Study of Stereoselectivity in Organometallic Additions to 1,2-O-Isopropylidene-O-R-α-D-xylopentodialdo-1,4-furanose

Department of Organic Chemistry, Faculty of Chemical Technology, Slovak University of Technology, Sk-812 37 Bratislava, Slovakia
*
Author to whom correspondence should be addressed.
Molecules 2000, 5(12), 1386-1398; https://doi.org/10.3390/51201386
Submission received: 2 August 2000
/
Accepted: 8 December 2000
/
Published: 21 December 2000
Abstract
:Diastereofacial selectivity of the addition of organometallic reagents to 1,2-O-isopropylidene-O-R-α-D-xylopentodialdo-1,4-furanoses (6) was studied.
Introduction
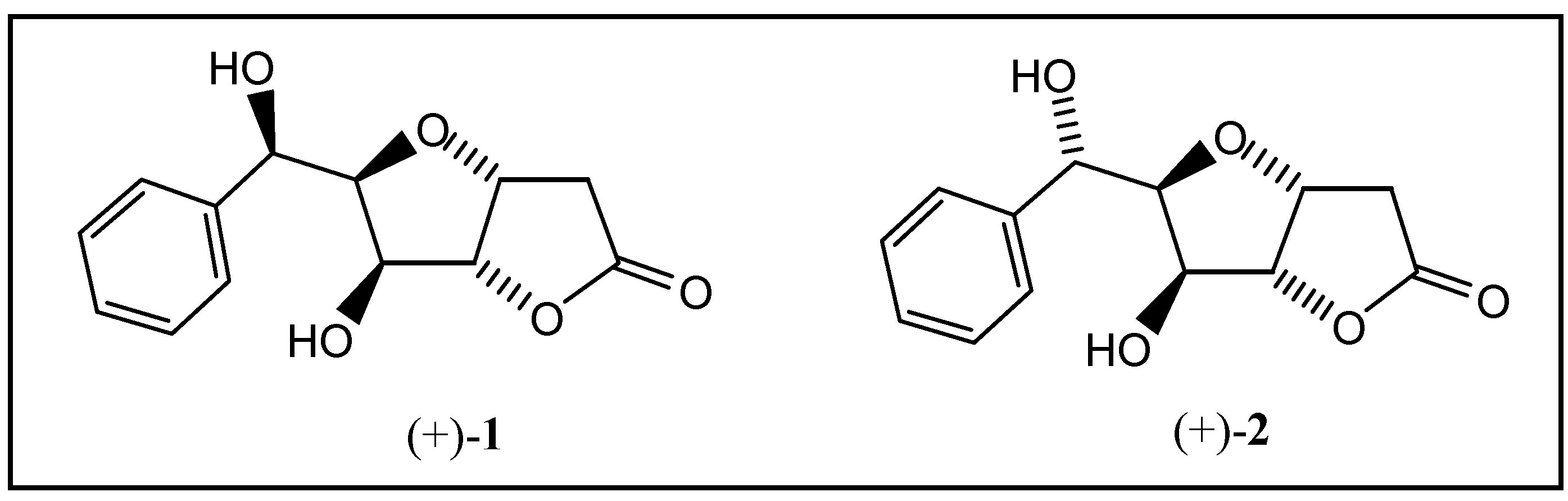
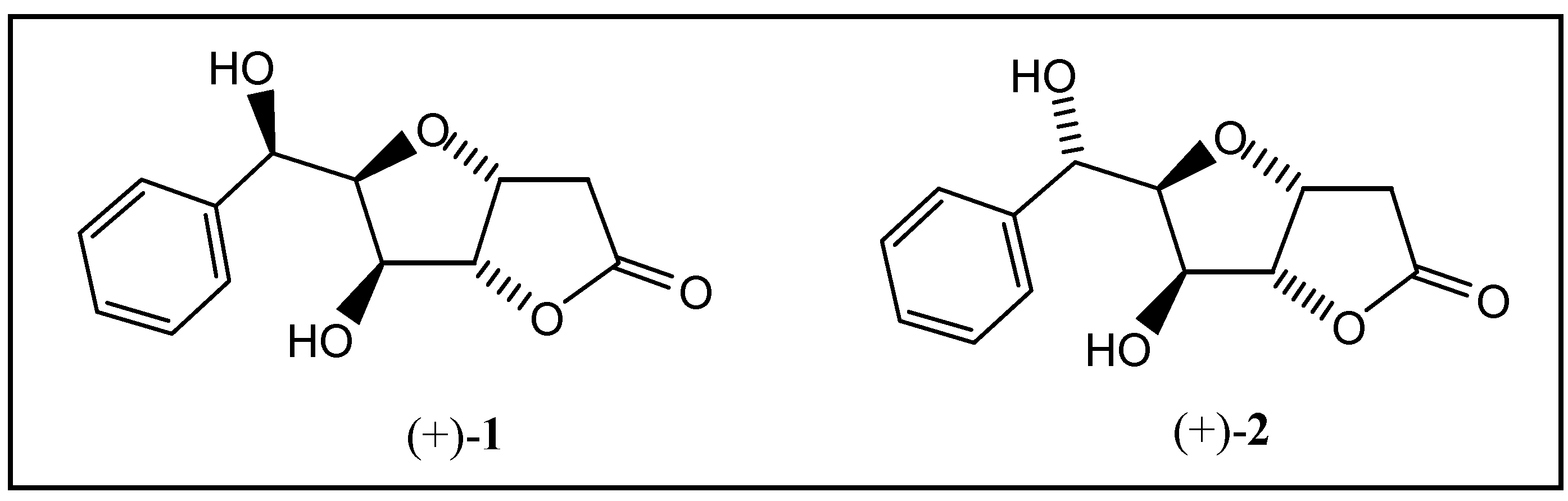
Goniofufurone (+)-1 and 7-epi-goniofufurone (+)-2 were recently isolated from the stem bark of Thai Goniothalamus giganteus Hook f., Thomas (Annonaceae) and shown to exhibit cytotoxic activity in tests with several human tumor cell lines [1,2]. We have developed a total synthesis of both diastereomers (+)-1 and (+)-2 starting from D-glucose [3]. The key steps are the phenyl magnesium bromide addition to 1,2-O-isopropylidene-α-D-xylopentodialdo-1,4-furanose and palladium(II)- catalyzed oxycarbonylation of the corresponding 1-phenyl-5-hexene-1,2,3,4-tetrols. Whilst the Pd(II)- catalyzed formal O-cyclization and carbon monoxide addition yielded the required bicyclic skeletons with high regio-preference and excellent threo-selectivity (concerning the newly formed stereocentre at C-3) [4,5], Grignard addition of phenyl magnesium bromide to 6a in tetrahydrofuran led to two diastereomeric alcohols 7a and 8a in a 1:3 ratio favouring formation of the L-ido diastereomer 8a, possessing the correct stereochemistry for less cytotoxic 7-epi-goniofufurone (+)-2 (Scheme 1).
Results and Discussion
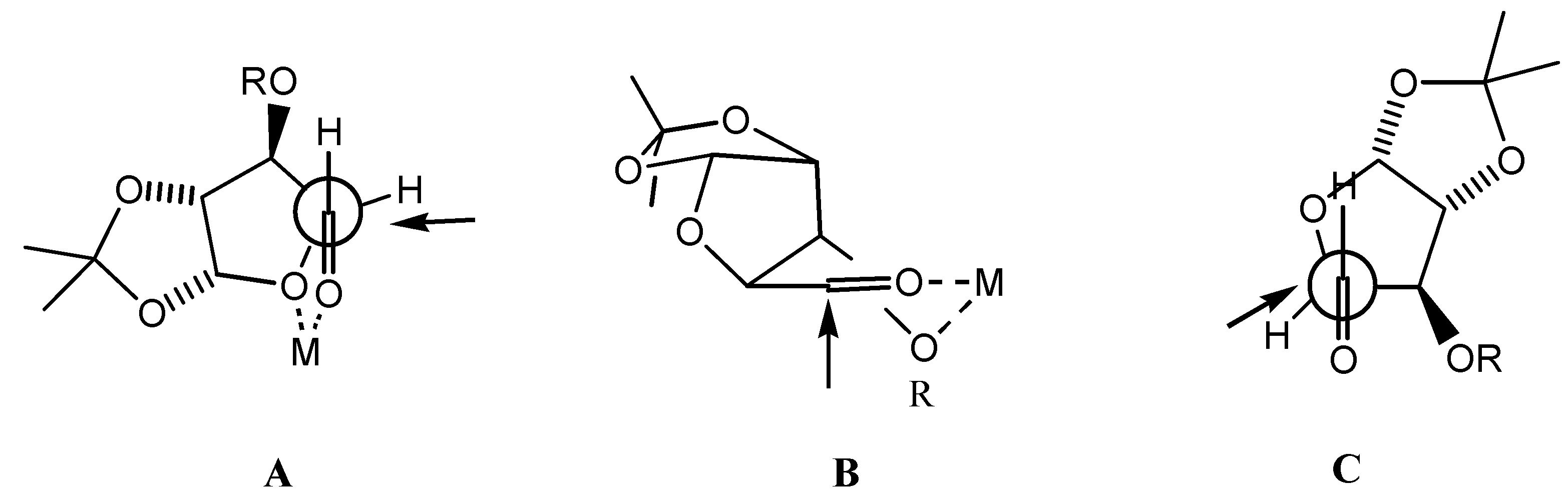
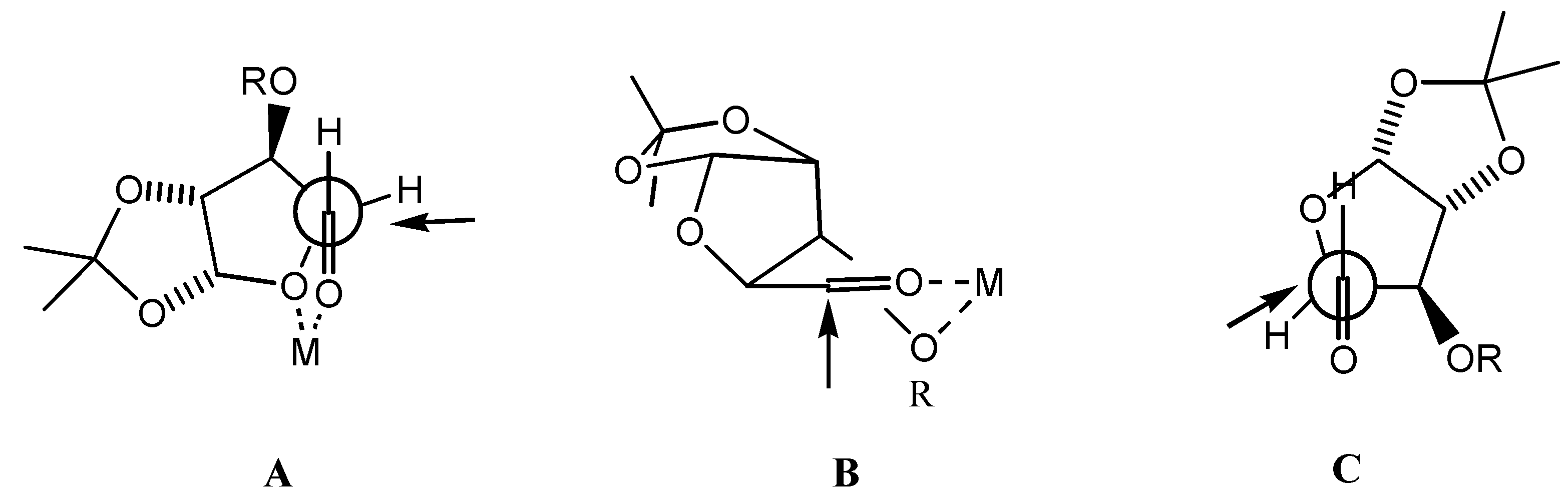
In connection with the above-mentioned synthesis, we were interested in obtaining a more reliable access to alcohol 7 while at the same time improving the ratio of this component to 8, therefore we decided to investigate the diastereoselectivity of additions of organometallic reagents to aldehydes 6 in a more detail. The design of addition of C-nucleophiles to aldehydes 6 could be based on models of either chelation-control: 1,2- (A) versus 1,3- (B) asymmetric induction or non-chelation-control (Felkin-Anh model C), leading to alcohols 8 (L-ido) (model A) or 7 (D-gluco) (models B, C) (Scheme 2).
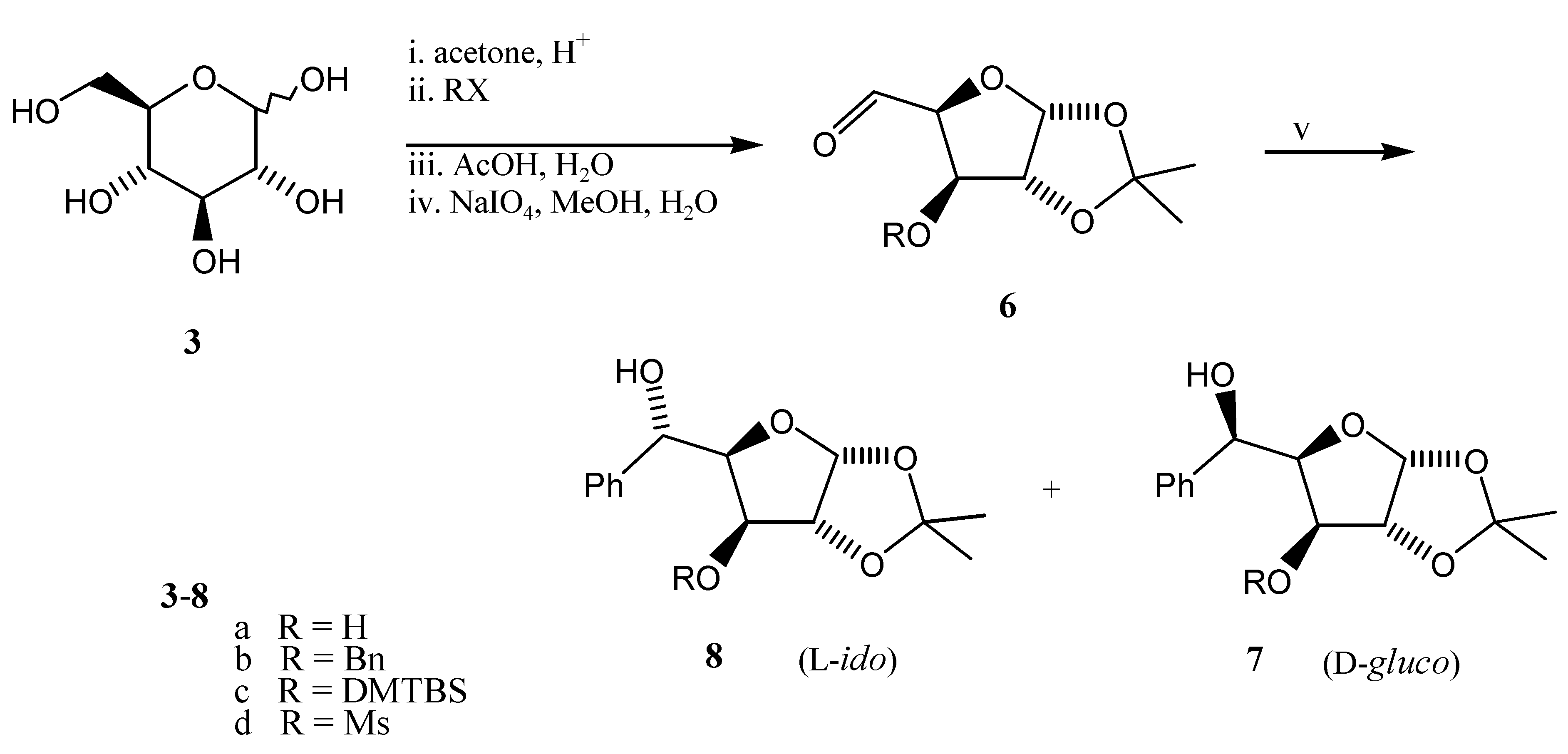
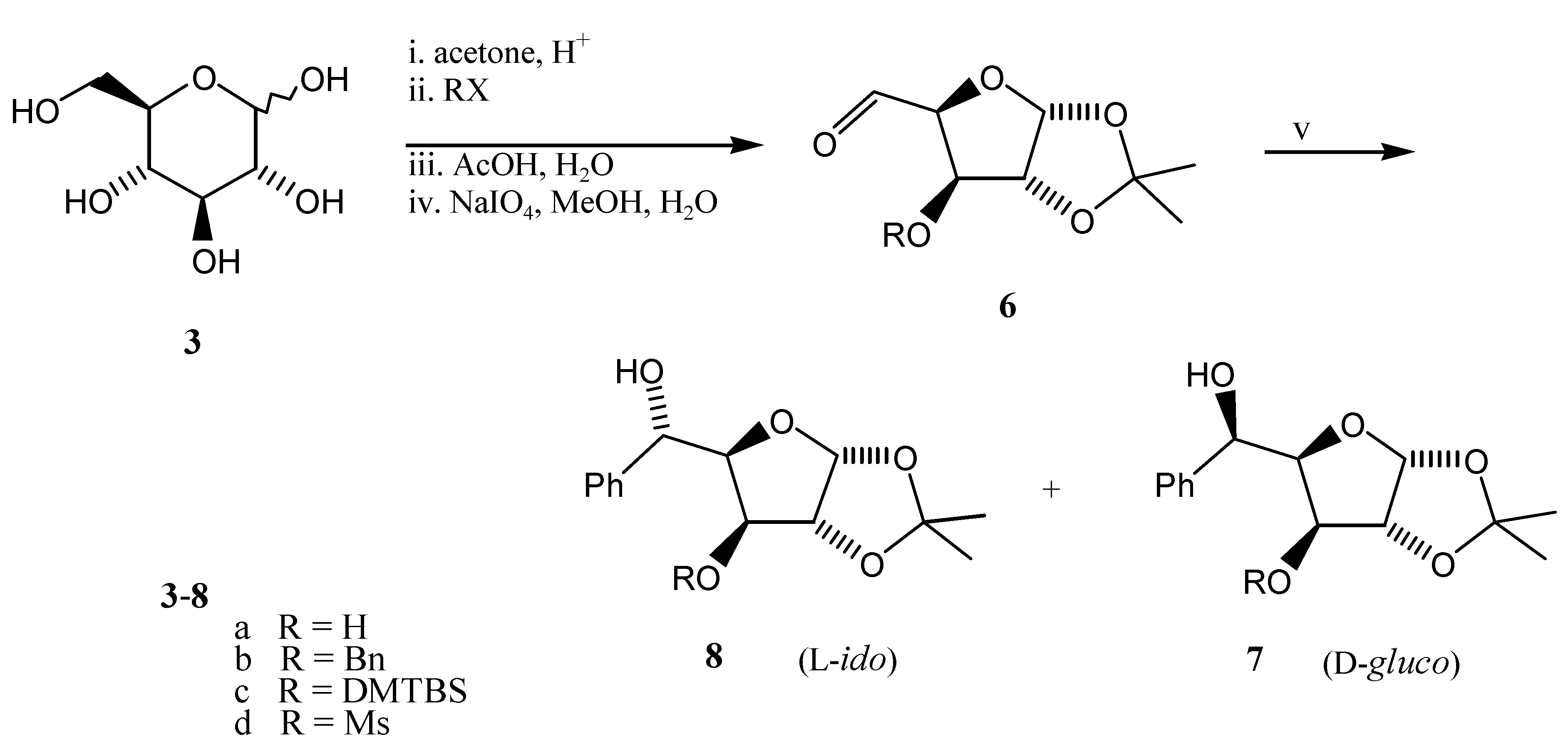
We have prepared 3-O-R-substituted aldehydes 6a-d from D-glucose (Scheme 3) with diverse protecting groups (R = H [6,7], Bn [6,8], TBDMS, Ms) in order to examine the effects of changes in the nature of the alkoxy group on the selectivity of the reaction. Treatment of aldehydes 6 with phenyl magnesium bromide in THF (see Table 1, Entry 1) or diethylether (Entries 2-4) gave in all cases the L-ido diastereomer 8 as a major product as a result of 1,2-chelation-control. The highest degree of selectivity found for 6b is in accordance with the literature [6,9,10,11,12], whilst aldehydes 6c and 6d with silyl (known for its non-chelation nature) and mesyl group (with opposite effect) gave about equal amounts of both diastereomers 7 and 8.
Generally, addition of polyethers to the reaction mixture to suppress chelation usually causes a reversal in the stereoselectivity, yielding a Cram,s product [13]. Addition of 18-C-6 and. dibenzo-18-C-6 crown-ether to the reaction of aldehyde 6b with phenylmagnesium bromide inhibits chelation leading to a decrease of selectivity from 14:1 to 1:1.2 (comparison of entries 5 and 6).
The reversal of diastereofacial selectivity was also achieved by use of phenylceriumdichloride [14,15] (Entry 7), indicating the probability that model B was operating in this case.
Turning from chelation- to non-chelation-control, we reacted 6b with phenyltitanium-tris-O-isopropoxide [16,17], which is known to have only weakly Lewis acid properties [18,19]. Obviously, phenyltitanium-tris-O-isopropoxide is incapable of effective chelation, so that carbonyl group in 6 is free to rotate. The fact that diastereoselectivity is nevertheless observed may be related to Anh’s model of 1,2-asymmetric induction in addition reactions of chiral aldehydes having electronegative substituents at the α-position. Accordingly, the most reactive conformation is the one having the alkoxy group positioned in such a way that the σC-OR orbital overlaps with the πC=O orbital, providing a low laying LUMO. Antiperiplanar attack at an angle larger than 90° (Burgi-Dunitz trajectory) according to model C results in the Felkin-Anh-product.
Analogously to methyltitanium-tris-O-isopropoxide addition to aldehyde 6b [20] we have carried out the reaction of 6b with phenyltitanium-tris-O-isopropoxide. Indeed, the D-gluco diastereomer 7b was preferentially formed (Entry 8).
The prepared compounds were identified by elemental analyses, optical rotations, 1H-NMR, 13C-NMR, and IR spectra (see Experimental). The absolute configuration at the newly formed stereocentre, and hence the configuration of alcohols 7b and 8b, was established by comparison of 1H-, 13C-NMR data, and specific rotations with those described in the literature [6,9,10,11,12]. The structures of 7a/ 8a were established by total synthesis of (+)-1, (+)-2 [3]. Assignment of the absolute configuration at C-5 in 7c,d/ 8c,d was not deemed of interest in view of low degree of selectivity observed.
Conclusions
Experimental
General
Solvents and reagents were purified and dried according to standard procedures. TLC analyses were carried out with Si60F254-coated aluminium sheets (E.Merck) using ethylacetate/ i-hexane mixtures; detection by UV at 254 nm, phosphomolybdic acid (10% in ethanol) or sulfuric acid (40% in water). Silica gel 32-63 μm (Woelm) was used for flash chromatography, eluents as above. Melting points were determined on a Kofler hot block and are uncorrected. The optical rotations were measured on a POLAR L-μP (IBZ Messtechnik) polarimeter at 589 nm. IR spectra were recorded on a PU 9800 FTIR spectrometer (Philips Analytical) in film or KBr discs (0.5 mg of sample and 300 mg of KBr). 1H- NMR and 13C NMR spectra were obtained on a Varian model VXR 300 spectrometer (at 300.3 MHz and 75.12 MHz, respectively). In the NMR experiments deuterochloroform solutions with tetramethylsilane as internal standard were measured; evaluation of 1H-NMR spectra was according to 1st order interpretation; and multiplicity of 13C-NMR signals was deduced from broad-decoupled or DEPT spectra. The ratios of diastereomers 7 and 8 were established by comparison of the integrals of H-1 signals in 1H NMR experiments performed on crude reaction mixtures.
3-O-[(tButyl)-dimethylsilyl]-1,2:5,6-di-O-isopropylidene-α-D-glucofuranose 4c
To a stirred solution of bisacetonide glucose (4a, 4g, 15 mmol) and triethylamine (2.4 mL,17 mmol, 1.1 equiv) in dry dimethylformamide (20 mL), kept at 0-5 °C, a solution of (tbutyl)- dimethylsilylchloride (2.562 g, 17 mmol, 1.1 equiv) in dry dimethylformamide (30 mL) was added dropwise over 30 min. The mixture was stirred at room temperature for another 30 h, then poured on ice/ water (100 mL) and extracted with chloroform (3 x 50 mL). The organic solutes were combined, dried (Na2SO4) and concentrated (rotary evaporator). The residue was treated with isohexane and unsoluble starting 4a (1 g) filtered off. The crude product after solvent removal was purified by flash chromatography (silica gel 80 g, eluent ethylacetate - isohexane 1:1). Yield 4.15 g (74%), Rf=0.68 (AcOEt - i-hexane 1:1), =-17.2 (c=0.43, CHCl3).
Spectral Data
1H-NMR (CDCl3) δ: 5.80 (d, 1H, H-1, J1,2=3.5 Hz), 4.28 (d, 1H, H-2, J1,2=3.5 Hz), 4.16 (m, 2H, H-3, H-5), 4.04 (dd, 1H, H-4, J3,4=6.1 Hz, J4,5=8.4 Hz), 3.96 (dd, 1H, H-6, B of ABX, JA,B=8.3 Hz, JB,X=2.7 Hz), 3.88 (dd, 1H, H-6, A of ABX, JA,B=8.3 Hz, JA,X=5.9 Hz), 0.85 (s, 9H, C(CH3)3), 0.07, 0.08 (all s, 3H, Si(CH3)2).
13C-NMR ( CDCl3) δ: 111.7 (s, C(CH3)2), 108.8 (s, C(CH3)2), 105.2 (d, C-1), 85.6, 82.2, 75.4, 72.1 (all d, C-2, C-3, C-4, C-5), 67.7 (t, C-6), 29.9, 26.7, 26.3, 25.2 (all q, C(CH3)2), 25.6 (q, SiC(CH3)3), 18.0 (s, SiC(CH3)3), -5.3, -5.1 (all q, Si(CH3)2).
IR (thin film) cm-1: 2988 w, 2955 s, 2934 s, 2893 w, 2859 w, 1381 m, 1372 s, 1255 s, 1217 s, 1132 s, 1078 s, 1022 s, 855 s, 779 m.
For C18H34O6Si (Mr = 374.55) Calcd.: 57.72 % C, 9.15 % H; Found: 57.51 % C, 9.18 % H.
3-O-[(tButyl)-dimethylsilyl]-1,2-O-isopropylidene-α-D-glucofuranose 5c
The glucofuranose bisacetonide (4c, 2.9 g, 7.7 mmol), dissolved in aq. acetic acid (75%, 30 mL), was stirred at room temperature for 48 h (TLC-monitoring). Removal of solvents in vacuo (20 mbar) leaves a yellow oil, which was dried in desiccator (NaOH, 20 mbar) for 2 days and purified by chromatography on silica gel 100g (eluent AcOEt - i-hexane 1:1). The furanose monoacetonide 5c was obtained as a colourless oil; yield 1.9 g (76%), Rf =0.3 (AcOEt - i-hexane 1:1), =-24.3 (c= 0.77, CHCl3).
Spectral Data
1H-NMR (CDCl3) δ: 5.82 (d, 1H, H-1, J1,2=3.6 Hz), 4.34 (d, 1H, H-2, J1,2=3.7 Hz), 4.29 (d, 1H, H-3, J3,4=2.7 Hz), 4.03 (dd, 1H, H-4, J3,4 =2.7 Hz, J4,5=8.2 Hz), 3.87 (ddd, 1H, H-5, dX of ABX, JA,X=5.3 Hz, JB,X=3.2 Hz, J4,5=8.2 Hz), 3.81 (dd, 1H, H-6, B of ABX, JA,B=11.5 Hz, JB,X=3.2 Hz), 3.73 (dd, 1H, H-6, A of ABX, JA,B=11.5 Hz, JA,X=5.3 Hz), 1.27, 1.46 (all s, 3H, C(CH3)3), 0.88 (s, 9H, SiC(CH3)3), 0.14, 0.12 (all s, 3H, Si(CH3)2).
13C-NMR (CDCl3) δ: 118.8 (s, C(CH3)2), 104.9 (d, C-1), 85.3, 80.8, 75.6, 64.4 (all d, C-2, C-3, C-4, C-5), 68.6 (t, C-6), 26.2, 26.7 (all q, C(CH3)2), 25.6 (q, SiC(CH3)3), 18.0 (s, SiC(CH3)3), -5.1, -4.9 (all q, Si(CH3)2).
IR (thin film) cm-1: 3436 br s ( OH), 2955 s, 2932 s, 2888 s, 2859 s, 1383 m, 1375 m, 1217 s, 1134 s, 1082 s, 1018 s, 835 s, 779 m.
For C15H30O6Si (Mr = 334.49) Calcd.: 53.86 % C, 9.04 % H; Found: 53.97 % C, 8.95 % H.
1,2:5,6-di-O-Isopropylidene-3-O-mesyl-α-D-glucofuranose 4d
Prepared from bisacetone D-glucose (4a, 4 g, 15 mmol), mesylchloride (1.40 mL, 18 mmol, 1.2 equiv.) and pyridine (20 mL) under Ar atmosphere according to lit. [21]. Yield 3.771 g (74%), m.p.= 78 – 80 °C, Rf = 0.54 (AcOEt - i-hexane 1:1), = -47.5 (c=0.53, CHCl3), which was used without further purification {Lit. [21]: m.p.=80 – 82 °C, =- 49 -1 (c=1.0, CHCl3)}.
Spectral Data
1H-NMR (CDCl3) δ: 5.88 (d, 1H, H-1, J1,2=3.6 Hz), 4.91 (d, 1H, H-3, J3,4=2.8 Hz), 4.72 (d, 1H, H-2, J1,2=3.6 Hz), 4.11 (m, 3H, H-4, H-5, H-6), 3.94 (dd, 1H, H-6, A of ABX, JA,B=8.9 Hz, JA,X=4.3 Hz), 3.03 (s, 3H, SO2CH3), 1.44, 1.36, 1.26, 1.25 (all s, 3H, CH3).
13C-NMR (CDCl3) δ: 112.7, 109.6 (all s, C(CH3)2), 105.2 (d, C-1), 83.7, 82.7, 79.8, 72.1 (all d, C-2, C-3, C-4, C-5), 67.6 (t, C-6), 38.0 (q, SO2CH3), 26.9, 26.6, 26.2, 25.2 (all s, CH3).
For C13H22O8S (Mr=338.38).
1,2-O-Isopropylidene-3-O-mesyl-α-D-glucofuranose 5d
Following the procedure described for preparation of 5c; the bisacetonide (4d, 3.5 g,10.3 mmol) was stirred in aq. acetic acid (75%, 30 mL) at room temperature for 24 h (TLC - monitoring). The monoacetonide 5d was obtained as a yellow, but analytically pure oil; yield 2.93 g (95%), Rf=0.3 (AcOEt - i-hexane 1:1), =-21 (c=0.42, CHCl3). {Lit. [22]: [α]D=-20.6 (c=1.0, CHCl3)}.
Spectral Data
1H-NMR (CDCl3) δ: 5.93 (d, 1H, H-1, J1,2=3.7 Hz), 5.10 (d, 1H, H-3, J3,4=2.5 Hz), 4.76 (d, 1H, H-2, J1,2=3.7 Hz), 4.22 (dd, 1H, H-4, J3,4=2.5 Hz), 3.86 (bd, 2H, H-6, AB of ABX, JA,B=9.4 Hz), 3.73 (m, 1H, H-5, X of ABX), 3.14 (s, 3H, SO2CH3), 1.49, 1.39 (all s, 3H, CH3).
13C-NMR (CDCl3) δ: 112.7 (s, C(CH3)2), 105.0 (d, C-1), 83.3, 82.0, 78.4, 68.2 (all d, C-2, C-3, C-4, C-5), 63.9 (t, C-6), 38.1 (q, SO2CH3), 26.4, 26.1 (all s, CH3).
For C10H18O8S (Mr=298.31).
1,2-O-Isopropylidene-3-O-R-α-D-xylopentodialdo-1,4-furanose 6a-d
Prepared from monoacetone glucoses 4a-d by modified procedures of Inch [6] and Lichtenthaler [7]. To a cooled solution (0 °C) of 4 in water/methanol (1:2) was added in one portion NaIO4 and stirring was continued at 0-20 °C for 2 h (TLC - monitoring). The solid precipitate ( NaIO3) was filtered off and the solvent was removed on a rotavapory (20 mbar, 25 °C), the residue was partioned between chloroform (3 x 50 mL) and water (15 mL), dried ( Na2SO4), and concentrated at reduced pressure to a colourless oil, which was additionally dried in a desiccator (P2O5, 0.01 mbar) for 2 h. The crude products of diol - cleavage were used in the next reactions without another purification.
1,2-O-Isopropylidene-α-D-xylopentodialdo-1,4-furanose 4a
Prepared from 5a (1 g, 4.54 mmol) and NaIO4 (1.1 g, 5.14 mmol) in water/methanol (1:2, 20 mL). Yield 580 mg (68%), Rf=0.6 (AcOEt).
Spectral Data
1H-NMR (CDCl3) δ: 7.40 (bs, 1H, H-5), 5.98 (bs, 1H, H-1), 3.97-4.55 (m, 3H, H-2, H-3, H-4), 1.47, 1.31 (all s, 3H, CH3).
For C8H12O5 (Mr=188.18).
3-O-Benzyl-1,2-O-isopropylidene-α-D-xylopentodialdo-1,4-furanose 4b
Prepared from 5b (1.0 g, 3 mmol), NaIO4 (1.28 g, 6 mmol, 2 equiv) in water/methanol (1:2, 15 mL). Yield 803 mg (96%), Rf=0.6 (AcOEt- i-hexane 1:1).
Spectral Data
1H-NMR (CDCl3) δ: 9.68 (s, 1H, H-5), 7.26-7.35 (m, 5H, C6H5), 6.12 (s, 1H, H-1, J1,2=3.5 Hz), 4.63 (m, 4H, H-2, H-4, CH2), 4.32 (d, 1H, H-3, J3,4=3.1 Hz), 1.46, 1.40 (all s, 3H, CH3).
For C15H18O5 (Mr=278.31).
3-O-[(tButyl)-dimethylsilyl]-1,2-O-Isopropylidene-α-D-xylopentodialdo-1,4-furanose 6c
From 5c (1.69 g, 5.0 mmol), NaIO4 (2.13 g, 10 mmol, 2 equiv) in water/methanol (1:2, 30 mL). Yield 1.305 g (86%), Rf =0.68 (AcOEt).
For C14H26O5Si (Mr=302.44).
1,2-O-Isopropylidene-3-O-mesyl-α-D-xylopentodialdo-1,4-furanose 6d
From 5d (3.28 g, 11 mmol), NaIO4 (4.705 g, 22 mmol, 2 equiv.) in water/methanol (1:2, 30 mL). Yield 2.41 g (82%), Rf=0.27 (AcOEt - i-hexane 1:1).
For C9H14O7S (Mr=266.27).
1,2-O-Isopropylidene-3-O-R-5-C-phenyl-β-L-ido- 8a-d and α-D-glucopentofuranose 7a-d
Prepared from aldehydes 6a-d according to lit. [4,6]. The crude aldehydes 6a-d were dissolved in dry ether and added dropwise to a solution of phenylmagnesium bromide in ether, prepared from bromobenzene and magnesium at –10 °C during 2 h. The mixtures were stirred at 0 °C for 4 h, and at r.t. for 24 h, then quenched with cold, saturated aqueous ammonium chloride and extracted with ether. After drying (Na2SO4) and solvent removal a yellow oils were obtained, whose were purified by flash chromatography on silica gel.
3-O-Benzyl-1,2-O-isopropylidene-5-C-phenyl-β-L-ido- 8b and α-D-glucopentofuranose 7b
Prepared from aldehyde 6b (1.113 g, 4 mmol), bromobenzene (1.05 mL, 10 mmol, 2.5 equiv) and magnesium (0.243 g, 10 mmol) in ether (35 mL). Yield 1.285 g (90%) of the mixture of diastereomers 8b and 7b (14:1) was purified by chromatography (silica gel, 40 g, AcOEt - i-hexane 1:2); fraction 1 compound 7b (D-gluco), 90 mg (6.3 %), yellow oil, Rf=0.35 (AcOEt - i-hexane 1:2), =-74 (c=1.0, CHCl3). {Lit. [12]: =-77 (c=4.02, CHCl3), lit. [9]: =-93 (CHCl3), lit. [6]: =-76 (c=1.0, CHCl3)}; fraction 2 compound 7b (L-ido); 580 mg (41 %), colourless oil, Rf=0.26 (AcOEt - i-hexane 1:2), =-32.1 (c=0.76, CHCl3), {lit. [12]: =-33.5 (c =2.0, CHCl3)}. In addition, 330 mg (23 %) of an intermediate fraction containig both 7b and 8b was collected.
Alcohol 8b (L-ido)
Spectral Data
1H-NMR (CDCl3) δ: 7.31- 7.48 (m, 10H, C6H5), 6.03 (d, 1H, J1,2=3.8 Hz, H-1), 5.07 (d, 1H, J4,5=7.6 Hz, H-5), 4.61 (d, 1H, J1,2=3.8 Hz, H-2), 4.56 (d, 1H, J=11.5 Hz, CH2), 4.34 (dd, 1H, J3,4=3.2 Hz, J4,5=7.6 Hz, H-4), 4.32 (d, 1H, J=11.7 Hz, CH2), 3.64 (d, 1H, J3,4=3.1Hz, H-3), 1.50 , 1.32 (all s, 3H, C(CH3)2).
13C-NMR (CDCl3) δ: 139.7, 137.0 (all s, i-C6H5), 128.5, 128.3, 128.1, 128.0, 127.7, 127.1 (all d, C6H5), 111.9 (s, C(CH3)2), 105.2 (d, C-1), 71.8 (t, CH2), 72.4, 82.2, 82.2, 84.5 (all d, C-2, C-3, C-4, C-5), 26.3, 26.8 (all q, C(CH3)2).
IR (thin film)/ cm-1: 3453 m (OH), 2979 m, 2932 m, 2892 m, 1497 m, 1455 s, 1375 s, 1217 s, 1076 s, 1022 s, 760 s, 700 s.
For C21 H24O5 (Mr =356.42) Calcd.: 70.77 %C, 6.97 %H; Found: 71.09 %C, 6.91 %H.
Alcohol 7b (D-gluco)
Spectral Data
1H NMR (CDCl3) δ: 7.34- 7.43 (m, 10H, C6H5), 6.12 (d, 1H, J1,2=3.8 Hz, H-1), 5.23 (d, 1H, J4,5=6.1 Hz, H-5), 4.72 (d, 1H, J=11.6 Hz, CH2), 4.70 (d, 1H, J1,2=3.9 Hz, H-2), 4.51 (d, 1H, J=11.4 Hz, CH2), 4.45 (dd, 1H, J3,4=3.3 Hz, J4,5=6.1 Hz, H-4), 4.10 (d, 1H, J3,4=3.2Hz, H-3), 1.55, 1.38 (all s, 3H, C(CH3)2).
13C-NMR (CDCl3) δ: 141.8, 137.6 (all s, i-C6H5), 129.9, 129.7, 128.9, 128.8, 127.7, 127.2 (all d, C6H5), 113.1 (s, C(CH3)2), 106.2 (d, C-1), 73.4 (t, CH2), 73.4, 82.8, 83.5, 83.9 (all d, C-2, C-3, C-4, C-5), 27.3, 27.8 (all q, C(CH3)2).
IR (thin film)/ cm-1: 3450 m (OH), 2980 m, 2930 m, 1497 m, 1455 s, 1456 s, 1376 s, 1220 s, 1070 s, 1024 s, 760 s, 700 s.
For C21 H24O5 (Mr=356.42) Calcd.: 70.77 %C, 6.97 %H; Found: 71.12 %C, 6.93 %H.
3-O-[(tButyl)-dimethylsilyl]-1,2-O-isopropylidene-5-C-phenyl-β-L-ido- 8c and α-D-glucopento- furanose 7c
Prepared from 6c (1.305 g, 4.3 mmol), bromobenzene (1.37 mL, 13 mmol, 3 equiv) and magnesium (0.316 g, 13 mmol) in ether (40 mL). Yield 1.382 g (84 %), yellow oil, Rf=0.26 (AcOEt - i-hexane 1:3). The product consists of a 2:1 mixture of L-ido/D-gluco diastereomers 8c/7c by 1H-NMR. The NMR data are given from this mixture and probable assignments may eventually be reversed.
Spectral Data for alcohol 8c (L-ido)
1H-NMR (CDCl3) δ: 7.26- 7.36 (m, 5H, C6H5), 6.01 (d, 1H, J1,2=3.6 Hz, H-1), 4.95 (d, 1H, J4,5=5.8 Hz, H-5),4.38 (m, 2H, J1,2=3.5 Hz, J3,4=3.2 Hz, H-2, H-4), 4.12 (d, 1H, J3,4=2.9 Hz, H-3), 1.48, 1.31 (all s, 3H, C(CH3)2), 0.94 (s, 9H, SiC(CH3)3), 0.10, -0.04 (all s, Si(CH3)2).
13C-NMR (CDCl3) δ: 139.8 (s, i-C6H5), 128.4, 128.1, 127.3 (all d, C6H5), 111.9 (s, C(CH3)2), 104.8 (d, C-1), 72.4, 76.9, 83.9, 85.7 (all d, C-2, C-3, C-4, C-5), 26.4, 26.9 (all q, C(CH3)2) 25.7 (q, C(CH3)3), 17.9 (s, C(CH3)3), -4.4, -5.2 (all q, Si(CH3)2).
Spectral Data for alcohol 7c (D-gluco)
1H-NMR (CDCl3) δ: 7.26- 7.38 (m, 5H, C6H5), 5.94 (d, 1H, J1,2=3.8 Hz, H-1), 4.98 (m, 1H, H-5), 4.35 (m, 3H, H-2, H-3, H-4), 1.46 , 1.30 (all s, 3H, C(CH3)2), 0.94 (s, 9H, SiC(CH3)3), 0.16, 0.10 (all s, Si(CH3)2).
13C-NMR (CDCl3) δ: 141.7 (s, i-C6H5), 128.4, 127.8, 126.5 (all d, C6H5), 111.6 (s, C(CH3)2), 105.0 (d, C-1), 72.1, 76.3, 83.0, 85.3 (all d, C-2, C-3, C-4, C-5), 26.4, 26.9 (all q, C(CH3)2) 25.7 (q, C(CH3)3), 17.9 (s, C(CH3)3), -4.6, -5.1 (all q, Si(CH3)2).
IR (thin film)/ cm-1: 3439 br m (OH), 2955 s, 2932 s, 1383 w, 1375 w, 1254 m, 1219 m, 1165 m, 1132 m, 1078 s, 844 s, 835 s, 777 m, 764 m.
For C20 H32O5Si (Mr=356.42) Calcd.: 63.12 %C, 8.48 %H; Found: 63.39 %C, 8.46 %H.
1,2-O-Isopropylidene-3-O-mesyl-5-C-phenyl-β-L-ido- 8d and α-D-gluco-pentofuranose 7d
From 6d (1.602 g, 6 mmol), bromobenzene (1.9 mL, 18 mmol, 3 equiv) and magnesium (437 mg, 18 mmol) in ether (45 mL). Yield 1.612 g (78 %) as a yellow oil, Rf=0.28 (AcOEt - i-hexane 1:1). The product consists of a 1.6:1 mixture of L-ido/D-gluco diastereomers 8d/7d. The NMR data are given from this mixture and probable assignments may eventually be reversed.
Spectral Data for alcohol 8d (L-ido)
13C-NMR (CDCl3) δ: 133.7 (s, i-C6H5), 129.0, 128.6, 127.3 (all d, C6H5), 112.7 (s, C(CH3)2), 104.6 (d, C-1), 72.2, 81.7, 82.7, 83.6 (all d, C-2, C-3, C-4, C-5), 26.6, 26.3 (all q, C(CH3)2) 37.9 (q, SO2CH3).
Spectral Data for alcohol 7d (D-gluco)
13C-NMR (CDCl3) δ: 138.4 (s, i-C6H5), 129.3, 128.9, 127.5 (all d, C6H5), 111.9 (s, C(CH3)2), 105.0 (d, C-1), 73.4, 75.3, 82.2, 85.1 (all s, C-2, C-3, C-4, C-5), 26.7, 26.1 (all q, C(CH3)2) 37.9 (q, SO2CH3).
IR (thin film)/ cm-1: 3486 br m (OH), 2990 m, 2936 m, 1363 s (SO), 1341 s, 1306 s, 1170 s (SO), 1163 w, 1152 s, 1088 s, 1024 s, 972 s, 957 s, 909 s, 845 s, 835 s.
For C15 H20O7S (Mr=344.39) Calcd.: 52.31 %C, 5.85 %H; Found: 52.21 %C, 5.94 %H.
3-O-Benzyl-1,2-O-isopropylidene-5-C-phenyl-α-D-gluco 7b and β-L-ido-pentofuranose 8b
To a freshly prepared solution of phenylmagnesium bromide [prepared from bromobenzene (0.8 mL, 7.6 mmol, 2.5 equiv) and magnesium (0.185 g, 7.6 mmol) in dry ether (15 mL) by standard procedure] a solution of 18-C-6 (2.009 g, 7.6 mmol, 2.5 equiv) and dibenzo 18-C-6 crown-ether (2.739 g, 7.6 mmol) in ether (10 mL) was added dropwise at room temperature. The mixture was additionally treated in ultrasound bath for 30 min. Then the aldehyde 6b (865 mg, 3 mmol) was added at –10 °C. The mixture was stirred at 0 °C for 4 h, and at r.t. for 24 h, then quenched with saturated aqueous ammonium chloride (30 mL) and extracted with ether. After drying (Na2SO4) and solvent removal a yellow oil was obtained; yield 950 mg (89 %) resp. 920 mg (86 %) as a 1.2:1 mixture of D-gluco/L-ido 7b/8b diastereomers.
To a stirred solution of aldehyde 6b (1.0 g, 3.59 mmol) in dry ether (20 mL) was added a solution of PhTi(OiPr)3 (1.195 g, 3.95 mmol, 1.1 equiv) in ether (20 mL) at –30 °C during 20 min. The mixture was stirred at –30 °C for 4 h and at r.t. for 48 h, then quenched with saturated aq. ammonium chloride (20 mL), extracted with ether (3 x 20 mL), dried with Na2SO4. After removal of solvent a yellow oil was obtained. Yield 383 mg (30 %). The product consists of a 14:1 mixture of 7b/ 8b diastereomers according to 1H-NMR data.
1,2-O-Isopropylidene-5-C-phenyl-α-D-gluco 7a and β-L-idopentofuranose 8a
Anhydrous cerium chloride (740 mg, 3 mmol) prepared by heating of CeCl3.7 H2O (1.12 g, 3 mmol) at 130-140 °C for 5 h in vacuo [23], was suspended in tetrahydrofuran (10 mL) at room temperature. The suspension was sonicated for 1 h, then stirred for 2 h at r.t. A solution of phenyllithium (3 mL, 1M solution in ether) was added at –10 °C and stirred for 45 min. A solution of crude 6a (580 mg, 3 mmol) in ether (5 mL) at –5 °C was added and stirring was continued at r.t. for 48 h, then quenched with saturated aqueous NaF (25 mL), extracted with ether (3 x 20 mL), dried with Na2SO4. After removal of solvent a yellow oil was obtained. Yield 544 mg (68 %). The product consists of a 1.6:1 mixture of D-gluco/L-ido diastereomers 7a/8a according to 1H-NMR data.
Alcohol 8a (L-ido)
Spectral Data
1H-NMR (CD3OD) δ: 7.31- 7.57 (m, 5H, C6H5), 6.01 (d, 1H, J1,2=3.7 Hz, H-1), 4.98 (d, 1H, J4,5=8.4 Hz, H-5), 4.46 (dd, 1H, J1,2=3.7 Hz, J2,3=0.8 Hz, H-2), 4.24 (dd, 1H, J3,4=2.8 Hz, J4,5=8.4 Hz, H-4), 3.61 (bd, 1H, J3,4=3.1Hz, H-3), 1.49 , 1.31 (all s, 3H, C(CH3)2).
13C-NMR (CD3OD) δ: 143.4 (s, i-C6H5), 130.2, 129.9, 129.2 (all d, C6H5), 113.7 (s, C(CH3)2), 107.4 (d, C-1), 79.6, 76.6, 86.9, 87.8 (all d, C-2, C-3, C-4, C-5), 28.0, 27.4 (all q, C(CH3)2).
Alcohol 7a (D-gluco)
Spectral Data
1H-NMR (CDCl3) δ: 7.34- 7.43 (m, 10H, C6H5), 6.12 (d, 1H, J1,2=3.8 Hz, H-1), 5.23 (d, 1H, J4,5=6.1 Hz, H-5), 4.72 (d, 1H, J=11.6 Hz, CH2), 4.70 (d, 1H, J1,2=3.9 Hz, H-2), 4.51 (d, 1H, J=11.4 Hz, CH2), 4.45 (dd, 1H, J3,4=3.3 Hz, J4,5=6.1 Hz, H-4), 4.10 (d, 1H, J3,4=3.2Hz, H-3), 1.55, 1.38 (all s, 3H, C(CH3)2).
13C-NMR (CD3OD) δ: 144.6 (s, i-C6H5), 130.2, 129.6, 129.0 (all d, C6H5), 113.6 (s, C(CH3)2), 107.2 (d, C-1), 73.1, 76.5, 85.6, 87.4 (all d, C-2, C-3, C-4, C-5), 28.0, 27.4 (all q, C(CH3)2).
References and Notes
- Fang, X.-p.; Anderson, J.E.; Chang, C.-j.; Fanwick, P.E.; McLaughlin, J.L. Novel Bioactive Styryl-lactones: Goniofufurone, Goniopypyrone, and 8-Acetylgoniotriol from Goniothalamus giganteus (Annonaceae). X-Ray Molecular Structure of Goniofufurone and of Goniopypyrone. J. Chem. Soc. Perkin 1 1990, 1655–1661. [Google Scholar] [CrossRef]
- Fang, X.-p.; Anderson, J.E.; Chang, C.-j.; McLaughlin, J.L. Two New Styryllactones, 9- deoxygoniopypyrone and 7-Epigoniofufurone, from Goniothalamus Giganteus. J. Nat. Prod. 1991, 54, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Gracza, T.; Jäger, V. Synthesis of Natural and Unnatural Enantiomers of Goniofufurone and Its 7- Epimers from D-Glucose. Application of Palladium(II)-Catalyzed Oxycarbonylation of Unsaturated Polyols. Synthesis 1994, 1359–1368. [Google Scholar] [CrossRef]
- Gracza, T.; Hasenöhrl, T.; Stahl, U.; Jäger, V. Synthesis of 3,5-Anhydro-2-deoxy-1,4- glyconolactones by Palladium(II)-Catalyzed, Regioselective Oxycarbonylation of C5- and C6- Enitols. ω-Homologation of Aldoses to Produce Intermediates for C-Glycoside/C-Nucleoside Synthesis. Synthesis 1991, 1108–1118. [Google Scholar] [CrossRef]
- Gracza, T.; Jäger, V. Palladium(II)-Catalyzed Oxycarbonylation of Unsaturated Polyols: Synthesis of (-)-Goniofufurone and Assigment of Absolute Configuration to the Natural (+)- Enantiomer, a Cytotoxic Styryllactone. Synlett 1992, 191–193. [Google Scholar] [CrossRef]
- Inch, T.D. Asymmetric Synthesis. Part I. A stereoselective synthesis of benzylic centres. Derivatives of 5-C –phenyl-D-gluco-pentose and 5-C –phenyl-L-ido-pentose. Carbohydr. Res. 1967, 5, 45–52. [Google Scholar] [CrossRef]
- Lichtenthaler, F.W.; Jargelis, P.; Lorenz, K. Convenient One-Pot Conversion of Alcohols into Esters via Hemiacetal Intermediates. Synthesis 1988, 790–794. [Google Scholar] [CrossRef]
- Wolfrom, M.L.; Hanessian, S. The Reaction of Free Carbonyl Sugar Derivatives with Organometallic Reagents. I. 6-Deoxy-L-idose and Derivatives. J. Org. Chem. 1962, 27, 1800–1804. [Google Scholar] [CrossRef]
- Prakash, K.R.C.; Prahlada Rao, S. Ethoxycarbonylmethylenetriphenylphosphorane in Carbohydrate Chemistry, Part II: A Short and Efficient Synthesis of (+)-Goniofufurone. Tetrahedron 1993, 49, 1505–1510. [Google Scholar] [CrossRef]
- Prakash, K.R.C.; Prahlada Rao, S. Ethoxycarbonylmethylenetriphenylphosphorane in Carbohydrate Chemistry, Part III: A Short and Highly Efficient Synthesis of (+)-Epigoniofufurone. Synlett 1993, 123–124. [Google Scholar] [CrossRef]
- Murphy, P.J. A Synthesis of Goniofufurone. J. Chem. Soc., Chem. Commun. 1992, 1096–1097. [Google Scholar] [CrossRef]
- Murphy, P.J.; Dennison, S.T. The Total Synthesis of Goniofufurone. Tetrahedron 1993, 49, 6695–6700. [Google Scholar] [CrossRef]
- Reitstoen, B.; Kilaas, L.; Anthonsen, T. Inverted Stereoselectivity in Grignard Additions to Chiral Aldehydes by Use of Polyethers. Acta Chem. Scand., Ser. B 40 1986, 440–443. [Google Scholar] [CrossRef]
- Marks, T.J.; Ernst, F.D. Comprehensive Organometallic Chemistry; Sir Geoffrey Wilkinson, F., Stone, Gordon A., Abel, Edward W., Eds.; Pergamon Press: Oxford, 1982; Volume 3, pp. 173–270. [Google Scholar]
- Imamoto, T.; Sugiura, Y.; Takiyama, N. Organocerium reagents. Nucleophilic addition to easily enolizable ketones. Tetrahedron Lett. 1984, 25, 4233–4236. [Google Scholar] [CrossRef]
- Waites, P.C.; Coutss, R.S.P.; Weigold, H. Organometallic Chemistry of Titanium, Zirconium, and Hafnium; Academic Press: New York, 1974. [Google Scholar]
- Holloway, H. Reaction of Phenyl Lithium with Titanium Monochloride Tri-isopropoxide. Chemistry and Industry 1962, 214–215. [Google Scholar]
- Reetz, M.T. Chelat- oder Nicht-Chelat-Kontrolle bei Additionsreaktionen von chiralen α- und β-Alkoxycarbonyl-Verbindungen. Angew. Chem. 1984, 96, 542–555, and references therein. [Google Scholar]
- Weidmann, B.; Seebach, D. Organometallverbindungen von Titan und Zirconium als selektive nucleophile Reagentien für die Organische Synthese. Angew. Chem. 1983, 95, 12–26. [Google Scholar] [CrossRef]
- Reetz, M.T.; Kesseler, K.; Schmidtberger, S.; Wenderoth, B.; Steinbach, R. Chelation- or Non- chelation-Control in Stereoselective Reactions of Titanium Reagents with Chiral Alkoxy Carbonyl Compounds. Angew. Chem. Suppl. 1983, 1511–1526. [Google Scholar] [CrossRef]
- Horton, D.; Jewell, J.S.; Prihar, H.S. Reaction of 3-O-p-bromophenylsulfonyl-1,2:5,6-di-O- isopropylidene-α-D-glucofuranose with dimethylamine. Can. J. Chem. 1968, 46, 1580–1582. [Google Scholar] [CrossRef]
- Helferich, B.; Dressler, H.; Griebel, R. Ester der Methansulfonsäure in der Zuckergruppe. J. Prakt. Chem. 1939, 153, 285–299. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Matsumoto, T.; Takemoto, Y.; Nakatani, K.; Ito, Y.; Kamijo, T.; Harada, H.; Terashima, S. Novel Synthesis of Three Types of C-Terminal Compounds of Renin Inhibitors from Unnatural (2S,3S)-Tartaric. Acid. Chem Pharm. Bull. 1991, 39, 2550–2554. [Google Scholar]
- Sample Availability: Available from the authors.
Scheme 1.
Goniofufurone (+)-1 and 7-epi-goniofufurone (+)-2.
Scheme 2.
Chelation models for 1,2- (A) and 1,3- (B) asymmetric induction, Felkin-Anh model (C).
Scheme 3.
Reagents and conditions: (i)-(iv) see lit. [6,7,8], (v) PhM, solvent and reaction conditions see Table 1.

{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | PhM | Conditions | Yield (%)b | L ido/D glucoa |
|---|---|---|---|---|---|
| 1 | 6a | PhMgBr | THF | 69 | 3.1 |
| 2 | 6b | PhMgBr | Et2O | 70 | 14.1 |
| 3 | 6c | PhMgBr | Et2O | 84 | 2.1c |
| 4 | 6d | PhMgBr | Et2O | 78 | 1.6.1c |
| 5 | 6b | PhMgBr | Et2O,18-C-6 | 89 | 1.1.2 |
| 6 | 6b | PhMgBr | Et2O,18-C-6 | 86 | 1.1.2 |
| 7 | 6a | PhCeCl2 | Et2O/ THF | 68 | 1.1.6 |
| 8 | 6b | PhTi(OiPr)2 | Et2O | 30 | 1.14 |
aAssignments of reaction ratios are based on the1H-NMR spectra of crude reaction mixtures.bThe yields refer to pure isolated products.cProbable assignments, may eventually be reversed.
© 2000 by MDPI (http://www.mdpi.org).
Share and Cite
MDPI and ACS Style
Gracza, T.; Szolcsányi, P. Study of Stereoselectivity in Organometallic Additions to 1,2-O-Isopropylidene-O-R-α-D-xylopentodialdo-1,4-furanose. Molecules 2000, 5, 1386-1398. https://doi.org/10.3390/51201386
AMA Style
Gracza T, Szolcsányi P. Study of Stereoselectivity in Organometallic Additions to 1,2-O-Isopropylidene-O-R-α-D-xylopentodialdo-1,4-furanose. Molecules. 2000; 5(12):1386-1398. https://doi.org/10.3390/51201386
Chicago/Turabian StyleGracza, Tibor, and Peter Szolcsányi. 2000. "Study of Stereoselectivity in Organometallic Additions to 1,2-O-Isopropylidene-O-R-α-D-xylopentodialdo-1,4-furanose" Molecules 5, no. 12: 1386-1398. https://doi.org/10.3390/51201386