Synthesis of 4-O-Methylcedrusin. Selective Protection of Catechols with Diphenyl Carbonate

Abstract
:Introduction
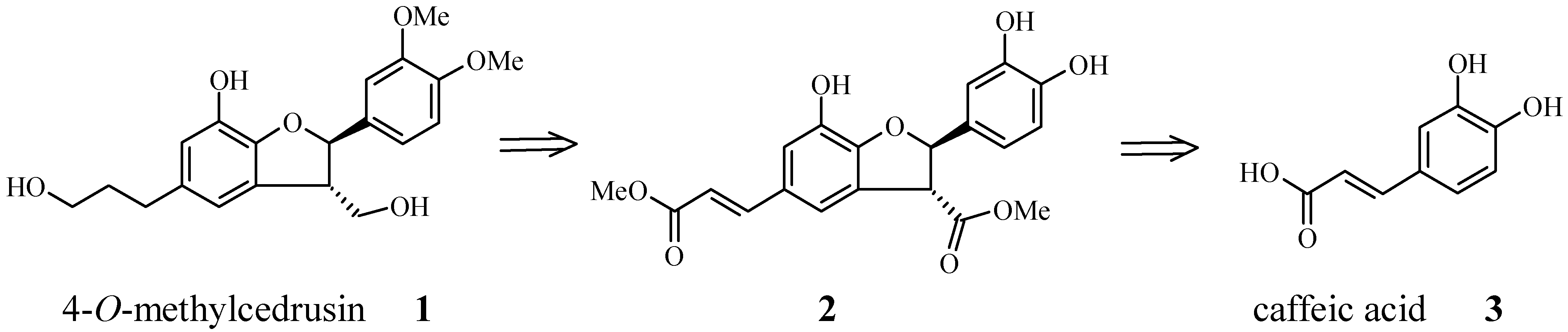
Results and Discussion
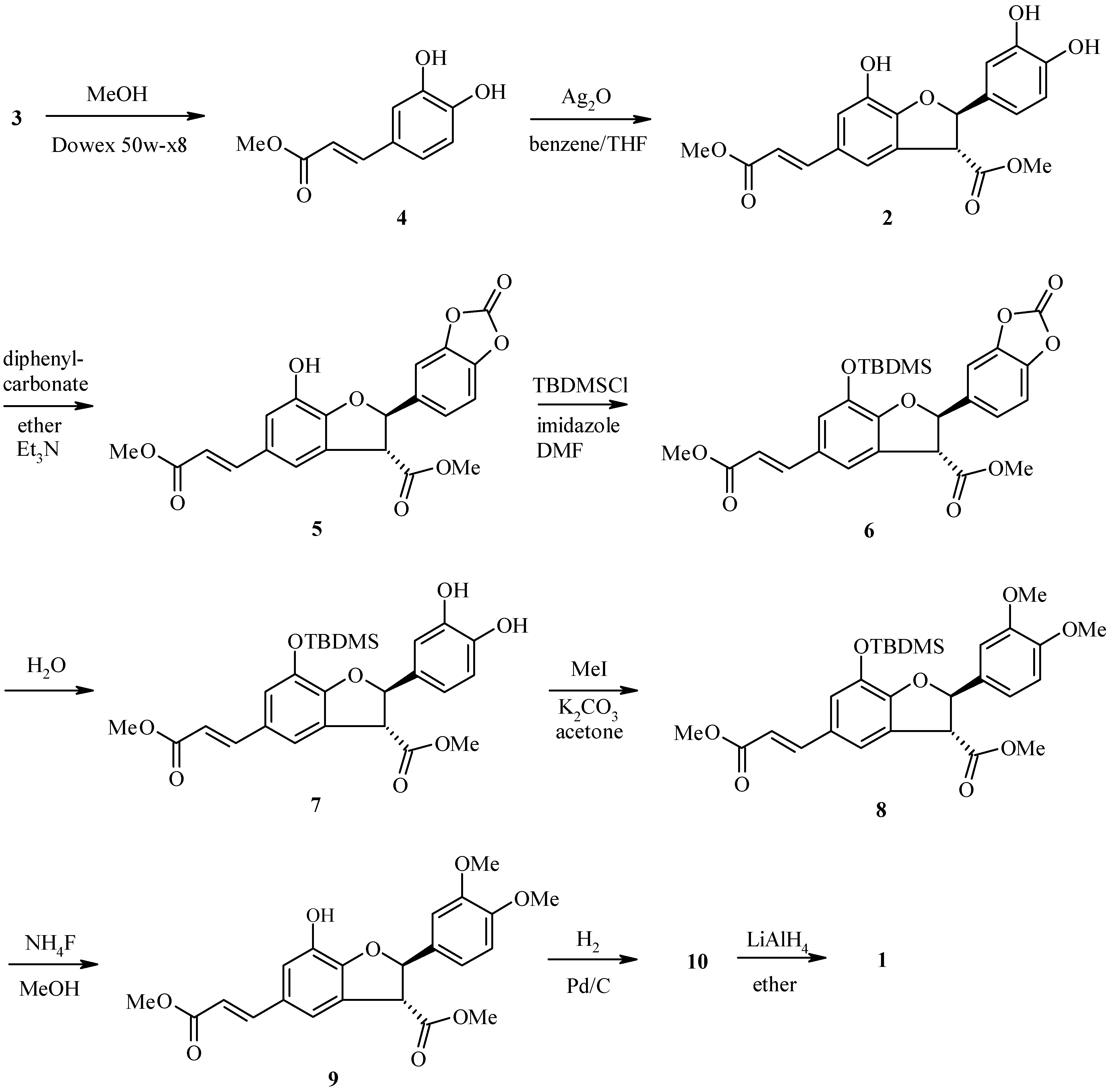
Conclusion
Experimental
General
Methyl caffeate (4)
Methyl (E)–3-[2-(3,4-dihydroxyphenyl)-7-hydroxy-3-methoxycarbonyl-2,3-dihydro-1-benzofuran-5-yl]prop-2-enoate (2)
Methyl (E)-3-[2-(2-oxo-1,3-benzodioxol-5-yl)-7-hydroxy-3-methoxycarbonyl-2,3-dihydro-1-benzofu-ran-5-yl]prop-2-enoate (5)
Methyl (E)-3-[2-(2-oxo-1,3-benzodioxol-5-yl)-7-(tert-butyldimethylsilyloxy)-3-methoxycarbonyl-2,3-dihydro-1-benzofuran-5-yl]prop-2-enoate (6)
Methyl (E)-3-[2-(3,4-dihyroxyphenyl)-7-(tert-butyldimethylsilyloxy)-3-methoxycarbonyl-2,3-dihydro-1-benzofuran-5-yl]prop-2-enoate (7)
Methyl (E)-3-[2-(3,4-dimethoxyphenyl)-7-(tert-butyldimethylsilyloxy)-3-methoxycarbonyl-2,3-dihydro-1-benzofuran-5-yl]prop-2-enoate (8)
Methyl (E)–3-[2-(3,4-dimethoxyphenyl)-7-hydroxy-3-methoxycarbonyl-2,3-dihydro-1-benzofuran-5-yl]prop-2-enoate (9)
Methyl 3-[2-(3,4-dimethoxyphenyl)-7-hydroxy-3-methoxycarbonyl-2,3-dihydro-1-benzofuran-5-yl]propanoate) (10)
3-[2-(3,4-Dimethoxyphenyl)-3-hydroxymethyl-7-hydroxy-2,3-dihydro-1-benzofuran-5-yl]propan-1-ol (1)
Acknowledgements
References and Notes
- Pieters, L.; De Bruyne, T.; Van Poel, B.; Vingerhoets, B.; Totté, J.; Vanden Berghe, D.; Vlietinck, A. Phytomed. 1995, 2, 17–22.
- Pieters, L.; Van Dyck, S.; Gao, M.; Bai, R.; Hamel, E.; Vlietinck, A.; Lemière, G. accepted for publication in J. Med. Chem.
- Pieters, L.; De Bruyne, T.; Claeys, M.; Vlietinck, A. J. Nat. Prod. 1993, 56(6), 899–906. [PubMed]
- The relative configurations in scheme 1 and scheme 2 are depicted according to a proposal of Maerhr, H. J. Chem. Ed. 1985, 62, 114–120.
- Antus, S.; Bauer, R.; Gottsegen, A.; Seligmann, O.; Wagner, H. Liebigs Ann. Chem. 1987, 357–360.
- Bolzacchini, E.; Brunow, G.; Meinardi, S.; Orlandi, M.; Rindone, B.; Rumakko, P.; Setela, H. Tetrahedron Lett. 1998, 39, 3291–3294.
- For a broad review on these and other protective groups see: Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Chemistry; John Wiley: New York, 1974. [Google Scholar]
- Raaijmakers, H.; Zwanenburg, B.; Chittenden, G. J. F. J. Carbohydrate Chemistry 1993, 12(8), 1117–1125.
- Einhorn, A.; Cobliner, J.; Pfeifer, H. Ber. 1904, 37, 100–128.
- The rate of hydrolysis is very dependent to the type of solvent. In acetone or dioxane the hydrolysis is significantly slower needing reaction times of about 18h.
- Lemière, G.; Gao, M.; De Groot, A.; Dommisse, R.; Lepoivre, J.; Pieters, L.; Buss, V. J. Chem. Soc. Perkin I 1995, 1775–1779.
- The numbering used for the assignment of 1H and 13C-NMR signals is as shown in the following structure. This numbering is used for easy comparison of the signals of the compounds (1)-(10) with earlier work [11] and is in accordance with a recent IUPAC recommendation [13]
![Molecules 05 00153 i001]()
- Provisional recommendation, IUBMB-IUPAC, Joint Commision on Biochemical Nomenclature (JCBN), Nomenclature of lignans an neolignans, 30 June 1999.

{kind=link}
{kind=link}
- Samples Availability: Available from the authors.
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Van Dyck, S.M.O.; Lemière, G.L.F.; Jonckers, T.H.M.; Dommisse, R. Synthesis of 4-O-Methylcedrusin. Selective Protection of Catechols with Diphenyl Carbonate. Molecules 2000, 5, 153-161. https://doi.org/10.3390/50200153
Van Dyck SMO, Lemière GLF, Jonckers THM, Dommisse R. Synthesis of 4-O-Methylcedrusin. Selective Protection of Catechols with Diphenyl Carbonate. Molecules. 2000; 5(2):153-161. https://doi.org/10.3390/50200153
Chicago/Turabian StyleVan Dyck, Stefaan M. O., Guy L. F. Lemière, Tim H. M. Jonckers, and Roger Dommisse. 2000. "Synthesis of 4-O-Methylcedrusin. Selective Protection of Catechols with Diphenyl Carbonate" Molecules 5, no. 2: 153-161. https://doi.org/10.3390/50200153