Synthesis and Reactions of New 4-Oxo-4H-benzopyran-3-carboxaldehydes Containing Hydroxy Groups or 2-Oxopyran Cycles

Abstract
:Introduction
Results and Discussion

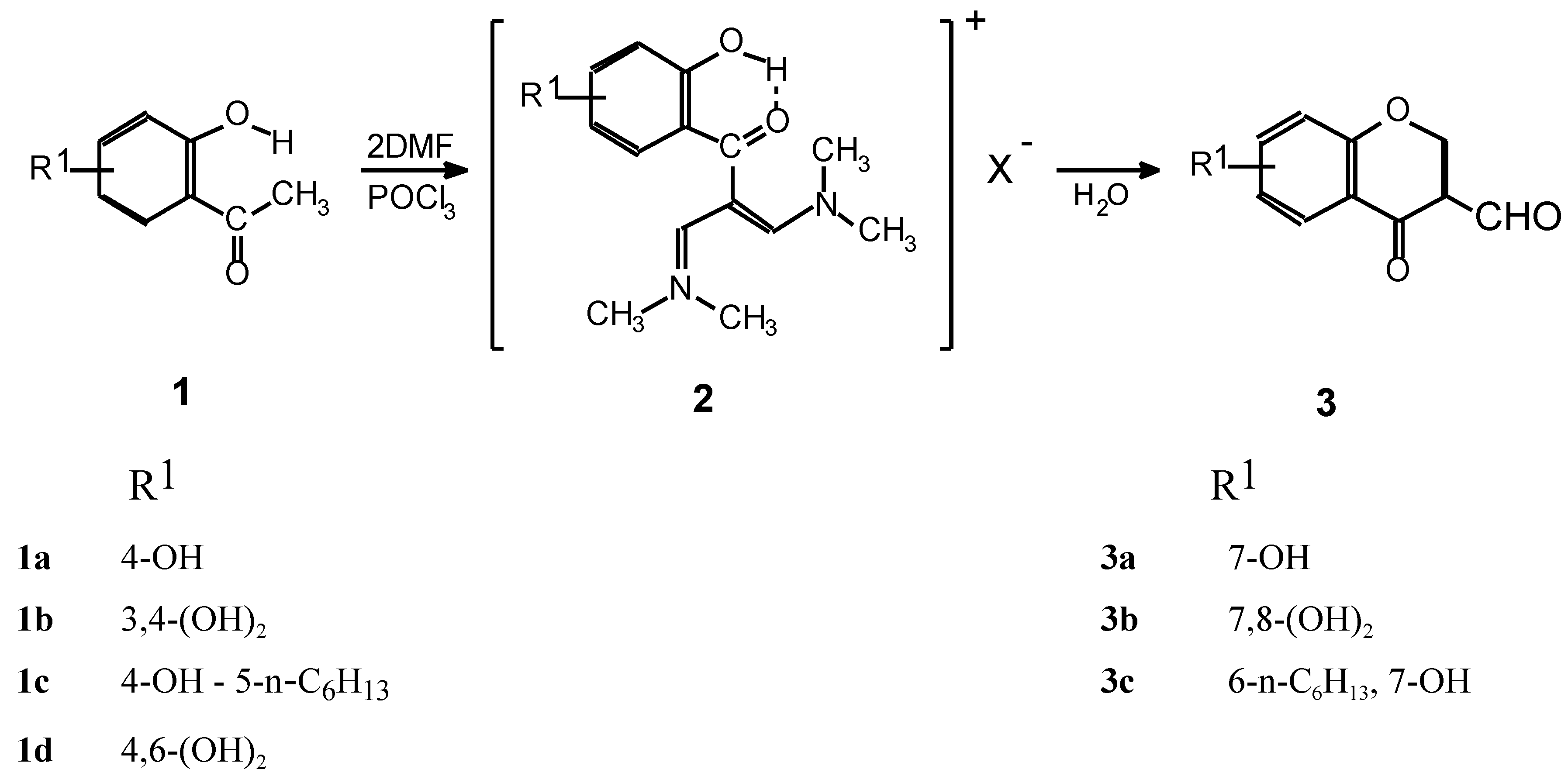
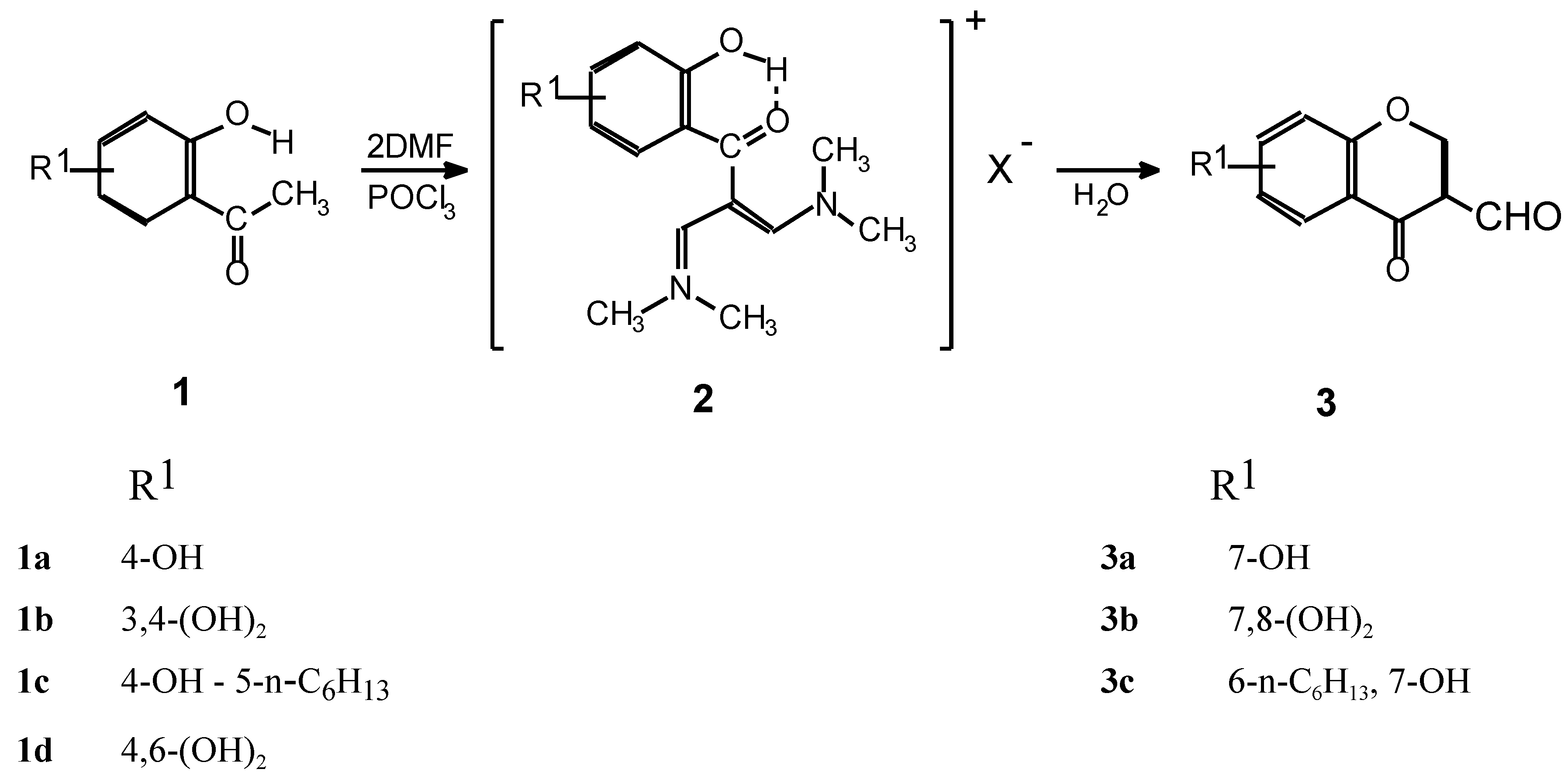{kind=link}
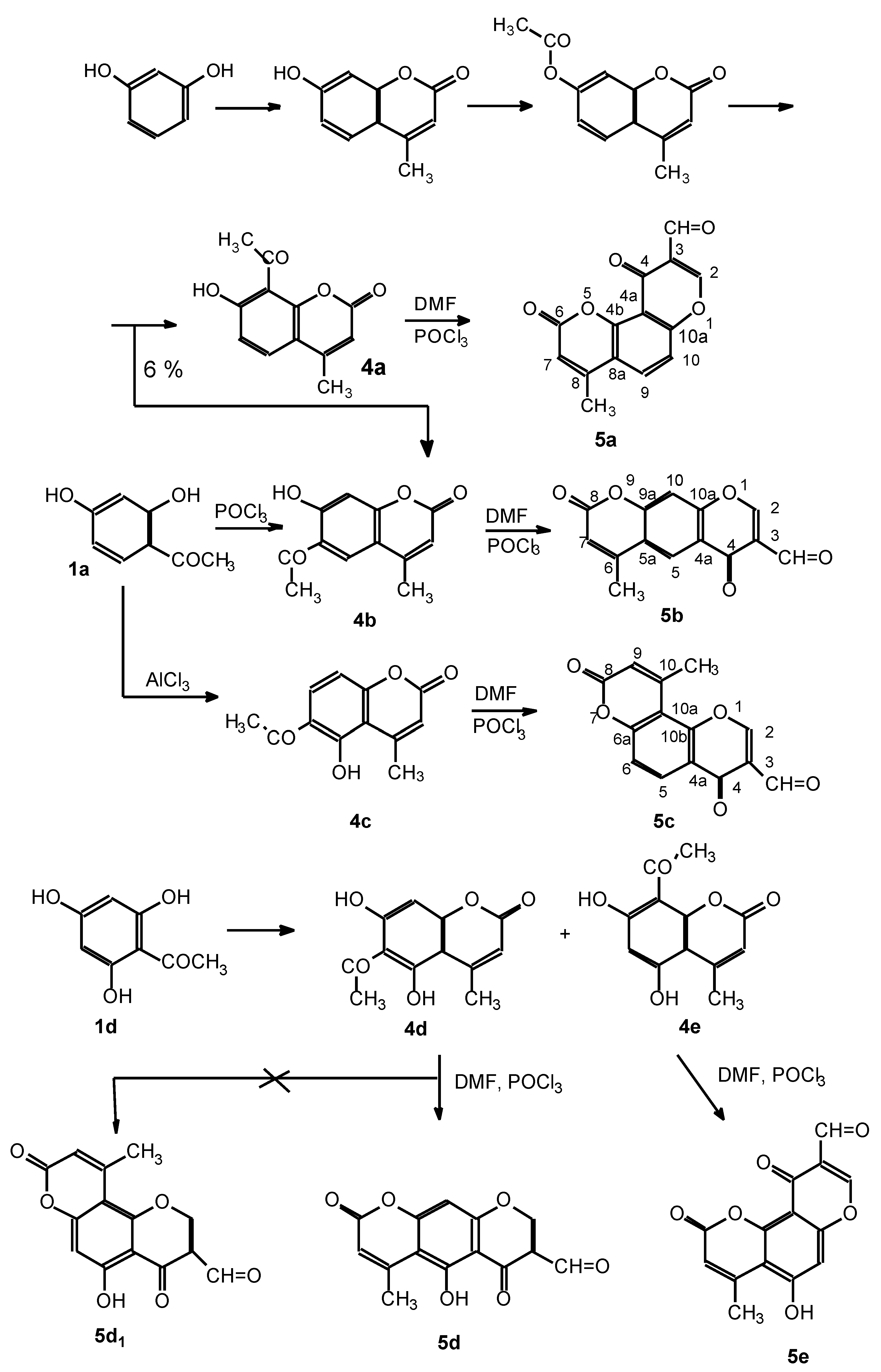
{kind=link}
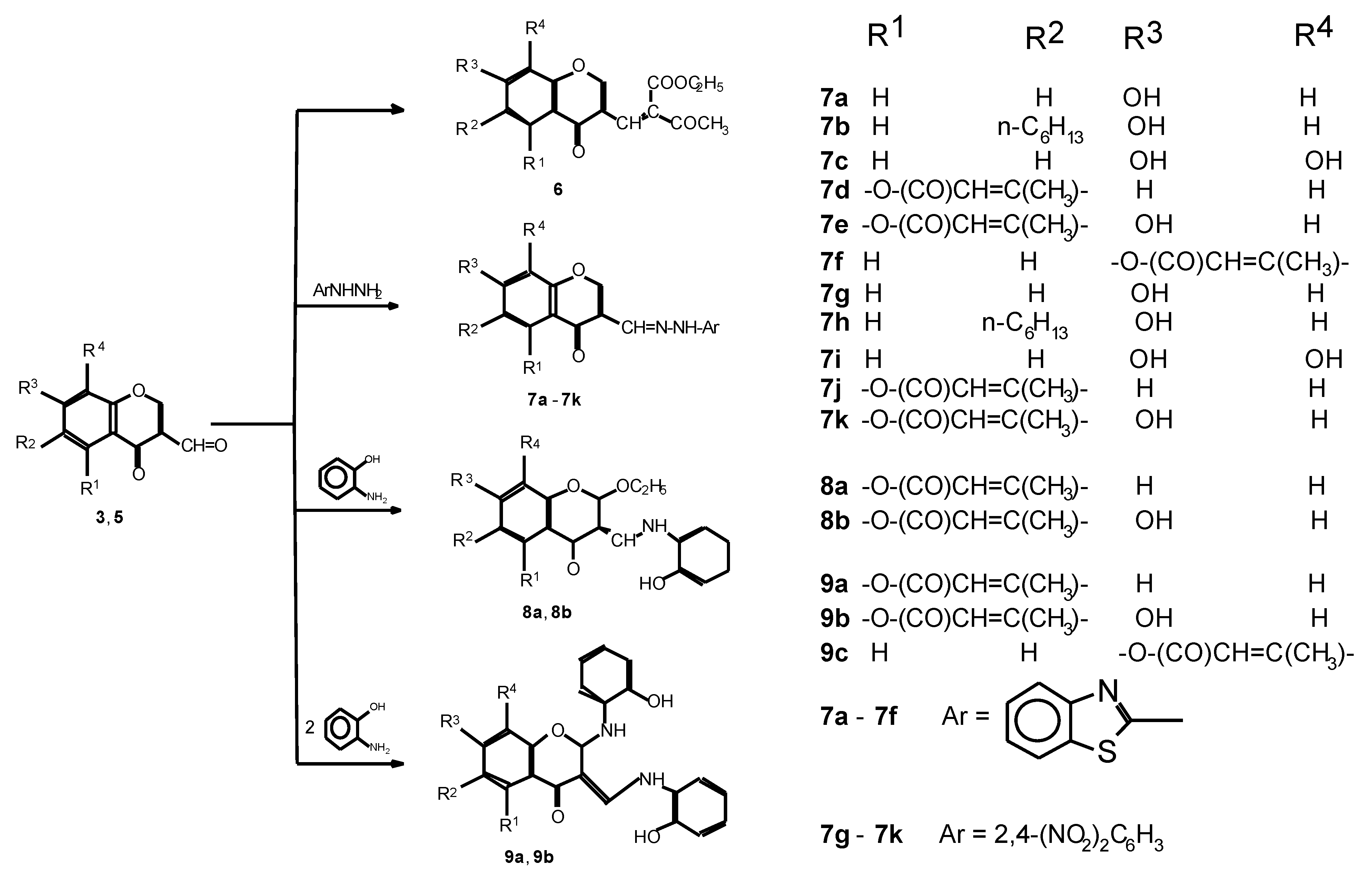{kind=link}
| Compound | Formula | Wi (calc.) % | M.p. (°C) | ||
| Mr | Wi (found) % | ||||
| C | H | N | |||
| 1c | C14H20O3 | 71.18 | 8.51 | 75-77 | |
| 236.2 | 71.13 | 8.47 | |||
| 3a | C10H6O4 | 63.14 | 3.17 | 268-270 | |
| 190.2 | 63.31 | 3.1 | |||
| 3b | C10H6O5 | 58.3 | 2.91 | 264-266 | |
| 206.2 | 58.26 | 2.98 | |||
| 3c | C16H18 | 70.07 | 6.57 | 233-234 | |
| 274.2 | 70.01 | 6.6 | |||
| 5a | C14H8O5 | 65.62 | 3.13 | 310-312 | |
| 256.2 | 65.33 | 3.12 | |||
| 5b | C14H8O5 | 65.62 | 3.13 | 255-260 | |
| 256.2 | 65.48 | 3.01 | |||
| 5c | C14H8O | 65.62 | 6.57 | 233-234 | |
| 256.2 | 65.32 | 3.07 | |||
| 5d | C14H8O6 | 61.79 | 2.94 | 273-274 | |
| 272.2 | 61.62 | 2.99 | |||
| 5e | C14H8O6 | 61.79 | 2.94 | 291-293 | |
| 272.2 | 61.77 | 2.92 | |||
| 7a | C17H11N3O3S | 60.53 | 3.26 | 12.46 | 248-250 |
| 337.3 | 60.37 | 3.25 | 12.27 | ||
| 7b | C23H23N3O3S | 65.6 | 5.46 | 9.97 | 219-220 |
| 421.4 | 65.35 | 5.33 | 9.54 | ||
| 7c | C17H11N3O4S | 57.79 | 3.12 | 11.9 | 259-261 |
| 403.3 | 57.48 | 3.11 | 11.76 | ||
| 7d | C21H13N3O4S | 62.5 | 3.24 | 10.41 | 253-255 |
| 403.3 | 62.37 | 3.23 | 10.29 | ||
| 7e | C21H13N3O5S | 60.13 | 3.12 | 10.01 | 325-8 |
| 419.3 | 60.22 | 3.19 | 9.71 | ||
| 7f | C21H13N3O4S | 62.5 | 3.24 | 10.41 | 240-242 |
| 403.3 | 62.38 | 3.2 | 10.39 | ||
| 7g | C16H10O7N4 | 51.9 | 2.72 | 15.13 | 297-9 |
| 378.3 | 51.62 | 2.76 | 14.89 | decomp. | |
| 7h | C22H22O7N4 | 58.15 | 4.88 | 12.33 | 296-8 |
| 454.4 | 57.86 | 4.84 | 12.09 | decomp. | |
| 7i | C16H10O8N4 | 49.75 | 2.61 | 14.5 | 173-6 |
| 386.3 | 49.36 | 2.66 | 14.28 | decomp. | |
| 7j | C20H12O8N4 | 55.05 | 2.77 | 12.84 | 289-94 |
| 436.3 | 54.89 | 2.77 | 12.75 | ||
| 7k | C20H12O9N4 | 53.11 | 2.67 | 12.38 | 300-2 |
| 452.3 | 52.84 | 2.8 | 12.06 | decomp. | |
| 8a | C22H19NO6 | 67.18 | 4.83 | 3.56 | 275-6 |
| 393.4 | 66.89 | 4.59 | 3.12 | ||
| 8b | C22H19NO7 | 64.55 | 4.65 | 3.42 | 259-60 |
| 409.4 | 64.36 | 4 | 3.3 | ||
| 9a | C26H20N2O6 | 68.42 | 4.39 | 6.13 | 180-5 |
| 456.4 | 68.22 | 4.51 | 6.02 | ||
| 9b | C26H20N2O6 | 66.1 | 4.24 | 5.92 | 158-62 |
| 472.4 | 66.05 | 4.24 | 5.74 | ||
| 9c | C26H20N2O6 | 68.42 | 4.39 | 6.13 | 188-90 |
| 456.4 | 68.51 | 4.37 | 6.19 | ||
| Compound |  | CH=O |  | νs(NO2) | νas(NO2) |
| 3a | 1620 | 1695 | - | - | - |
| 3b | 1630 | 1682 | - | - | - |
| 3c | 1630 | 1696 | - | - | - |
| 5a | 1657 | 1700 | 1726 | - | - |
| 5b | 1655 | 1693 | 1748 | - | - |
| 5c | 1637 | 1693 | 1700 | - | - |
| 5d | 1640 | 1702 | 1734 | - | - |
| 5e | 1640 | 1704 | 1724 | - | - |
| 7a | 1634 | - | - | - | - |
| 7b | 1630 | - | - | - | - |
| 7d | 1630 | - | 1720 | - | - |
| 7g | 1640 | - | - | 1318 | 1580 |
| 7h | 1612 | - | - | 1350 | 1580 |
| 7i | 1610 | - | - | 1345 | 1580 |
| 7j | 1640 | - | 1722 | 1345 | 1580 |
| 7k | 1606 | - | 1748 | 1310 | 1580 |
| 8a | 1642 | - | 1718 | - | - |
| 8b | 1642 | - | 1708 | - | - |
| 9a | 1648 | - | 1700 | - | - |
| compound | solvent | spectra δ (ppm) |
| 1a | CDCl3 | 12.52 (1H,s,OH), 7.42 (1H,s,H-6), 6.34 (1H,s,H-3), 1.65-0.87 |
| (13Hm) | ||
| 3a | DMSO | 10.11 (1H,s,CHO), 8.78 (1H,s,H-2), 7.99 (1H,d,H-5), 7.04-6.94 |
| (2H,t,H-6,8) | ||
| 3b | DMSO | 10.12 (1H,s,CHO), 8.77 (1H,s,H-2), 7.48 (1H,d,H-5), 7.00 |
| (1H,d,H-6) | ||
| 3c | DMSO | 10.12 (1H,s.CHO), 8.73 (1H,s,H-2), 7.79 (1H,s,H-5), 6.93 |
| (1H,s,H-8), 2.9 (2H,t), 1.30 (8H,m), 0.86 (3H,t) | ||
| 4a | CDCl3 | 7.68 (1H,d,H-5), 6.90 (1H,d,H-6), 6.12 (1H,s,H-3), 2.95 |
| (3H,s,CH CO), 2.41 (3H,s,CH ), 13.54 (1H,s,OH) | ||
| 4b | CDCl3 | 7.96 (1H,s,H-5), 6.84 (1H,s,H-8), 6.17 (1H,s,H-3), 2.70 |
| (3H,s,CH CO), 2.44 (3H,s,CH ), 12.61 (1H,s,OH) | ||
| 4c | CDCl3 | 7.85 (1H,d,H-7), 6.83 (1H,d,H-8), 6.13 (1H,s,H-3), 2.66 |
| (6H,s,CHCO), 14.07 (1H,s,OH) | ||
| 4d | CDCl3 | 6.26 (1H,s,H-3), 5.99 (1H,d,H-8), 2.68 (3H,s,CH CO), 2.51 |
| (3H,s,CH ) | ||
| 4e | CDCl3 | 6.37 (1H,s,H-3), 5.94 (1H,s,H-6), 2.68 (3H,s,CH CO), 2.51 |
| (3H,s,CH ) | ||
| 5aa | DMSO | 10.12 (1H,s,CHO), 8.86 (1H,s,H-2), 8.18 (1H,d,H-10), 7.67 |
| (1H,d,H-9), 6.53 (1H,s,H-7) | ||
| 5ba | DMSO | 10.12 (1H,s,CHO), 8.97 (1H,s,H-2), 8.39 (1H,s,H-5), 7.87 |
| (1H,s,H-10), 6.56 (1H,s,H-7), 2.54 (3H,s,CH ) | ||
| 5ca | DMSO | 10.14 (1H,s,CHO), 9.02 (1H,s,H-2), 8.31 (1H,d,H-5), 7.58 |
| (1H,d,H-6), 6.57 (1H,s,H-9), 2.74 (3H,s,CH ) | ||
| 5da | DMSO | 10.05 (1H,s,CHO), 8.63 (1H,s,h-2), 8.12 (1H,s,H-10), 6.78 |
| (1H,s,H-7), 6.26 (1H,s,OH), 2.54 (3H,s,CH ) | ||
| 5e | DMSO | 10.07 (1H,s,CHO), 9.06 (1H,s,H-2), 7.30 (1H,s,H-10), 6.31 |
| (1H,s,H-7), 2.62 (3H,s,CH ) | ||
| 6 | CDCl3 | 8.26 (2H,t,H-2,CH), 7.10-7.56 (3H,m,arom), 4.31 (2H,q,CH ), |
| 2.47 (3H,s,CH COO), 2.35 (3H,s,CH CO), 1.35 (3H,t,CH ) |
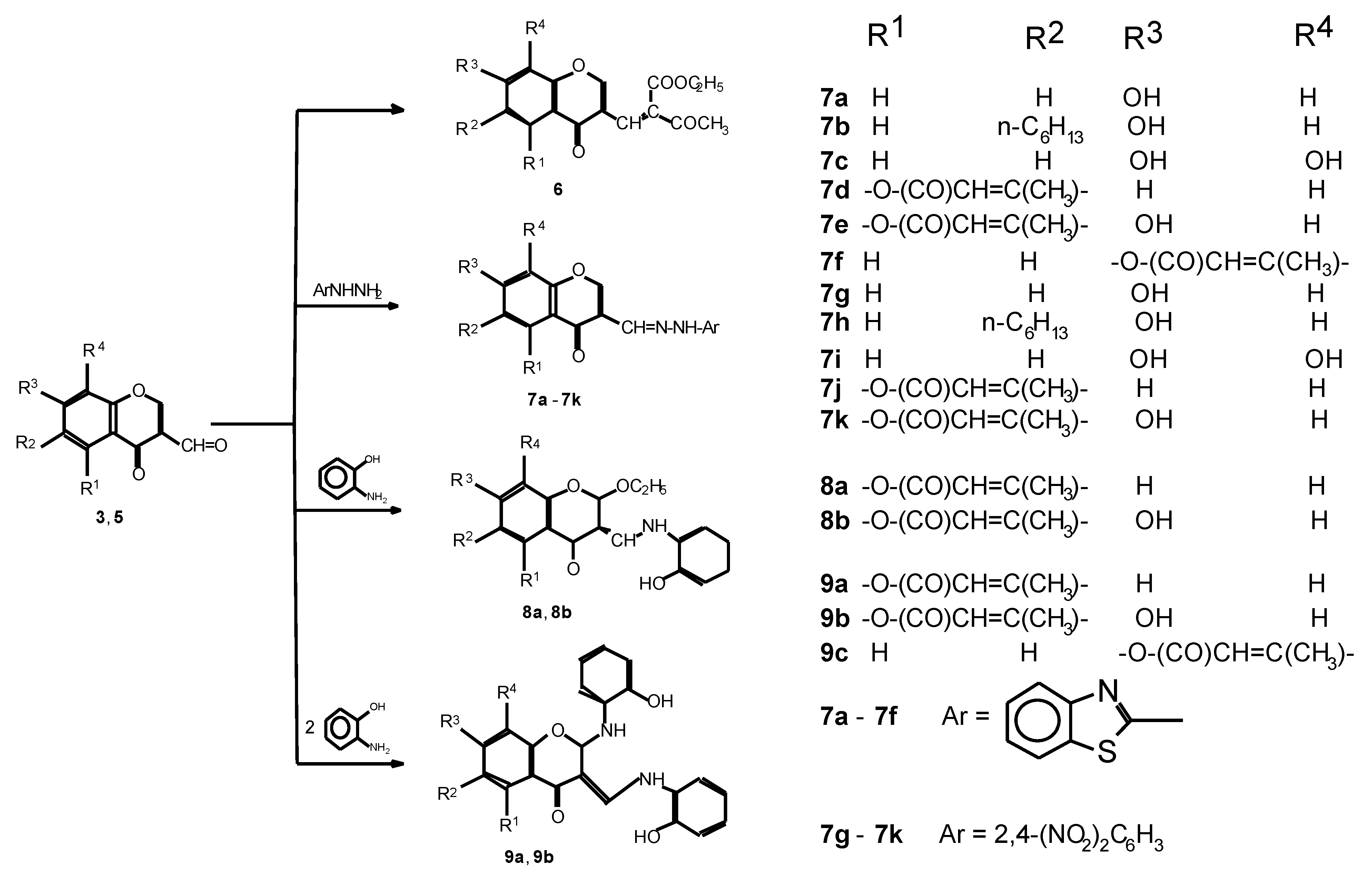
Experimental Section
General details
2, 4-Dihydroxy-5-n-hexylacetophenone 1c
3-Formylchromones 3, 5. General procedure
3-(4-Oxo-7-acetoxy-4H-1-benzopyran-3-yl)-2-(1- oxoethyl)-2-ethylpropenoate 6
2-Benzothiazolylhydrazone-3-formylchromone 7a - 7f, 2,4-dinitrophenylhydrazone-3-formylchromone 7g - 7k and 2-ethoxy-3-(2-hydroxyphenylaminomethylene)chroman-4-ones 8a, 8b
2-(2-hydroxyphenylamino)-3-(2-hydroxyphenylaminometylene)chroman-4-ones 9a - 9c
Acknowledgements
References
- El-Shaer, H. M.; Zahradnik, P.; Lacova, M.; Matulova, M. Collect. Czech. Chem. Commun. 1994, 59, 1673.
- El-Shaaer, H.M.; Lacova, M.; Odlerova, Z.; Furdik, M. Chem. Papers 1994, 59, 1673.
- El-Schaaer, H.M.; Perjessy, A.; Zahradnik, P.; Lacova, M.; Sustekova, Z. Monatsh. Chem. 1993, 124, 539.
- Gasparova, R.; Lacova, M. Collect. Czech. Chem. Commun. 1995, 60, 1178.
- Lacova, M.; Stankovicova, H.; Odlerova, Z. Il - farmaco 1995, 50, 885. [PubMed]
- Kralova, K.; Sersen, F.; Lacova, M.; Stankovicova, H. Biol. Plant. 1995, 38, 397.
- El-Schaaer, H.M.; Foltinova, P.; Lacova, M.; Chovancova, J.; Stankovicova, H. Il - farmaco. (in press).
- Nohara, A.; Umetani, T.; Sanno, Y. Tetrahedron 1974, 30, 3553.
- Nohara, A.; Umetani, T.; Sanno, Y. Tetrahedron Lett. 1973, (22), 1995-8.
- Dewar, M. J. S.; Zoebisch, E. G.; Healy, E. F.; Stewart, J. J. P. J. Am. Chem. Soc. 1985, 57, 3698.
- Brown, R.C.; Harard, R. U.S. 1980, 4238, 606, Chem. Abstr. 1981, 94p, 156755.
- Klutchko, S.; Kaniansky, D. U.S. 1977, 4008, 232, Chem. Abstr. 1977, 87p, 5808.
- Cooper, R. S. Org. Synth. Coll.; Vol. III.761, John Wiley and Sons: New York, 1967. [Google Scholar]
- Desai, R.D.; Ekhlas, M. Proc Indian Acad SCi (A) 1938, 567. Chem. Abstr. 1939, 33, 3356.
- Sethna, S.M.; Shah, R.C. J. Chem. Soc. 1938, 228.
- Hoesch, K. Ber. 1942, 48, 1125.
- Samples Availability: Samples are available from MDPI and the authors.
© 1998 MDPI. All rights reserved. Molecules http://www.mdpi.org/molecules/
Share and Cite
Lacova, M.; Loos, D.; Furdik, M.; Matulova, M.; El-Shaaer, H.M. Synthesis and Reactions of New 4-Oxo-4H-benzopyran-3-carboxaldehydes Containing Hydroxy Groups or 2-Oxopyran Cycles. Molecules 1998, 3, 149-158. https://doi.org/10.3390/30600149
Lacova M, Loos D, Furdik M, Matulova M, El-Shaaer HM. Synthesis and Reactions of New 4-Oxo-4H-benzopyran-3-carboxaldehydes Containing Hydroxy Groups or 2-Oxopyran Cycles. Molecules. 1998; 3(6):149-158. https://doi.org/10.3390/30600149
Chicago/Turabian StyleLacova, Margita, Dusan Loos, Mikulas Furdik, Maria Matulova, and Hafez M. El-Shaaer. 1998. "Synthesis and Reactions of New 4-Oxo-4H-benzopyran-3-carboxaldehydes Containing Hydroxy Groups or 2-Oxopyran Cycles" Molecules 3, no. 6: 149-158. https://doi.org/10.3390/30600149