A Straightforward Route to Enantiopure Pyrrolizidines and Indolizidines by Cycloaddition to Pyrroline N-Oxides Derived from the Chiral Pool

Abstract
:Introduction
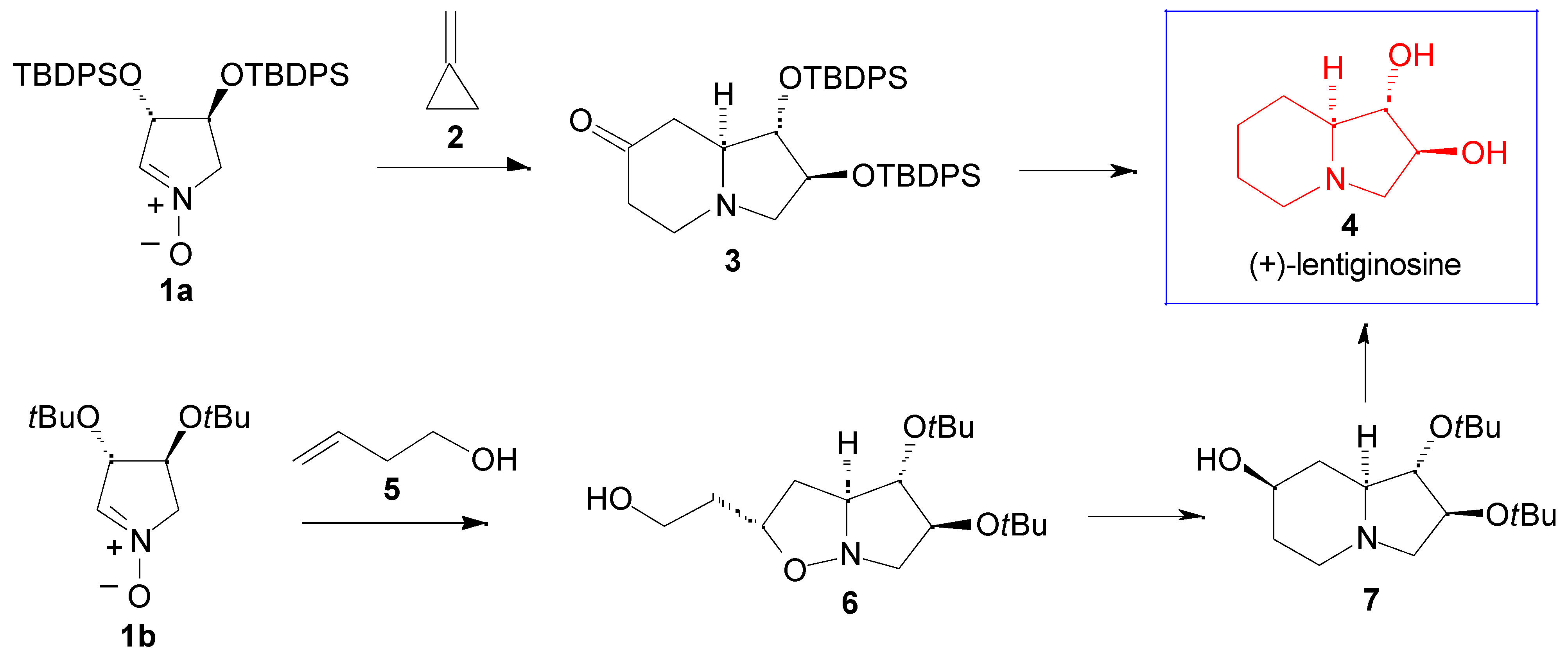
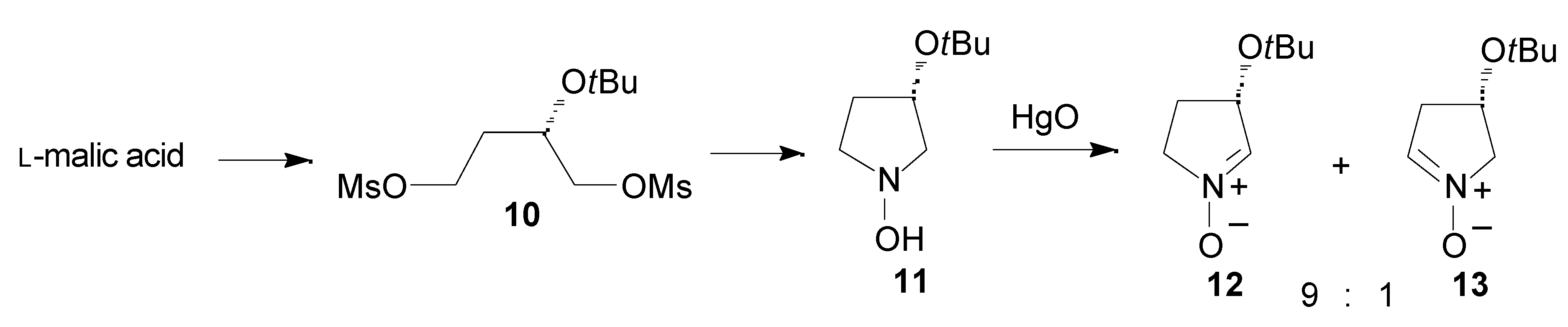
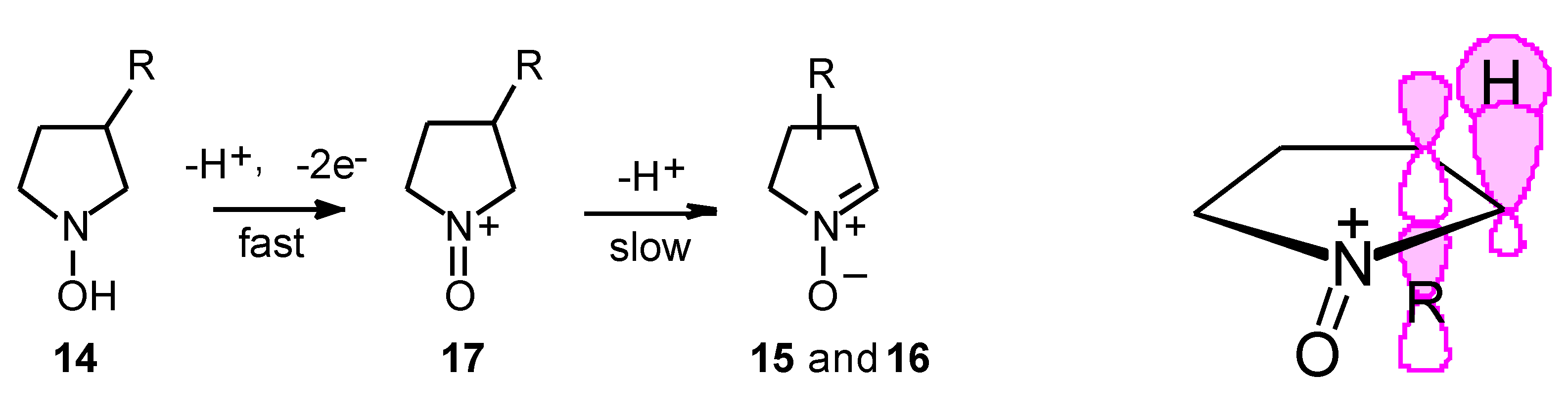
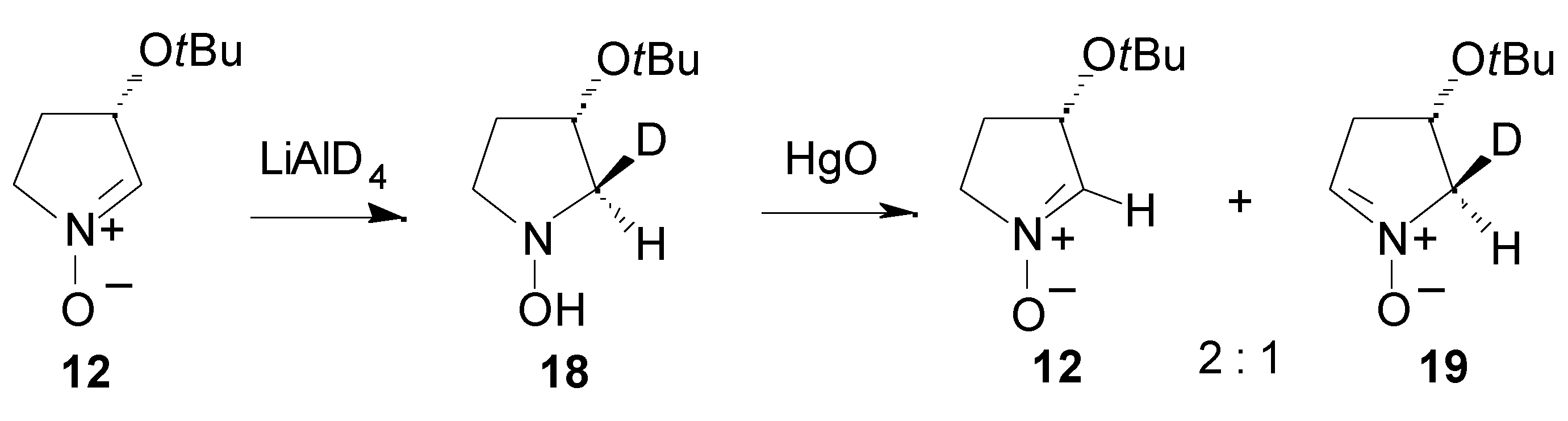
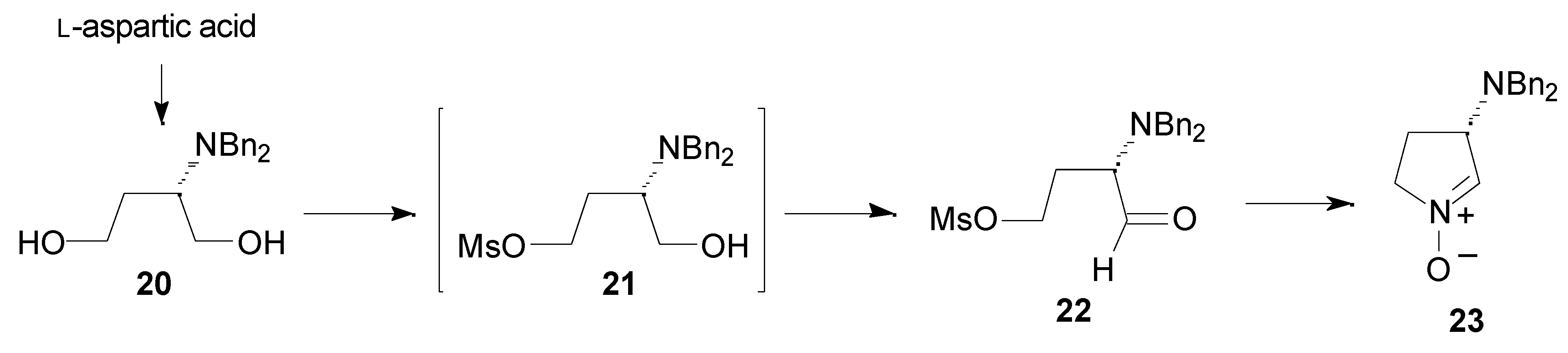
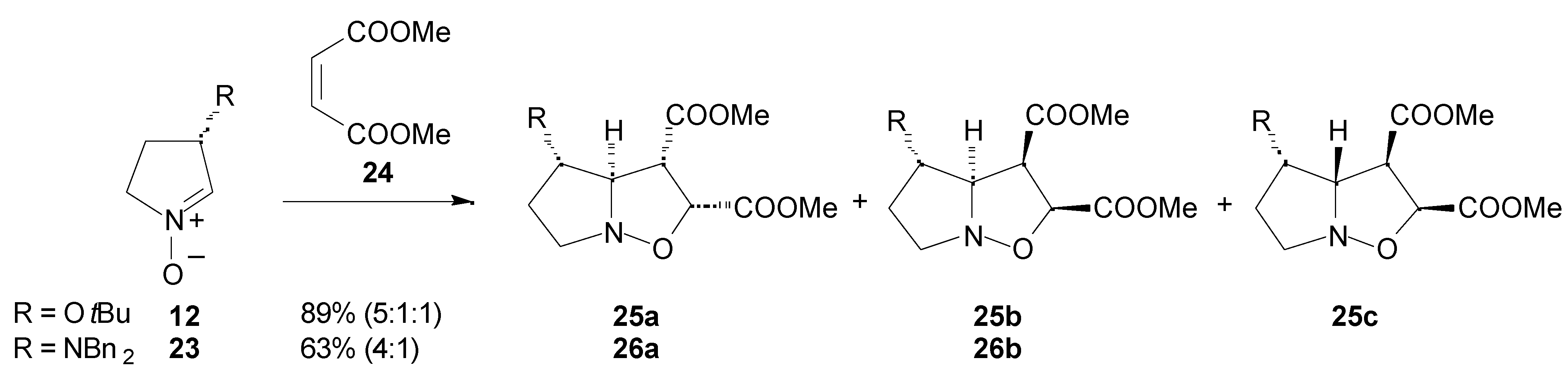
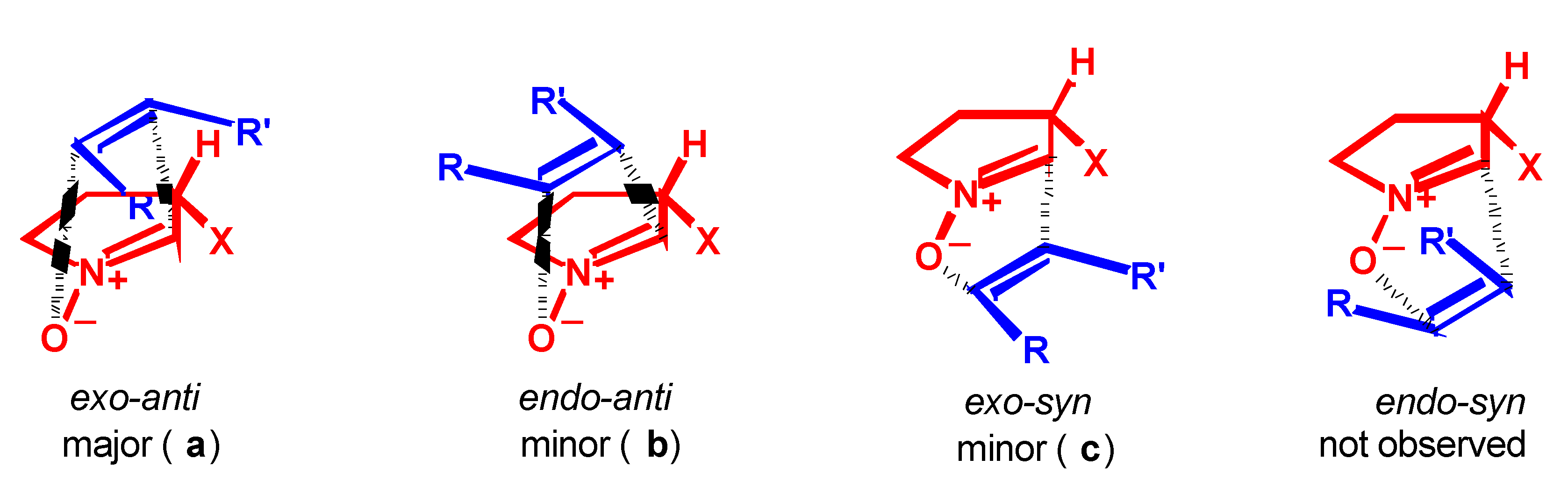
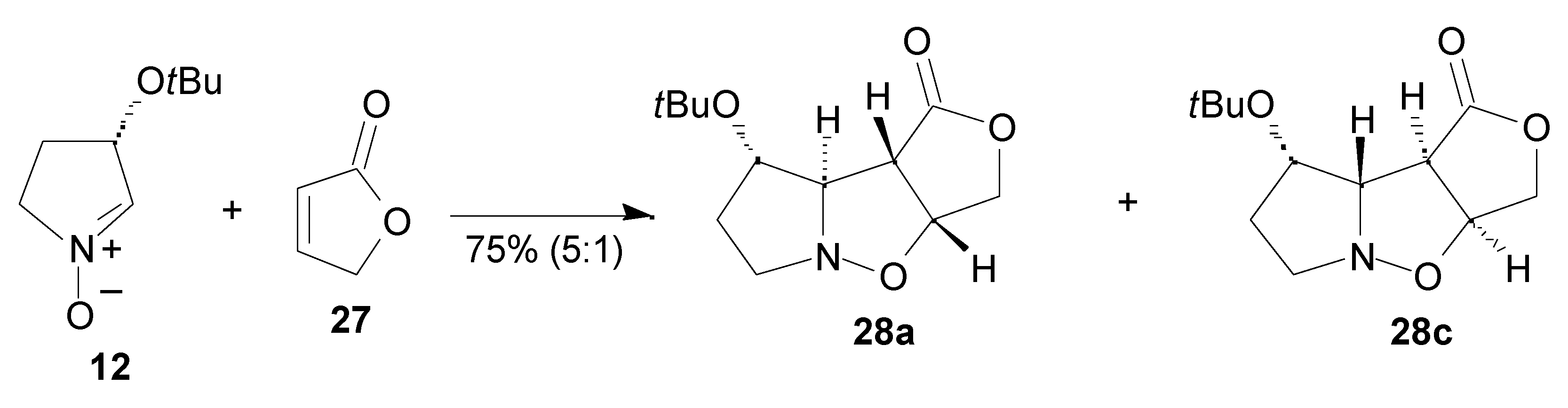
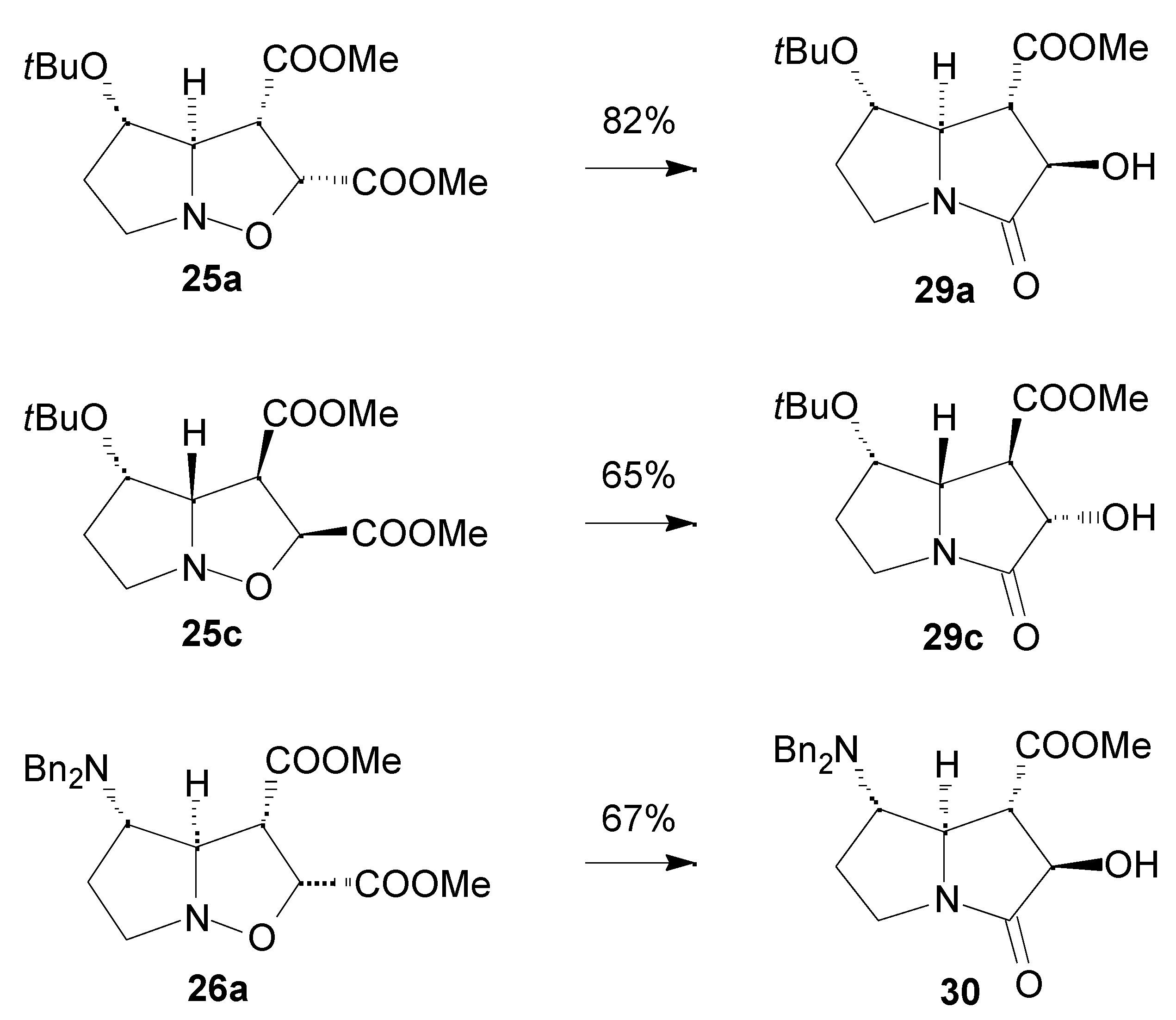
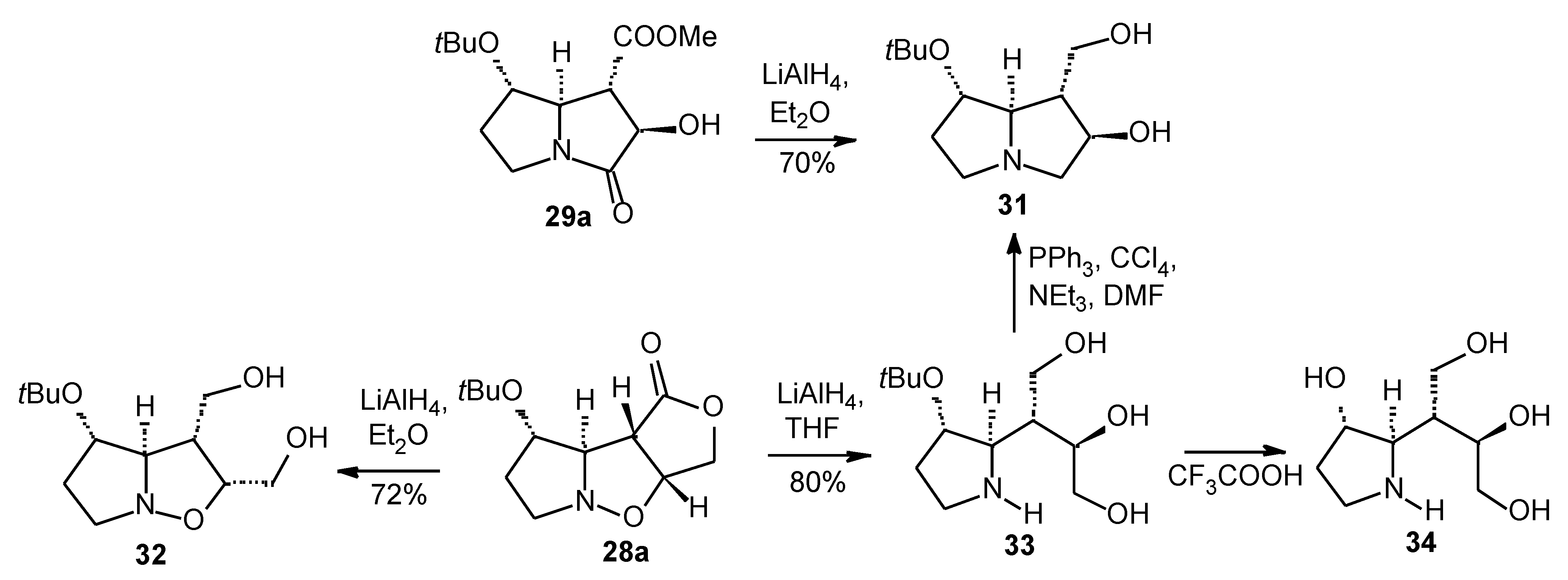
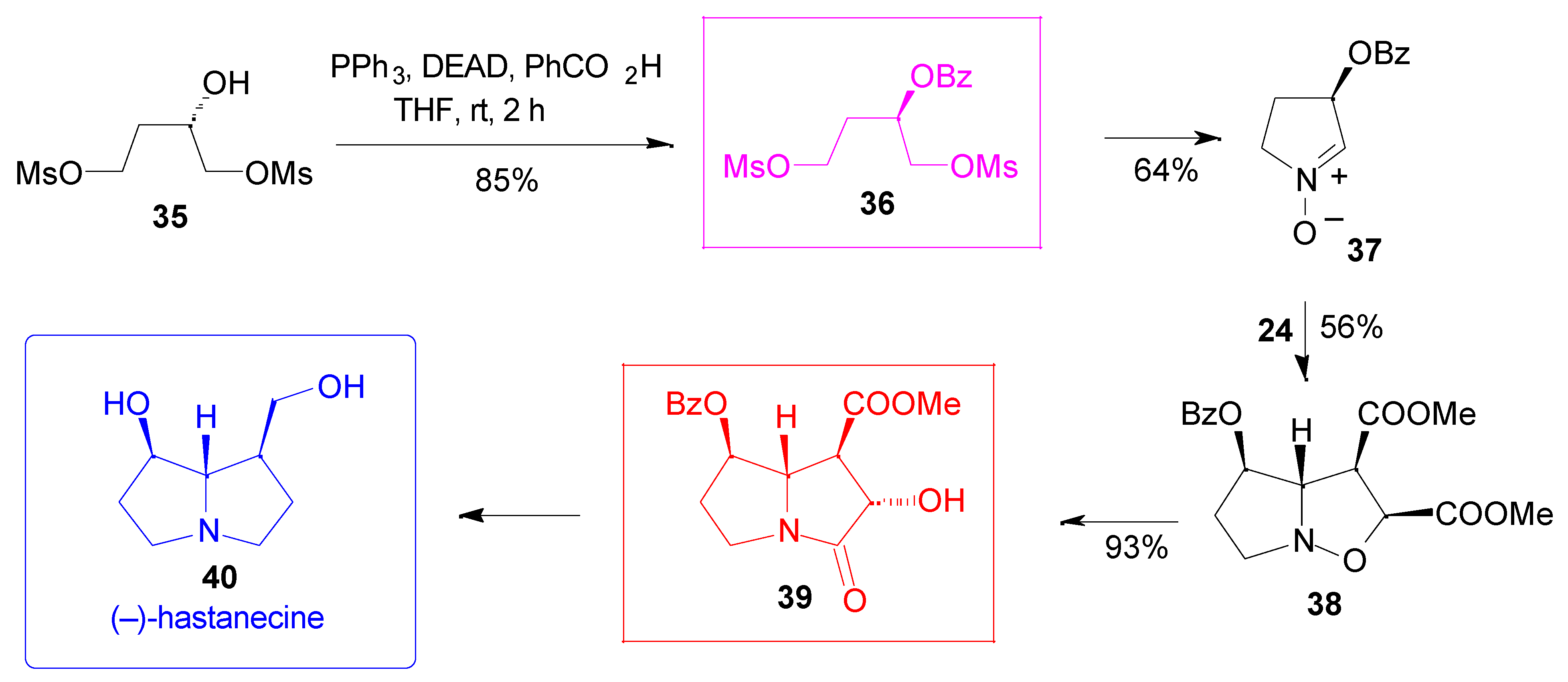
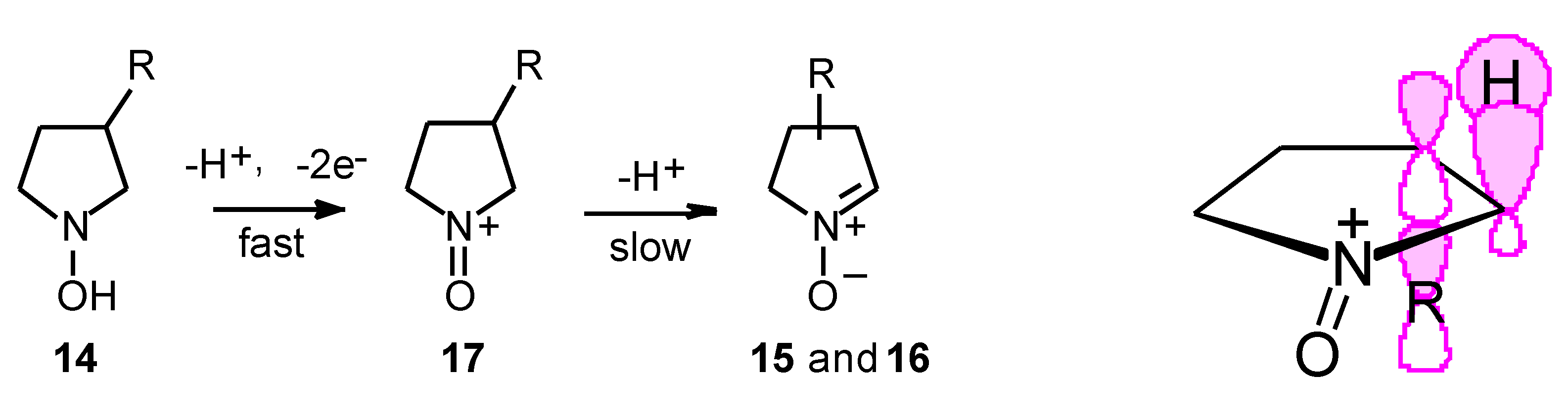
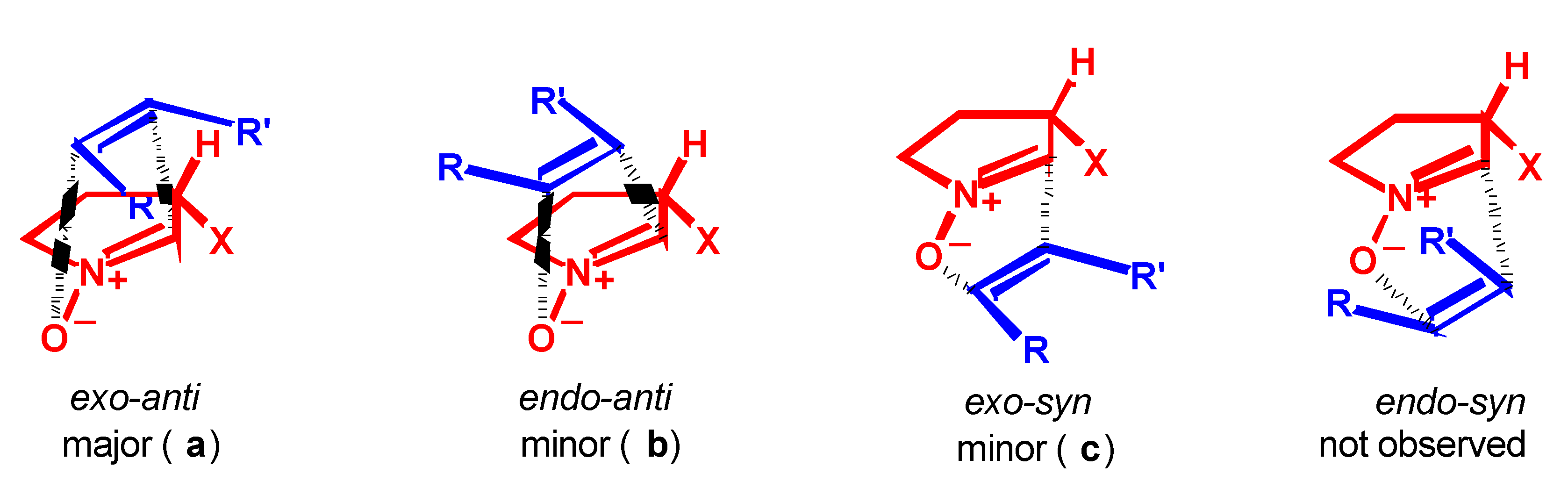
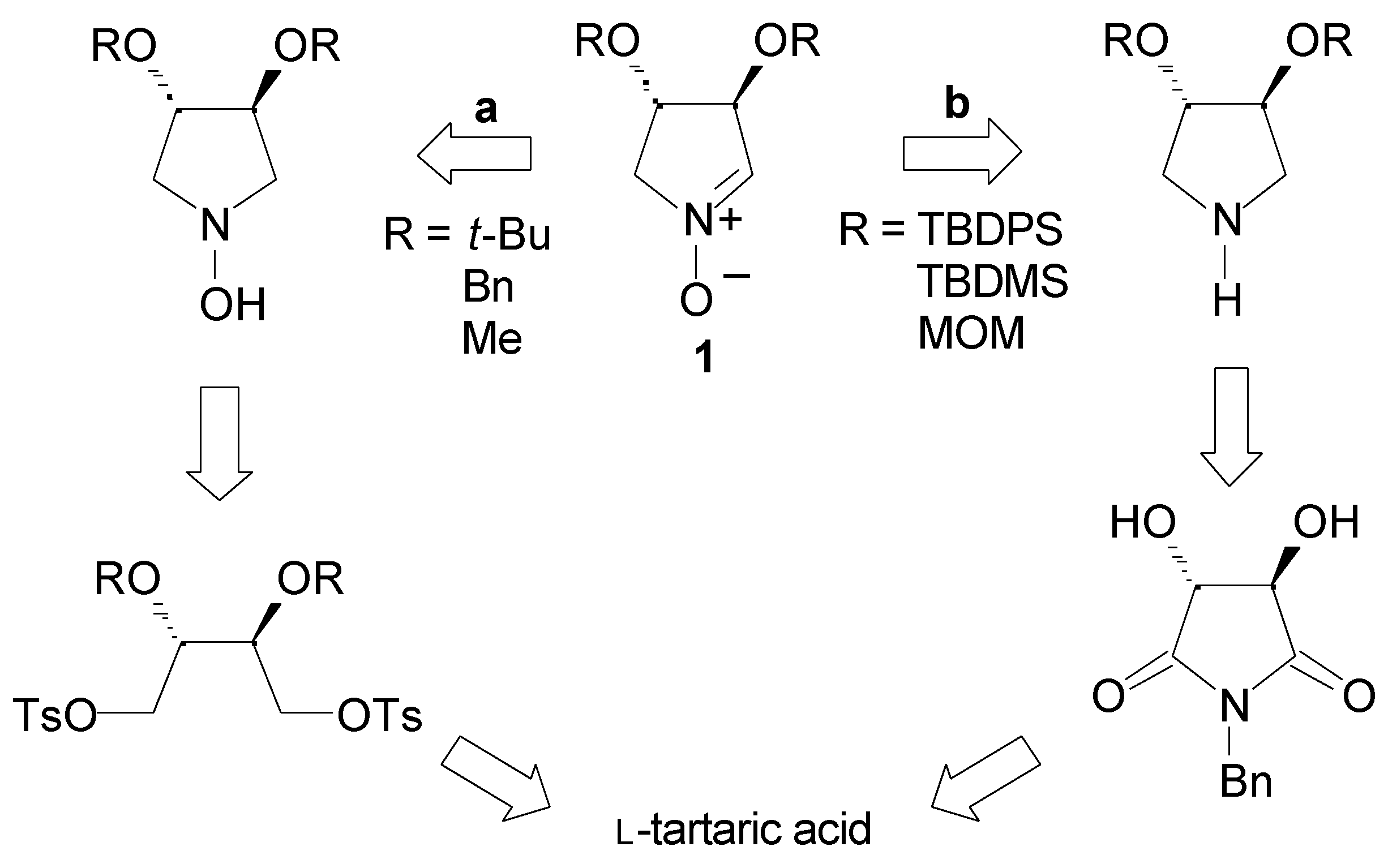
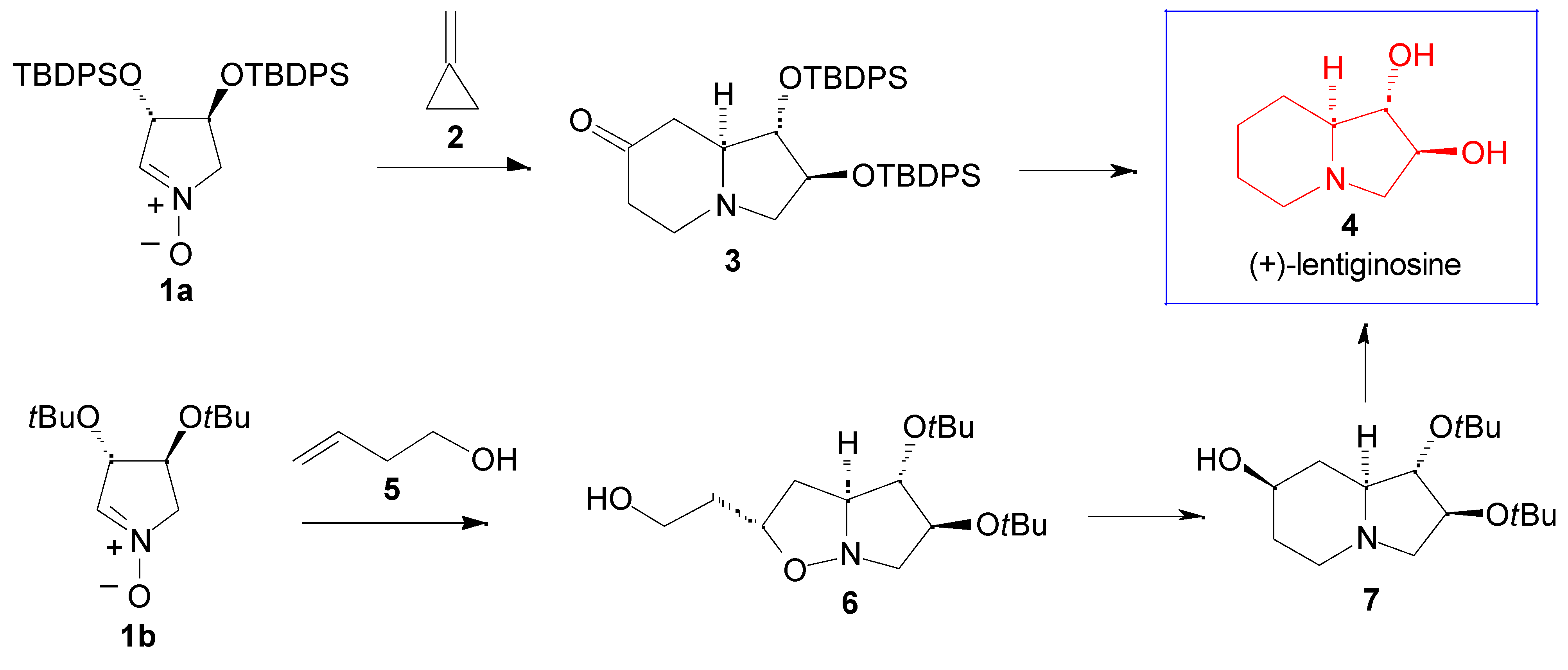
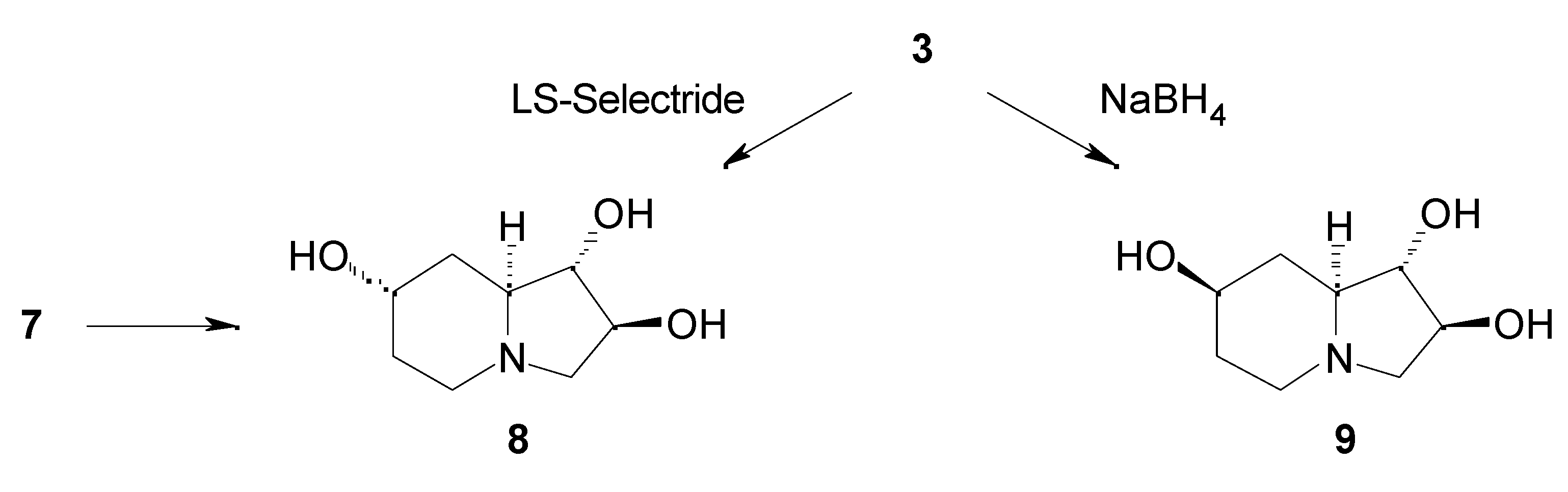
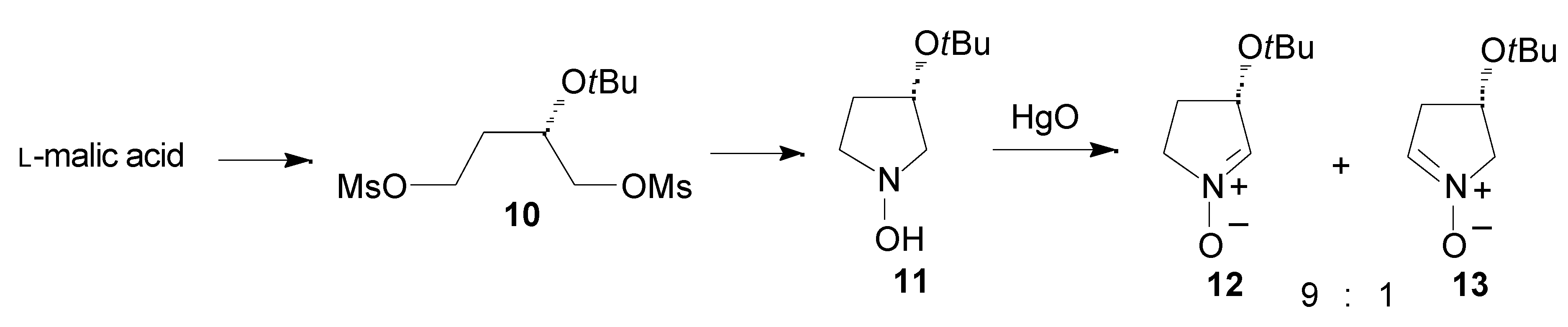
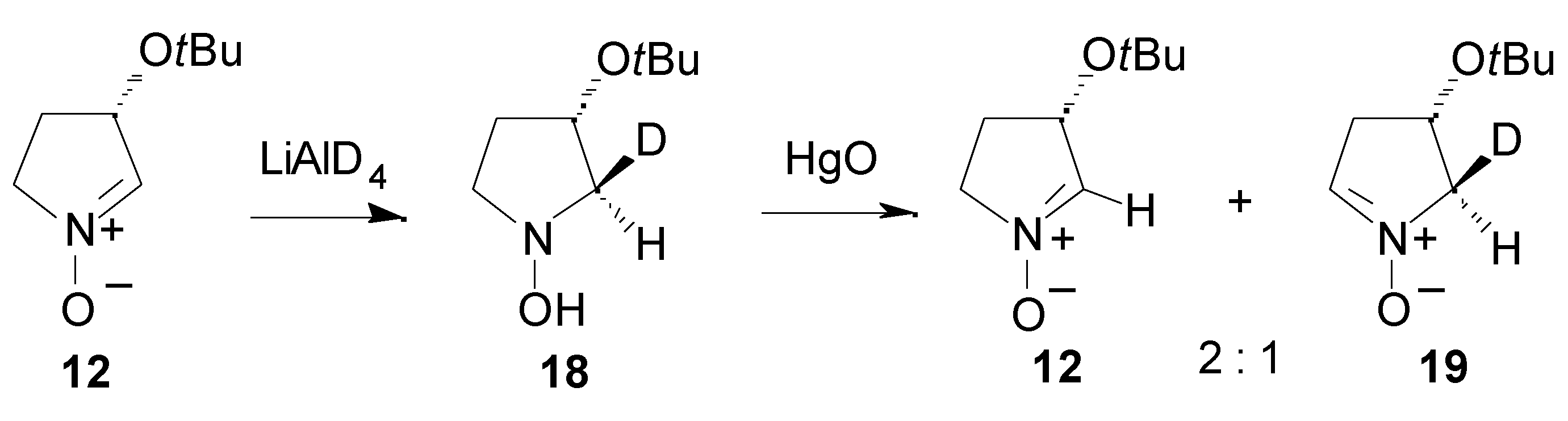
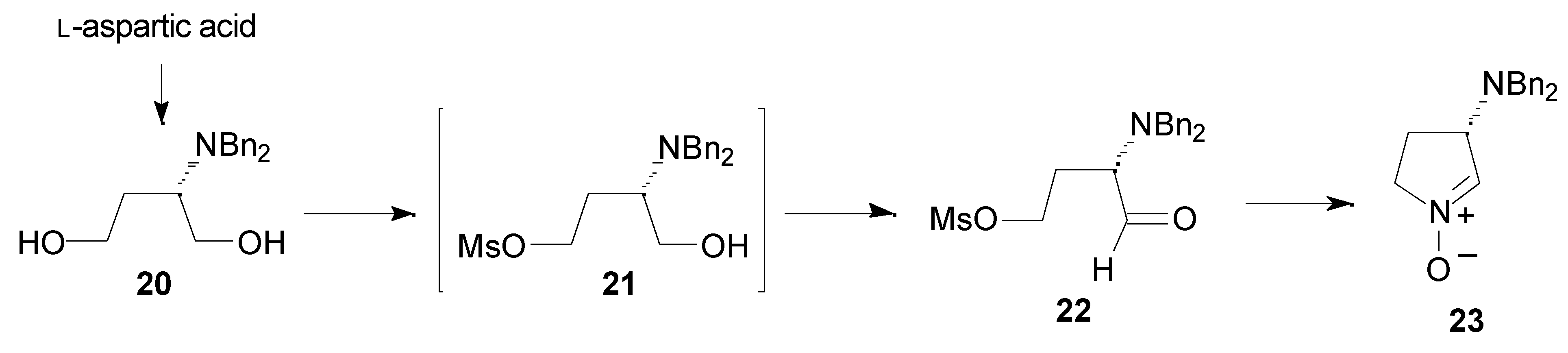
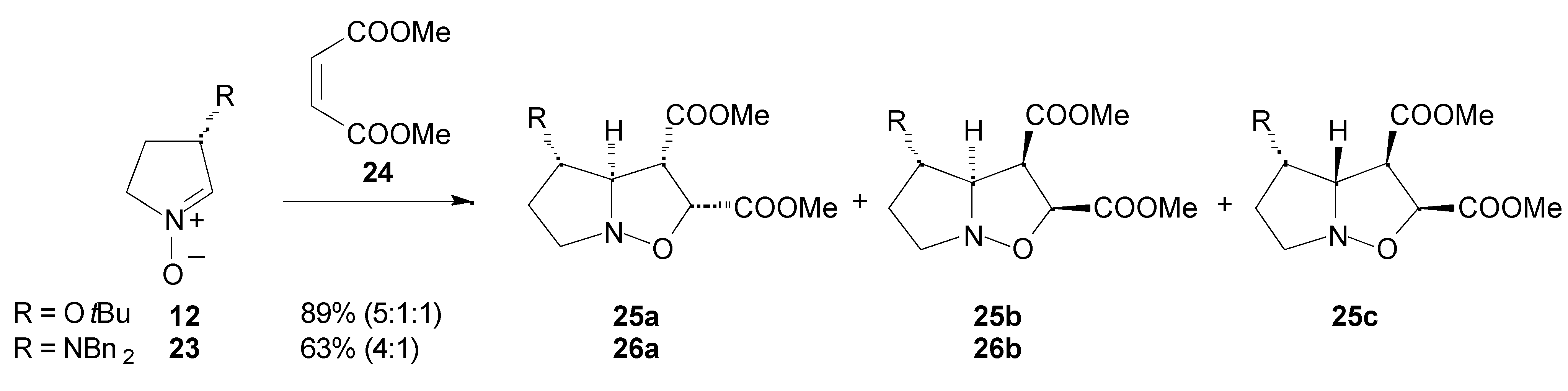
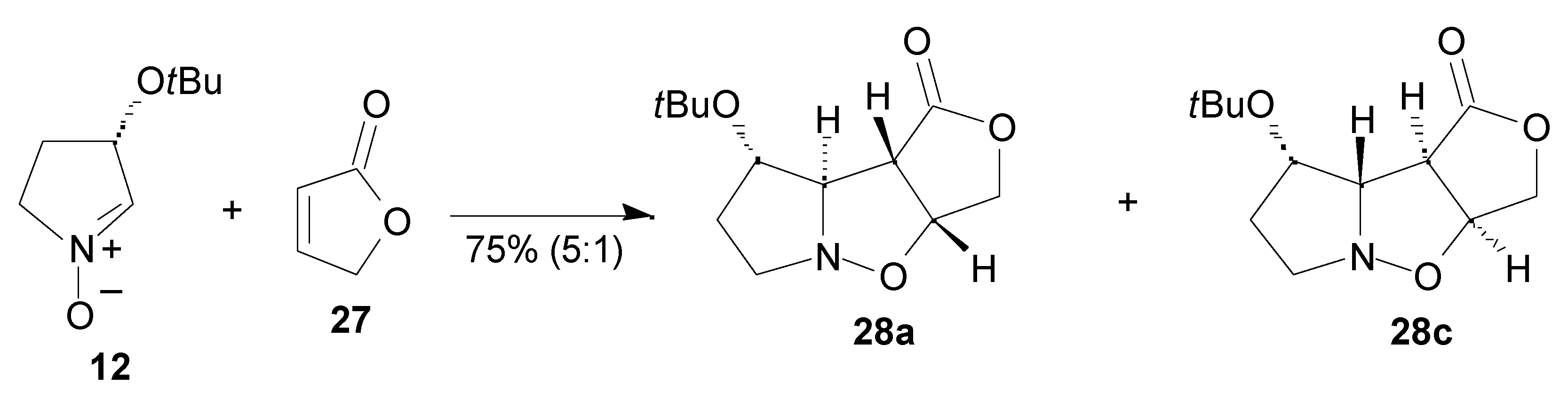
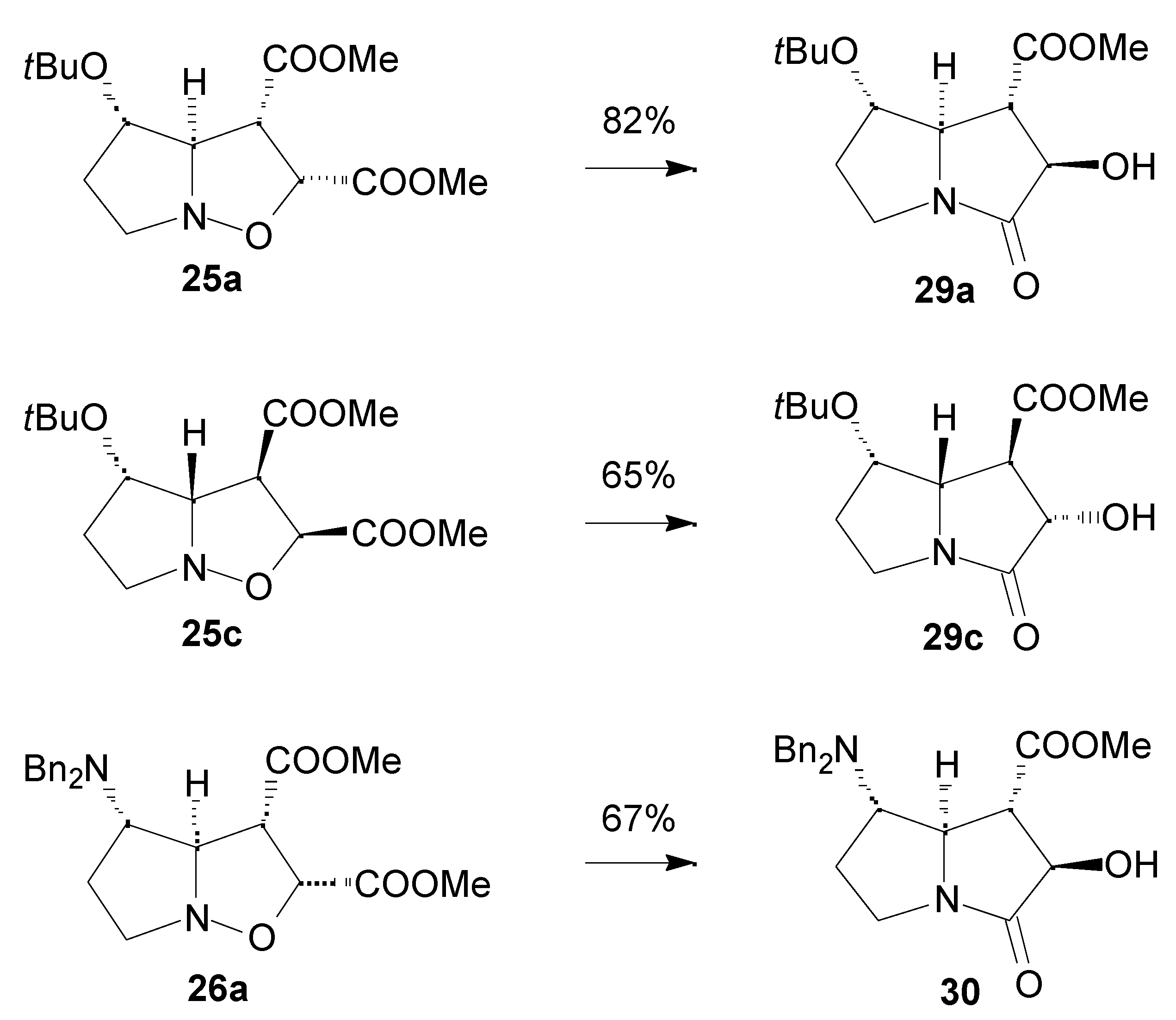
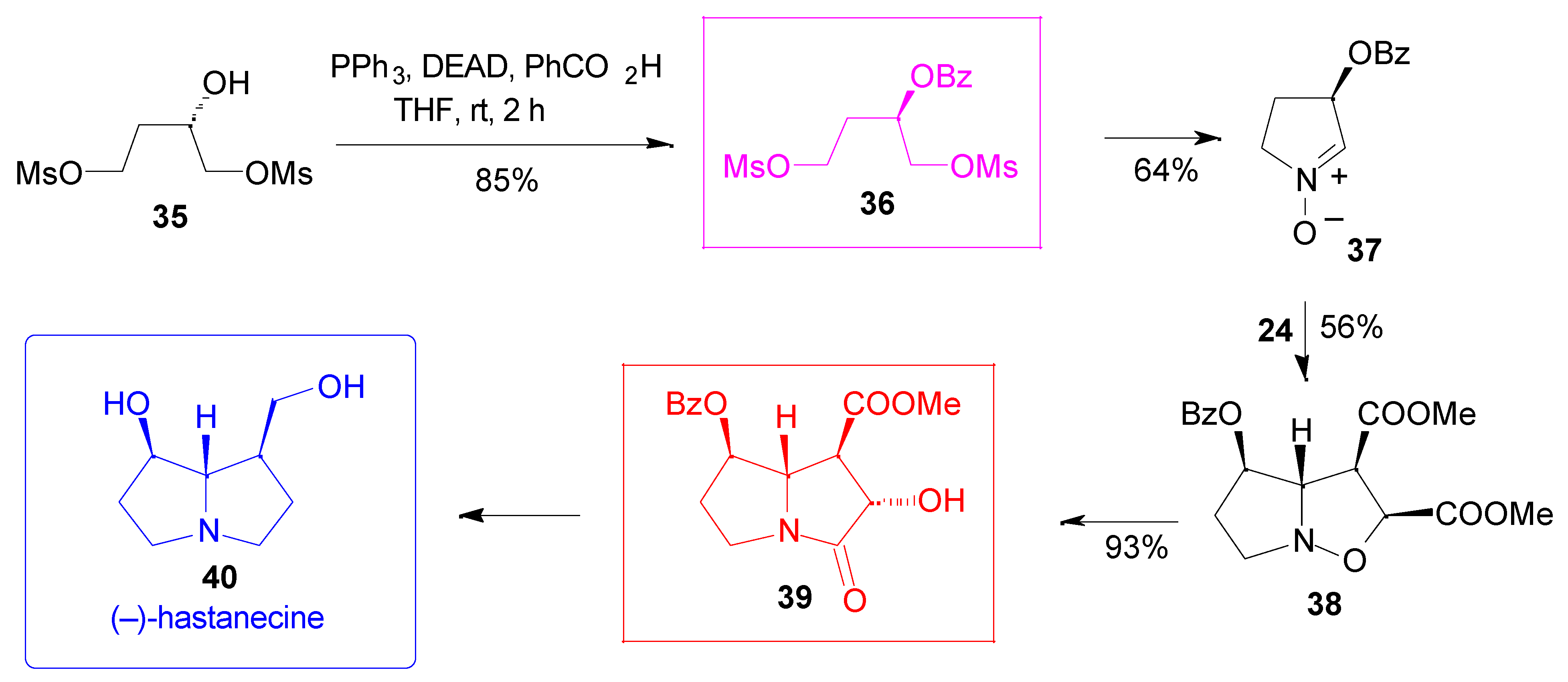
Results
Conclusion
References and Notes
- Tufariello, J. J. 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; John Wiley & Sons: New York, 1984. [Google Scholar] Confalone, P. N.; Huie, E. M. Org. React. 1988, 36, 1–173. Torssell, K. B. G. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; Feuer, H., Ed.; VCH Publishers: New York, 1988. [Google Scholar] Döpp, D.; Döpp, H. Houben-Weyl - Methoden der organischen Chemie; vol. E14b, Klamann, D., Hagemann, H., Eds.; Georg Thieme Verlag: Stuttgart, 1990. [Google Scholar] Breuer, E. The Chemistry of Amino, Nitroso and Nitro Compounds and Their Derivatives; Patai, S., Ed.; Wiley Interscience: New York, 1982. [Google Scholar] Frederickson, M. Tetrahedron 1997, 53, 403–425.
- Enders, D.; Reinhold, U. Tetrahedron: Asymmetry 1997, 8, 1895–1946, and references cited therein. Dondoni, A.; Junquera, F.; Merchán, F. L.; Merino, P.; Scherrmann, M.-C.; Tejero, T. J. Org. Chem. 1997, 62, 5484–5496. Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T.; Bertolasi, V. Chem. Eur. J. 1995, 1, 505–520. Merino, P.; Lanaspa, A.; Merchán, F. L.; Tejero, T. Tetrahedron: Asymmetry 1997, 8, 2381–2401. Merino, P.; Franco, S.; Merchán, F. L.; Tejero, T. Tetrahedron: Asymmetry 1997, 8, 3489–3496. Degiorgis, F.; Lombardo, M.; Trombini, C. Synthesis 1997, 1243–1245. Mancini, F.; Piazza, M. G.; Trombini, C. J. Org. Chem. 1991, 56, 4246–4252. Giovannini, R.; Marcantoni, E.; Petrini, M. J. Org. Chem. 1995, 60, 5706–5707. Ballini, R.; Marcantoni, E.; Petrini, M. J. Org. Chem. 1992, 57, 1316–1318.
- Hamer, J.; Macaluso, A. Chem. Rev. 1964, 64, 473–495.
- Desimoni, G.; Tacconi, G.; Barco, A; Pollini, G. P. Natural Products Synthesis Through Pericyclic Reactions; ACS Monograph n. 180; Caserio, M. C., Ed.; American Chemical Society: Washington, 1983. [Google Scholar] Tufariello, J. J. Acc. Chem. Res. 1979, 12, 396–403.
- For some recent examples on the synthesis and use of different chiral, enantiomerically pure, endocyclic nitrones, see: Golik, J.; Wong, H.; Krishnan, B.; Vyas, D. M.; Doyle, T. W. Tetrahedron Lett. 1991, 32, 1851–1854. Tronchet, J. M. J.; Zosimo-Landolfo, G.; Balkadjian, M.; Ricca, A.; Zsély, M.; Barbalat-Rey, F.; Cabrini, D.; Lichtle, P.; Geoffroy, M. Tetrahedron Lett. 1991, 32, 4129–4132. Grigg, R.; Markandu, J.; Perrior, T.; Surendrakumar, S.; Warnock, W. J. Tetrahedron 1992, 48, 6929–6952. Herczegh, P.; Kovács, I.; Szilágyi, L.; Varga, T.; Dinya, T.; Sztaricskai, F. Tetrahedron Lett. 1993, 34, 1211–1214. Berranger, T.; Langlois, Y. J. Org. Chem. 1995, 60, 1720–1726. [CrossRef] de March, P.; Figueredo, M.; Font, J.; Gallagher, T.; Milán, S. J. Chem. Soc., Chem. Commun. 1995, 2097–2098. van den Broek, L. A. G. M. Tetrahedron 1996, 52, 4461–4478. Ishikawa, T.; Tajima, Y.; Fukui, M.; Saito, S. Angew. Chem. Int. Ed. Engl. 1996, 35, 1863–1864. Tamura, O.; Gotanda, K.; Terashima, R.; Kikuchi, M.; Miyawaki, T.; Sakamoto, M. J. Chem. Soc., Chem. Commun. 1996, 1861–1862. Hall, A.; Meldrum, K. P.; Therond, P. R.; Wightman, R. H. Synlett 1997, 123–125. Closa, M.; de March, P.; Figueredo, M.; Font, J. Tetrahedron: Asymmetry 1997, 8, 1031–1037. Katagiri, N.; Okada, M.; Morishita, Y.; Kaneko, C. Tetrahedron 1997, 53, 5725–5746. Chackalamannil, S.; Wang, Y. Tetrahedron 1997, 53, 11203–11210.
- Cicchi, S.; Höld, I.; Brandi, A. J. Org. Chem. 1993, 58, 5274–5275. Brandi, A.; Cicchi, S.; Goti, A.; Koprowski, A.; Pietrusiewicz, K. M. J. Org. Chem. 1994, 59, 1315–1318. Goti, A.; Cardona, F.; Brandi, A.; Picasso, S.; Vogel, P. Tetrahedron: Asymmetry 1996, 7, 1659–1674.
- Reviews: Elbein, A. D. Ann. Rev. Biochem. 1987, 56, 497–534. [PubMed]Vogel, P. Chimica Oggi 1992, 10(August–September), 9–15. Winchester, B.; Fleet, G. W. J. Glycobiology 1992, 2, 199–210. [PubMed]Legler, G. Adv. Carbohydr. Chem. Biochem. 1990, 48, 319–384. [PubMed]Elbein, A. D.; Molyneux, R. J. Alkaloids: Chemical and Biological Perspectives; Pelletier, S. W., Ed.; Wiley-Interscience: New York, 1987; vol. 5, ch. 1. [Google Scholar] Howard, A. S.; Michael, J. P. The Alkaloids; Brossi, A., Ed.; Academic Press: New York, 1986; vol. 28, ch. 3. [Google Scholar] Cossy, J.; Vogel, P. Studies in Natural Product Chemistry; Atta-ur-Rahman, Ed.; Elsevier: New York, 1993; vol. 12, p. 275. [Google Scholar]
- Isolation: Pastuszak, I.; Molyneux, R. J.; James, L. F.; Elbein, A. D. Biochemistry 1990, 29. [PubMed]
- Brandi, A.; Cicchi, S.; Cordero, F. M.; Frignoli, R.; Goti, A.; Picasso, S.; Vogel, P. J. Org. Chem. 1995, 60, 6806–6812.
- Cardona, F.; Goti, A.; Brandi, A.; Scarselli, M.; Niccolai, N.; Mangani, S. J. Mol. Model. 1997, 3, 249–260.
- Cordero, F. M.; Cicchi, S.; Goti, A.; Brandi, A. Tetrahedron Lett. 1994, 35, 949–952.
- Brandi, A.; Cordero, F. M.; Goti, A.; De Sarlo, F.; Guarna, A. Synlett 1993, 1–8. Goti, A.; Cordero, F. M.; Brandi, A. Top. Curr. Chem. 1996, 178, 1–97.
- Goti, A.; Cardona, F.; Brandi, A. Synlett 1996, 761–763.
- Tufariello, J. J.; Tegeler, J. J. Tetrahedron Lett. 1976, 4037–4040.
- Cicchi, S.; Goti, A.; Brandi, A. J. Org. Chem. 1995, 60, 4743–4748.
- Goti, A.; Cicchi, S.; Fedi, V.; Nannelli, L.; Brandi, A. J. Org. Chem. 1997, 62, 3119–3125. [PubMed]
- Goti, A.; De Sarlo, F.; Romani, M. Tetrahedron Lett. 1994, 35, 6517–6574.
- Murahashi, S.-I.; Imada, Y.; Ohtake, H. J. Org. Chem. 1994, 59, 6170–6172.
- For a synthesis of hydroxyindolizidines from nitrone 12, see ref. 15 and: Mc Caig, A. E.; Wightman, R. H. Tetrahedron Lett. 1993, 34, 3939–3942.
- Syn and anti refer to the approaches of dipolarophile from the same or the opposite face of the substituent at C-3 of the nitrone, respectively.
- Tufariello, J. J.; Tette, J. P. J. Org. Chem. 1975, 40, 3866–3869. [PubMed]
- Cid, P.; de March, P.; Figueredo, M.; Font, J.; Milán, S.; Soria, Á.; Virgili, A. Tetrahedron 1993, 49, 3857–3870. de Lange, B.; Feringa, B. L. Tetrahedron Lett. 1988, 29, 5317–5320. Banerji, A.; Basu, S. Tetrahedron 1992, 48, 3335–3344. Saito, S.; Ishikawa, T.; Kishimoto, N.; Kohara, T.; Moriwake, T. Synlett 1994, 282–284.
- Huisgen, R.; Hauck, H.; Grashey, R.; Seidl, H. Chem. Ber. 1968, 101, 2568–2584.
- Cicchi, S.; Goti, A.; Brandi, A.; Guarna, A.; De Sarlo, F. Tetrahedron Lett. 1990, 31, 3351–3354.
- Hartmann, T.; Witte, L. Chemistry, Biology and Chemoecology of the Pyrrolizidine Alkaloids. In Alkaloids: Chemical & Biological Perspectives; Pelletier, S. W., Ed.; Pergamon: Oxford, 1995; vol. 9, pp. 155–233. [Google Scholar] Mattocks, A. R. Chemistry and Toxicology of Pyrrolizidine Alkaloids; Academic Press: New York, 1986. [Google Scholar]
- Goti, A.; Cicchi, S.; Fedi, V.; Nannelli, L.; Brandi, A. Synlett 1997, 577–579.
- Denmark, S. E.; Thorarensen, A. J. Org. Chem. 1994, 59, 5672–5680.
- Samples Availability: Available from the authors.




| R | Regioisomeric Ratio | Yield (%) |
|---|---|---|
| Me | 1.8 | 90 |
| Ph | 2 | 95 |
| (S)-NBn2 | 4 | 12 |
| (S)-OH | 5 | 24 |
| (S)-OtBu | 9 | 80 |
| (S)-OAllyl | 9 | 100 |
| (S)-OTBDMS | 12 | 64 |
| (S)-OCOPh | >20 | 50 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 1999 MDPI. All rights reserved.
Share and Cite
Goti, A.; Cicchi, S.; Cordero, F.M.; Fedi, V.; Brandi, A. A Straightforward Route to Enantiopure Pyrrolizidines and Indolizidines by Cycloaddition to Pyrroline N-Oxides Derived from the Chiral Pool. Molecules 1999, 4, 1-12. https://doi.org/10.3390/40100001
Goti A, Cicchi S, Cordero FM, Fedi V, Brandi A. A Straightforward Route to Enantiopure Pyrrolizidines and Indolizidines by Cycloaddition to Pyrroline N-Oxides Derived from the Chiral Pool. Molecules. 1999; 4(1):1-12. https://doi.org/10.3390/40100001
Chicago/Turabian StyleGoti, Andrea, Stefano Cicchi, Franca M. Cordero, Valentina Fedi, and Alberto Brandi. 1999. "A Straightforward Route to Enantiopure Pyrrolizidines and Indolizidines by Cycloaddition to Pyrroline N-Oxides Derived from the Chiral Pool" Molecules 4, no. 1: 1-12. https://doi.org/10.3390/40100001