Design and Synthesis of a Conformationally Rigid Mimic of the Dihydropyrimidine Calcium Channel Modulator SQ 32,926

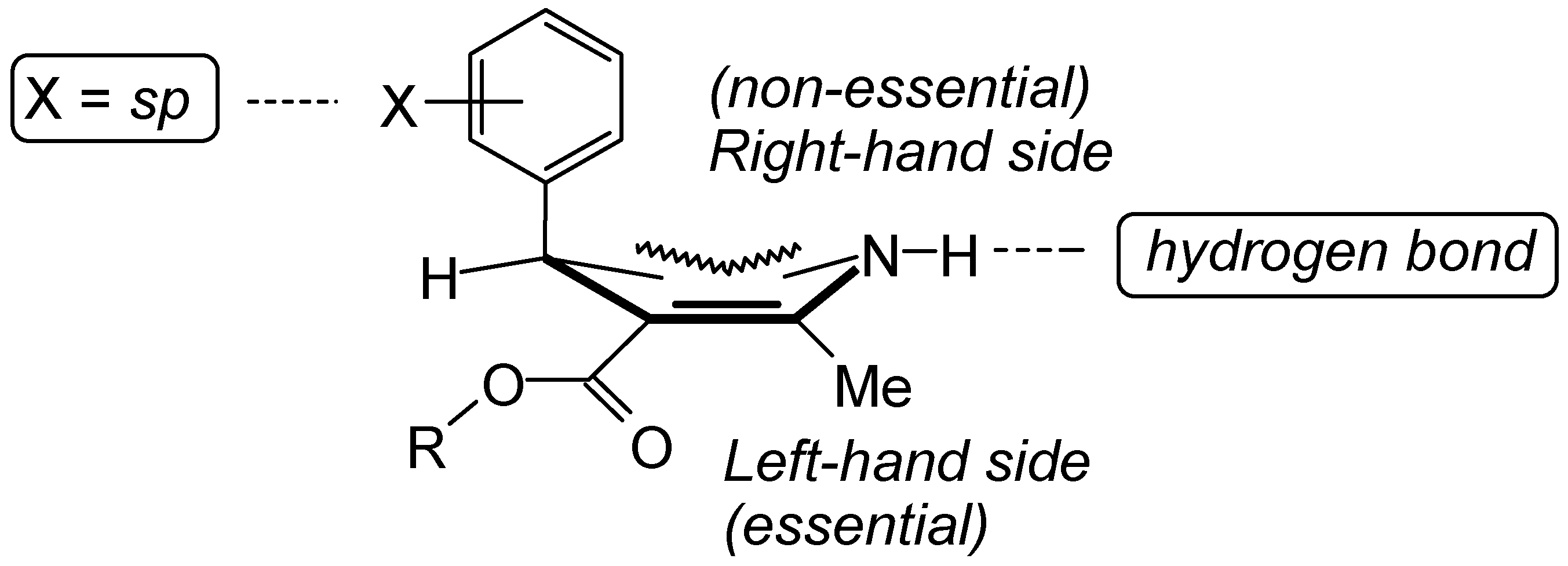{kind=link}
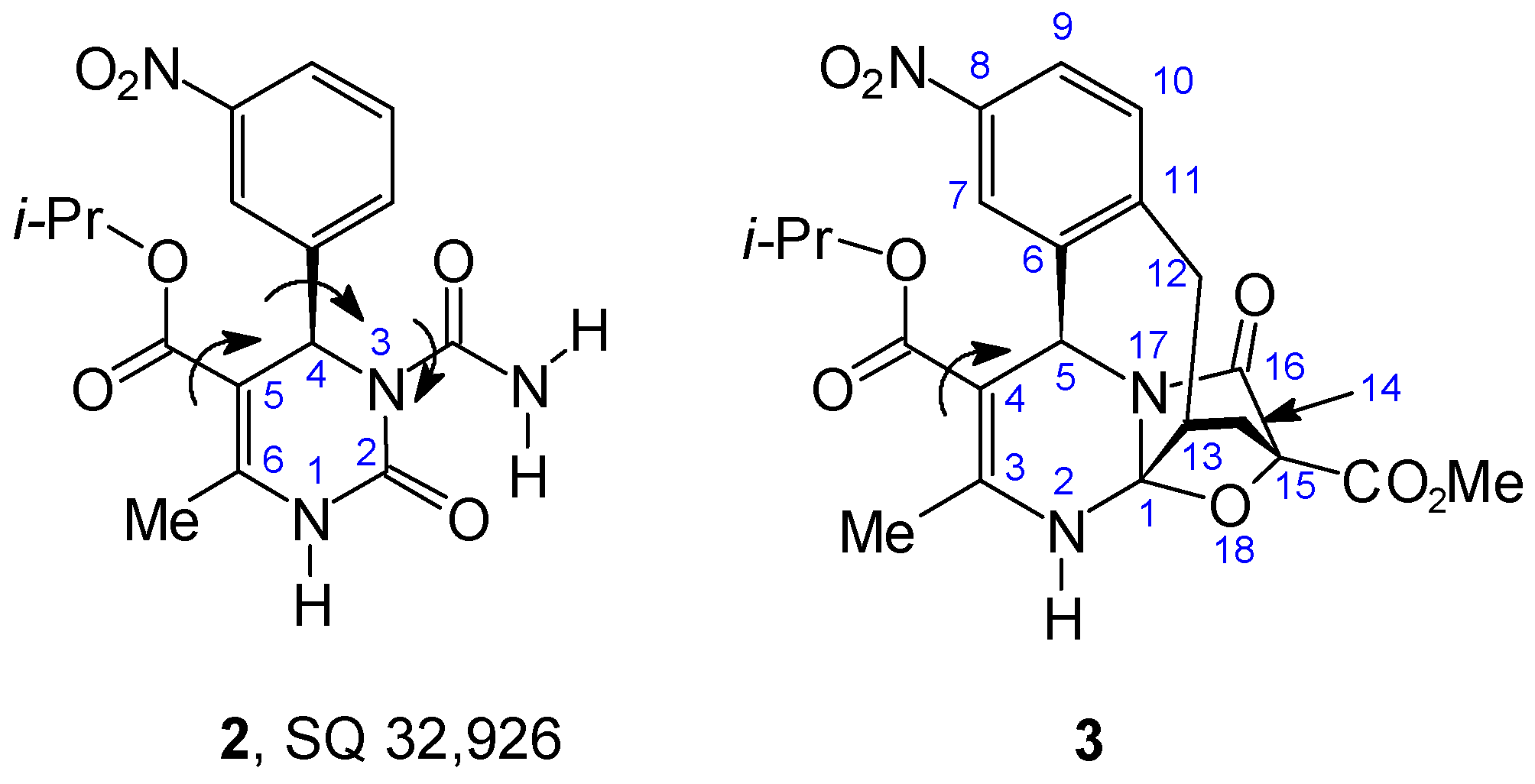{kind=link}
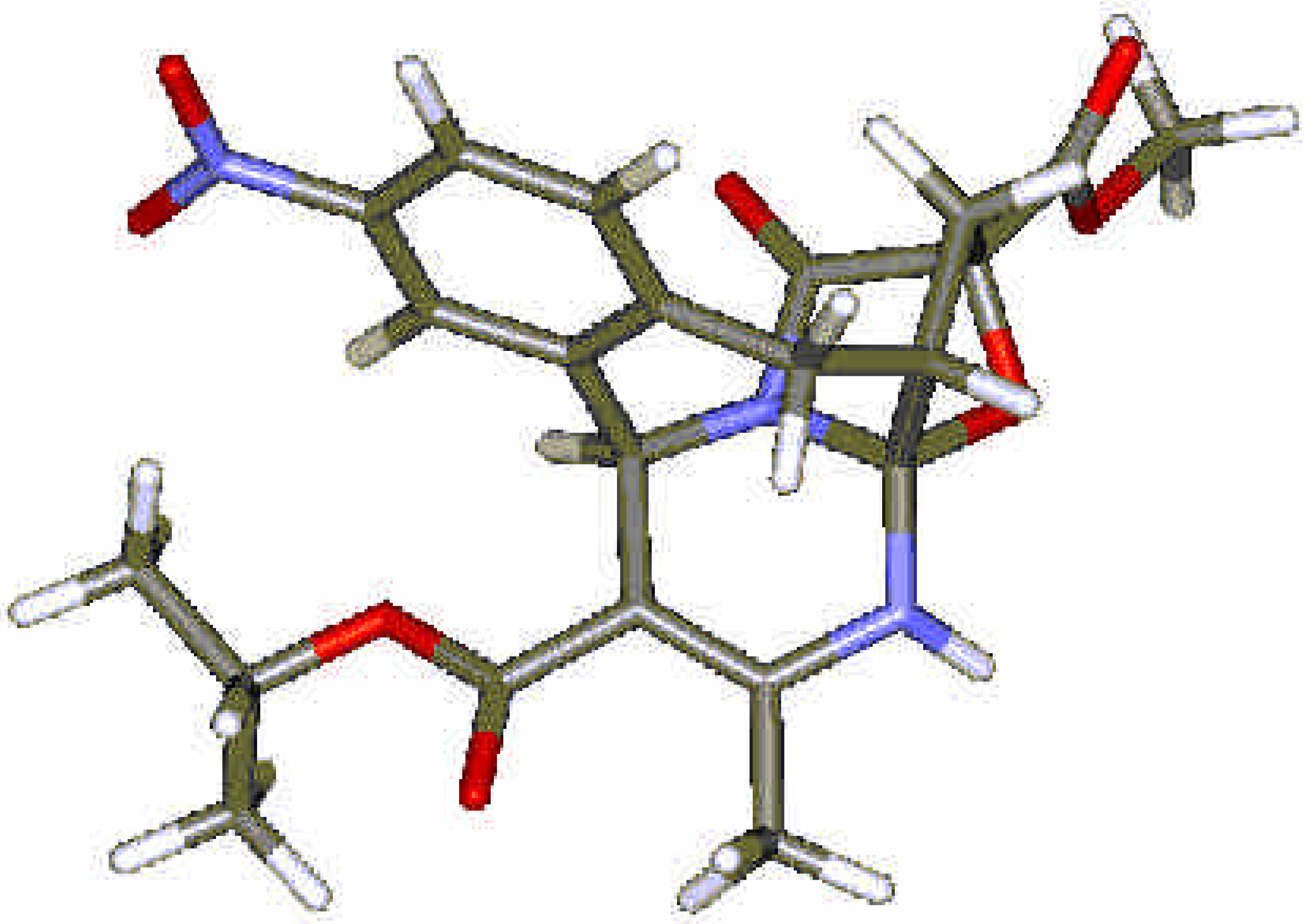{kind=link}
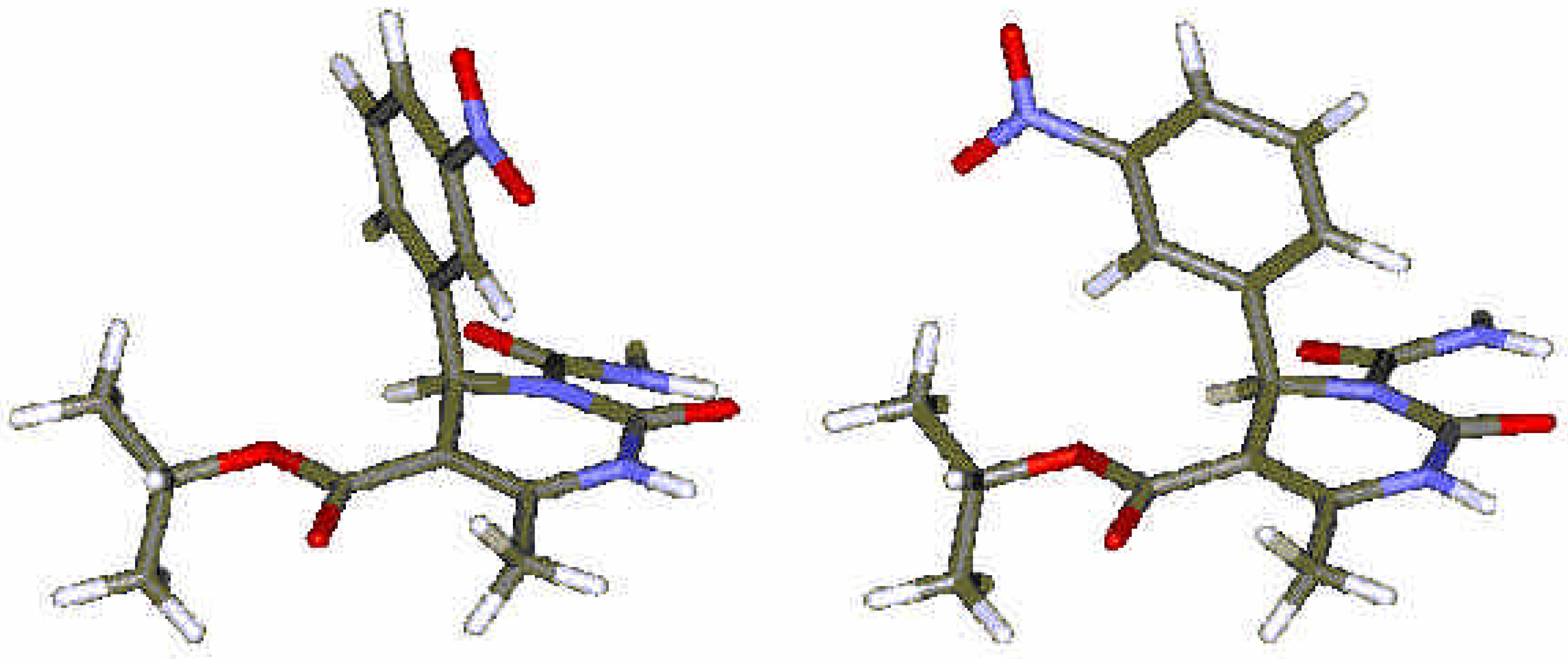{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
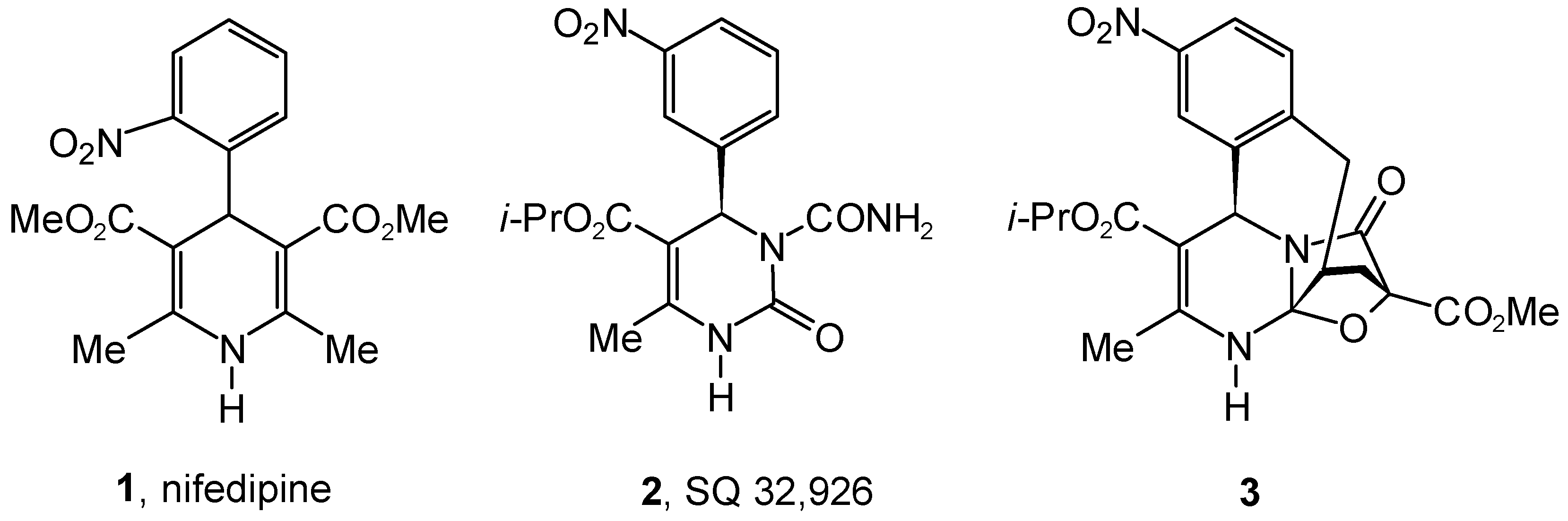

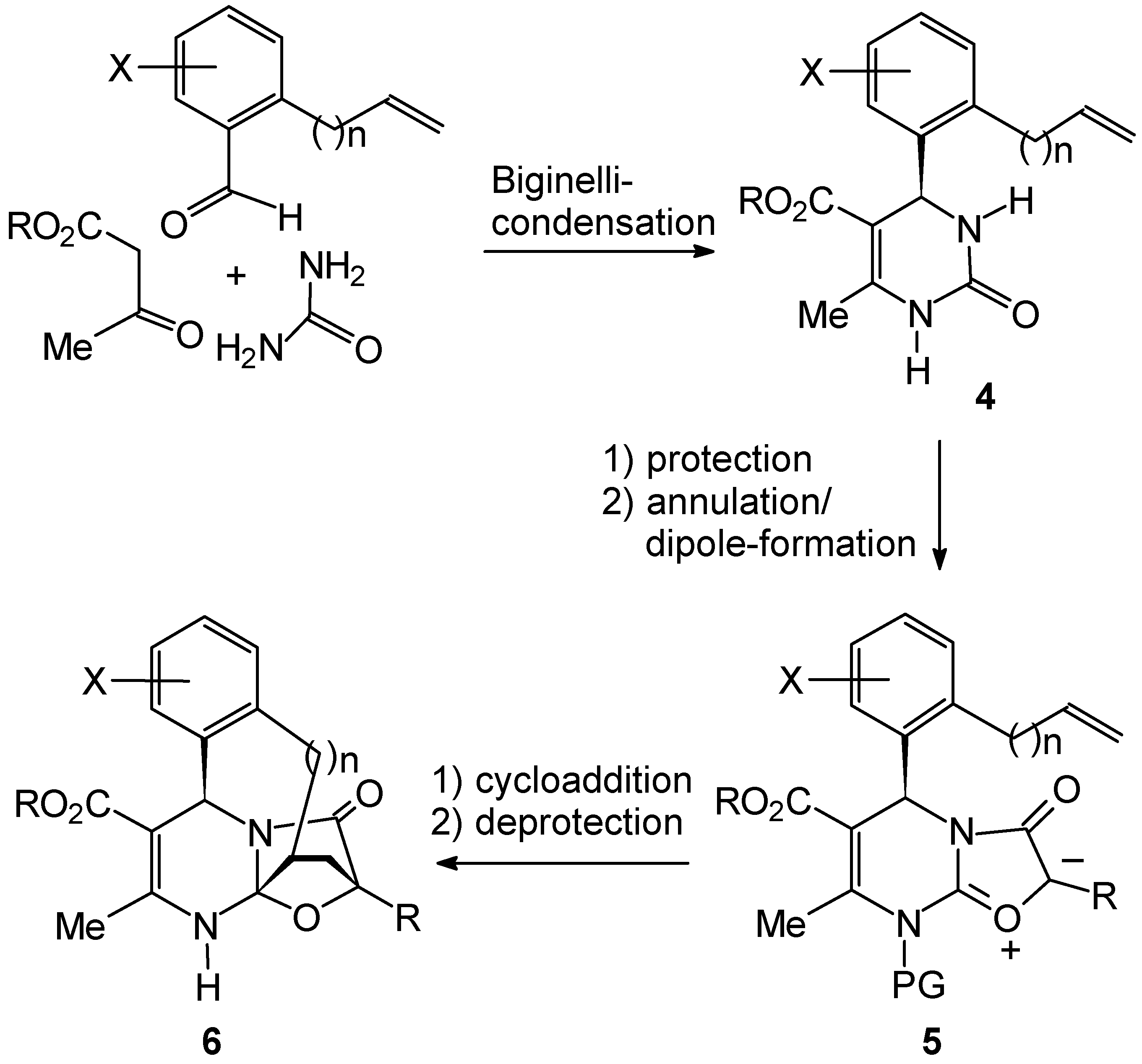
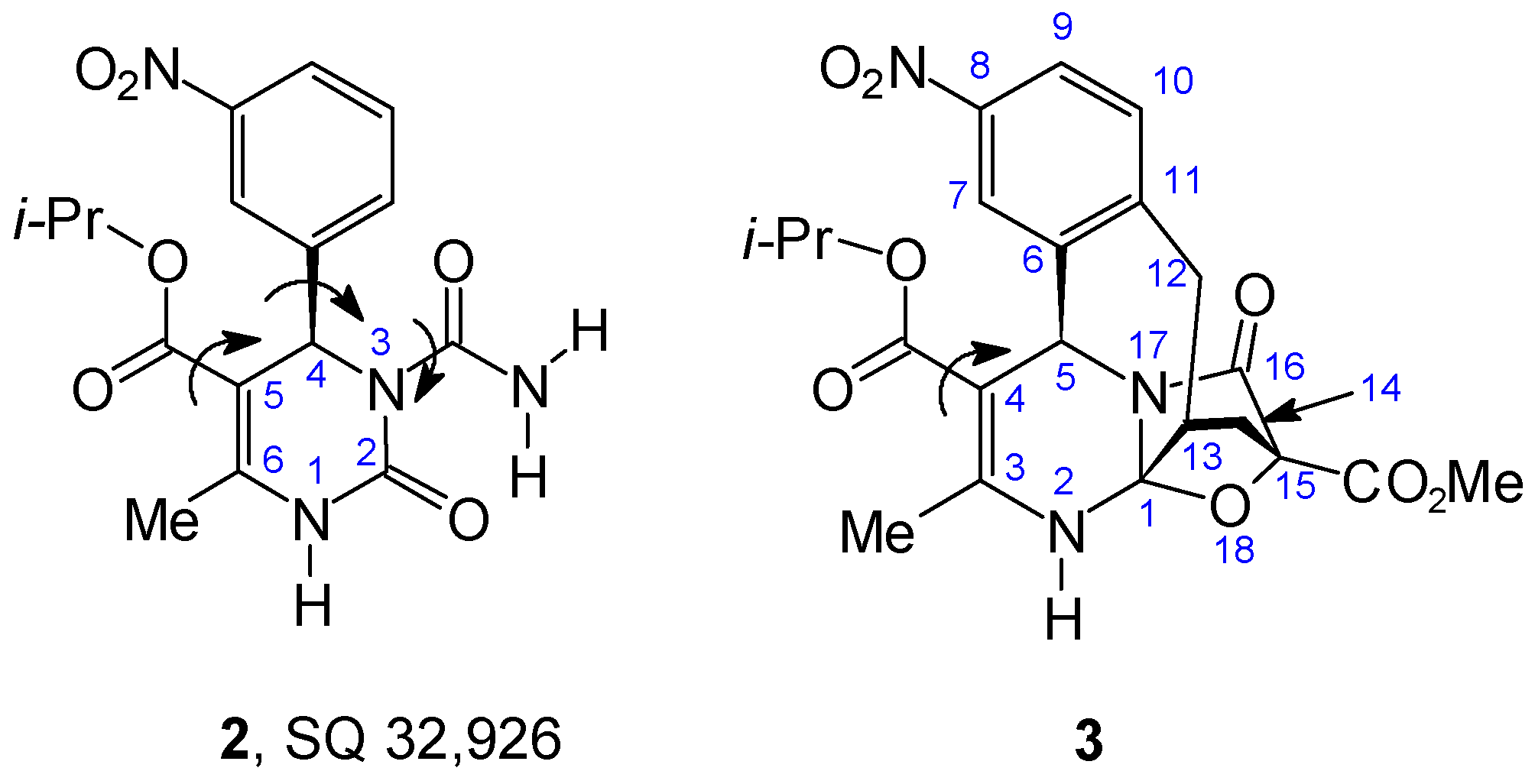
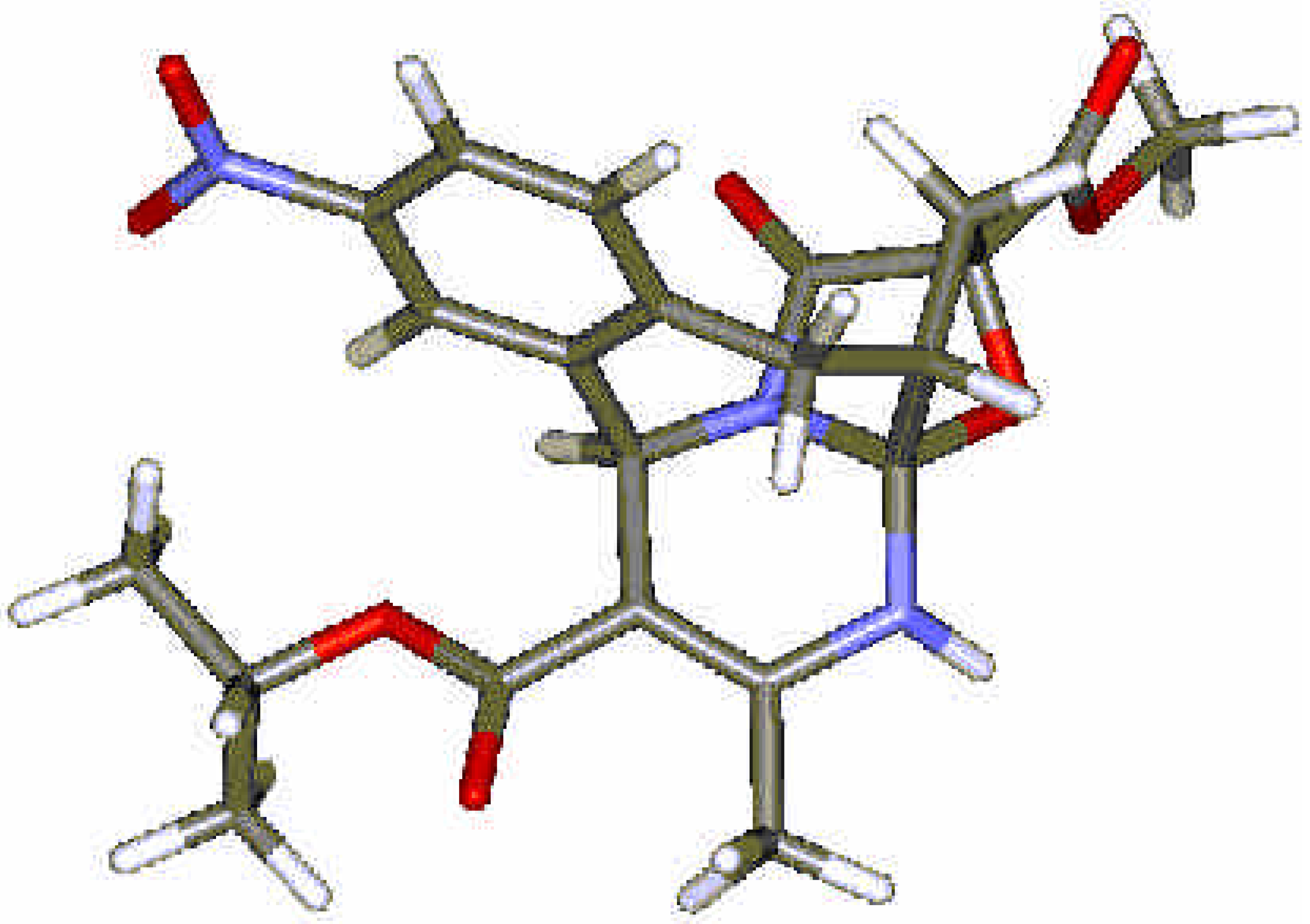
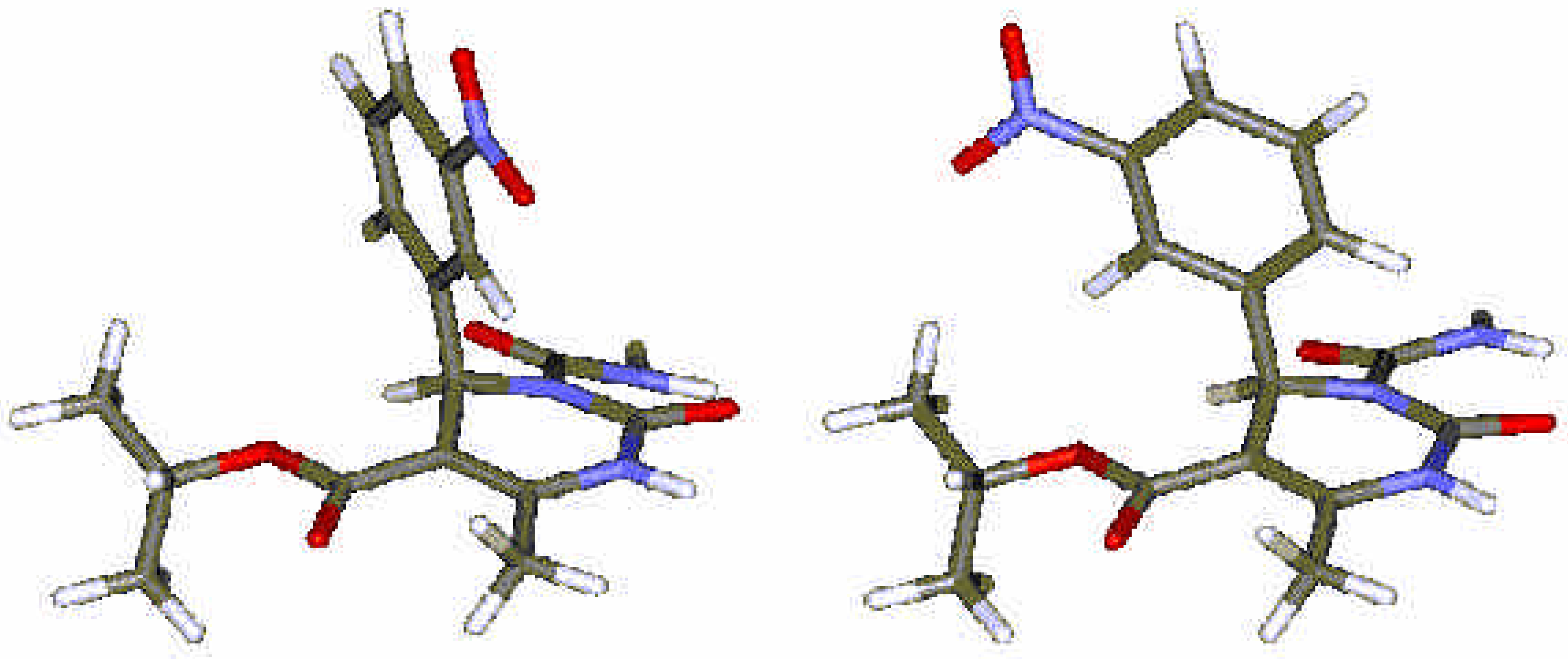
Results and Discussion
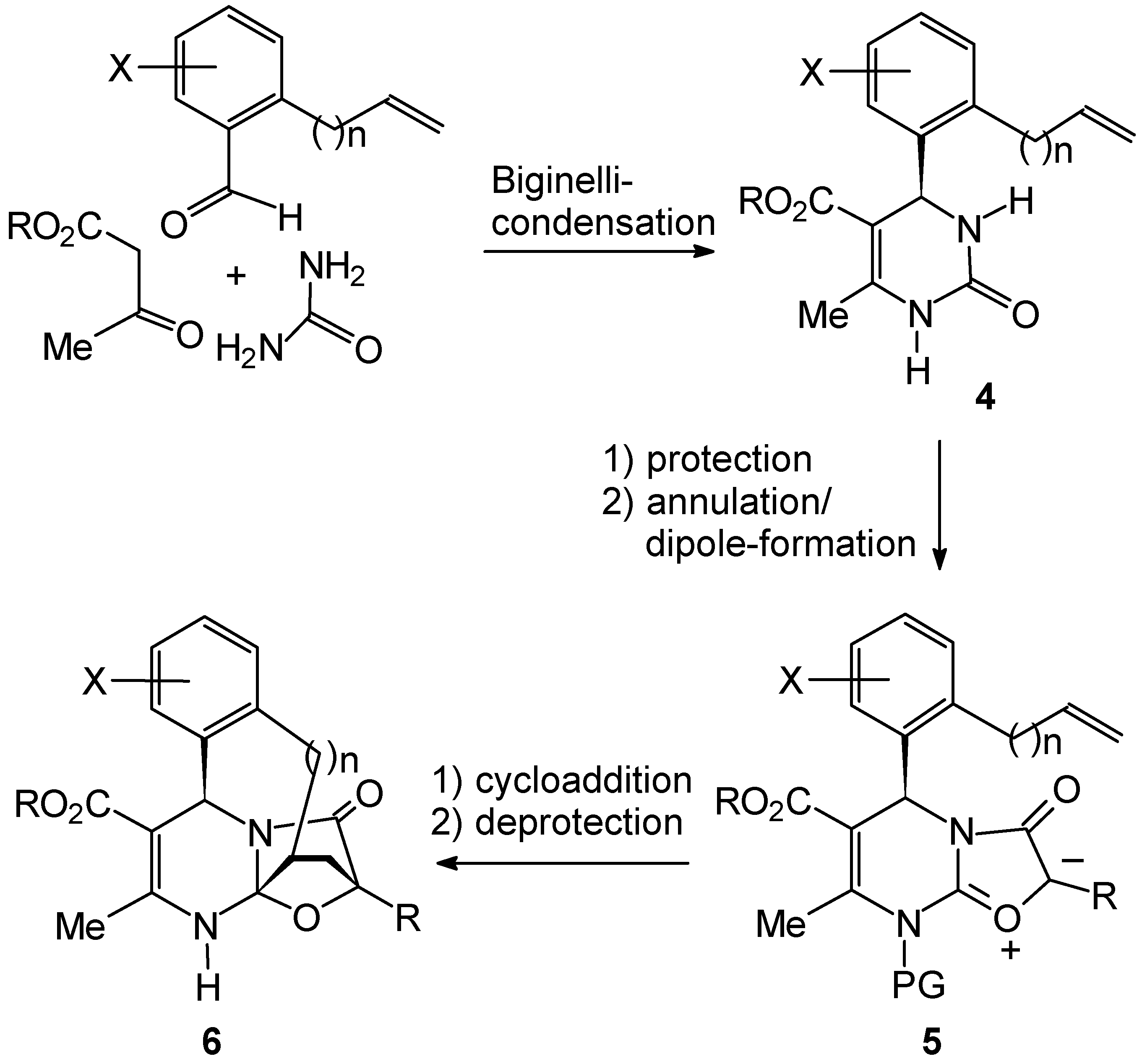
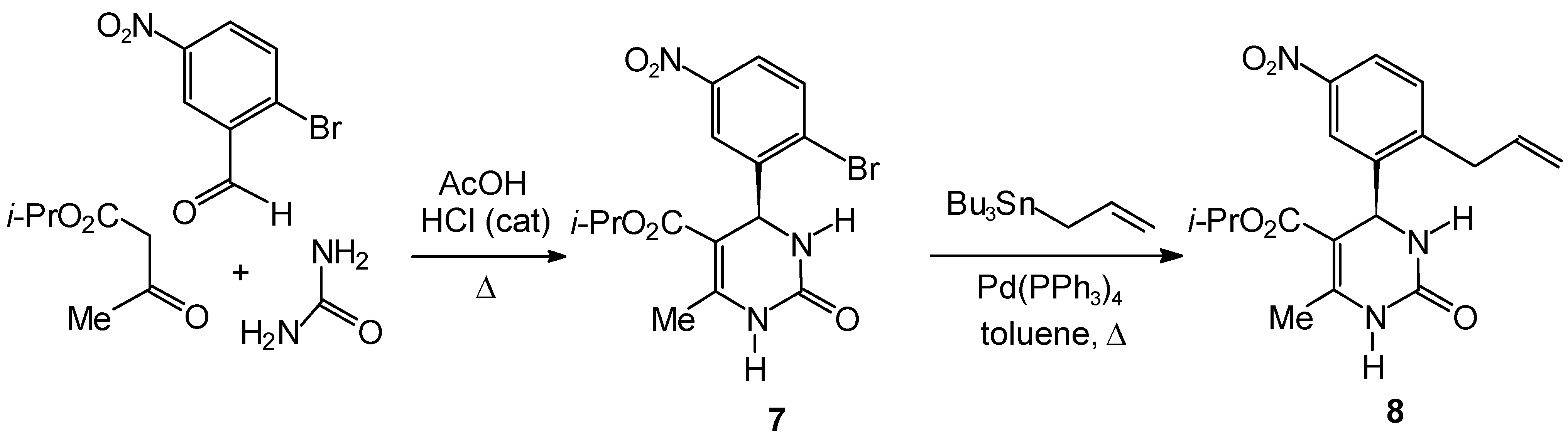

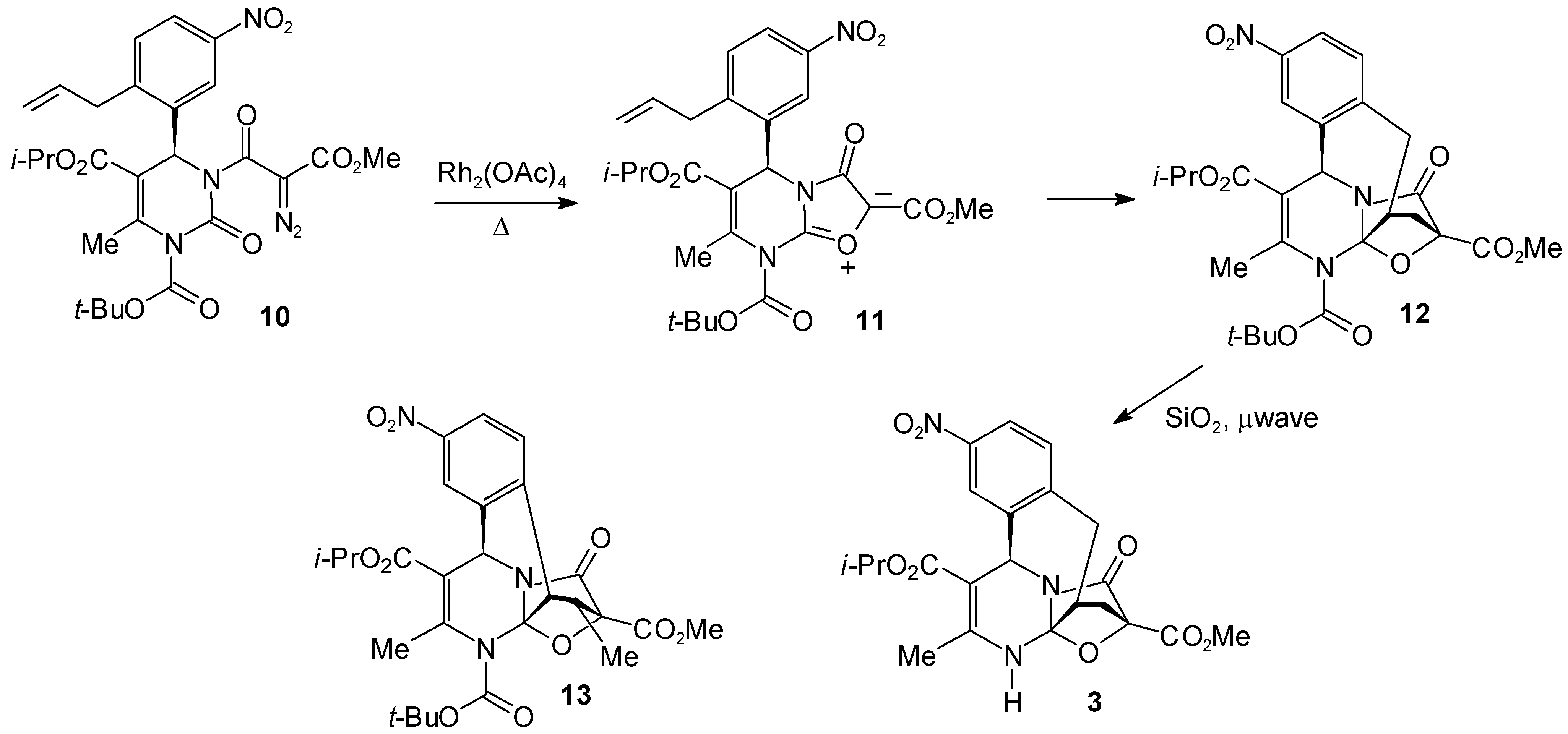
Experimental
General Procedures and Materials
Computational Methods
2-Bromo-5-nitrobenzaldehyde
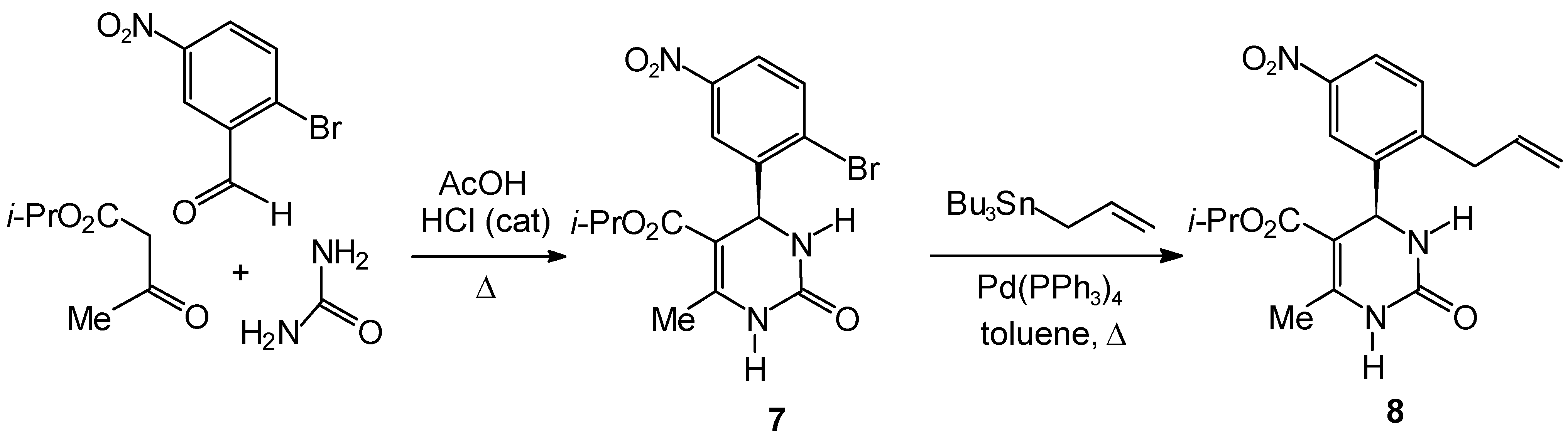
Isopropyl 4-(2-Bromo-5-nitrophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (7)
Isopropyl 4-(2-Allyl-5-nitrophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8)
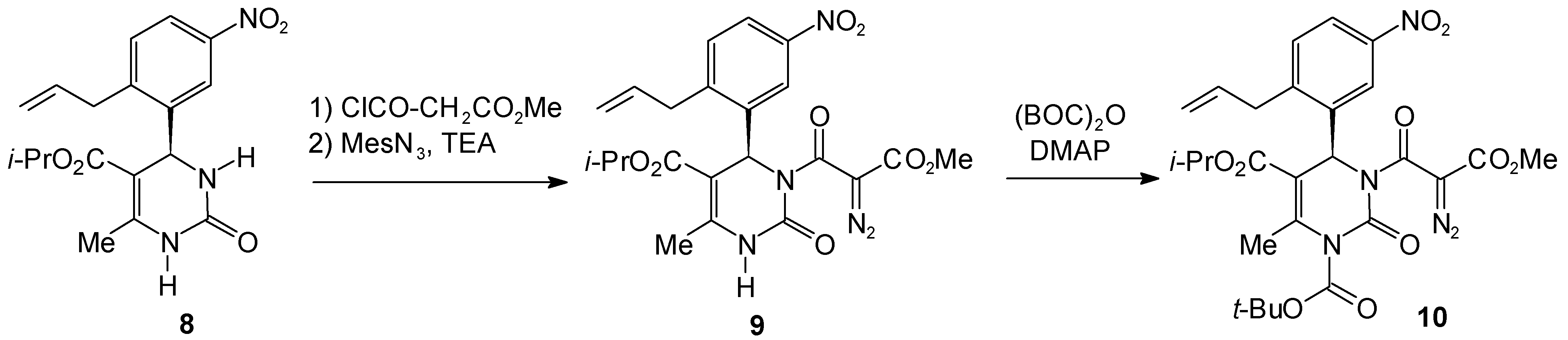
Isopropyl 4-(2-Allyl-5-nitrophenyl)-3-[2-diazo-2-[(methyloxy)carbonyl]acetyl]-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (9)
1-tert.-Butyl 5-Isopropyl 4-(2-Allyl-5-nitrophenyl)-6-methyl-3-[2-diazo-2-[(methyloxy)carbonyl]-acetyl]-2-oxo-1,2,3,4-tetrahydro-1,5-pyrimidinedicarboxylate (10)
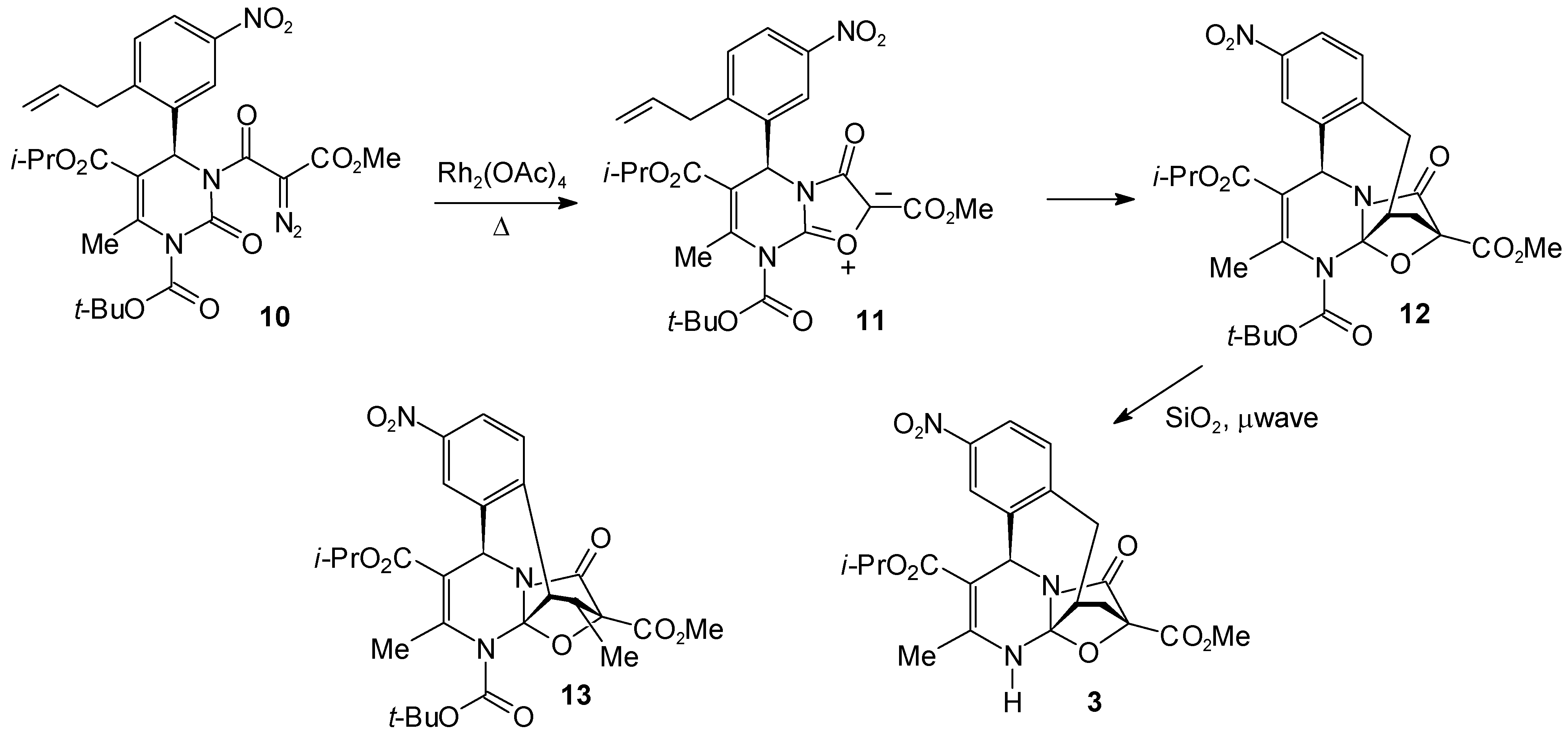
4-Isopropyl 15-Methyl 2-tert.-Butyl 3-Methyl-8-nitro-16-oxo-18-oxa-2,17-diazapentacyclo[13.2.1.01,13.05,17.06,11]octadeca-3,6,8,10-tetraene-2,4,15-tricarboxylate (12)
4-Isopropyl 15-Methyl 3-Methyl-8-nitro-16-oxo-18-oxa-2,17-diazapentacyclo[13.2.1.01,13.05,17.06,11]octadeca-3,6,8,10-tetraene-4,15-dicarboxylate (3)
Acknowledgements
References and Notes
- Janis, R. A.; Silver, P. J.; Triggle, D. J. Adv. Drug Res. 1987, 16, 309.
- Bossert, F.; Vater, W. Med. Res. Rev. 1989, 9, 291. [PubMed]
- Cho, H.; Ueda, M.; Shima, K.; Mizuno, A.; Hayashimatsu, M.; Ohnaka, Y.; Takeuchi, Y.; Hamaguchi, M.; Aisaka, K.; Hidaka, T.; Kawai, M.; Takeda, M.; Ishihara, T.; Funahashi, K.; Satah, F.; Morita, M.; Noguchi, T. J. Med. Chem. 1989, 32, 2399. [PubMed]
- Atwal, K.; Rovnyak, G. C.; Schwartz, J.; Moreland, S.; Hedberg, A.; Gougoutas, J. Z.; Malley, M. F.; Floyd, D. M. J. Med. Chem. 1990, 33, 1510. [PubMed]
- Atwal, K. S.; Rovnyak, G. C.; Kimball, S. D.; Floyd, D. M.; Moreland, S.; Swanson, B. N.; Gougoutas, J. Z.; Schwartz, J.; Smillie, K. M.; Malley, M. F. J. Med. Chem. 1990, 33, 2629. [PubMed]
- Atwal, K. S.; Swanson, B. N.; Unger, S. E.; Floyd, D. M.; Moreland, S.; Hedberg, A.; O’Reilly, B. C. J. Med. Chem. 1991, 34, 806. [PubMed]
- Rovnyak, G. C.; Atwal, K. S.; Hedberg, A.; Kimball, S. D.; Moreland, S.; Gougoutas, J. Z.; O’Reilly, B. C.; Schwartz, J.; Malley, M. F. J. Med. Chem. 1992, 35, 3254. [PubMed]
- Grover, G. J.; Dzwonczyk, S.; McMullen, D. M.; Normadinam, C. S.; Sleph, P. G.; Moreland, S. J. J. Cardiovasc. Pharmacol. 1995, 26, 289. [PubMed]
- (a) Rovnyak, G. C.; Kimball, S. D.; Beyer, B.; Cucinotta, G.; DiMarco, J. D.; Gougoutas, J.; Hedberg, A.; Malley, M.; McCarthy, J. P.; Zhang, R.; Moreland, S. J. Med. Chem. 1995, 38, 119. [PubMed] (b) Triggle, D. J.; Padmanabhan, S. Chemtracts: Org. Chem. 1995, 8, 191.
- Review: Goldman, S.; Stoltefuss, J. Angew. Chem. 1991, 103, 1587, Angew. Chem., Int. Ed. Engl. 1991, 30, 1559 and references therein.
- Schleifer, K.-J. J. Med. Chem. 1999, 42, 2204. [PubMed]
- Striessnig, J.; Grabner, M.; Mitterdorfer, J.; Hering, S.; Sinnegger, M.; Glossmann, H. Trends Pharmacol. Sci. 1998, 19. 108 and refs cited therein. [PubMed]
- For a review on this methodology, see: Osterhout, M. H.; Nadler, W. R.; Padwa, A. Synthesis 1994, 123.
- Kappe, C. O.; Peters, K.; Peters, E. M. J. Org. Chem. 1997, 62, 3109. [PubMed]
- Jauk, B.; Belaj, F.; Kappe, C. O. J. Chem. Soc., Perkin Trans. 1 1999, 307.
- For a review of the Biginelli reaction, see: Kappe, C. O. Tetrahedron 1993, 49, 6937.
- (a) Hu, E. H.; Sidler, D. R.; Dolling, U.-H. J. Org. Chem. 1998, 63, 3454. (b) Kappe, C. O.; Falsone, F. S. Synlett 1998, 718. (c) Bigi, F.; Carloni, S.; Frullanti, B.; Maggi, R.; Sartori, G. Tetrahedron Lett. 1999, 40, 3465. (d) Singh, K.; Singh, J.; Deb, P. K.; Singh, H. Tetrahedron 1999, 55, 12873. (e) Kappe, C. O.; Kumar, D.; Varma, R. S. Synthesis 1999, 1799. (f) Kappe, C. O. Bioorg. Med. Chem. Lett. 2000, 10, 49. (g) Lewandowski, K.; Murer, P.; Svec, F.; Fréchet, J. M. J. J. Comb. Chem. 1999, 1, 105.
- Folkers, K.; Harwood, H. J.; Johnson, T. B. J. Am. Chem. Soc. 1932, 54, 3751.
- Meegalla, S. K.; Taylor, N. J.; Rodrigo, R. J. Org. Chem. 1992, 57, 2422.
- Farina, V.; Krishnamurthy, V.; Scott, W. J. The Stille Reaction; Wiley: New York, 1998. [Google Scholar]
- Padwa, A.; Austin, D. J.; Price, A. T.; Weingarten, M. D. Tetrahedron 1996, 52, 3247.
- Taber, D. F.; Ruckle, R. E., Jr.; Hennesy, M. J. Org. Chem. 1986, 51, 4077.
- Spry, D. D.; Snyder, N. J.; Bhala, A. R.; Pasini, C. E.; Indelicato, J. M. Heterocycles 1987, 26, 2911.
- Kappe, C. O. Tetrahedron Lett. 1997, 38, 3323.
- Siro, J. G.; Martin, J.; Garcia-Navio, J. L.; Remuinan, M. J.; Vaquero, J. J. Synlett 1998, 147.
- Kappe, C. O.; Fabian, W. M. F.; Semones, M. A. Tetrahedron 1997, 53, 2803.
- Shishkin, O. V.; Solomovich, E. V.; Vakula, V. M.; Yaremenko, F. G. Russ. Chem. Bull. 1997, 46, 1938.
- Fabian, W. M. F.; Semones, M. A.; Kappe, C. O. J. Mol. Struct. (Theochem) 1998, 432, 219.
- Krenn, W.; Verdino, P.; Uray, G.; Faber, K.; Kappe, C. O. Chirality 1999, 11, 659. [PubMed]
- Schnell, B.; Strauss, U. T.; Verdino, P.; Faber, K.; Kappe, C. O. Tetrahedron: Asymmetry 2000, 11. in press.
- Samples Availability: Available from the authors.
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Jauk, B.; Pernat, T.; Kappe, C.O. Design and Synthesis of a Conformationally Rigid Mimic of the Dihydropyrimidine Calcium Channel Modulator SQ 32,926. Molecules 2000, 5, 227-239. https://doi.org/10.3390/50300227
Jauk B, Pernat T, Kappe CO. Design and Synthesis of a Conformationally Rigid Mimic of the Dihydropyrimidine Calcium Channel Modulator SQ 32,926. Molecules. 2000; 5(3):227-239. https://doi.org/10.3390/50300227
Chicago/Turabian StyleJauk, Birgit, Tetiana Pernat, and C. Oliver Kappe. 2000. "Design and Synthesis of a Conformationally Rigid Mimic of the Dihydropyrimidine Calcium Channel Modulator SQ 32,926" Molecules 5, no. 3: 227-239. https://doi.org/10.3390/50300227