaryanan, S.; Kulkarni, B. A.; Chattopadhyay, S. J. Org. Chem. 1991, 56, 6823. Pawar, A. S; Sankaranarayanan, S.; Chattopadhyay, S. Tetrahedron Asymm. 1995, 6, 2219. Sharma, A.; Sankaranarayanan, S.; Chattopadhyay, S. J. Org. Chem. 1996, 61, 1814. Sankaranarayanan, S.; Sharma, A.; Chattopadhyay, S. Tetrahedron Asymm. 1996, 7, 2639. Sharma, A.; Sankaranarayanan, S.; Chattopadhyay, S. Enantiomer. (in press).
- Sharma, A.; Pawar, A. S.; Chattopadhyay, S. Synth. Commun. 1996, 26, 19.
- Moore, B. P.; Brown, W. V. Aust. J. Chem. 1976, 29, 1365.
- Kitahara, T.; Koseki, K.; Mori, K. Agric. Biol. Chem. 1983, 47, 389.
- Saikia, A. K.; Hazarika, M. J.; Barua, N. C.; Bezbarua, M. S.; Sharma, R. P.; Ghosh, A. C. Synthesis 1996, 981. and ref. cited therin.
- Kitahara, T.; Koseki, K.; Mori, K. Agric. Biol. Chem. 1983, 47, 389. Naoshima, Y.; Hasegama, H.; Nishiyama, J.; Nakamura, A. Bull Chem. Soc. Jpn. 1989, 62, 608. Nagumo, S.; Suemune, H.; Sakai, K. Tetrahedron 1992, 48, 8667.
- Kang, S. -K.; Kim, W. -S.; Moon, B. -H. Synthesis 1985, 1161. [CrossRef]
- Ishihara, T.; Yamamoto, A. Agric Biol. Chem . 1984, 48, 211.
- Keinan, E.; Hafeli, E. K.; Seth, K. K.; Lamed, R. J. Am. Chem. Soc. 1986, 108, 162.
- Sample Availability: Available from the authors.
© 1998 MDPI. All rights reserved. Molecules http://www.mdpi.org/molecules/
Share and Cite
MDPI and ACS Style
Sharma, A.; Chattopadhyay, S. A Short and Efficient Synthesis of (R)-Phoracantholide I. Molecules 1998, 3, 44-47. https://doi.org/10.3390/30200044
AMA Style
Sharma A, Chattopadhyay S. A Short and Efficient Synthesis of (R)-Phoracantholide I. Molecules. 1998; 3(2):44-47. https://doi.org/10.3390/30200044
Chicago/Turabian StyleSharma, A., and S. Chattopadhyay. 1998. "A Short and Efficient Synthesis of (R)-Phoracantholide I" Molecules 3, no. 2: 44-47. https://doi.org/10.3390/30200044