Effect of Microwave Irradiation on the Condensation of 6-Substituted 3-Formylchromones with Some Five-membered Heterocyclic Compounds

Abstract
:Introduction
Results and Discussion
Experimental
General
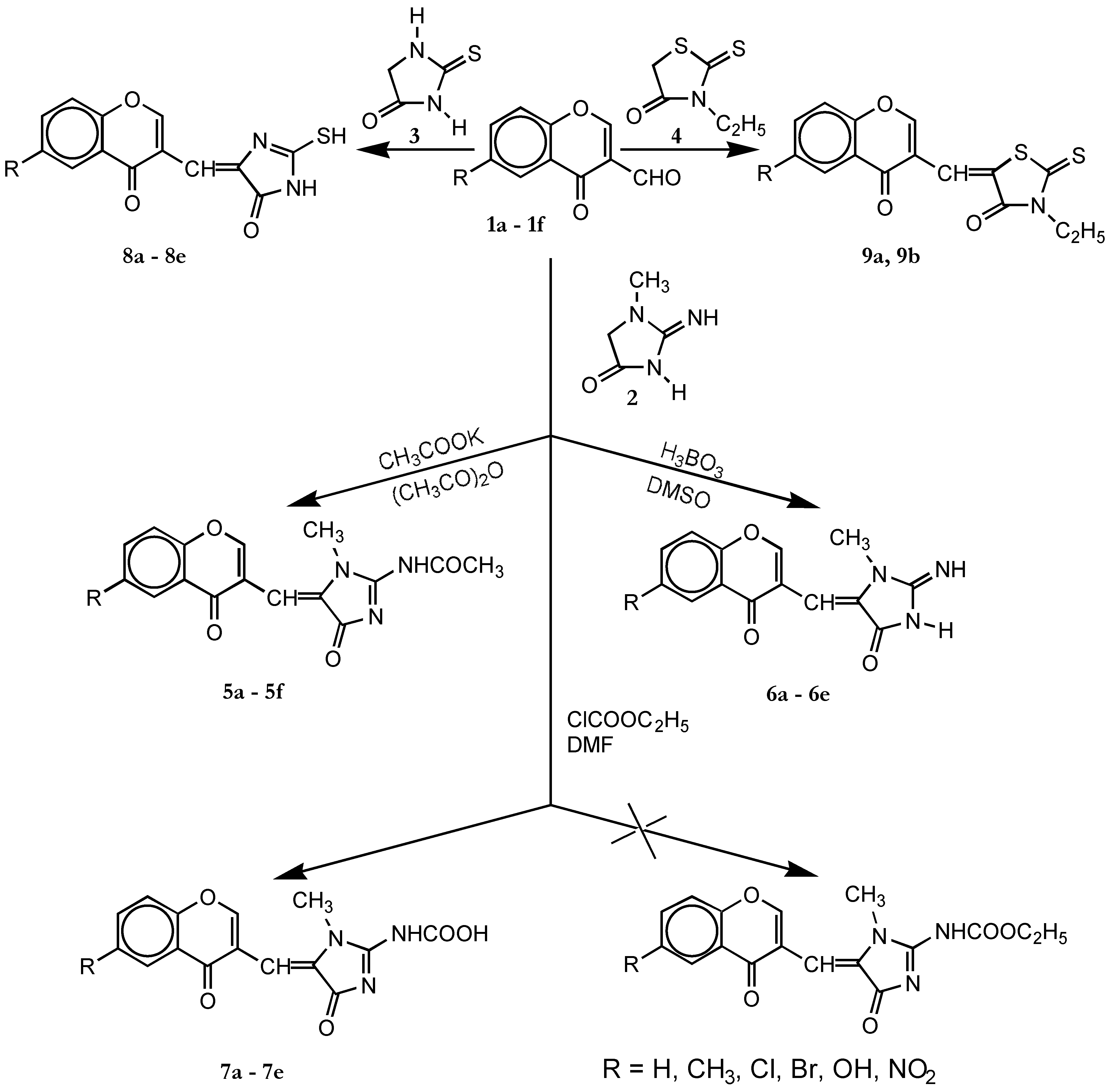
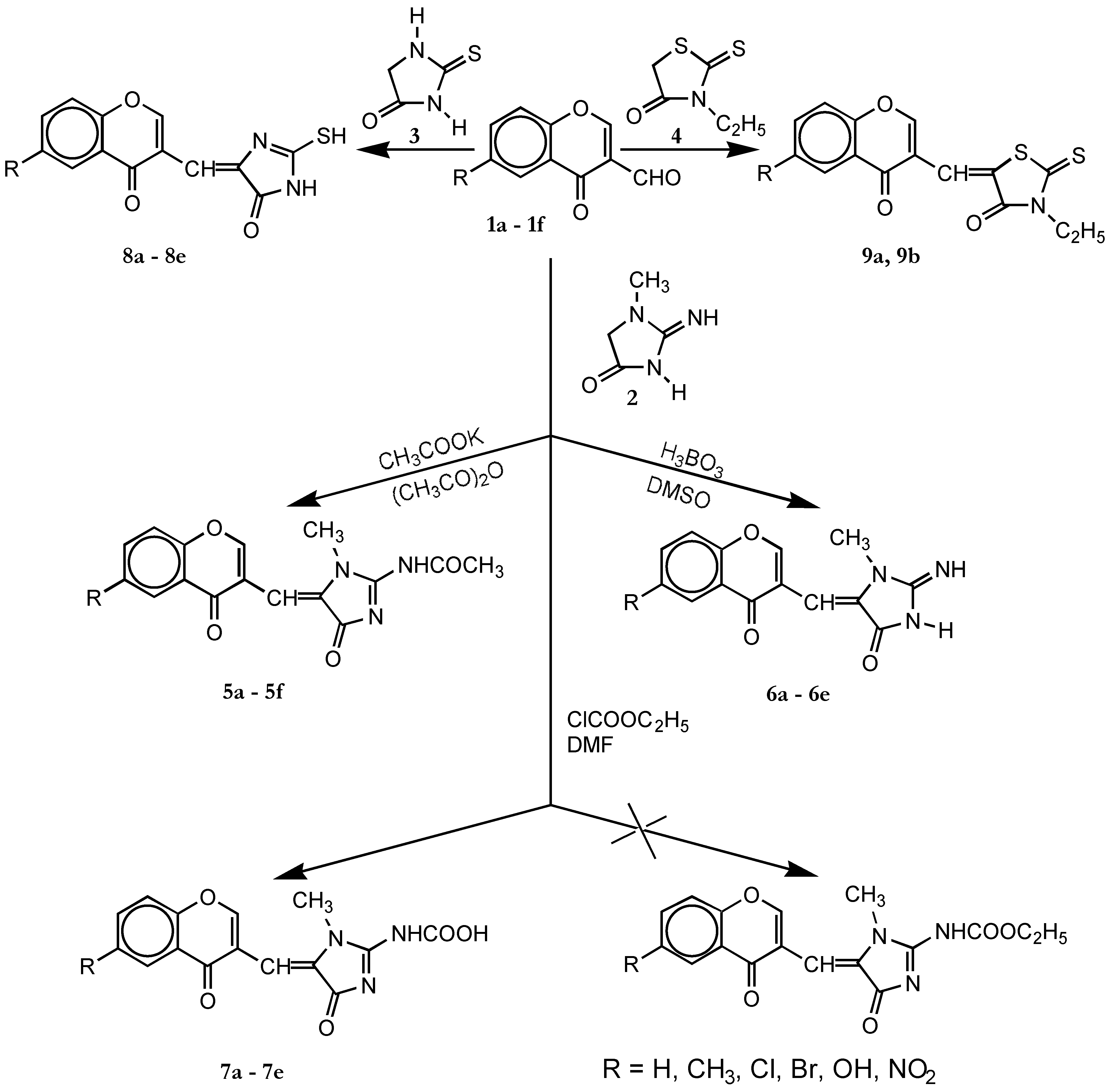
Synthesis of 5a-5f , 8a-8e and 9a, 9b
Method A
Method B
Synthesis of 6a–6e
Method A
Method B
Synthesis of 7a, 7b
Method A
Method B
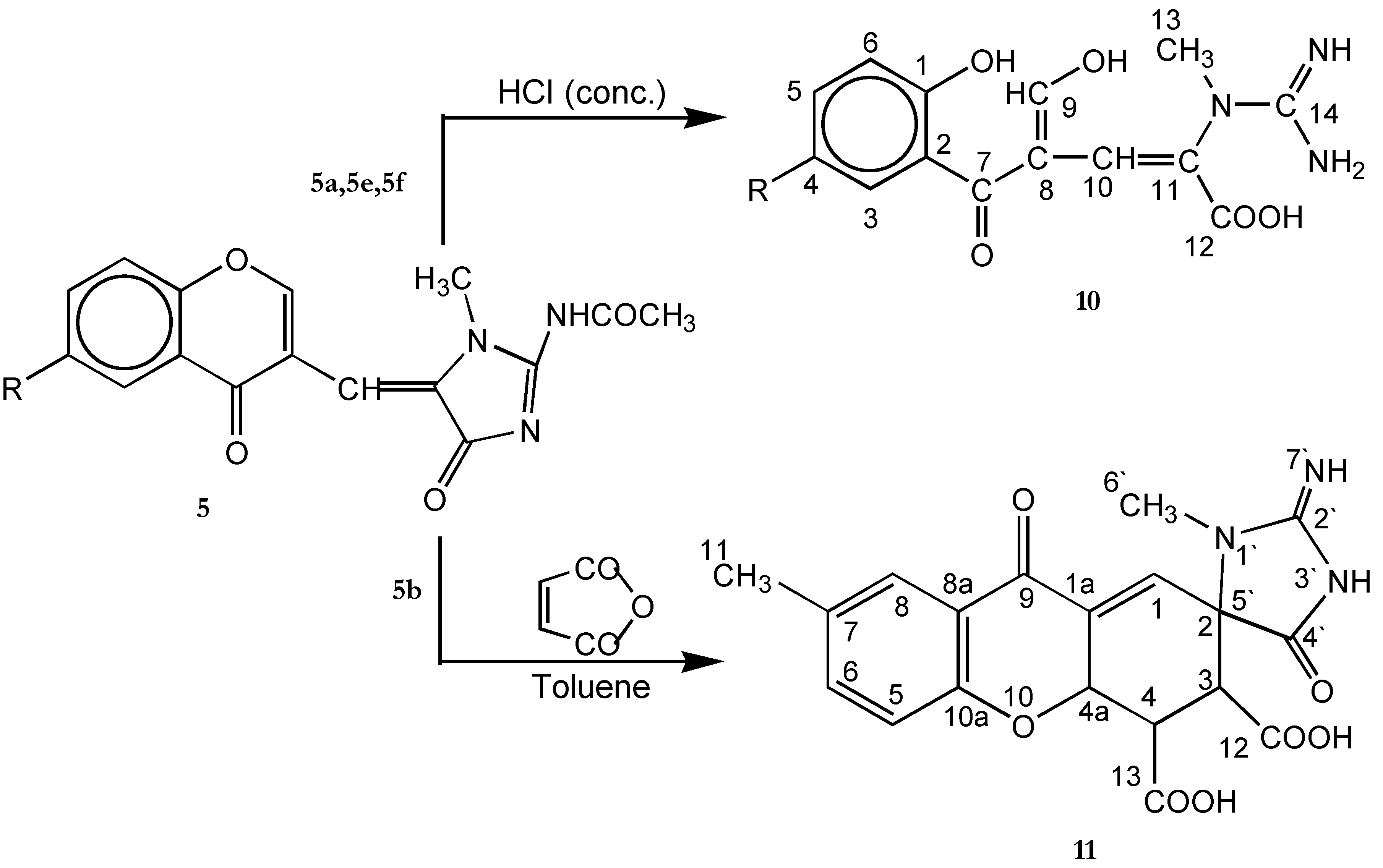
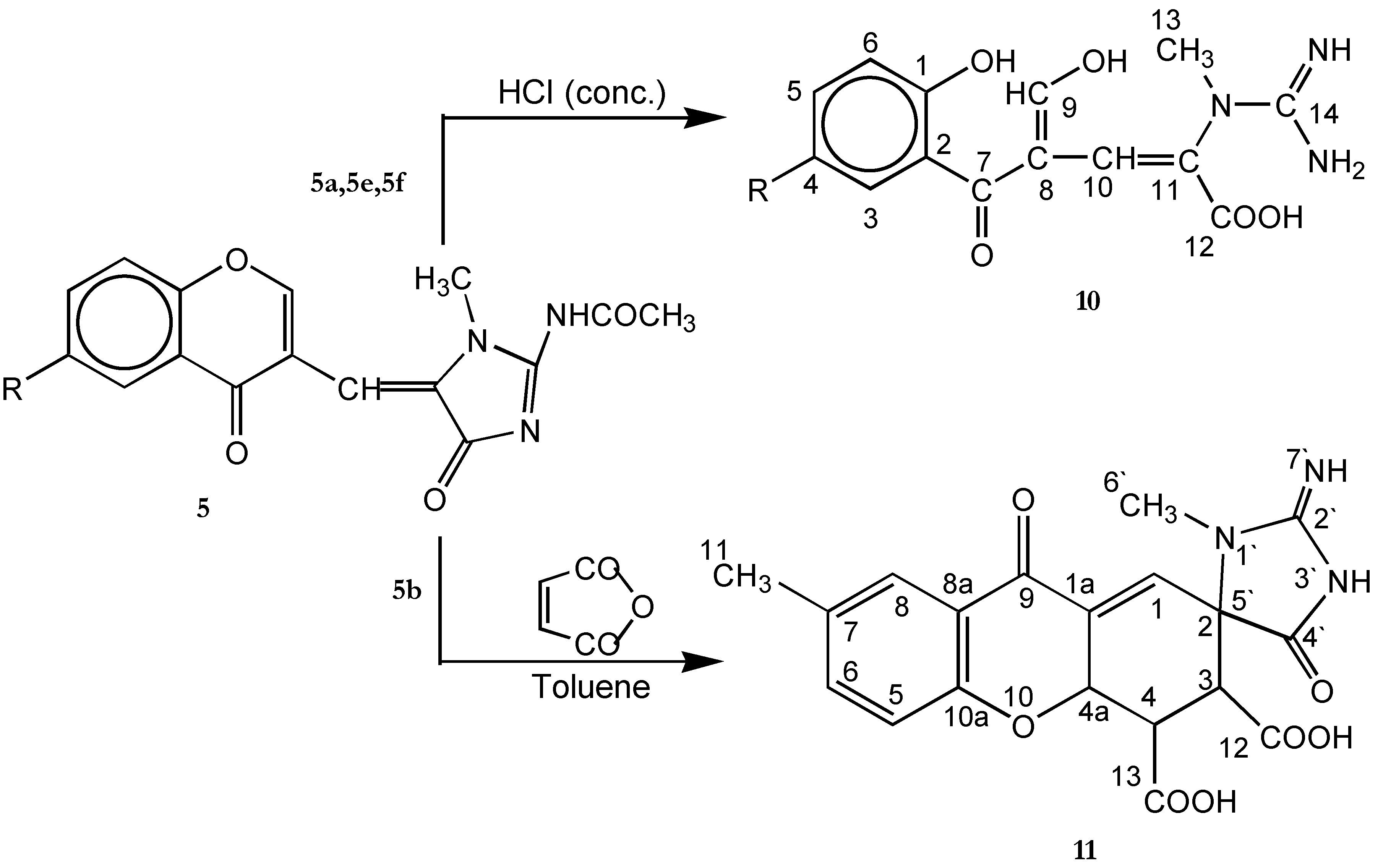
Acid hydrolysis of compounds 5a, 5e and 5f
| 13C NMR spectral data for compound 10e [δ(ppm); DMSO - d6, 300 MHz] | ||||||
| C-1 | C-2 | C-3 | C-4 | C-5 | C-6 | C-7 |
| 155.45 | 123.77 | 123.65 | 154.87 | 107.95 | 120.11 | 174.53 |
| C-8 | C-9 | C-10 | C-11 | C-12 | C-13 | C-14 |
| 114.78 | 157.60 | 109.79 | 130.04 | 163.32 | 28.69 | 149.23 |
Diels - Alder reaction of compound 5b with maleic anhydride (11)
| 13C NMR spectral data for compound 11 [δ(ppm); DMSO - d6, 300 MHz] | ||||||
| C-1 | C-1a | C-2 | C-3 | C-4 | C-4a | C-5 |
| 131.1 | 131.6 | 107.8 | 39.2 | 39.2 | 135.8 | 116.0 |
| C-6 | C-7 | C-8 | C-8a | C-9 | C-10a | C-11 |
| 135.7 | 130.9 | 124.6 | 153.8 | 190.0 | 118.5 | 20.5 |
| C-12 | C-13 | C-2′ | C-4′ | C-6′ | ||
| 166.8 | 166.8 | 157.6 | 174.8 | 28.4 | ||
Analytical Data
References and Notes
- Stankovicova, H.; Fabian, W. M.; Lacova, M. Molecules 1996, 1, 223.
- Stankovicova, H.; Gasparova, R.; Lacova, M.; Chovancova, J. Collect. Czech. Chem. Commun. 1997, 62, 781.
- Lacova, M.; Stankovicova, H.; Odlerova, Z. Il Pharmaco 1995, 50, 885.
- El-Shaaer, H.M.; Perjessy, A.; Zahradnik, P.; Lacova, M.; Matulova, M. Monatsh. Chem 1993, 124, 539.
- Lacova, M.; El-Shaaer, H.M.; Loos, D.; Matulova, M.; Chovancova, J.; FurdIk, M. Molecules 1998, 3, 120.
- Gasparova, R.; Lacova, M.; El-Shaaer, H.M.; Odlerova, Z. Il Pharmaco 1997, 52, 251.
- Lacova, M.; Chovancova, J.; Veverkova, J.; Toma, S. Tetrahedron 1996, 52, 14995.
- Nohara, A.; Sugihara, H.; Ukawa, K. Japan. Kokai Tokyo Koho 1978, 78, 111,070. ; Chem. Abstr. 1979, 90, 54828z.
- Klutchko, S.; Kaminski, D.; Von Strandtmann, M. U.S. Patent 4,008,252, 1977. ; Chem. Abstr. 1977, 87, 5808a.
- Lacova, M.; El-Shaaer, H. M.; Odlerova, Z.; Furdik, M. Chem. Papers 1994, 48, 344.
- Deulofeu, V.; Guerrero, T. J. Org. Synth. Coll. 1955, 3, 586.
- Coirnwaite, W. R.; Lazarus, S.; Snelling, R. H.; Denoon, C. E. J. Am. Chem. Soc. 1936, 58, 628.
- Fitton, A. O.; Frost, J. R.; Suschitsky, H.; Houghton, P. G. Synthesis 1977, 133.
- Gasparova, R.; Lacova, M. Collect. Czech. Chem. Commun. 1995, 60, 1178.
- Villemin, D.; Martin, B. Synth. Commun. 1995, 25, 3135.
- Villemin, D.; Ricard, M. Synth. Commun. 1987, 17, 283.
- Nohara, A.; Ishiguto, T.; Sanno, Y. Tetrahedron Lett. 1974, 13, 1183.
- Silva, A. M. S.; Silva, A. M. G.; Tome, A. C.; Cavaleiro, J. A. S. J. Org. Chem. 1999, 135.
- Samples Availability: Available from the authors and MDPI.

{kind=link}
{kind=link}
| Comp. | R | Formula Mr | m.p. °C | wi (calcd)/ % wI (found)/ % | Yield % | tr min | ||
| C | H | N | ||||||
| 5a | H | C16H13 N3 O4 | 246 - 248 | 61.73 | 4.21 | 13.50 | 75 | 3 |
| 311.29 | 61.31 | 4.14 | 13.41 | 71 | 60 | |||
| 5b | CH3 | C17H15 N3O4 | 277 - 279 | 62.76 | 4.65 | 12.92 | 60 | 4 |
| 325.32 | 62.15 | 4.66 | 12.59 | 57 | 120 | |||
| 5c | Cl | C16H12ClN3O4 | 268 - 270 | 55.58 | 3.50 | 12.15 | 76 | 2 |
| 345.74 | 55.58 | 3.59 | 12.13 | 72 | 60 | |||
| 5d | Br | C16H12BrN3O4 | 270 - 272 | 49.25 | 3.10 | 10.77 | 84 | 1 |
| 390.19 | 48.91 | 3.02 | 10.54 | - | - | |||
| 5e | AcO | C18H15N3O6 | 259 - 262 | 58.54 | 4.09 | 11.38 | 46 | 3 |
| 369.33 | 58.06 | 4.07 | 11.28 | 40 | 90 | |||
| 5f | NO2 | C16H12N4O6 | 285 - 286 | 53.94 | 3.39 | 15.72 | 84 | 3 |
| 356.29 | 53.83 | 3.29 | 15.43 | - | - | |||
| 6a | H | C14H11N3O3 | 250 - 252 | 62.45 | 4.12 | 15.61 | 70 | 3 |
| 269.26 | 62.38 | 3.96 | 15.72 | |||||
| 6b | CH3 | C15H13N3O3 | 294 - 297 | 63.60 | 4.63 | 14.83 | 67 | 3 |
| 283.28 | 63.24 | 4.68 | 14.06 | |||||
| 6c | Cl | C14H10ClN3O3 | 356 - 360 | 55.36 | 3.32 | 13.83 | 92 | 3 |
| 303.7 | 55.21 | 3.49 | 13.22 | |||||
| 6d | Br | C14H10BrN3O3 | 276 - 278 | 48.30 | 2.89 | 12.07 | 68 | 3 |
| 348.16 | 48.25 | 2.96 | 12.44 | |||||
| 6e | NO2 | C14H10N4O5 | 237 - 240 | 53.51 | 3.21 | 17.83 | 98 | 3 |
| 314.26 | 52.94 | 2.99 | 17.39 | |||||
| 7a | H | C15H11N3O5 | 253 - 255 | 57.82 | 3.51 | 13.41 | 69 | 6 |
| 313.3 | 57.50 | 3.90 | 13.08 | |||||
| 7b | Cl | C15H10ClN3O5 | 356 - 360 | 54.34 | 3.76 | 11.18 | 71 | 4 |
| 375.76 | 54.69 | 3.23 | 11.23 | |||||
| 7c | CH3 | C16H13N3O5 | 299 - 301 | 58.71 | 3.97 | 12.84 | 58 | 7 |
| 327.3 | 58.44 | 4.30 | 12.75 | |||||
| 7d | Br | C15H10BrN3O5 | 350 | 45.92 | 2.55 | 10.71 | 60 | 7 |
| 392.2 | 46.28 | 2.36 | 10.48 | |||||
| 8a | H | C13H8N2O3S | 292 - 295 | 57.35 | 2.96 | 10.29 | 79 | 8 |
| 272.3 | 56.98 | 2.91 | 10.05 | 72 | 60 | |||
| 8b | CH3 | C14H10N2O3S | 315 - 317 | 58.73 | 3.52 | 9.78 | 70 | 6 |
| 286.3 | 58.50 | 3.52 | 9.04 | 66 | 30 | |||
| 8c | Cl | C13H7ClN2O3S | 319 - 321 | 50.91 | 2.30 | 9.13 | 74 | 4 |
| 306.7 | 51.03 | 2.34 | 9.05 | 71 | 30 | |||
| 8d | Br | C13H7BrN2O3S | 329-331 | 44.46 | 2.01 | 7.98 | 96 | 9 |
| 351.2 | 44.53 | 2.02 | 7.16 | 88 | 60 | |||
| 8e | AcO | C15H10N2O5S | 303 - 305 | 54.54 | 3.05 | 8.48 | 62 | 10 |
| 330.3 | 53.82 | 2.98 | 7.99 | 59 | 120 | |||
| 9a | H | C15H11NO3S2 | 215 - 217 | 56.77 | 3.49 | 4.41 | 70 | 5 |
| 317.4 | 57.07 | 3.48 | 4.45 | 74 | 60 | |||
| 9b | Cl | C15H10ClNO3S2 | 231 - 233 | 51.21 | 2.86 | 3.98 | 67 | 5 |
| 351.8 | 50.96 | 2.82 | 3.79 | 65 | 60 | |||
| 10a | H | C14H15N5O5 | 325 - 327 | 55.08 | 4.95 | 13.76 | 0 | 20 |
| 305.3 | decomp. | 55.35 | 4.59 | 13.65 | 52 | 240 | ||
| 10e | OH | C14H15N3O6 | 315 - 317 | 52.33 | 4.70 | 13.08 | 0 | 20 |
| 321.3 | decomp. | 52.12 | 4.59 | 12.72 | 47 | 260 | ||
| 10f | NO2 | C14H14N4O7 | >360 | 48.00 | 4.02 | 15.98 | 0 | 20 |
| 350.3 | decomp. | 48.09 | 3.855 | 15.77 | 56 | 250 | ||
| 11 | CH3 | C19H17N3O7 | 221 - 223 | 57.16 | 4.25 | 10.51 | 0 | 20 - 30 |
| 399.3 | 57.10 | 4.22 | 10.41 | 89 | 15 hr. | |||
| Compound | Solvent | 1H NMR spectrum δ (ppm) |
| 5a | CDCl3 | 2.25 (s, 3H, CH3); 3.39 (s, 3H, CH3-N); 6.86 - 8.32 (m, 5H, H-Ar); 9.68 (s, 1H, H-2); 10.84 (s, 1H, NH). |
| 5b | DMSO-d6 | 2.50 (s, 3H, CH3); 2.54(s, 3H, CH3); 3.57(s, 3H, CH3-N); 6.56 (s, 1H, H-9); 7.60 - 7.70 (m, 5H, H-Ar); 7.97 (s, 1H, H-5); 9.56 (s, 1H, H-2). |
| 5c | DMSO-d6 | 2.76 (s, 3H, CH3); 3.52 (s, 3H, CH3-N); 6.72 (s, 1H, H-9); 8.02 (d, 1H, H-8, 3J=9Hz); 8.11 (d, 1H, H-7, 3J=9Hz); 8.32 (d, 1H, H-5, 4J=2Hz); 9.54 (s, 1H, H-2). |
| 5d | DMSO-d6 | 2.78 (s, 3H, CH3); 3.53 (s, 3H, CH3-N); 6.74 (s, 1H, H-9); 7.95 (d, 1H, H-8, 3J=9Hz); 8.24 (d, 1H. H-7, 3J=9Hz); 8.49 (d, 1H, H-5, 4J=1.8Hz), 9.55 (s, 1H, H-2). |
| 5e | DMSO-d6 | 2.13 (s, 3H, CH3O); 2.3 (s, 3H, CH3O); 3.45 (s, 3H, CH3-N); 6.47 (s, 1H, H-9); 7.6 - 7.85 (m, 3H, H-8, 7, 5); 9.30 (s, 1H, H-2); 11.41 (s (broad), 1H, N-H). |
| 5f | DMSO-d6 | 2.13 (s, 3H, CH3O); 3.21 (s, 3H, CH3-N); 6.43 (s, 1H, H-9); 8.00(s, 1H, H-8); 8.64 (s, 1H, H-7); 8.85 (s, 1H, H-7); 8.85 (s, 1H, H-5); 9.353 (s, 1H, H-2). |
| 6a | DMSO-d6 | 3.35 (s, 3H, CH3-N); 6.24 (s, 1H, H-9); 7.52 (dd, 1H, H- 7,3J=7.8Hz); 7.69 (dd, 1H, H-8, 3J=7.8Hz); 7.81 (dd, 1H, H-6, 3J=7.8Hz); 8.12 (dd, 1H, H-5, 3J=7.8Hz); 9.88 (s, 1H, H-2); 7.4 - 8.4 (broad, 1H, NH). |
| 6b | DMSO-d6 | 2.45 (s, 3H, CH3); 3.38 (s, 3H, CH3N); 6.25 (s, 1H, H-9); 7.60 - 7.95 (m, 3H, H-8, 7, 5); 9.86 (s, 1H, H-2); 7.4 - 8.4 (broad, 1H, NH). |
| 7a | DMSO-d6 | 3.39 (s, 3H, CH3CN); 6.19 (s, 1H, H-9); 7.53 (dd, 1H, H-6, 3J=7Hz); 7.69 (d, 1H, H-8, 3J=7Hz); 7.84 (dd, 1H, H-7, 3J=7Hz); 8.12 (d, 1H, H-5, 3J=7Hz); 9.8 (s, 1H, H-2). |
| 7d | DMSO-d6 | 3.39 (s, 3H, CH3CN); 6.19 (s, 1H, H-9); 7.70 (d, 1H, H-8, 3J=8Hz); 7.97 (dd, 1H, H-7, 3J=8Hz, 4J=2.2Hz); 8.17 (d, 1H, H-5, 4J=2.2Hz); 9.895 (s, 1H, H-2). |
| 7e | DMSO-d6 | 3.44 (s, 3H, CH3-N); 6.59 (s, 1H, H-9); 8.00 (d, 1H, H-8, 3J=9Hz); 8.61 (dd, 1H, H-7, 3J=9Hz, 4J=2.5Hz); 8.79 (d, 1H, H-5, 4J=2.54Hz); 9.395 (s, 1H, H-2); 9.64 (s( broad), 1H, NH). |
| 8c | DMSO-d6 | 6.36 (s, 1H, H - 9); 7.78 (d, 1H, H-8, 3J=10Hz); 7.87 (dd, 1H, H - 7, 3J=10Hz, 4J=2.2Hz); 8.23 (d, 1H, 4J=2.2Hz); 8.94 (s, 1H, H-5); 11.65 (s, 1H, NH); 12.46 (s, 1H, OH). |
| 8d | DMSO-d6 | 6.35 (s, 1H, H - 9); 7.74 (d, 1H, H-8, 3J=8.8Hz); 8.02 (dd, 1H, H - 7, 3J=8.8Hz, 4J=2.2Hz); 8.22 (d, 1H, 4J=2.2Hz); 8.92 (s, 1H, H-2); |
| 8e | DMSO-d6 | 2.33 (s, 3H, CH3); 6.38 (s, 1H, H - 9); 7.60 (dd, 1H, H-7, 3J=9Hz, 4J=1.9Hz); 7.83 (d, 1H, H-8, 3J=9Hz); 7.88 (d, 1H, H-5, 4J=1.9Hz); 8.94 (s, 1H, H-2); 11.80 (s, 1H, NH); 12.45 (s, 1H, OH). |
| 9b | DMSO-d6 | 2.490 (t, 3H, CH3); 5.346 (q, 2H, CH2); 8.84 (s, 1h, H-2); 9.07 (d, 1H, H-8, 3J=9.1Hz); 9.072 (dd, 1H, H-7, 3J=9.1Hz, 4J=2Hz); 9.36 (d, 1H, H-5, 4J=2Hz); 10.27 (s, 1H, H-9). |
| 10a | DMSO-d6 | 6.27 (s, 1H, CH); 7.23 - 7.66 (m, H, Ar - H); 9.25 (s, 1H, CH); 10.26 (b., 6H, NH2, NH, OH). |
| 10e | DMSO-d6 | 3.38 (s, 3H, CH3); 6.70 (s, 1H, CH); 7.29 - 7.39 (dd, 1H, H-5, J=9Hz); 7.39 - 7.41 (d, 1H, H-3, J=3Hz); 7.57 - 7.60 (d, 1H, H-6, J=9Hz); 9.23 (s, 1H, CH); 10.28 (b., 7H, OH, NH, NH2). |
| 10f | DMSO-d6 | 3.68 (s, 3H, CH3); 7.95 - 8.81 (m, 3H, Ar-H); 9.32 (s, 1H, CH); 10.00 (b., 6H, OH, NH, NH2). |
| 11 | DMSO-d6 | 2.46 (s, 3H, CH3); 2.48 (s, 3H, CH3); 4.71 (broad, 5H, NH amide, CO2H, CH); 6.23 - 6.56 (m, 4H, Ar-H, H-2); 7.64 (s, 1H, =CH); 9.47 (s, 1H, NH-imine). |
| ν (cm−1) | ||||
| Comp. | ν(NH) | ν(C=O)heterocycl | ν(C=O)pyrone | ν(C=O)other |
| 5a | 3078 - 3162 | 1739 | 1650 | 1643 |
| 5b | 3070 - 3175 | 1740 | 1650 | 1642 |
| 5c | 3075 - 3179 | 1735 | 1659 | 1636 |
| 5d | 3075 - 3180 | 1729 | 1658 | 1640 |
| 5e | 3081 - 3181 | 1739 | 1652 | 1630, 1758 |
| 5f | 3080 - 3177 | 1745 | 1660 | 1641 |
| 6a | 3070 - 3185 | 1720 | 1650 | |
| 6b | 3095 - 3180 | 1720 | 1645 | |
| 6c | 3070 - 3175 | 1735 | 1655 | |
| 6d | 3090 - 3180 | 1721 | 1655 | |
| 6e | 3094 - 3186 | 1719 | 1658 | |
| 7a | 3080 - 3230 | 1719 | 1657 | 1740 |
| 7b | 3075 - 3225 | 1721 | 1658 | 1742 |
| 8a | 3038 - 3180 | 1740 | 1665 | |
| 8b | 3086 - 3185 | 1739 | 1668 | |
| 8c | 3070 - 3190 | 1742 | 1665 | |
| 8d | 3060 - 3180 | 1736 | 1668 | |
| 8e | 3078 - 3185 | 1745 | 1663 | 1750 |
| 9a | 3069 - 3110 | 1688 | 1660 | |
| 9b | 3060 - 3110 | 1688 | 1662 | |
| ν (cm−1) | |||||
| Comp. | ν(OH) | ν(NH) | ν(C=O) | ν(C=O)acid | ν(C=C) |
| 10a | 3480-3400 | 3080-3040 | 1740 | 1700 | 1646 |
| 10e | 3540-3440 | 3080-3020 | 1748 | 1700 | 1642 |
| 10f | 3560-3480 | 3110-3010 | 1723 | 1700 | 1660 |
| 11 | 3460 | 3190-3120 | 1647a | 1720-1722 | 1620 |
| - | - | 1708b | - | - | |
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Lacova, M.; Gasparova, R.; Loos, D.; Liptay, T.; Pronayova, N. Effect of Microwave Irradiation on the Condensation of 6-Substituted 3-Formylchromones with Some Five-membered Heterocyclic Compounds. Molecules 2000, 5, 167-178. https://doi.org/10.3390/50200167
Lacova M, Gasparova R, Loos D, Liptay T, Pronayova N. Effect of Microwave Irradiation on the Condensation of 6-Substituted 3-Formylchromones with Some Five-membered Heterocyclic Compounds. Molecules. 2000; 5(2):167-178. https://doi.org/10.3390/50200167
Chicago/Turabian StyleLacova, Margita, Renata Gasparova, Dusan Loos, Tibor Liptay, and Nada Pronayova. 2000. "Effect of Microwave Irradiation on the Condensation of 6-Substituted 3-Formylchromones with Some Five-membered Heterocyclic Compounds" Molecules 5, no. 2: 167-178. https://doi.org/10.3390/50200167