Results and Discussion
The α-tosyl substituted thioureas
1a-c and ureas
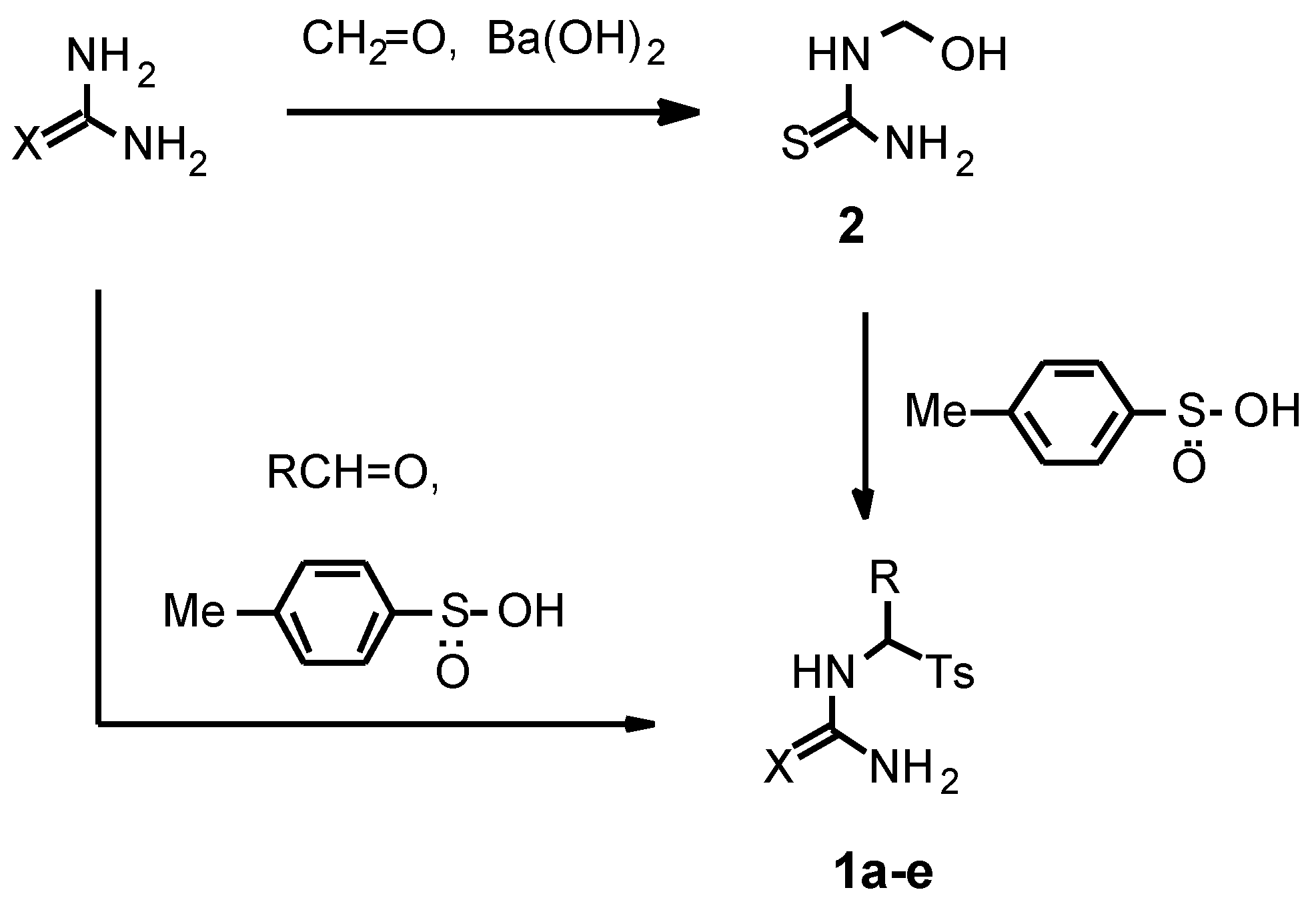
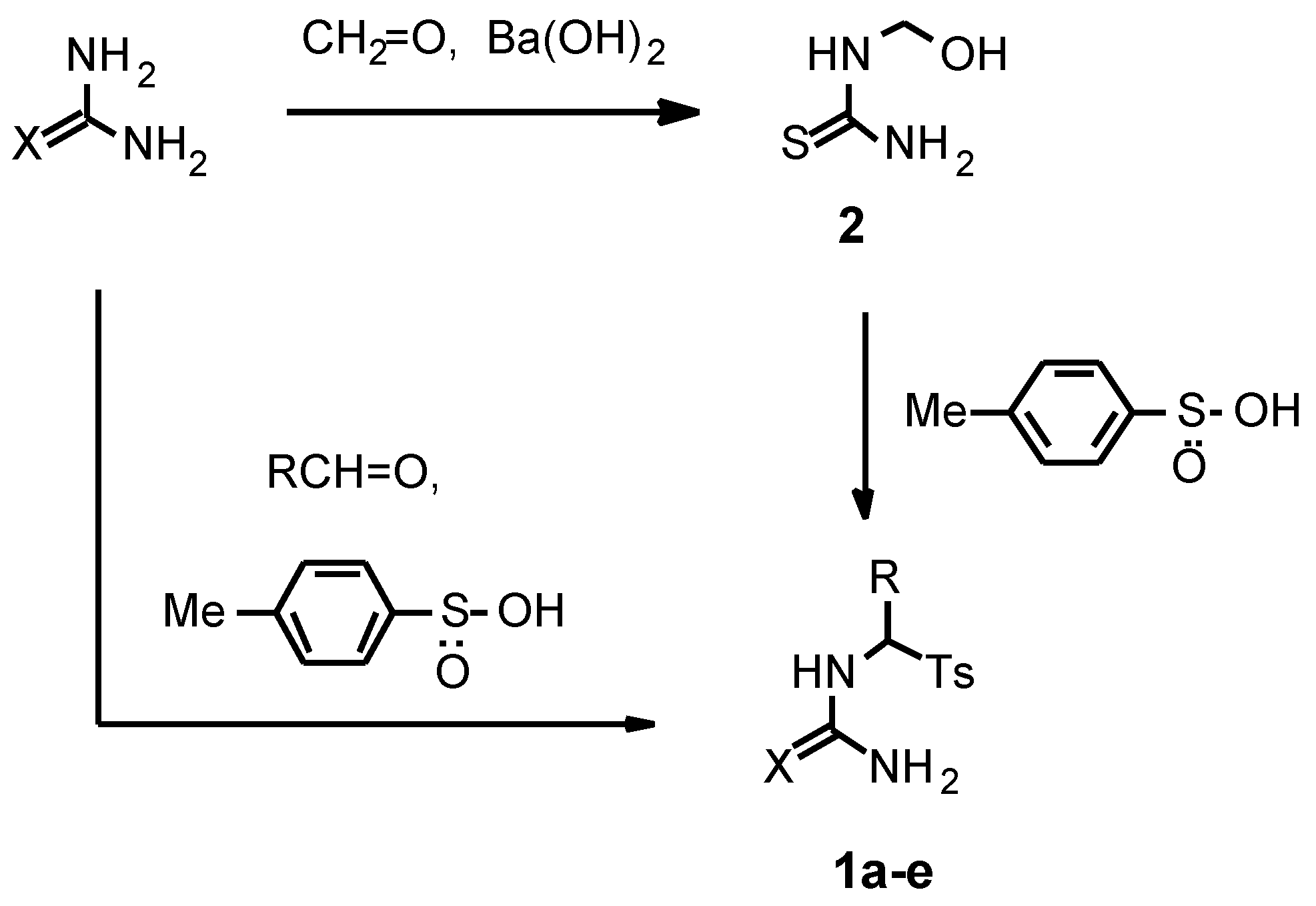
1d,e were chosen as starting compounds for the present investigation. These compounds were conveniently prepared in 1-2 steps from thiourea or urea in good yields using two different procedures (
Scheme 1).
N-(Tosylmethyl)thiourea 1a was synthesized by reaction of readily available hydroxymethylthiourea 2 with p-toluenesulfinic acid in water (r.t., 24 h) in 94 % yield.
For the synthesis of α-substituted
N-(tosylmethyl)-thioureas and ureas we used methods based on a three-component condensation of thiourea or urea with aldehydes and
p-toluenesulfinic acid. The main problem of the synthesis was the formation not only of the desired
N-monosubstituted (thio)ureas of the type
1 but also of
N,N’-disubstituted products. We showed that the amount of the latter depends on the molar ratio of the reagents, reaction conditions and the solvent [
8]. Thus
N-(1-tosyl-propyl)thiourea
1b and
N-(α-tosylbenzyl)thiourea
1c were synthesized by treatment of thiourea with
p-toluenesulfinic acid and propionic aldehyde or benzaldehyde in water at r.t. for 21 h in good yields using an equimolar ratio of the reagents (
Table 1). According to NMR spectra of the obtained products, the corresponding
N,N’-bis(1-tosylpropyl)thiourea and
N,N’-bis(α-tosylbenzyl)thiourea were also formed as by-products in these reactions. However, the amount of these disubstituted thioureas in relation to the monosubstituted products
1b,c under the above conditions was only 1-2.5 mol%.
We found that the reaction of urea with p-toluenesulfinic acid and propionic aldehyde or benzaldehyde in water afforded significantly more N,N’-bis-products than in the case of thiourea under the described above conditions. In order to decrease the formation of the bis-products we used a three-fold molar excess of urea and a short reaction time (2 h). Thus we prepared N-(1-tosylpropyl)urea 1d and N-(α-tosylbenzyl)urea 1e in 85-90 % yields. According to NMR spectra, the amount of N,N’-bis(1-tosylpropyl)urea and N,N’-bis(α-tosylbenzyl)urea in relation to the compounds 1d,e was 5.5 mol% and less than 1 mol%, respectively. The obtained α-tosyl substituted (thio)ureas 1a-e owing to their good purity (> 94 %) were used in the pyrimidine synthesis without further purification.
It should be noted, that earlier Engberts and co-workers demonstrated [
9] that the reaction of thiourea or urea with benzaldehyde and sodium
p-toluenesulfinate in the presence of an excess of formic acid (water-ethanol, r.t.) gave, respectively, a mixture of
1c and
N,N’-bis(α-tosylbenzyl)thiourea or a mixture of
1e (37 %) and
N,N’-bis(α-tosylbenzyl)urea (43 %). The authors were only able to isolate a
1c from these mixtures.
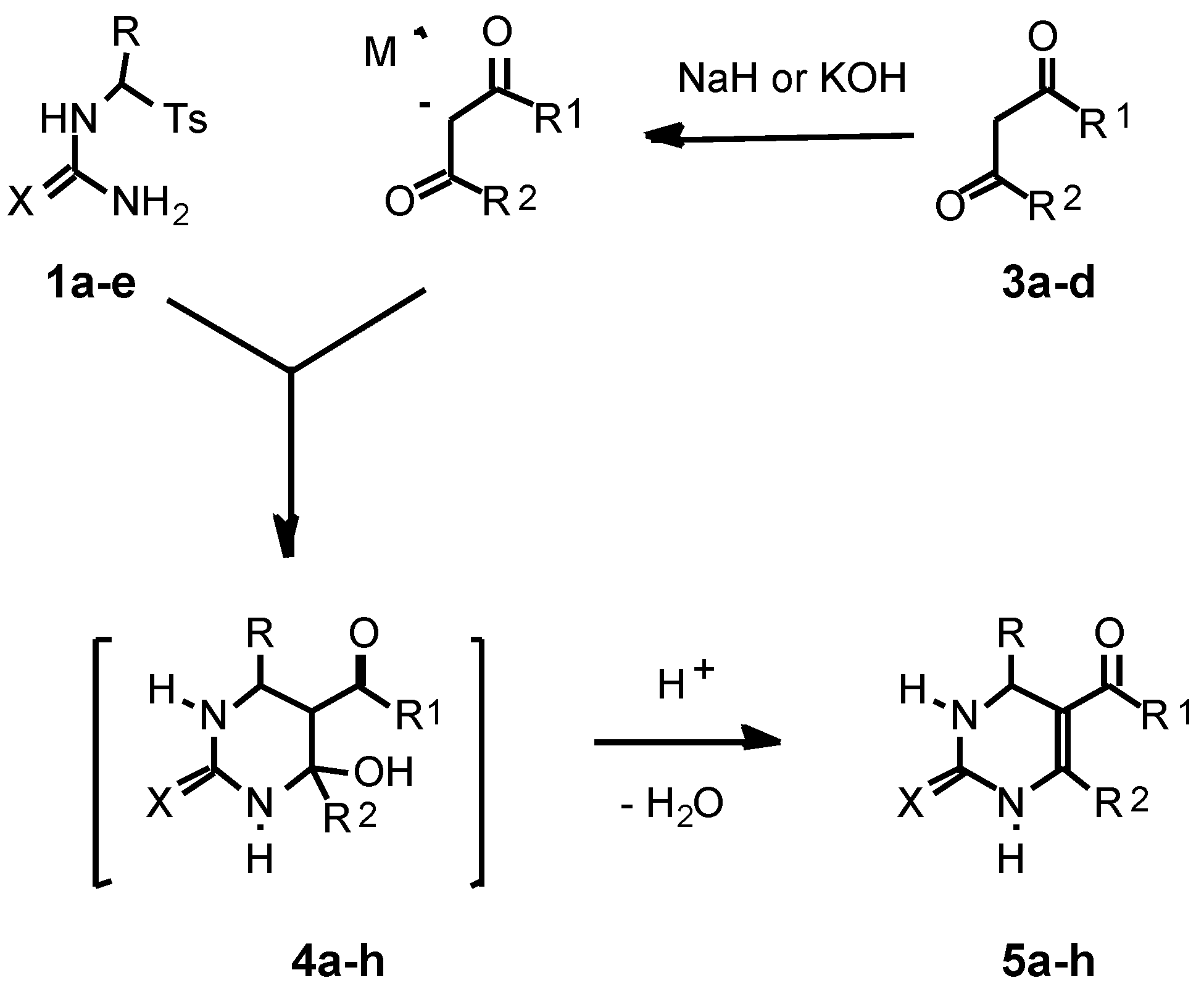
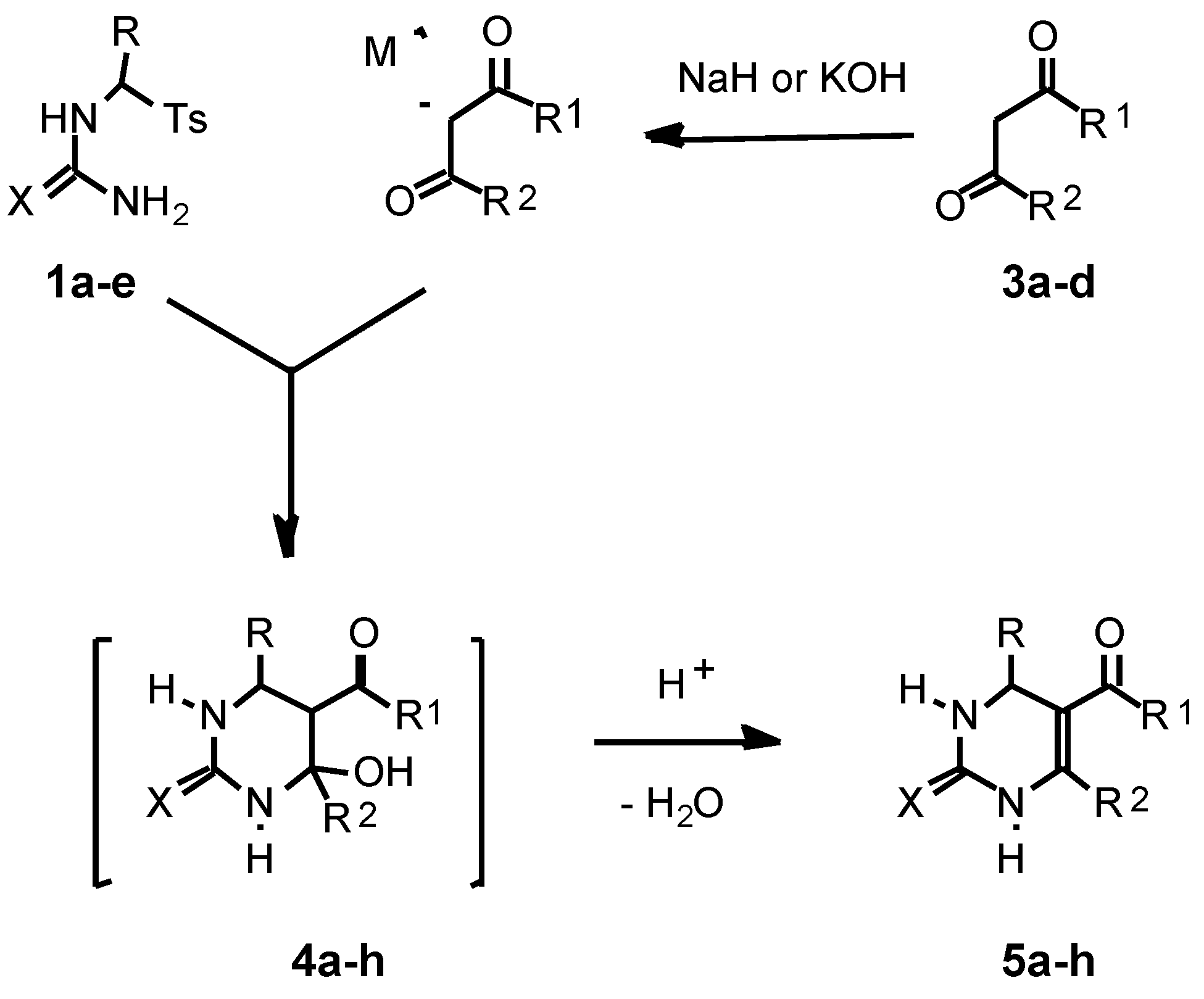
As a second building block for the pyrimidine synthesis in the present work we used β-oxoesters 3a,b or 1,3-dicar-bonyl compounds 3c,d.
For the first time we studied the reaction of the thioureas
1a-c with the sodium enolate of ethyl acetoacetate obtained previously from
3a or generated
in situ by treatment of
3a with NaH in acetonitrile. We found that the reaction of
1a with the sodium enolate of
3a (1.15 equiv.) easily proceeded in acetonitrile at r.t. for 3 h to afford 4-hydroxyhexahydropyrimidine-2-thione
4a which was dehydrated after the addition of TsOH (0.3 equiv.) followed by refluxing of the reaction mixture for 1.5 h. Thus we obtained the target 1,2,3,4-tetrahydro-pyrimidine-2-thione
5a in 86 % yield and in a spectroscopically and TLC pure form (Method A) (
Scheme 2). It should be noted that a workable catalyst for the dehydration is
p-toluenesulfinic acid which is generated by the reaction of TsOH with sodium
p-toluenesulfinate obtained in the first stage of the synthesis.
Similarly, the thioureas 1b,c reacted with the sodium enolate of 3a in acetonitrile (r.t., 3 h for 1b and r.t., 3h, then reflux, 2 h for 1c) to produce the hydroxypyrimidines 4e,f which without their isolation were dehydrated into 1,2,3,4-tetrahydropyrimidine-2-thiones 5e,f in 74-86% yields after addition of TsOH to the reaction mixtures (reflux, 1.5 h).
The described approach was also applied to the one-pot synthesis of 1,2,3,4-tetrahydropyrimidine-2-ones
5g,h by reaction of the ureas
1d,e with the sodium enolate of
3a in acetonitrile followed by acidification and refluxing of the reaction mixtures. The yield of the pyrimidines
5g,h was 73 and 65 %, correspondingly (
Table 2).
We also found hat for the pyrimidine synthesis potassium hydroxide in ethanol can be effectively used for generation of enolates from CH acids
3a-d instead of NaH in acetonitrile. Thus reaction of the tosylthiourea
1a with the potassium enolates of
3a-d (1.2 equiv. for
3a,c and 1.05 equiv. for
3b,d) obtained by treatment of
3a-d with KOH in ethanol afforded (r.t., 4.5 h) the corresponding hydroxypyrimidine-2-thiones
4a-d which were dehydrated after addition of TsOH (0.27 equiv. for
4a,c,d and 1.08 equiv. for
4b) and refluxing of the reaction mixtures for 1.5 h to give the tetrahydropyrimidine-2-thiones
5a-d in 71-87% yields (
Table 2) (Method B). Analogously, we prepared the pyrimidine
5e starting from
1b and
3a in 77 % yield.
A comparison of Method A and Method B shows that, even though both methods give similar yields of the target pyrimidines (
Table 2), Method B is preparatively more convenient.
Experimental
General
IR spectra (in Nujol) were recorded on a Shimadzu IR 435 spectrophotometer. Peak intensities in the IR spectra are defined as strong (s), medium (m) or weak (w). UV spectra (in methanol) were obtained on a Beckman DU 6 spectrophotometer. 1H NMR (200.13 MHz) spectra were recorded on a Bruker MSL 200 spectrometer using DMSO-d6 or CDCl3 as solvents. Chemical shifts (δ) are given in ppm relative to TMS. Multiplicities are reported as singlet (s), doublet (d), triplet (t), quartet (q), some combination of these, multiplet (m). Thin layer chromatography (TLC) was performed on silica gel plates Silufol UV 254 (Czech Republic) or Kieselgel 60 F254 (Merck) in chloroform-methanol (20:1, v/v) and chloroform-methanol (9:1, v/v) as solvent systems. Plates were visualised with iodine vapor or UV light.
p-Toluenesulfinic acid was synthesized by reduction of tosyl chloride with sodium sulfite in water [
13], dried over P
2O
5 and stored at 0°C. Hydroxymethylthiourea 2 was prepared according to the literature procedure [
14]. Propionic aldehyde and benzaldehyde were distilled before use. For the pyrimidine synthesis we used commercially available ethanol (96 %) and anhydrous acetonitrile dried by distillation from P
2O
5 and then from CaH
2. Ethyl acetoacetate and acetylacetone were dried over CaCl
2 and distilled under vacuum, ethyl benzoylacetate and benzoylacetone were commercially available products. Sodium hydride (80 % suspension in mineral oil) was washed with dry hexane, dried in vacuum desiccator prior to use. The sodium enolate of ethyl acetoacetate was obtained by reaction of ethyl acetoacetate with NaH in dry acetonitrile followed by filtration of the resulting product. α-Tosyl substituted (thio)ureas
1a-e were used in the syntheses freshly prepared because these compounds (especially
1b-e) decomposed slightly during prolonged storage.
The obtained tetrahydropyrimidine-2-thiones/ones
5a-h were identical (TLC, IR,
1H NMR) with authentic samples prepared according to the literature procedures [
4,
6,
7]. All yields refer to isolated, spectroscopically and TLC pure material.
N-(Tosylmethyl)thiourea (1a)
A mixture of hydroxymethylthiourea 2 (1.356 g, 12.77 mmol) and p-toluenesulfinic acid (2.399 g, 15.36 mmol) in 15 mL of water was stirred at r.t. for 24 h, then cooled to 0 °C. The solid was collected by filtration, washed carefully with ice water, hexane and dried to give 2.943 g (94 %) of 1a as a white powder. The obtained product was used in the pyrimidine syntheses without further purification. Analytically pure material was obtained by recrystallisation from acetone, mp 156.5-157 °C.
IR 3392 br m, 3291 br s, 3180 br m, 3077 w, 3041 w, 1608 s, 1548 s, 1271 s, 1133 s, 802 m cm-1.
1H NMR (DMSO-d6) 2.41 (3H, s, CH3), 5.13 (2H, br d, J ~ 6 Hz, CH2), 7.08-7.75 (2H, NH2, the signals are covered under Harom), 7.43 (2H, d, J = 8.3 Hz, Harom), 7.71 (2H, d, J = 8.3 Hz, Harom), 8.30 (1H, br t, J ~ 6 Hz, NH).
Anal. Calcd for C9H12N2O2S2: C, 44.22; H, 4.95; N, 11.51. Found: C, 44.19; H, 4.96; N, 11.62.
N-(1-Tosylpropyl)thiourea (1b)
To a mixture of propionic aldehyde (0.519 g, 8.94 mmol) and water (7 mL) was added thiourea (0.648 g, 8.51 mmol) and then p-toluenesulfinic acid (1.397 g, 8.94 mmol). The resulting suspension was stirred at r.t. for 21 h, cooled to 0 °C, the solid was collected by filtration, washed carefully with ice water, hexane and dried to give 2.192 g (95 %) of 1b as a white solid. According to the NMR spectrum, the crude product also contained about 1 mol% of N,N’-bis(1-tosylpropyl)thiourea. The obtained product was stored at 0°C and used in the pyrimidine syntheses without further purification. Analytically pure material was obtained by recrystallisation from acetonitrile, mp 123.5-124.5 °C dec.
IR 3413 br s, 3315 br s, 3185 br s, 3044 br s, 1606 s, 1570 s, 1492 w, 1301 s, 1139 s, 808 m cm-1.
1H NMR (DMSO-d6) 0.93 (3H, t, J = 7.3 Hz, CH2CH3), 1.69 (1H, m, J = 14.0, 10.2, 7.3 Hz, CH2CH3), 2.08 (1H, m, J = 14.0, 7.3, 3.3 Hz, CH2CH3), 2.39 (3H, s, CH3), 5.71 (1H, dt, J = 10.2, 10.2, 3.3 Hz, N-CH), 6.98 (1H, br s, NH2), 7.41 (2H, d, J = 8.1 Hz, Harom), 7.68 (2H, d, J = 8.1 Hz, Harom), 7.72 (1H, br s, NH2), 8.33 (1H, d, J = 10.2 Hz, NH).
Anal. Calcd for C11H16N2O2S2: C, 48.51; H, 5.92; N, 10.29. Found: C, 48.61; H, 5.93; N, 10.41.
N-(α-Tosylbenzyl)thiourea (1c)
This compound (2.750 g, 83 %) was obtained as a white powder by the reaction of thiourea (0.786 g, 10.32 mmol), benzaldehyde (1.095 g, 10.32 mmol) and
p-toluenesulfinic acid (1.612 g, 10.32 mmol) in 8 mL of water according to the procedure described for
1b. According to the NMR spectrum, the crude product also contained 2.5 mol% of
N,N’-bis(α-tosylbenzyl)thiourea. The obtained product was stored at 0 °C and used in the pyrimidine syntheses without further purification. Analytically pure material was obtained by recrystallisation from acetonitrile, mp 139.5-140.5 °C (lit. mp 153-154 °C (ethanol) [
9]).
IR 3347 br m, 3270 br s, 3168 br m, 1609 s, 1530 s, 1493 w, 1285 s, 1135 s, 802 s, 736 s, 697 s cm-1.
1H NMR (DMSO-d6) 2.40 (3H, s, CH3), 6.89 (1H, d, J = 10.5 Hz, N-CH), 7.43 (2H, d, J = 8.1 Hz, Harom), 7.73 (2H, d, J = 8.1 Hz, Harom), 6.95-8.00 (5H, m, C6H5), ~ 7.08 (1H, br s, NH2), ~ 7.87 (1H, br s, NH2), 9.14 (1H, d, J = 10.5 Hz, NH).
Anal. Calcd for C15H16N2O2S2: C, 56.23; H, 5.03; N, 8.74. Found: C, 55.86; H, 4.98; N, 8.98.
N-(1-Tosylpropyl)urea (1d)
To a mixture of propionic aldehyde (2.124 g, 36.57 mmol) and water (40 mL) was added p-toluenesulfinic acid (5.719 g, 36.61 mmol) followed by the addition of urea (6.594 g, 109.80 mmol). The resulting suspension was stirred at r.t. for 2 h, cooled to 0 °C, the solid was filtered, washed with ice water, hexane and dried to yield 7.942 g (85 %) of 1d as a white powder. According to the NMR spectrum, the crude product also contained 5.5 mol% of N,N’-bis(1-tosylpropyl)urea. The obtained product was stored at 0 °C and used in the pyrimidine syntheses without further purification.
IR 3415 s, 3381 m, 3191 br s, 1676 s, 1622 m, 1596 m, 1518 s, 1279 s, 1133 s, 811 m cm-1.
1H NMR (DMSO-d6) 0.91 (3H, t, J = 7.3 Hz, CH2CH3), 1.54 (1H, m, J = 13.8, 10.5, 7.3 Hz, CH2CH3), 2.01 (1H, m, J = 13.8, 7.3, 3.4 Hz, CH2CH3), 2.39 (3H, s, CH3), 4.81(1H, dt, J = 10.3, 10.5, 3.4 Hz, N-CH), 5.65 (2H, br s, NH2), 6.84 (1H, d, J = 10.3 Hz, NH), 7.40 (2H, d, J = 8.2 Hz, Harom), 7.68 (2H, d, J = 8.2 Hz, Harom).
N-(α-Tosylbenzyl)urea (1e)
This compound (10.611 g, 90 %) was obtained as a white powder by the reaction of urea (6.995 g, 116.48 mmol), benzaldehyde (4.119 g, 38.81 mmol) and p-toluenesulfinic acid (6.074 g, 38.88 mmol) in 40 mL of water according to the procedure described for 1d. According to the NMR spectrum, the crude product contained less than 1 mol% of N,N’-bis(α-tosylbenzyl)urea. The obtained product was stored at 0 °C and used in the pyrimidine syntheses without further purification.
IR 3465 br m, 3338 br s, 3180 br m, 1667 s, 1595 s, 1533 s, 1493 w, 1283 s, 1142 s, 812 m, 692 m cm-1.
1H NMR (DMSO-d6) 2.40 (3H, s, CH3), 5.77 (2H, br s, NH2), 6.10 (1H, d, J = 10.6 Hz, N-CH), 7.41 (2H, d, J = 8.1 Hz, Harom), 7.70 (2H, d, J = 8.1 Hz, Harom), 7.30-7.82 (5H, m, C6H5), 7.73 (1H, d, J = 10.6 Hz, NH).
5-Acyl-1,2,3,4-tetrahydropyrimidine-2-thiones/ones; General Procedures
Method A
To a stirred suspension of NaH (0.257 g, 10.71 mmol) in dry acetonitrile (10 mL) at 0 °C was added a solution of ethyl acetoacetate
3a (1.394 g, 10.71 mmol) in dry acetonitrile (10 mL) dropwise over a period of 5 min and the resulting mixture was stirred at r.t. for 30 min. To the obtained suspension of the sodium enolate of ethyl acetoacetate was added the appropriate (thio)urea
1 (9.31 mmol) all at once. After stirring for 3 h at r.t. (for
1a,b,d) or for 3 h at r.t. and then 2 h under reflux (for
1c,e), the reaction mixture was acidified by addition of TsOH.H
2O (0.532 g, 2.80 mmol) and refluxed for 1.5 h. The solvent was removed under reduced pressure and the solid residue was treated with water (10 mL). The mixture was cooled to 0 °C over 1 h, the precipitate was filtered, washed carefully with ice water, hexane and dried to give the corresponding pyrimidine
5 (
Table 2).
Analogously and in similar yields the pyrimidines 5a,e-h were also obtained by modification of Method A based on the reaction of an appropriate (thio)urea 1 (3.71 mmol) with the previously prepared sodium enolate of ethyl acetoacetate (0.648 g, 4.26 mmol) in acetonitrile (18 mL) followed by addition of TsOH.H2O (0.285 g, 1.50 mmol).
Method B
To a stirred solution of KOH (0.308 g, 5.49 mmol) in ethanol (10 mL) was added a solution of CH acid
3 (5.49 mmol for
3a,c or 4.81 mmol for
3b,d) in ethanol (15 mL) at r.t. in one portion and the resulting mixture was stirred for 15 min. Tosylthiourea
1a or
1b (4.58 mmol) was added all at once. After stirring for 4.5 h at r.t., the reaction mixture was acidified by addition of TsOH.H
2O (0.235 g, 1.24 mmol in the case of
3a,c,d or 0.942 g, 4.95 mmol in the case of
3b) and refluxed for 1.5 h. The solvent was removed under reduced pressure and the solid residue was treated with water (5 mL). The mixture was cooled to 0 °C over 1 h, the precipitate was filtered, washed carefully with ice water, hexane and dried to give the corresponding pyrimidine
5 (
Table 2).
Ethyl 6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5a)
Yield 86 % (Method A) and 87 % (Method B), colorless crystals, mp 236-237 °C dec (methanol).
IR 3194 br s, 3152 br s, 1716 s, 1662 s, 1615 s, 1595 m, 1505 s, 1274 s, 1205 s, 1104 s cm-1.
UV λmax (log ε) 206 (4.11), ~280 sh, 306 nm (4.18).
1H NMR (DMSO-d6) 1.19 (3H, t, J = 6.9 Hz, OCH2CH3), 2.18 (3H, s, CH3), 3.89 (2H, s, N-CH2), 4.08 (2H, q, J = 6.9 Hz, OCH2CH3), 8.79 (1H, br s, NH), 9.78 (1H, br s, NH).
Anal. Calcd for C8H12N2O2S: C, 47.98; H, 6.04; N, 13.99; S, 16.01. Found: C, 47.73; H, 5.92; N, 14.01; S, 16.00.
Ethyl 6-phenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5b)
The crude product (Method B) was washed with a small portion of cold diethyl ether (-15 °C), yield 71 %, pale yellow crystals, mp 196-197 °C (ethanol).
IR 3303 br m, 3174 br s, 1665 s, 1581 s, 1292 s, 1187 s, 1138 s, 760 s, 695 s cm-1.
UV λmax (log ε) 208 (4.43), 244 sh, ~284 sh, 310 nm (4.19).
1H NMR (CDCl3) 0.92 (3H, t, J = 7.1 Hz, OCH2CH3), 3.93 (2H, q, J = 7.1 Hz, OCH2CH3), 4.26 (2H, d, J = 1.8 Hz, N-CH2), 7.26-7.44 (5H, m, C6H5), 7.49 (1H, br s, NH), 7.75 (1H, br s, NH).
Anal. Calcd for C13H14N2O2S: C, 59.52; H, 5.38; N, 10.68; S, 12.22. Found: C, 59.03; H, 5.42; N, 10.48; S, 11.88.
5-Acetyl-6-methyl-1,2,3,4-tetrahydropyrimidine-2-thione (5c)
Yield 74 % (Method B), colorless crystals, mp 230-230.5 °C dec (ethanol).
IR 3274 br s, 3180 br s, 3128 br s, 1647 sh, 1613 s, 1592 m, 1189 s, 1035 s cm-1.
UV λmax (log ε) 207 (3.99), ~290 sh, 325 nm (4.19).
1H NMR (DMSO-d6) 2.17 (6H, s, CH3 and CH3C=O), 3.96 (2H, s, N-CH2), 8.90 (1H, br s, NH), 9.88 (1H, br s, NH).
Anal. Calcd for C7H10N2OS: C, 49.39; H, 5.92; N, 16.46. Found: C, 49.00; H, 5.82; N, 16.52.
5-Benzoyl-6-methyl-1,2,3,4-tetrahydropyrimidine-2-thione (5d)
The crude product (Method B) was washed with a small portion of cold diethyl ether (-15 °C), yield 74 %, pale yellow crystals, mp 227.5-228 °C dec (ethanol).
IR 3282 br s, 3172 br s, 3108 br s, 1651 m, 1606 s, 1590 s, 1200 s, 732 m, 702 m cm-1.
UV λmax (log ε) 207 (4.37), 253 (4.05), ~288 sh, 336 nm (4.22).
1H NMR (DMSO-d6) 1.72 (3H, s, CH3), 3.91 (2H, s, N-CH2), 7.40-7.60 (5H, m, C6H5), 8.97 (1H, br s, NH), 10.00 (1H, br s, NH).
Anal. Calcd for C12H12N2OS: C, 62.05; H, 5.21; N, 12.06. Found: C, 62.00; H, 5.18; N, 12.22.
Ethyl 4-ethyl-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5e)
Yield 74 % (Method A) and 77 % (Method B), colorless crystals, mp 150.5-151.5 °C (ethanol).
IR 3316 br s, 3182 br s, 3109 br m, 1656 s, 1575 s, 1276 s, 1188 s, 1122 s cm-1.
UV λmax (log ε) 207 (4.04), 304 nm (4.22).
1H NMR (CDCl3) 0.91 (3H, t, J = 7.5 Hz, CH2CH3), 1.27 (3H, t, J = 7.2 Hz, OCH2CH3), 1.45-1.72 (2H, m, CH2CH3), 2.30 (3H, s, CH3), 4.16 (1H, dq, J = 11.0, 7.2 Hz, OCH2CH3), 4.19 (1H, dq, J = 11.0, 7.2 Hz, OCH2CH3), 4.32 (1H, ddd, N-CH), 7.60 (1H, br s, NH), 8.10 (1H, br s, NH).
Anal. Calcd for C10H16N2O2S: C, 52.61; H, 7.06; N, 12.27; S, 14.04. Found: C, 52.52; H, 7.15; N, 12.34; S, 13.90.
Ethyl 6-methyl-4-phenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5f)
Yield 86 % (Method A), colorless crystals, mp 212-213 °C (ethanol) (lit. mp 207 °C (ethanol) [
10]).
UV λmax (log ε) 209 (4.31), 308 nm (4.33).
IR 3324 br s, 3159 br s, 3088 br m, 1667 s, 1573 s, 1281 s, 1198 s, 1178 s, 1117 s, 758 s, 718 s, 688 s cm-1.
1H NMR (CDCl3) 1.14 (3H, t, J = 7.2 Hz, OCH2CH3), 2.34 (3H, s, CH3), 4.05 (1H, dq, J = 10.8, 7.2 Hz, OCH2CH3), 4.08 (1H, dq, J = 10.8, 7.2 Hz, OCH2CH3), 5.38 (1H, d, J = 3.2 Hz, N-CH), 7.22-7.37 (5H, m, C6H5), 7.86 (1H, br s, NH), 7.86 (1H, br s, NH).
Anal. Calcd for C14H16N2O2S: C, 60.85; H, 5.84; N, 10.14; S, 11.60. Found: C, 60.65; H, 5.84; N, 10.28; S, 11.36.
Ethyl 4-ethyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5g)
Yield 73 % (Method A), colorless crystals, mp 184-185 °C (ethanol) (lit. mp 184-185 °C (ethanol)[
11]).
IR 3248 br s, 3118 br s, 1730 s, 1706 s, 1676 s, 1647 s, 1285 s, 1237 s, 1119 s cm-1.
1H NMR (DMSO-d6) 0.80 (3H, t, J = 7.3 Hz, CH2CH3), 1.19 (3H, t, J = 7.0 Hz, OCH2CH3), 1.43 (2H, m, CH2CH3), 2.17 (3H, s, CH3), 3.99-4.10 (1H, m, N-CH, the signals are partly overlapped with OCH2), 4.05 (1H, dq, J = 10.8, 7.0 Hz, OCH2CH3), 4.08 (1H, dq, J = 10.8, 7.0 Hz, OCH2CH3), 7.20 (1H, br s, NH), 8.84 (1H, br s, NH).
Anal. Calcd for C10H16N2O3: C, 56.59; H, 7.60; N, 13.20. Found: C, 56.72; H, 7.38; N, 13.27.
Ethyl 6-methyl-4-phenyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5h)
Yield 65 % (Method A), colorless crystals, mp 212.5-213.5 °C (ethanol) (lit. mp 203-204 °C [
11], 202-204 °C [
12], 206-208 °C [
15]).
IR 3228 br s, 3100 br s, 1728 s, 1702 s, 1645 s, 1600 w, 1287 s, 1226 s, 1091 s, 754 s, 695 s, cm-1.


{kind=link}
{kind=link}