Hydroxyquinones: Synthesis and Reactivity

Laboratory of Organic Chemistry, Chemistry Department, University of Thessaloniki, Thessaloniki 54006, Greece
Molecules 2000, 5(12), 1291-1330; https://doi.org/10.3390/51201291
Submission received: 30 June 2000
/
Revised: 1 October 2000
/
Accepted: 6 October 2000
/
Published: 20 December 2000
{kind=link}
{kind=link}
{kind=link}
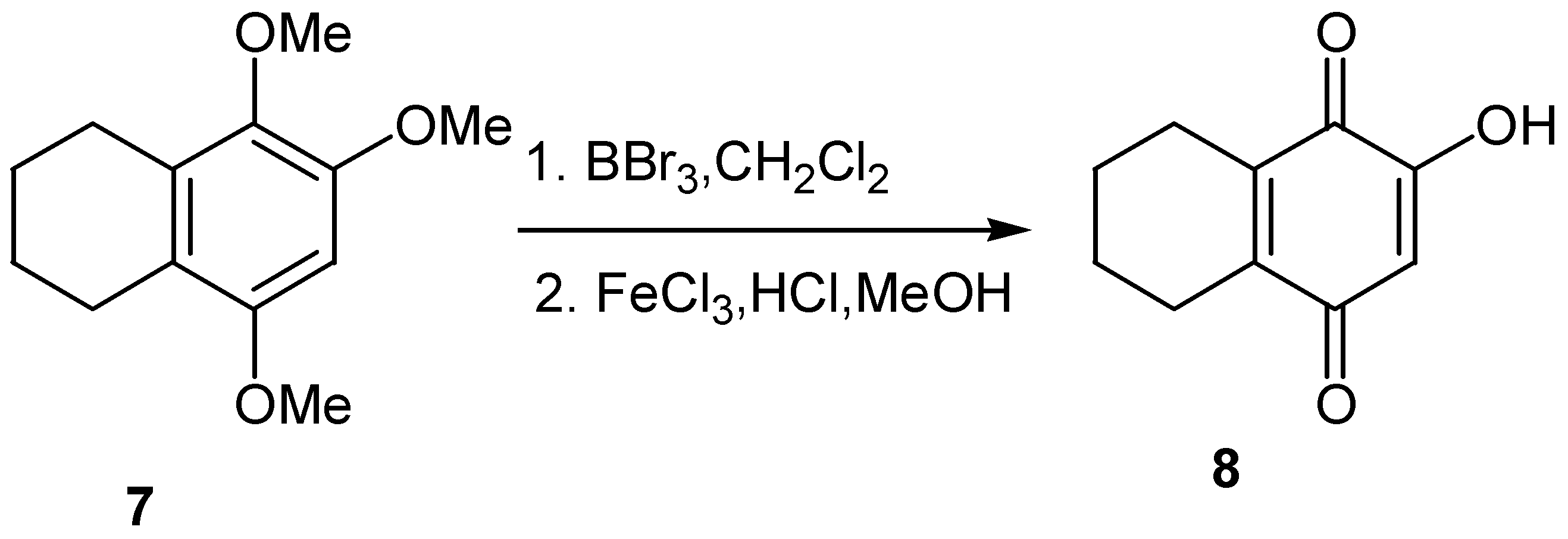{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
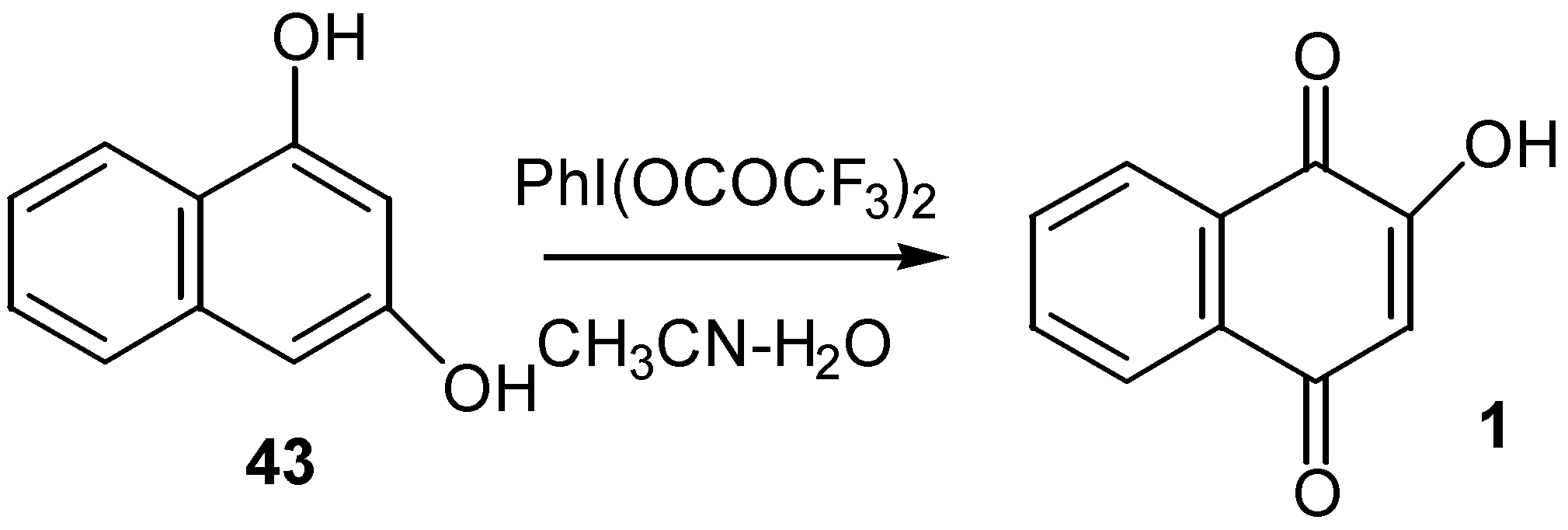{kind=link}
{kind=link}
{kind=link}
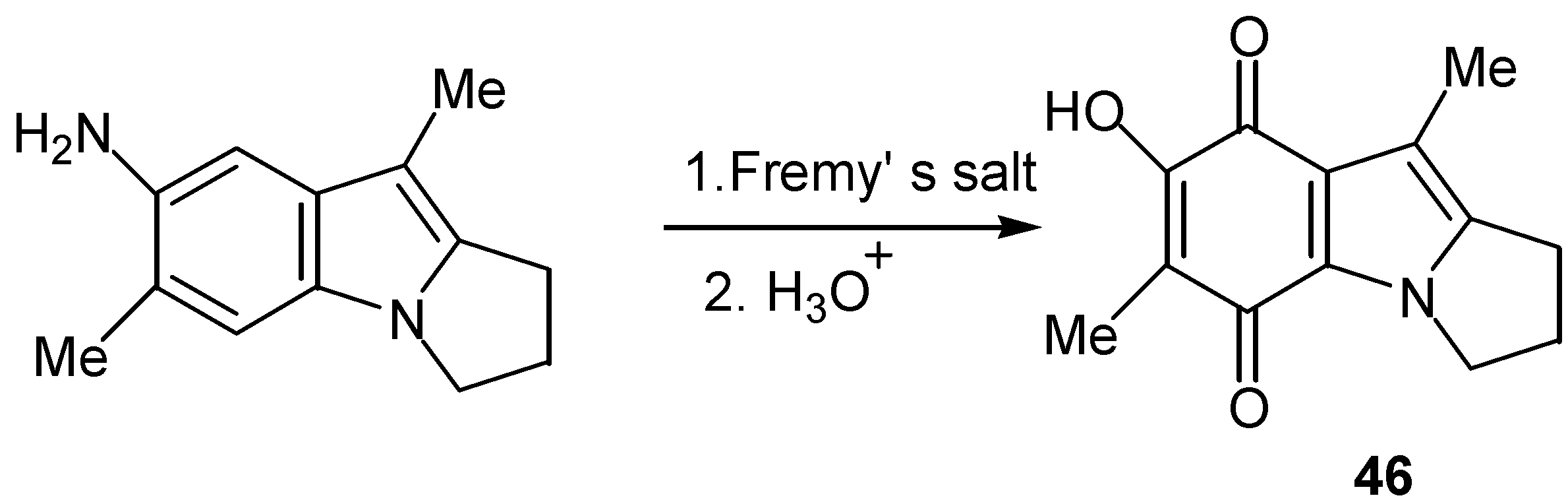{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
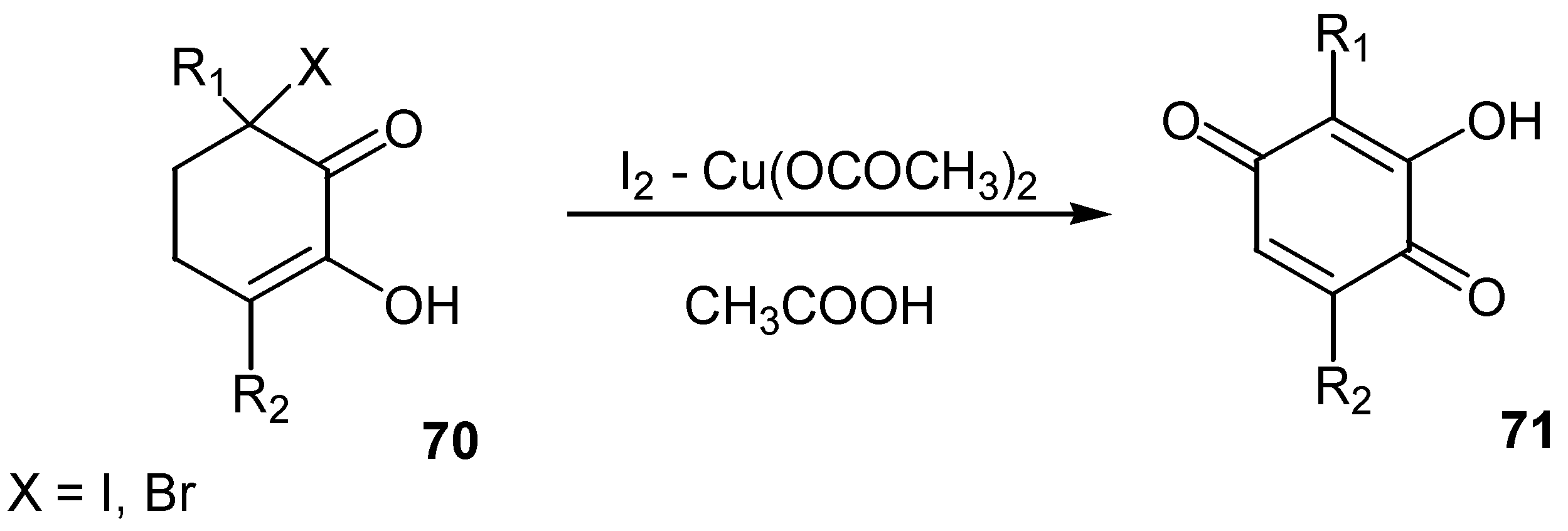{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
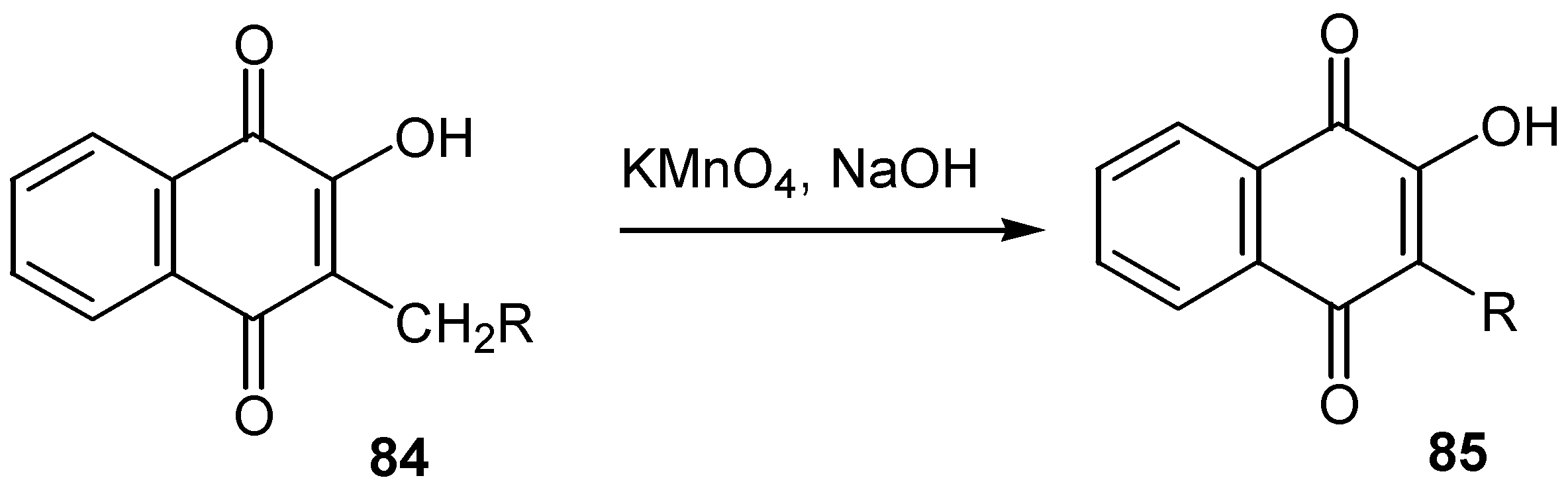{kind=link}
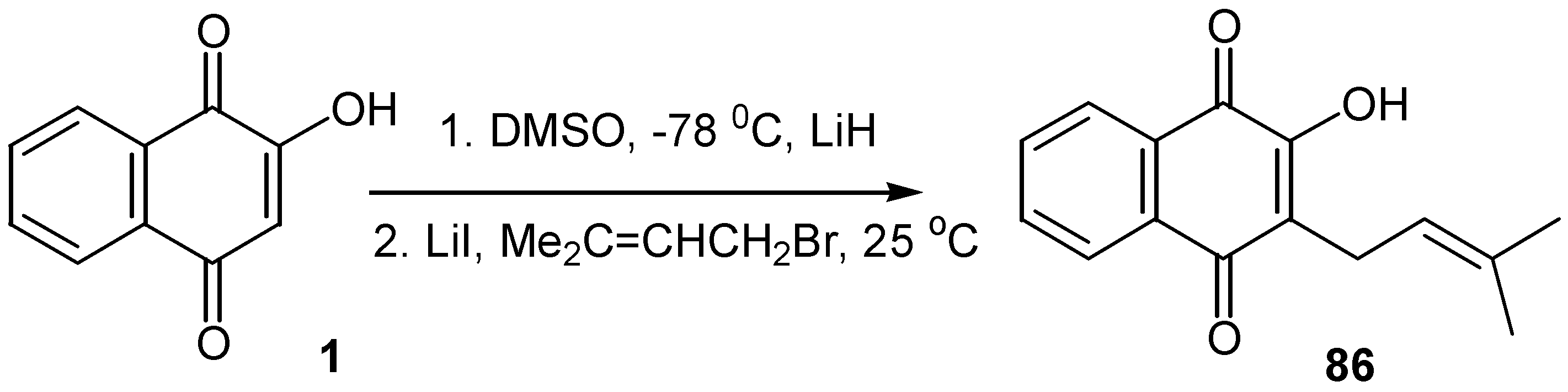{kind=link}
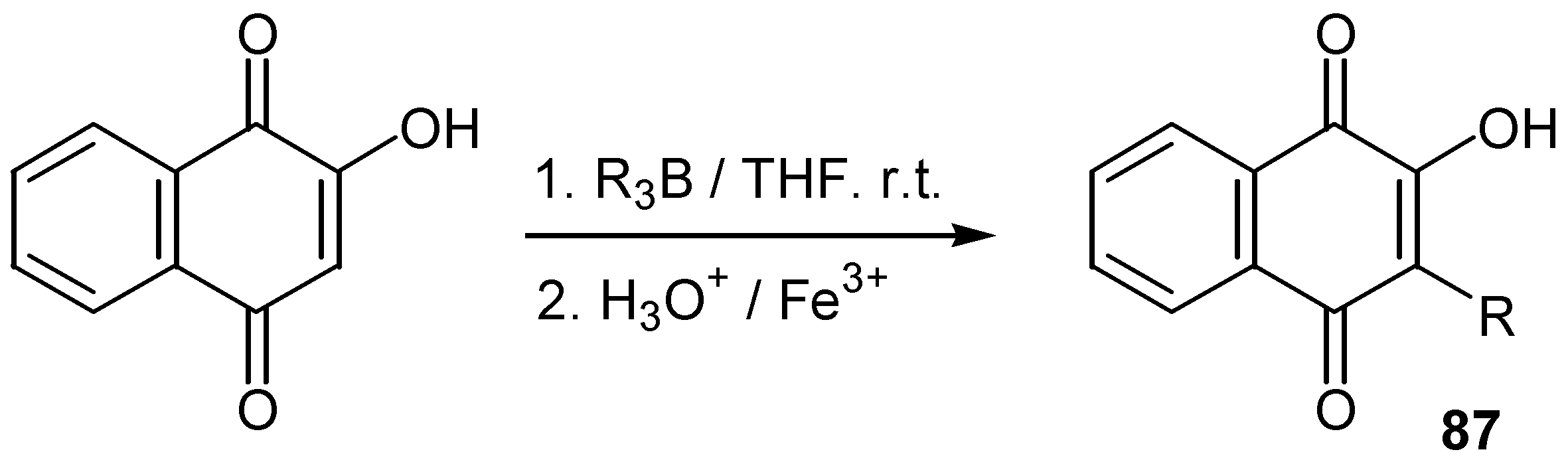{kind=link}
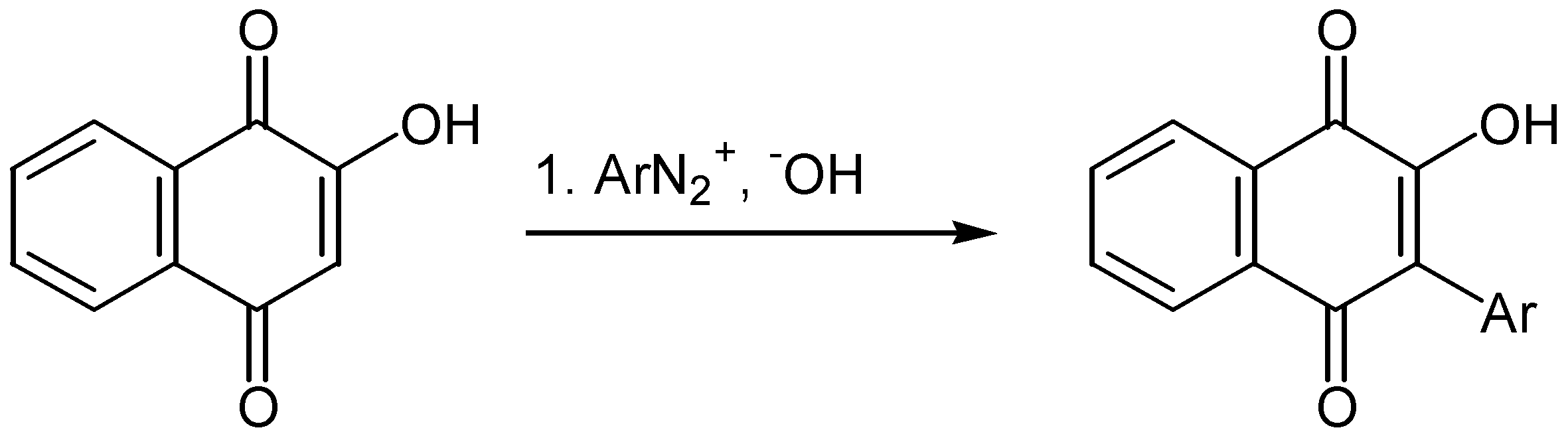{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
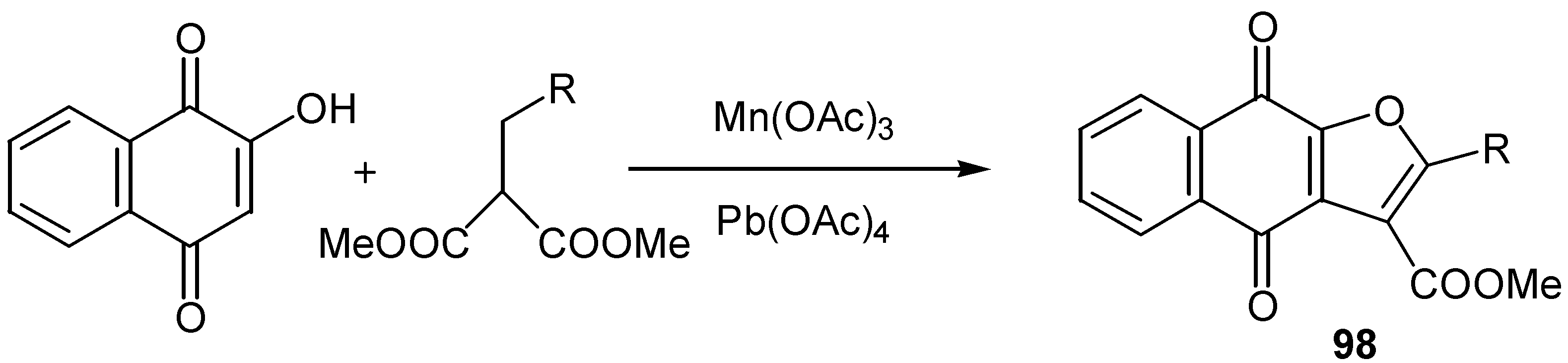{kind=link}
{kind=link}
{kind=link}
{kind=link}
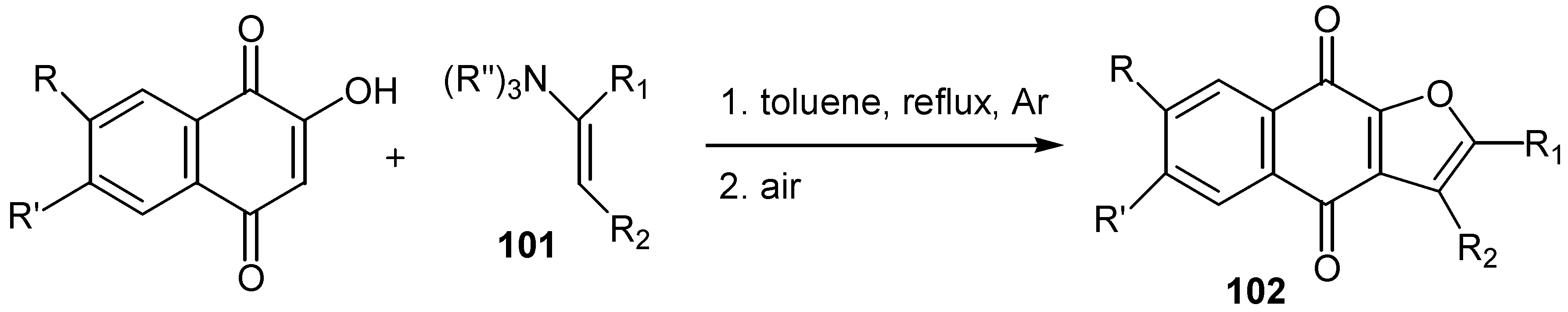{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
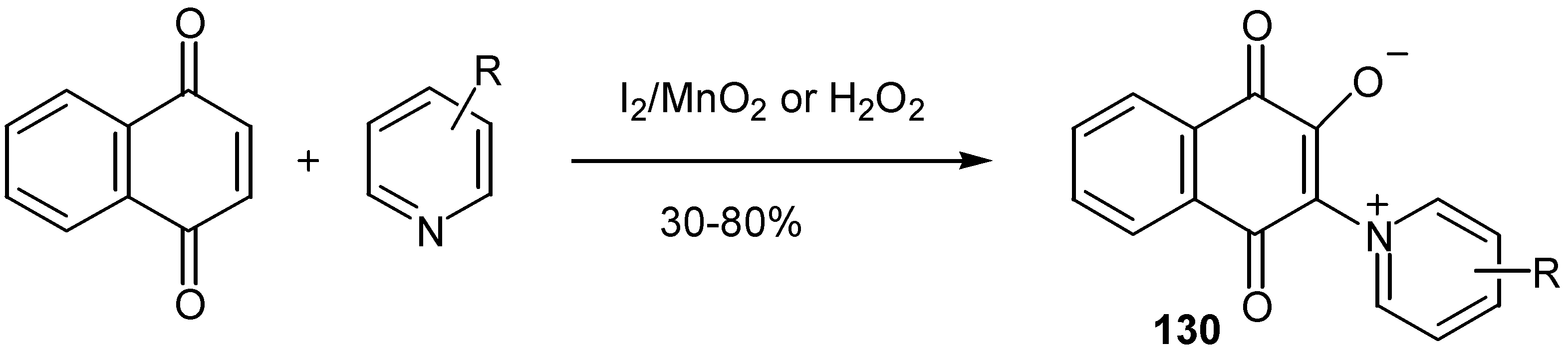{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
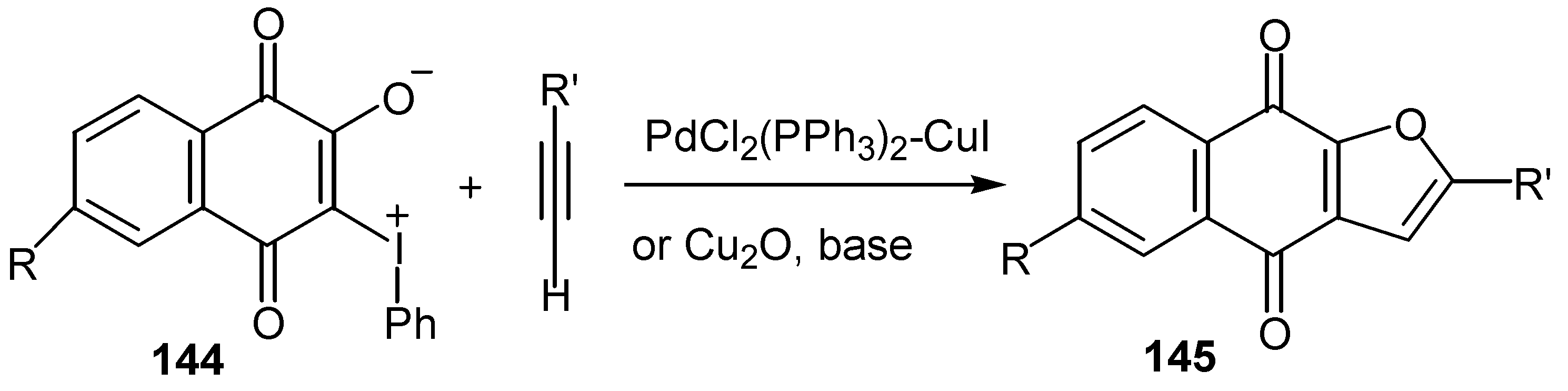{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
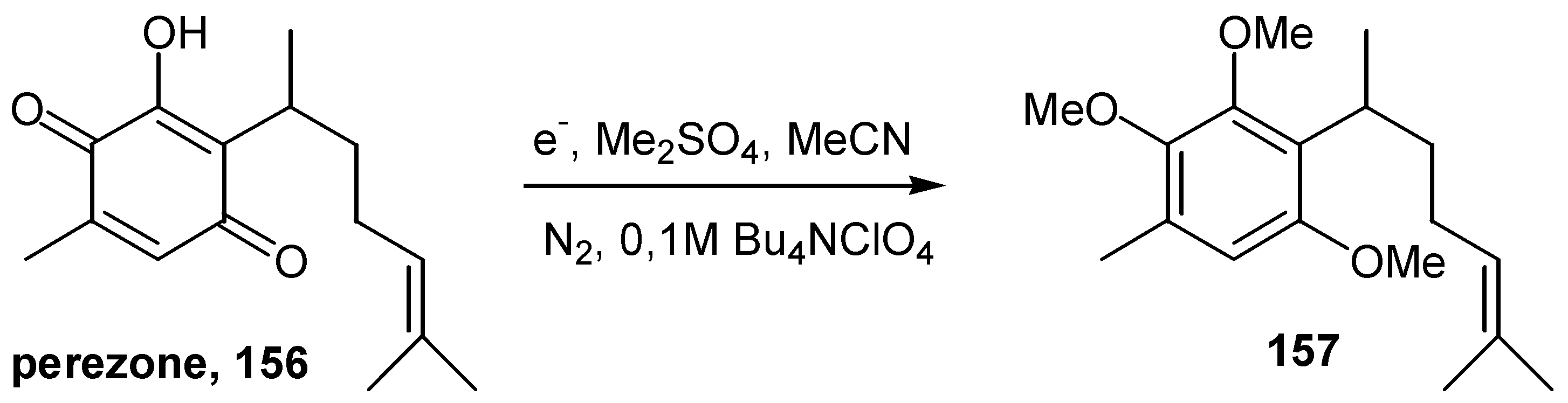{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Quinones having hydroxy groups directly attached to the quinone ring constitute a very interesting class of quinoid compounds. A great number of hydroxyquinones are found in nature and the majority of them exhibit unique biological activity. Their syntheses and their main reactivity patterns are reviewed in this paper.
Contents
- Introduction
- Synthesis
- Reactivity
- Conclusion
- References
1. Introduction
Molecules with the quinoid structure constitute one of the most interesting classes of compounds in organic chemistry. Their syntheses as well as their diverse chemical and physical properties have been compiled in the two volumes of Patai's series The Chemistry of Functional Groups [1].
The chemistry of quinones is largely dependent on the substituents being either on the quinonic or on adjacent rings. This is reflected in their chemical reactivity, especially in heterocyclic quinones [2].
Hydroxylated quinones that have one or more hydroxy groups attached directly to the quinone moiety are found in nature in great variety. As most of them exhibit interesting biological activity, there are an increasing number of publications annually about their isolation, characterization and their synthesis in the laboratory.
Natural hydroxyquinones vary in structural complexity from the simple hydroxynaphthoquinone, lawsone (1), the main component of a natural dye [3], to complex structures such as the trimeric hydroxynaphthoquinone conocurvone (2), a potential anti-HIV agent [4].

This review is focused mostly on the chemistry of the hydroxyquinone moiety, with emphasis on the literature of the last two decades. Methods for hydroxyquinone preparation are reviewed in the next section, followed by their reactivity patterns.
2. Synthesis
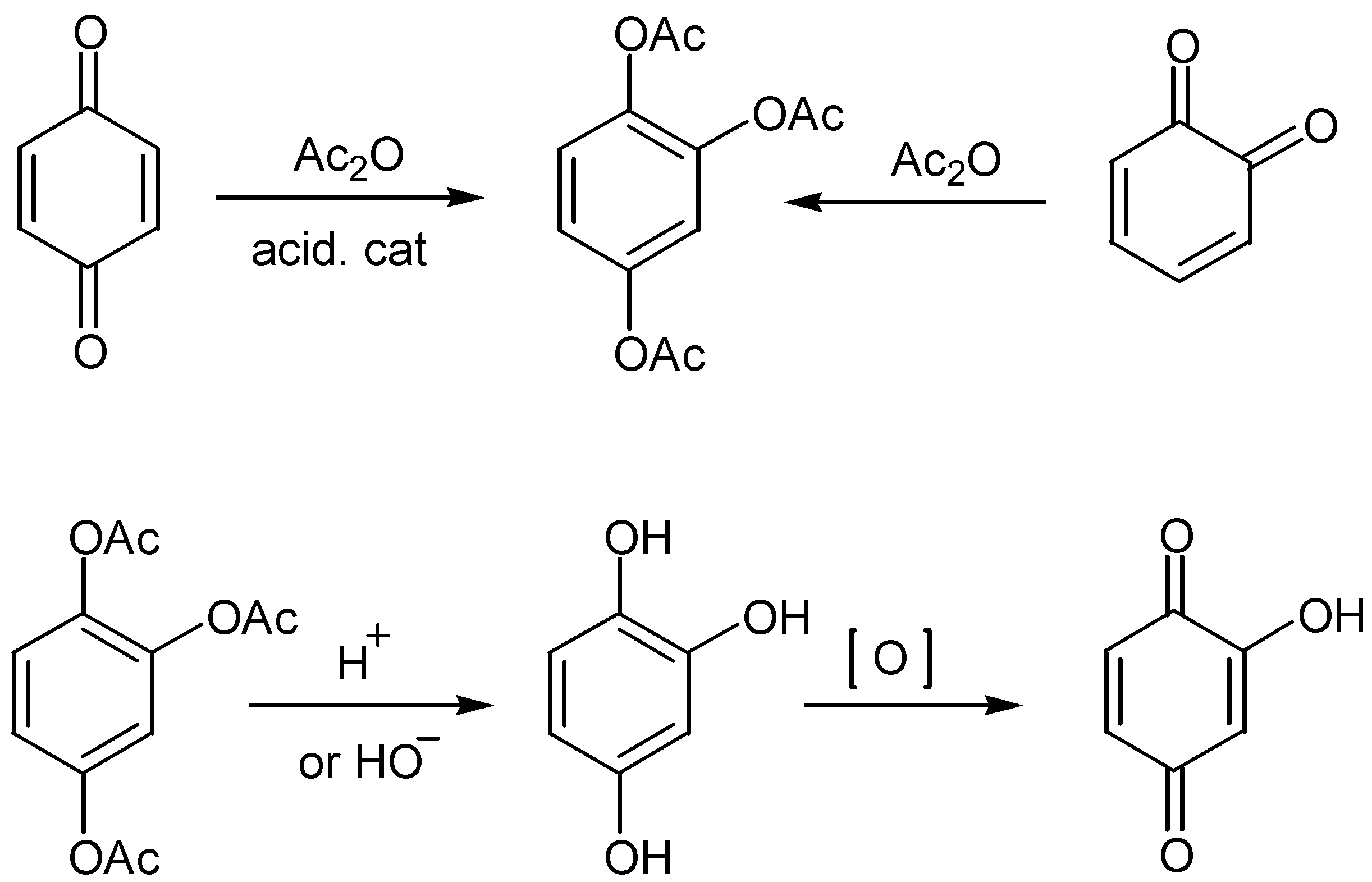
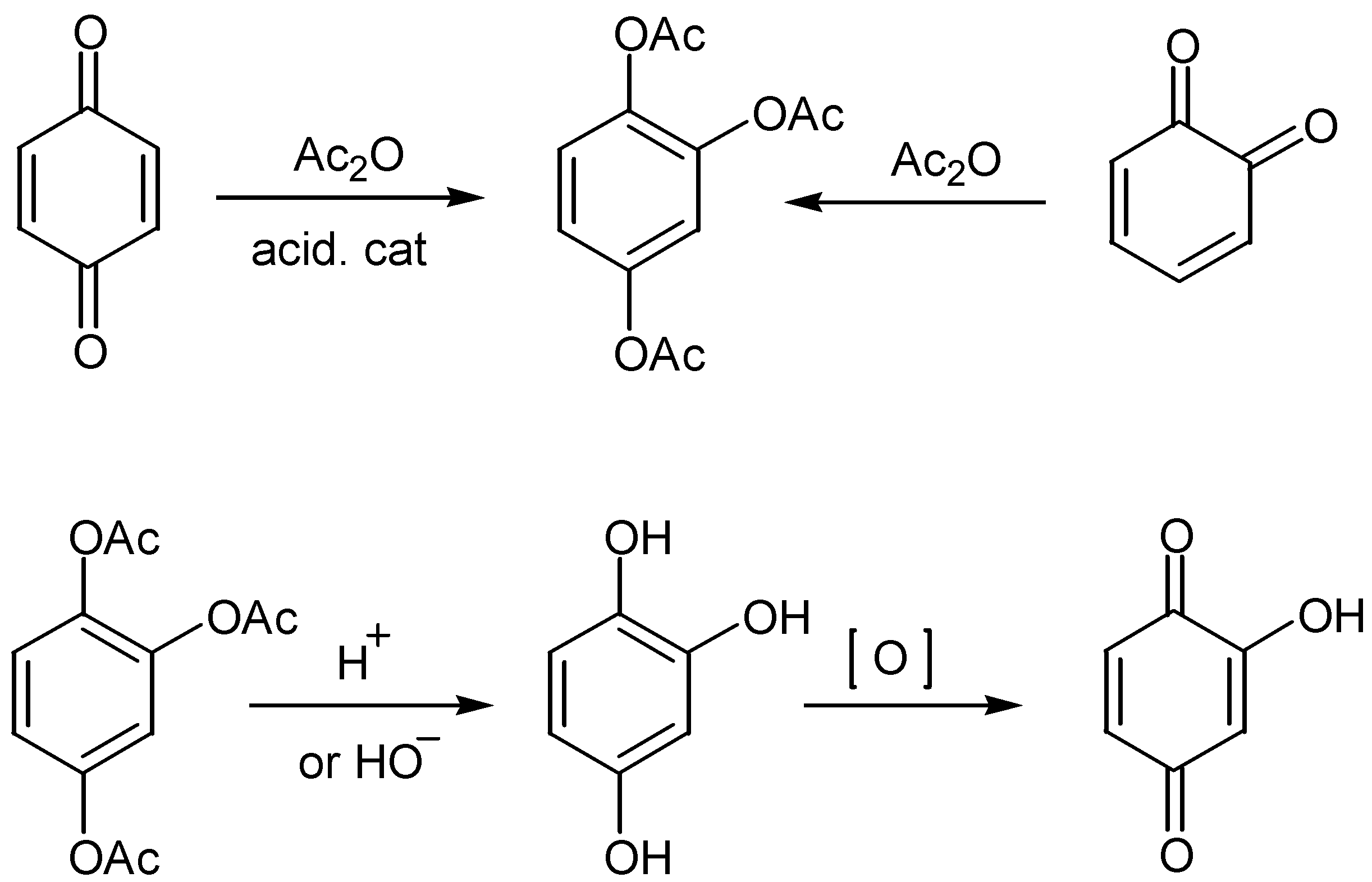
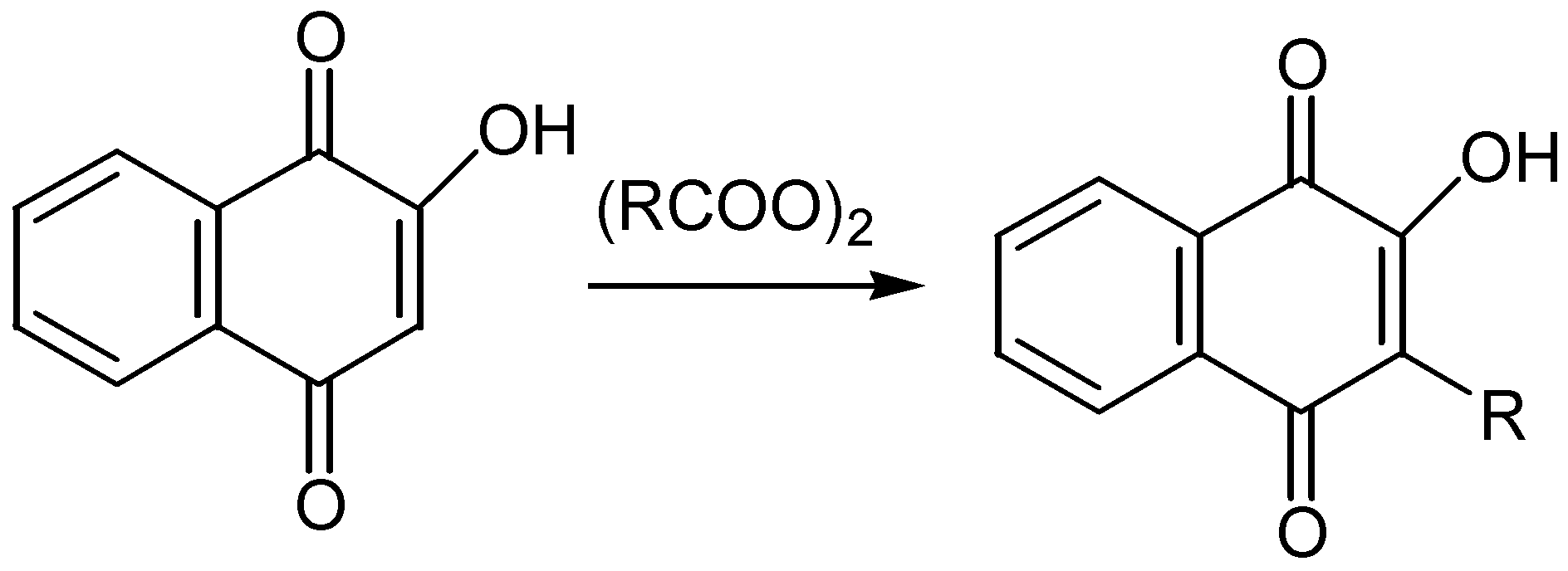
A method of broad applicability for the preparation of the hydroxyquinone moiety is through Thiele-Winter acetoxylation. The method involves the reaction of 1,4- or 1,2-quinone derivatives with acetic anhydride, in the presence of an acidic catalyst. The triacetoxy derivatives - isolated in fair to excellent yields - are hydrolysed to the corresponding hydroxyhydroquinone derivatives under either acidic or basic conditions. The latter, as a rule without isolation, are then oxidized to the desired hydroxyquinone compounds. In many cases, especially under basic conditions, the oxidation proceeds also with atmospheric oxygen. The course of the reaction for hydroxy-p-benzoquinone, is given in Scheme 1.
Synthetic and mechanistic aspects of the reaction were reviewed in detail by McOmie and Blatchly in 1972 [5], whereas more recent results concerning the orientation of the third acetoxy group relatively to bulky t-butyl groups are found in Patai's series [6]. Best results for the Thiele-Winter acetoxylation are obtained when the acid catalyst is H2SO4 or BF3-etherate [5,6]. In a recent paper the use of CF3SO3H as a more effective catalyst is suggested in certain cases [7].
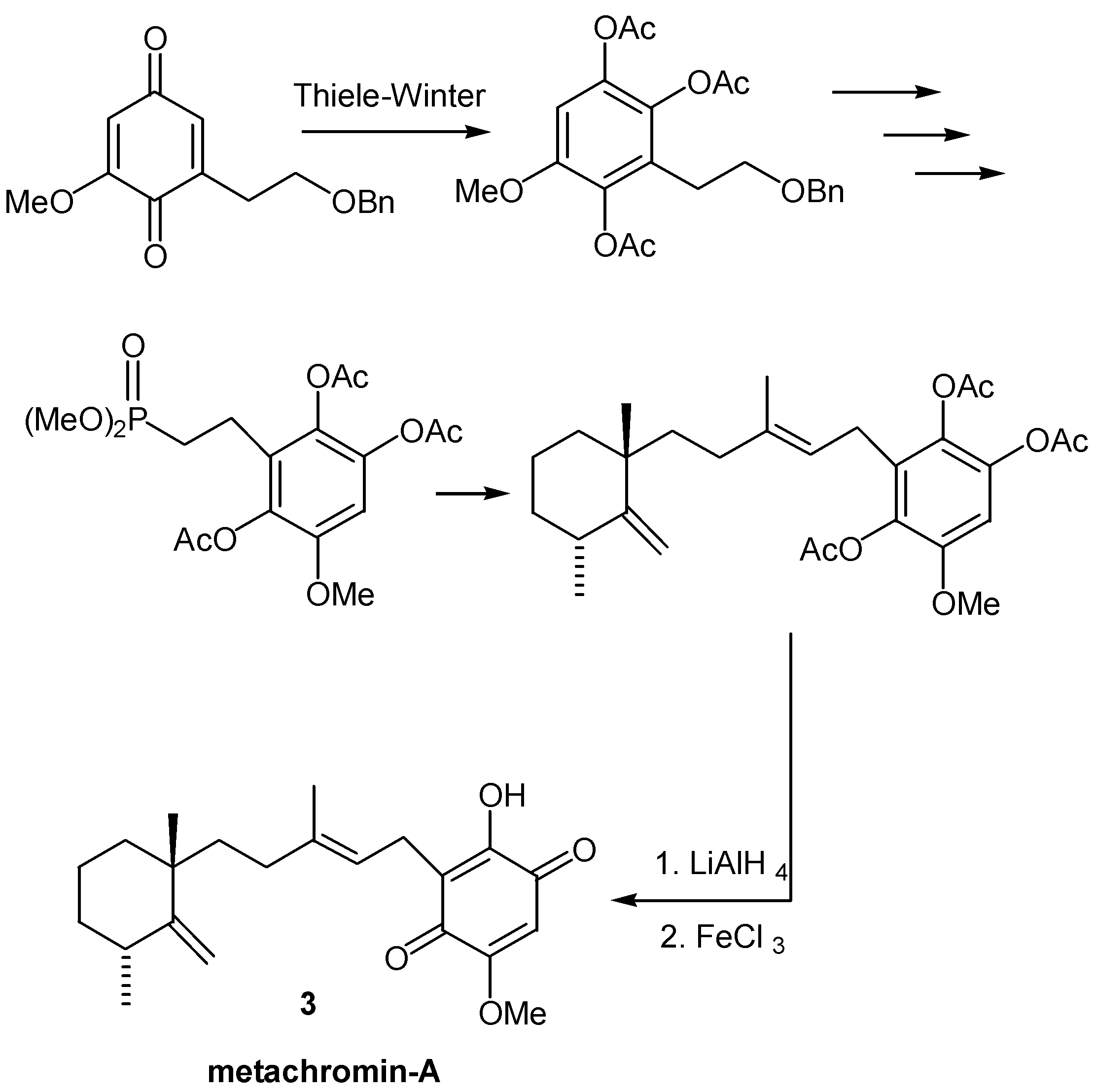
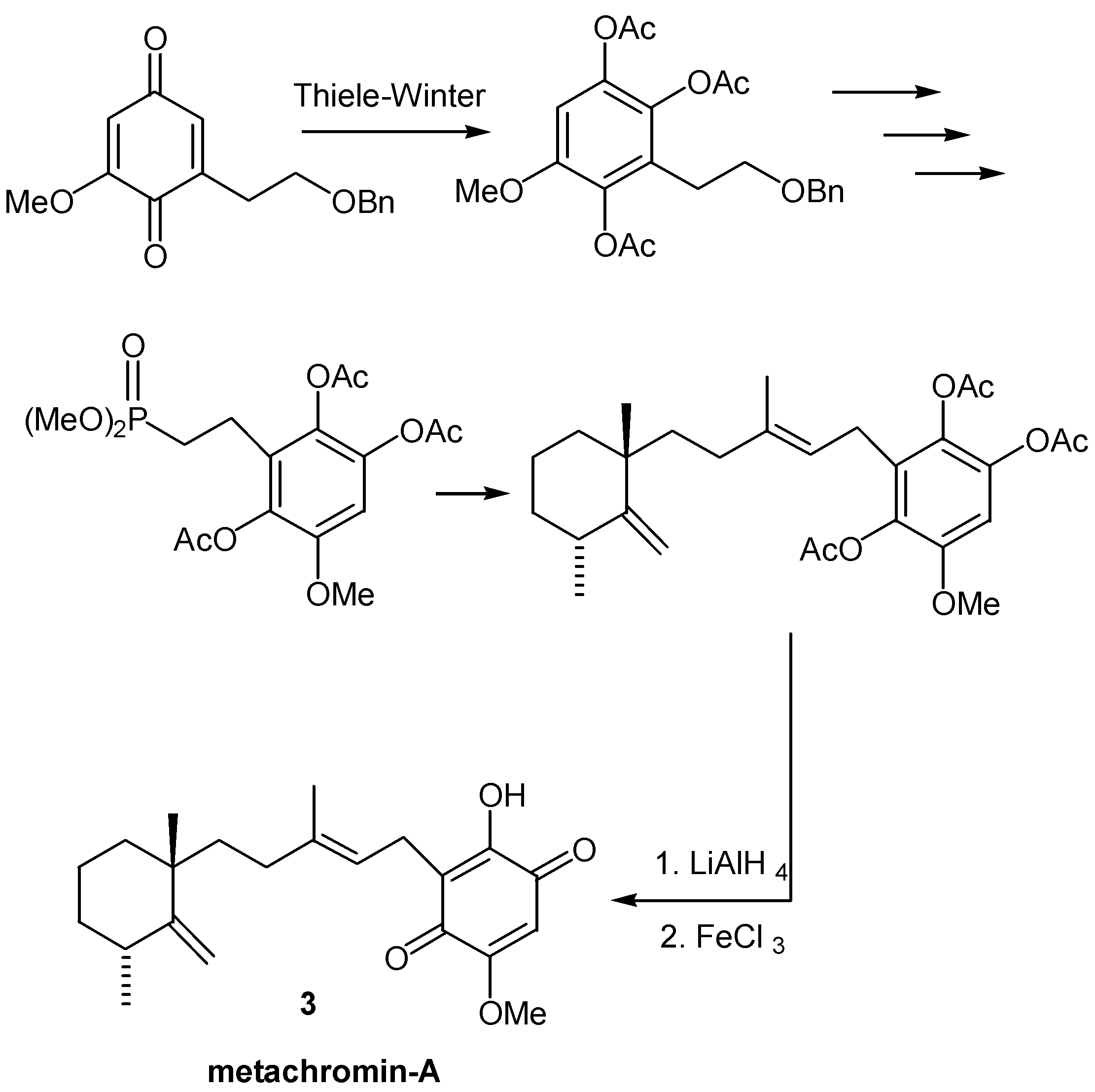
Sometimes, in order to avoid undesirable side reactions of sensitive groups during Thiele-Winter acetoxylation, the timing of events is changed: The acetoxylation takes place in a quinonic precursor, the moiety with the sensitive group (usually a double bond) is attached to the triacetoxybenzene ring, followed by hydrolysis and oxidation of the resulting hydroxyhydroquinone. This procedure was used for the preparation of the sesquiterpene quinone metachromin-A (3) [8], as indicated in Scheme 2.
Analogous methodology was successfully employed for the synthesis of 2-hydroxy-5-methoxy-3-(8', Z, 11' Z)-pentadeca-8', 11',14',-trienyl-1,4-benzoquinone, a host germination stimulant for striga asiatica (witchweed) [9]. Experimental modifications of the method for the preparation of more conventional hydroxyquinones, such as 2-hydroxyphenanthrene-1,4-quinone, can be found in later publications [10,11].
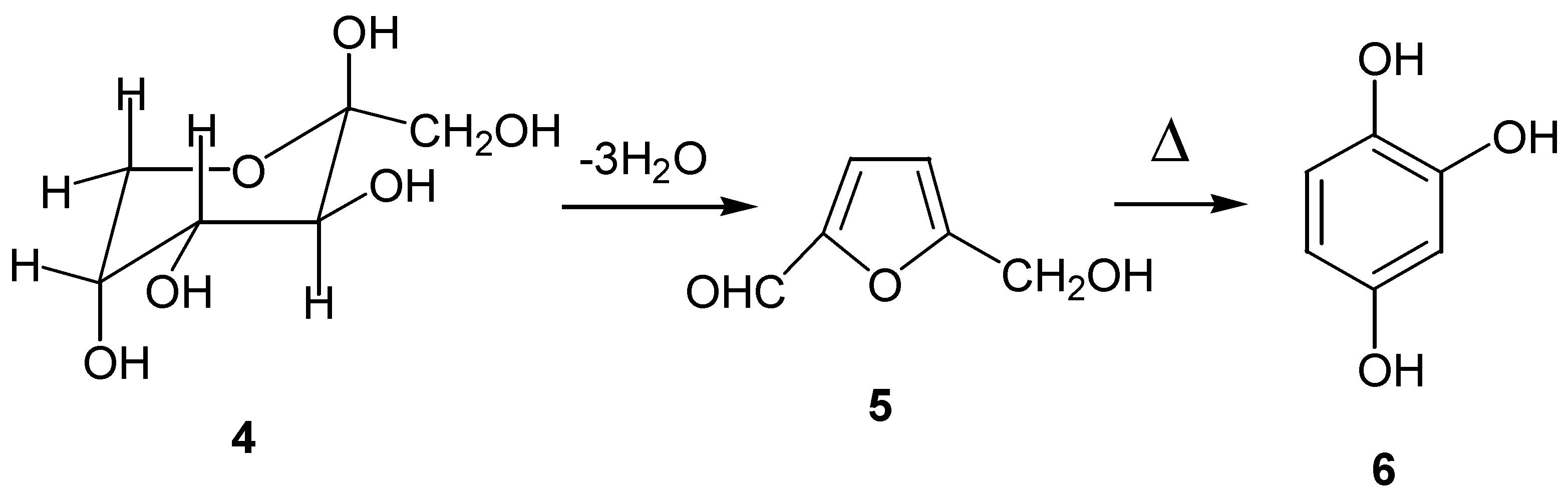
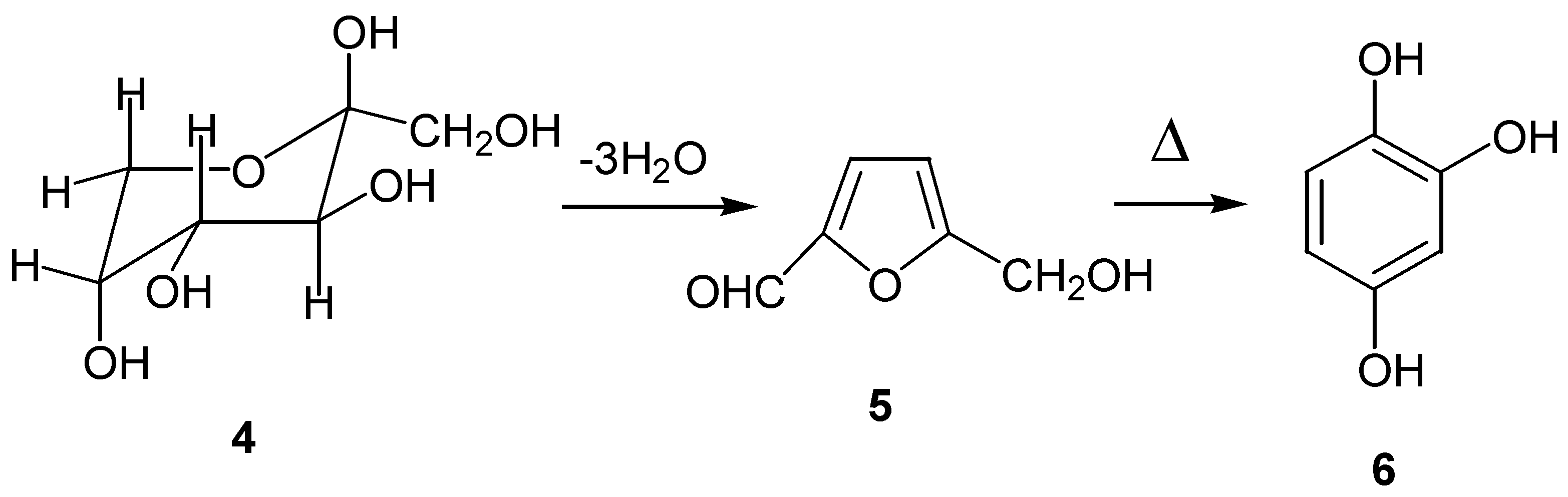
The parent hydroxybenzoquinone can also be obtained from oxidation of hydroxyhydroquinone (6), prepared by an entirely different route: thermal dehydration of carbohydrates such as â-D-fructofuranose (4), through the intermediate 5-hydroxymethylfurfural (5) [12]. (Scheme 3)
Another approach to hydroxyquinones is through demethylation of available trimethoxy derivatives, such as 7, and subsequent oxidation of the resulting hydroxyhydroquinone; this approach was used for the preparation of 6-hydroxy-1,2,3,4-tetrahydronaphthalene-5,8-dione (8) [13] (Scheme 4).
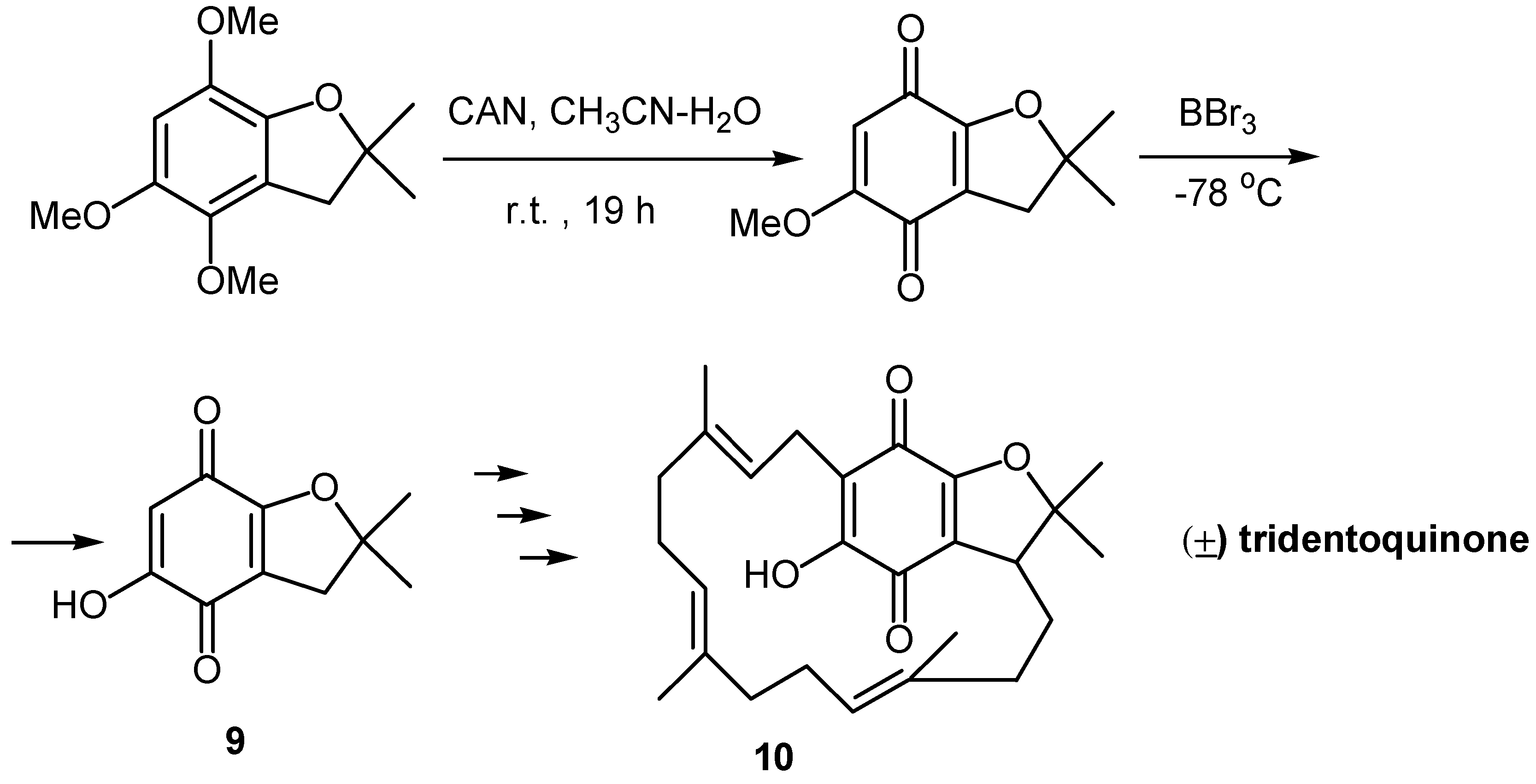
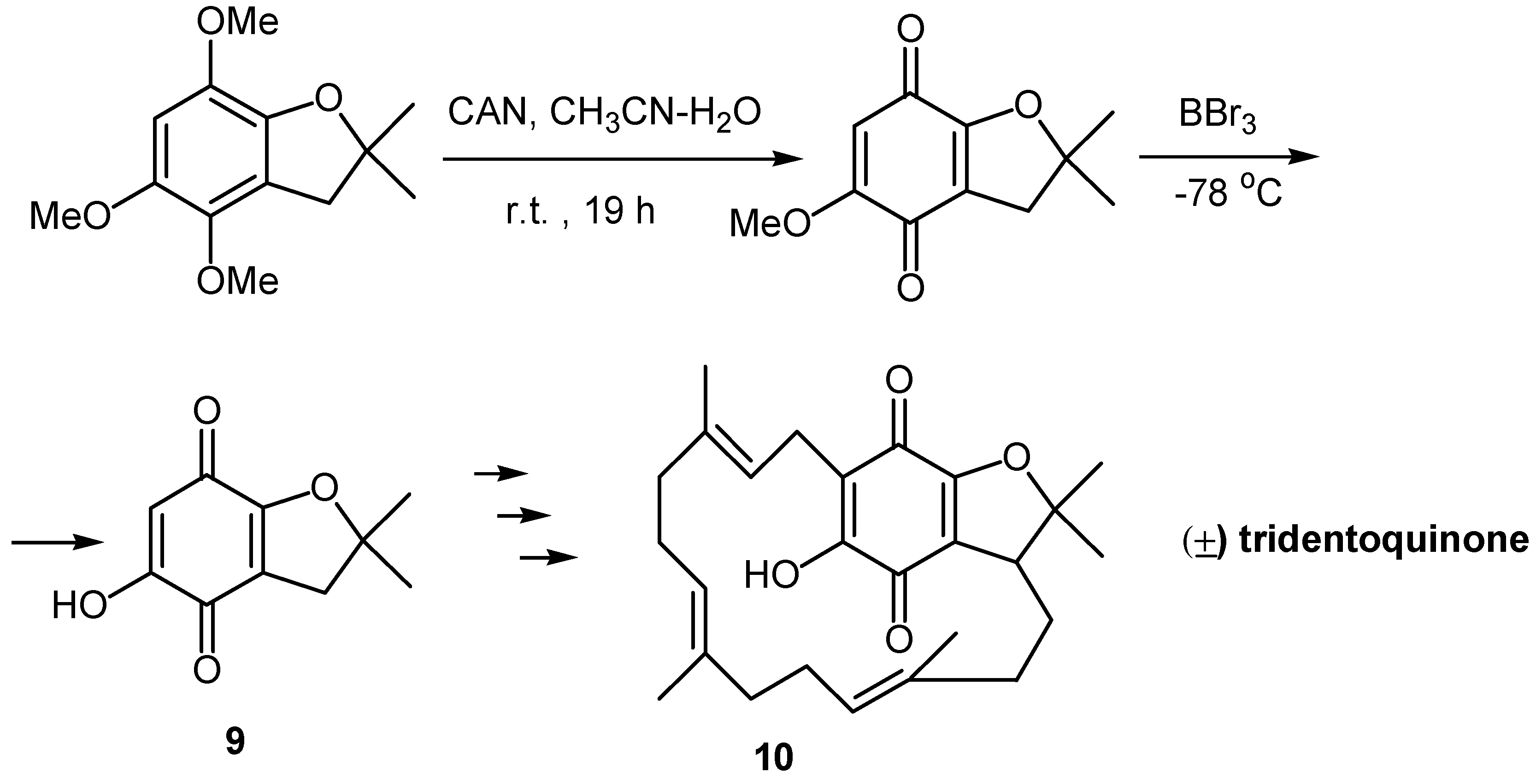
Alternatively, oxidative demethylation of a trimethoxy derivative and demethylation of the resulting methoxyquinone proved to be effective in the case of 5-hydroxy-2,2-dimethyl-2,3-dihydrobenzofuran-4,7-dione (9), a key compound towards the synthesis of (±) tridentoquinone (10) [14]. (Scheme 5)
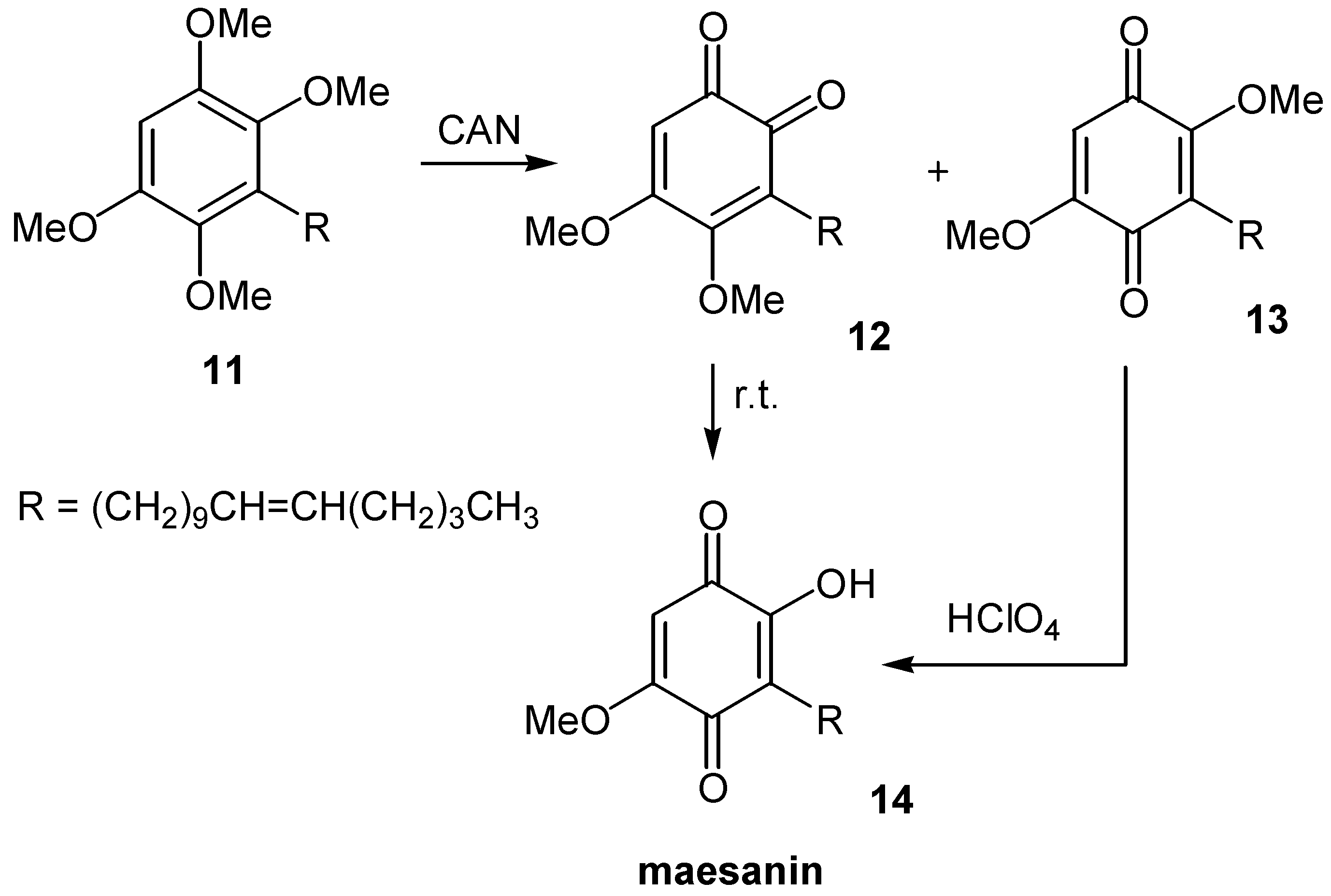
Samadi and co-workers suggested a most simple and effective approach for the synthesis of naturally occurring maesanin (14), and analogues [15]. 1,2,4,5-Tetramethoxybenzene is subjected to alkylation with BuLi and the appropriate alkyl bromide, and the resulting derivative, 11, is oxidatively demethyated by cerium(IV) ammonium nitrate (CAN) to a mixture of dimethoxy ortho- and paraquinones. Whereas ortho-quinone 12, is thermally transformed to the desired maesanin (14), the paraquinone 13 is selectively demethylated at the more hindered methyl group by the use of perchloric acid. (Scheme 6)
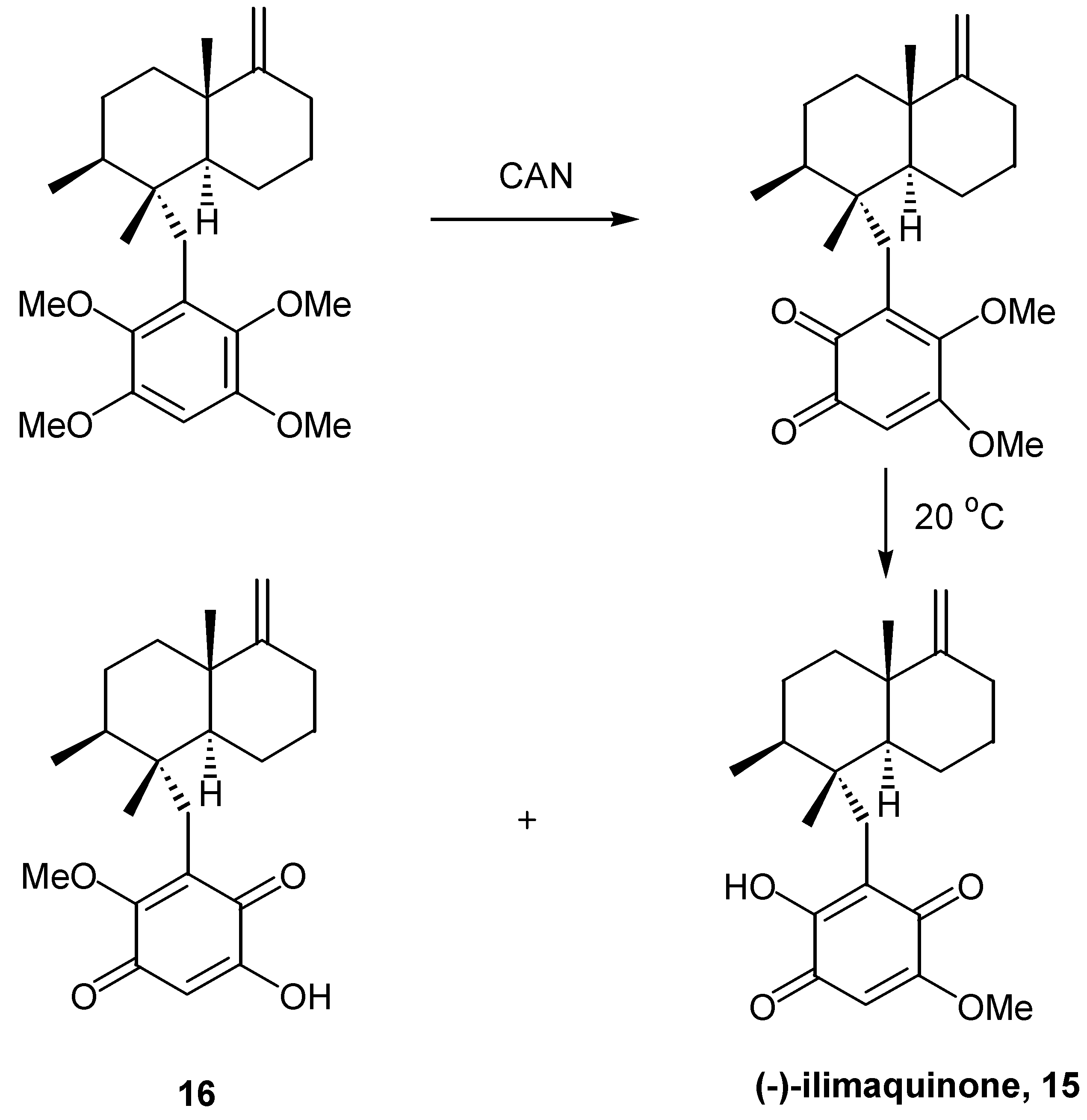
An analogous methodology was used by the same authors [16] for the synthesis of the marine natural product (-)-illimaquinone (15), which exhibits most interesting biological activity. The 6- methoxy isomer of ilimaquinone, 16, also formed, and was easily separated chromatographically. (Scheme 7)
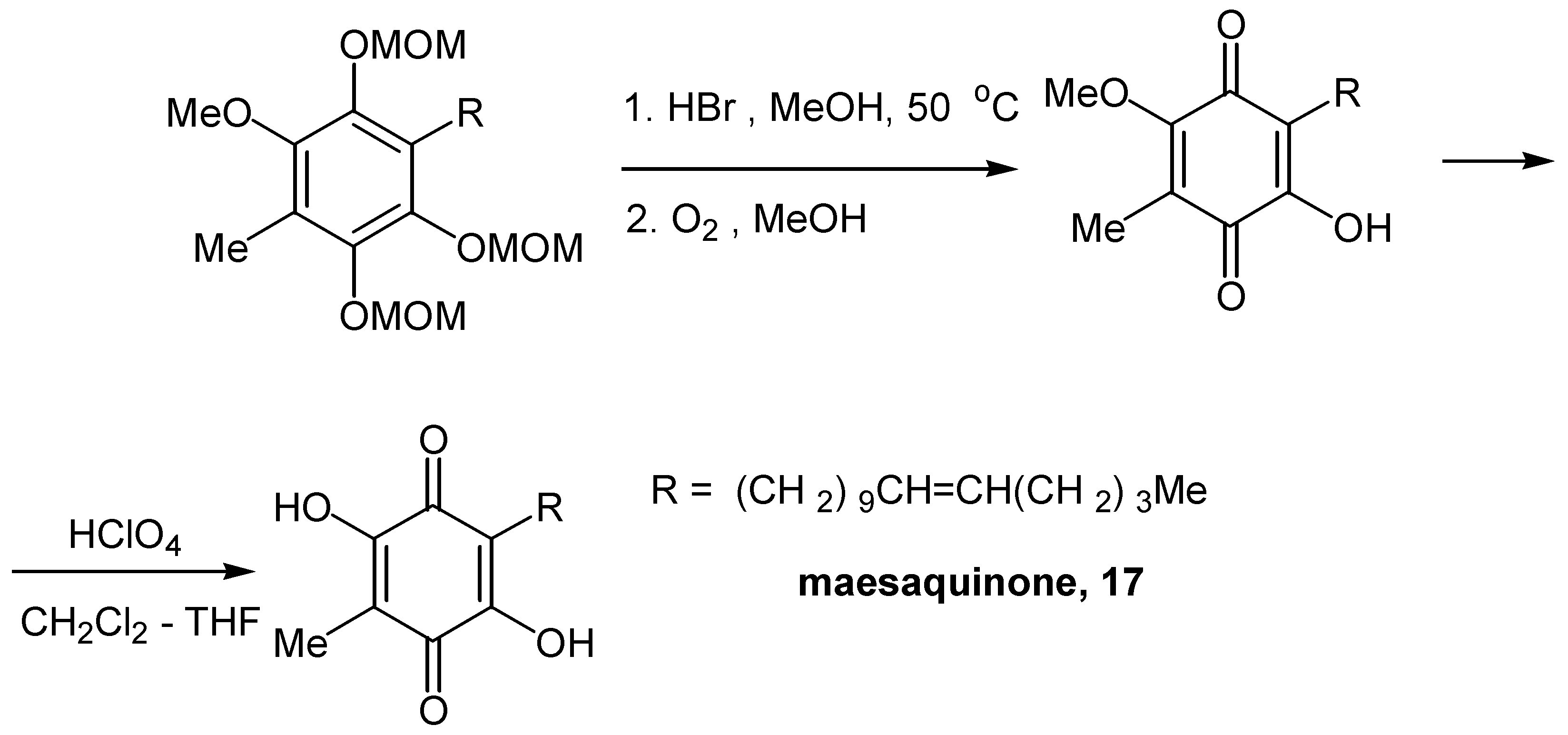
The synthesis of cytotoxic maesaquinone (17), a quinone with two hydroxy groups, proved more complicated. Finally the problem was solved by protection of the second hydroxyl with a MOM (methoxymethyl) group and deprotection in two steps, HBr for the OMOM and HClO4 for the OMe group [17]. The two other OMOM groups were effectively oxidized to the quinonic moiety. (Scheme 8)
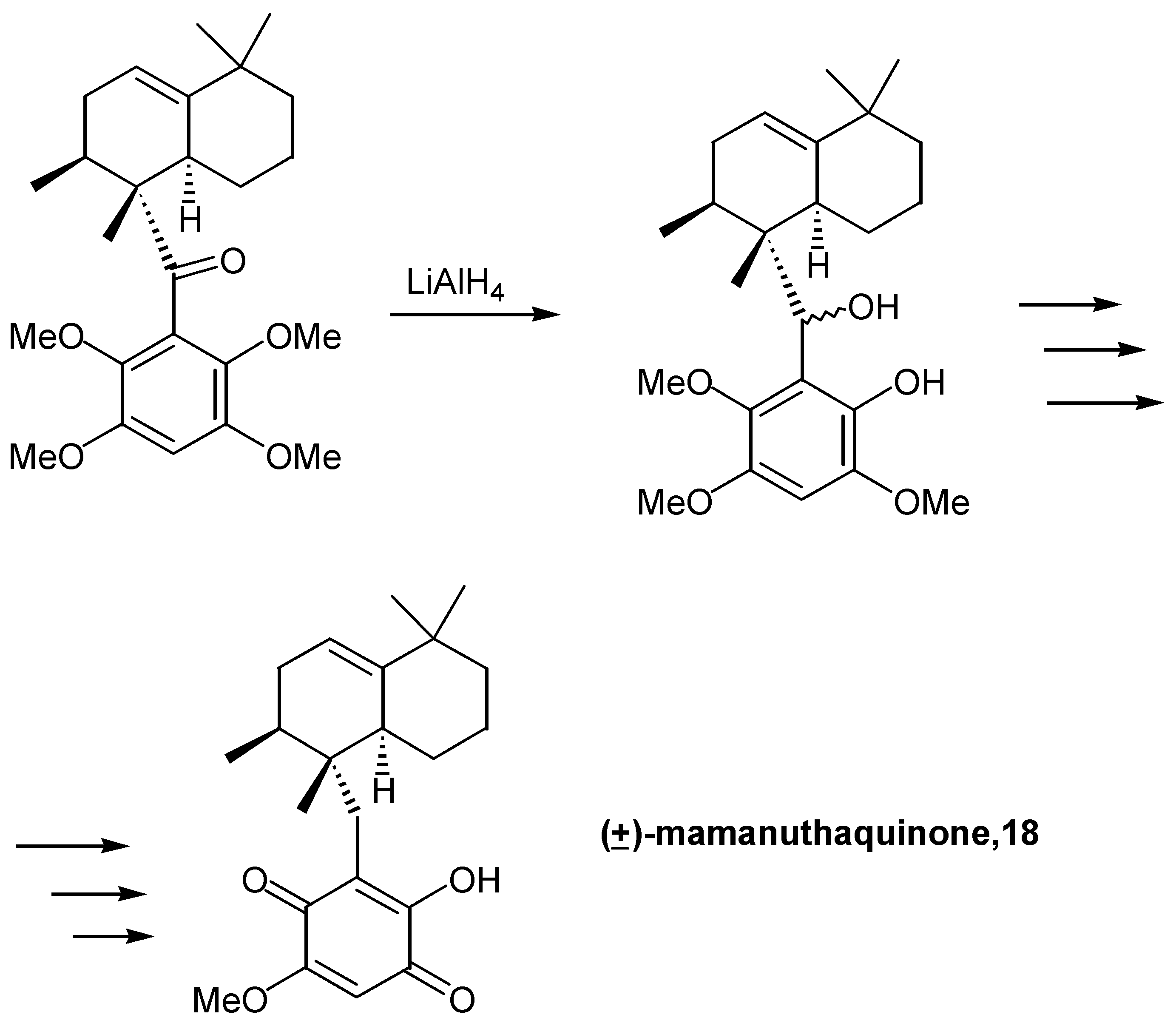
Danishefsky reported a concise total synthesis of (±)-mamanuthaquinone (18), a hydroxy sesquiterpenoid quinone with cytotoxicity towards human colon tumour cell lines [18]. The sesquiterpenoid part of the molecule was constructed by a Diels-Alder reaction and, interestingly enough, on reduction of the ketone with lithium aluminium hydride one of the ortho methoxy groups was also demethylated. Esterification of this group, reduction of the hydroxy group, oxidative demethylation with CAN and, finally, deprotection of the phenolic hydroxyl afforded racemic mamanuthaquinone (18). (Scheme 9)
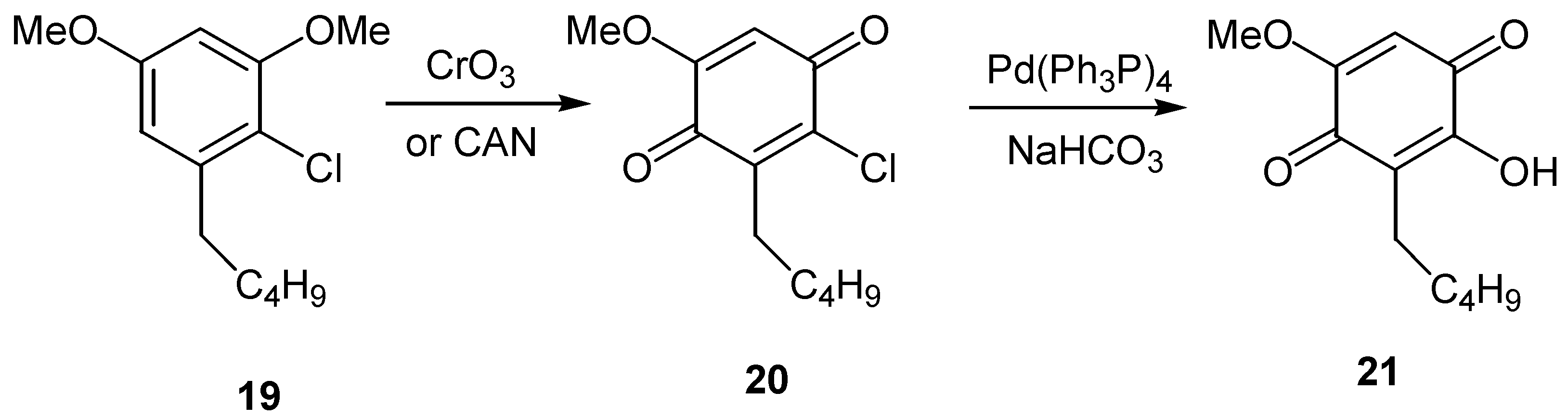
Another approach to hydroxyquinones is through conversion of chloride to a hydroxyl group. Snapper and co-workers reported that a chloromethoxyquinone moiety could be converted to the corresponding hydroxy functionality (eg.- 21), by palladium-mediated exchange of the chloride for the hydroxyl group [19]. The oxidation of initial chlorodimethoxybenzene derivative, 19, to chloromethoxyquinone, 20, was achieved either by CrO3 or by CAN, as shown in Scheme 10 for a model compound.
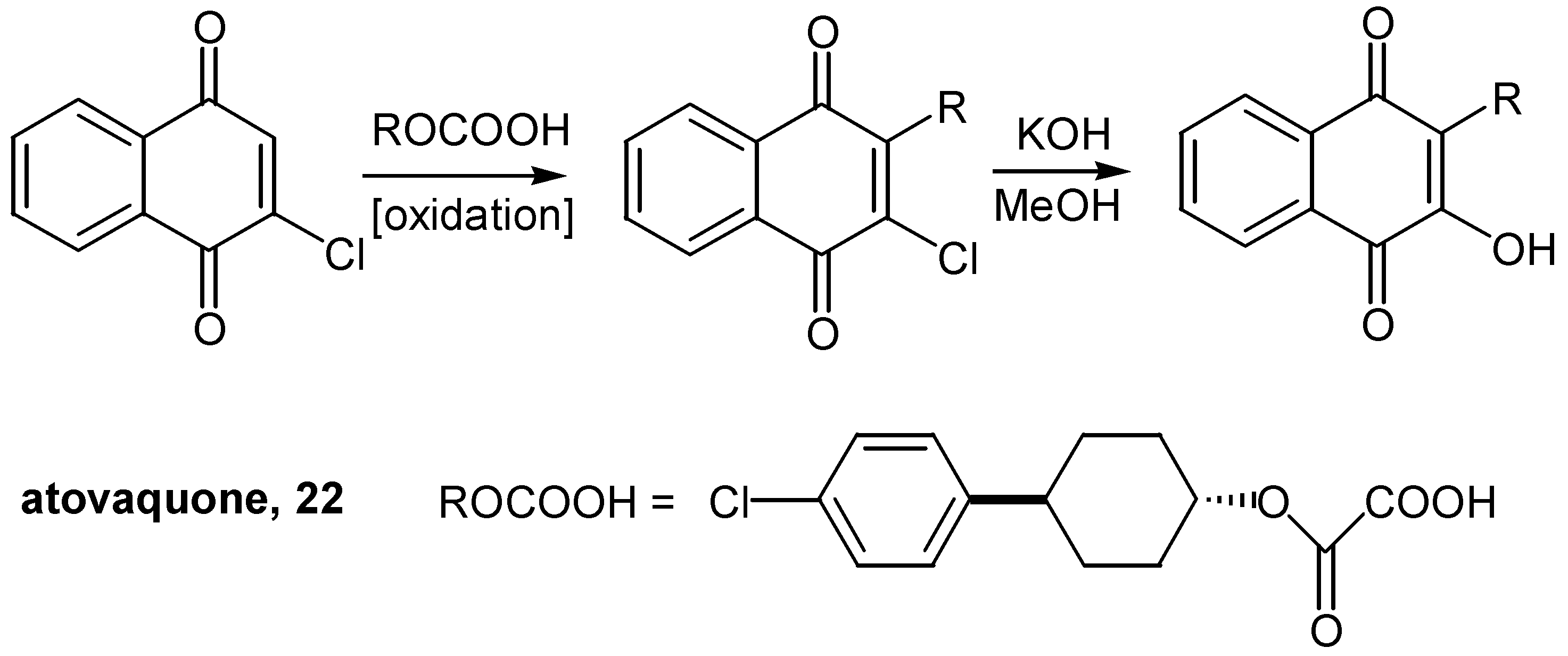
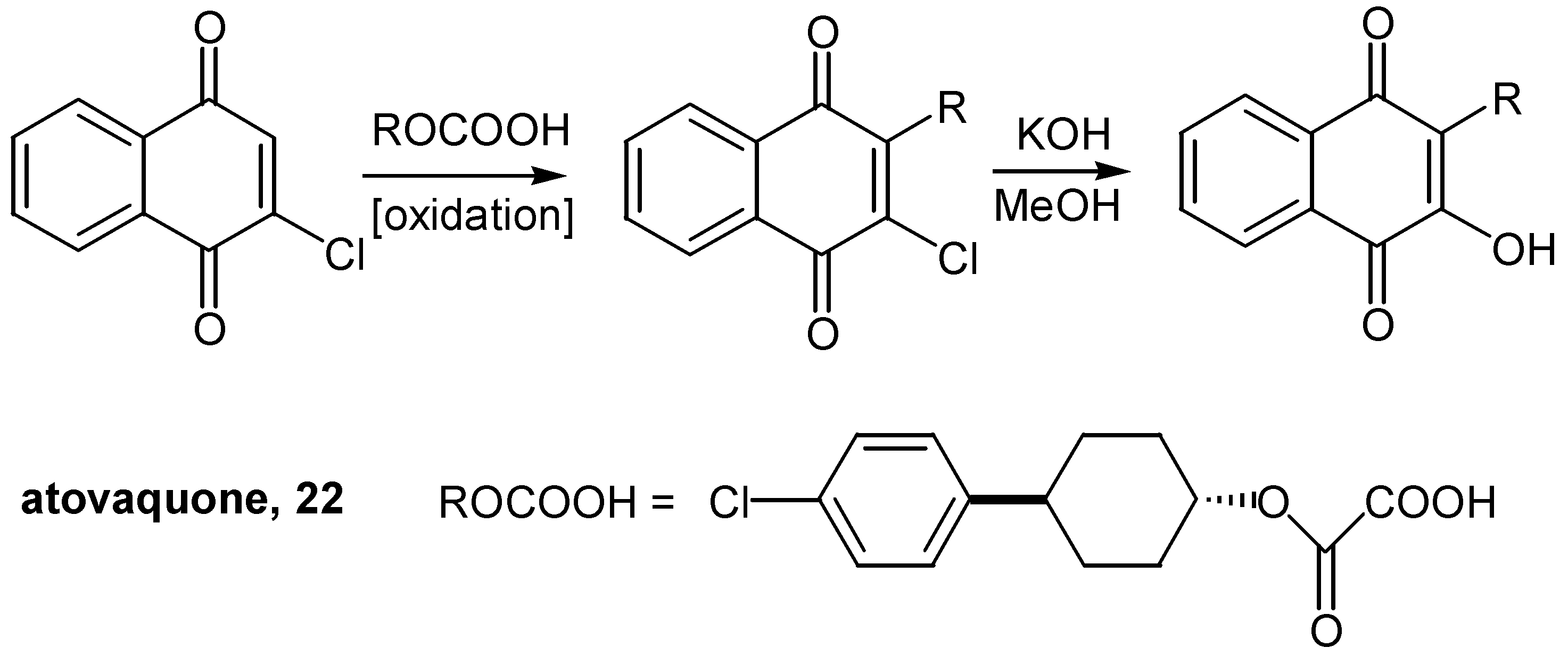
This methodology was successfully employed for an alternative synthesis of (-)-ilimaquinone [19] and its analogues [20]. In contrast, the conversion of chloride to hydroxyl was most easily achieved in 94% yield by simply refluxing the proper choronaphthoquinone with KOH in methanol, in the case of atovaquone, [21]. (Scheme 11) The insertion of the complicated R group into the quinonic ring proceeded through radical coupling. Atovaquone (22), is marketed as a prescription drug for the treatment of a special case of pneumonia.
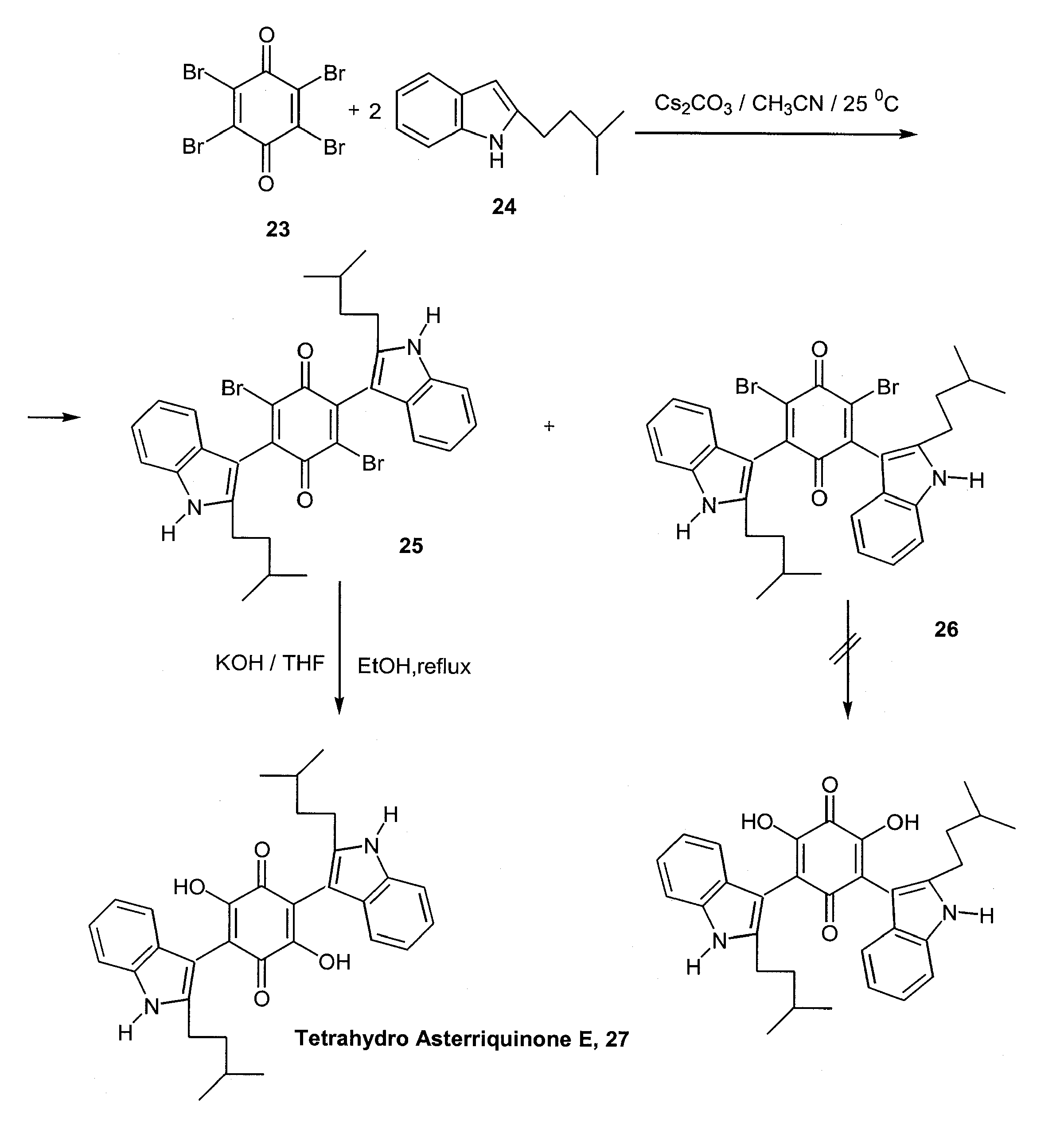
Analogous methodology was applied for a convenient one-pot, two-step synthesis of tetrahydro asterriquinone E (27), which exhibits the same biological activity as the naturally occurring unsaturated analogue [22]. Starting from commercially available para-bromanil (23), coupling with 2 equiv. of the appropriate indole derivative, 24, leads to a mixture of 2,5- and 2,6-bis(indolyl)-dibromoquinone regioisomers, 25 and 26. Only the former is converted to the desired dihydroxyquinone derivative by a KOH mediated replacement of the two bromides by hydroxy groups. The overall yield is 36%. (Scheme 12)
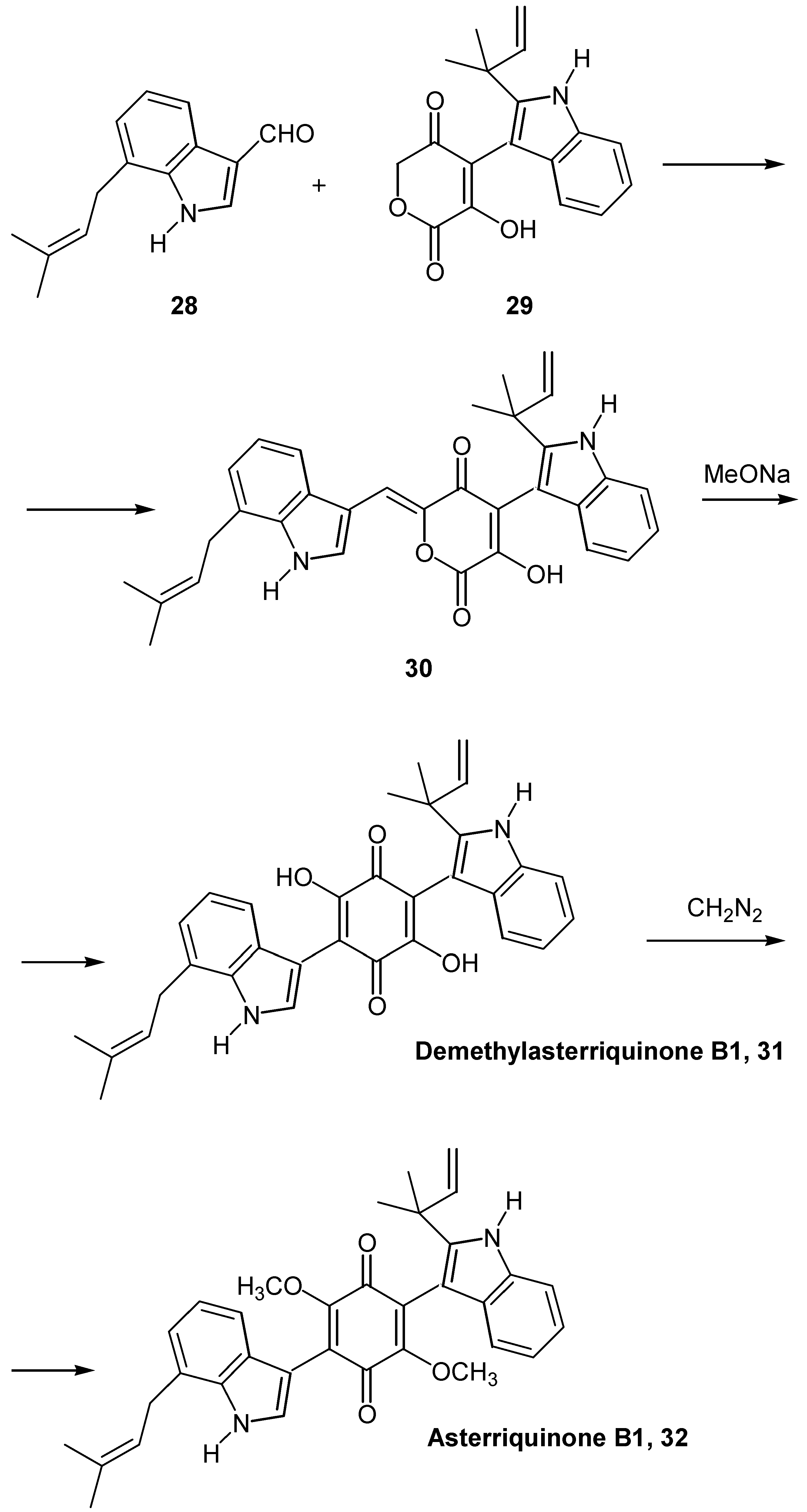
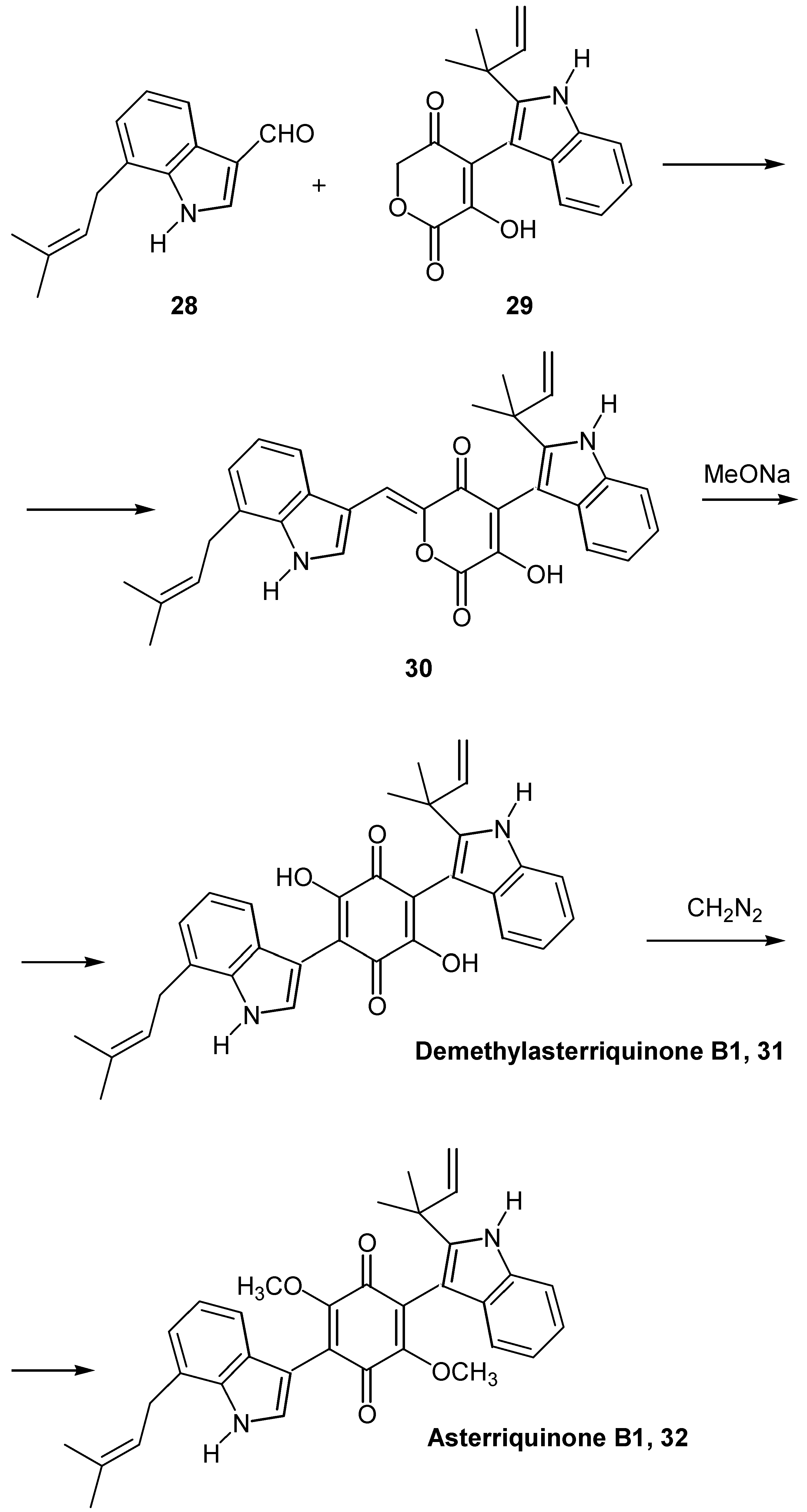
In the case of asterriquinone B1 (32), a dimethoxybenzoquinone asymmetrically substituted with different indole substituents, a lengthier route was followed [23a].
The formyl prenyl indole derivative 28, prepared from 2-iodoaniline in five steps, was condensed with pyrandione derivative 29, prepared from indole-3-carboxaldehyde in six steps. The condensation product 30 was quantitatively rearranged to demethylasterriquinone B1 (31), by a catalytic amount of sodium methoxide. The latter was finally methylated to the desired dimethoxy derivative, asterriquinone B1 (32) (Scheme 13). This synthesis of asymmetrically substituted asterriquinones is of great importance, since demethylasterriquinone B1, initially isolated as a fungal metabolite with the code name L-783, 281, was found to act as an insulin mimetic with anti-diabetic activity [23b]. Another route to the hydroxyquinone functionality is by replacement of an amino group by a hydroxy group, considering the fact that the insertion of the amino group into the quinonic ring is comparatively easy [24]. This replacement is not a general reaction and takes place mostly in polysubstituted quinones.
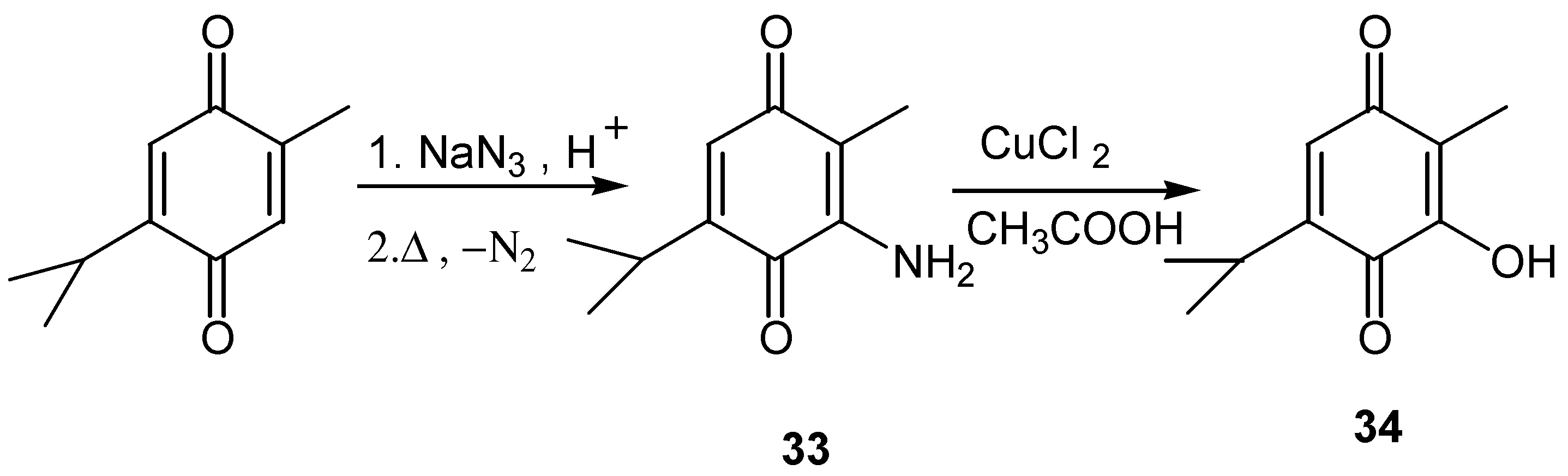
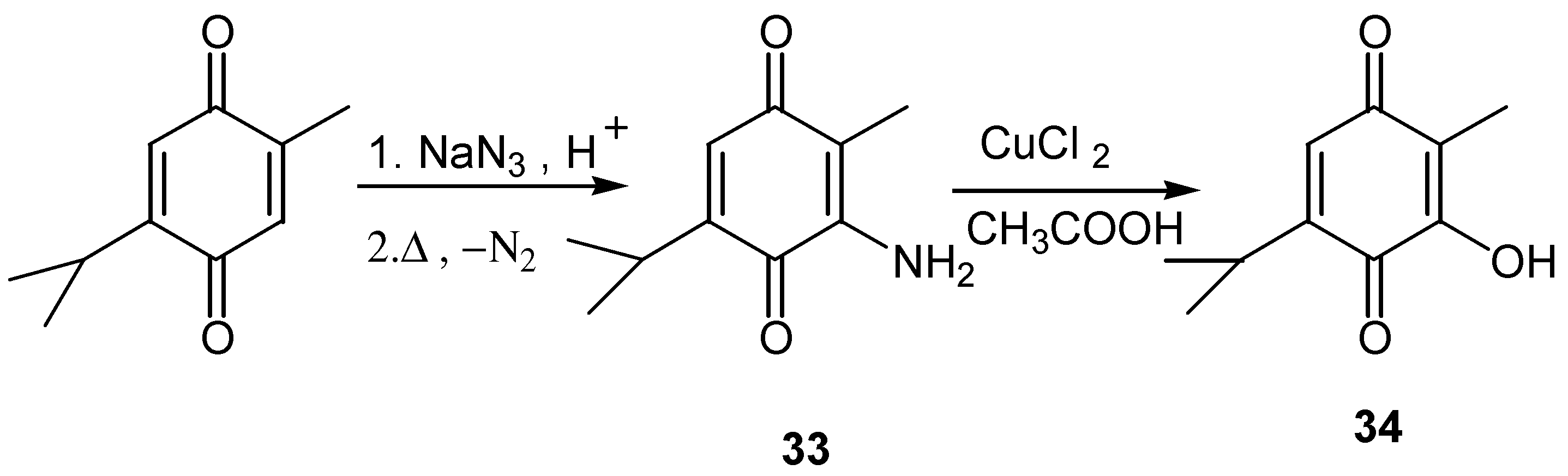
In the case of 3-aminothymoquinone, conversion to 3-hydroxy-2-methyl-5-isopropyl-1,4-benzoquinone is effected by the CuCl2/CH3COOH [25]. (Scheme 14)
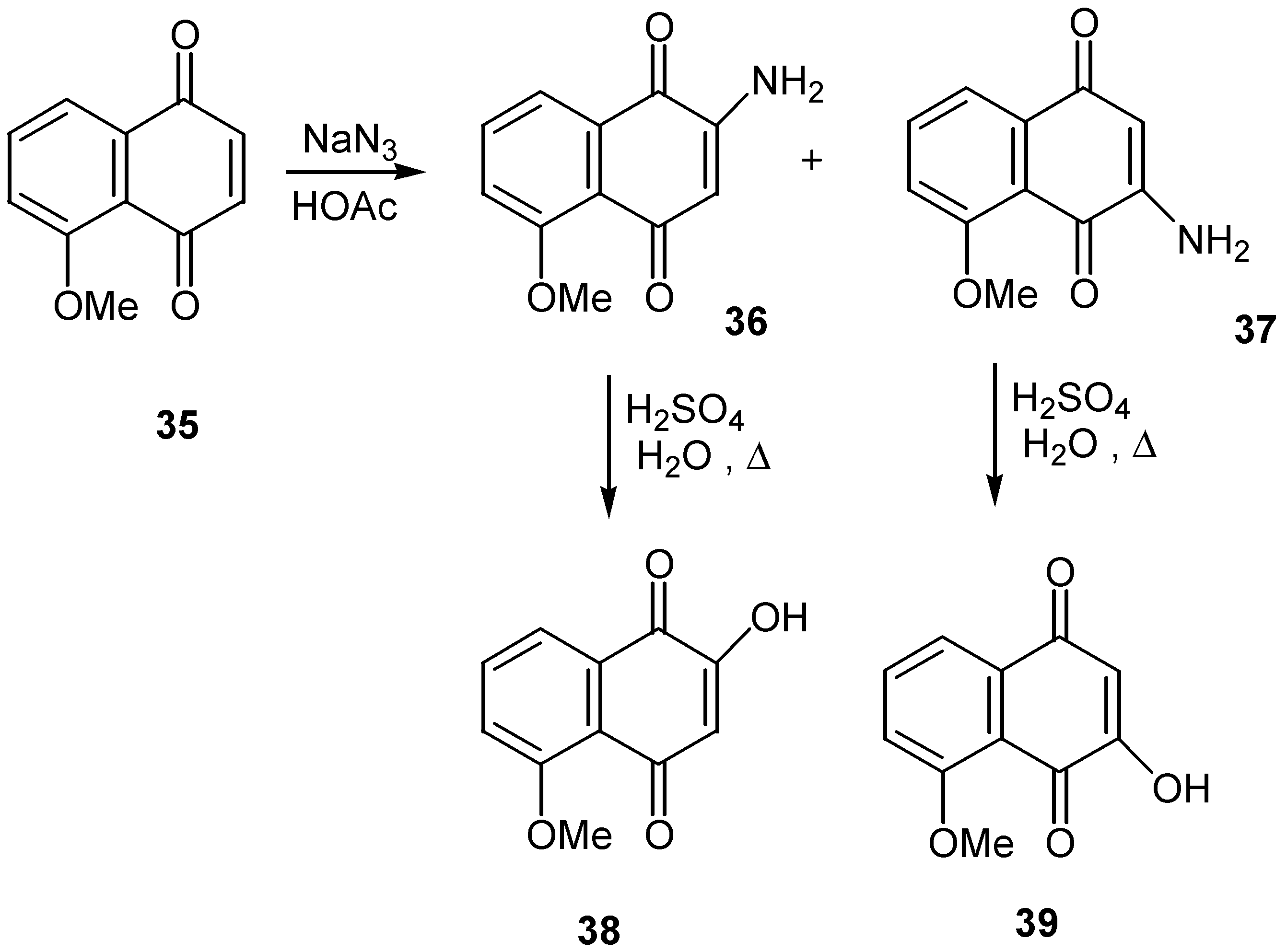
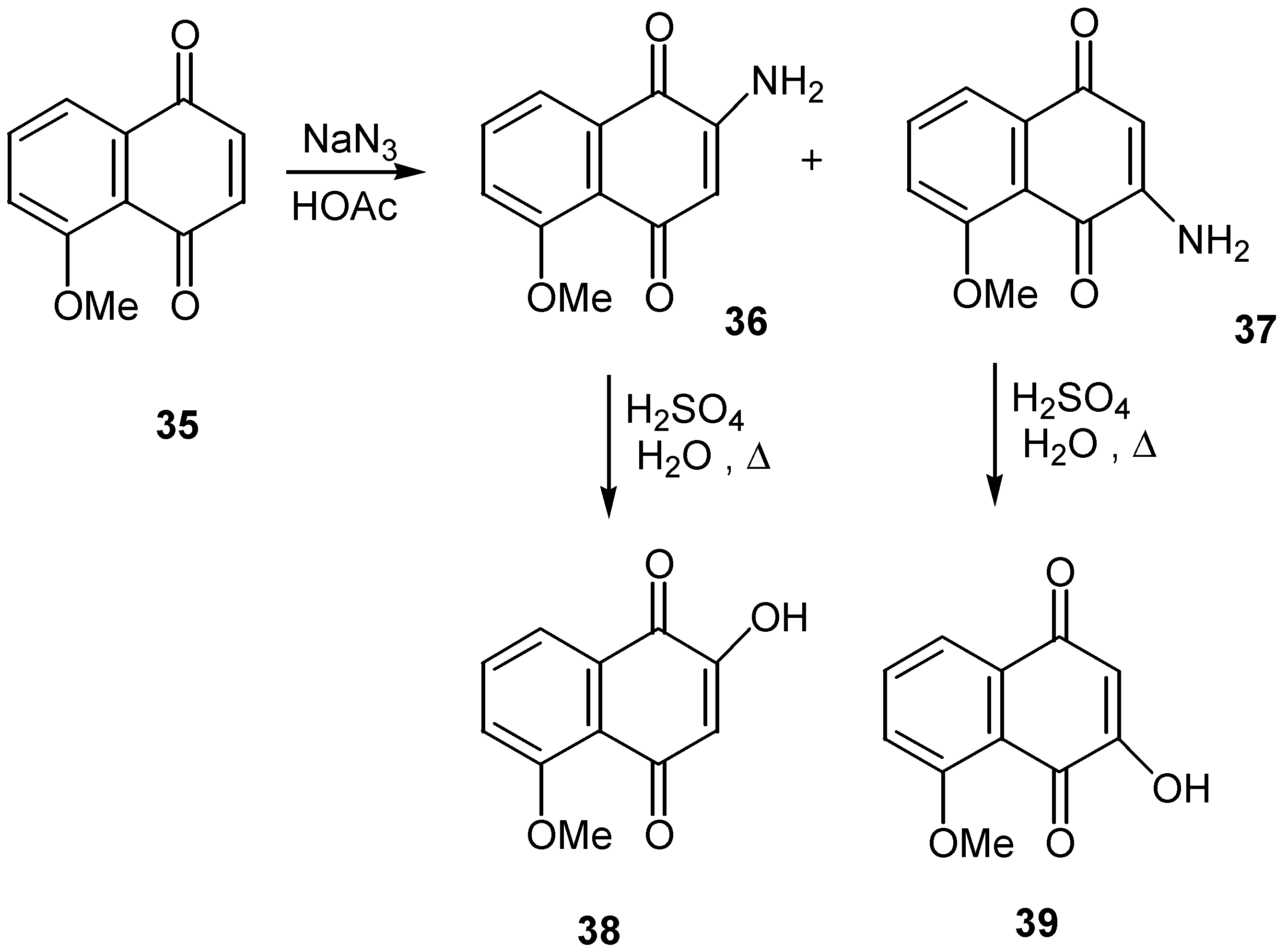
Refluxing in aqueous sulfuric acid was enough to convert the two isomeric aminojuglone ethers, 36 and 37, to the corresponding hydroxy derivatives, 38 and 39 [26]. The former were obtained as a mixture from the reaction of juglone methyl ether (35) with sodium azide and were separated by crystallization. (Scheme 15)
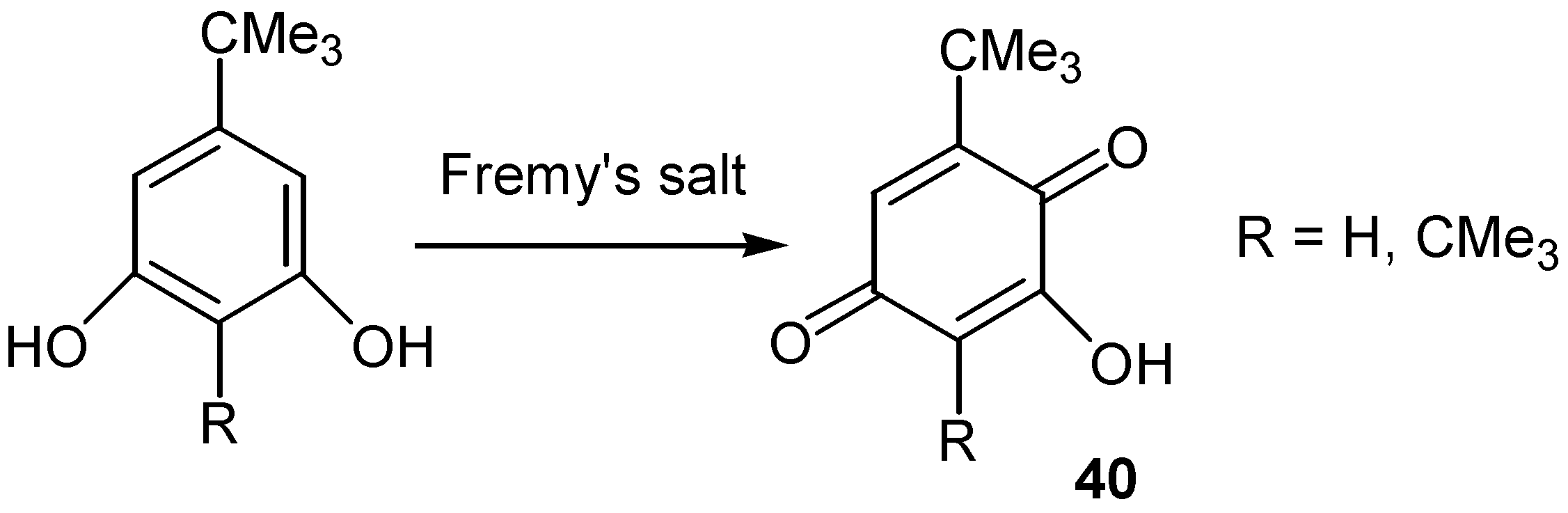
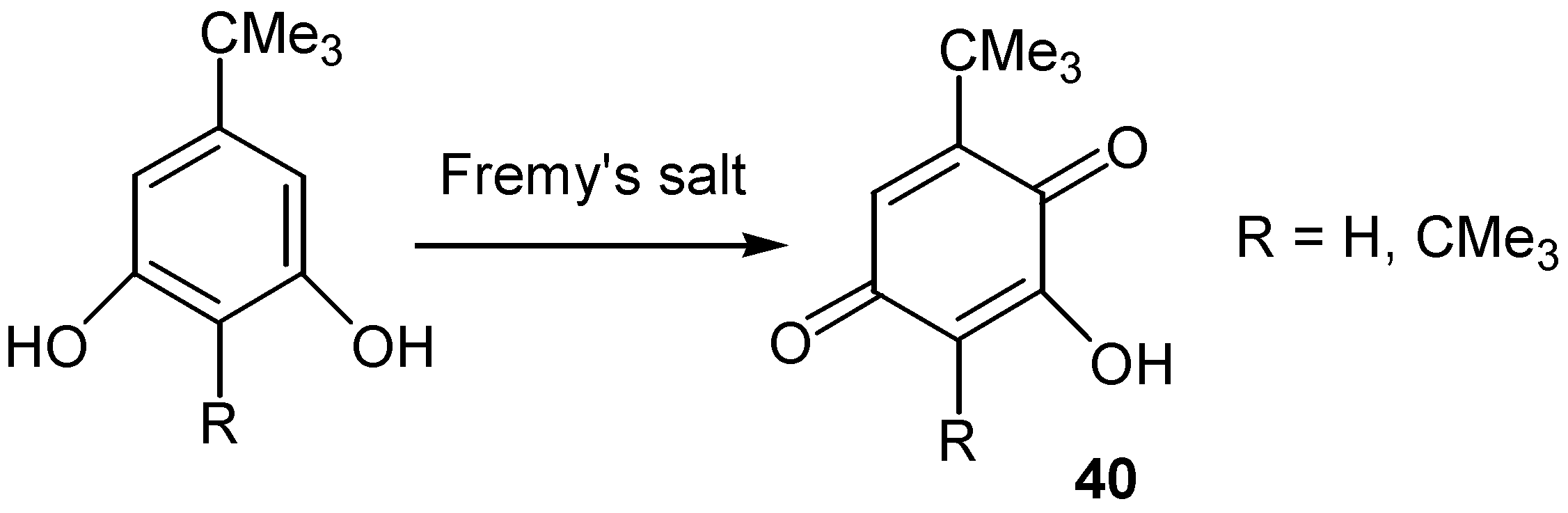
A different approach for the construction of hydroxyquinone moiety is through oxidation of phenol, resorcinol, catechol or hydroquinone derivatives. The reaction proceeds with insertion of hydroxy groups into the aromatic ring followed by oxidation to the quinone moiety. This approach gives better results with simple molecules, since strong oxidants that might affect other sensitive groups are used for this conversion. Fremy' s salt was used for the preparation of hydroxyquinones with bulky t-butyl groups, such as 40, [27]. (Scheme 16)
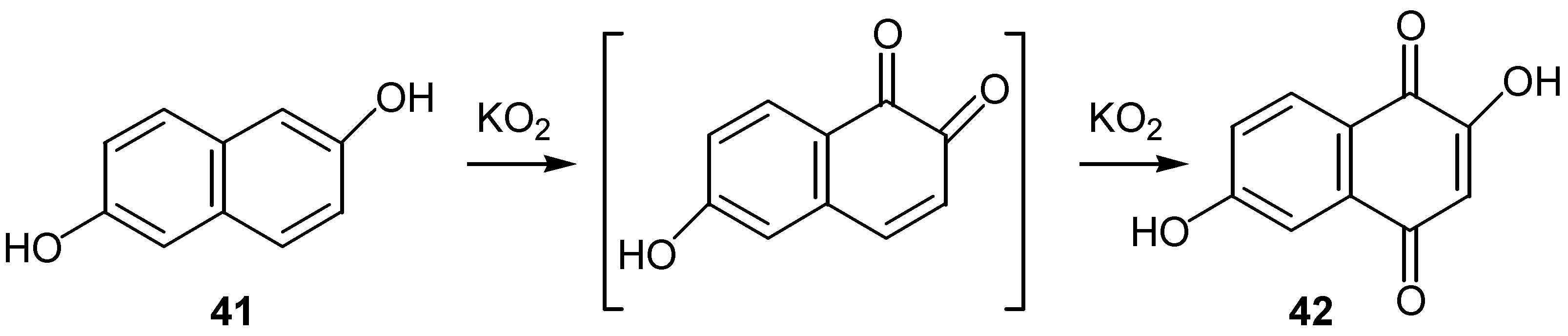
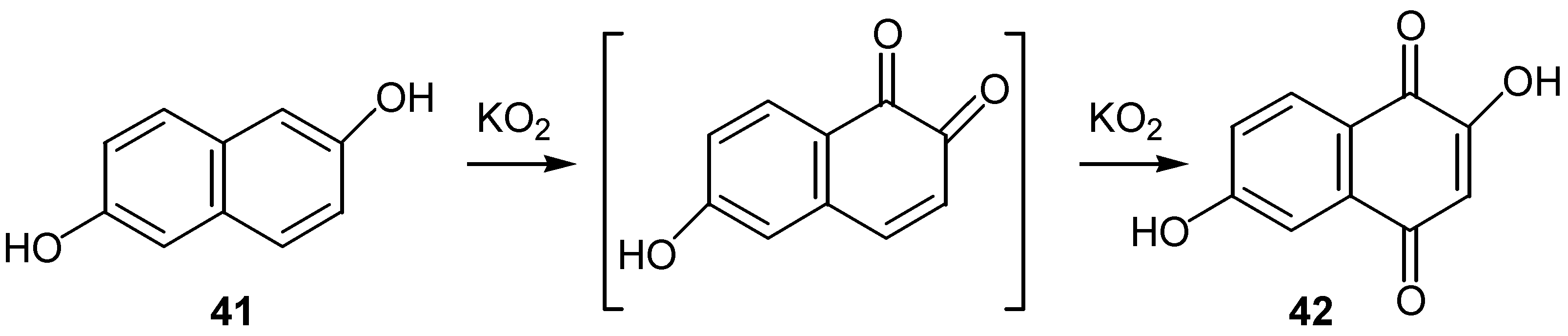
Oliveros and co-workers used singlet oxygen for the conversion of a series of naphthalenediols into the corresponding hydroxynaphthoquinone derivatives [28], although better results are obtained using solid KO2 , as reported by the same authors in a later study [29]. The oxidation of 2,6-naphthalenediol 41 to dihydroxyquinone 42 is a characteristic example. (Scheme 17)
Analogous oxidation of 1,3-dihydroxynaphthalene (43) to lawsone (1) was performed by [bis(trifluoroacetoxy)iodo]benzene in CH3CN-H2O [30]. (Scheme 18)
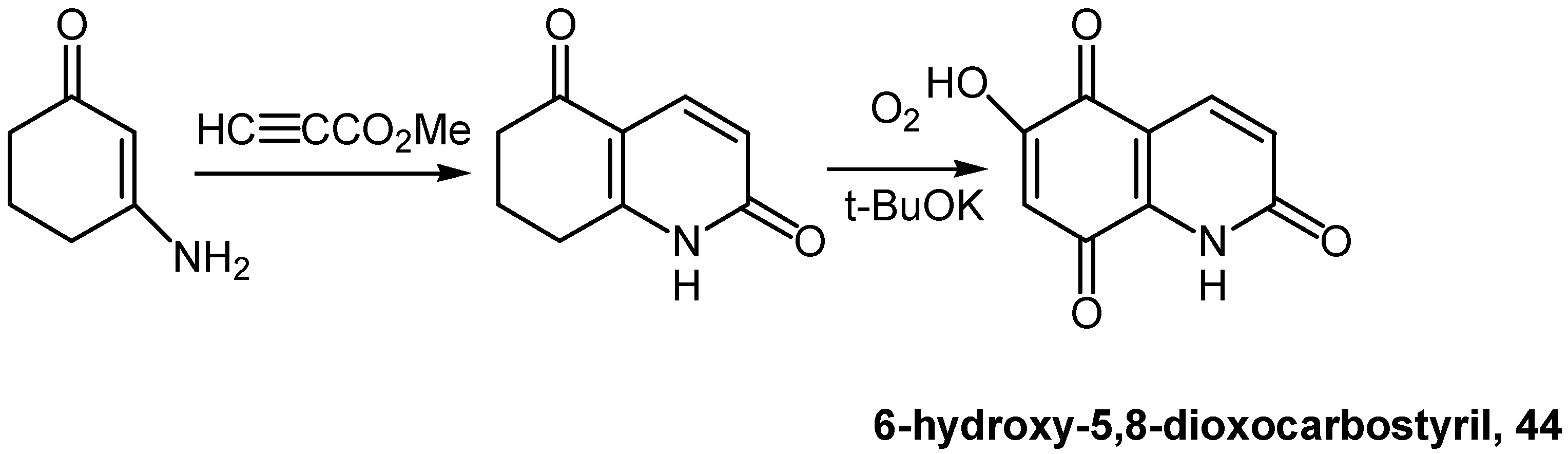
á-Tetralones can be effectively oxidized to hydroxynaphthoquinones by oxygen and potassium t-butoxide in dimethyl sulfoxide [31]. This methodology was applied for the preparation of 6-hydroxy5,8-dioxocarbostyril (44) (Scheme 19) whereas the corresponding 7-hydroxy derivative was prepared by a lengthier route starting from 8-hydroxy-quinoline (oxine), involving amino to hydroxyl conversion as the final step [32].
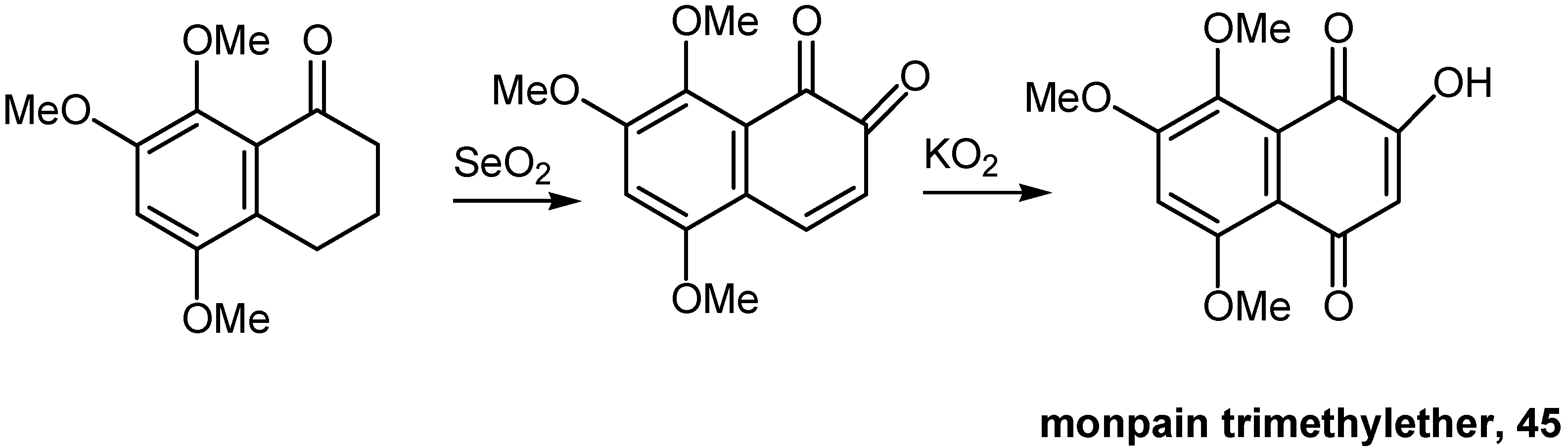
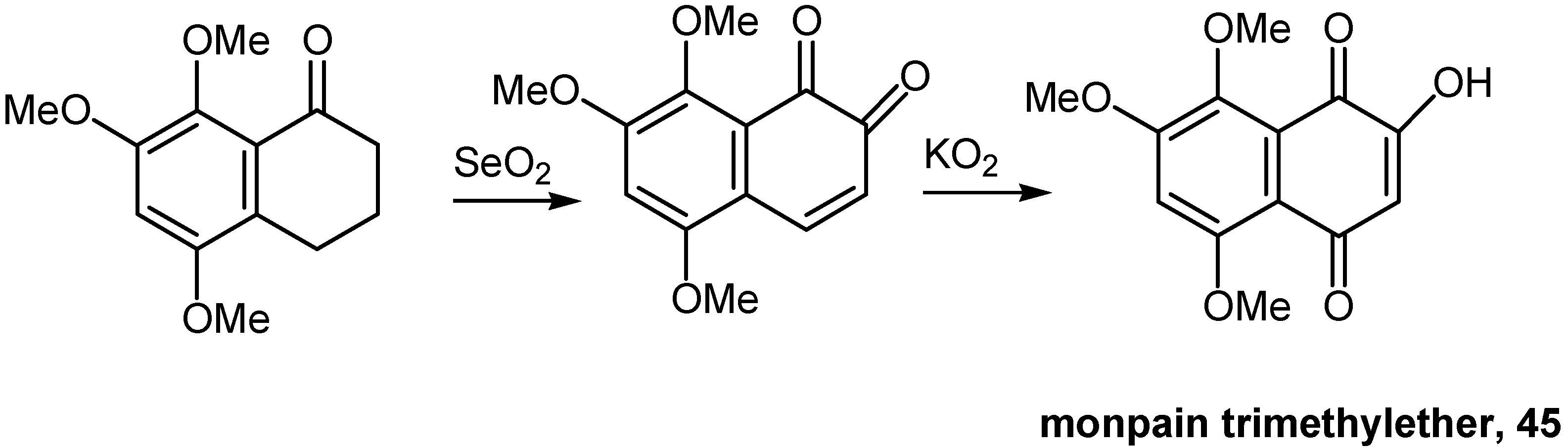
Tetralone oxidation in two steps proved to be a most effective approach to highly polymethoxylated hydroxynaphthoquinones. SeO2 oxidized the proper tetralone derivative to the corresponding orthoquinone and solid KO2 converted the latter to the desired hydroxynaphthoquinone. This approach found application to the synthesis of 5,7,8-trimethoxy-1,4-naphthoquinone (45), monpain trimethyl ether [33]. (Scheme 20)
Fremy’s salt can oxidize aniline derivatives to quinones under proper conditions [1]. In some cases direct conversion to hydroxy quinone is possible, as indicated in Scheme 21 for the preparation of hydroxy pyrrolo[1,2-a]-indoloquinone (46) of the mitosene type [34].
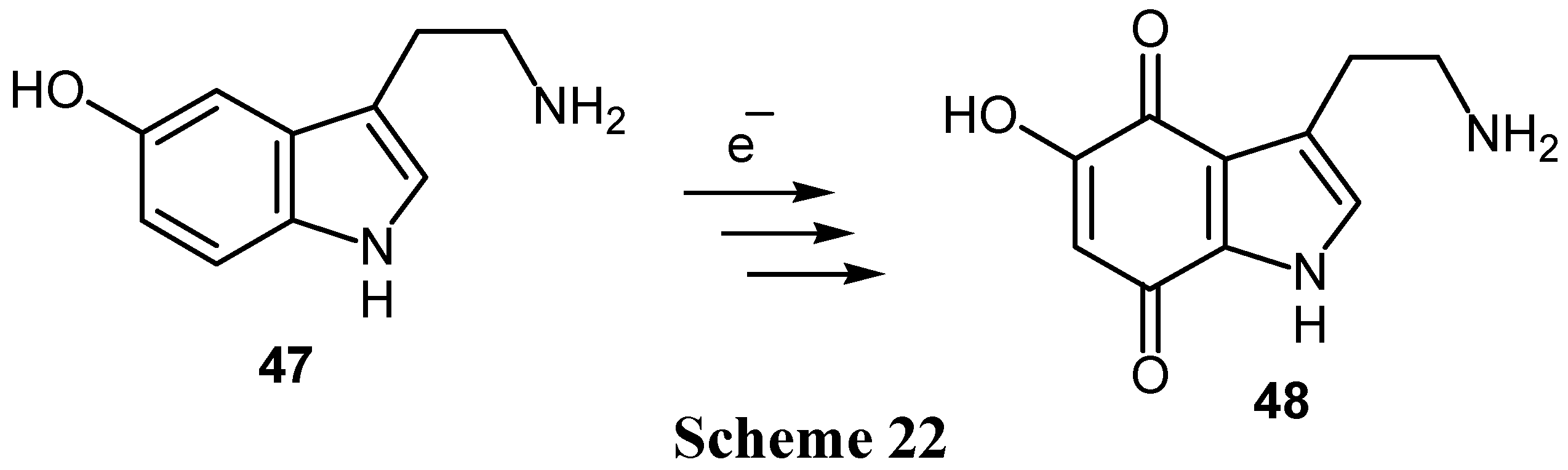
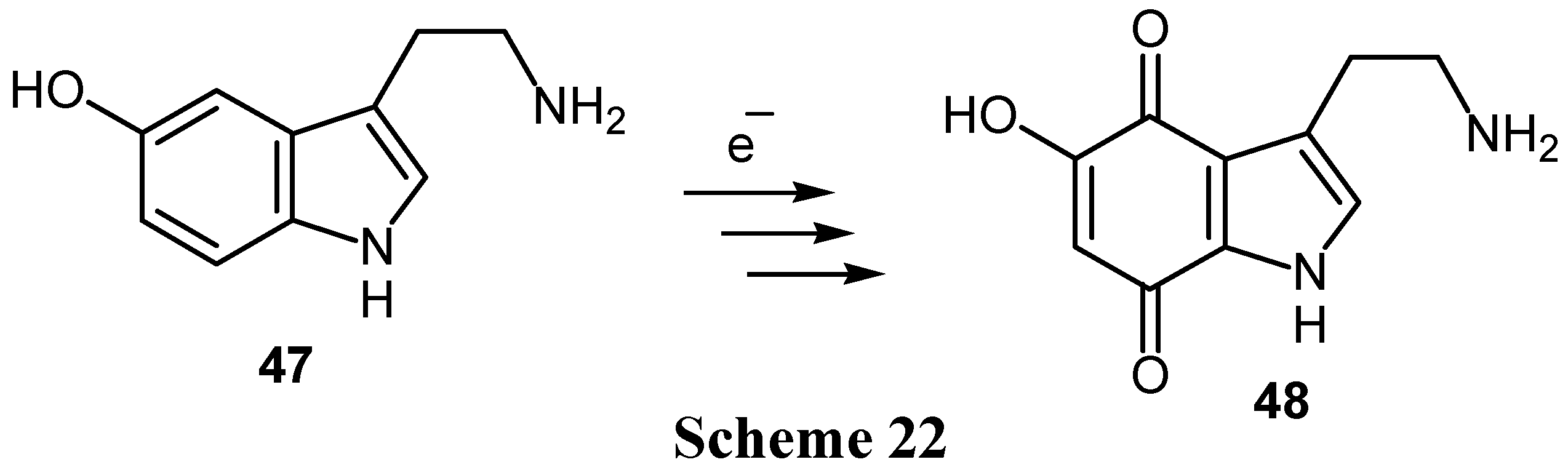
Finally, the biologically interesting hydroxyamines can be oxidized electrochemically to hydroxyquinone derivatives without oxidation of the amino group. Thus 5-hydroxytryptamine (47) can be converted to 5-hydroxytryptamine-4,7-dione (48), a potent nervous system toxin. (Scheme 22) The reaction proceeds via 5,7-dihydroxy and 4,5,7-trihydroxytryptamines [2].
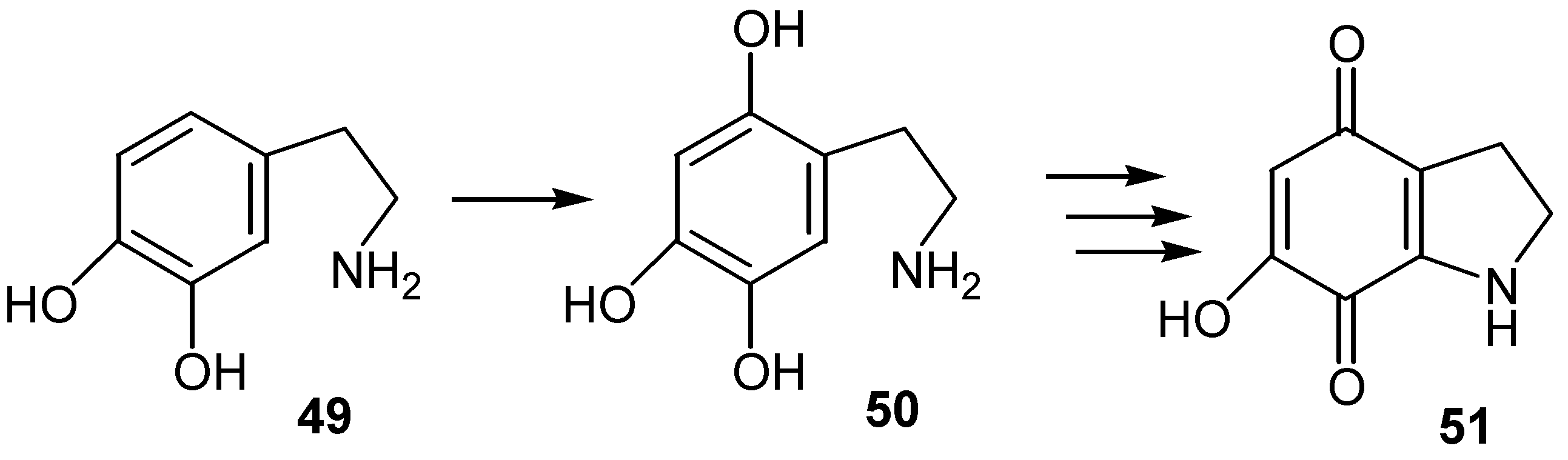
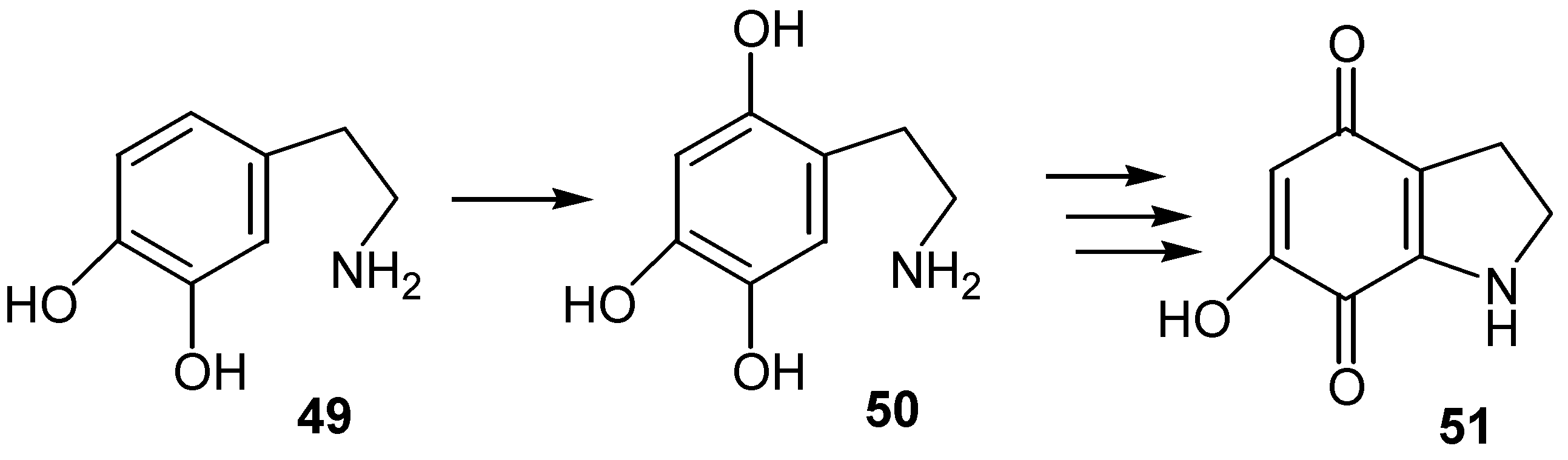
In an analogous reaction dopamine, 49, is oxidized to 2,4,5-trixydroxyphenylethylamine, 50, and the latter by subsequent oxidation, cyclization and reoxidation is converted to hydroxydihydroindolequinone derivative, 51, [35]. (Scheme 23)
The oxidation of dopamine to 6-hydroxydopamine quinone has been the subject of many studies, since the formation of the latter in vivo is related to neurodegenerative diseases. One of the most effective oxidation systems is fatty acid hydroperoxides in the presence of ferrous ions [36].
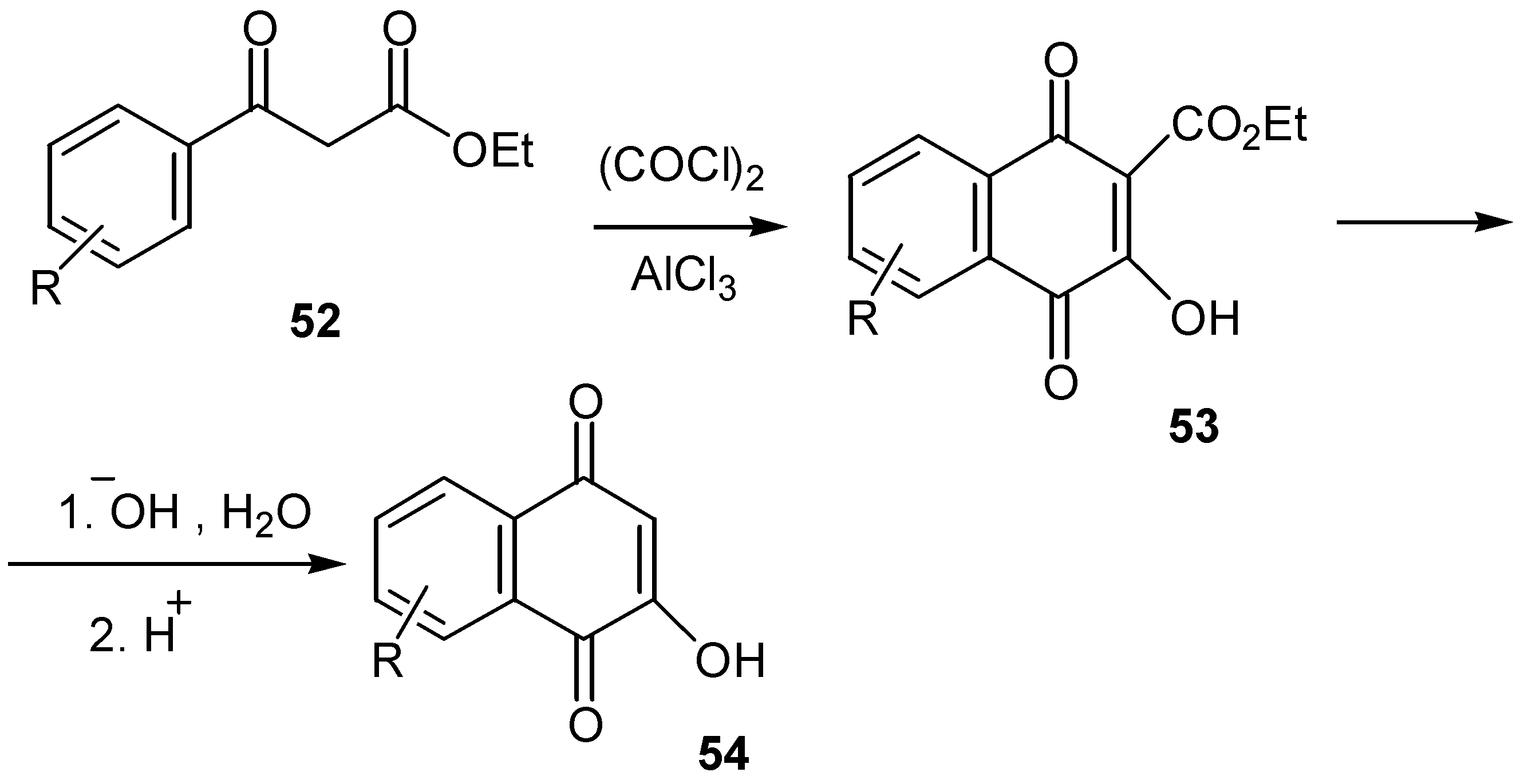
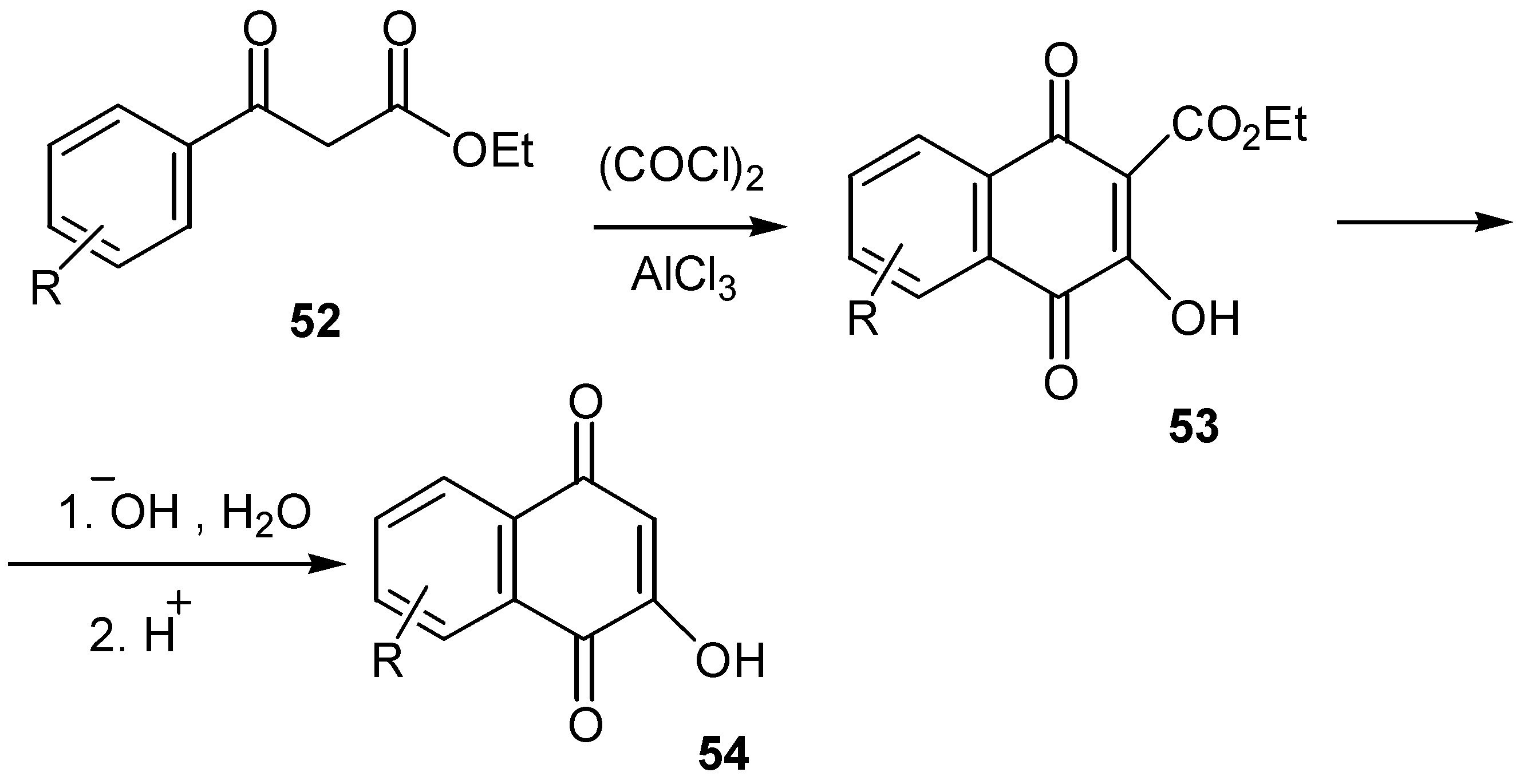
Substituted hydroxynaphthoquinones can be obtained via a Friedel-Crafts cyclization process. Oxalyl chloride addition to an aromatic â-keto ester, 52, in the presence of aluminium chloride led to the corresponding 3-hydroxy-1,4-naphthoquinone-4-carboxylate, 53. The latter was hydrolysed and decarboxylated to the desired 2-hydroxy-1,4-naphthoquinone derivative 54. (Scheme 24) The method can also be applied for the preparation of heterocyclic quinones, which are not easily available by the other methods mentioned so far [37].
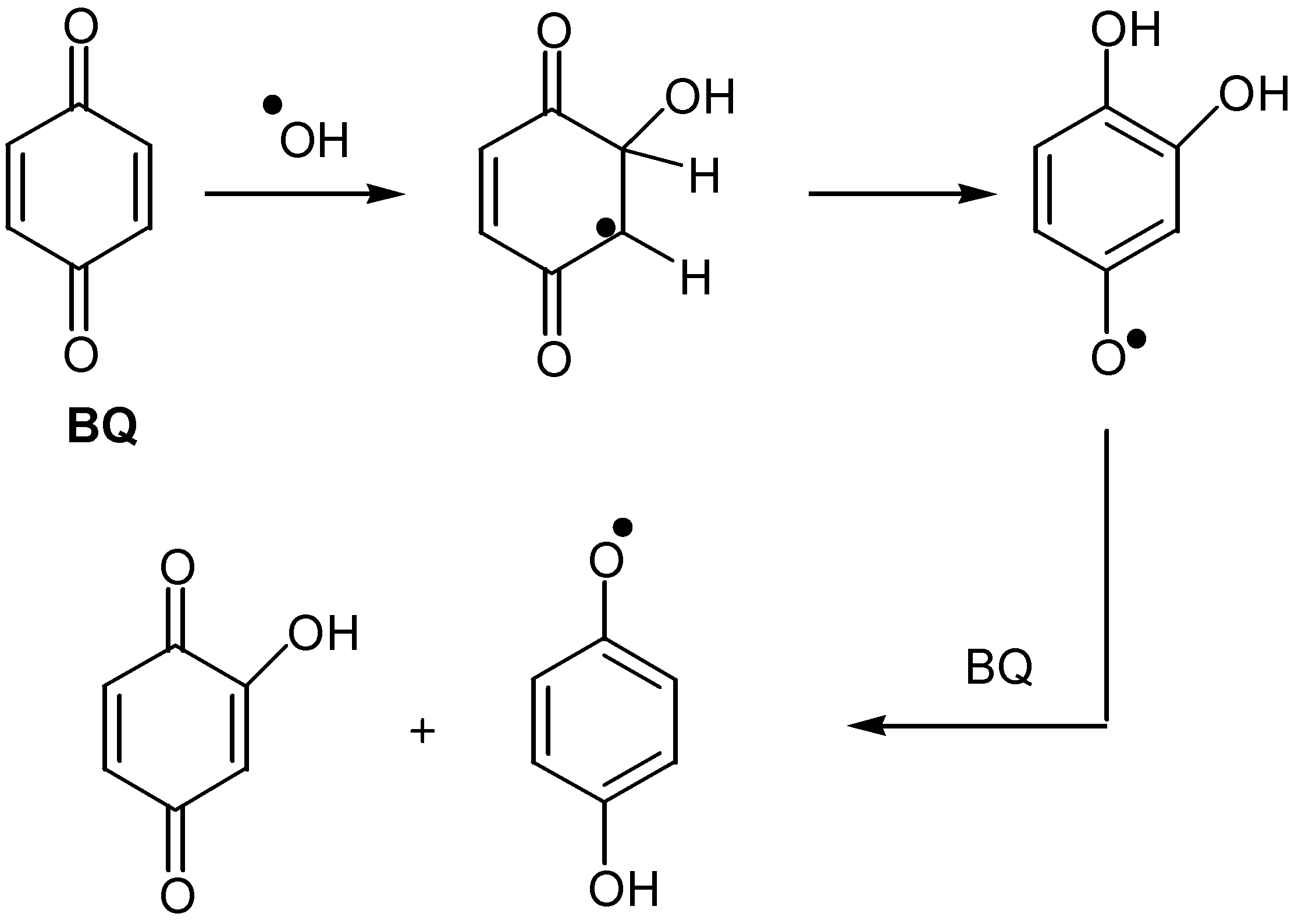
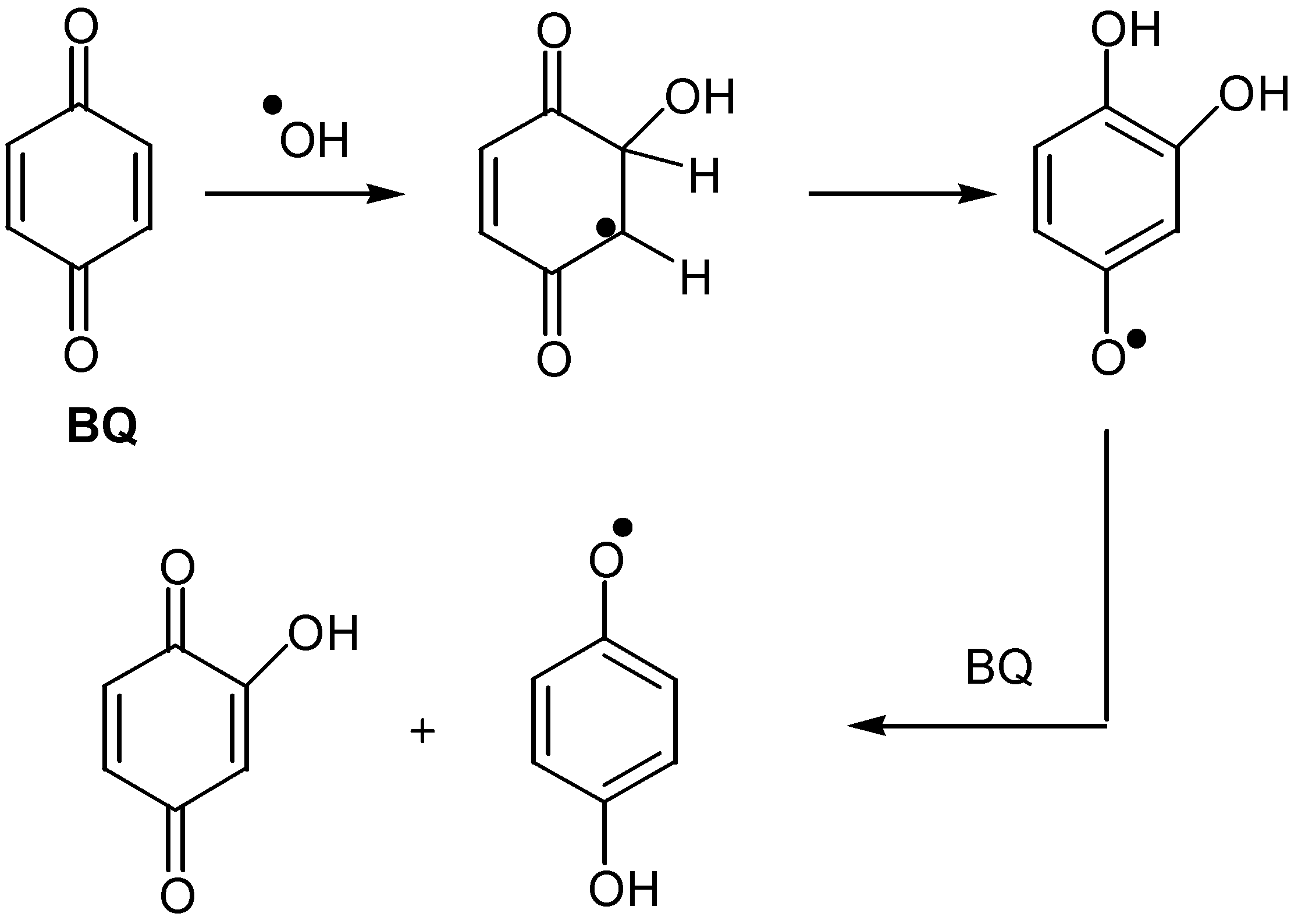
It is worth mentioning the formation of hydroxyquinones by the direct insertion of hydroxyl radicals into the quinonic ring. The radicals are produced by pulse radiolysis in aqueous solutions of benzoquinone [38], but so far this reaction has not found synthetic application.
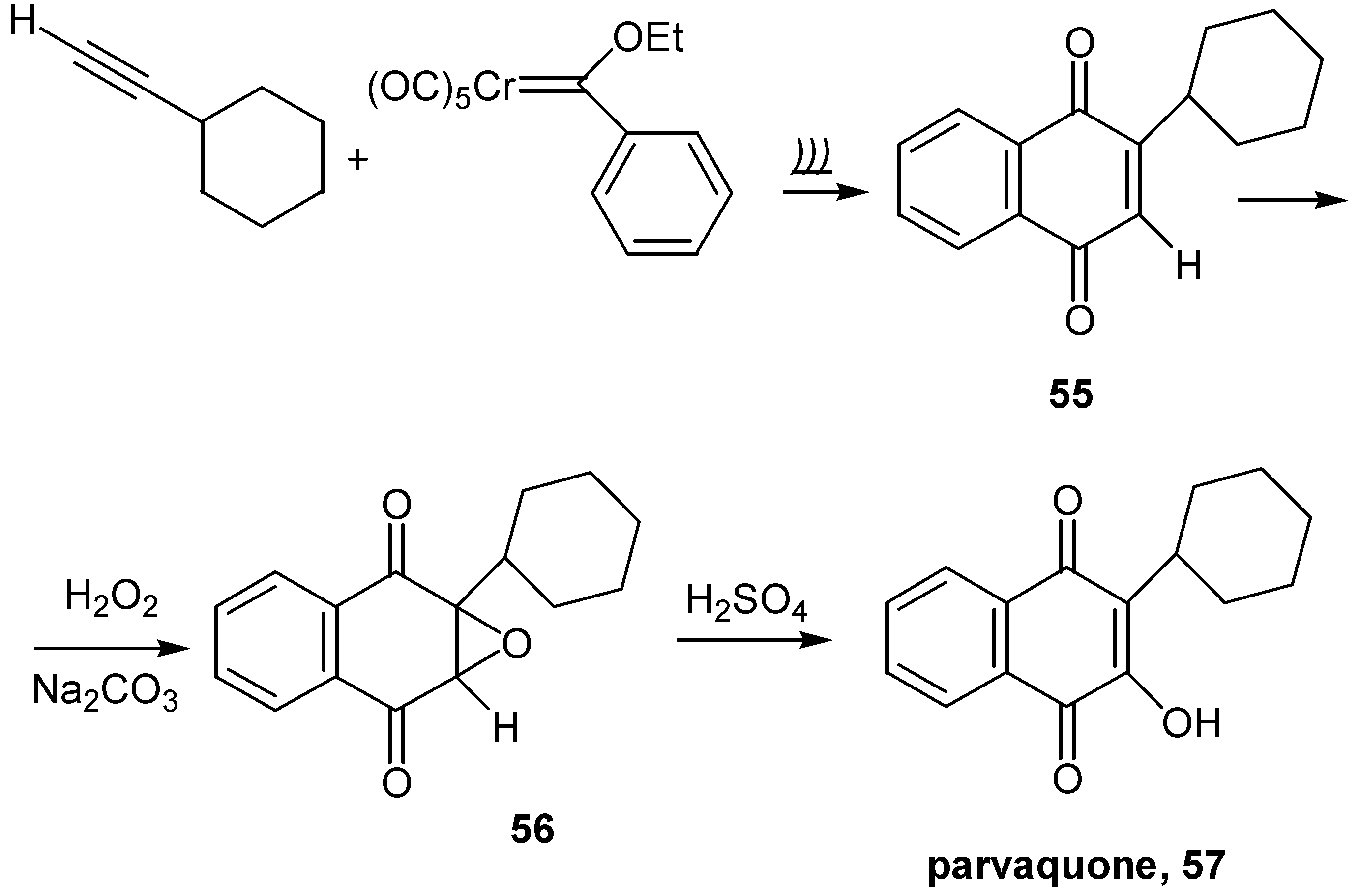
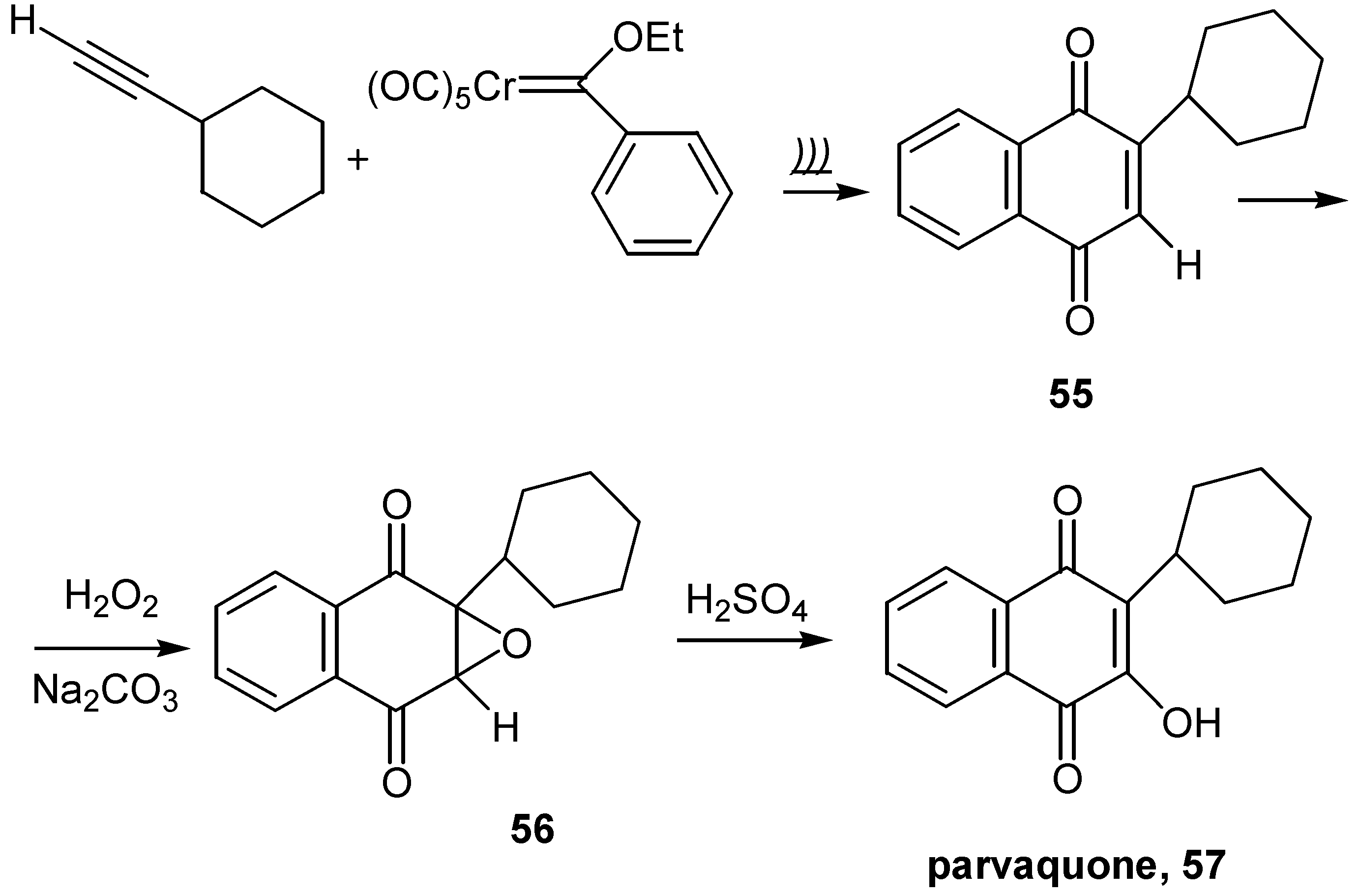
Hydroxyl insertion to the quinone ring can be effected by the ring opening of the initially formed oxirane, 56, obtained from the reaction of quinone and hydrogen peroxide in alkaline solution. This approach was applied to the synthesis of 3-cyclohexyl-2-hydroxy-1,4-naphthoquinone, parvaquone (57), known to display antiprotozoal activity. The parent quinone, 55, was prepared by modification of the Dötz annulation reaction, involving chromium-carbene complexes and use of ultrasound [39]. (Scheme 26)
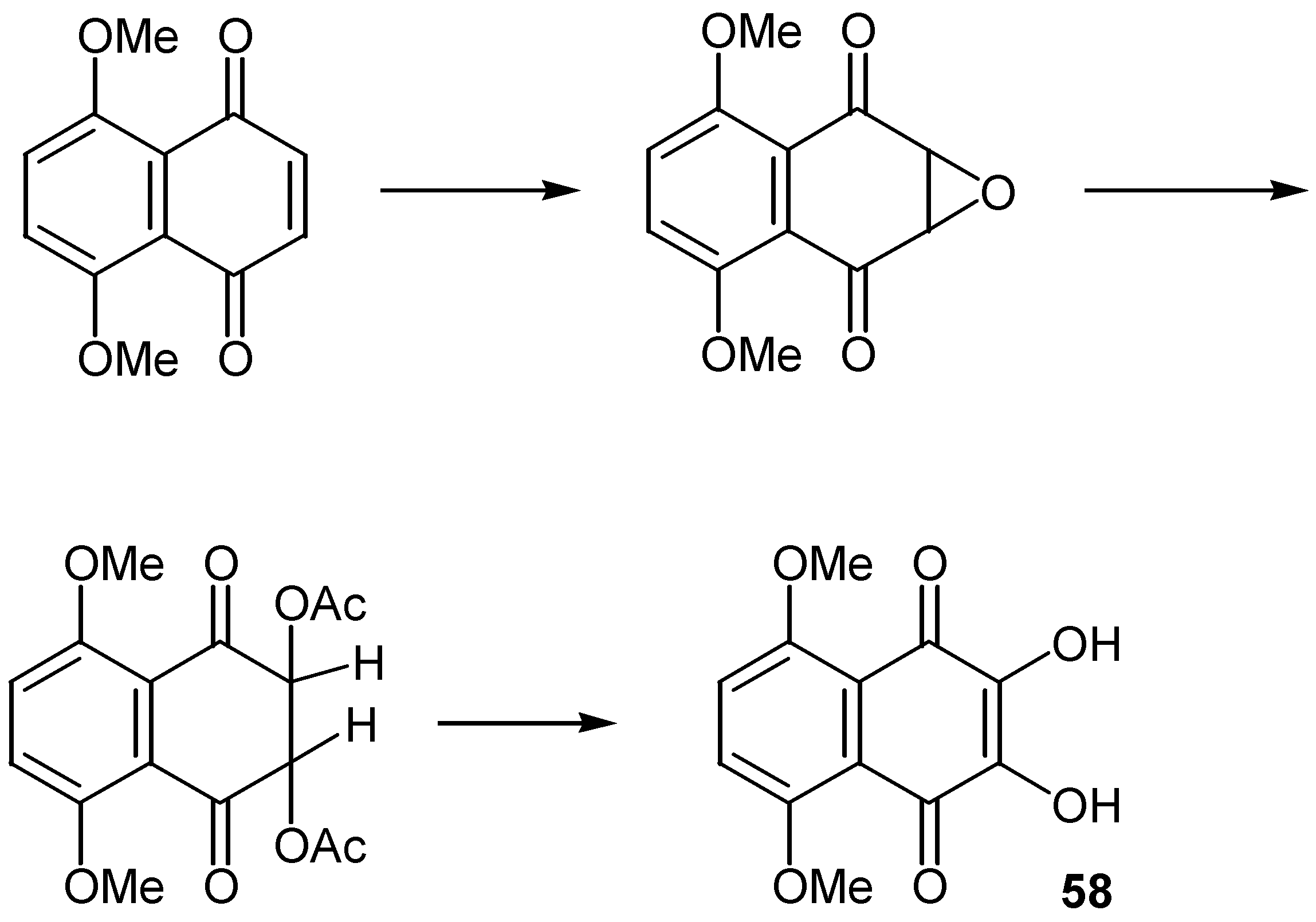
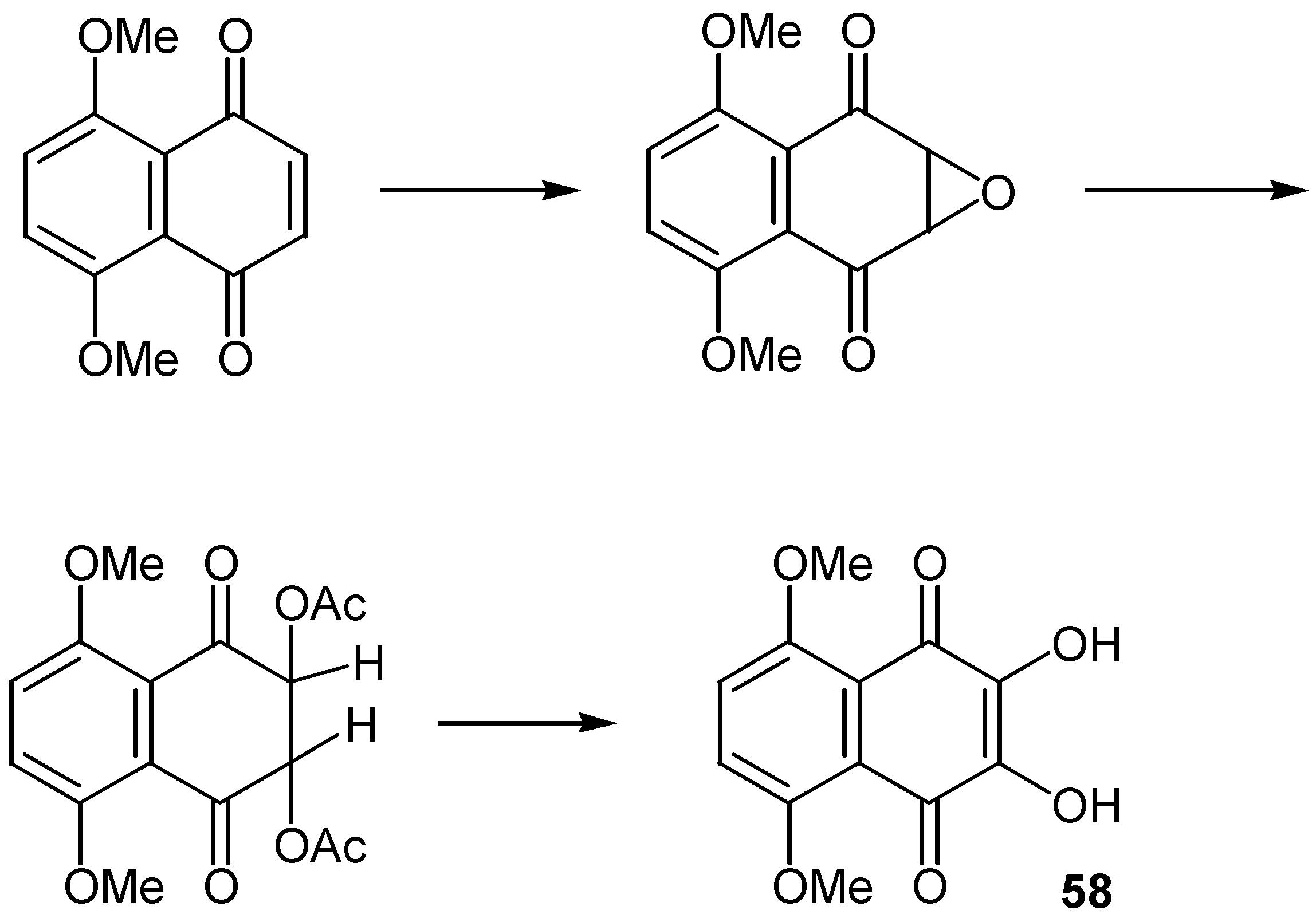
The sequence quinone→oxirane→hydroxyquinone was used for the preparation of dihydroxynaphthoquinones, 58, as shown in Scheme 27 [40].
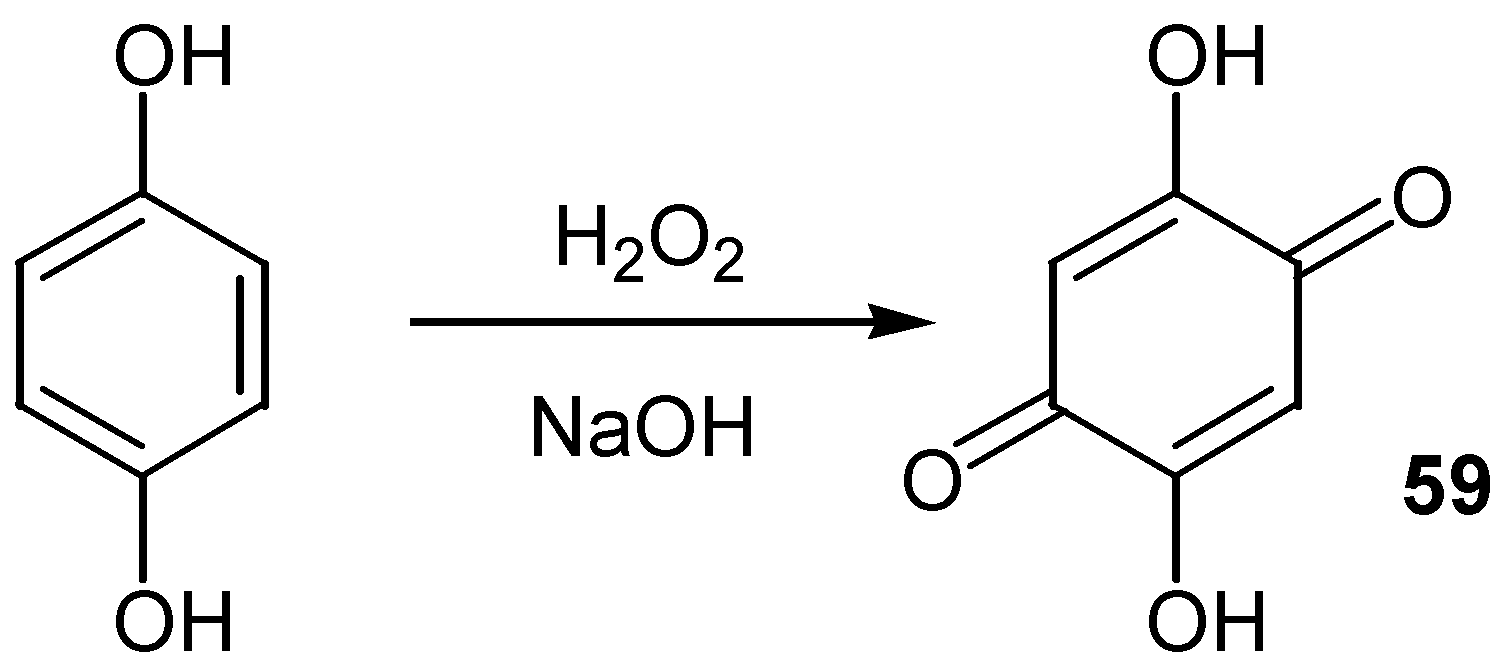
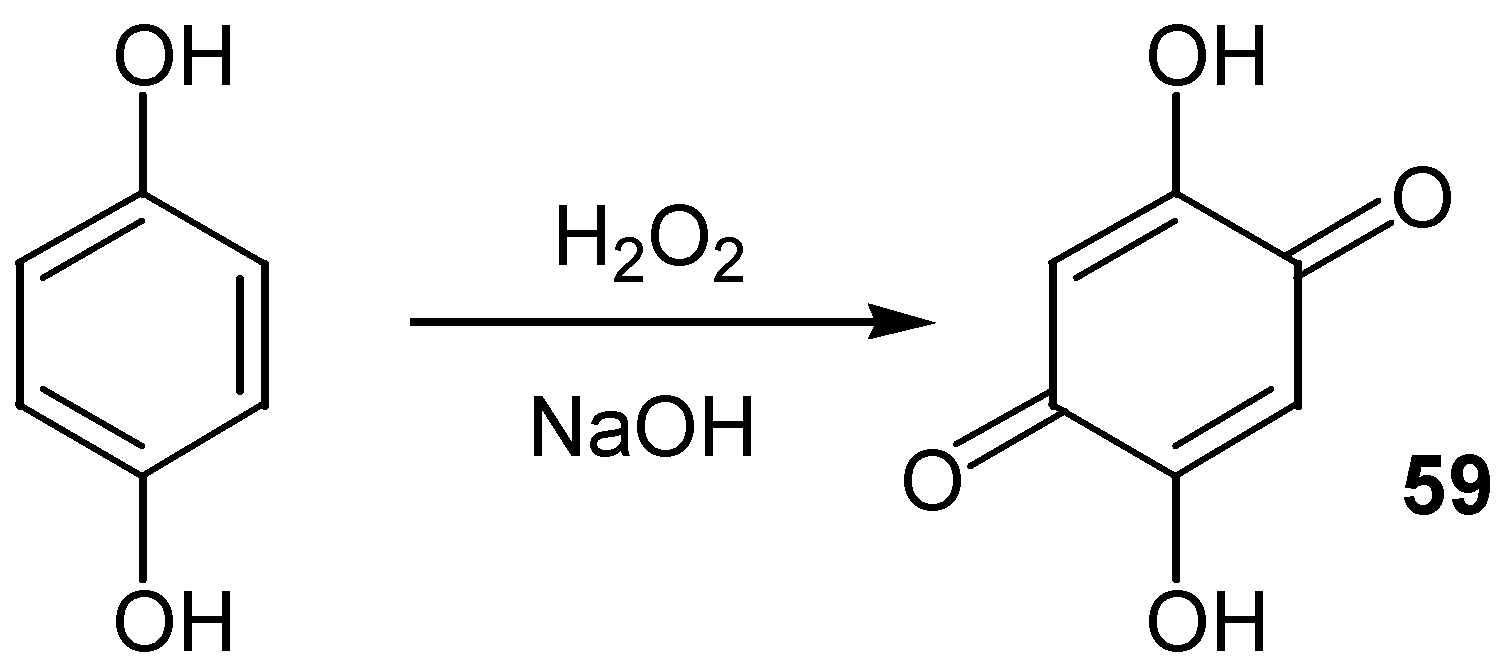
Analogously, 2,5-dihydroxy-1,4-benzoquinone, 59, an interesting commercially available building block, was most easily prepared [41] from the oxidation of hydroquinone by hydrogen peroxide in alkaline solution. (Scheme 28)
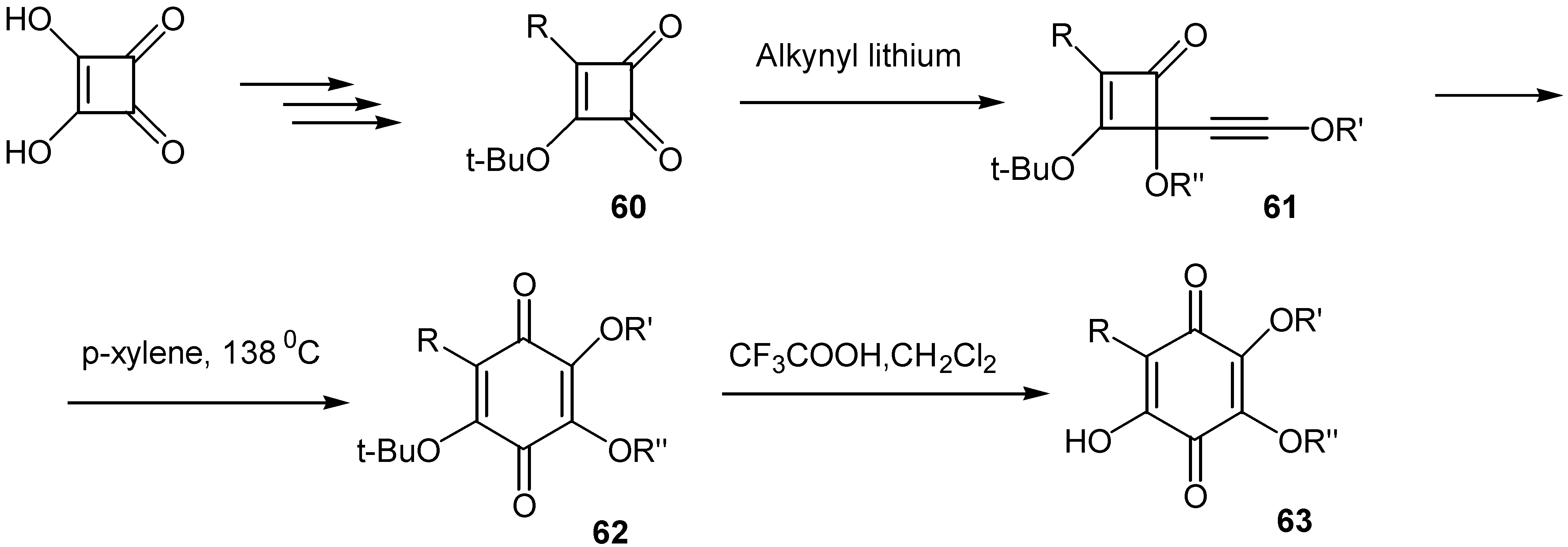
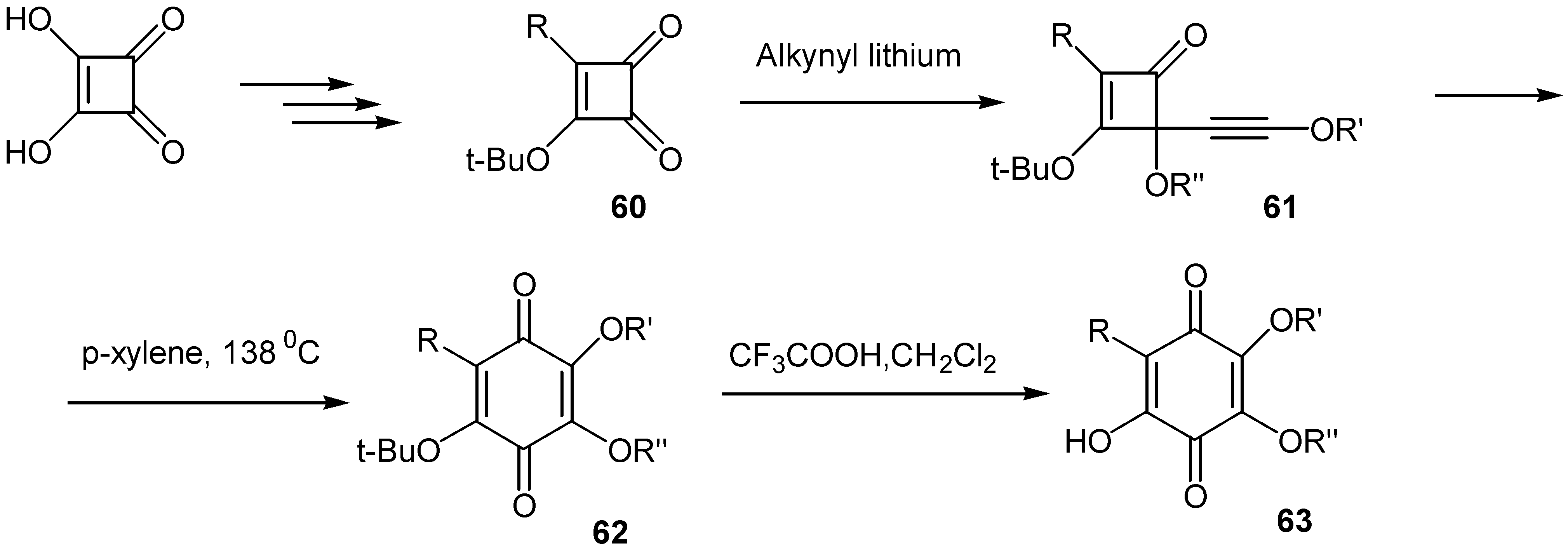
Moore and co-workers suggested an interesting method for the preparation of a great variety of hydroxyquinones [42a,b]: t-butoxy-cyclobutanedione (60), readily available from squaric acid, is converted to the corresponding alkynyl cyclobutanedione, 61, by the action of the proper alkynyl lithium reagent. The latter through a thermal ring expansion affords the corresponding t-butoxy- quinone, 62, and finally removal of the t-butoxy group by trifluoracetic acid under mild conditions leads to the desired hydroxyquinone, 63.(Scheme 29)
This methodology offers an easy access to hydroxy quinone functionality and finds application for the preparation of either simple quinones or complicated natural analogs. From this point it can be compared to the Thiele-Winter route to hydroxyquinones.
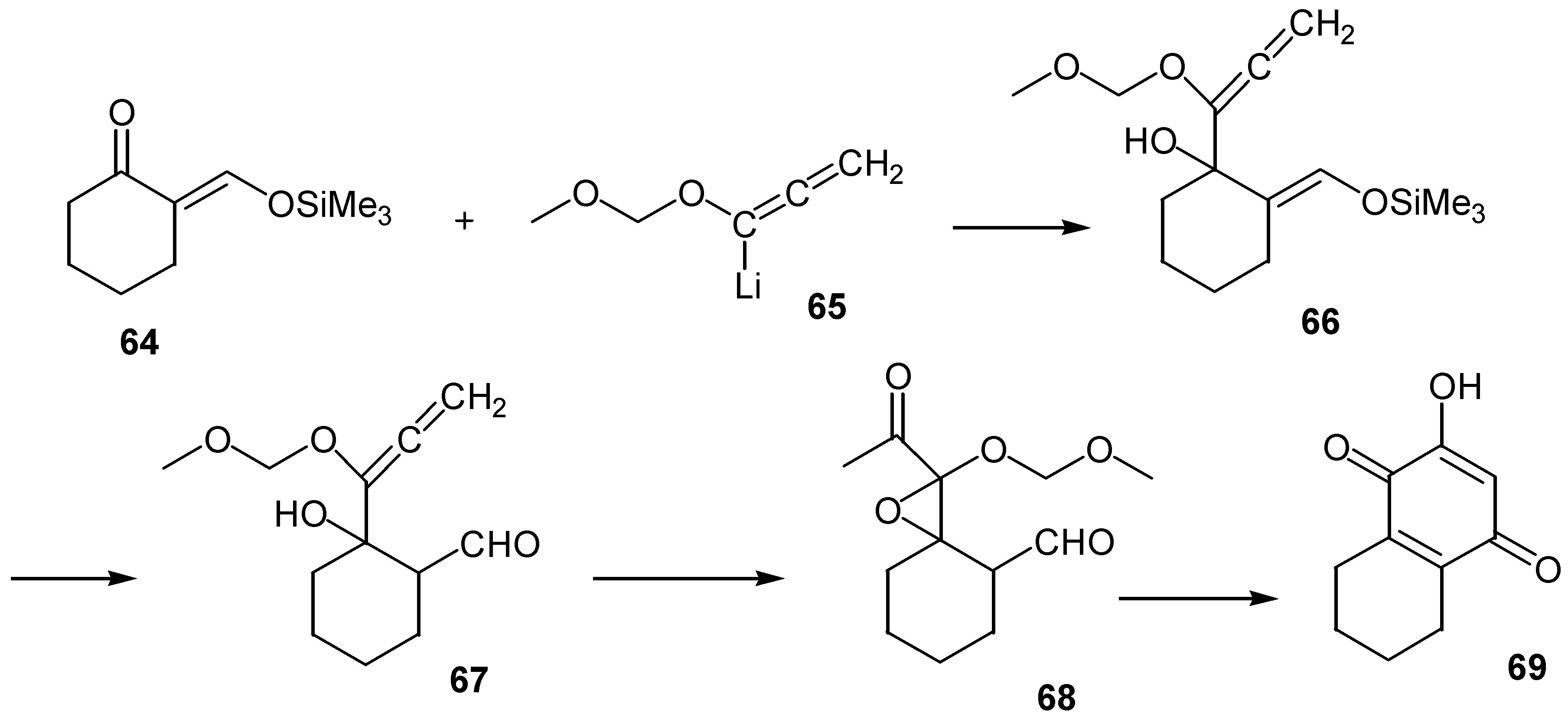
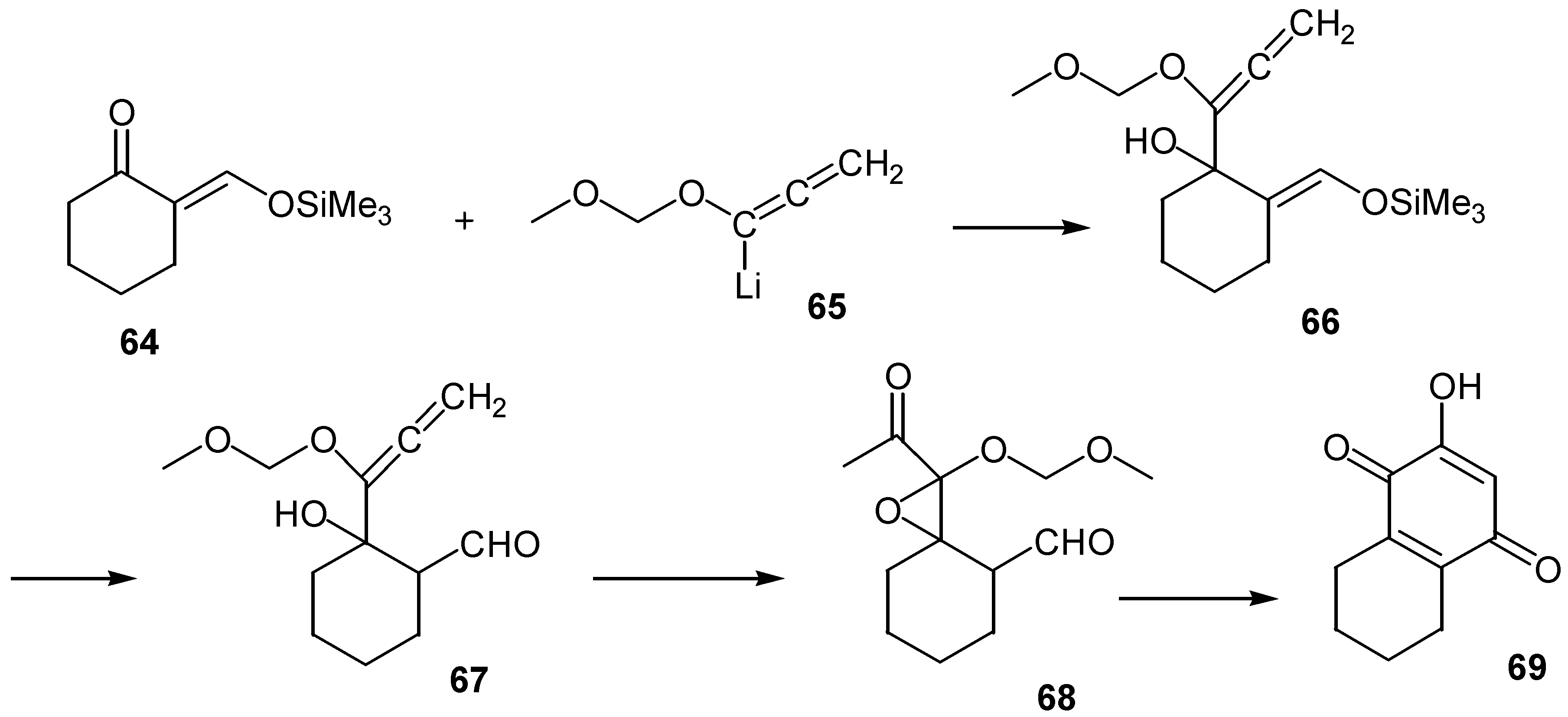
Another rather unusual annelation gives rise to various ring fused hydroxybenzoquinones [43]. The method for the preparation of the cyclohexane-fused derivative, 69, is outlined in Scheme 30.
The preparation starts with the reaction of the proper vinylogous silyl ether, 64, with the lithio anion of the (methoxy)methoxyallene, 65. The intermediate silyl or formyl compounds, 66 or 67, are epoxidized with m-chloroperoxybenzoic acid with simultaneous rearrangement of an initially formed zwiterionic intermediate. Finally, the oxirane derivative is converted to the fused hydroxybenzoquinone, 69, through a base-catalysed intramolecular aldol reaction. Yields are satisfactory for a variety of cyclopentanones and cyclohexanones, as well as for some aromatic ketones.
The reaction of 3-halo-3,6-dialkyl-1,2-cyclohexanediones (under their enolic form, 70) with iodine – copper(II) acetate afforded the corresponding 3-hydroxy-2,5-dialkyl-1,4-benzoquinones, 71, in 40-80% yield [44]. (Scheme 31)
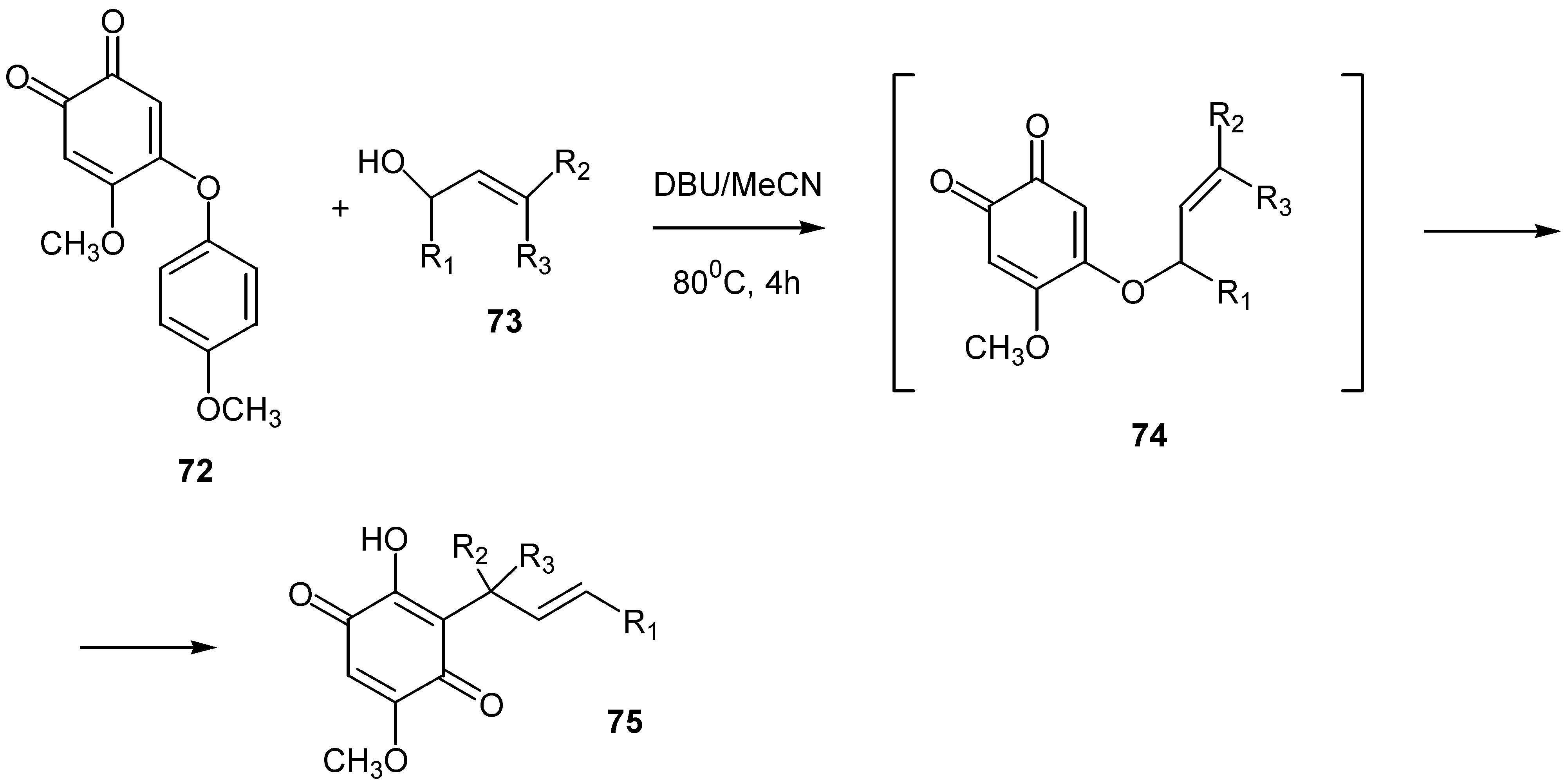
Finally, alkenyl substituted methoxy-hydroxybenzoquinones, 75, are obtained via Claisen rearrangement of the corresponding propenyloxy-o-benzoquinones,74, which are easily prepared from 5-methoxy-4-(4’-methoxy-phenyl)-1,2-benzoquinone, 72, and the proper allylic alcohol, 73, [45]. Propenyloxy-o-benzoquinones can be isolated, but the one pot reaction gives also satisfactory yields (35-87%). (Scheme 32)
3. Reactivity
The reactivity of hydroxyquinones in general is related to the reactivity of quinones bearing electron-donor substituents. In addition, the enol-enone moiety offers some interesting synthetic possibilities. The main patterns of hydroxyquinone reactivity are summarized below.
a) C - C bond formation
A large number of biologically active compounds related to hydroxyquinone structure bear a carbon group to the position next to hydroxyl. In some compounds the hydroxyl is free, retaining thus the hydroxyquinone moiety, whereas in others the oxygen of the hydroxy group participates in a heterocyclic ring. This explains the fact that a lot of synthetic effort has been put on the general reaction scheme outlined below.

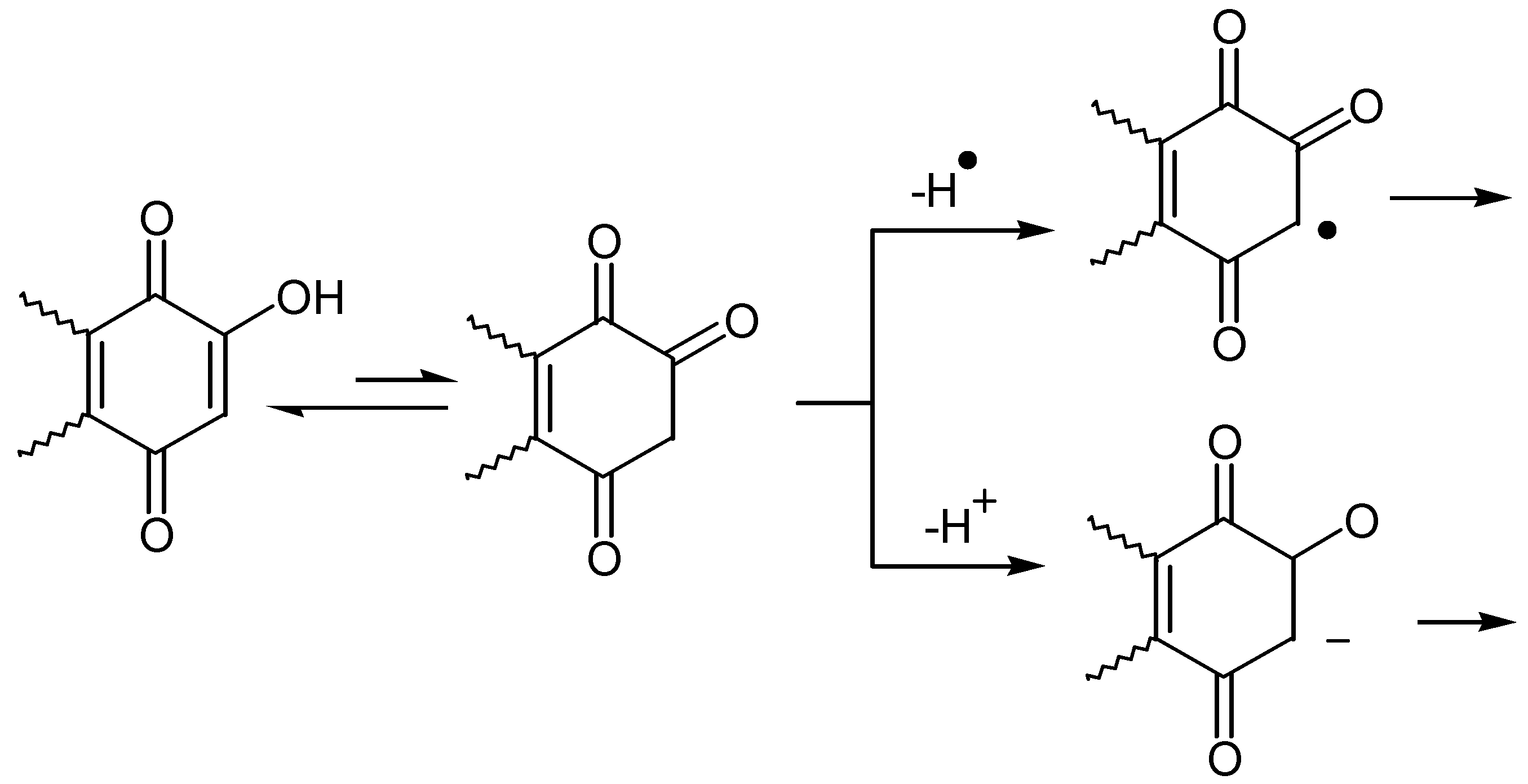
Sometimes the alkyl derivative is the synthetic target, whereas in other cases the cyclization proceeds either with or without the isolation of the intermediate alkyl hydroxyquinone. The C - C bond formation proceeds through two main reaction pathways: The hydroxyquinone, being in equilibrium with its keto form, can be alkylated either through a radical mechanism or via conjugated addition to the enolate formed by addition of base to hydroxyquinone solution. (Scheme 33)
I. Alkyl substitution at carbon
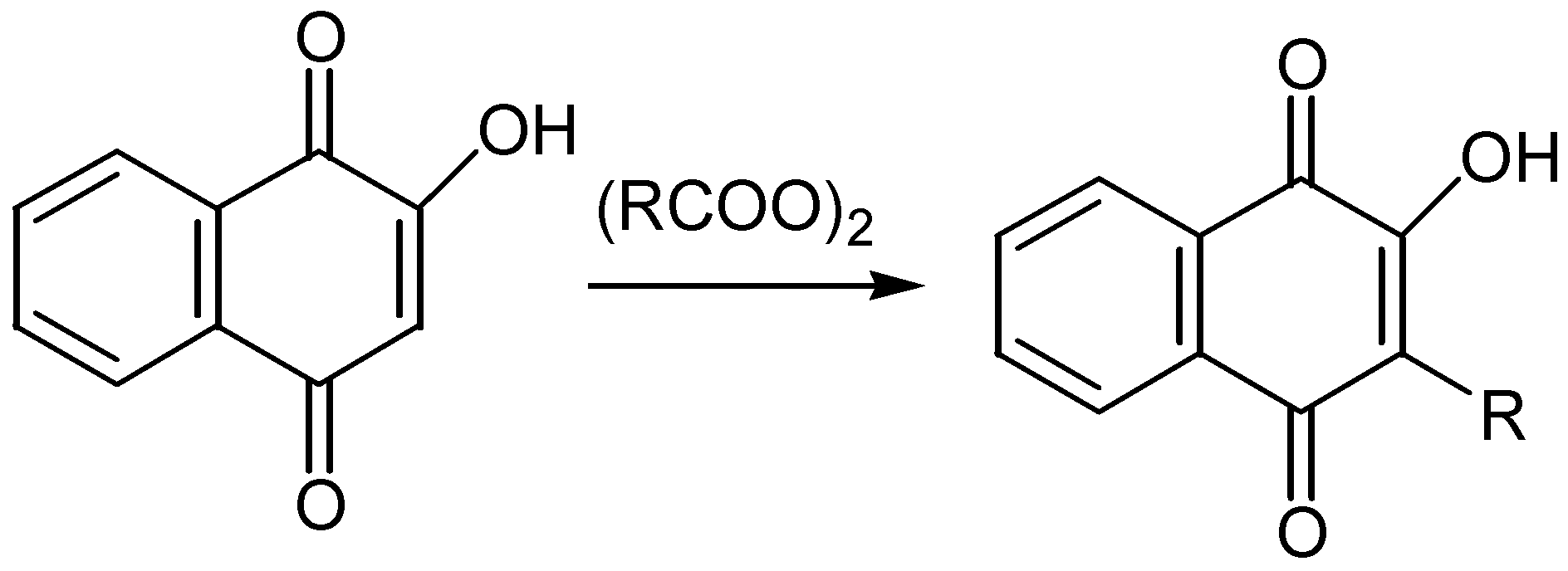
Although the alkylation of quinones has been examined in some detail [46], relatively few reports exist on the alkylation of hydroxyquinones. One of the first methods was the reaction of diacyl peroxides with hydroxynaphthoquinone (lawsone) to produce a great variety of naphthoquinones possesing antimalarial activity [47]. The reaction proceeds through a radical mechanism but usually yields are not satisfactory. (Scheme 34)
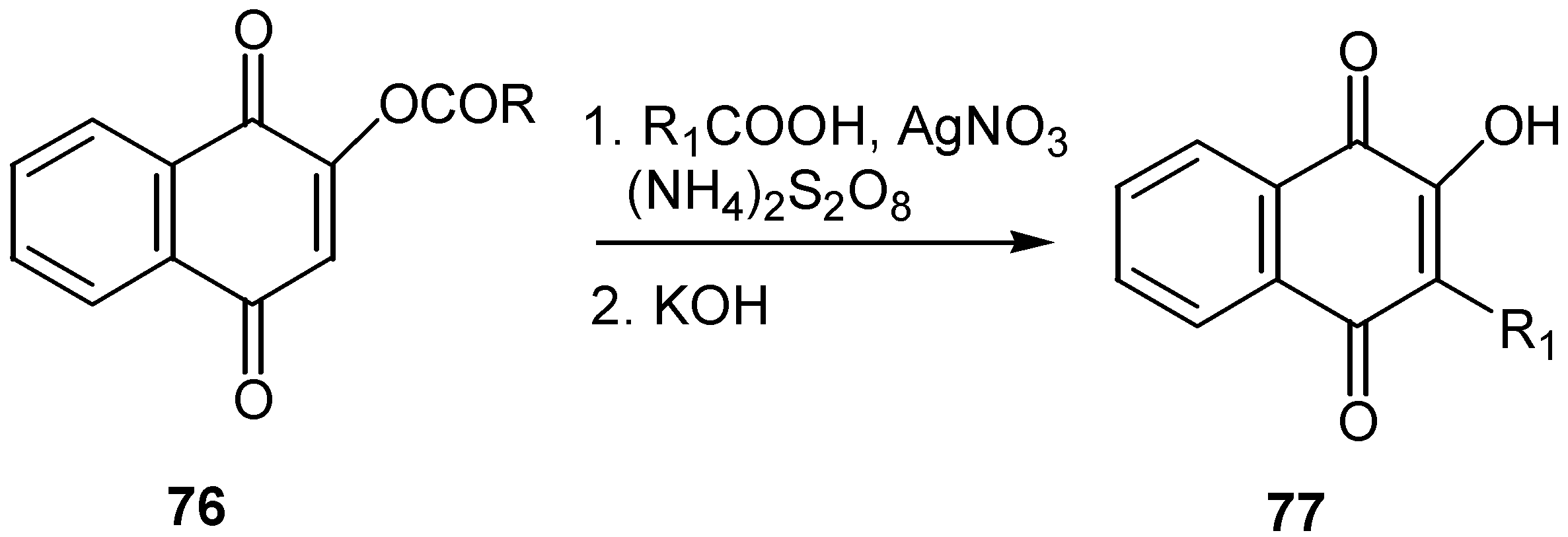
Better results are obtained from the reaction of acylated lawsone, 76, with a carboxylic acid in the presence of peroxysulfate-mediated radical decarboxylation reaction [48]. (Scheme 35)
The method was employed for the preparation of naphthoquinone derivatives of the general structures 78, 79, and 80, exhibiting potential pesticidal activities.

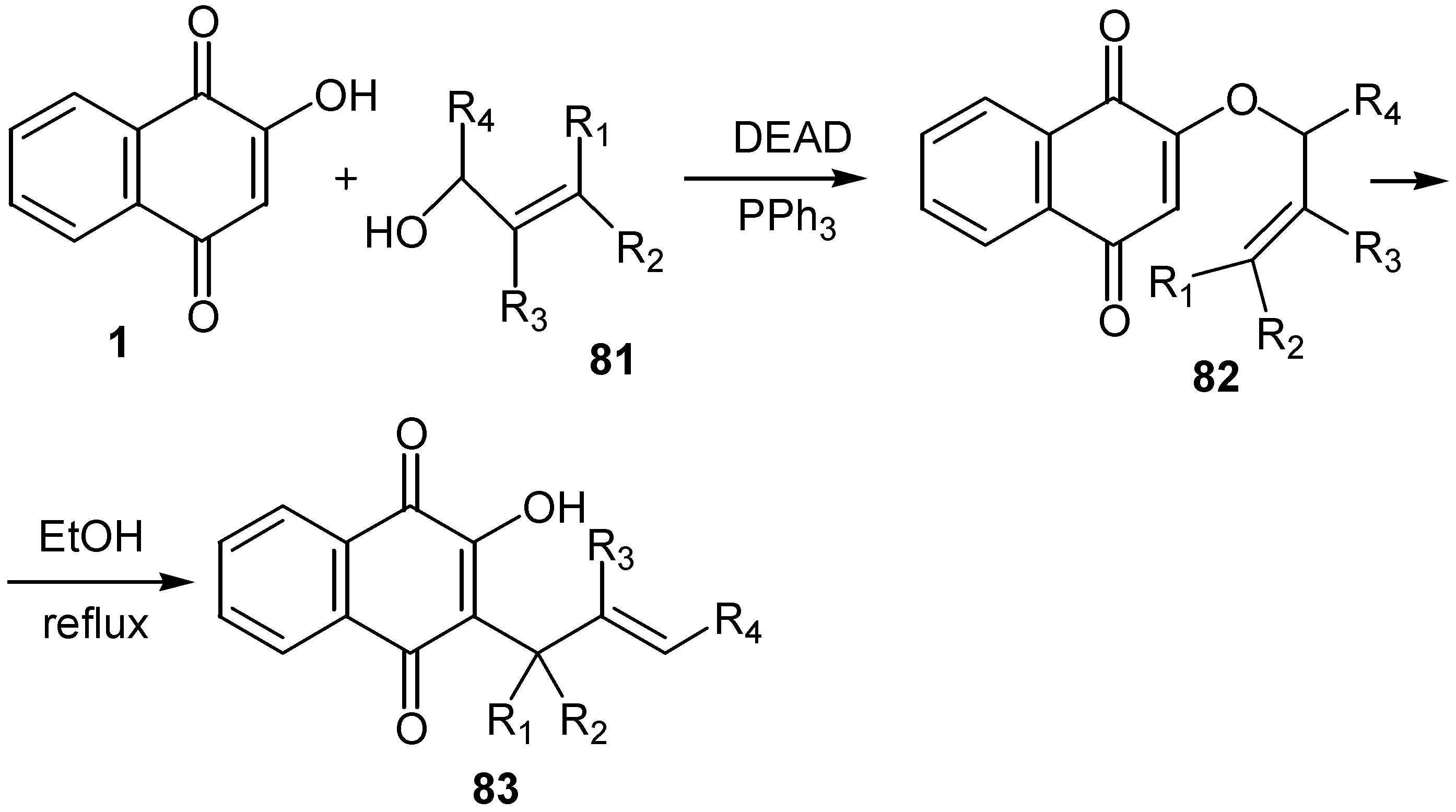
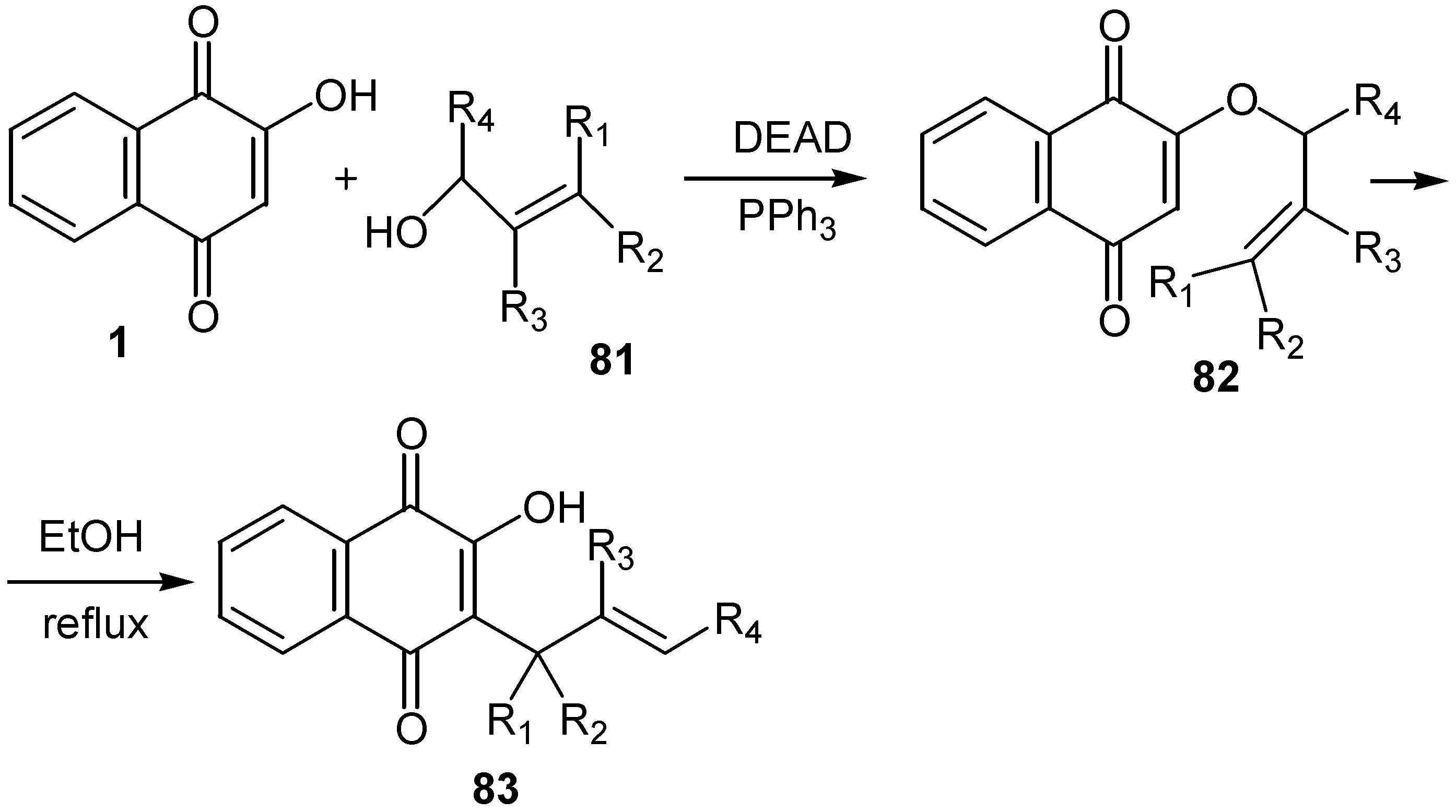
The same authors prepared series of allyl substituted hydroxynaphthoquinones, 83, from the reaction of lawsone, 1, with the corresponding allylic alcohol, 81, under Mitsonobu reaction conditions and subsequent Claisen rearrangement. (Scheme 36)
In a minor extension of the methodology, the Hooker oxidation with alkaline permanganate or hydrogen peroxide was used for the preparation of alkyl homologues with one less carbon at C-1 at the side chain, 85, exhibiting the same pesticide activity. (Scheme 37)
An interesting alkylation method of lawsone towards a preparative synthesis of lapachone in a large scale was recently reported [49]: The lithium salt of lawsone, 1, was prepared in situ by addition of lithium hydride to a frozen solution of the quinone in dimethylsulfoxide. As the solution thawed, the lithium salt was slowly formed and it was then alkylated with 3,3-dimethylallyl bromide, to afford the desired alkyl derivative, 86, in 30% yield. (Scheme 38)
Finally, Bieber and co-workers [50] showed that lawsone, as well as alkoxy quinones, can be directly alkylated by alkylboranes at C-3 position. The initially formed product, a reduced boranequinone complex, was oxidatively hydrolysed to alkyl lawsone, 87, (Scheme39).
The direct arylation of hydroxyquinones is not so common. In an older method lawsone was arylated by diazonium salts under alkaline conditions [51] (Scheme 40).
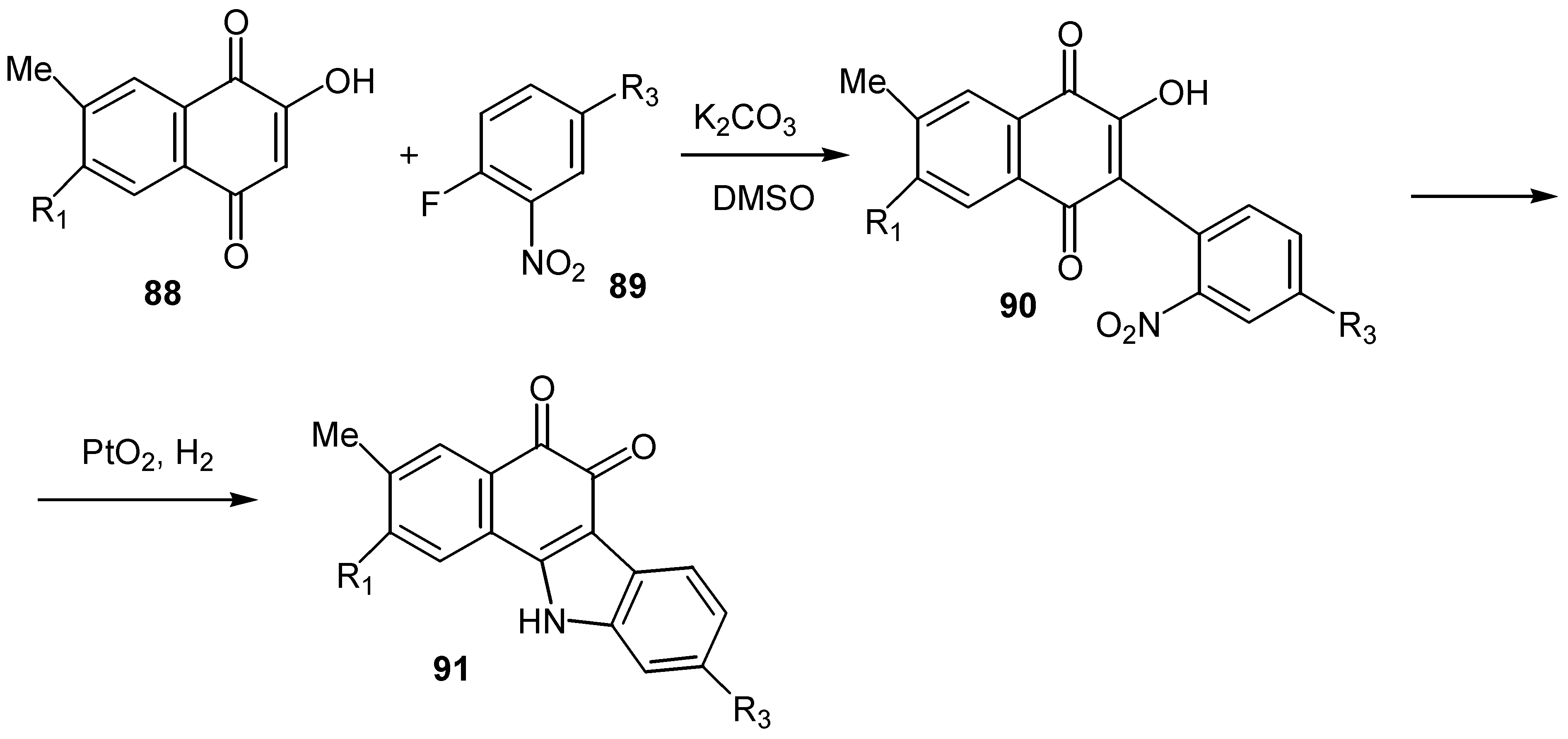
Recently the reaction of substituted lawsone derivatives, 88, with o-fluoronitrobenzenes, 89, gave 2-hydroxy-3-(2-nitroaryl)-1,4-naphthoquinones, 90, in fair yields. The latter were transformed into the corresponding benzocarbazolequinones, 91, through reduction of the nitro group to an amino group and subsequent cyclisation [52] (Scheme 41).
II. Cyclization to furan derivatives
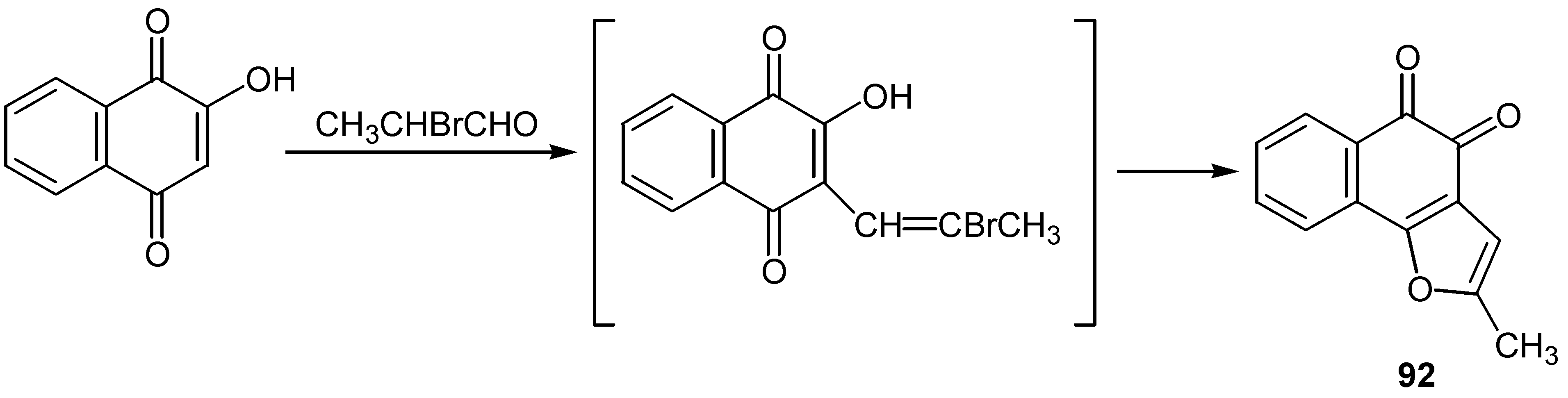
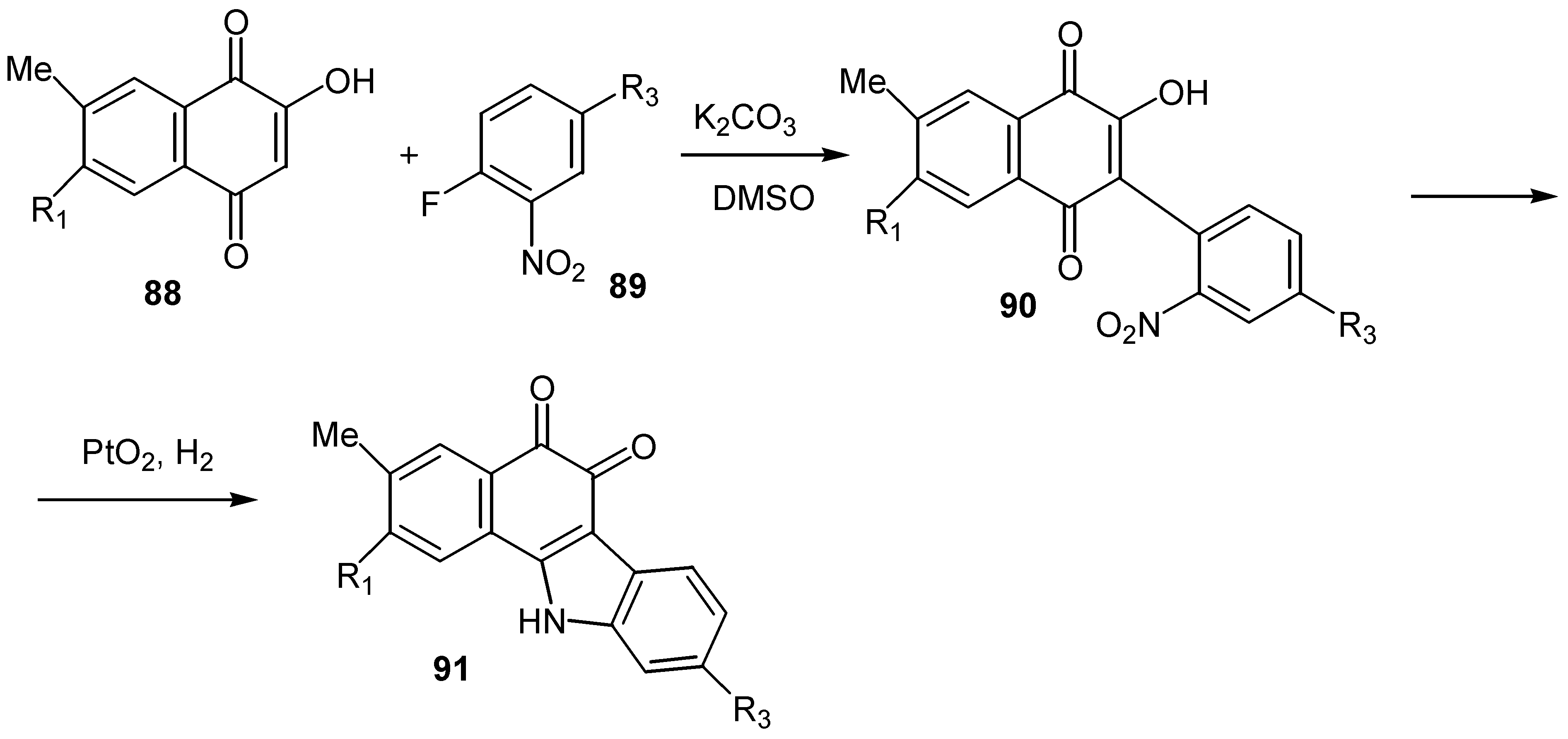
A great number of furoquinones, especially naphthofuranodiones, are natural products exhibiting a broad spectrum of biological activity. For this reason the cyclization of hydroxyquinones to the corresponding furan derivatives consists one of the most interesting features of their chemistry and several cyclization methods have been developed. The reaction of lawsone with 2-bromopropanal afforded the ortho-quinone furo derivative, 92, through the initial alkylation of C-2 [11]. (Scheme 42)
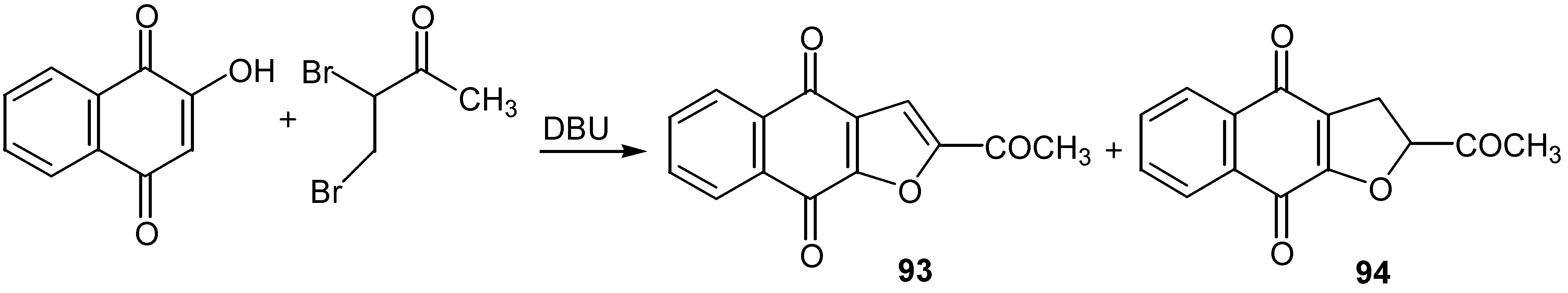
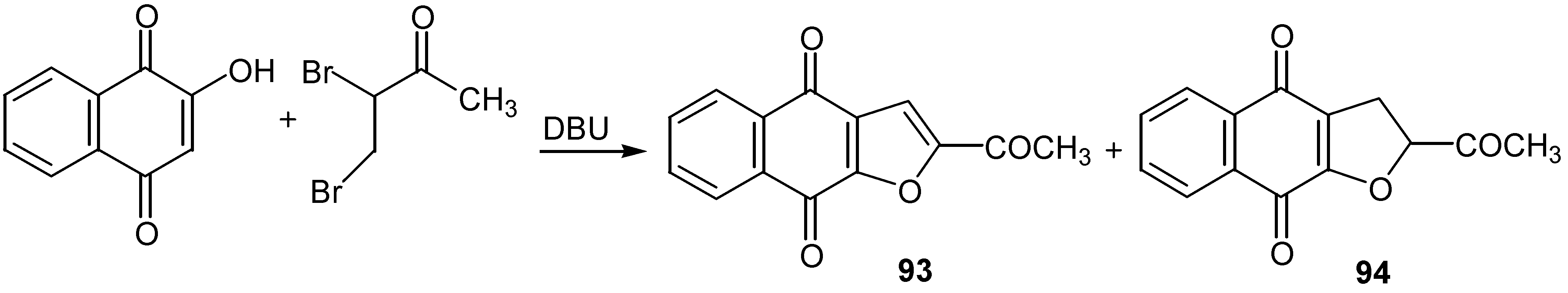
An analogous reaction using 3,4-dibromo-2-butanone led to a mixture of furan, 93, and dehydrofuran, 94, derivatives. (Scheme 43). The former are natural products with significant antineoplastic activity [53].
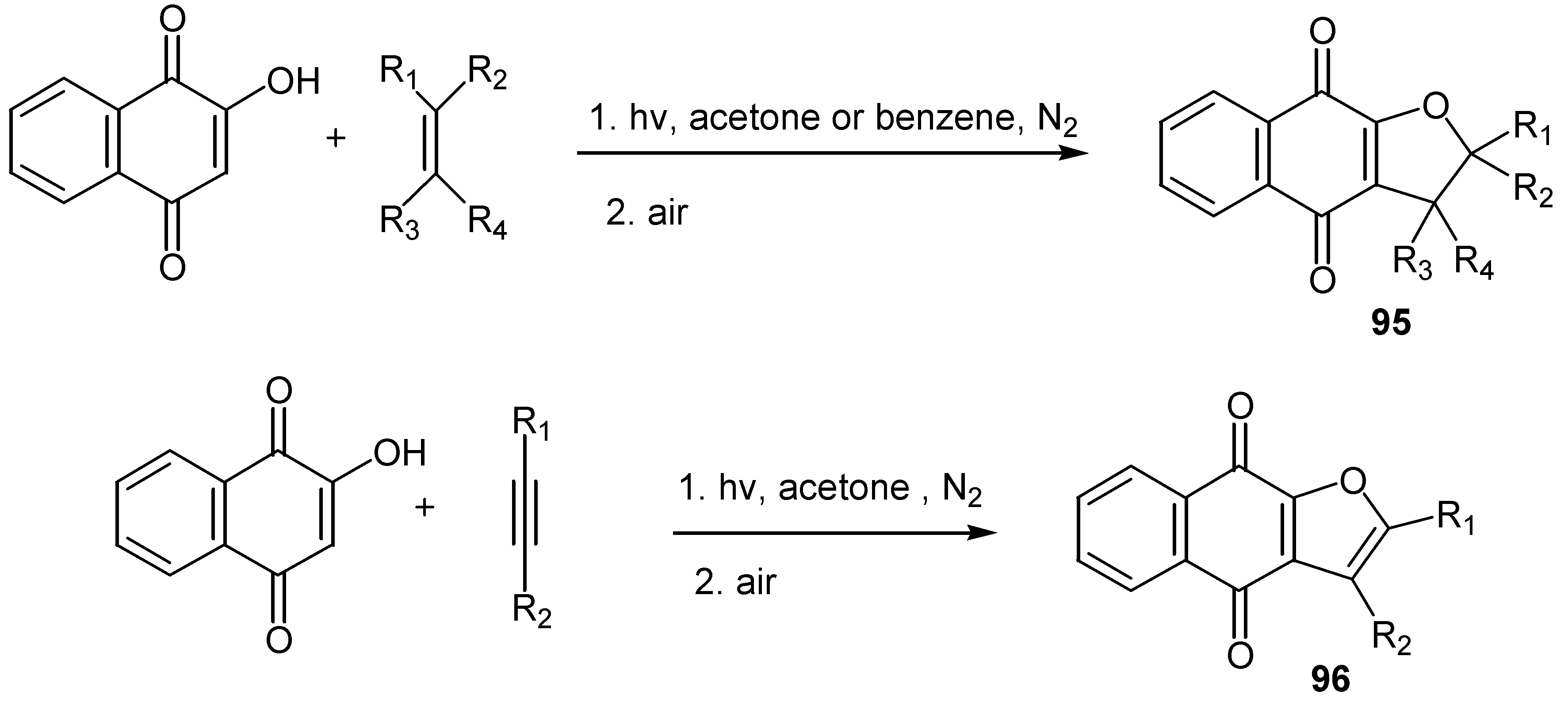
The regioselective [3+2] photoaddition of 2-hydroxy-napthoquinones with a variety of alkenes offers an easy one-step access to dihydrofuran derivatives, 95, whereas the reaction with alkynes led to the furan analogues, 96, [54,55] (Scheme 44). Similar results were obtained from the reaction with 2-hydroxy-benzoquinones [13].
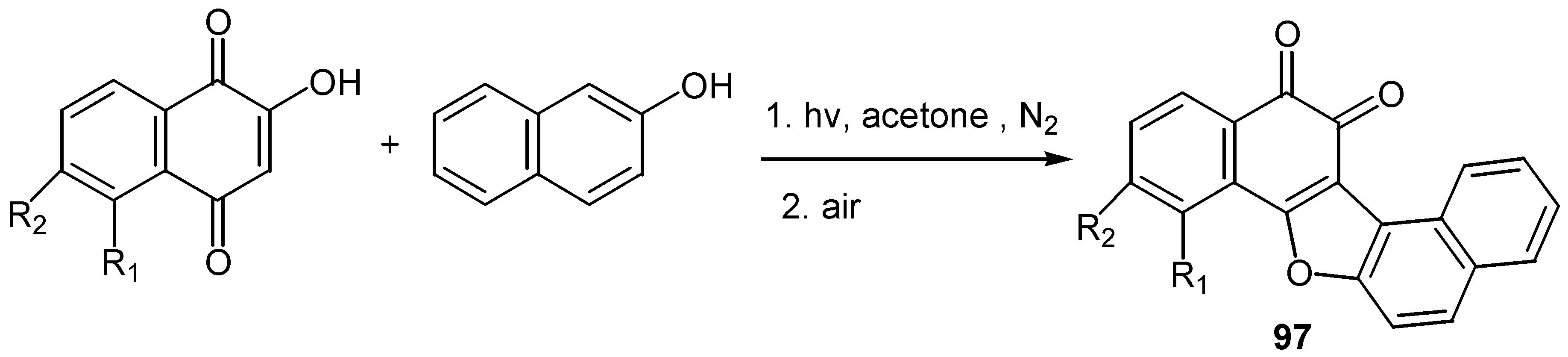
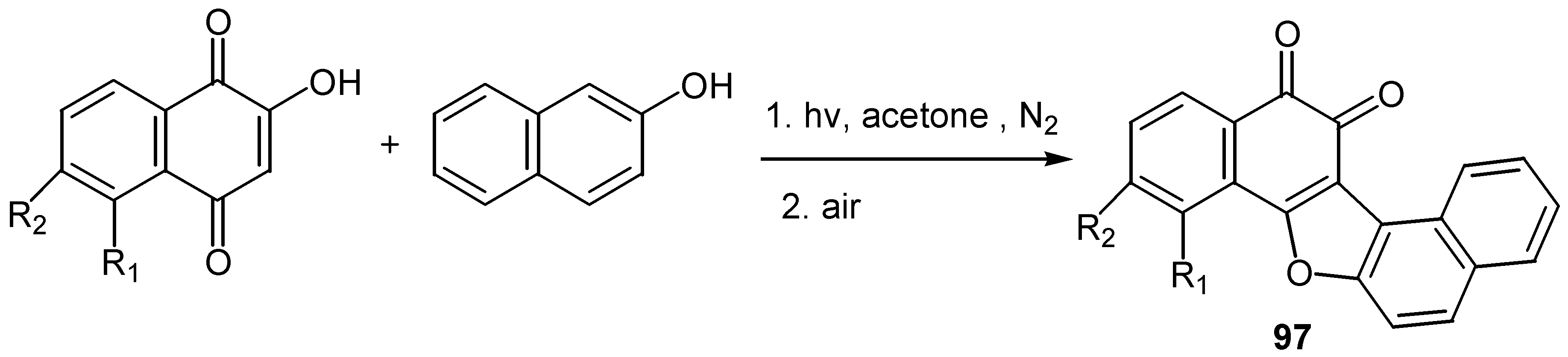
The reaction proceeds through radical formation, the proposed reaction pathway explains the regioselectivity of the cyclization and yields are from fair to very good. A radical mechanism also explains the products from the reaction of lawsone and its derivatives with â-naphthol: the cyclization takes place with carbonyl-4 and ortho-quinone furodinaphthoderivatives, 97, are isolated [56]. (Scheme 45)
Since in many cases the first step of the reaction involves radical formation, one-electron oxidant reagents were used for the cyclization of hydroxyquinones to furan derivatives. The reaction of lawsone with á-alkyl malonates in the presence of manganese(III)acetate gave poor yields of the corresponding furan derivatives, 98 (Scheme 46). The use of lead tetraacetate as cooxidant improved yields but the method is of little synthetic value [57].
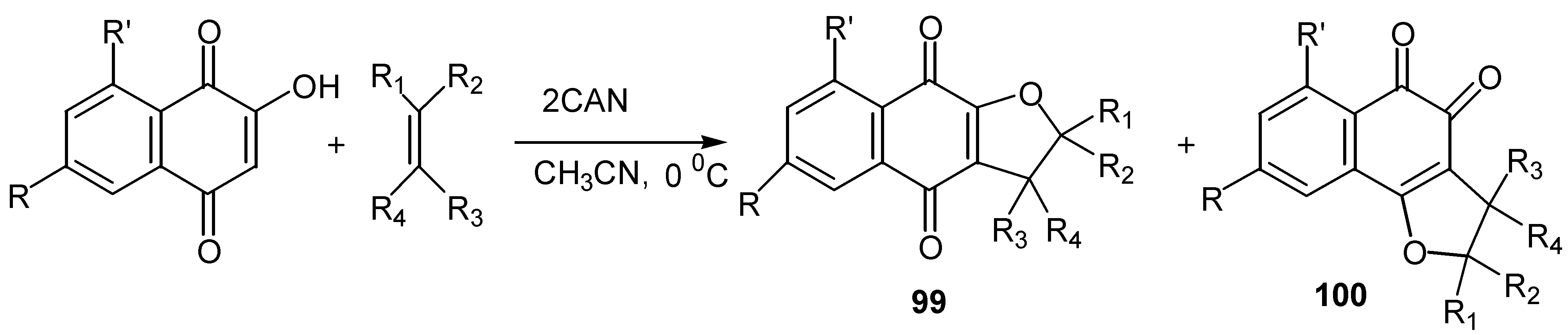
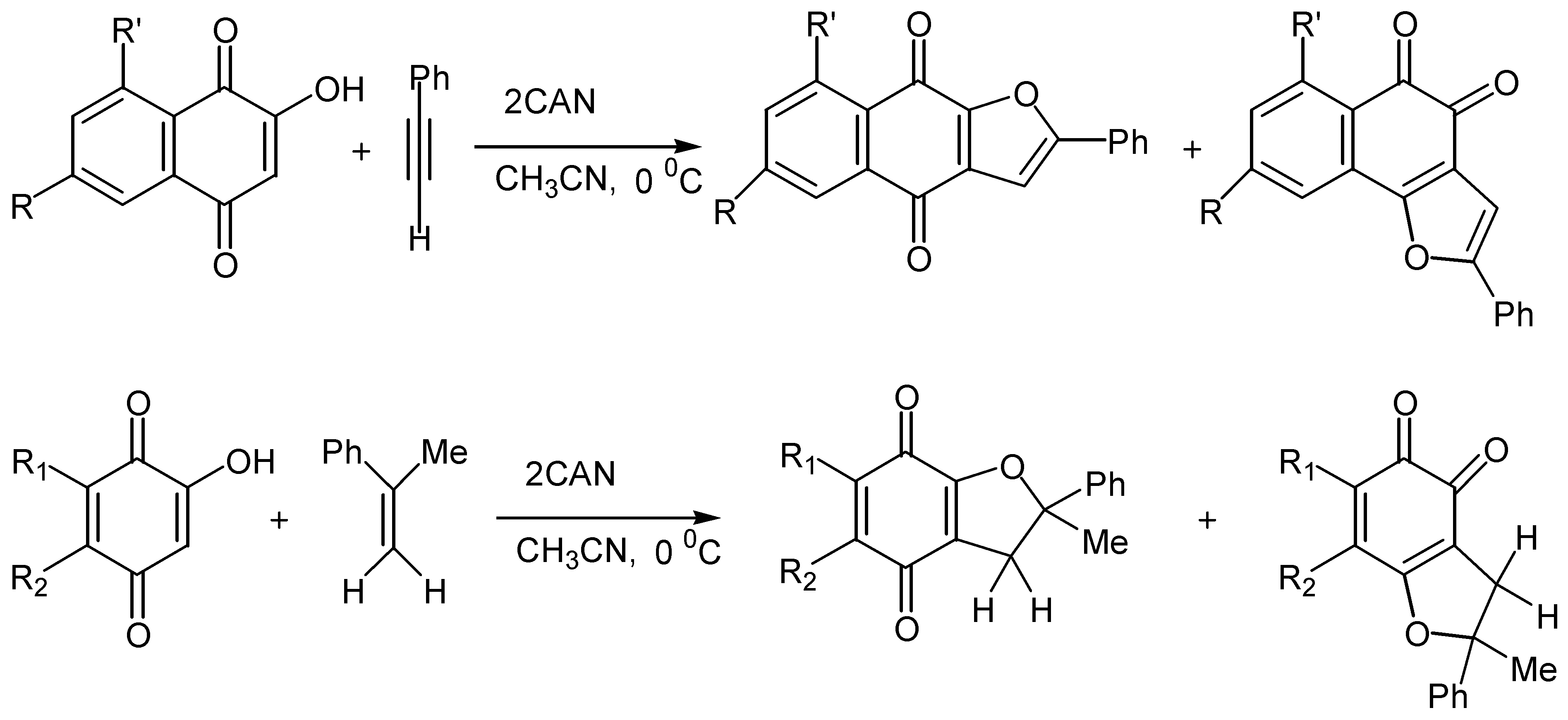
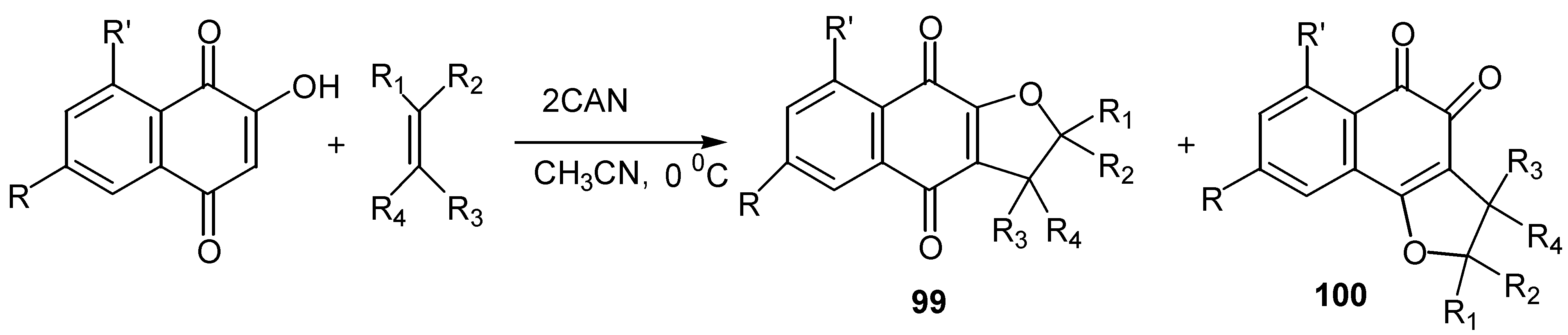
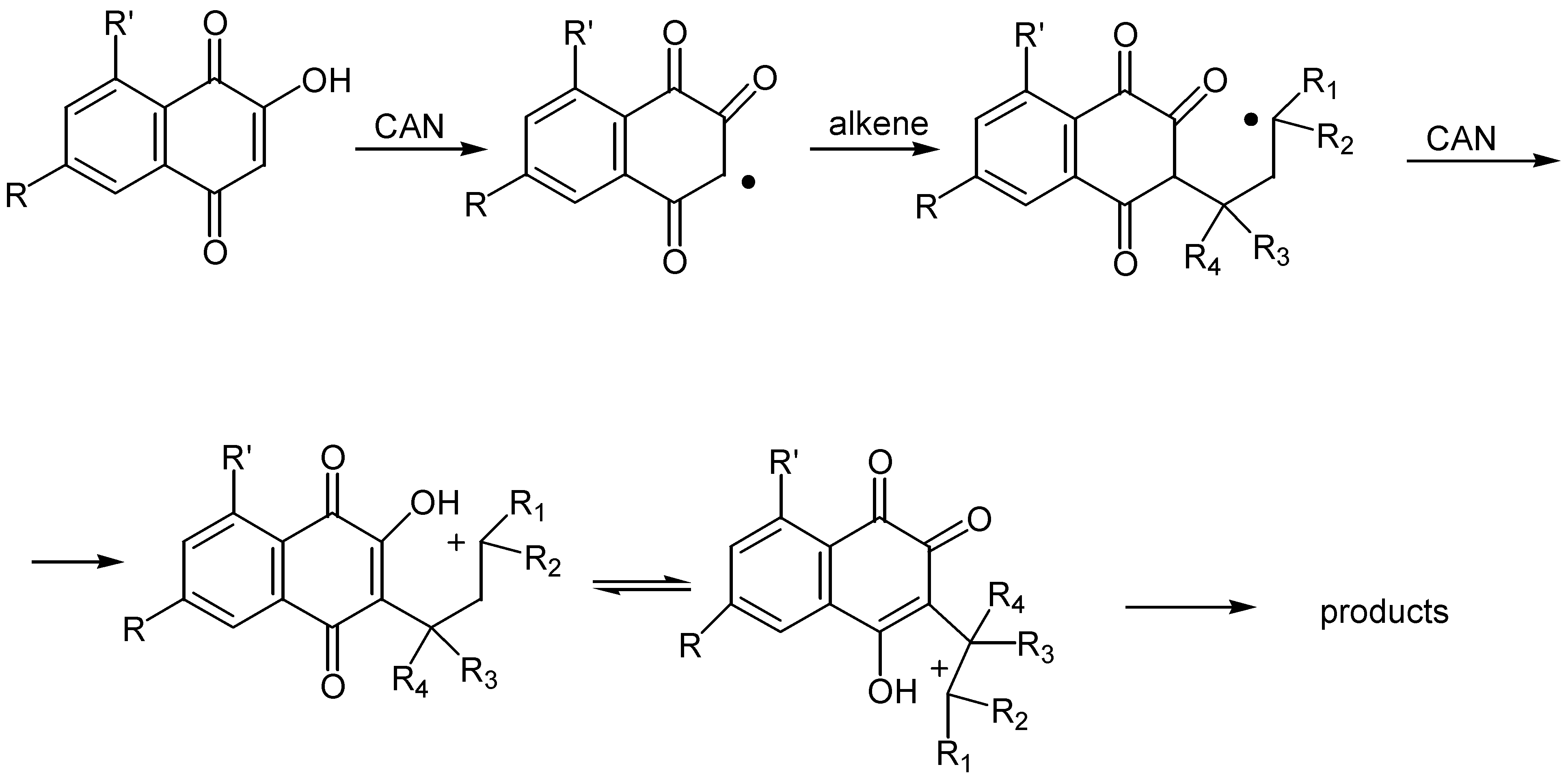
On the contrary, cerium(IV) ammonium nitrate, CAN, mediated cycloaddition of a variety of alkenes and alkynes with hydroxy benzo and naphthoquinones offered some quite remarkable results [56]. Thus, lawsone and its derivatives reacted with alkenes in the presence of two equivalents of CAN to afford mixtures of ortho and para-quinone furo derivatives, 99 and 100. (Scheme 47).
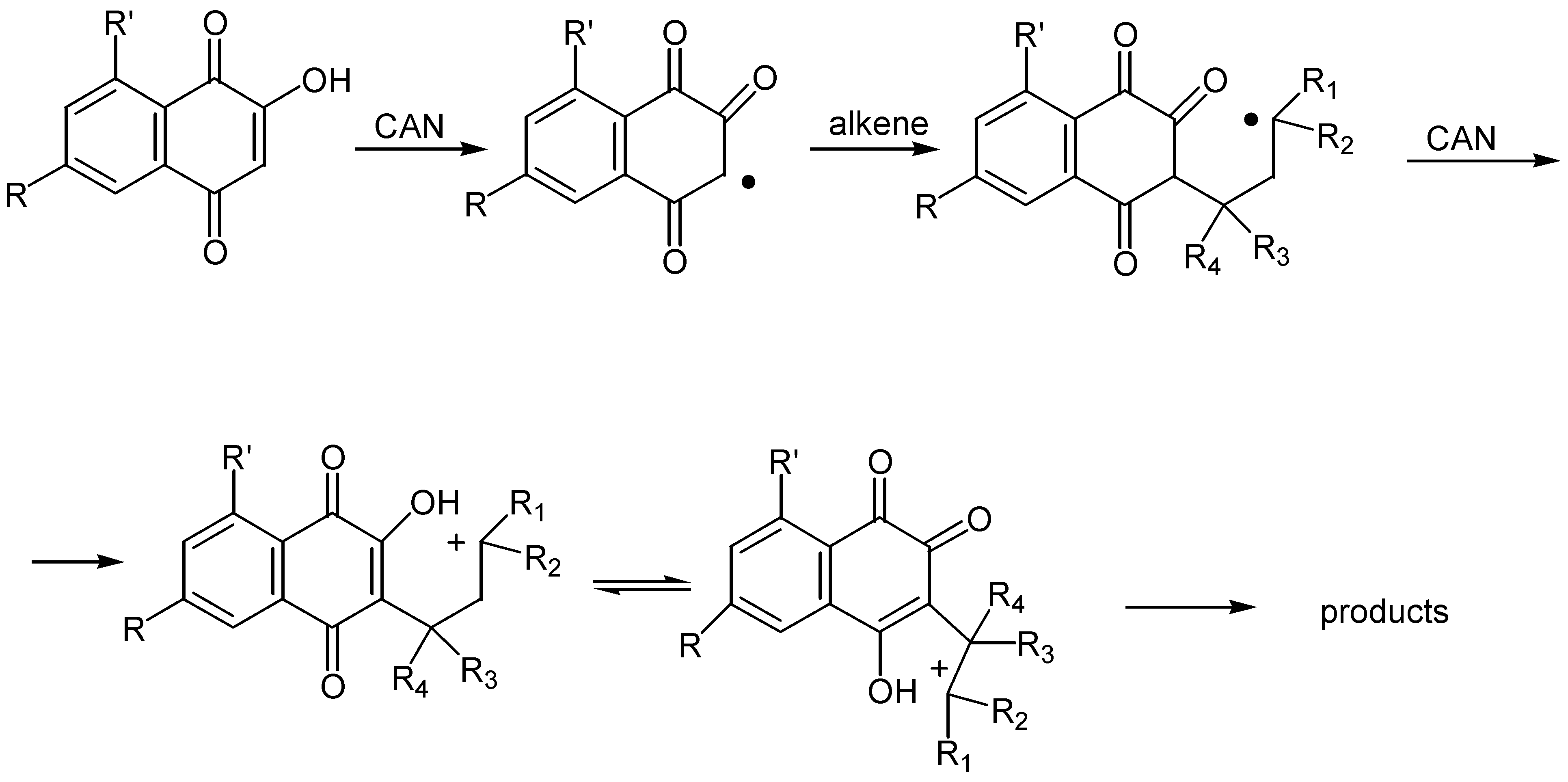
Yields are satisfactory and the para-quinone isomer usually predominates, while acetonitrile was found the best solvent for the reaction. The regioselectivity of the cyclization, as regards the substituents on the alkene, is high and easily explainable by the proposed reaction pathway: initial formation of reactive radicals, followed by oxidation to tautomeric carbenium ion intermediates. The latter are trapped intermolecularly by the hydroxy groups to afford the ortho and para-quinone isomers. (Scheme 48)
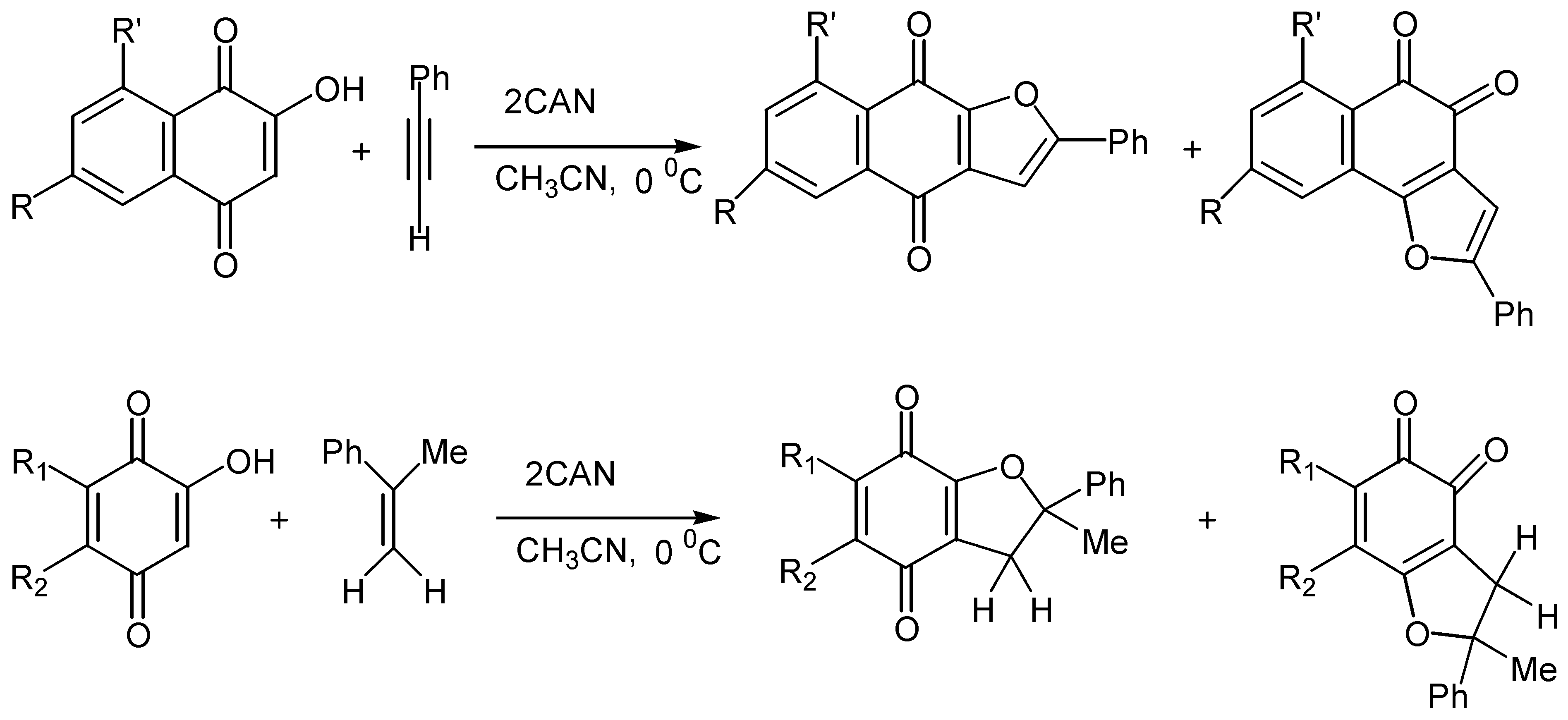
The reaction is also effective with phenyl acetylene, whereas other alkynes gave intractable mixtures. Finally, cyclisation proceeds satisfactorily with substituted hydroxybenzoquinones. (Scheme 49)
All the above methods of cyclisation involve the initial formation of free radicals. Kobayashi and co-workers reported [59] that the reaction of hydroxy naphthoquinones with enamines, 101, which starts with the nucleophilic attack of quinone to enamine, affords furo para-quinones, 102, in satisfactory yields. (Scheme 50) In this case no formation of the corresponding ortho-quinone isomers was observed.
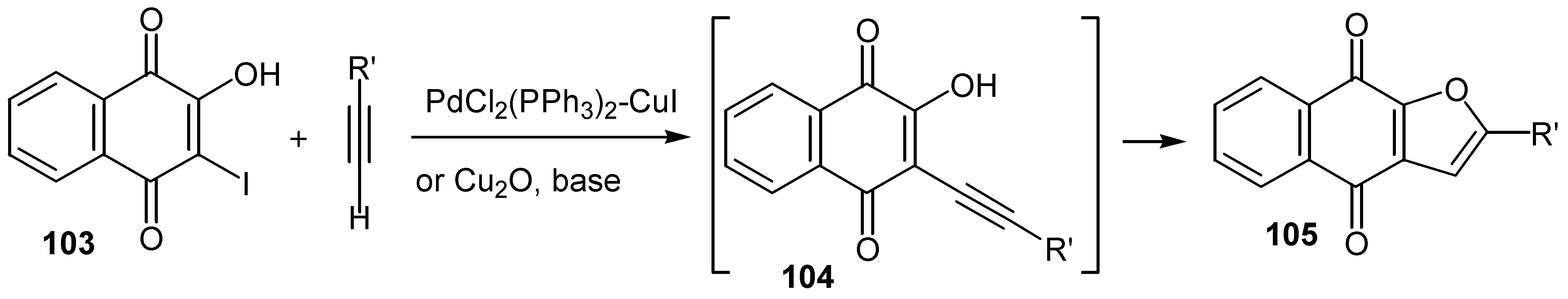
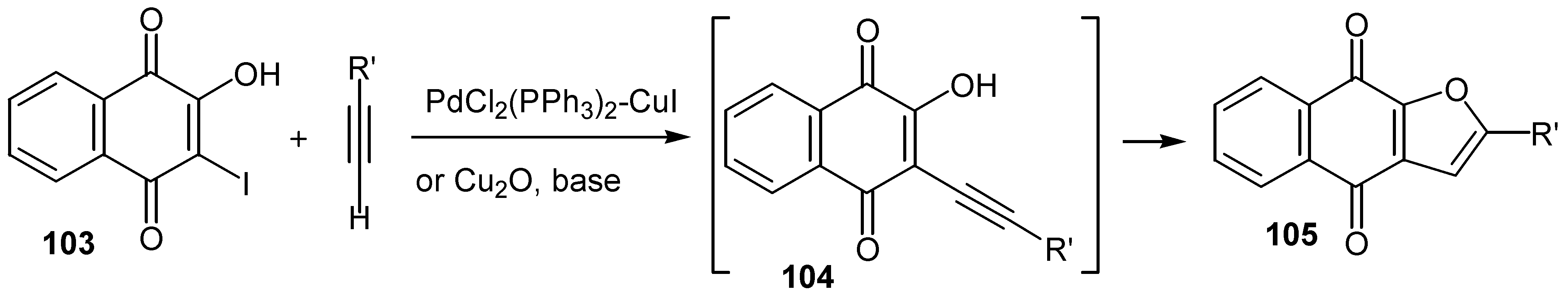
The same group reported another one pot synthesis of naphtho[2,3-b]furan-4,9-diones by sequential coupling/ring closure reactions [60]. Sonagashira coupling of the proper terminal alkyne with iodo hydroxy naphthoquinone, 103, leads to the corresponding non-isolable alkyne derivative, 104, which is cyclized to the desired furo quinone, 105, in moderate yield. (Scheme 51)
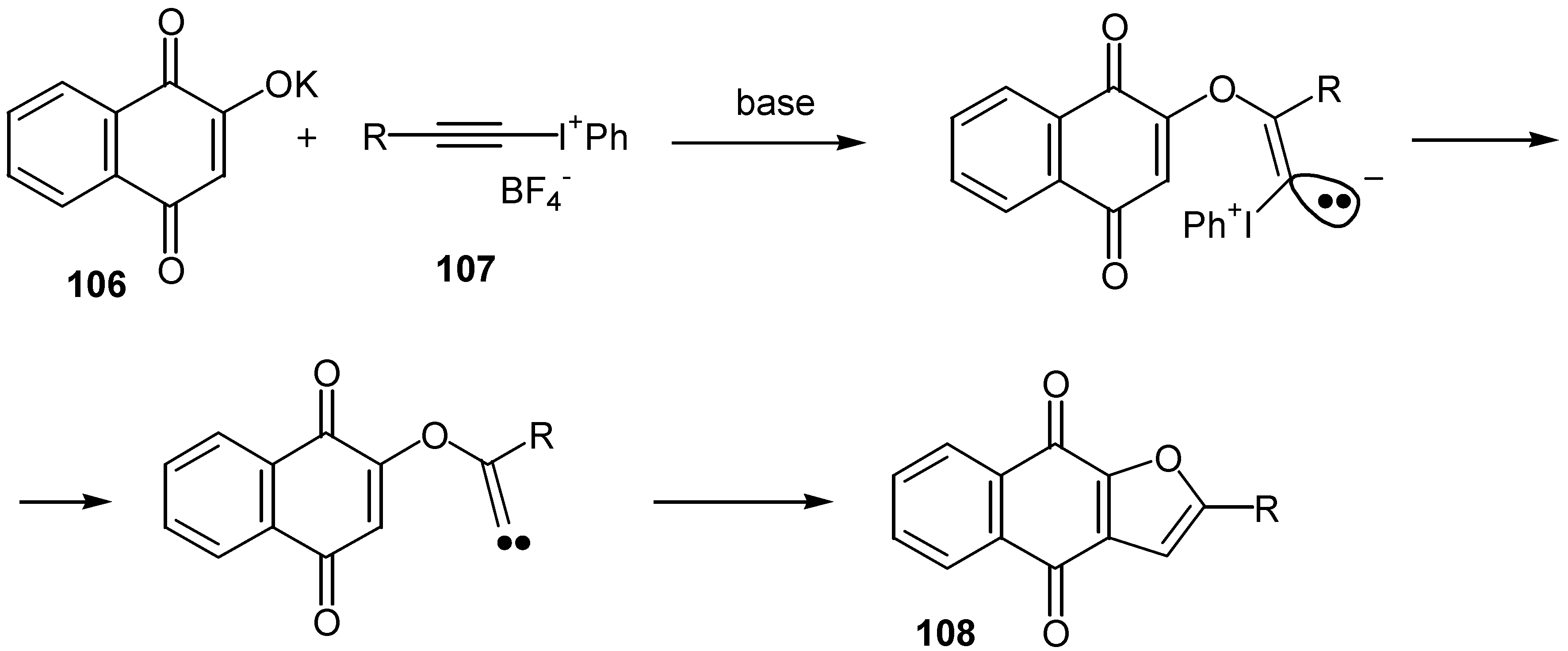
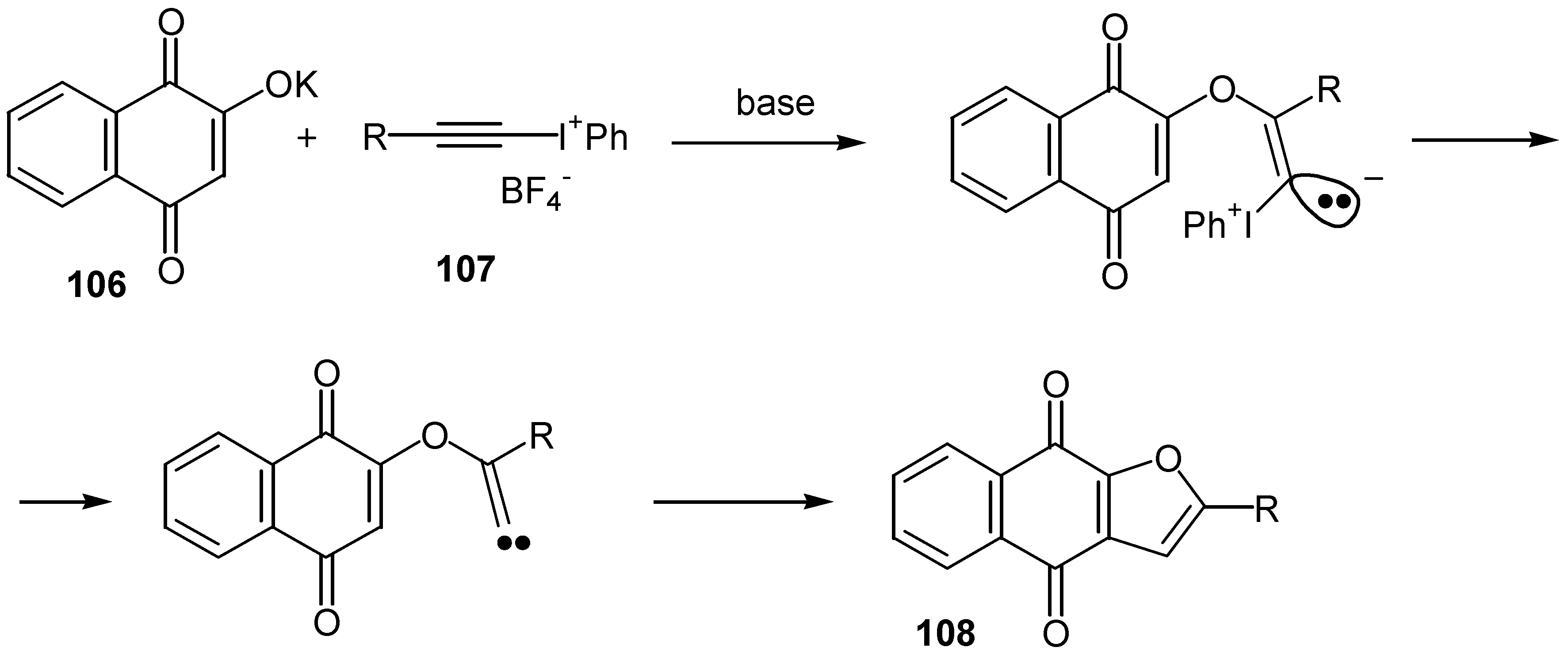
A very interesting approach to the same furo quinones was suggested by Ochiai [61]. Exposure of the potassium salt of 2-hydroxy-1,4-naphthoquinone, 106, to alkynyliodonium salts, 107, undergoes the tandem Michael-carbene insertion to afford the cyclization products, 108, again in moderate yields. (Scheme 52)
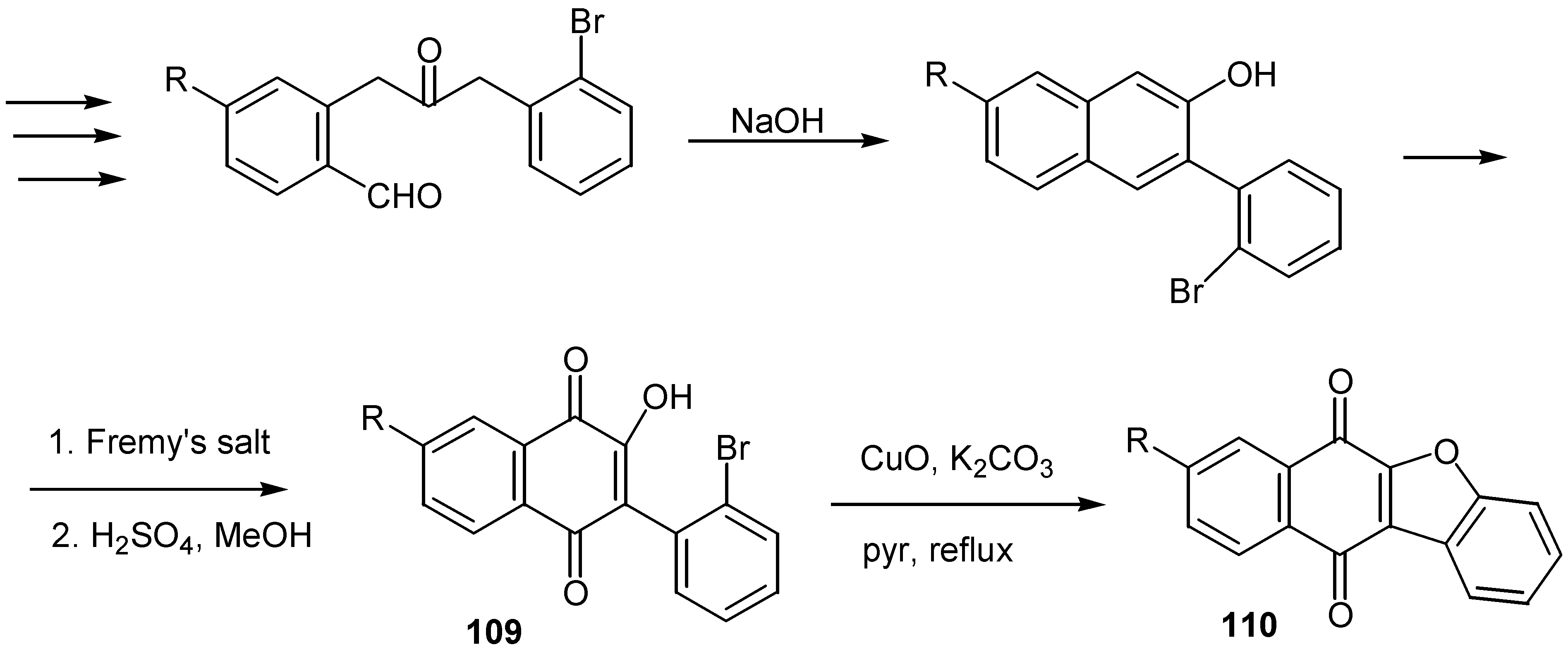
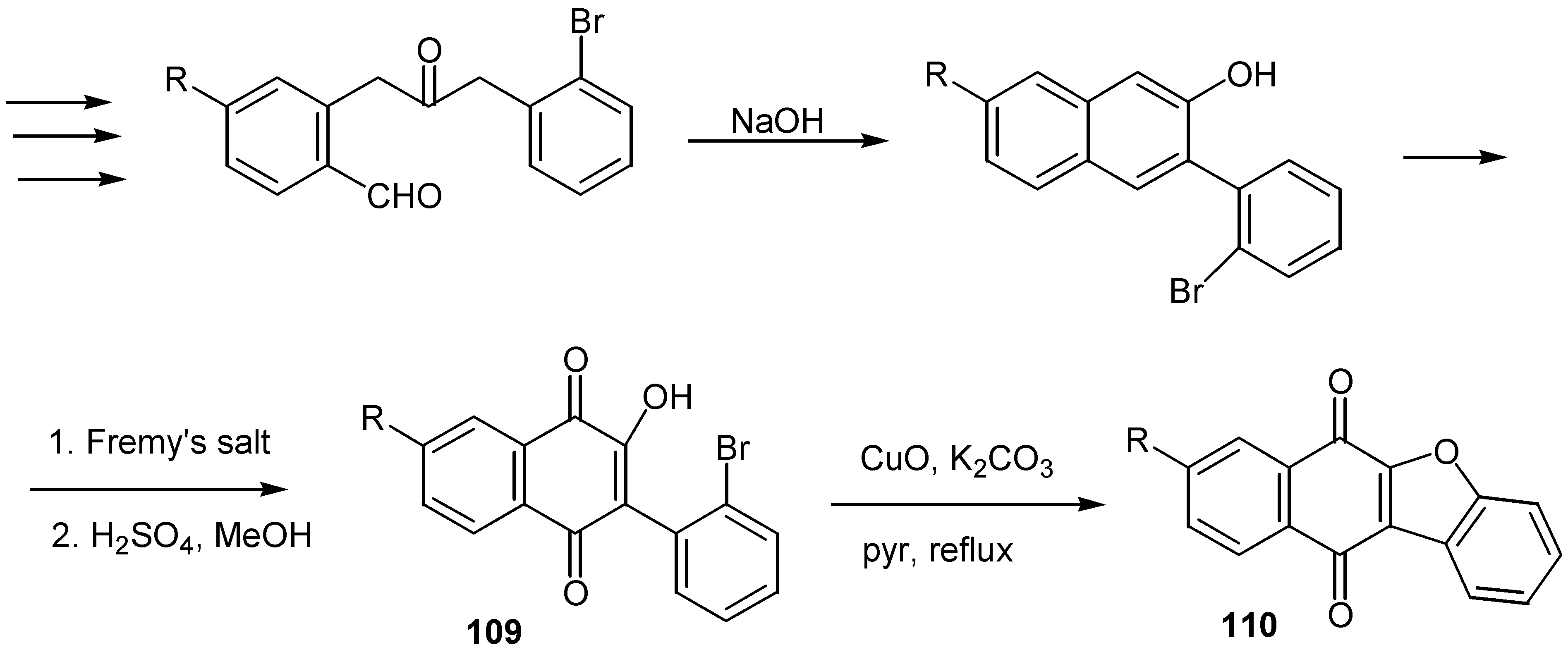
Finally, a general synthesis of benzo[b]naphtho[2,3-d]furan-6,11-diones, 110, was reported recently [62a,b]. The final step is the internal coupling of an aryl bromide with the hydroxy group of a naphthoquinone under standard Ullmann coupling conditions. The 3-(2'-bromophenyl)-2-hydroxy-1,4-naphthoquinone, 109, is prepared in six steps from commercially available materials. Overall yields are in the range of 30-40%. (Scheme 53)
III. Other cyclizations
Cyclization of hydroxyquinones to the corresponding six-member pyran derivatives is also of importance, since a lot of the latter are natural products [63]. The usual methodology is the oxidative cyclization of the proper 3-alkenyl derivative of 2-hydroxy-1,4-quinone, leading to ortho or para-quinone pyran derivative [4]. Some recent examples are illustrated below.
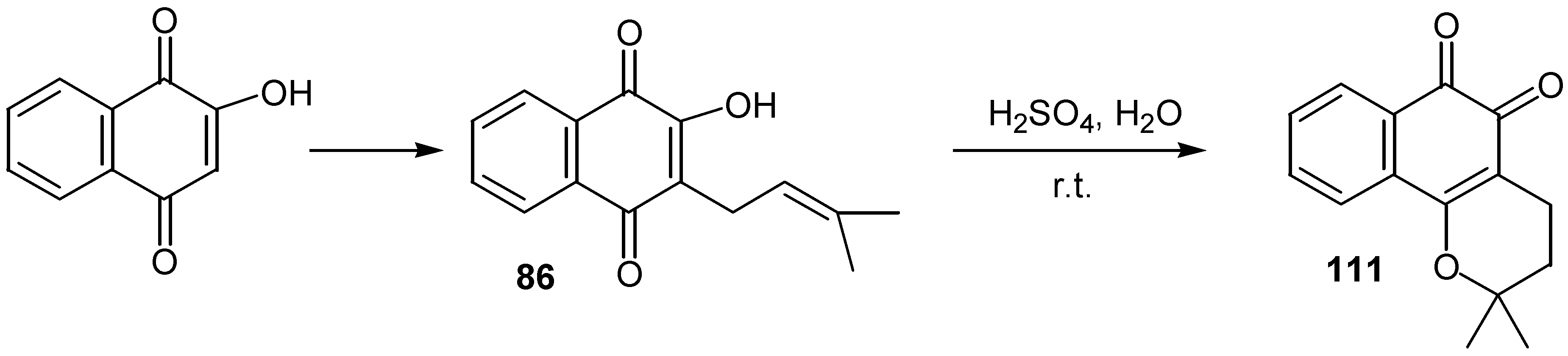
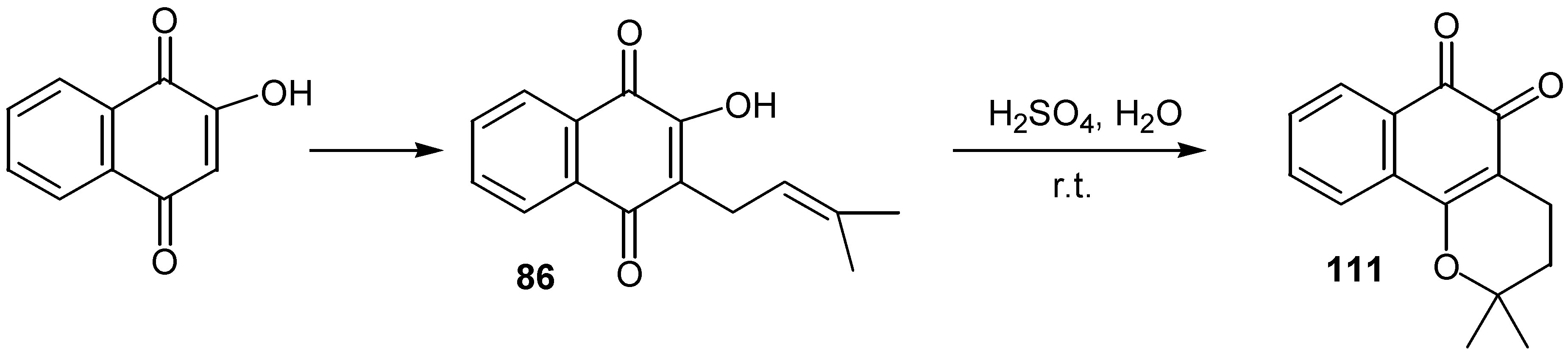
Lapachole, 86, prepared in a large scale by alkylation of lawsone, as it was mentioned earlier (Scheme 38), was converted to â-lapachone, 111, by sulfuric acid at room temperature [49]. (Scheme 54)
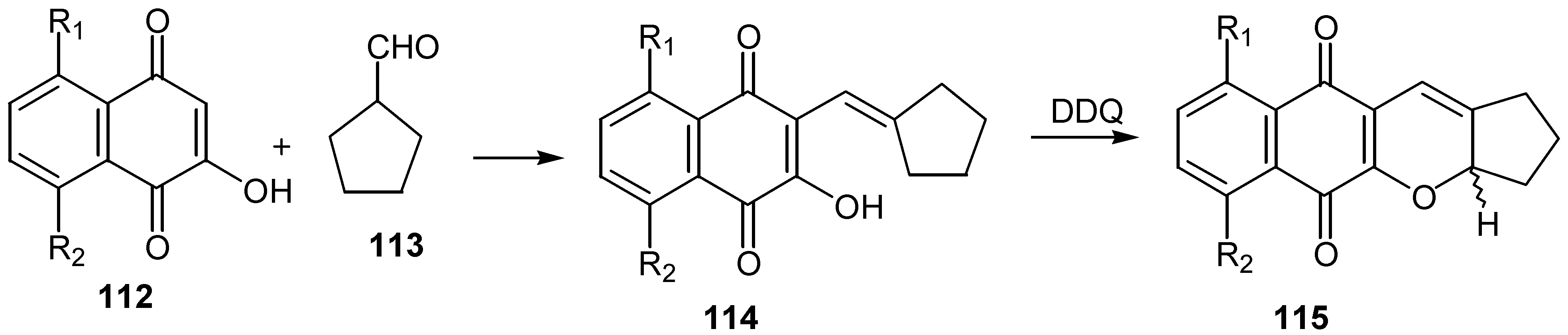
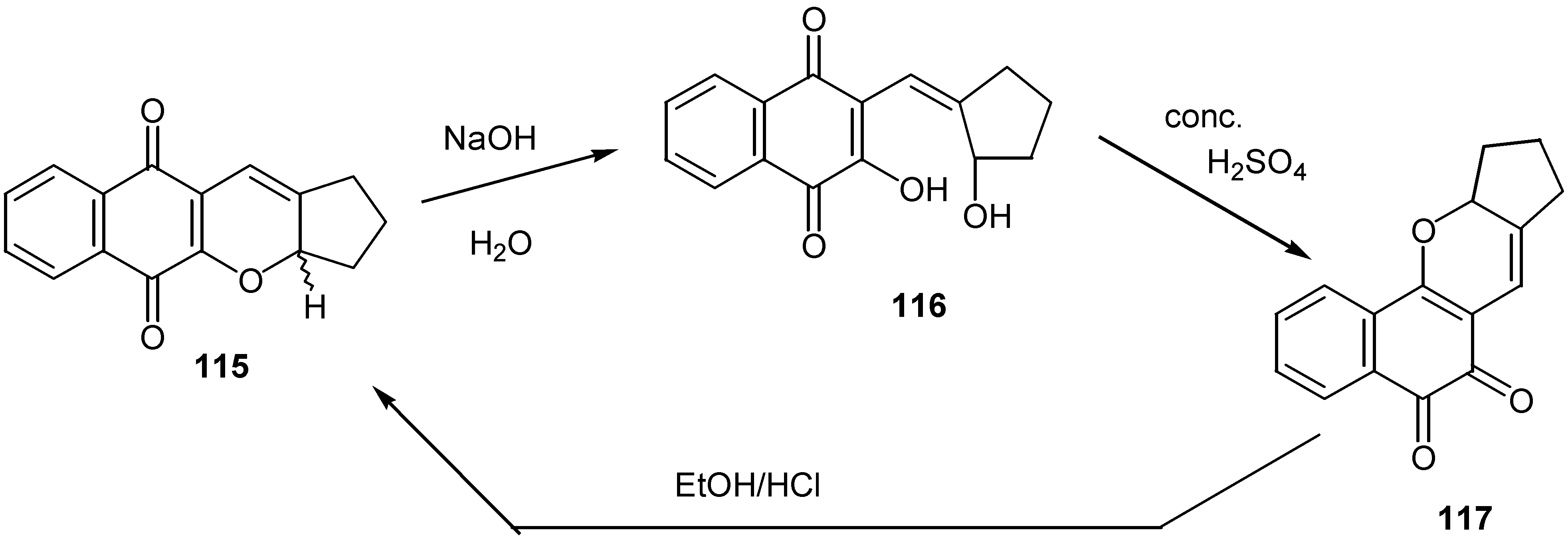
In an analogous reaction condensation of lawsone and derivatives, 112, with cyclopentancarboxaldehyde, 113, afforded the corresponding alkenyl compounds, 114, and the latter were cyclised to the para-quinone pyran derivatives, 115, [64], using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as oxidative agent. (Scheme 55)
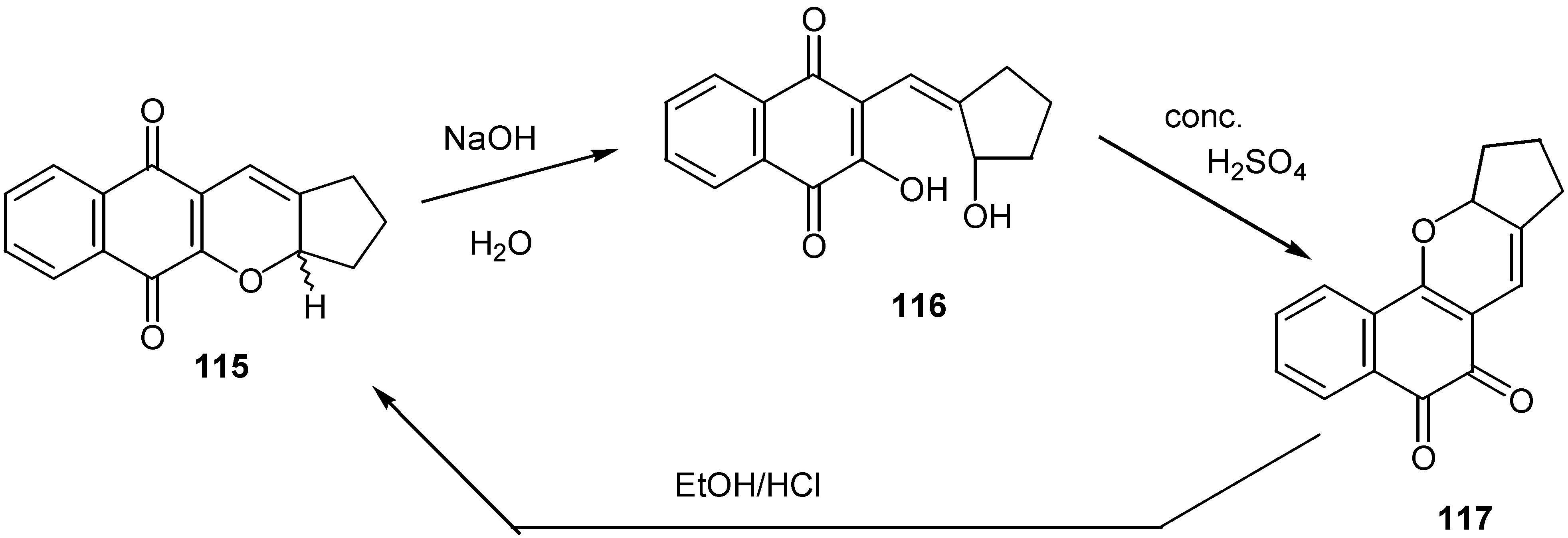
Interestingly enough, the para-quinone derivative, 115, is converted to the corresponding orthoquinone isomer, 117, through opening to the hydroxyquinone derivative, 116, as it is shown in Scheme 56. The ortho-quinone isomer again is converted to the para-quinone isomer, by the action of ethanolic HCl.
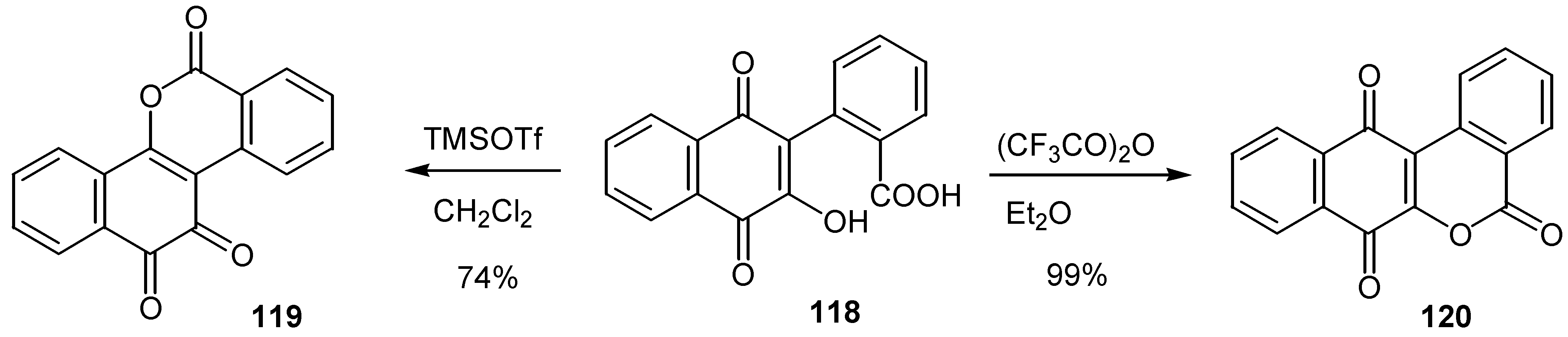
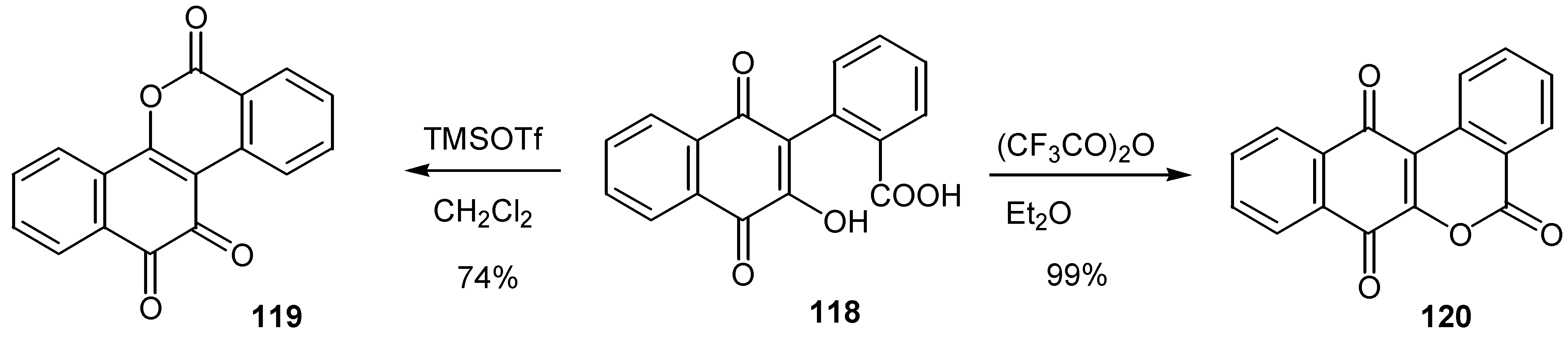
An acid promoted quinolactonization of hydroxynaphthoquinones, has been developed, providing direct access to compounds with potential antitumoral activity [65]. Again selection of the acid reagent leads exclusively the desired ortho or para-quinone isomer, 119 or 120, as it is shown in Scheme 57 for a model compound.
b) Ylide formation
Hydroxy quinones form stable zwiterionic compounds of the general type:

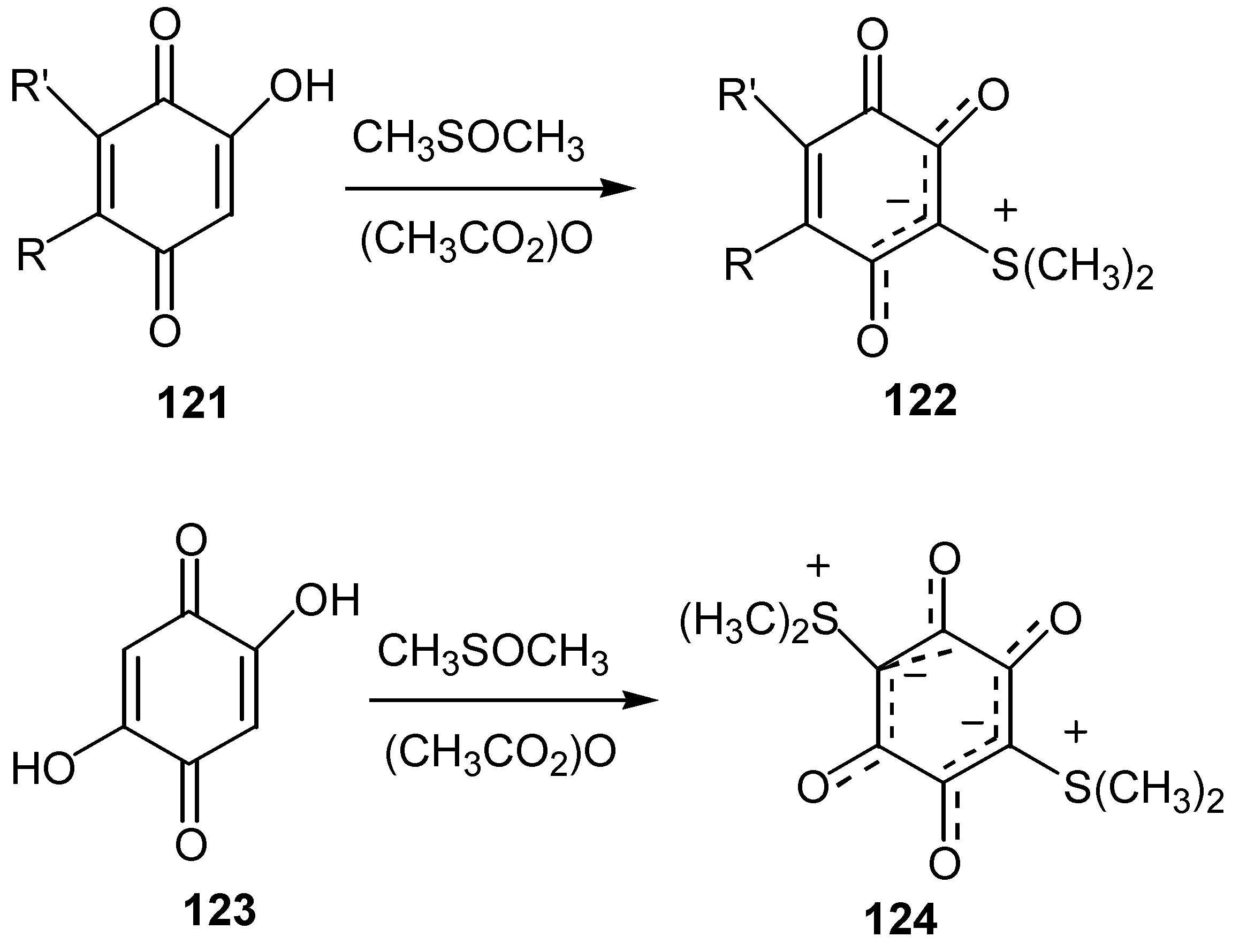
These compounds can be described as hybrids of 1,4 or 1,2-dipoles or as ylides, whereas Z is a moiety of the elements P, S, N or I. All these ylides exhibit significant stability, and hence low reactivity, with the exception of iodonium ylides: The easy rupture of the C-I bond leads to interesting derivatives. Sulfonium ylides, 122, 124, can easily be prepared from the reaction of hydroxy, 121, or dihydroxyquinones, 123, with dimethyl sulfoxide-acetic anhydride [66] (Scheme 58).
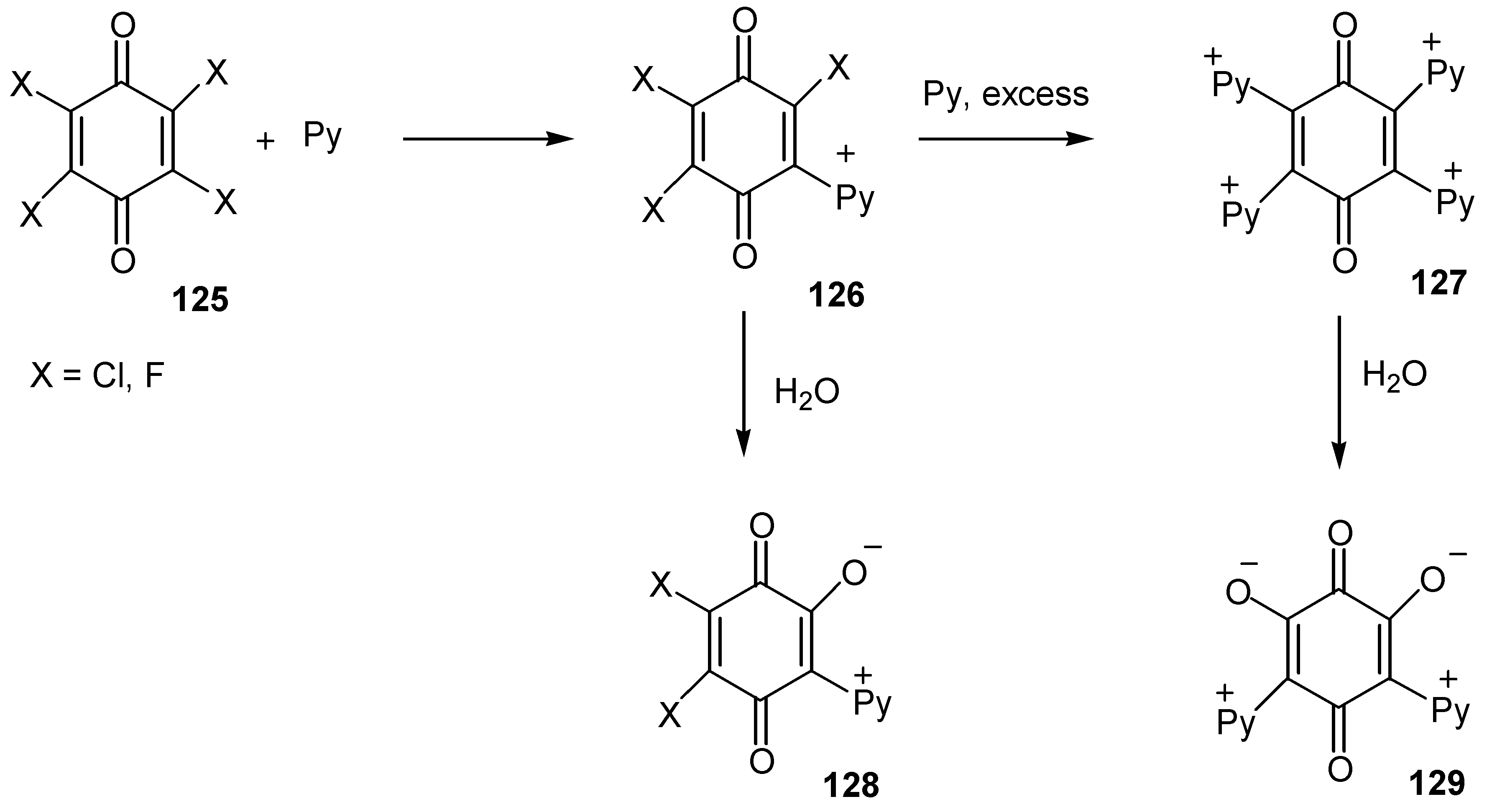
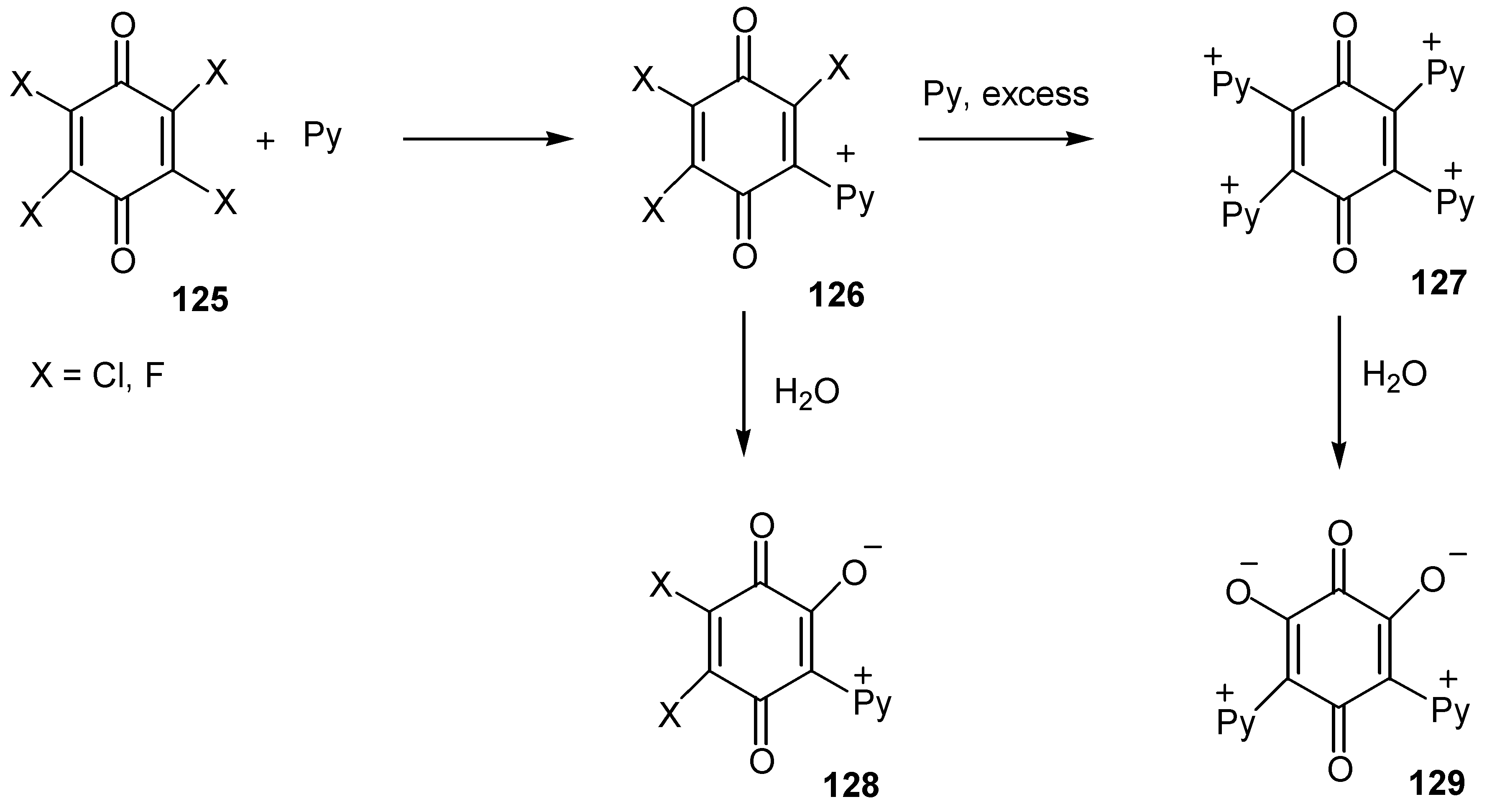
The corresponding pyridinium-oxy zwitterionic quinones, 128 and 129, were prepared either by the action of pyridine on polyhalogen quinones, 125, and hydrolysis of the resulting pyridinium salts, 126 and 127 [67], (Scheme 59).
or by oxidation of 1,4-naphthoquinones with iodine or hydrogen peroxide in the presence of substituted pyridines [68] (Scheme 60).
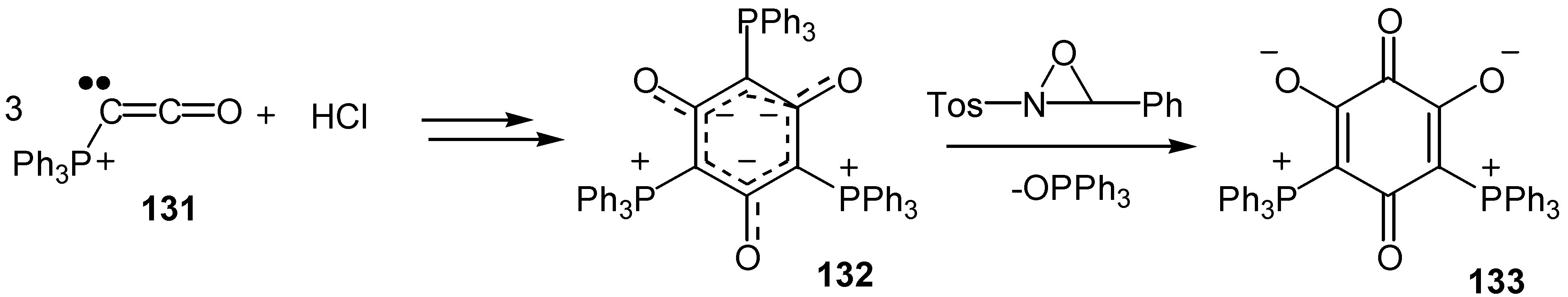
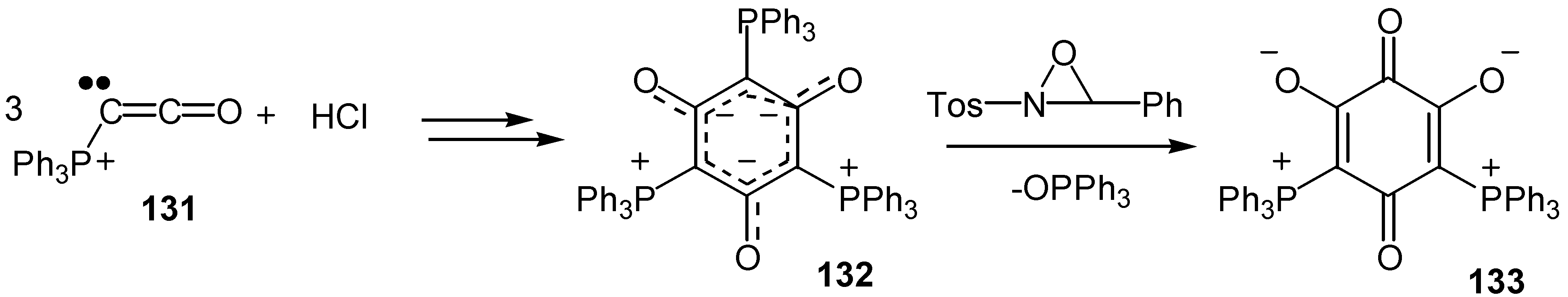
A bis-phosphonium-bis-oxy zwitterionic quinone, 133, was prepared by a lengthier route: Ketenylidene(triphenyl)phosphorane, 131, was dimerized and finally trimerized to form a symmetric tris-ylide, 132, and the latter was converted to the quinone derivative by elimination of one mole of triphenylphosphinoxide [69] (Scheme 61).
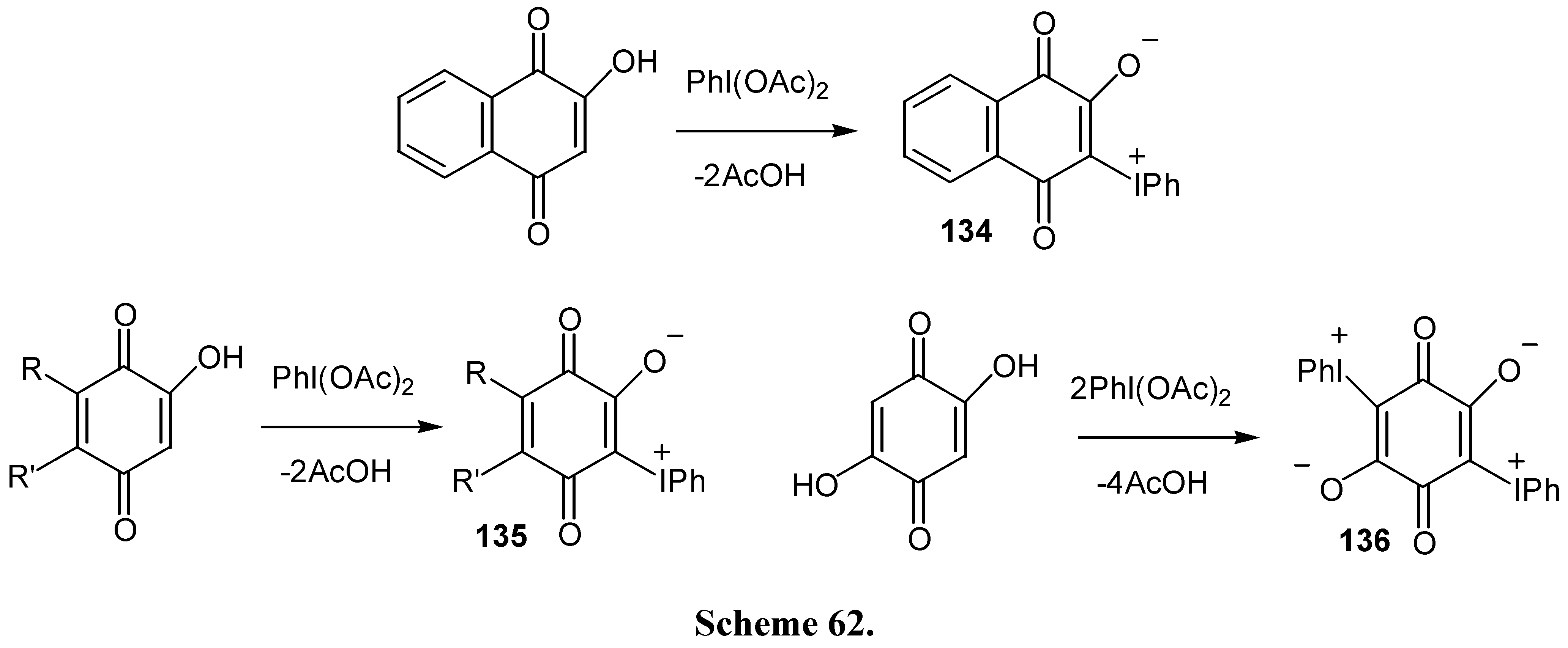
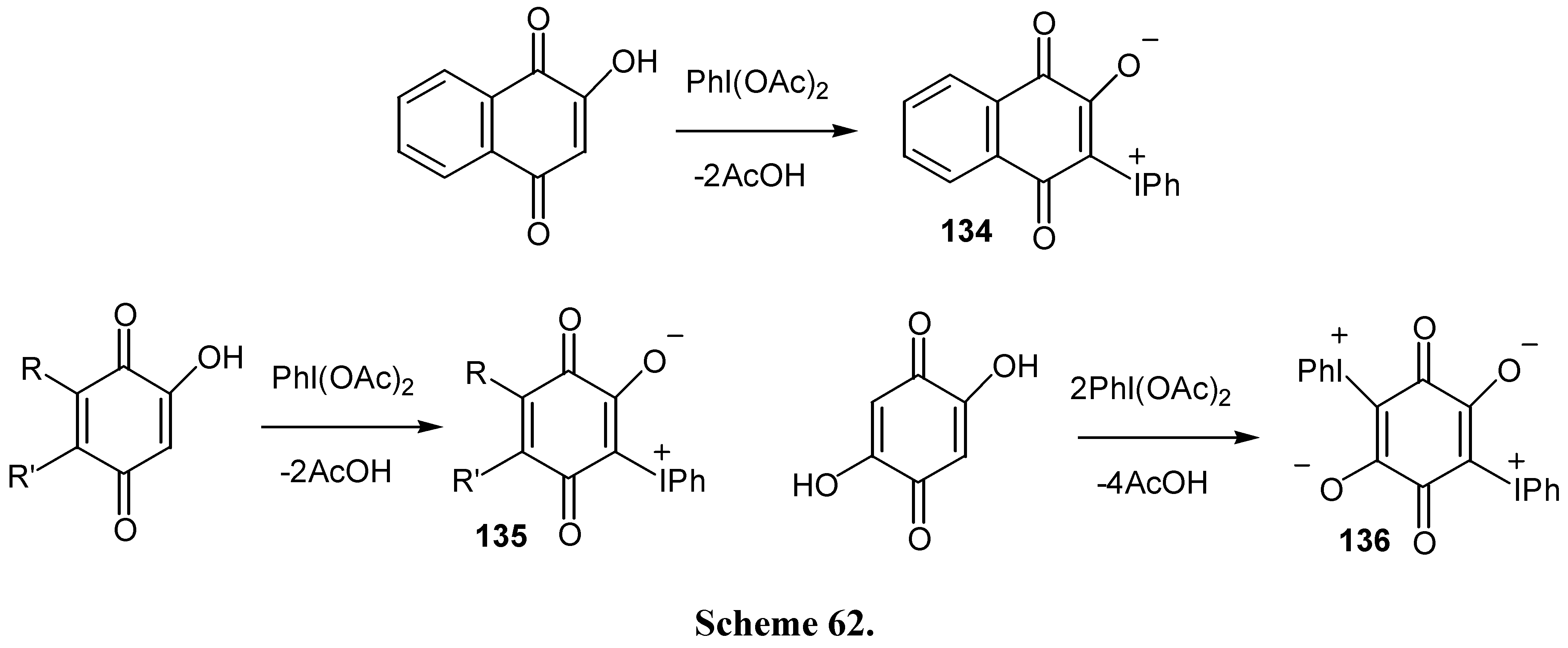
As it was already mentioned, iodonium ylides of hydroxy quinones offer interesting synthetic possibilities. These ylides are prepared in good yields from the reaction of hydroxybenzo- or naphthoquinones with (diacetoxyiodo)benzene [70,71] (Scheme 62) .
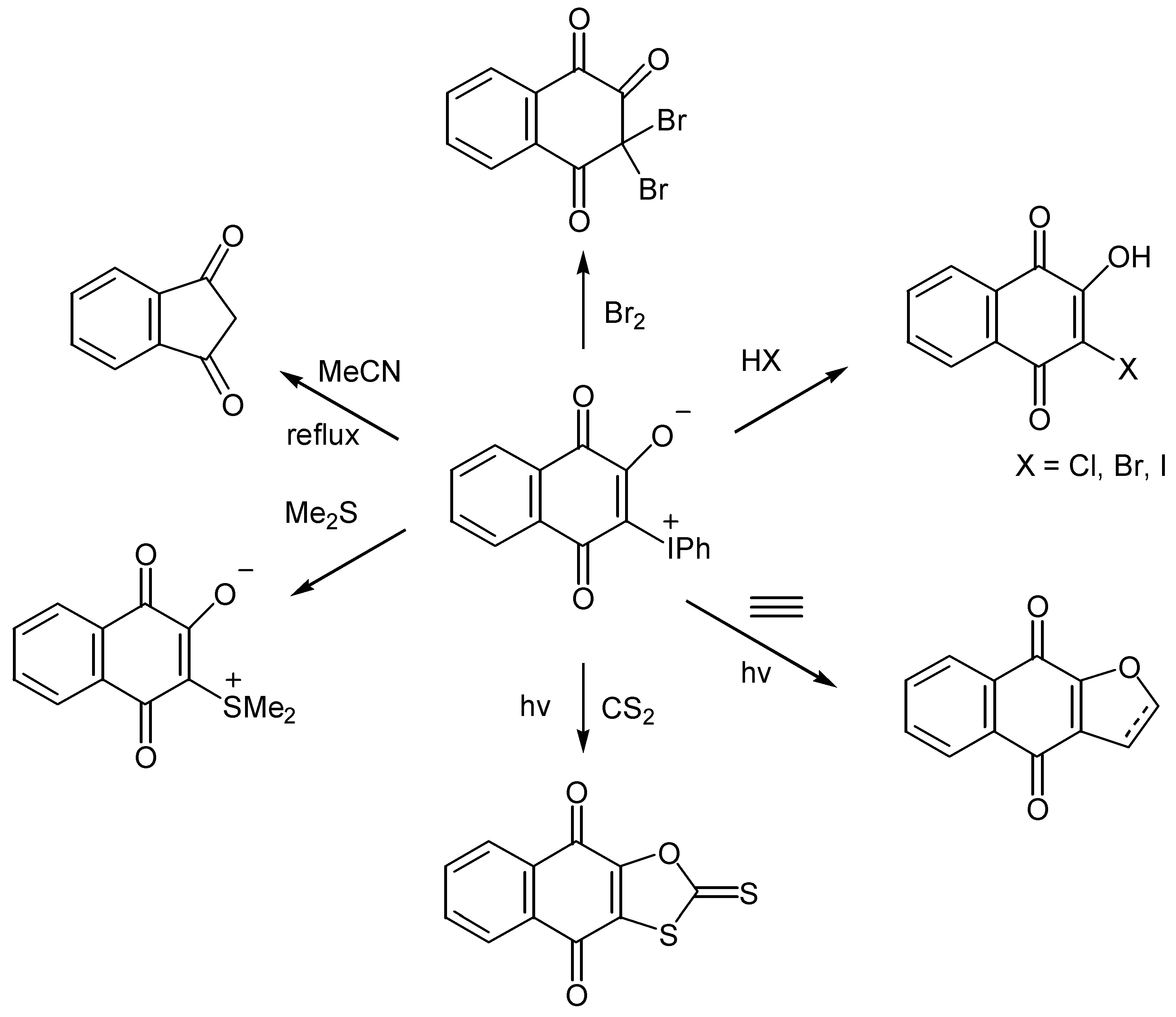
Iodonium ylides are labile compounds, react with both electrophiles and nucleophiles and afford cyclization products under photochemical conditions, as illustrated in Scheme 63 [70].
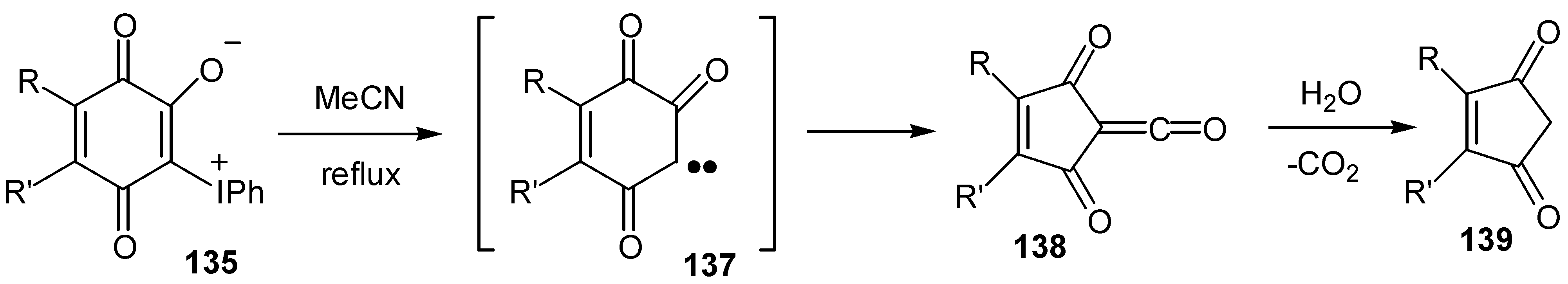
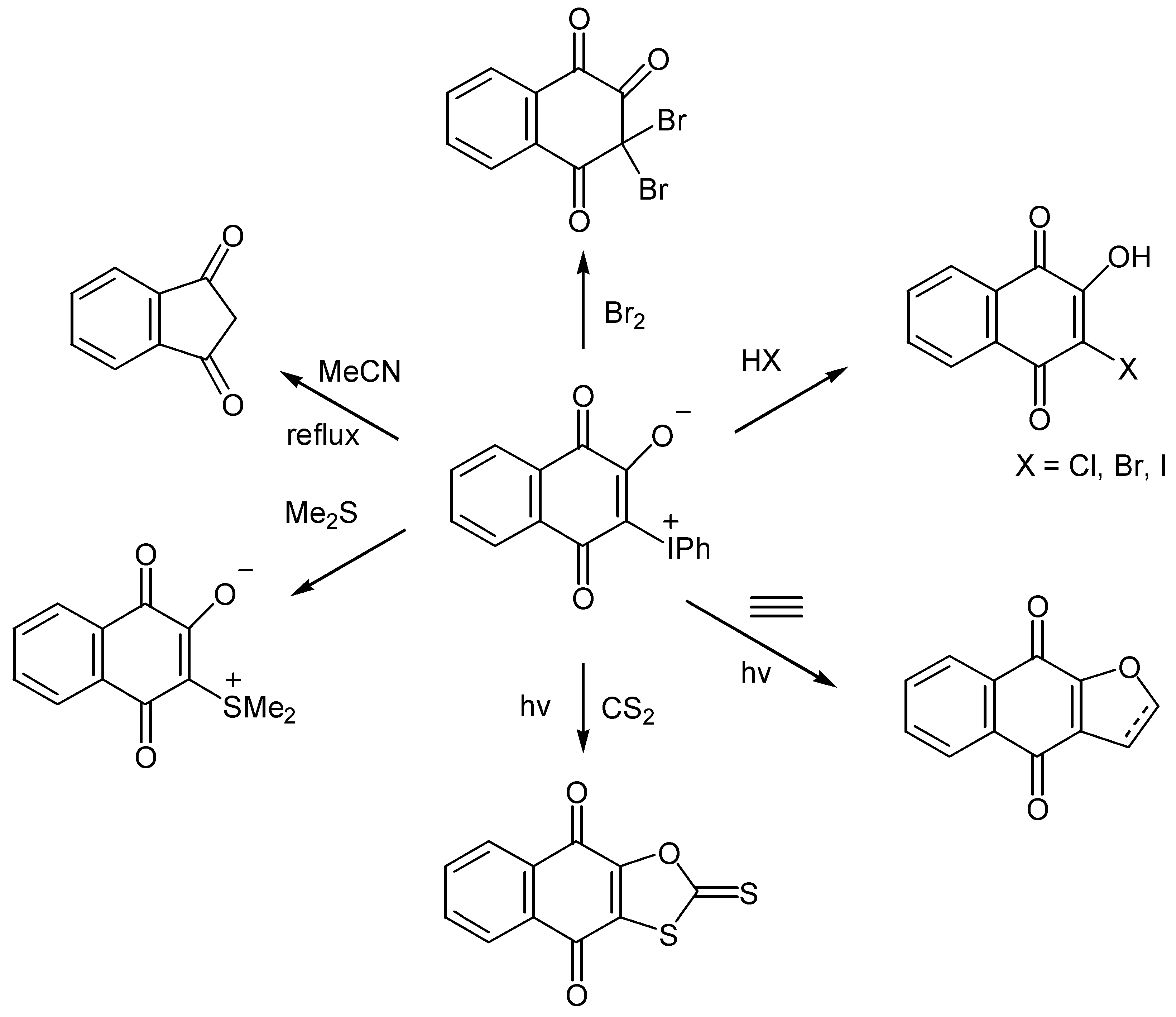
Especially noteworthy is the thermal ring contraction to indandione in high yield. This contraction takes place also with ylides of hydroxybenzoquinones, 135, proceeds through formation of carbenes, 137, and Wolf-rearrangement products, 138, and offers an easy one-pot synthesis of substituted cyclopentenediones, 139, [71](Scheme 64).
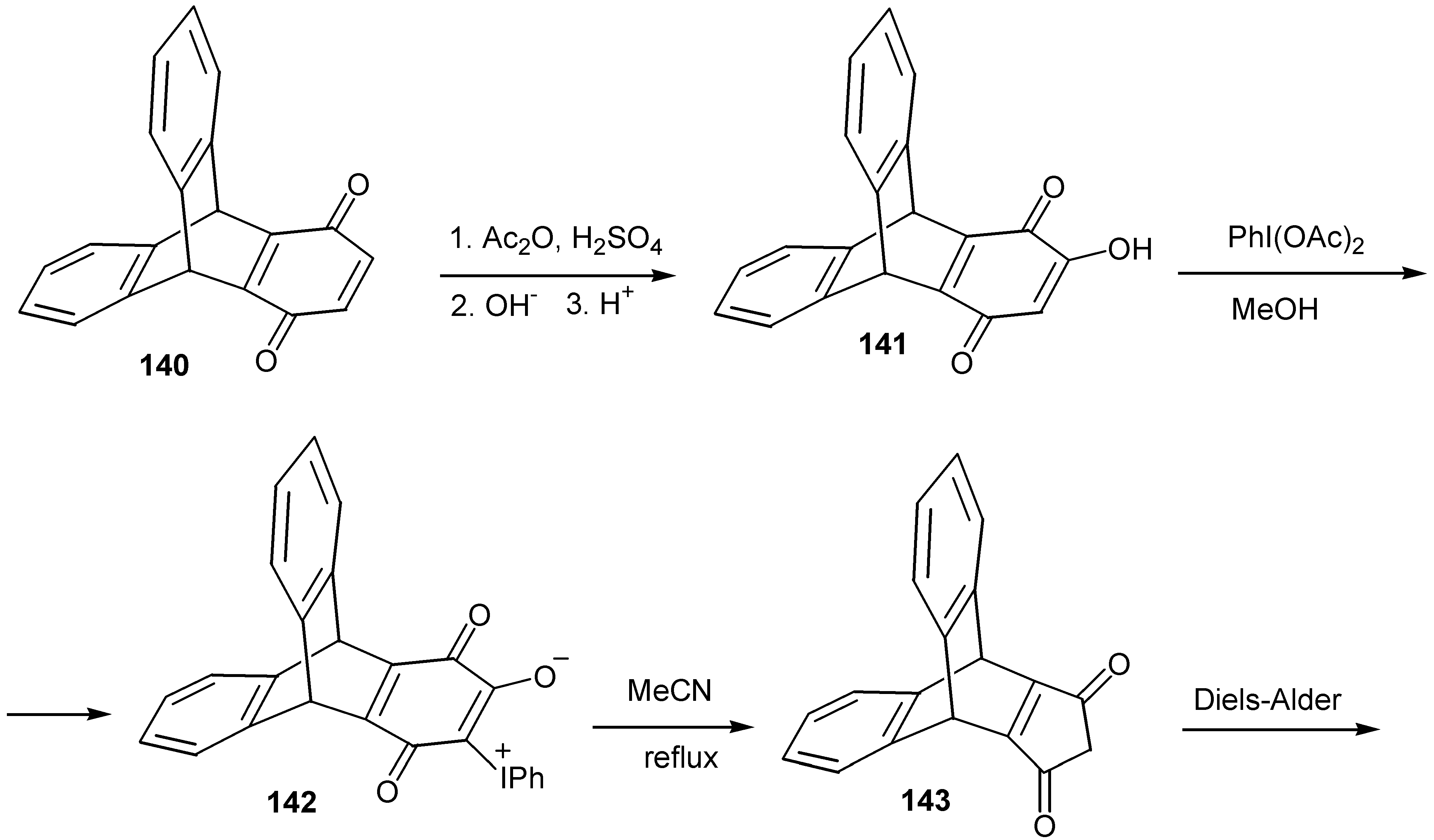
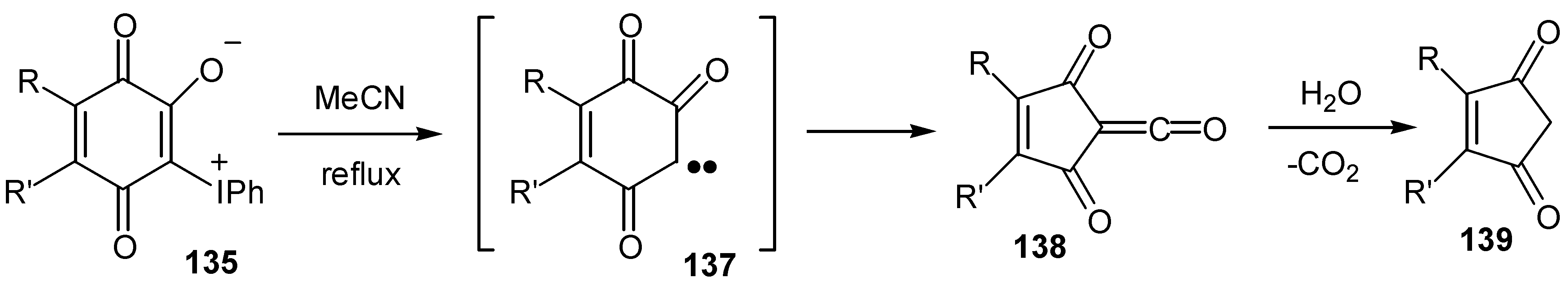
This reaction can find application for the preparation of more complicated cyclopentenediones, which are potential dienophiles. The case of hydroxytriptycenequinone, 141, which affords triptycenecylopentenedione, 143, and triptycene-tricyclic products through Diels-Alder reactions of the latter, is a characteristic example [72] (Scheme 65).
3-Phenyliodonium ylides of lawsone and derivatives, 144, give satisfactory yields of furoquinones, 145, through Pd- or Cu-catalyzed cyclization reaction with terminal acetylenes [60]. (Scheme 66) The same cyclization takes place, as it was already mentioned, with 3-iodo-lawsone and derivatives but yields are considerably lower.
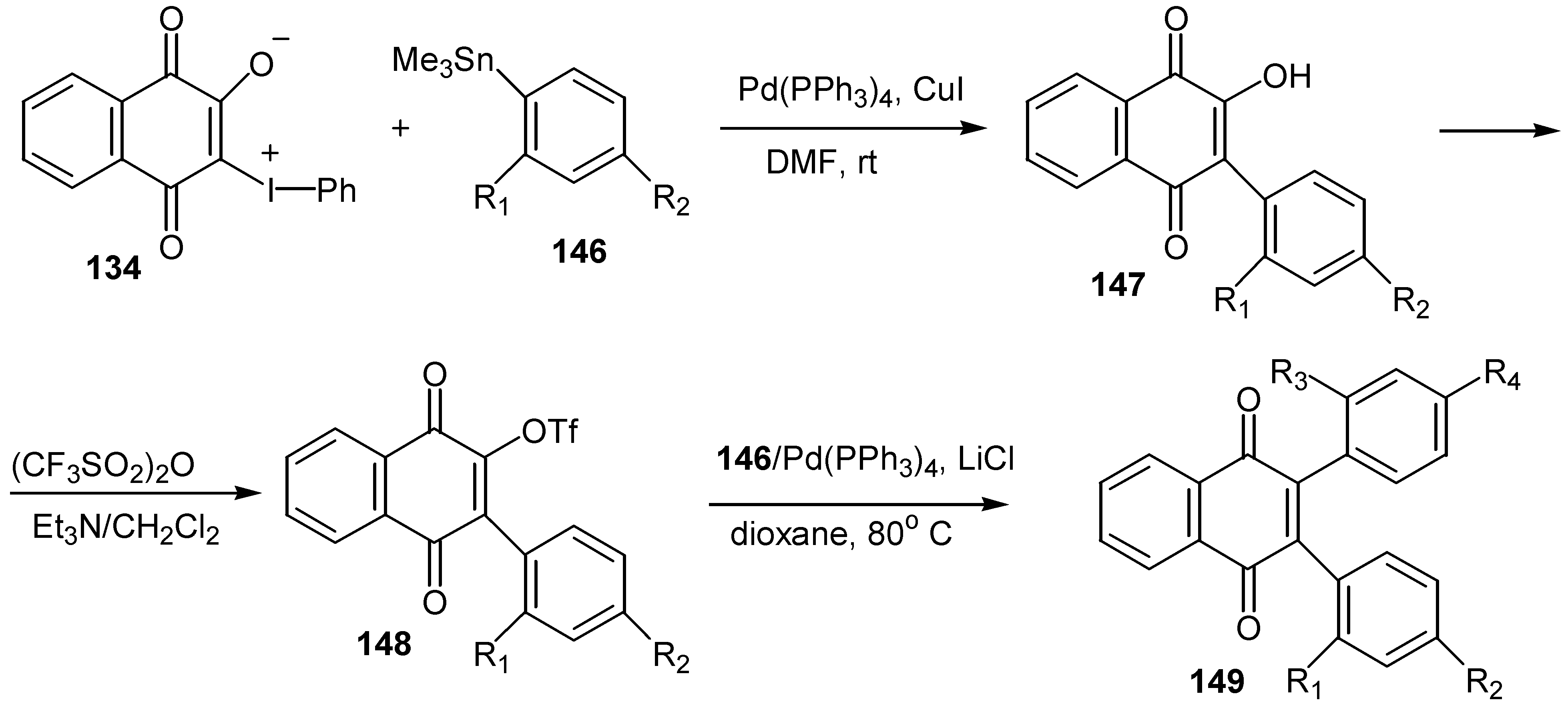
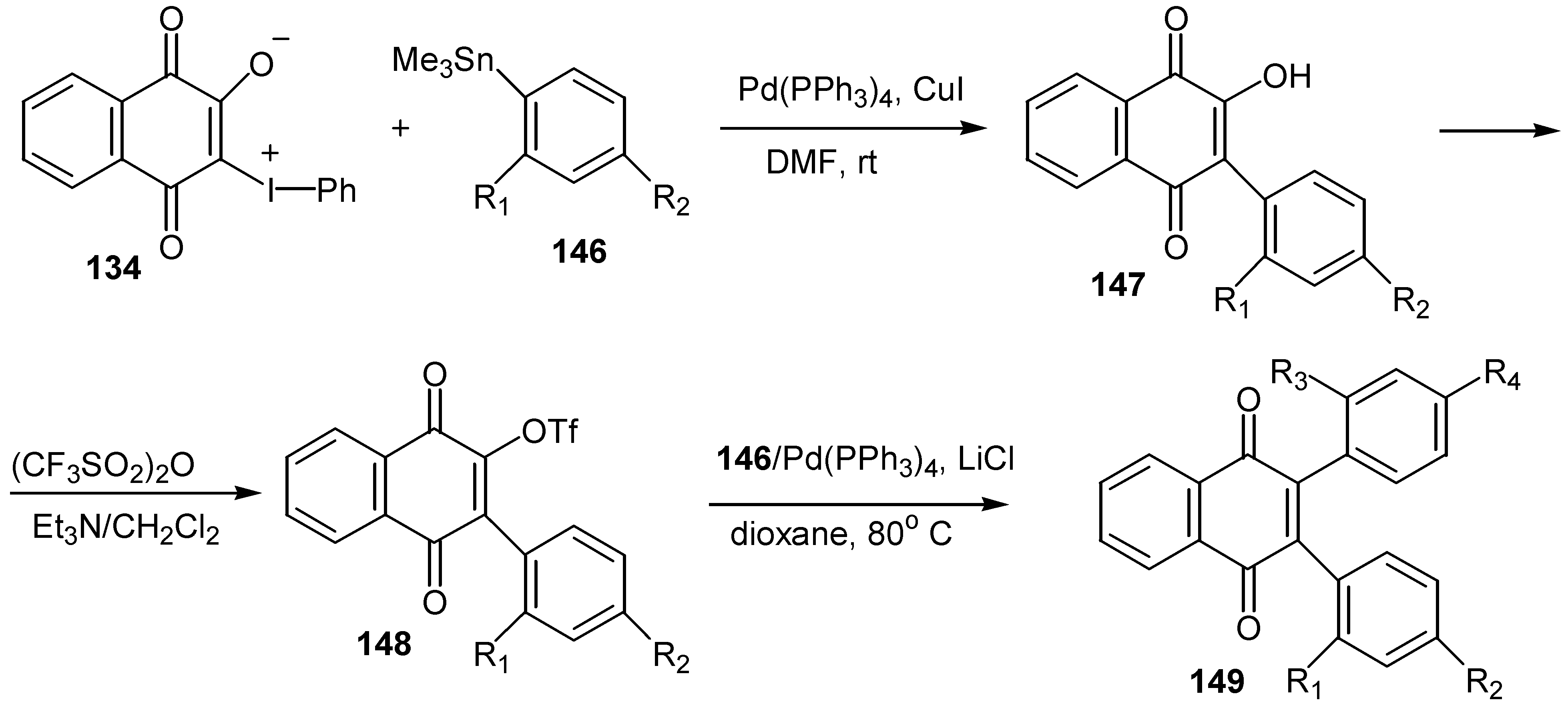
The same ylides were used for the regiospecific synthesis of unsymmetrical 2,3-diarylquinones, 149, via stepwise Pd(0)-catalyzed coupling with arylstannanes [73]. The ylide serves as a doubly activated quinone equivalent, one activator being the phenyliodonio group and the other the anionic oxygen that is a masked triflate activator (Scheme 67).
The stepwise synthesis, with overall yields ranging 50-55%, is outlined in Scheme 68 below.
The same authors reported the application of this procedure for the preparation of 2,3-bisnaphthopyranyl quinones related to concurvone [74], offering thus a key-step for the synthesis of this plant-derived trimeric quinone with potent anti-HIV activity.
c) Miscellaneous.
In many cases the conversion of the hydroxy group of the quinone ring to a corresponding ester or ether group is necessary for a further transformation or for monitoring the biological activity of the certain derivatives. The formation of the triflate, 148, in the previous scheme is a characteristic example. Although these esterifications and ethers formation take place using the conventional methods, some problems arise in the case of dihydroxyquinones.
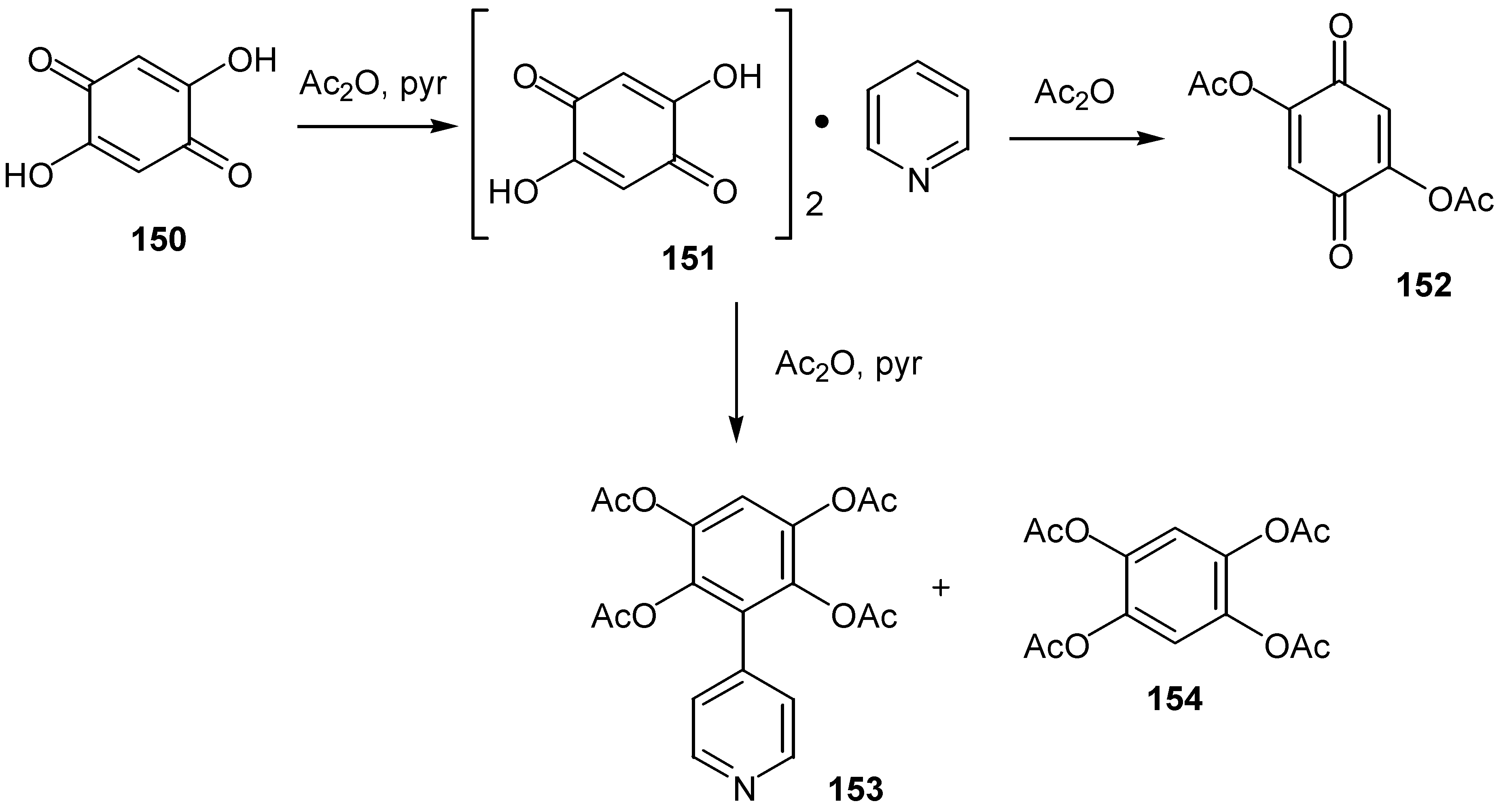
2,5-Diacetoxy-1,4-benzoquinone, 152, is prepared from the parent dihydroxy compound, 150, by a two-step procedure: The first step is the formation of an isolable quinone-pyridine complex, 151,which with further addition of acetic anhydride yields the desired diacetoxy compound. Instead, 4-(1',2',4',5'-tetraacetoxyphenyl)-pyridine, 153, and 1,2,4,5-tetraacetoxybenzene, 154, are the products when the reaction takes place in the presence of pyridine [75] (Scheme 69) .
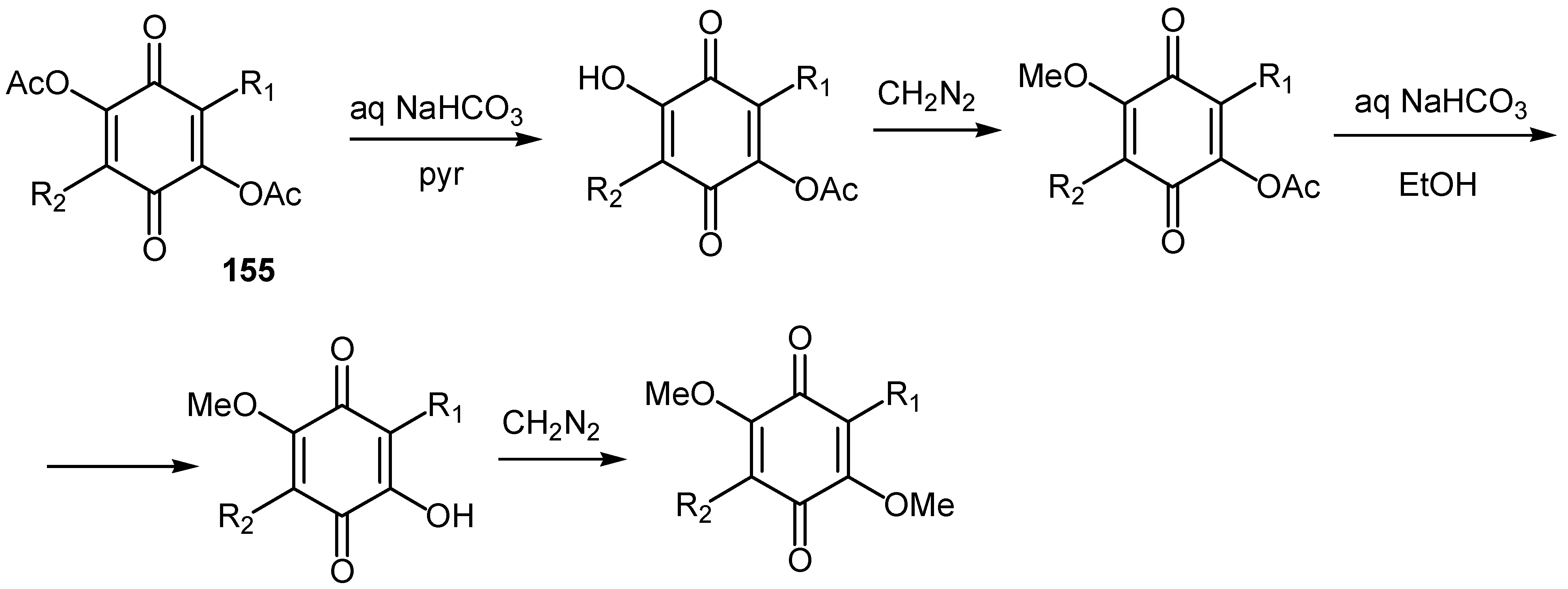
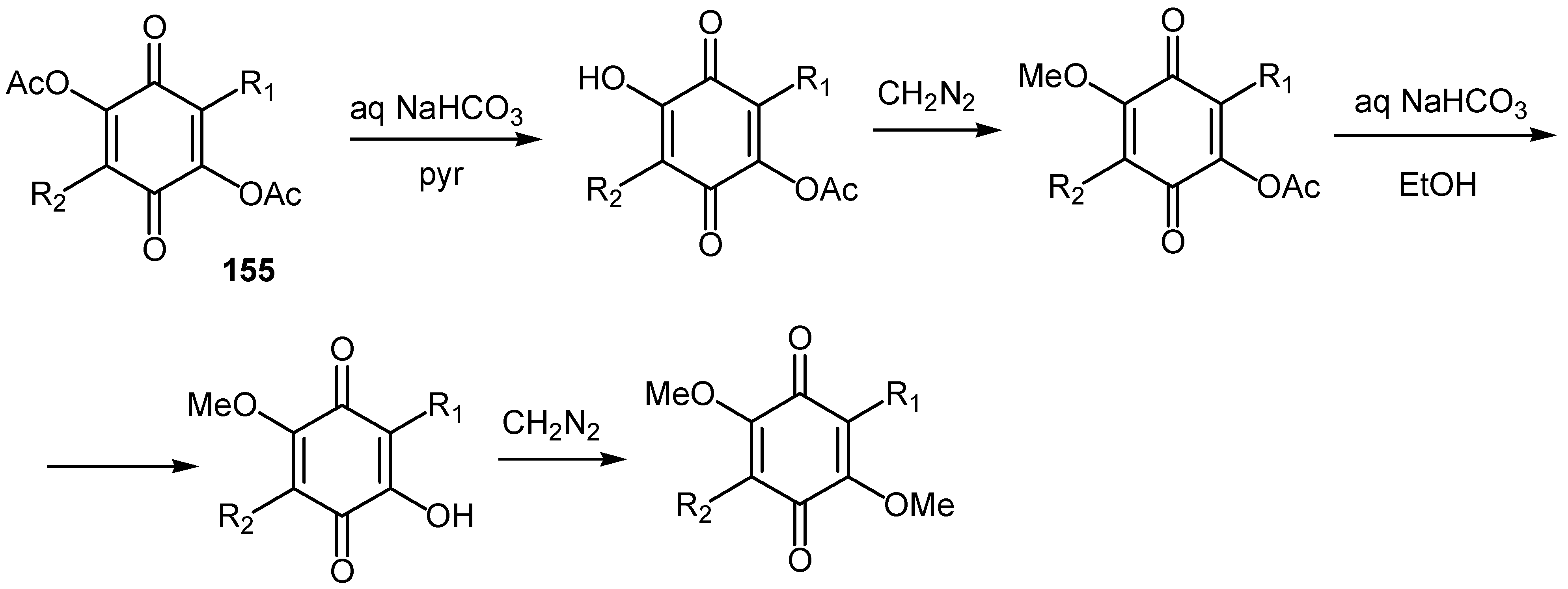
Derivatives of 2,5-diacetoxy-1,4-benzoquinone, 155, can be converted to various mono- or disubstituted hydroxy, methoxy and acetoxy quinones by the sequence of reactions shown in Scheme 70.
It must be noted that R1 and R2 are, except H and CH3, substituted indole derivatives and the asterriquinones prepared in this manner are used for an interesting structure-activity relationship study [76].
The preparative electrochemical reductive methylation of 2-hydroxy-1,4-benzoquinones was proposed as a most effective methodology for the protection of this functionality [77]. The products are aromatic methyl ethers of the reduced quinone function, such as 157, and the method finds application in a variety of naturally occurring hydroxyquinones, such as perezone, 156, in the example outlined in Scheme 71 below.
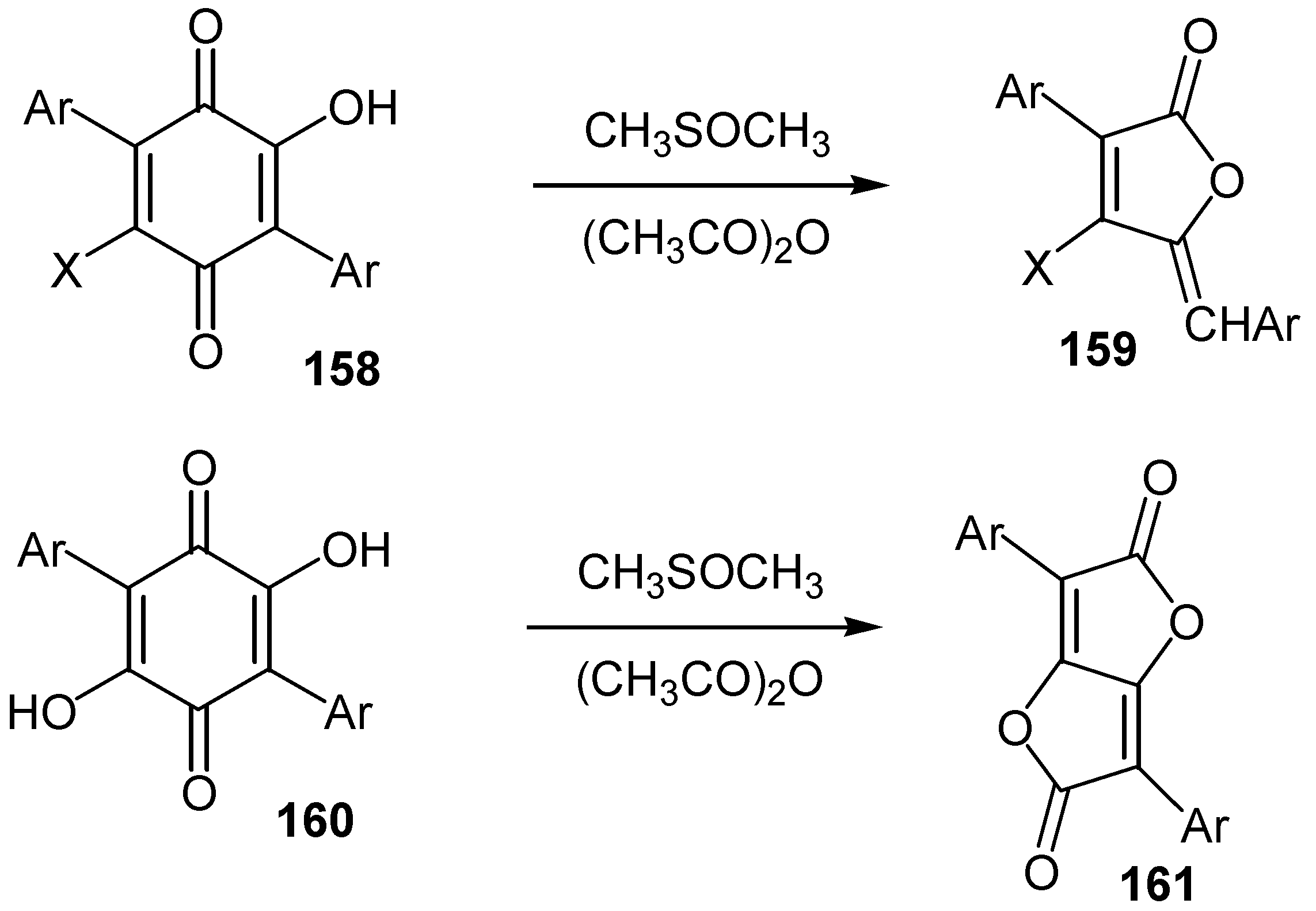
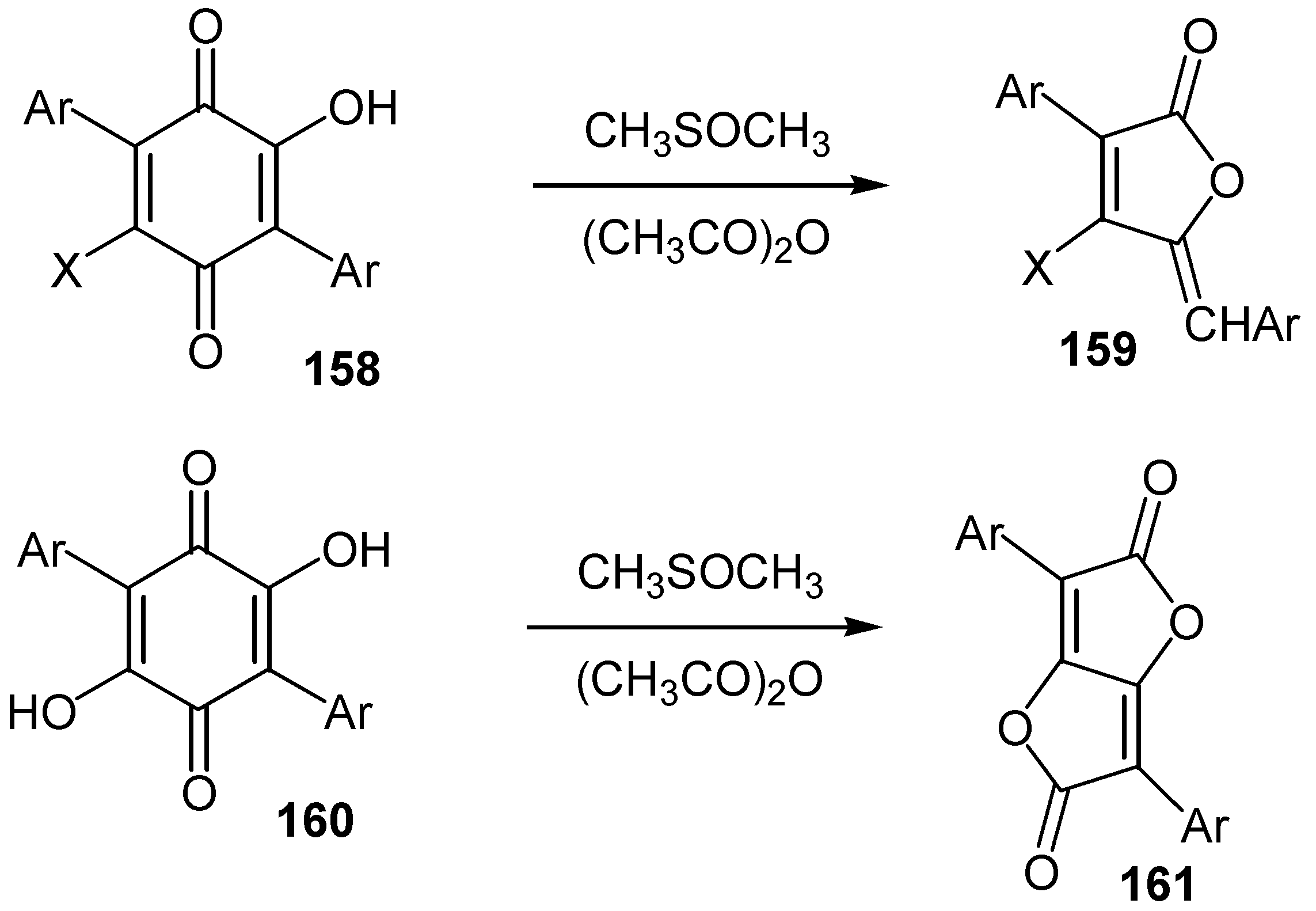
In some cases an oxidative rearrangement, followed by ring contraction, is observed from the reaction of hydroxyquinones with the proper oxidative reagents. Thus 3,5,6-trisubstituted-2-hydroxy- and 3,6-disubstituted-2,5-dihydroxy-1,4-benzoquinones, 158 and 160, give good yields of the corresponding lactone derivatives, 159 and 161, upon heating in dimethyl sulfoxide-acetic anhydride [66] (Scheme 72).
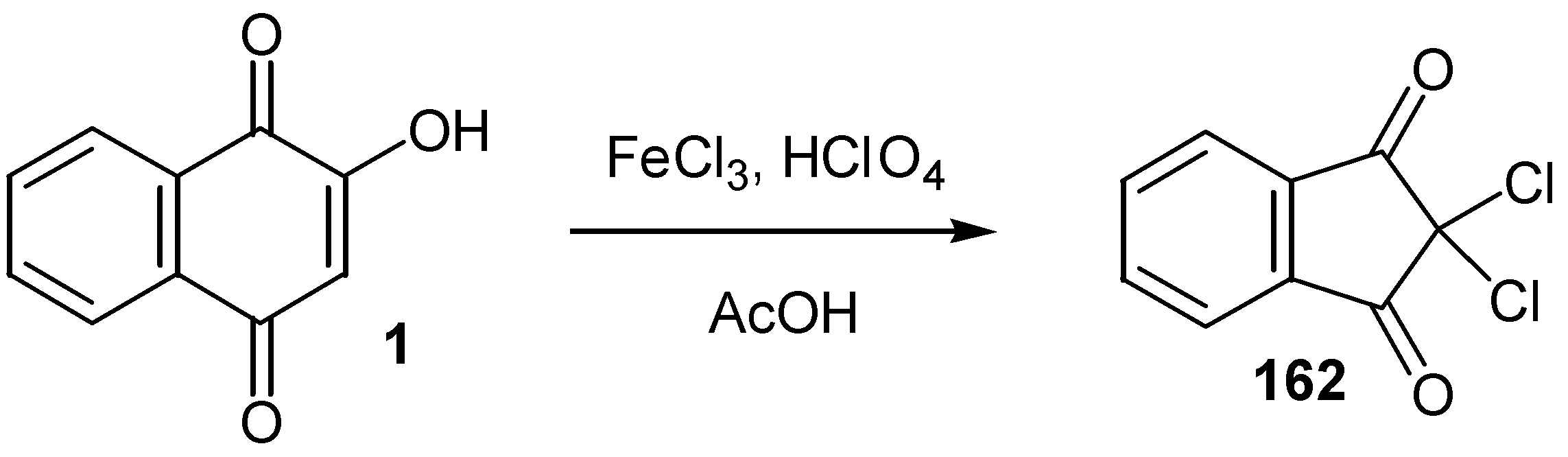
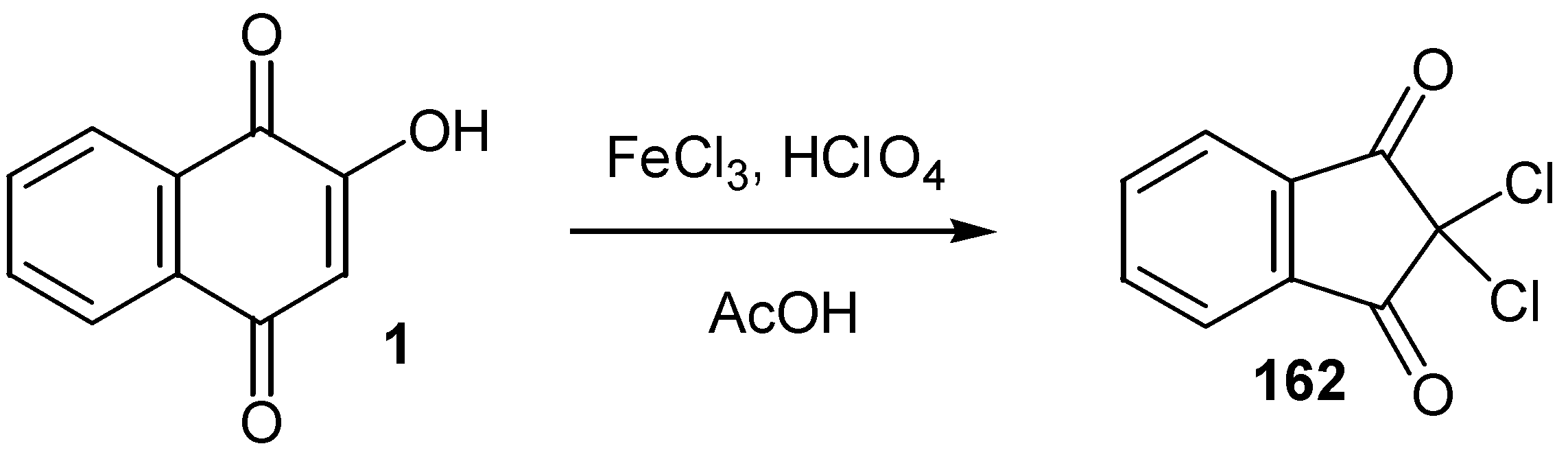
An analogous oxidative transformation of lawsone (1) to 2,2-dicloroindane-1,3-dione (162), was also reported [78] (Scheme 73).
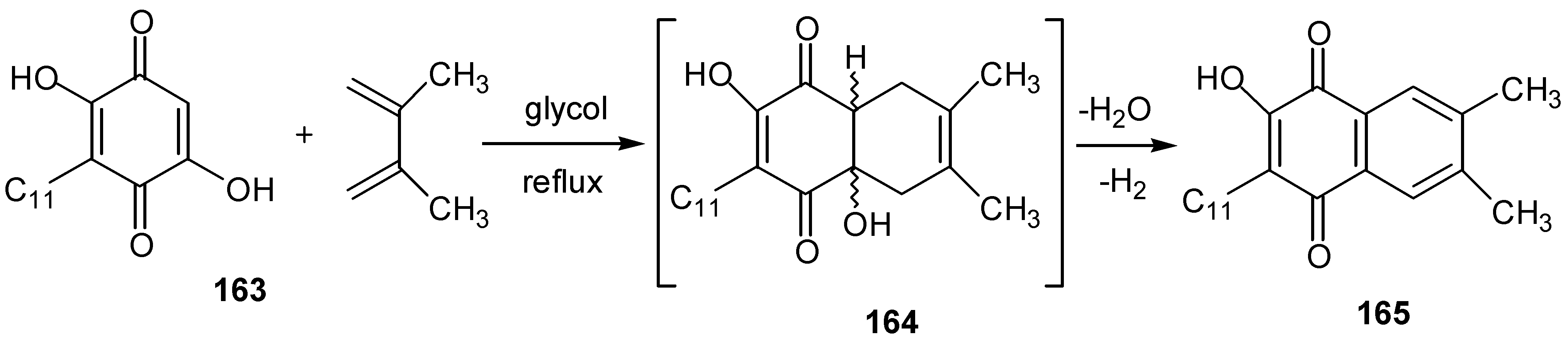
The presence of the hydroxy group on the quinone ring diminishes the dienophilic character of the double bond that bears this group and Diels-Alder cyclizations take place from the other bond. When two hydroxy groups are present, Diels-Alder cyclization proceeds with participation of the less hindered bond, under vigorous conditions [79] (Scheme 74).
The initially formed Diels-Alder adduct, 164, is dehydrated and oxidized to the corresponding hydroxynaphthoquinone derivative, 165, under the conditions of the reaction.
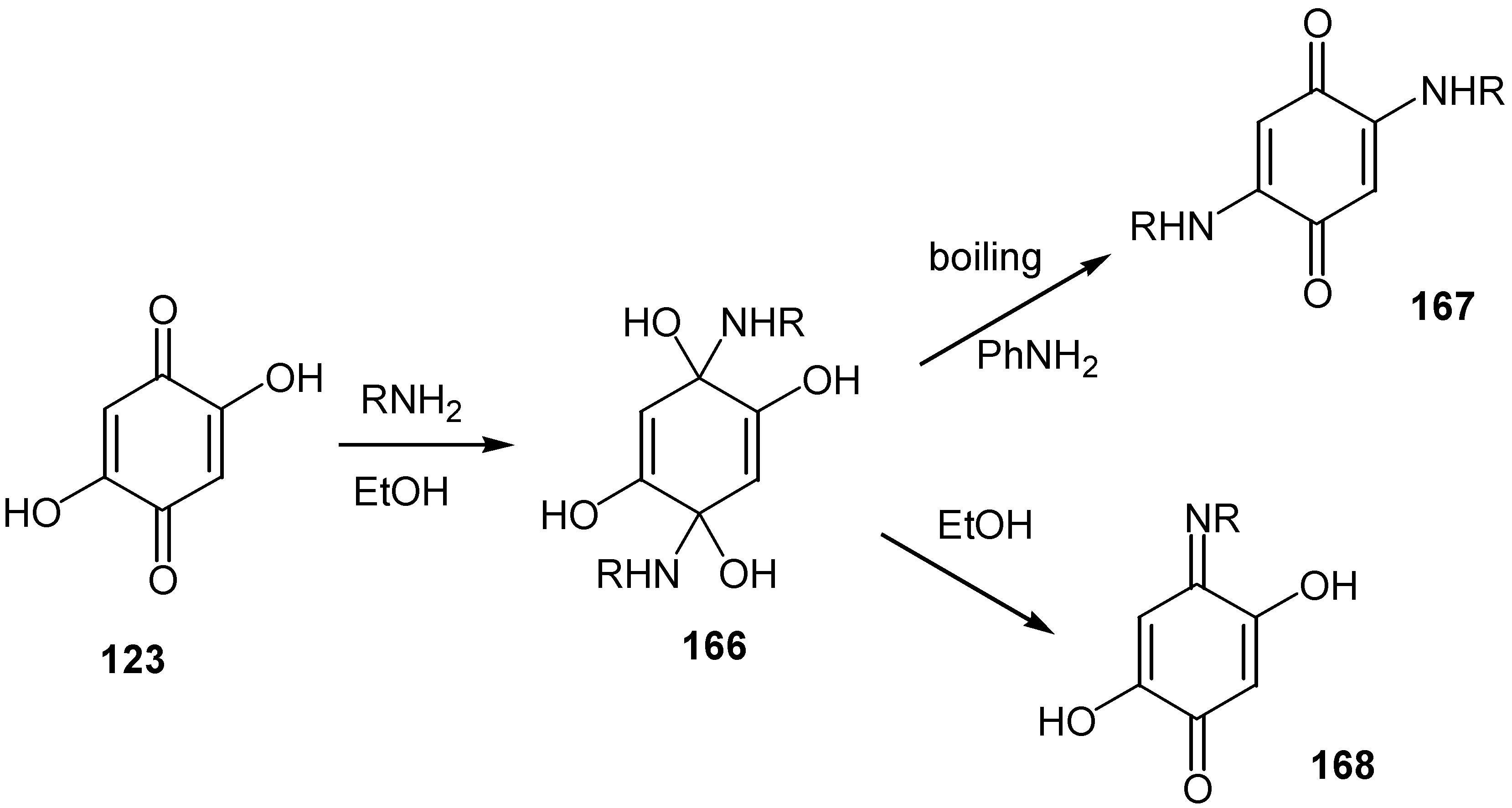
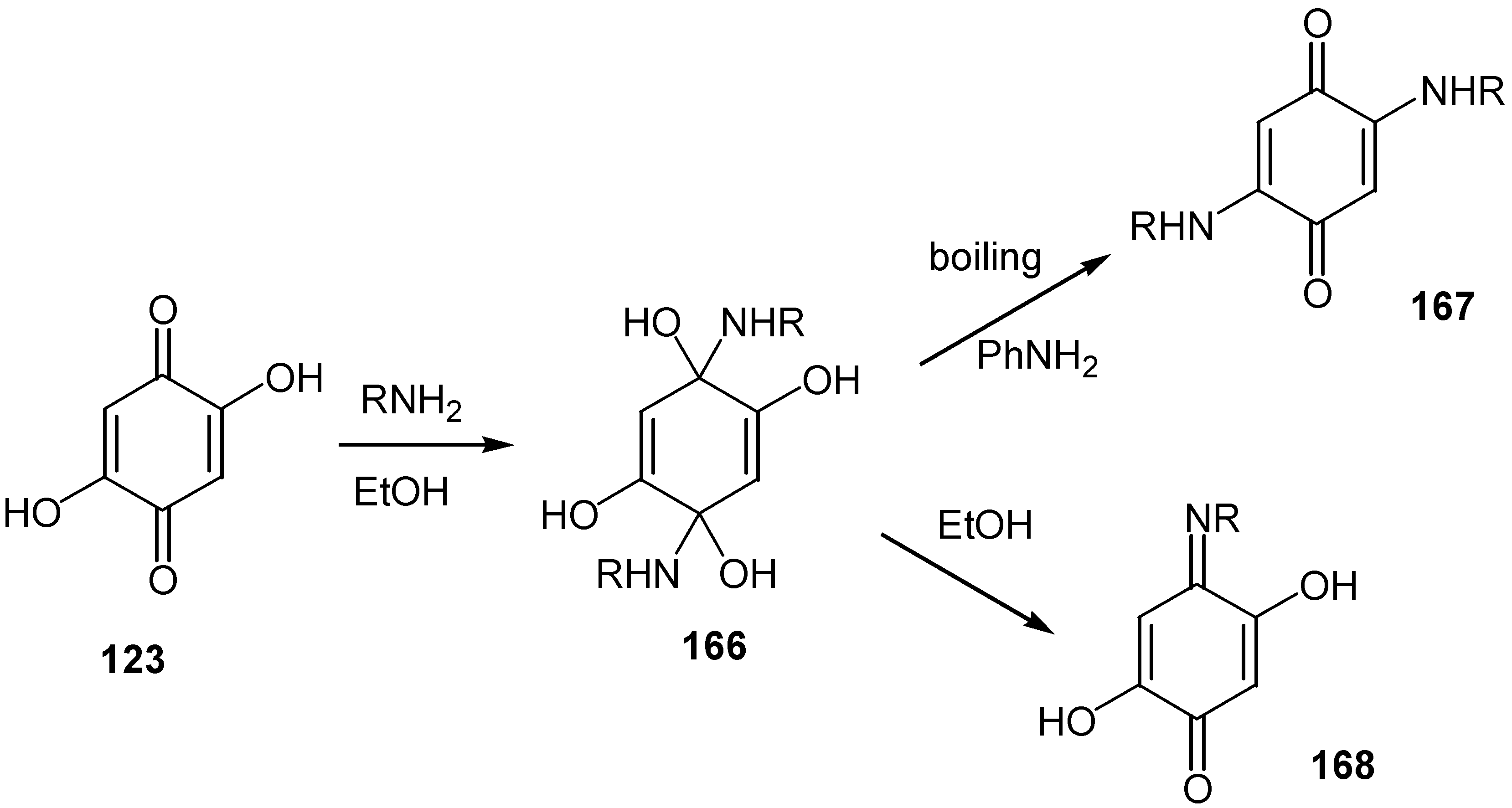
Finally, 2,5-dihydroxy-1,4-benzoquinone, 123, reacts with biogenic amines like 2-phenylethylamine, p-tyramine and histamine to an isolable tetrol derivative, 166. The latter is converted either to 2,5-di-alkylamino-1,4-benzoquinone, 167, or to the monoimine of the parent compound, 168, depending on the reaction conditions [80] (Scheme 75).
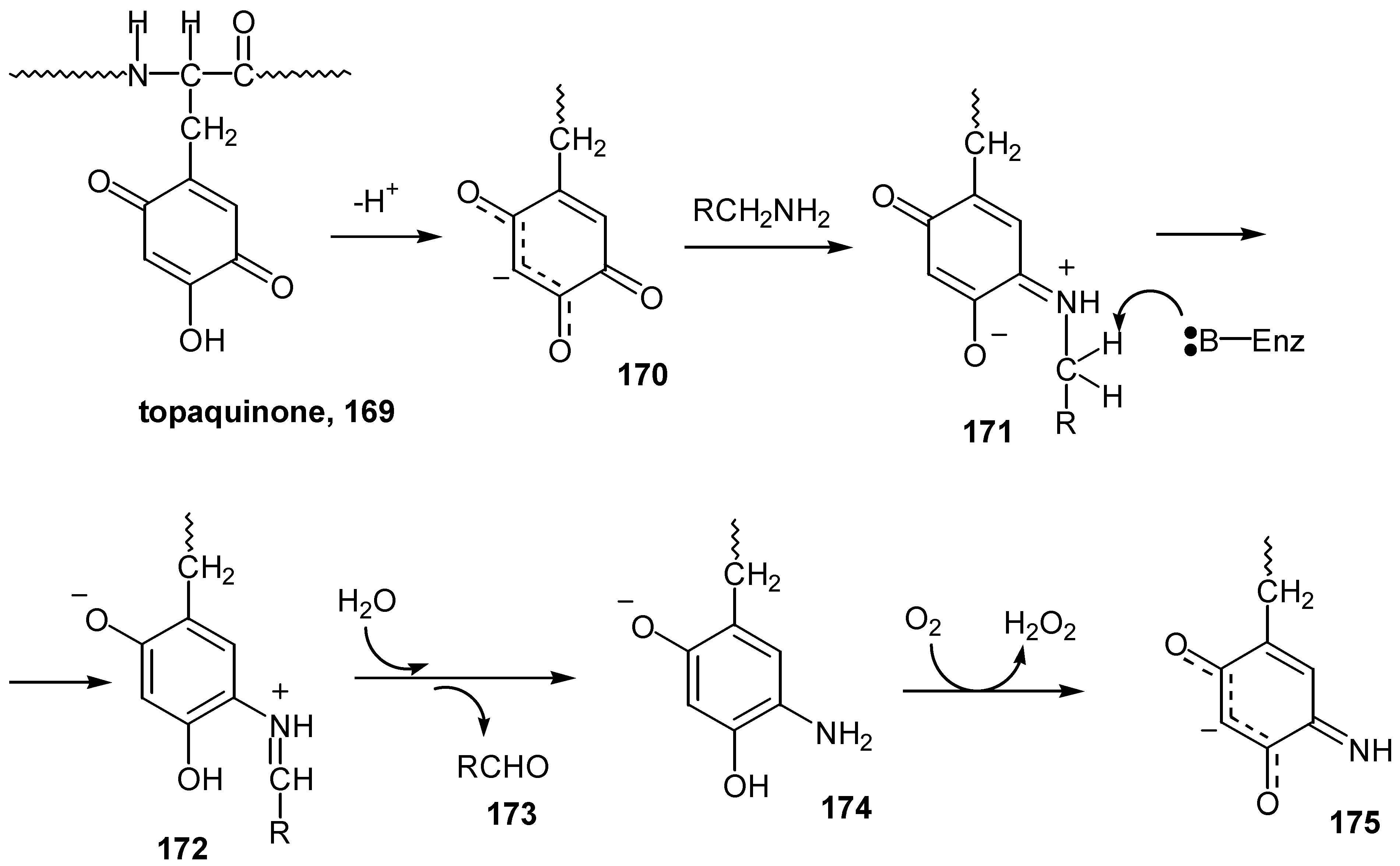
The formation of a Schiff base analogue is the first step of a biochemical process: the enzymatic oxidative deamination of a primary amine by topaquinone-dependent amine oxidases. Topaquinone (6-hydroxydopaquinone, 169), was identified as the covalently bound active site cofactor of bovine serum amine oxidase and some copper containing amine oxidases have been demonstrated to be topaquinonedependent enzymes. In a detailed study with model compounds [81a,b] it was shown that the initial formation of a covalent Schiff base complex between topaquinone and amine, 171, is followed by enzymatic hydrogen abstraction. Hydrolysis of the resulting Shiff base 172, leads to aldehyde 173, and a reduced cofactor (such as aminoresorcinol, 174), which undergoes reoxidation to quinone-imine, 175, coupled to a two-electron reduction of dioxygen to hydrogen peroxide (Scheme 76).
4. Conclusions
As a conclusion, hydroxyquinones is an important class of the quinone family, with interesting and diverse reactivity. The naturally occurring hydroxyquinones are attractive synthetic targets, due to their biological activity and their participation in biochemical processes. It is hoped that more interesting results will be achieved in the future and that this review will be a useful tool for people interested in this field.
References
- The Chemistry of the Quinoid Compounds; Patai, S.; Rappoport, Z. (Eds.) Wiley-Interscience: New York, 1988.
- Tisler, M. Heterocyclic Quinones. In Advances in Heterocyclic Chemistry; Katritzky, A. R., Ed.; Academic Press: London, 1989; Volume 45, p. 37. [Google Scholar]
- Mehendale, A.R.; Thomson, R.H. Binaphthoquinones in lomatia ferruginea. Phytochemistry 1975, 14, 801–802. [Google Scholar] [CrossRef]
- Yin, J.; Liebeskind, L. S. A synthesis of trisquinones. J. Org. Chem. 1998, 63, 5726–5727, and references cited therein. [Google Scholar] [CrossRef] [PubMed]
- McOmie, J. F. W.; Blatchly, J.M. The Thiele-Winter acetoxylation of quinones in Organic Reactions. Vol. 19, Wiley: New York, 1972; p. 199. [Google Scholar]
- Finley, K. T. in ref. 1, Vol. 2, p. 537.
- Villemin, D.; Bar, N.; Hammadi, M. Triflic acid an efficient catalyst for the Thiele-Winter reaction. Tetrahedron Lett. 1997, 38, 4777–4778. [Google Scholar] [CrossRef]
- Almeida, W. P.; Correia, C. R. D. A total synthesis of the sesquiterpene quinone metachromin-A. Tetrahedron Lett. 1994, 35, 1367–1370. [Google Scholar] [CrossRef]
- Sargent, M. V.; Wangchareontrakul, S. The synthesis of the first natural host germination stimulant for Striga asiatica (witchweed). J. Chem. Soc. Perkin Trans. 1 1990, 1429–1434. [Google Scholar] [CrossRef]
- Suginome, H.; Kamekawa, H.; Sakurai, H.; Konishi, A.; Senboku, H.; Kobayashi, K. Photoinduced molecular transformations. Part 145. Regioselective [3+2] photoadditions of 2-hydroxyphenanthrene-1,4-dione with electron rich alkenes and phenylacetylene: New one-step synthesis of 9,10-dihydrophenanthro [2,3-b]furan-7,11-diones and 2-phenylphenanthro[2,3-b] furan-7,11-dione. J. Chem. Soc. Perkin Trans. 1 1994, 471–475. [Google Scholar]
- Huot, R.; Brassard, P. Synthèse de méthyl-3 furoquinones. Can. J. Chem. 1974, 52, 88–94. [Google Scholar] [CrossRef]
- (a) Waldhör, E.; Schwederski, B.; Kaim, W. Ruthenium(II) coordination to a model for the topasemiquinone cofactor of amine oxidases. Resolution of 1H and 99,101Ru EPR hyperfine structure. J. Chem. Soc. Perkin Trans. 2 1993, 2109–2111. [Google Scholar] ; (b) Luijkx, G. C. A.; Rantwijk, van F.; Bekkum, van H. Hydrothermal formation of 1,2,4-benzenetriol from 5-hydroxymethyl-2-furaldehyde and D-fructose. Carbohydr. Res. 1993, 242, 131–140. [Google Scholar]
- Kobayashi, K.; Kanno, Y.; Suginome, H. Photoinduced molecular transformations. Part 141. New one-step general synthesis of benzofuran-4,7-diones by the regioselective (3+2) photo-addition of 2-hydroxy-1,4-benzoquinones with various alkenes. J. Chem. Soc. Perkin Trans. 1 1993, 1449–1452. [Google Scholar] [CrossRef]
- Brown, R. F. C.; Robinson, A. J. A synthetic approach to (±)-tridentoquinone. Aust. J. Chem. 1995, 48, 515–529. [Google Scholar] [CrossRef]
- Poigny, S.; Guyot, M.; Samadi, M. Total synthesis of maesanin and analogues. Tetrahedron 1998, 54, 14791–14802. [Google Scholar] [CrossRef]
- Poigny, S.; Guyot, M.; Samadi, M. Efficient synthesis of (-)-illimaquinone. J. Org. Chem. 1998, 63, 5890–5894. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Yaso, H.; Kiriyama, H.; Takahashi, H.; Minami, H.; Kamikawa, T. Synthesis of cytotoxic maesaquinone bearing a 2,5-dihydroxy-6-methyl-1,4-benzoquinone nucleus. Tetrahedron 1997, 53, 16969–19976. [Google Scholar] [CrossRef]
- Yoon, T.; Danishefski, S. J.; Cala de, S. A concise total synthesis of (±)-mamanuthaquinone by using an exo-Diels-Alder reaction. Angew. Chem. Int. Ed. Engl. 1994, 33, 853–855. [Google Scholar] [CrossRef]
- Bruner, S.D.; Radeke, H. S.; Tallarico, J. A.; Snapper, M. L. Total synthesis of (-)-illimaquinone. J. Org. Chem. 1995, 60, 1114–1115. [Google Scholar] [CrossRef]
- Radeke, H. S.; Digits, C.A.; Bruner, S. D.; Snapper, M. L. New tools for studying vesicularmediated protein trafficking: Synthesis and evaluation of illimaquinone analogs in a nonradioisotope-based antisecretory assay. J. Org. Chem. 1997, 62, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Williams, D. R.; Clark, M. P. Synthesis of Atovaquone. Tetrahedron Lett. 1998, 39, 7629–7632. [Google Scholar] [CrossRef]
- Harris, G. D., Jr.; Nguen, A.; App, H.; Hirth, P.; McMahon, G.; Tang, C. A one-pot, two-step synthesis of tetrahydro asterriquinone E. Organic Lett. 1999, 1, 434–436. [Google Scholar] [CrossRef]
- (a) Liu, K.; Wood, H. B.; Jones, A. B. Total synthesis of asterriquinone B1. Tetrahedron Lett. 1999, 40, 5119–5122. [Google Scholar] ; (b) Zhang, B.; Zhang, G.; Szalkowski, D.; Li, Z.; Zhang, Y.; Royo, I.; Villela, D.; Diez, M.T.; Pelaez, F.; Ruby, C.; Kendall, R. L.; Mao, X.; Griffin, P.; Calaycay, J.; Zierath, J. R.; Heck, J. V.; Smith, R. G.; Moller, D. E. Discovery of a small molecule insulin mimetic with antidiabetic activity in mice. Science 1999, 284, 284–977. [Google Scholar]
- Ref. 6, p. 552.
- Moore, H. W.; Shelden, H. R. Rearrangement of azidoquinones. Reaction of thymoquinone and 2-5-dimethyl-1,4-benzoquinone with sodium azide in trichloroacetic acid. J. Org. Chem. 1968, 33, 4019–4024. [Google Scholar] [CrossRef]
- Parker, K. A.; Sworin, M. E. Assignment of regiochemistry to substituted naphthoquinones by chemical and spectroscopic methods. Amino-, hydroxy-, and bromojuglone derivatives. J. Org. Chem. 1981, 46, 3218–3223. [Google Scholar]
- Cuntze, U.; Maasen, D.; Musso, H. Uber die bildung von hydroxy-tert.-butyl-chinonen und deren abbau zu cyclopentenon-derivaten durch alkali. Chem. Ber. 1969, 102, 2851–2861. [Google Scholar] [CrossRef]
- Croux, S.; Maurette, M.-T.; Hocquaux, M.; Ananides, A.; Braun, A. M.; Oliveros, E. Kinetic parameters of the reactivity of dihydroxynaphthalenes with singlet oxygen. New J. Chem. 1990, 14, 161–167. [Google Scholar]
- Min, D. M.; Croux, S.; Tournair, C.; Hocquaux, M.; Jacquet, B.; Oliveros, E.; Maurette, M.-T. Réactivité du superoxyde de potassium en phase hétérogène: Oxydation de naphtalénediols en Naphtoquinones hydroxylées. Tetrahedron 1990, 48, 1869–1882. [Google Scholar] [CrossRef]
- Spyroudis, S.; Xanthopoulou, N. unpublished results.
- (a) Kasturi, T. R.; Arunachalam, T. General method of preparation of substituted 2-hydroxy-1,4-quinones. Can. J. Chem 1966, 44, 1086. [Google Scholar] ; (b) Baillie, A. C.; Thomson, R. H. Quinones. Part VII. New routes to 2-hydroxy-1,4-naphthaquinones. J. Chem. Soc. Sec. C 1966, 2184–2186. [Google Scholar]
- Pettit, G. R.; Fleming, W. C.; Paull, K. D. Synthesis of the 6- and 7-hydroxy-5,8-dioxocarbostyrils. J. Org. Chem. 1968, 33, 1089–1092. [Google Scholar] [CrossRef]
- Bekaert, A.; Andrieux, J.; Plat, M.; Brion, J-D. A convenient synthesis of monpain trimethylether. Tetrahedron Lett. 1997, 38, 4219–4220. [Google Scholar] [CrossRef]
- Takada, T.; Akiba, M. Synthesis of 1H-pyrrolo[1,2-a]indole derivatives. III. Synthesis of 2,3-dihydro-7-hydroxy-6,9-dimethyl-5,8-dioxo-1H-pyrrolo[1,2-a]indole. Chem. Pharm. Bull. 1972, 20, 1785–1792. [Google Scholar] [CrossRef]
- Senoh, S.; Witkop, B. Formation and rearrangements of aminochromes from a new metabolite of dopamine and some of its derivatives. J. Am. Chem. Soc. 1959, 81, 6231–6235. [Google Scholar] [CrossRef]
- Pezzella, A.; Ischia, d' M.; Napolitano, A.; Misuraca, G.; Prota, G. Iron-Mediated generation of the neurotoxin 6-hydroxydopamine quinone in the reaction of fatty acid hydroperoxides with dopamine: A possible contributory mechanism for neuronal degeneration in Parkinson’s disease. J. Med. Chem. 1997, 40, 2211–2216. [Google Scholar] [PubMed]
- Sartori, G.; Bigi, F.; Canali, G.; Maggi, R.; Casnati, G.; Tao, X. Friedel-Crafts coordinated processes: Highly selective synthesis of hydroxynaphthoquinones. J. Org. Chem. 1993, 58, 840–843. [Google Scholar] [CrossRef]
- Schuchman, M. N.; Bothe, E.; Sonntag, v. J.; Sonntag, v. C. Reaction of OH radicals withbenzoquinone in aqueous solutions. A pulse radiolysis study. J. Chem. Soc. Perkin II 1998, 791–796. [Google Scholar] [CrossRef]
- Harrity, J. P. A.; Kerr, W. J.; Middlemiss, D.; Scott, J. S. Total synthesis of parvaquone and the serendipitous discovery of a novel chromium-mediated method for a-lactone formation. J. Organomet. Chem. 1997, 532, 219–227. [Google Scholar] [CrossRef]
- Singh, I.; Moore, R. E.; Chang, C. W. J.; Ogata, R. T.; Scheuer, P. J. Spinochrome synthesis. Tetrahedron 1968, 24, 2969–2978. [Google Scholar] [CrossRef]
- Jones, R. G.; Shonle, H. A. The preparation of 2,5-dihydroxyquinone. J. Am. Chem. Soc. 1945, 67, 1034–1035. [Google Scholar] [CrossRef]
- (a) Enhsen, A.; Karabelas, K.; Heerding, J. M.; Moore, H. W. Synthesis of hydroxyquinones and related compounds: Semisquaric acids, (±)-terreic acid, (±)-perezone, and (±)-isoperezone. J. Org. Chem. 1990, 55, 1177–1185. [Google Scholar] ; (b) Heerding, J. M.; Moore, H. W. Regiospecific synthesis of hydroxyquinones and related compounds from 3-tert-butoxycyclobutene-1,2-dione. J. Org. Chem. 1991, 56, 4048–4050. [Google Scholar]
- Tius, M. A.; Cullingham, J. M.; Ali, S. Hydroxyquinone annelation. J. Chem. Soc. Chem. Commun. 1989, 867–869. [Google Scholar] [CrossRef]
- Horiuchi, C. A.; Suzuki, Y. A new synthesis of 3-hydroxy-2,5-dialkyl-1,4-benzoquinone from 3-halo-3,6-dialkyl-1,2-cyclohexanedione using iodine-copper(II) acetate. Bull. Chem. Soc. Jpn. 1989, 62, 2919–2922. [Google Scholar] [CrossRef]
- Reinaud, O.; Capdevielle, P.; Maumy, M. Synthesis of new 3-(-2-alkenyl)-2-hydroxy-5-methoxy-p-benzoquinones via Claisen rearrangement of original 5-methoxy-4-(2-propenyloxy)-o-benzoquinones. Synthesis 1988, 293–300. [Google Scholar] [CrossRef]
- Finley, K. T. in ref. 1, Vol 2, p 603.
- Fieser, L.F.; Berliner, E.; Bondhus, F.J.; Chang, F.C.; Dauben, W.G.; Ettlinger, M.G.; Fawaz, G.; Fields, M.; Heidelberger, C.; Heymann, H.; Vaughan, W.R.; Wilson, E.; Moore, E.E.; Moore, M.B.; Zaugg, H.E. Naphthoquinone antimalarials. IV-XI. Synthesis. J. Am. Chem. Soc. 1948, 70, 3174. [Google Scholar] [CrossRef] [PubMed]
- Khambay, B.P.S.; Batty, D.; Beddie, D.G.; Denholm, I.; Cahill, M.R. A new group of plant-derived naphthoquinone pesticides. Pestic. Sci. 1997, 50, 291–296. [Google Scholar] [CrossRef]
- Sun, J.S.; Geiser, A.H.; Frydman, B. A preparative synthesis of lapachol and related naphthoquinones. Tetrahedron Lett. 1998, 39, 8221–8224. [Google Scholar] [CrossRef]
- Bieber, L.W.; Neto, P.J.R.; Generino, R.M. Regioselective alkylation of substituted quinones by trialkylboranes. Tetrahedron Lett. 1999, 40, 4473–4476. [Google Scholar] [CrossRef]
- Brassard, P.; L’ Ecuyer, P. Arylation of quinones by diazonium salts. IV. Reaction of these salts with 2,5-dihydroxy-p-benzoquinone and the synthesis of 3-hydroxy-2,5-diphenyl-p-benzoquinone. Can. J. Chem. 1958, 36, 1346. [Google Scholar] [CrossRef]
- Kobayashi, K.; Taki, T.; Kawakita, M.; Uchida, M.; Morikawa, O.; Konishi, H. A simple synthesis of benzocarbazolequinones via o-nitroarylation of 2-hydroxy-1,4-naphthoquinones. Heterocycles 1999, 51, 349–354. [Google Scholar] [CrossRef]
- Hagiwara, H.; Sato, K.; Suzuki, T.; Ando, M. Tandem nucleophilic reaction leading to hydrofurans. Application to one-pot synthesis of antitumor naphthofuran natural product. Heterocycles 1999, 51, 497–500. [Google Scholar] [CrossRef]
- Kobayashi, K.; Shimizu, H.; Sasaki, A.; Suginome, H. New one-step synthesis of 2,3-dihydronaphtho[2,3-b]furan-4,9-diones by regioselective [3+2] photoaddition of 2-hydroxy-1,4-naphthoquinones with various alkenes and its application to a two-step synthesis of maturinone. J. Org. Chem. 1991, 56, 3204–3206. [Google Scholar] [CrossRef]
- Kobayashi, K.; Shimizu, H.; Sasaki, A.; Suginome, H. Photoinduced molecular transformations. 140. New one-step general synthesis of naphtho[2,3-b]furan-4,9-diones and their 2,3-dihydro derivatives by the regioselective [3+2] photoaddition of 2-hydroxy-1,4-naphthoquinones with various alkynes and alkenes: Application of the photoaddition to a two-step synthesis of maturinone. J. Org. Chem. 1993, 58, 4614–4618. [Google Scholar]
- Suginome, H.; Konishi, A.; Sakurai, H.; Minakawa, H.; Takeda, T.; Senboku, H.; Tokuda, M.; Kobayashi, K. Photoinduced molecular transformations. Part 156. New photoadditions of 2-hydroxy-1,4-naphthoquinones with naphthols and their derivatives. Tetrahedron 1995, 51, 1377–1386. [Google Scholar] [CrossRef]
- Chuang, C.-P.; Wang, S.-F. Manganese(III) acetate initiated free radical reaction between 1,4-naphthoquinones and a-alkylmalonates. Tetrahedron 1998, 54, 10043–10052. [Google Scholar] [CrossRef]
- (a) Kobayashi, K.; Mori, M.; Uneda, T.; Morikawa, O.; Konishi, H. Ceric ammonium nitrate mediated cycloaddition of hydroxyquinones with alkenes for the one-step construction of furoquinone derivatives. Chem. Lett. 1996, 451–452. [Google Scholar] ; (b) Kobayashi, K.; Uneda, T.; Uneda, K.; Mori, M.; Tanaka, H.; Morikawa,, O.; Konishi, H. One-step synthesis of naphthofurandione, benzofurandione, and phenalenofuranone derivatives by the CAN-mediated cycloaddition. Bull. Chem. Soc. Jpn. 1998, 71, 1691–1697. [Google Scholar]
- Kobayashi, K.; Tanaka, K.; Uneda, T.; Maeda, K.; Morikawa, O.; Konishi, H. A direct one-pot preparation of naphtho[2,3-b]furan-4,9-diones from 2-hydroxy-1,4-naphthoquinones and enamines. Synthesis 1998, 1243–1245. [Google Scholar] [CrossRef]
- Kobayashi, K.; Uneda, T.; Kawakita, M.; Morikawa, O.; Konishi, H. One-pot synthesis of naphtho[2,3-b]furan-4,9-diones by sequential coupling/ring-closing reactions. Tetrahedron Lett. 1997, 38, 837–840. [Google Scholar] [CrossRef]
- Shu, T.; Chen, D.-W.; Ochiai, M. Direct synthesis of 2-substituted furotropones fromtropolones utilizing alkynyl(phenyl)iodonium salts. Tetrahedron Lett. 1996, 37, 5539–5542. [Google Scholar] [CrossRef]
- (a) Martínez, E.; Martínez, L.; Estévez, J.C.; Estévez, R.J.; Castedo, L. New, simple total syntheses of benzo[b]naphtho[2,3-d]furan-6,11-diones and benzo[b]naphtho[2,1-d]furans. Tetrahedron Lett. 1998, 39, 2175–2176. [Google Scholar] ; (b) Martínez, A.; Estévez, J.C.; Estévez, R.J.; Castedo, L. 1,2- and 1,4-naphthoquinones: general synthesis of benzo[b]naphtho[2,3-d]furan-6,11-diones. Tetrahedron Lett. 2000, 41, 2365–2367. [Google Scholar]
- Thomson, R.H. Naturally occurring quinones; Blackie Academic and Professional: London, 1997. [Google Scholar]
- Kopanski, L.; Karbach, D.; Selbitschka, G.; Steglich, W. Vesparion, ein naphtho[2,3-b] pyrandion-derivat aus dem schleimpilz metrtichia vesparium (myxomycetes). Liebigs Ann. Chem. 1987, 793–796. [Google Scholar] [CrossRef]
- Gabaja, C.; Perchelet, E.M.; Perchellet, J.-P.; Jones, G.B. Regioselective lactonization of naphtho-quinones: synthesis and antitumorial activity of the WS-5995 antibiotics. Tetrahedron Lett. 2000, 41, 3007–3010. [Google Scholar]
- Wilkholm, R.J.; Moore, H.W. Dimethyl sulfoxide-acetic anhydride oxidative rearrangements of hydroxyterphenylquinones. A possible biosynthetic model. J. Am. Chem. Soc. 1972, 94, 6152–6158. [Google Scholar] [CrossRef]
- Koch, A.S.; Harbison, W.G.; Hubbard, J. M.; de Kort, M.; Roe, B. A. Stability of pyridiniumylquinones to aqueous media: The formation of pyridinium-oxy zwitterionic quinones. J. Org. Chem. 1996, 61, 5959–5963. [Google Scholar] [CrossRef]
- Citterio, A.; Fichi, M.; Maronati, A.; Sebastiano, R.; Mele, A. Synthesis of 2-oxy-3-(pyridinium1’-yl)-1,4-naphthoquinone derivatives by iodine or hydrogen peroxide oxidation of 1,4-naphthoquinones in the presence of substituted pyridines. Synthesis 1997, 614–616. [Google Scholar] [CrossRef]
- Bestmann, H. J.; Furst, T. G.; Schier, A. Trimeric ketenylidene(triphenyl)phosphorane: A hybrid between an arene and an ylide. Angew. Chem. Int. Ed. Engl. 1993, 32, 1747–1750. [Google Scholar] [CrossRef]
- Hatzigrigoriou, E.; Spyroudis, S.; Varvoglis, A. Derivatives of 1,4-naphthoquinone via 3-(phenyliodonio)-1,2,4-trioxo-1,2,3,4-tetrahydronaphthalenide. Liebigs. Ann. Chem. 1988, 167–170. [Google Scholar] [CrossRef]
- Papoutsis, I.; Spyroudis, S.; Varvoglis, A. The chemistry of 2-oxido-3-phenyliodonio-1,4-benzoquinones: Transformation to 2-cyclopentene-1,4-diones and cycloadditions. Tetrahedron Lett. 1994, 35, 8449–8452. [Google Scholar] [CrossRef]
- Spyroudis, S.; Xanthopoulou, N. To be published.
- Stagliano, K.W.; Malinakova, H. C. Regiospecific synthesis of unsymmetrical 2,3-diaryl-quinones via stepwise Pd(0)-catalyzed couplings of arylstannanes to doubly activated quinone equivalents. Tetrahedron Lett. 1997, 38, 6617–6620. [Google Scholar] [CrossRef]
- Stagliano, K.W.; Malinakova, H. C. Regiospecific synthesis of 2,3-bisnaphthopyranyl quinones related to conocurvone. Effect of substituents on palladium-catalyzed cross coupling of organostannanes to naphthopyranyl hydroxyquinone triflates. J. Org. Chem. 1999, 64, 8034–8040. [Google Scholar] [CrossRef]
- Farfán, N.; Ortega, E.; Contreras, R. New heterocycles derived from a simultaneous substitution and reductive acetylation of 2,5-dihydroxy-1,4-benzoquinone by N-containing heterocycles. J. Heterocycl. Chem. 1985, 23, 1609–1612. [Google Scholar] [CrossRef]
- Kaji, A.; Kimura, K.; Teranishi, M.; Kiriyama, N.; Nomura, M.; Miyamoto, K. Preparation and structure-activity relationships of novel asterriquinone derivatives. Chem. Pharm. Bull. 1998, 46, 1325–1329. [Google Scholar] [CrossRef]
- Frontana, B.; Cárdenas, J.; Rodríguez-Hahn, L.; Baeza, A. Preparative electrochemicalreductive methylation of ortho-hydroxy-para-benzoquinones. Tetrahedron 1997, 53, 469–478. [Google Scholar] [CrossRef]
- Singh, P.; Khanna, R. Synthesis of 2,2-dichloroindane-1,3-diones from 1,4-naphthoquinones. Tetrahedron Lett. 1994, 35, 3753–3754. [Google Scholar] [CrossRef]
- Khajuria, R.; Jain, S.; Phar, K. Studies on cycloaddition reactions of 2,4-dihydroxy-3-undecyl-1,4-benzoquinone (embelin) and 2,3-dimethylbuta-1,3-diene. Indian J. Chem., Section B. 1996, 35B(8), 860–861. [Google Scholar]
- Tindale, C.R. Reactions of biogenic amines with quinones. Aust. J. Chem. 1984, 37, 611–617. [Google Scholar] [CrossRef]
- (a) Mure, M.; Klinman, J.P. Model studies of topaquinone-dependent amine oxidases. 1. Oxidation of benzylamine by topaquinone analogs. J. Am. Chem. Soc. 1995, 117, 8698–8706. [Google Scholar] ; (b) Mure, M.; Klinman, J.P. Model studies of topaquinone-dependent amine oxidases. 2. Characterization of reaction intermediates and mechanism. J. Am. Chem. Soc. 1995, 117, 8707–8718. [Google Scholar]
Scheme 1.
Scheme 2.
Scheme 3.
Scheme 4.
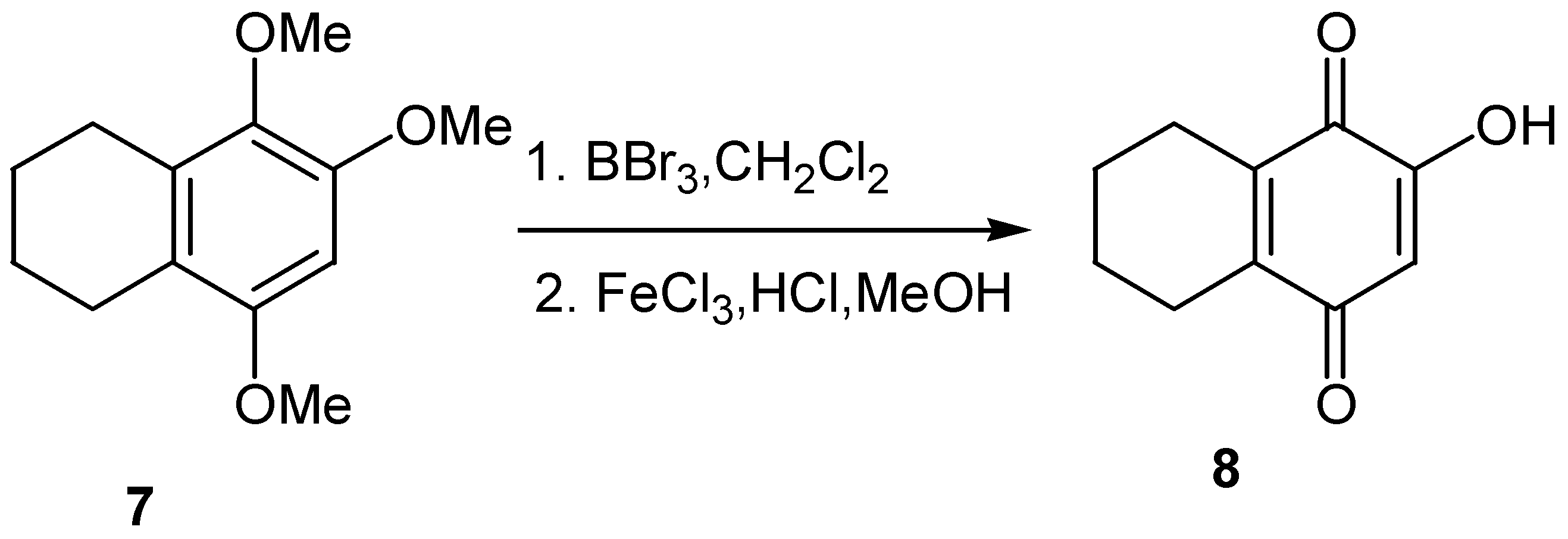
Scheme 5.
Scheme 6.
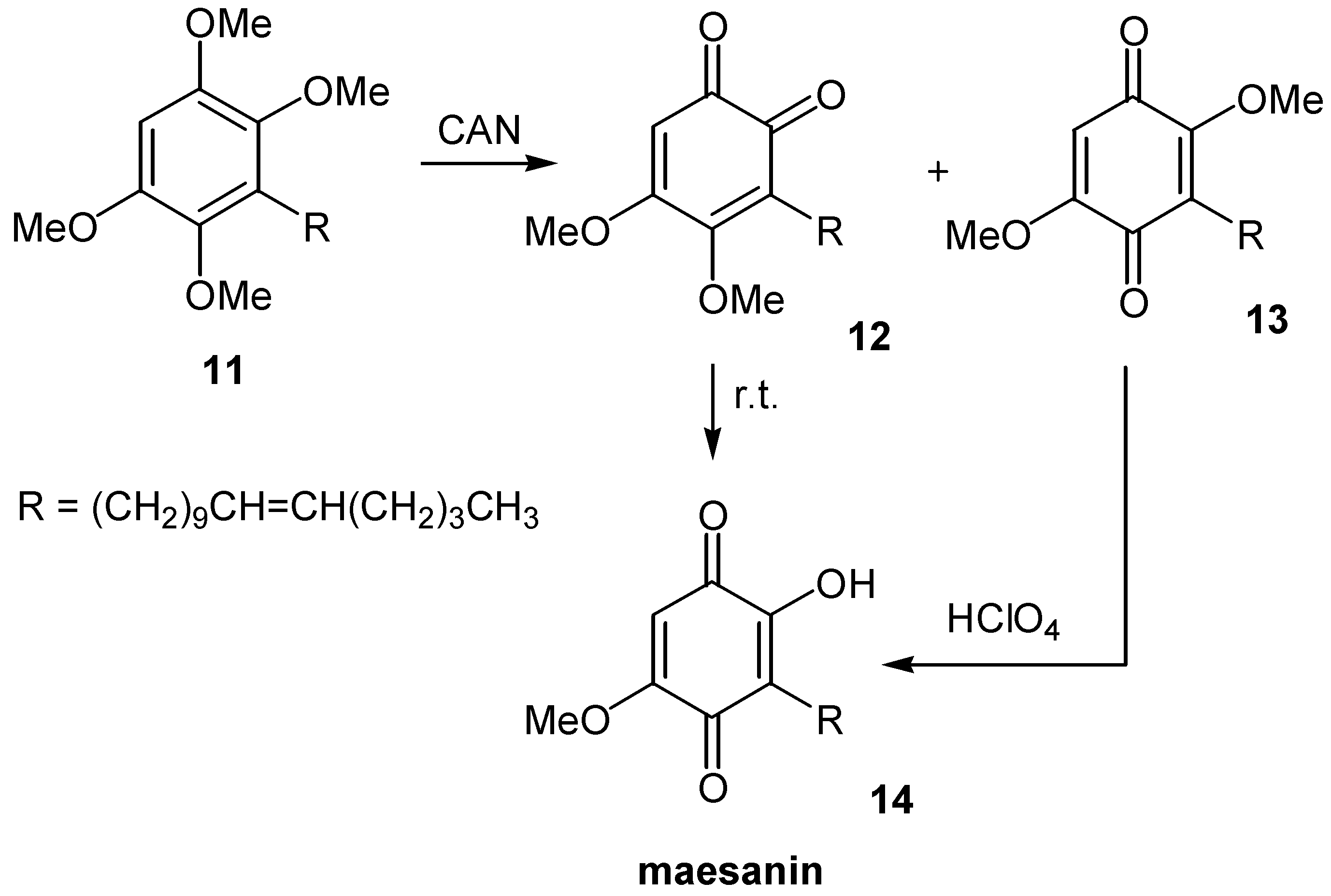
Scheme 7.

Scheme 8.

Scheme 9.
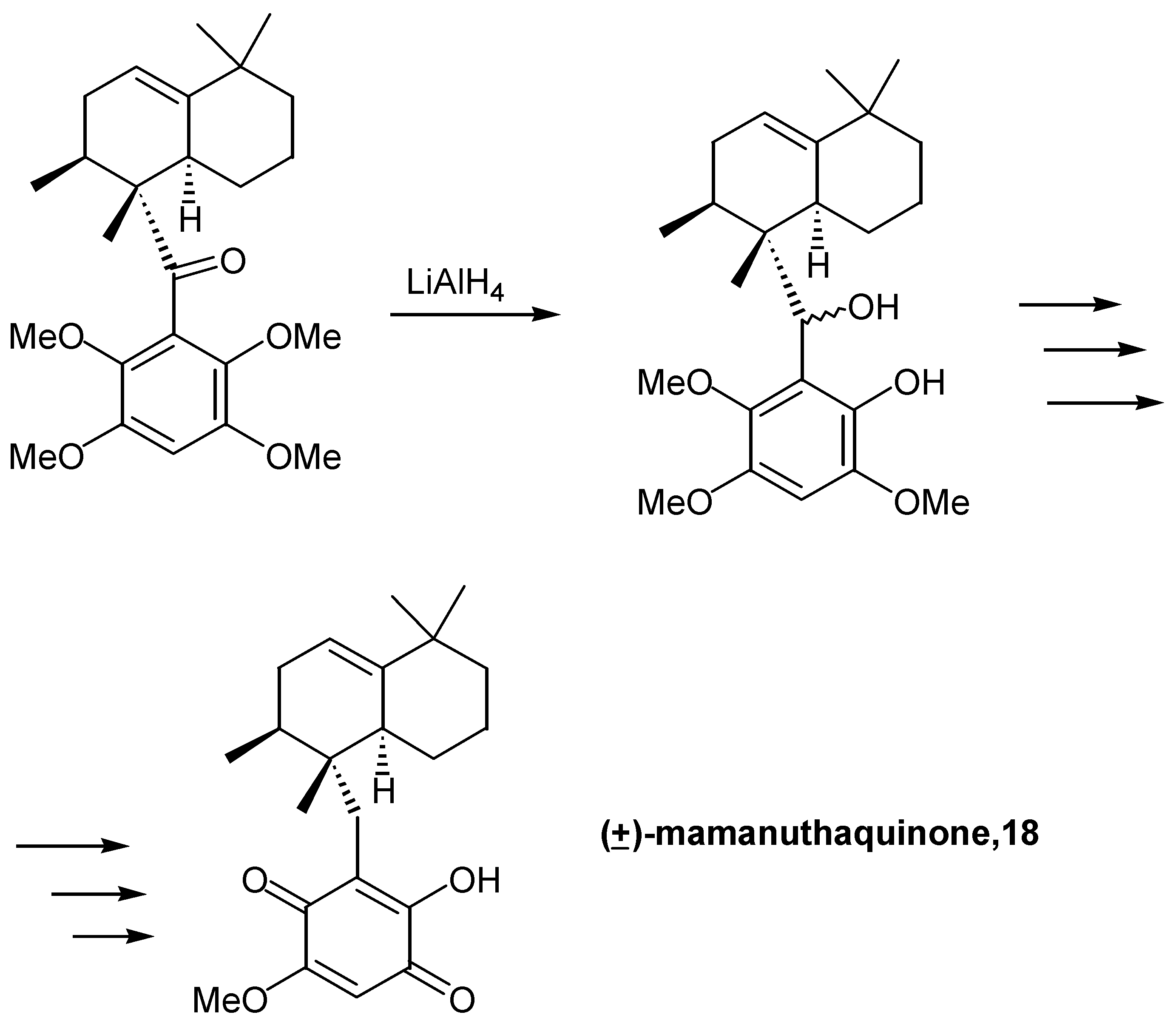
Scheme 10.

Scheme 11.
Scheme 12.
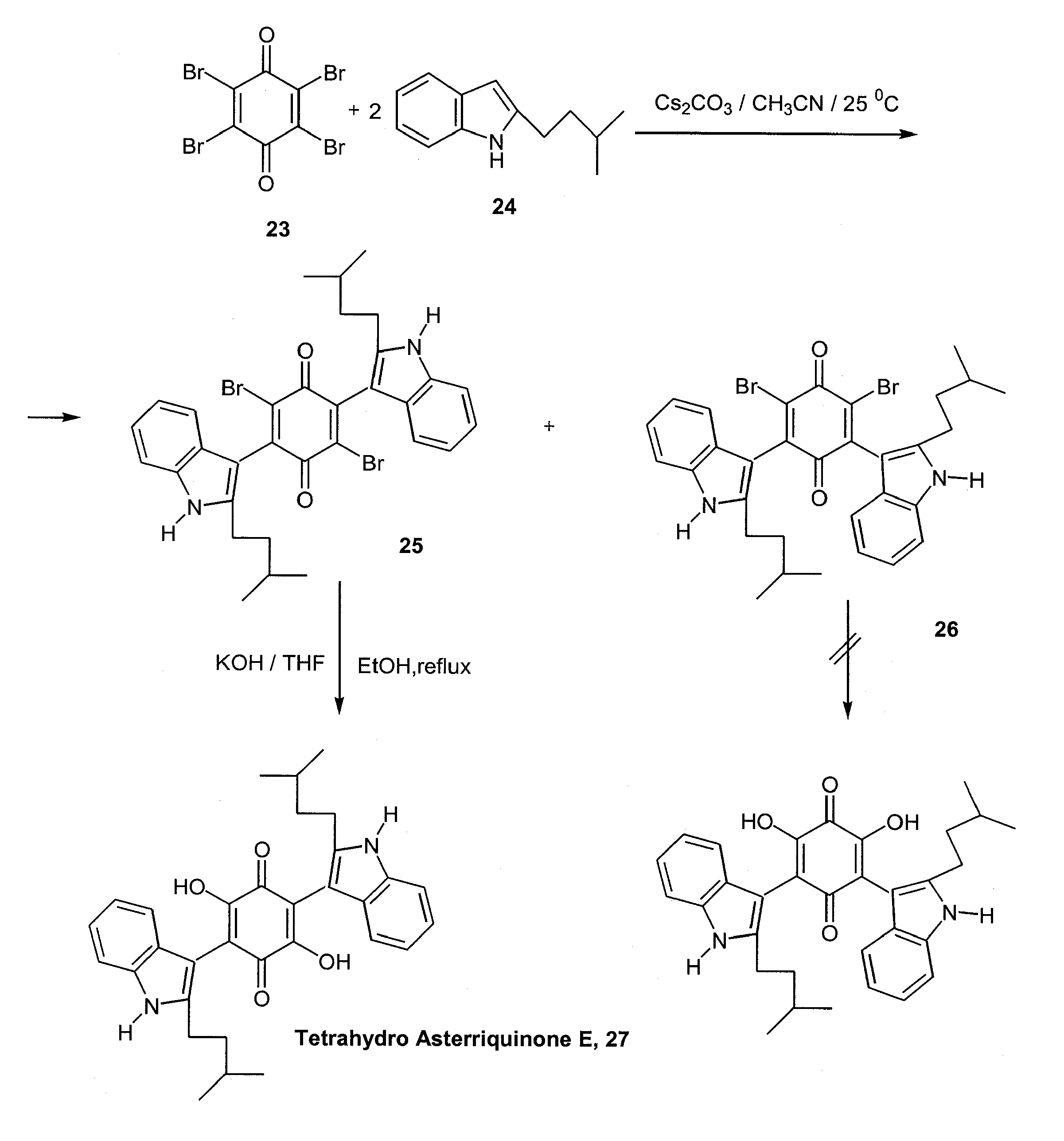
Scheme 13.
Scheme 14.
Scheme 15.
Scheme 16.
Scheme 17.
Scheme 18.
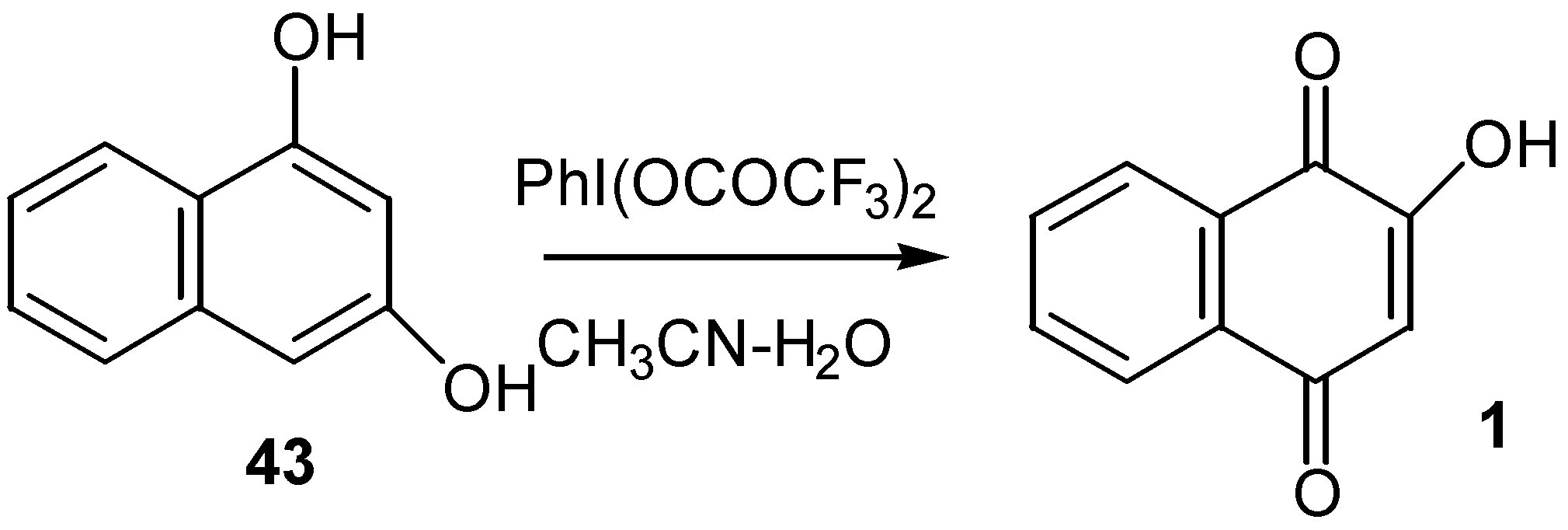
Scheme 19.

Scheme 20.
Scheme 21.
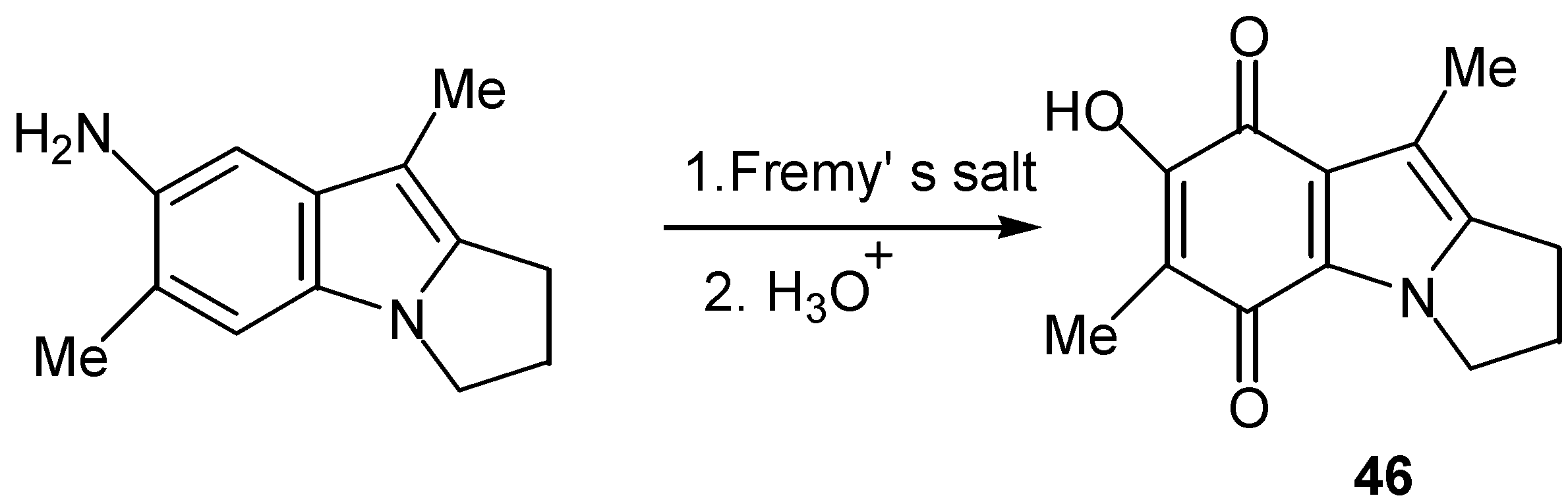
Scheme 22.
Scheme 23.
Scheme 24.
Scheme 25.
Scheme 26.
Scheme 27.
Scheme 28.
Scheme 29.
Scheme 30.
Scheme 31.
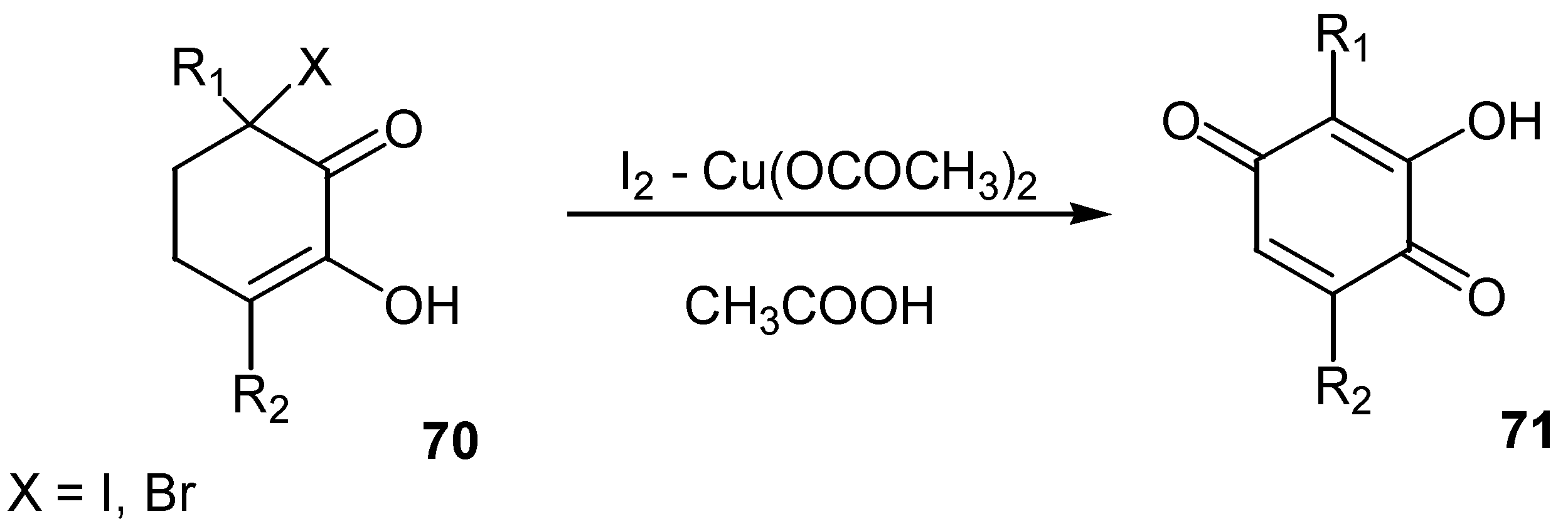
Scheme 32.

Scheme 33.

Scheme 34.
Scheme 35.

Scheme 36.
Scheme 37.
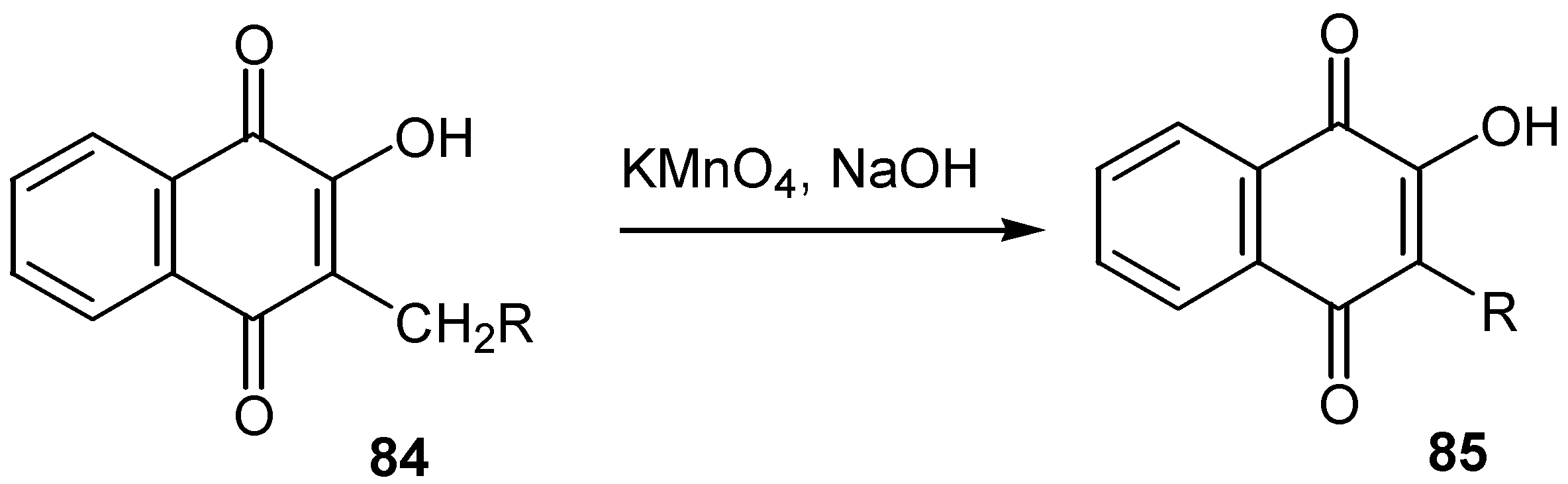
Scheme 38.

Scheme 39.
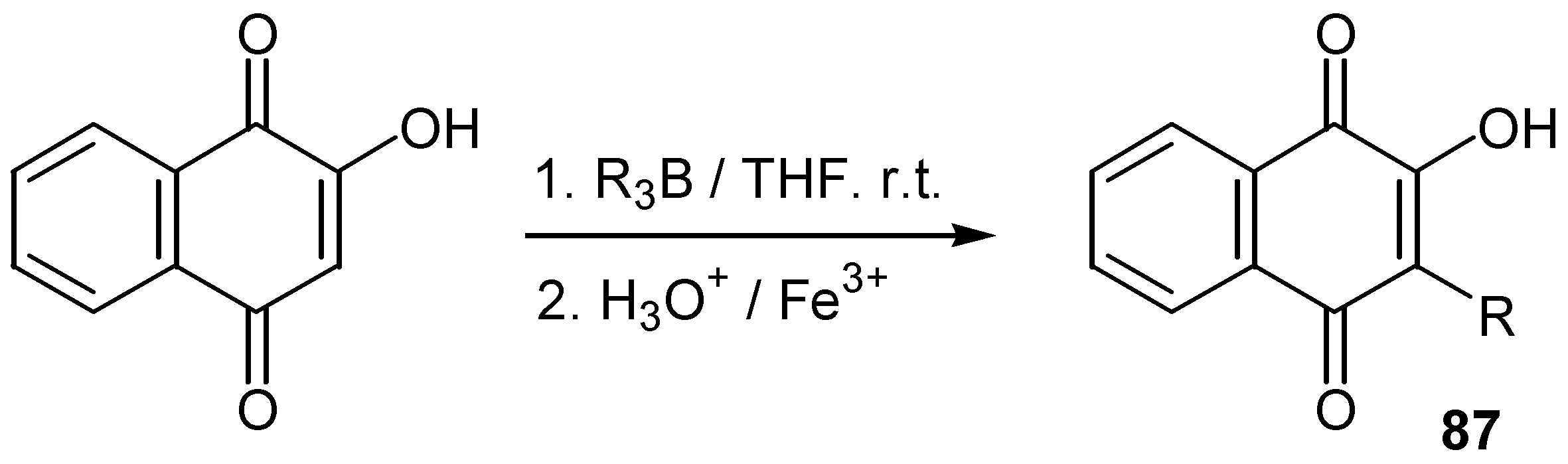
Scheme 40.
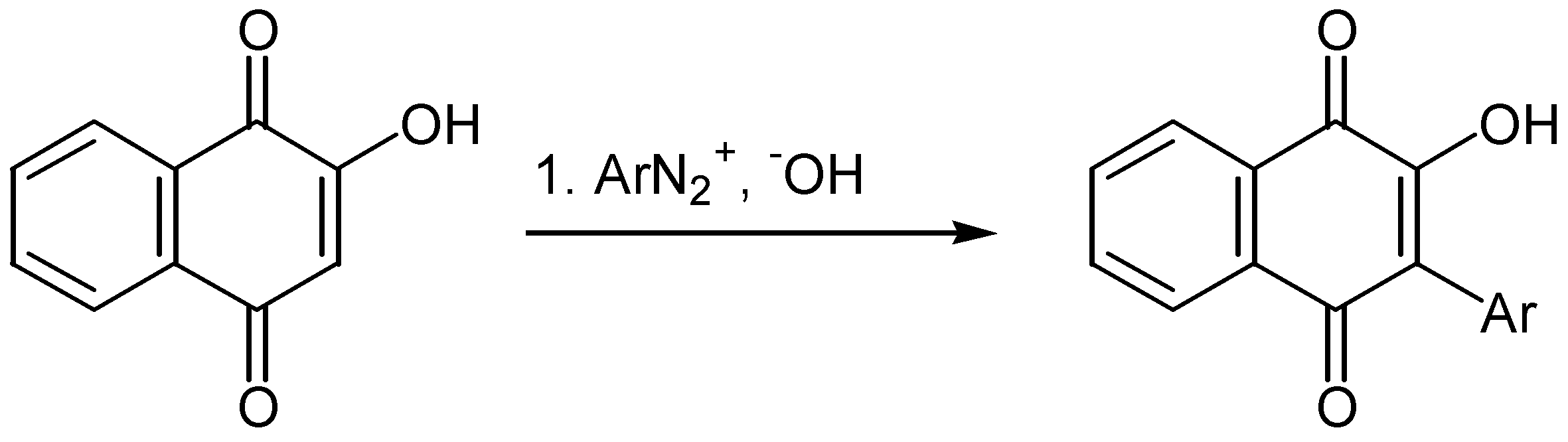
Scheme 41.
Scheme 42.
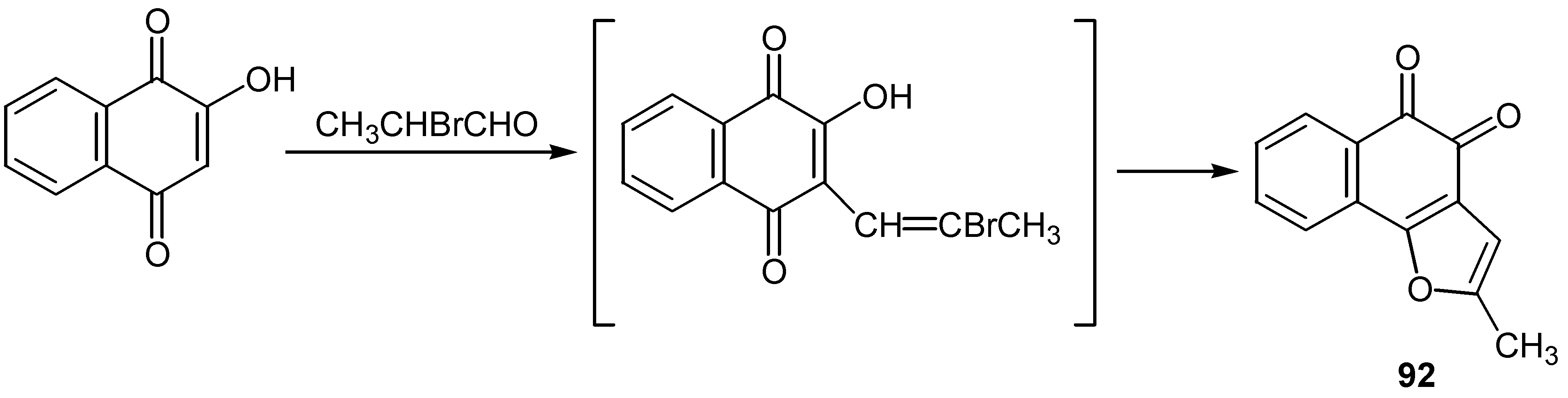
Scheme 43.
Scheme 44.

Scheme 45.
Scheme 46.

Scheme 47.
Scheme 48.
Scheme 49.
Scheme 50.
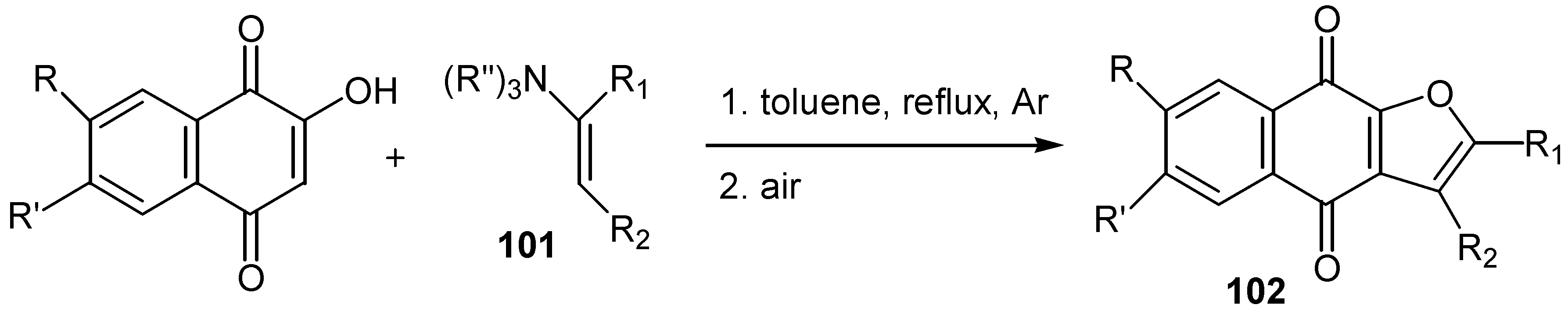
Scheme 51.
Scheme 52.
Scheme 53.
Scheme 54.
Scheme 55.

Scheme 56.
Scheme 57.
Scheme 58.

Scheme 59.
Scheme 60.
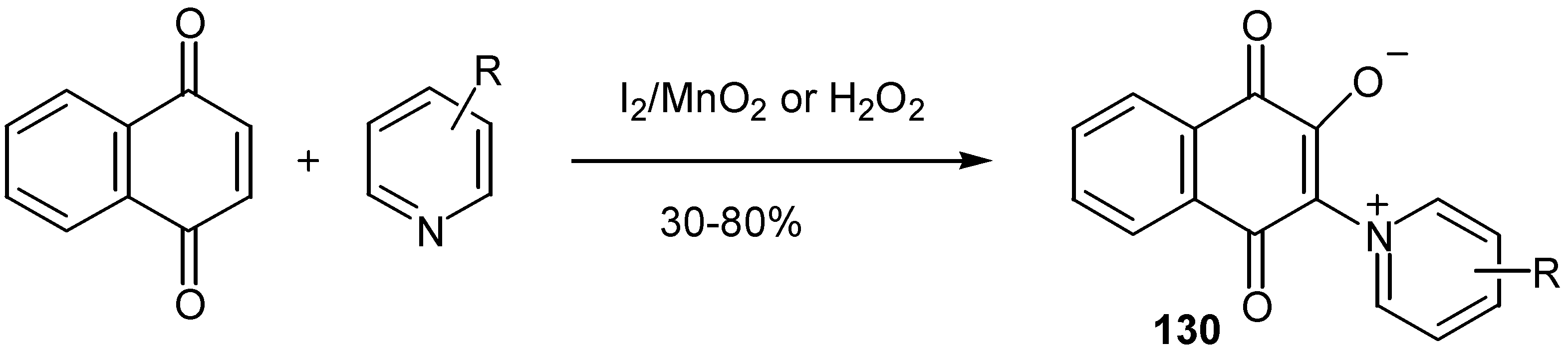
Scheme 61.
Scheme 62.
Scheme 63.
Scheme 64.
Scheme 65.
Scheme 66.
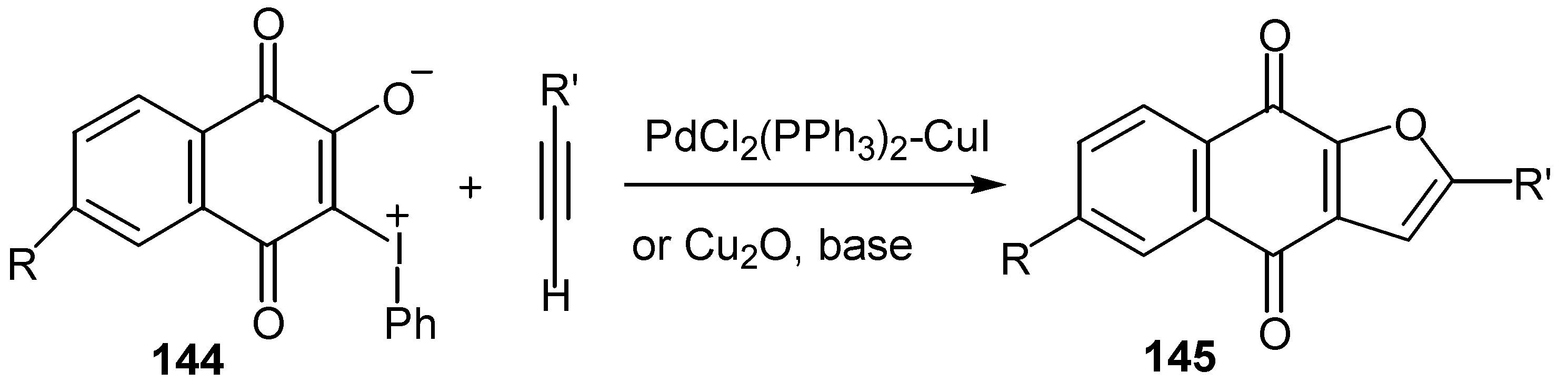
Scheme 67.

Scheme 68.
Scheme 69.

Scheme 70.
Scheme 71.
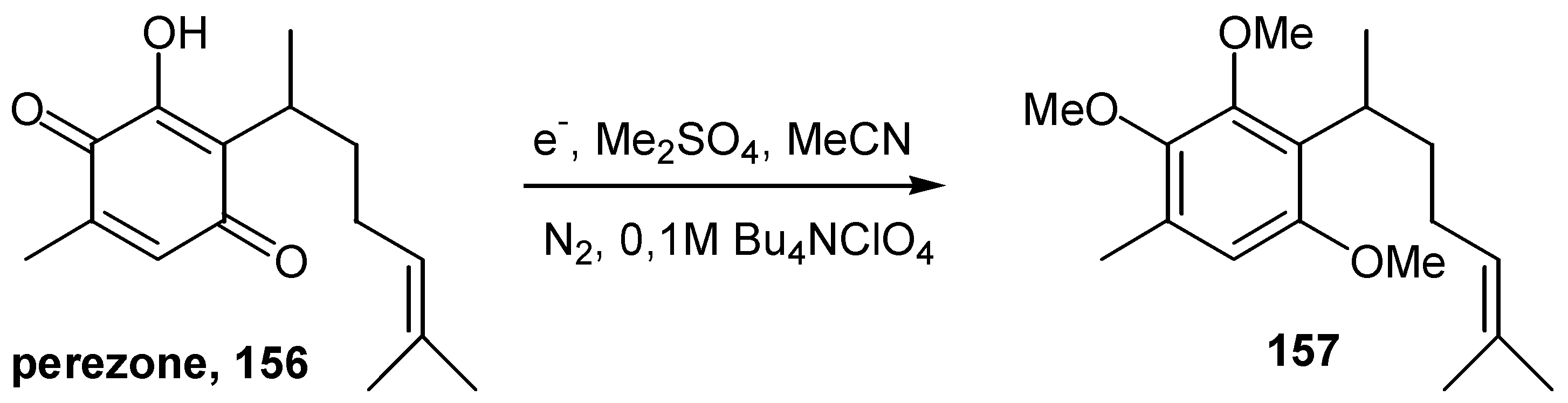
Scheme 72.
Scheme 73.
Scheme 74.

Scheme 75.
Scheme 76.

© 2000 by MDPI (http://www.mdpi.org).
Share and Cite
MDPI and ACS Style
Spyroudis, S. Hydroxyquinones: Synthesis and Reactivity. Molecules 2000, 5, 1291-1330. https://doi.org/10.3390/51201291
AMA Style
Spyroudis S. Hydroxyquinones: Synthesis and Reactivity. Molecules. 2000; 5(12):1291-1330. https://doi.org/10.3390/51201291
Chicago/Turabian StyleSpyroudis, Spyros. 2000. "Hydroxyquinones: Synthesis and Reactivity" Molecules 5, no. 12: 1291-1330. https://doi.org/10.3390/51201291