Synthesis of Novel Fused Heterocycles Based on Pyrano[2,3-c]pyrazole
National Research Centre, Dokki, Cairo, Egypt
Molecules 1998, 3(3), 71-79; https://doi.org/10.3390/30300071
Submission received: 30 October 1997
/
Accepted: 18 January 1998
/
Published: 23 February 1998
Abstract
:3-Substituted 1,2,4-triazole derivatives (5, 9, 13, 16b, 19), pyrimidinium salts (21), triazines (24), and aryl methylene hydrazono pyrazolopyrano-pyrimidine (27a,b) which are rearranged in basic medium to 30a,b, were afforded via the reaction of 6-amino 1,4,5,6-tetrahydro-5-imino-3-methyl-1-phenyl-4 (p-chlorophenyl) pyrazolo [4', 3' : 5,6]-pyrano [2,3-d] pyrimidine (3) with different reagents.
Introduction
Condensed pyrazoles are biologically interesting compounds and their chemistry has received considerable attention [1,2]. Several pyrano[2,3-c]pyrazoles are reported to have useful biological effects, such as analgesic and anti-inflammatory activities [3]. Moreover, the biological activity of fused azoles has led to intensive research on their synthesis [4,5,6].
Results and Discussion
In the present study, we report on the synthesis of pyrazolo [4`, 3` : 5,6] pyrano [3,2-e] [1,2,4]-triazolo [1,5-c] pyrimidine and pyrazolo [4``, 3`` : 4`,5`] pyrano [2` 3` : 4,5] pyrimido [1,6-b] [1,2,4]-triazine derivatives via the reaction of 6-amino-1,4,5,6-tetrahydro-5-imino-3-methyl-1-phenyl-4-(p-chlorophenyl) pyrazolo [4`, 3` : 5,6] pyrano [2,3-d] pyrimidine (3) with different reagents.
Scheme 1.
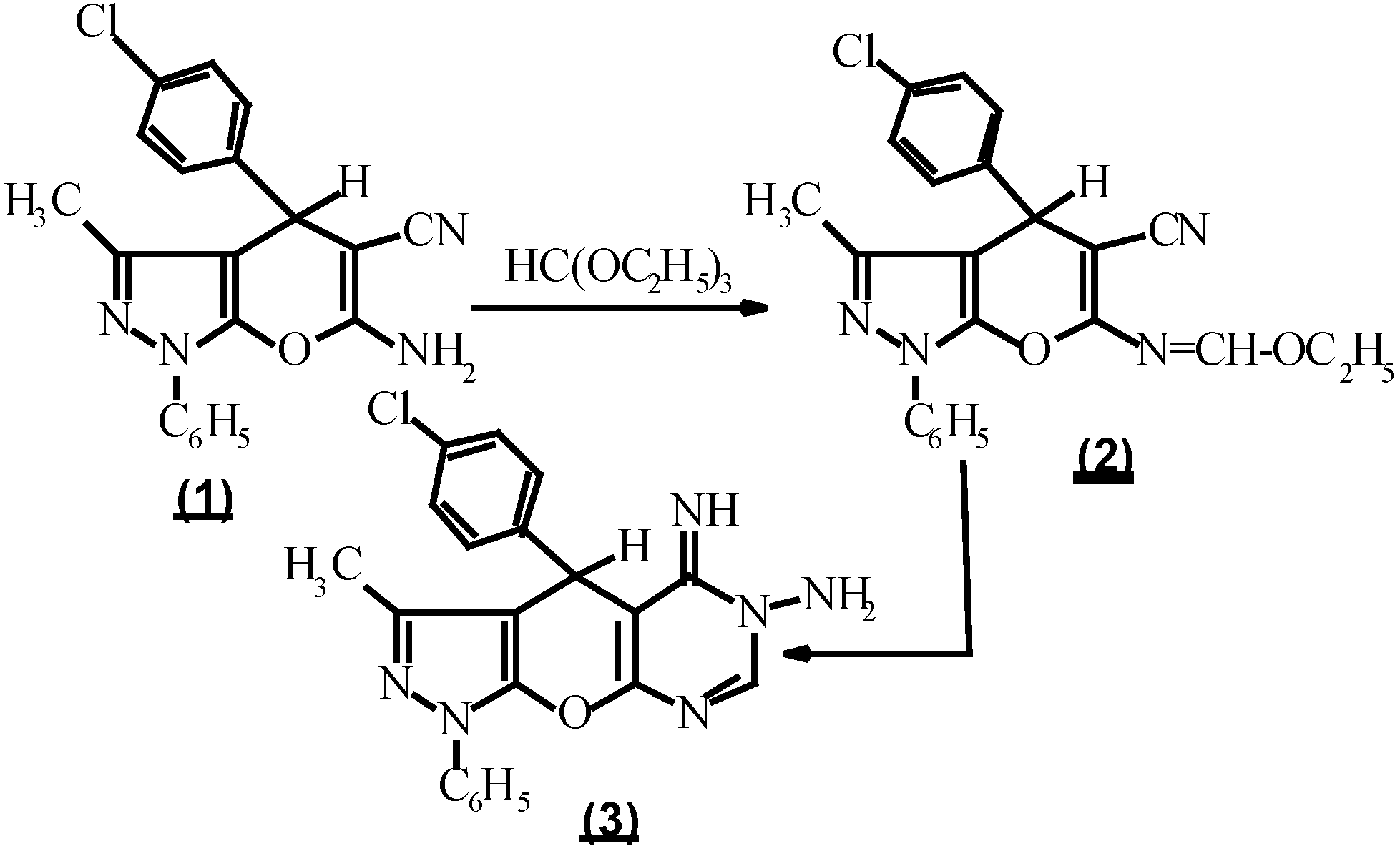
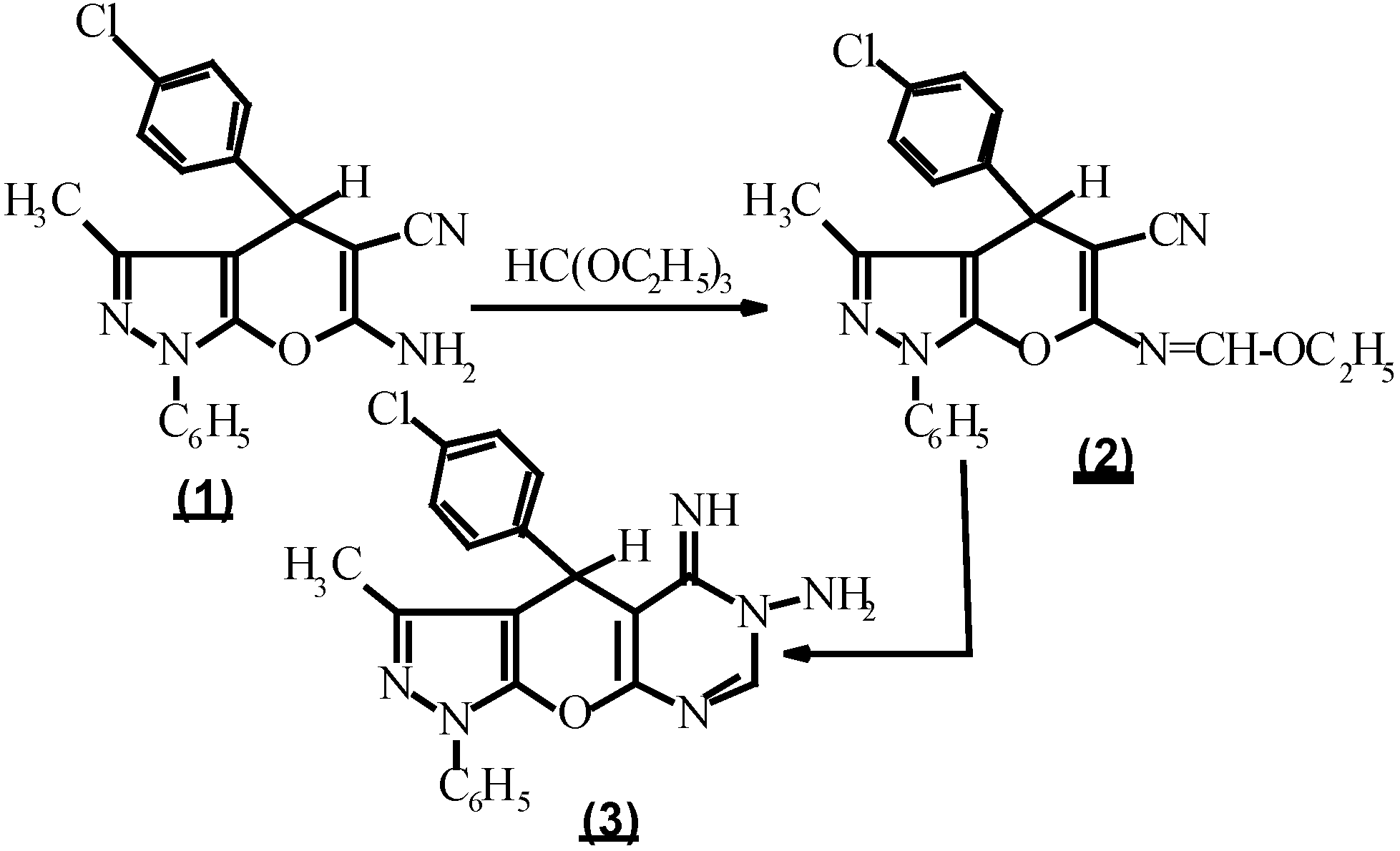
Compound 3, considered as a key intermediate to prepare fused heterocycles as triazolo [1,5-c] pyrimidines which may possess pharmacological properties similar to theophylline [7,8], was prepared by treating ethyl N-(5-cyano-1,4-dihydro-3-methyl-1-phenyl-4-(p-chlorophenyl)-pyrano-[2,3-c] pyrazol-6-yl-methan-imidate (2) with hydrazine hydrate [9,10]. On the other hand, compound 2 was prepared by the reaction of 6-amino-5-cyano-1,4-dihydro-3-methyl-1-phenyl-4 (p-chlorophenyl) pyrano [2,3-c] pyrazole (1) [11,12] with an equimolar amount of triethyl orthoformate and acetic anhydride [9,10] (Scheme 1).
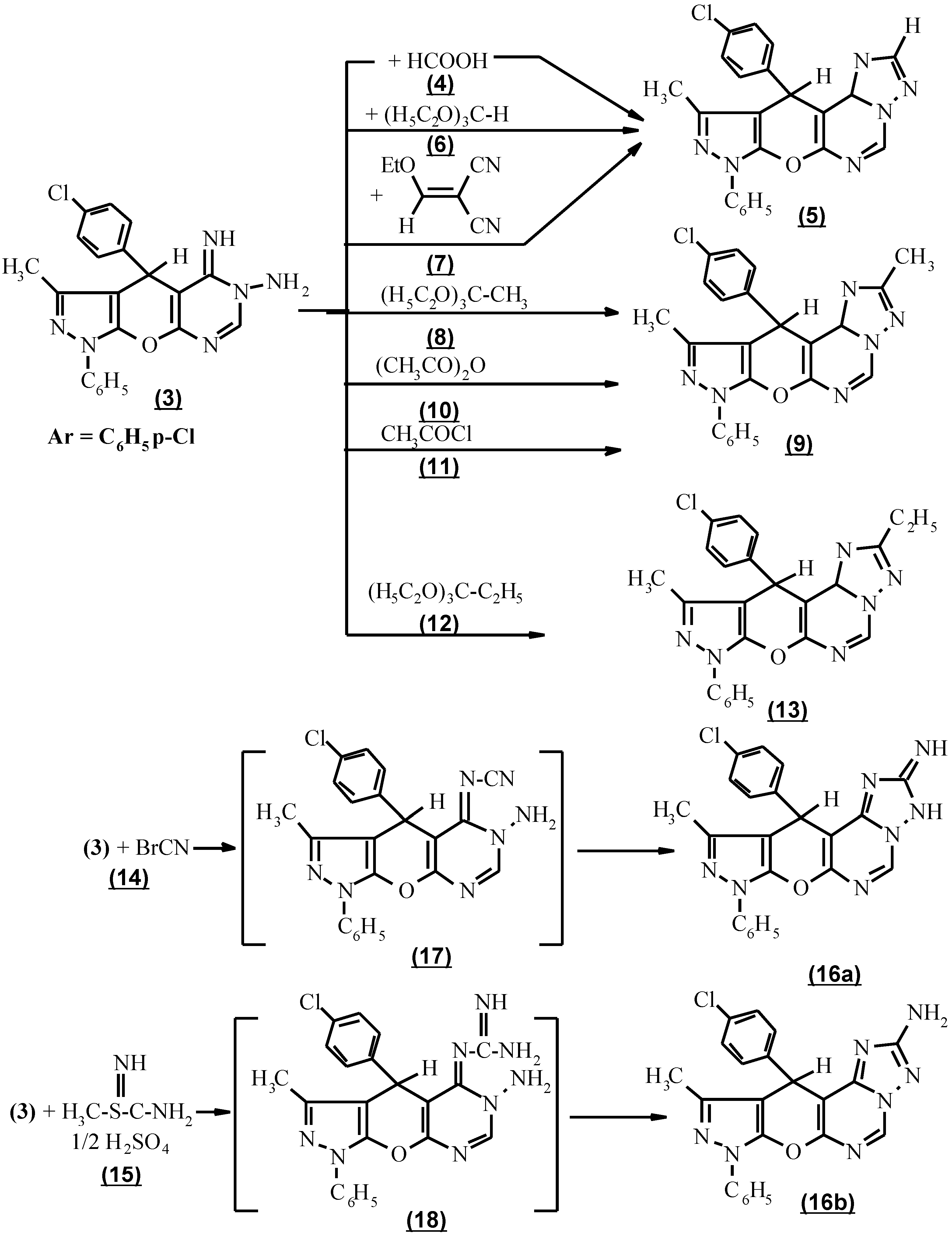
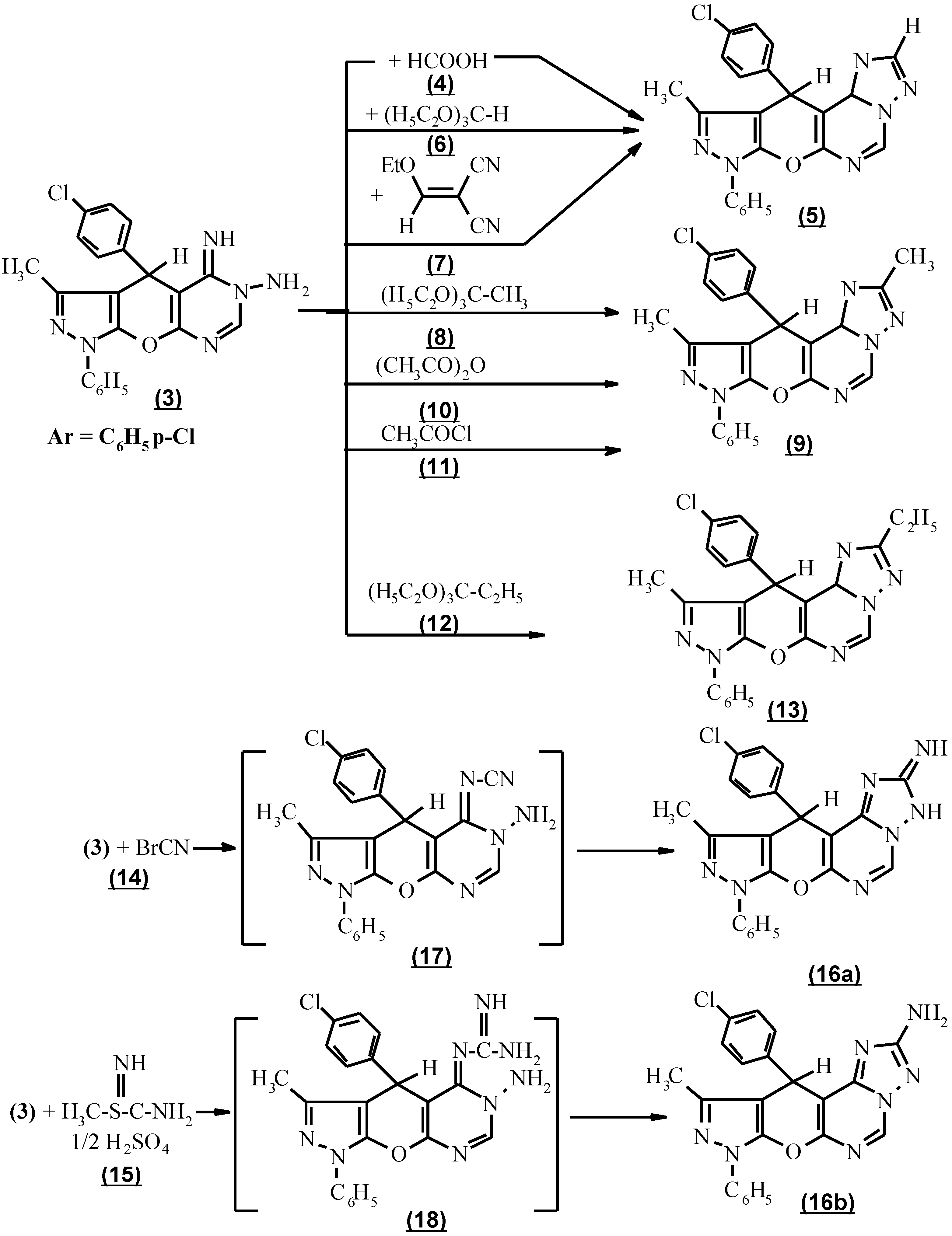
The cyclocondensation of compound 3 with the appropriate carboxylic acid derivatives was performed by heating compound 3 with an excess of neat formic acid 4 to afford 8,11-dihydro-10-methyl-8-phenyl-11-(p-chlorophenyl) [4`,3` : 5`,6`] pyrano [3,2-e] [1,2,4]-triazolo [1,5-c] pyrimidine (5) in a 65% combined yield. An improvement in the preparation of this compound was achieved using triethylorthoformate (6) by refluxing neat at 140°C, or by reacting of compound (3) with ethoxymethelyene malononitrile (7) to afford 80-82% yield (Scheme 2).
When triethylorthoacetate (8) was used in the above cyclocondensation, 3,10-dimethyl-8,11-dihydro-11-(p-chlorophenyl)-8-phenyl pyrazolo [4`,3`: 5,6] pyrano [3,2-e]-[1,2,4] triazolo [1,5-c] pyrimidine (9) was produced. Compound 9 was also produced via the reaction of 3 with acetic anhydride (10) as carboxylic acid anhydride and acetyl chloride (11) as acid chloride. The structure of 9 was based on the absence of NH and NH2 absorption in the i.r. spectrum. (Scheme 2).
Moreover, the interaction of triethylorthopropionate (12) with 3 afforded 3-ethyl -10-methyl-8,11-dihydro-11-(p-chlorophenyl)-8-phenyl-pyrazolo [4`, 3` : 5,6] pyrano [3,2-e] [1,2,4] triazolo [1,5-c] pyrimidine (13).
The subsequent incorporation of a one- carbon unit carrying an amino group into 3 to form the triazolo component was performed via two facile procedures. The first one consisted in applying cyanogen bromide (14) condensation to compound 3 and the second one involved refluxing the compound 3 and S-methyl isothiourea sulfate (15) [13] to afford 3-amino-10-methyl-8,11-dihydro-11-(p-chlorophenyl)-8-phenyl pyrazolo-[4`, 3` : 5,6] pyrano [3,2-e] [1,2,4] triazolo [1,5-c] pyrimidine (16b).That compound 16b exists in the 2-amino tautomeric form and not in the 2-iminoform was shown by the appearance of an intense absorption band at 3400 cm-1 and a two proton singlet as broad band at 3.7 ppm due to NH2 in the i.r. and 1H NMR spectra respectively. In the case involving the condensation of compound 3 with cyanogen bromide (14) and S-methyl isothiourea sulfate (15), the intermediates formed might bear a cyanimino (17) or guanidino function (18). These intermediates (17 and 18) were cyclized ) in an alkaline medium to give the target molecule 16b as expected (Scheme 2). However because of the different electronic influence on each reaction centre, 16b was obtained in different yields.
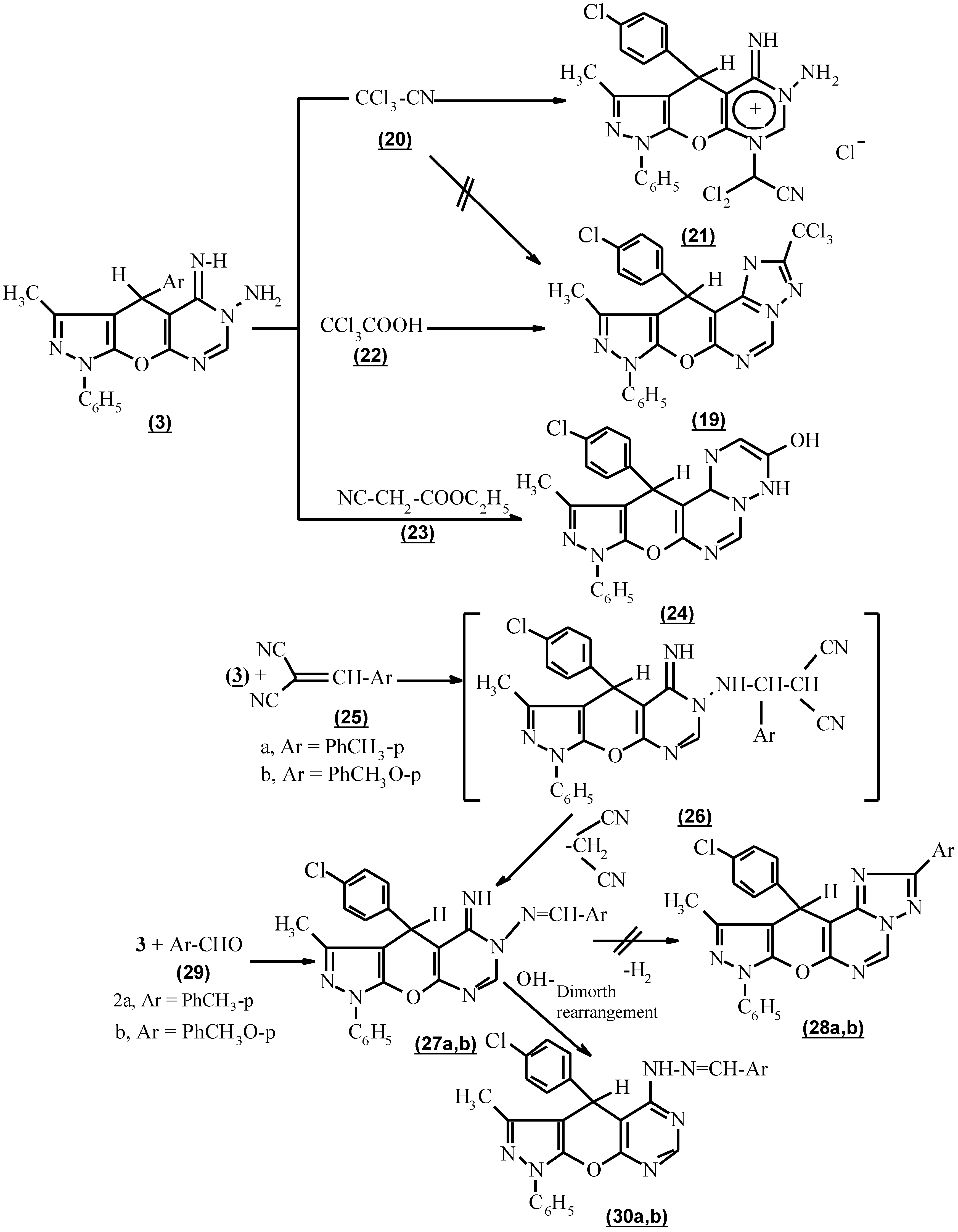
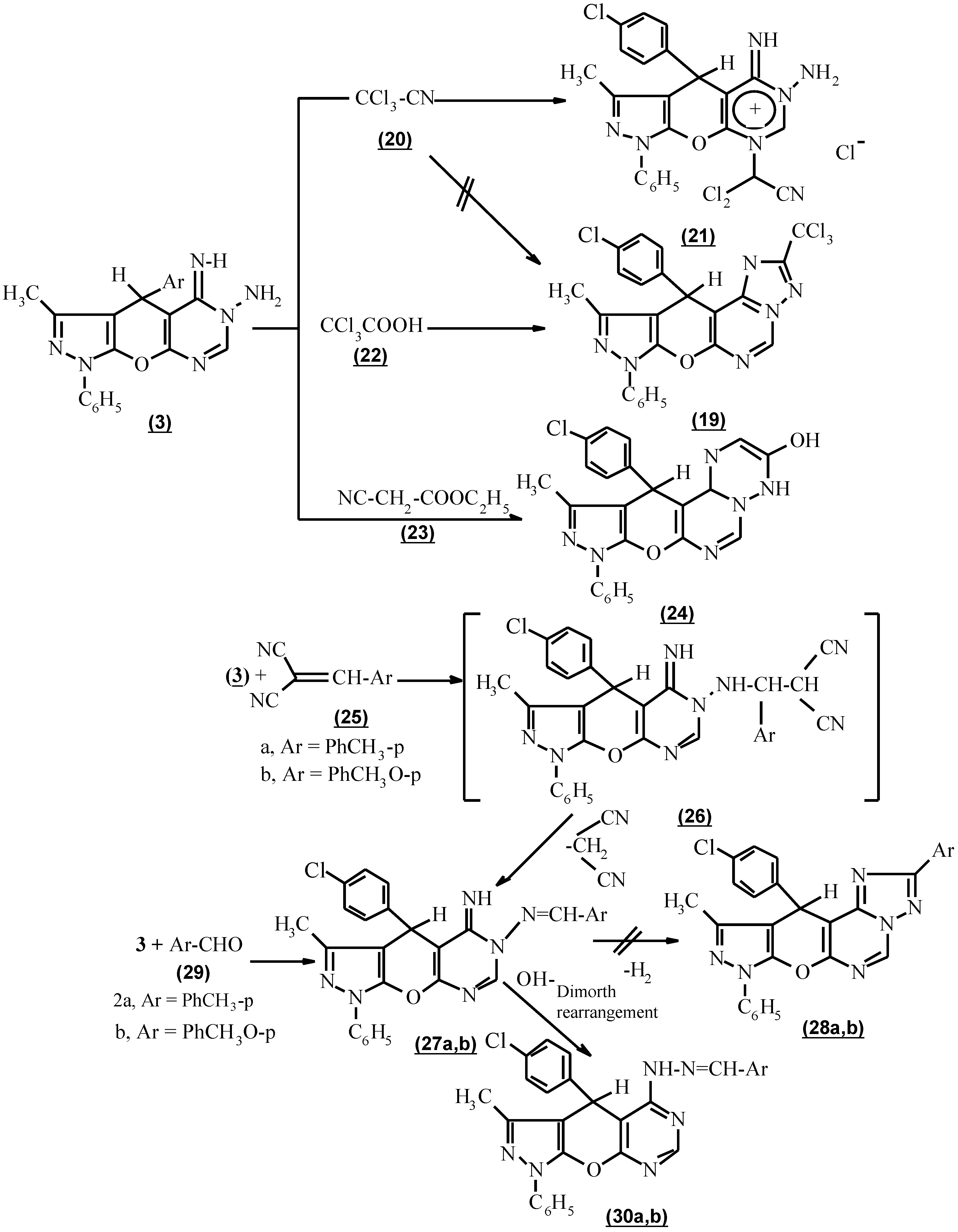
The formation of 3-trichloromethyl-10-methyl-8,11-dihydro-11-(p-chlorophenyl)-8-phenyl pyrazolo [4`, 3` : 5,6] pyrano [3,2-e] [1,2,4] triazolo [1,5-c] pyrimidine (19) via the interaction of compound 3 with trichloroacetonitrile (20) in the absence of solvent under reflux according to a reported procedure for the construction of an allied heterocyclic system [14] was unsuccessful (Scheme 3). The pyrimidinium salt (21) was formed [15,16a,b], based on the presence of NH and NH2 in its i.r. spectrum and the appearance of CN, its mass spectrum was (M+ - NH2, 532.2, 30%). The trichloro methyl derivatives (19) were achieved using trichloroacetic acid (22) in the presence of phosphoryl chloride under reflux for 3 hours. The structure (19) was confirmed by the absence of NH and NH2 absorption in its i.r. spectrum.
The activity of compound 3 towards the reaction product was active methylene was also tested [17]. Thus, with ethyl cyanoacetate (23), 9,12-dihydro-3-hydroxy-11-methyl-9-phenyl-12-(p-chlorophenyl) 2H-pyrazolo [4``, 3`` : 5`,6`] pyrano [2`,3`:5,6] pyrimido [1,6-b] [1,2,4] triazine (24) as confirmed by its spectral data. (Scheme 3).
Compound 3, when reacted with β-cyanocinnamonitrile derivatives, namely p-tolylmalononitrile and p-anisylmalononitrile (25a,b) respectively, in dioxane under reflux and in the presence of a catalytic amount of piperidine failed to afford 8,11-dihydro-11-(p-chlorophenyl)-3-aryl-8-methyl-10-phenylpyrazolo [4`,3` : 5,6] pyrano [3,2-e] [1,2,4] triazolo [1,5-c] pyrimidine (28a,b). In addition p-tolylmalononitrile and p-anisylmalononitrile (25a,b) respectively reacted with compound 3 to give 6-aryl methylene hydrazono-1,4,5,6-tetrahydro-3-methyl-1-phenyl-4-(p-chlorophenyl) pyrazolo-[4`, 3` : 5,6] pyrano [3,2-d] pyrimidine (27a,b) via the formation of 1,1 adduct (26a,b) followed by the loss of malononitrile [18]. Also the same product (27b) was isolated from the reaction of compound 3 with p-tolulaldehyde and p-anisaldehyde 29a,b respectively. Based on their spectral data no cyclization took place (Scheme 3). From this experiment, it can be concluded that a Taylor-Löeffler transformation has occured [9]. Because the reaction was carried out in basic medium as catalyst which facilitates the Dimorth rearrangement, the formal structure 30a,b is preferable (Scheme 3).
For convenience of the readers, the spectroscopic data are also collected in Table 1.
(Scheme 2.).
(Scheme 3.).
Experimental
General
Melting points are uncorrected and were taken on a Boetius melting point microscope. Microanalyses were performed by the Micro-analytical unit Cairo University I.R. spectra were recorded on a Mattson 5000 FIR spectrometer 1H NMR spectra were determined on a Varian EM NMR spectrometer using tetra-methylsilan as an internal standand. Mass spectra (MS) were recorded on a Finigan SSQ 7000 mass spectrometer.
Ethyl N-(5-cyano-1,4-dihydro-3-methyl-1-phenyl-4-(p-chlorophenyl) pyrano[2,3-c] [pyrazol -6-yl] methanimidate (2):
A mixture of 1 (1.79 g, 0.005 mol) triethylorthoformate (0.741 g, 0.005 mol) and acetic anhydride (16 ml) was refluxed for 5 h. The solvent was removed under reduced pressure and the resulting solid product is crystallized from benzene to give 1.84 g (92%) of 2 as colourless needles. M.p. 166-168°C. 1H NMR (DMSO-d6) δ 1.5 (t,3H, CH3), 2.00 (s, 3H, CH3), 4.41 (q, 2H, CH2), 4.9 (s, 1H, H-4), 7.45-7.84 (m, 9H, arom.) 8.3 (s, 1H, CHOEt). IR (kBr) 2220 (CN). Anal. Calcd. for C23H19N4OCl; C; 58.57; H, 4.75 Found: C; 68.44; H 4.59.
6-Amino-1,4,5,6-tetrahydro-5-imino-3-methyl-1-phenyl-4-(p-chlorophenyl) pyrazolo [4`,3`, :5,6] pyrano [2,3-d] pyrimidine (3).
To a solution of 2 (2g, 0.005 mol) in methanol (25 ml) a solution of hydrazine hydrate (5 ml) was added and the mixture stirred for 1 hr. Then it is allowed to stand overnight. The precipitate formed is filtered, dried and crystallized from methanol to afford. 55% of 3. M.p. 213-214°C: 1H NMR (DMSO-d 6): δ 1.95 (s, 3H, CH3), 5.00 (s, 1H, H-4), 5.6 (s, 1H, NH), 7.45-7.84 (m, 9H, arom.), 8.3 (s, 1H, H-7). IR: 3370, 3340 (NH2), 3150(NH). MS m/z (%), 404 (20) M+, 388 (60) 174(100), 77(65). Anal. Calcd. for C21H17N6OCl; C, 62.30; H, 4.23, Found C, 62.19, H; 4.18.
8,11-Dihydro-10-methyl-8-phenyl-4-(p-chlorophenyl) pyrazolo [4`,3`: 5,6] pyrano [3,2-e] [1,2,4] triazolo [1,5-c] pyrimidine (5).
Method A
A mixture of 3 (1.01g, 0.0025 mol) and (32 ml, 0.7 mol) of formic acid 4 was refluxed for 10 hrs. and then cooled poured onto ice-water to give a white precipitate which was filtered, washed several times by water, dried. and then crystallized from ethanol to afford 55% of 5. M.p. 324-325°C. 1H NMR (DMSO-d6): δ 1.9 (s, 3H, CH3) 5.7 (s, 1H, H-11), 7.4-7.8 (m, 9H, arom.), 8.65 (s, 1H, H-2), 9.7 (s, 1H, H-5). MS: m/z (%) 414 (47) M+, 303 (100), 278 (25), 77 (20). Anal. Calcd. for C22H15N6OCl; C; 63.69, H; 3.64 Found C; 63.49, H; 3.49.
Method B
A solution of 3 (1.01 g; 0.0025 mol) and (2.2 g, 0.02 mole) of triethylorthoformate 6 neat was refluxed for 8 hrs. A precipitate was formed, filtered while and recrystallized from ethanol to give 0.83g (80%) of 5.
Method C
To a solution of 3 (1.01g, 0.0025 mol) in absolute ethanol (30 ml) was added ethoxymethylene malononitrile 7 and the mixture was refluxed for 6 hr (TLC control). On cooling, a solid product separated, which was filtered and recrystallized from ethanol to give 0.85g (82%) of 5.
3,10-dimethyl-8,11-dihydro-11-(p-chlorophenyl)-8-phenyl pyrazolo [4`,3`,: 5,6] pyrano [3,2-e] [1,2,4,] triazolo [1,5-c] pyrimidine (9)
Method A
A solution of 3 (1.01g, 0.0025 mol) and (3.3g 0.2 mol) of triethylorthoacetate (8), neat was refluxed for 8 hrs, a precipitate was formed, filtered while hot, and recrystallized from ethanol to give 0.75g (70%) of 9. M.p. 264-265°C. 1H NMR (DMSO-d6): δ 1.9(s, 3H, CH3), 2.3 (s, 3H, CH3) 5.6 (s, 1H, H-11), 7.4-7.8 (m, 9H, arom.) 9.6 (s, 1H, H-5). MS, m/z (%) 428 (25) M+, 317 (100) 77 (17). Anal. Calcd. for C23H17N6OCl; C: 64.41, H; 3.99, Found C. 64.32 H, 3.79.
Method B
A solution of 3 (1.01g, .0025 mol) and 10 ml of acetic anhydride (10) was refluxed for 10 hrs. A precipitate was formed, filtered while hot, and recrystallized from ethanol to give 0.8g (75%) of 9.
Method C
A mixture of 3 (1.01g, 0.0025 mol) and 7 ml of acetyl chloride (11) in 10 ml dimethylformamide was heated for 4 hrs on a water bath and after cooling, poured into 100 ml of ice water. The precipitate was collected on a filter, washed with water and recrystallized from ethanol to give 0.9g (84%) of 9.
3-Ethyl 10-methyl-8,11-dihydro-11-(p-chlorophenyl)-8-phenyl pyrazolo [4`,3`:5,6]-pyrano[3,2-e][1,2,4]triazolo [1,5-c] pyrimidine (13).
A solution of 3 (1.01 g, 0.0025 mol) and (3.4 g, 0.02 mol) of triethy-lor-thopropionate 12 neat was refluxed for 8 hrs, and then worked out as described above to produce 0.83 g (75%) of 13. M.p. 310-312°C. 1H NMR (DMSO-d6): δ 1.35 (t, 3H, CH3), 1.95 (s, 3H, CH3), 3.4 (q, 2H, CH2), 5.6 (s, 1H, H-11) 7.4-7.8 (m, 9H, arom.), 9.5(s, 1H, H-5). MS: m/z (%) 428 (M+, 28), CH3, 317 (100). Anal. Calcd. for C24H19N6OCl, C: 65.08, H, 4.32, Found C: 64.89, H, 4.22.
3-amino-10-methyl-8,11-dihydro-11-(p-chlorophenyl)-8-phenyl pyrazolo [4`,3`:5,6] pyrano [3,2-e] [1,2,4] triazolo [1,5-c] pyrimidine(16b).



{kind=link}
{kind=link}
{kind=link}
Method A
A solution of 3 (1.01 g, 0.0025 mol) in 50 ml dimethyl-formamide was kept at 0°C and treated with cyanogen bromide 14 (0.6 g, 0.005 mol). The mixture was refluxed for 5 hrs, then cooled and poured onto water to give a white precipitate, filtered, washed with water and crystallized from dimethylformamide to yield 0.54 g (50%) of 16b. M.p.>300 C. 1H NMR (DMSO-d6): δ 1.6 (s, 3H, CH3), 3.6 (br, 2H, NH2 exchangeable with D2O), 6.2 (s, 1H, H-11) 7.3-7.8 (m, 9H, arom.), 9.3(s, 1H, H-5), I.R.: 3178, 3320 (NH2). MS: m/z (%) 429 (M+, 37), 388 (57), 318 (60), 278 (100). Anal. Calcd. for C22H16N7OCl, C: 61.47, H; 3.75, Found C: 61.37, H; 3.66.
Method B
A solution of 3 (1.01 g, 0.0025 mol) and (0.28 g, 0.001 mol) of S-methylisothiourea sulfate 15 in alcoholic potassium hydroxide (0.5 N) was heated under reflux for 8 hours. After cooling, the mixture was poured on to water to give a white precipitate, which was collected, washed with water and recrystallized from dimethylformamide to yield 0.6 g (60%) of 16b.
3-trichloromethyl-8,11-dihydro-11-(p-chlorophenyl)-8-phenyl pyrazolo [4`,3`:5.6] pyrano[3,2-e] [1,2,4] triazolo [1,5 -c] pyrimidine (19)
A mixture of 3 (1.01 g, 0.0025 mol) and 0.49 g (0.003 mol) of trichloroacetic acid (22) in 15 ml of freshly distilled phosphorus oxychloride, was refluxed on a water bath for 4 hrs, cooled then poured onto ice -water to give a precipitate which was filtered off, and washed with water. The solid that collected by filteration, was recrystallized in dioxane to afford 0.7 g (51%) of (19). m.p. 270-271°C. 1H NMR (DMSO-d6): 1.8 (s, 3H, CH3), 4.6 (s, 1H, H-11), 7.2-7.7 (m, 9H, arom.) 8.4 (s, 1H, H-pyrimidine). Anal., Calcd. for C23H14N6Ocl4 C: 51.91, H: 2.65, Found C: 51.81, H: 2.61.
Formation of pyrimidinum salt (21)
A mixture of 3 (1.01 g, 0.0025 mol) and 20 ml (0.2 mol) of trichloroacetonitrile (20) was heated under reflux for 15 hours. The precipitate formed, was filtered, washed with a small amount of benzene and recrystallized in anhydrous ethanol to afford 0.62 g (45%) of 21. M.p. 150-151°C. 1H NMR (DMSO-d6): 1.78 (s, 3H, CH3), 4.7 (s, 1H, H-11), 7.2-7.8 (m, 13H, arom. + NH2 + 1H pyrimidine). I.R.: 34.55, 3327 (NH2), 3197 (NH), 2199 (CN), 1659 (C=NH). MS, m/z (%) 532 (M+ NH3), 419 (100), 385 (25) 143 (5), 77 (33). Anal., Calcd. for C23H14N7OCl4 C: 50.57, H; 2.58, Found C: 50.66, H; 2.51.
9,12-dihydro-3-hydroxy-11-methyl-9-phenyl-12-(p-chlorophenyl)-2H-pyrazolo [4``,3``:5`,6`] pyrano [2`,3`:4.5] pyrimido [1,6-b] [1,2,4] triazine (24)
A mixture of (3) (1.01 g, 0.0025 mol) andethylcyanoacetate (23) (0.1 ml, 0.001 mol) in absolute ethanol (50 ml) was refluxed for 10 hr. A precipitate was formed, filtered off washed with ethanol, and recrystallized from dioxane to afford 0.78 g (70%) of (25). m.p. 276-277° C. 1H NMR (DMSO-d6): 1.95 (s, 3H, CH3), 4.4 (s, 1H, NH), 5.6 (s, 1H, H-12), 7.4-7.8 (m, 9H, arom.) 8.4 (s, 1H, H-2), 9.5 (s, 1H, H-6). IR: 3500 (OH, NH). MS m/z (%), 414 (40) (M+ -CH3,OH), 303 (100). Anal., Calcd. forC23H17N6O2Cl C: 62.09, H: 3.85, Found C: 61.89, H: 3.66.
5-arylmethylene hydrazono-1,4,5,6-tetrahydro-3-methyl-1-phenyl-4-(p-chlorophenyl) pyrazolo [4`,3`:5.6] pyrano [2,3 d] pyrimidine (30a,b).
Method A
A mixture of 3 (1.01 g, 0.0025 mol) and p-tolylmalononitrile (25a) or p-anisylmalononitrile (25b) (0.0025 mol) in dioxane, in the presence of piperidine as a catalyst was refluxed for 8 hours. After cooling, the solid product formed was collected and crystallized from dioxane to yield 70% and 80% of 30a,b respectively.
Method B
A mixture of 3 (1.01 g, 0.0025 mol) and p-tolulaldehyde (29a) or p-anisaldehyde (29b) (0.0025 mol) in dioxane in the presence of piperidine as a catalyst was refluxed for 8 hours and then worked out as described above to produce. 76%, 85% of (30a,b).
30a: m.p. 184-186°C. 1H NMR: 1.9 (s, 3H, CH3) 2.4 (s, 3H, CH3), 5.9 (s, 1H, H-11), 7.3-7.5 (m, 13H, arom.) 8.2 (s, 1H, N=CH), 8.5 (s, 1H, H-pyrimidine), 10.6 (br, 1H, NH, D2O exchangeable). IR: 1600 (C=N), 3450 (NH). MS m/z (%), 506 (M+, 14), 393 (40), 386, 100, 77 (15). Anal., Calcd. for C29H23N6OCl; C: 68.70, H: 4.57, Found C: 68.60, H: 4.49.
30b: m.p. 196-197°C. 1H NMR: 1.9 (s, 3H, CH3), 3.4 (s, 3H, OCH3), 5.9 (s, 1H, H-11), 7.3-7.8 (m, 13H, arom.) 8.4 (s, 1H, N=CH), 8.56 (s, 1H, H-pyrimidine), 10.8 (br, 1H, NH, D2O exchangeable). IR: 1620 (C=N), 3450 (NH). Anal., Calcd. for C29H23N6O3Cl C: 66.60, H: 4.43, Found C: 66.49, H: 4.33.
Acknowledgment
The author would like to acknowledge Prof. Dr. W. Zielinski, Institute of Organic Chemistry & Technology Gliwice, Poland for his encouragement. The author thanks National Research Centre (NRC-3/6/1-2/96 95-98) for support of this research.
References
- EL-Nagdi, M. H.; EL-Maoghayer, M. R. H.; EL-Gemeie, G. E. H. Adv. Heterocyclic Chem. 1987, 41, 320.
- EL-Nagdi, M. H.; EL-Maoghayer, M. R. H.; Sadek, K. U. Adv. Heterocyclic Chem. 1990, 48, 223.
- Kuo, S. G.; Huang, L. J.; Nakamura, H. J. Med. Chem. 1984, 27, 539. [PubMed]
- Hori, H.; Ito, E.; Jakta, T.; Koyama, G; Umezawa, H. J. Antibiot. 1964, 17A, 96.
- Koyama, G.; Umezawa, H. J. Antibiot. 1965, 18A, 175.
- Robins, R. K.; Beaman, A. G. J. Heterocyclic Chem. 1965, 3, 110.
- Miller, G. W.; Rose, F. L. J. Chem. Soc. 1963, 5642.
- Miller, G. W.; Rose, F. L. J. Chem. Soc. 1965, 3357.
- Taylor, E . C.; Loeffler, P. K. J. Amer. Chem. Soc. 1960, 82, 3147.
- San gapoure, S . S.; Agasimun din, Y. S. Indian J . Chem. 1980, 19b, 115.
- Tacconi, G.; Gatti, G.; Desimoni, G.; Messori, V. G. J. Prakt. Chem. 1980, 322, 831.
- Sharanina, L. G.; Promonenkov, V. K.; Marshtupa, V. P.; Pashchenko, A. V.; Puzanova, V. V.; Sharanin, Yu. A.; Klyuev, N. A.; Gusev, L. F.; Gnatusina, A. P. Khim. Geterotsikl. Soedin 1982, 801. C.A. 97, 10991.
- Lin, Y. I.; Fields, T .L.; Lang, S. A. J. Heterocyclic Chem. 1978, 15, 311.
- Lin, K. C.; Shih, B. J.; Tao, T. M. J. Heterocycl. Chem. 1984, 2, 1572.
- Duffin, G. F. "Advances in Heterocyclic Chemistry"; Vol. 3, Katritzky, A. R., Boulton, A. J., Eds.; Academic Press: New York; p. 21.
- a) Tominaga, Y.; Shiroshita, Y.; Hosomi, A. J. Heterocyclic Chem. 1988, 5, 1745. ; (b) Laus, G.; Klotzer, W. Synthesis 1990, 107.
- Younes, M. I.; Metwally, S. A. M.; Atta, A. H. Synthesis 1990, 704.
- Osman, S. A.; EL-Gemei, G. E. H.; Nawar, G. A.; EL-Nagdi, M. H. Monatsh Chem. 1986, 117, 105.
- Sample Availability: Available from MDPI
© 1998 MDPI. All rights reserved. Molecules http://www.mdpi.org/molecules/
Share and Cite
MDPI and ACS Style
Zaki, M.E.A. Synthesis of Novel Fused Heterocycles Based on Pyrano[2,3-c]pyrazole. Molecules 1998, 3, 71-79. https://doi.org/10.3390/30300071
AMA Style
Zaki MEA. Synthesis of Novel Fused Heterocycles Based on Pyrano[2,3-c]pyrazole. Molecules. 1998; 3(3):71-79. https://doi.org/10.3390/30300071
Chicago/Turabian StyleZaki, M. E. A. 1998. "Synthesis of Novel Fused Heterocycles Based on Pyrano[2,3-c]pyrazole" Molecules 3, no. 3: 71-79. https://doi.org/10.3390/30300071