Molecular Interactions Between the Active Sites of RGD (Arg-Gly-Asp) with its Receptor (Integrine)
Departamento de Química, Facultad de Química Bioquímica y Farmacia, Universidad Nacional de San Luis, Chacabuco y Pedernera, CP:5700, San Luis, Argentina
*
Author to whom correspondence should be addressed.
Molecules 2000, 5(3), 583-584; https://doi.org/10.3390/50300583
Published: 22 March 2000
{kind=link}
{kind=link}
Abstract
:A study of the molecular interactions between the active sites of RGD (Arg-Gly-Asp) with it receptor using simulations is reported. Our calculations indicate that the gua-nidine-carboxylate complex is energetically favoured with respect to the guanidine-methyl tetrazole complex.
Introduction
It is well documented that peptides and peptidomimetics containing the RGD (Arg-Gly-Asp) sequence are capable of effectively inhibiting the binding of fibrinogen to the GP IIb - IIIa receptor (1). Thus these compounds show promise for improving the immediate treatment of arterial occlusion.
On the other hand both guanidine and carboxylate groups play a determinant role in the recognition and dock process (2).
In the present work we report a simulation study for the mo-lecular interactions between model systems which are mi-metizing the guanidine-carboxylate and guanidine-tetrazole in-teractions.
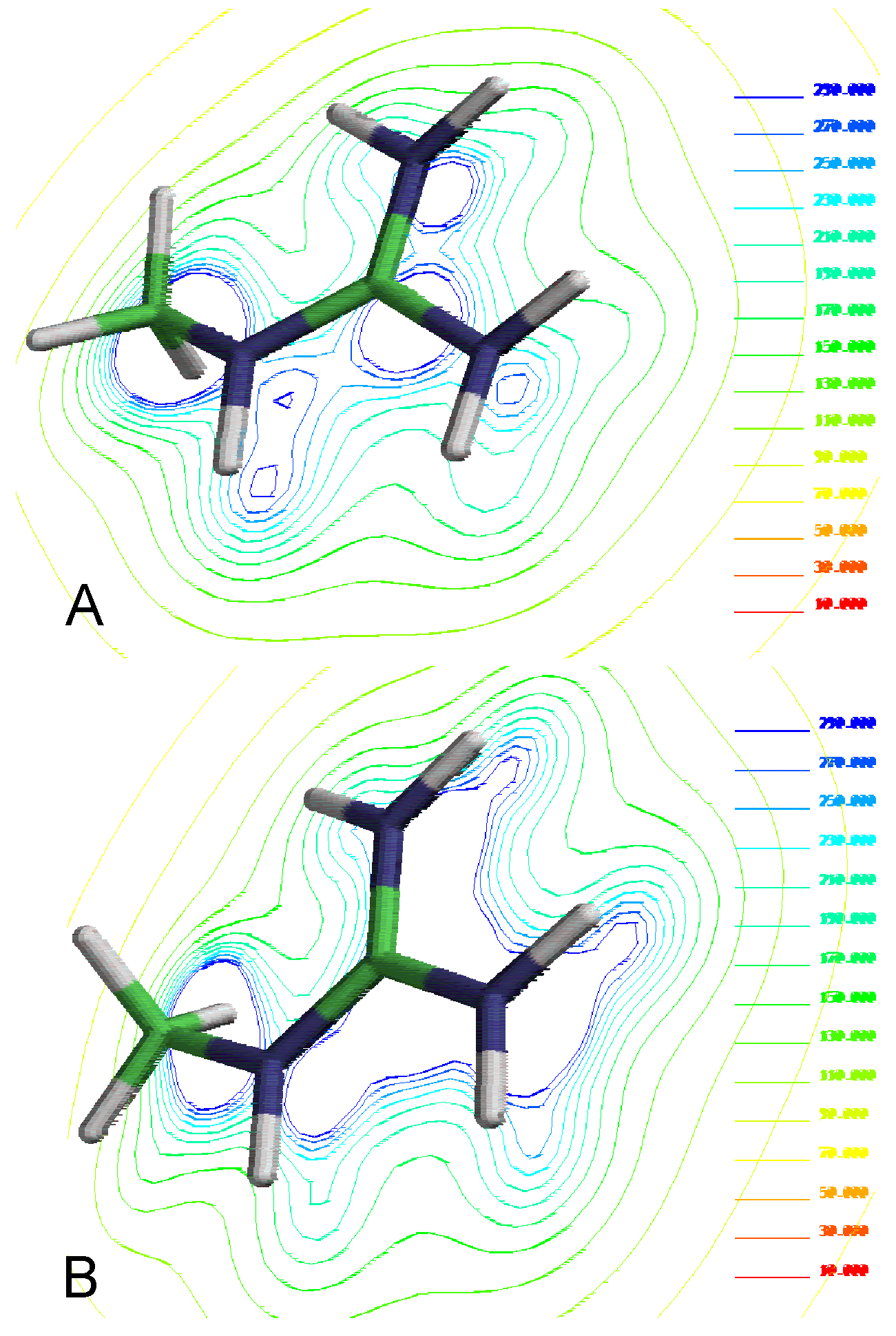
Figure 1.
Molecular electrostatic potential from different theory levels by methyl guanidine cations. A) Semiempirical method PM3 and B) Ab-initio HF/3-21G*
Figure 1.
Molecular electrostatic potential from different theory levels by methyl guanidine cations. A) Semiempirical method PM3 and B) Ab-initio HF/3-21G*

Calculation Methods
Guanidine-carboxylate and guanidine- tetrazole interactions were optimized using MM2, AM1 and PM3 calculations. These structures were finally optimized using ab-initio RHF/3-21G* calculations. All calculations were carried out using the SPAR- TAN program. With the aim to obtain a more suitable elec-tronic description we performed B3LYP / 6-31++G** single point calculations from the GAUSSIAN 94 program.
The used equation was: ΔHF = ΣΔH Products - ΣΔH Reactives
Results and Discussion
Our results show a significant dif-ference for the electronic description of the molecules using different levels of theory (Figure 1). It should be noted a believable electronic description of these compounds is an essential requirement in order to explain the molecular interactions involved in this proc-ess.
The ab-initio results indicate a positive charge distribution focused on acidic – protons the preference for the planar forms of the complex. In contrast the semiempirical results predict a positive charge distribution on the imine group indicating that no-planar forms are also available.
Figure 2.
General scheme of interaction in study. A) Between guanidine cation and anion carboxylate. B) Between of guanidine cation and anión tetrazole.
Figure 2.
General scheme of interaction in study. A) Between guanidine cation and anion carboxylate. B) Between of guanidine cation and anión tetrazole.
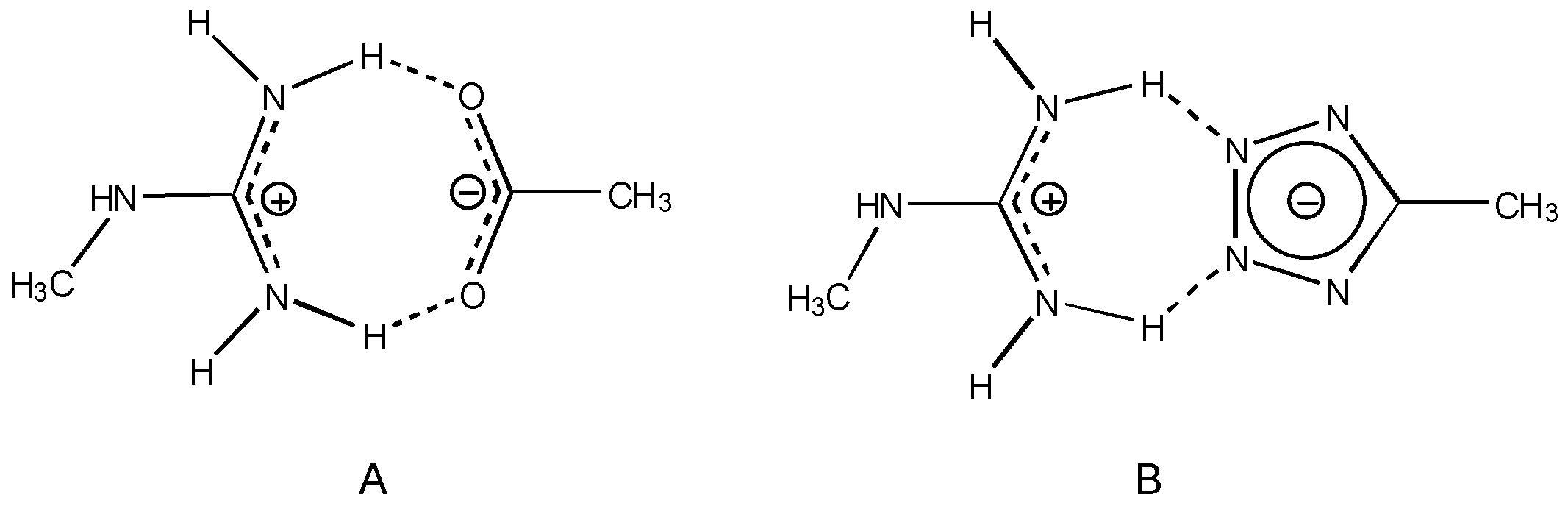
These results show that it is necessary to perform calculations at high level of theory (at least at RHF /3-21G) to obtain an acceptable electronic description which is essential to evaluate the complex formation process.
From the medicinal chemistry point of view it is interesting to note that our results suggest that the guanidine-carboxylate interactions are energetically favoured with respect to the guanidine-tetrazole interactions (Figure 2). These results indicate that tetrazole group it is not good enough to replace the carboxylate group and therefore they are in a complete agreement with the experimental results re-ported for the above groups.
Acknowledgements
The research was supported by grants from the Fundación Antorchas and Univer-sidad Nacional de San Luis (UNSL). Dr R.D. Enriz is carrier researcher of CONICET (Concejo Na-cional de Ciencia y Tecnología) Argentina.
References and Notes
- Plow, E. F.; Pierschbachers, M.N.; Ruaslahti, E.; Marguerie, G.; Ginsberg, M. V. Blood 1987, 70, 110. [PubMed]
- Sinoh, J; Thosnton, J.M.; Snarey, M.; Campbel, S. F. FEBS Lett. 1987, 224, 161.
Share and Cite
MDPI and ACS Style
Suivre, F.; Floridia, R.; Giannini, F.; Rodriguez, A.; Enriz, R.; Jauregui, E. Molecular Interactions Between the Active Sites of RGD (Arg-Gly-Asp) with its Receptor (Integrine). Molecules 2000, 5, 583-584. https://doi.org/10.3390/50300583
AMA Style
Suivre F, Floridia R, Giannini F, Rodriguez A, Enriz R, Jauregui E. Molecular Interactions Between the Active Sites of RGD (Arg-Gly-Asp) with its Receptor (Integrine). Molecules. 2000; 5(3):583-584. https://doi.org/10.3390/50300583
Chicago/Turabian StyleSuivre, F., R. Floridia, F. Giannini, A. Rodriguez, R. Enriz, and E. Jauregui. 2000. "Molecular Interactions Between the Active Sites of RGD (Arg-Gly-Asp) with its Receptor (Integrine)" Molecules 5, no. 3: 583-584. https://doi.org/10.3390/50300583