Synthesis of a Highly Functionalized Triquinane: Studies Towards a Total Synthesis of Subergorgic Acid and Its Analogues

Oceanographic Center, Nova Southeastern University, 8000 North Ocean Drive, Dania, 33004, USA
Molecules 2000, 5(4), 674-698; https://doi.org/10.3390/50400674
Submission received: 14 March 2000
/
Accepted: 14 April 2000
/
Published: 20 April 2000
Abstract
:Reaction of enone (11) with the bifunctional Grignard reagent (7) in the presence of copper(I) bromide – dimethyl sulfide, followed by intramolecular alkylation of the resultant chloroketone (15) gave the tricyclic ketone (12). The tricyclic ketone (12) was transformed into the angular triquinane dienedione (9) by means of an 8-step sequence.
Introduction
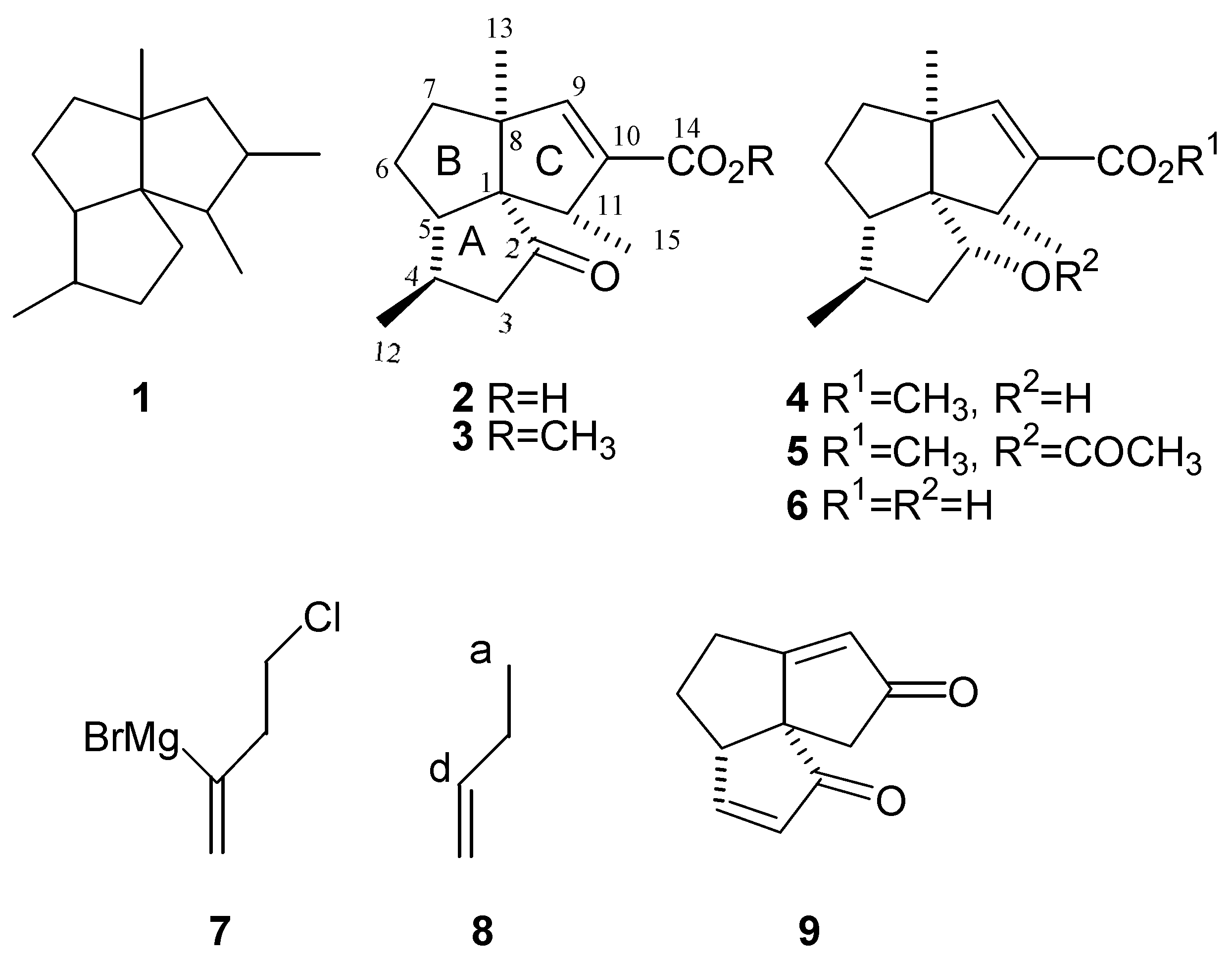
Chemistry of triquinane natural products has been an area of extensive synthetic studies since the mid-1970s [1,2,3,4]. Subergorgic acid is an angular sesquiterpene triquinane with a silphiperfolane skeleton (1) [5,6]. It has been shown that subergorgic acid (2) exhibits cardiotoxic activity [5], anticholinesterase activity [7], and activity against "Soman" toxicity in mice [8].
An access to triquinane natural products has been achieved by means of bifunctional conjunctive reagents [9]. The bifunctional reagent (7), developed by Piers’ group [10,11], corresponds to the 1- butene d2,a4-synthon (8) as it includes a nucleophilic center at the C-2 (d2-synthon) and an electrophilic center at the C-4 (a4-synthon). The reagent (7) is useful for carrying out methylenecyclopentane annulations as it undergoes efficient Cu(I)-catalyzed 1,4-addition to enones [10] followed by cyclization of the intermediate chloroketone. The naturally occurring triquinanes (±)-Δ9(12)-capnellene [10], (±)-penta-lenene [11], (±)-methyl cantabrenonate [12] and (±)-methyl epoxycantabronate [12] have already been prepared via routes in which the described annulation sequence played a key role.
Several synthetic approaches to subergorgic acid (2) (Figure 1) have appeared in the literature [13,14,15,16]. The principal difficulty encountered in the synthesis of subergorgic acid is related to the unusual functionality present in the molecule as compared with other silphiperfolanes. Subergorgic acid and its analogues have an oxygen-containing function at position C-2, a feature that is unknown in other silphiperfolane molecules. An application of the bifunctional reagent (7) toward the synthesis of the highly functionalized triquinane (9) is reported herein. Triquinane (9) has rings A and C functionalized. In the course of its preparation, the keto function was removed from ring B. Therefore, triquinane (9), along with its precursors, allows for the functionalization of any carbon atom on an angular triquinane skeleton. Thus (9) represents an important intermediate in synthesis of naturally occurring triquinanes such as subergorgic acid (2) and its analogues (3-6) [5,6].
Results and Discussion
Preparation of the Enone Ketal (11)
The keto ketal (10), prepared using an established procedure [17], was converted into the corresponding enol silyl ether, by treatment with trimethylsilyl iodide and triethylamine in dichloromethane [18]. The enol silyl ether was oxidized with palladium acetate in acetonitrile [19] to give the enone ketal (11) in 72% overall yield (Scheme 1).
Preparation of the Tricyclic Ketone (12)
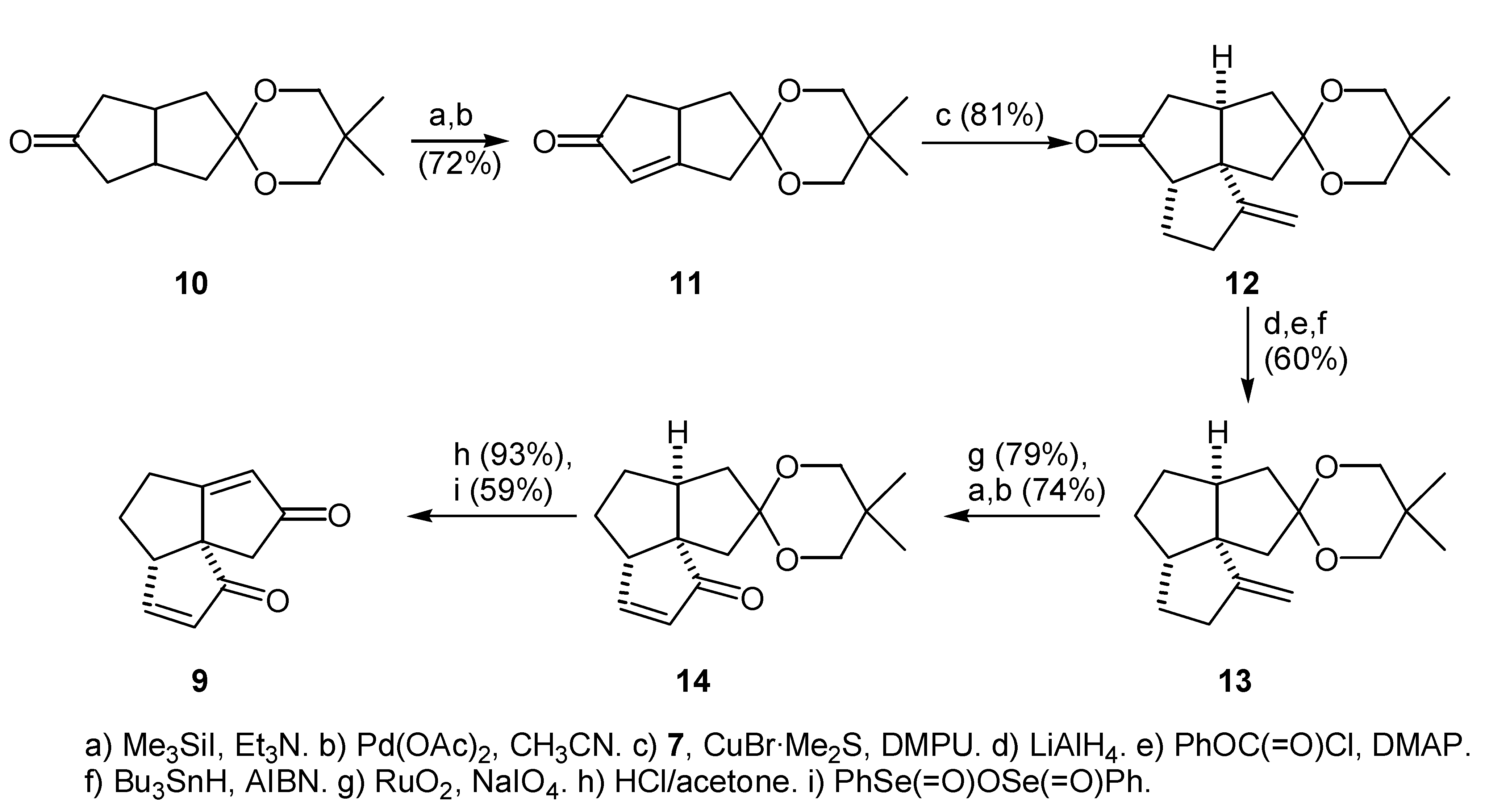
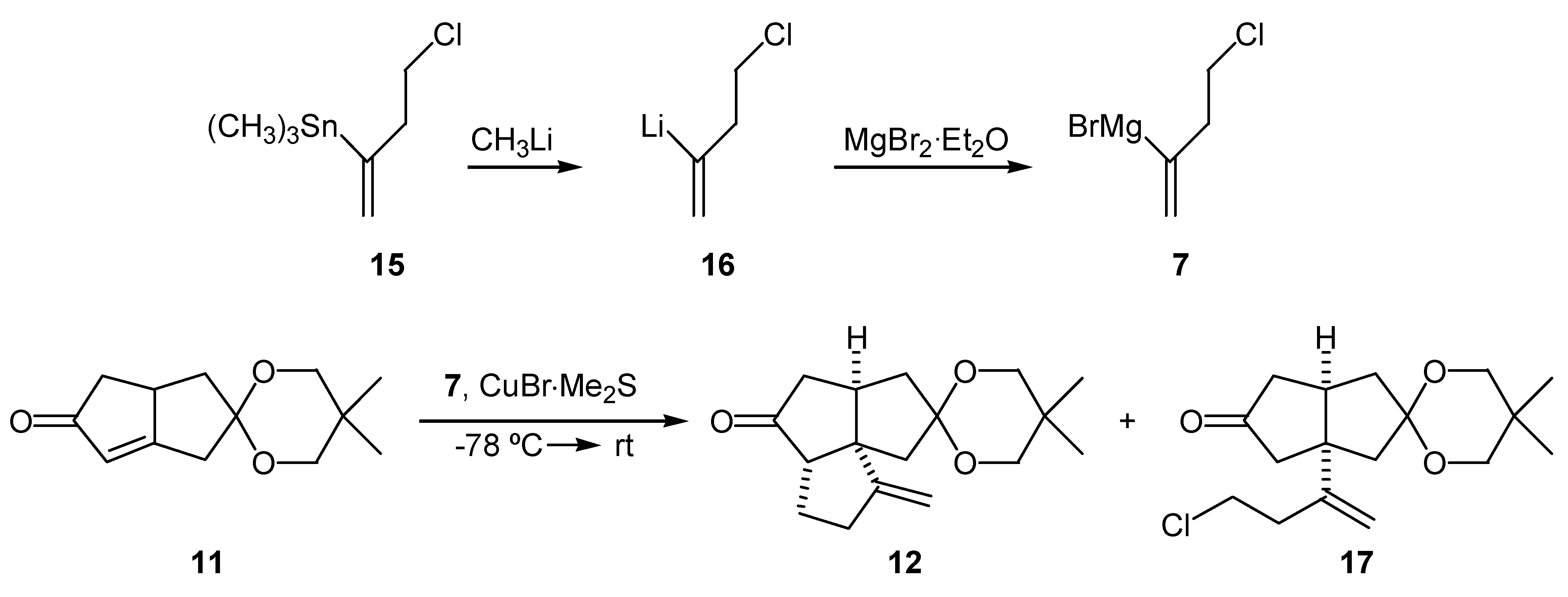
2-Lithio-4-chloro-1-butene (16) was generated by the transmetallation of 4-chloro-2- trimethylstannyl-1-butene (15) with methyllithium following a method developed by Piers’ group [20,21] (Scheme 2). Treatment of the vinyllithium (16) with magnesium bromide-etherate complex, provided the corresponding Grignard reagent (7). The Grignard reagent was employed in the copper bromide-dimethyl sulfide catalyzed 1,4-addition to the ketal enone (11), followed by addition of a complexing agent [22] to provide tricyclic keto ketal (12) (Scheme 2). The ratio of cyclized product (12) to the uncyclized product (17) depended on whether or not an additive was employed prior to warming up of the reaction mixture. Results are summarized in the Table 1. The best results were obtained when 2 equivalents of either HMPA or DMPU (1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)- pyrimidinone) were used as an additive. Due to the high toxicity of HMPA, DMPU was chosen as the additive in subsequent reactions. Under these conditions, the isolated yield of the cyclized product (12) was 81% and was accompanied by 4% of the uncyclized chloro ketone (17), which could be efficiently cyclized upon treatment with potassium hydride in THF. Alternatively, the chloro ketone (17) could be isolated in 77% yield, and then cyclized in 94% yield upon treatment with potassium hydride.
Deoxygenation of the tricyclic ketone (12)
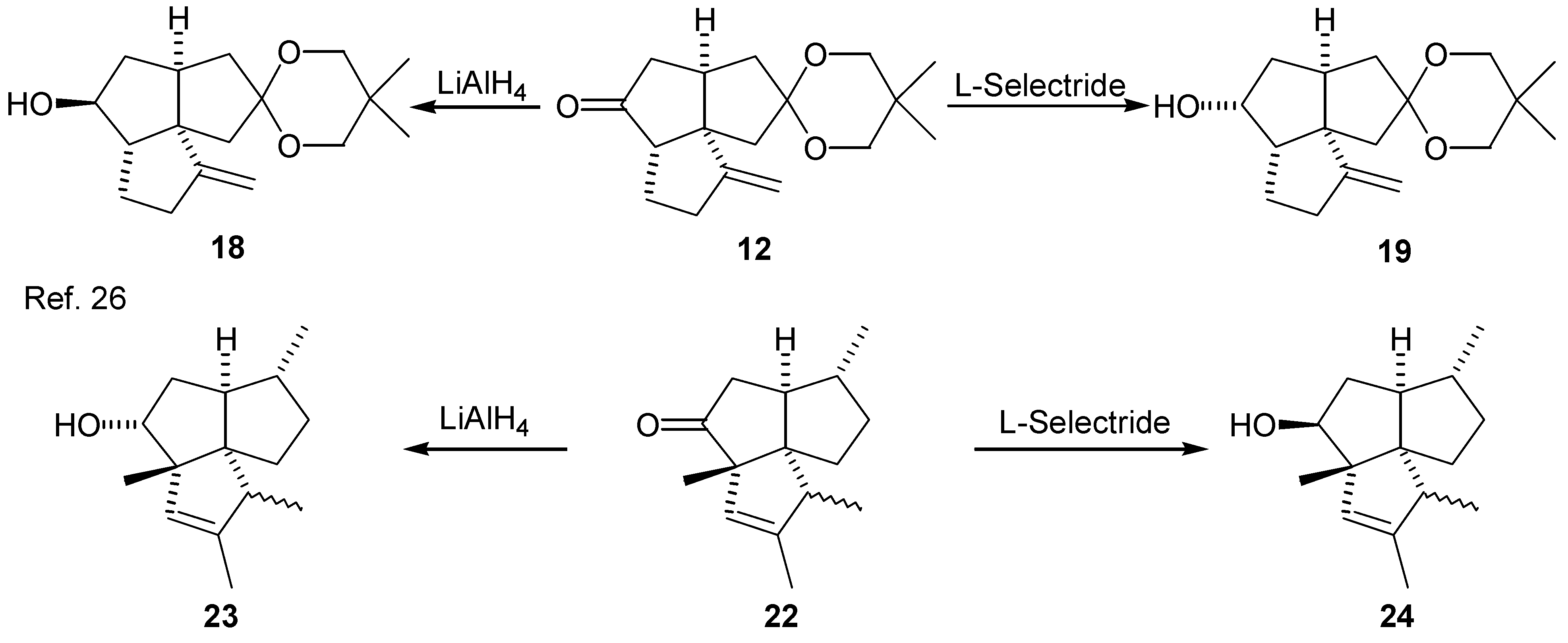
In order to deoxygenate the tricyclic keto ketal (12) according to procedure reported by Robins and coworkers [23,24], the corresponding alcohol (18) or (19) is needed as a precursor. Results from earlier work by Story indicated that the stereochemistry of alcohols may have an effect on the yield and ease of preparation of phenyl thionocarbonate derivatives, and subsequently on the yield of deoxygenated product [25]. Several reducing agents were investigated in order to obtain, in a pure form, each of the stereoisomeric alcohols (18) and (19). Results of the reductions are summarized in the Table 2.
The phenyl thionocarbonates (20) and (21), required by Robins’ procedure, were prepared in 76% and 71% yield, respectively, from the reaction between the corresponding alcohols (18) and (19) and phenyl chlorothionoformate, in the presence of DMAP [23,24]. The phenyl thionocarbonates (20) and (21) were reduced to the triquinane (13) in refluxing benzene with tributyltin hydride and a small amount of AIBN, as a radical initiator. The overall yield for the three step deoxygenation procedure via the phenyl thionocarbonate (20) was 60% (58% via (21)) (Scheme 1).
Stereochemistry of the Alcohols (18) and (19), and the Phenyl Thionocarbonates (20) and (21)
Both of the epimeric alcohols (18) and (19) underwent phenyl thionocarbonate formation, and subsequent deoxygenation with equal ease. Nonetheless, the relative stereochemistry of each epimer was of interest. It was not possible to selectively conduct NOE experiments on the alcohol (19) as the signals of interest (signals due to protons at C-4, C-5, C-7 and C-8) in the proton NMR spectrum were overlapping. Although results of NOE experiments on the alcohol (18) indicated that the stereochemistry of the hydroxyl group was β, further studies on the corresponding phenyl thionocarbonates were carried out. The proton NMR spectra of the corresponding phenyl thionocarbonates gave better signal dispersion and thus were more suitable for this study. The results of NOE difference experiments on the phenyl thionocarbonates (20) and (21) are shown in the Table 3. The NOE difference experiments on the phenyl thionocarbonate (20) have shown that irradiation of the signal due to H-6 caused enhancement of the signals assigned to H-3α, H-4α, H-5 and H-7α. Irradiation of the signals due to H- 4 α and H-5 caused enhancement of the signal due to H-6. The reciprocal enhancement between H-6 and H-4 α indicates that the stereochemistry of C-6 proton is α. The NOE enhancement between H-5 and H-6 protons is probably due to a conformation of the triquinane skeleton of (20) in which the H-5 and H-6 protons are relatively close to one another. However, the magnitude of the NOE (3.6%) at proton H-6 when proton H-5 was irradiated is much smaller compared with the corresponding enhancement in compound (21) (11.2%). The NOE difference experiments on the phenyl thionocarbonate (21) have shown that irradiation of the signal due to the H-6 proton caused enhancement of the H-5, H-7 β and H-9 β proton resonances. The irradiations of H-5 and H-9 β protons caused NOE enhancement of the H-6 proton. Thus the stereochemistry of the latter proton is β.
It is interesting to note the reversal in stereochemistry resulting from the reduction of ketone (12) with L-Selectride® and lithium aluminum hydride, as compared with that obtained by the reduction of ketone (22) [26] (Scheme 3). This can be attributed to the presence of an angular methyl group in the compound (22), as opposed to a proton in ketone (12). It has been proposed that the reduction of ketones with non-sterically demanding metal hydrides (such as lithium aluminum hydride) is controlled primarily by torsional strain in the transition state, while reduction with bulky reagents (such as L-Selectride®) is controlled primarily by the degree of steric hindrance of the carbonyl group [27,28,29,30,31,32].
Preparation of the Tricyclic Keto Ketal (25)
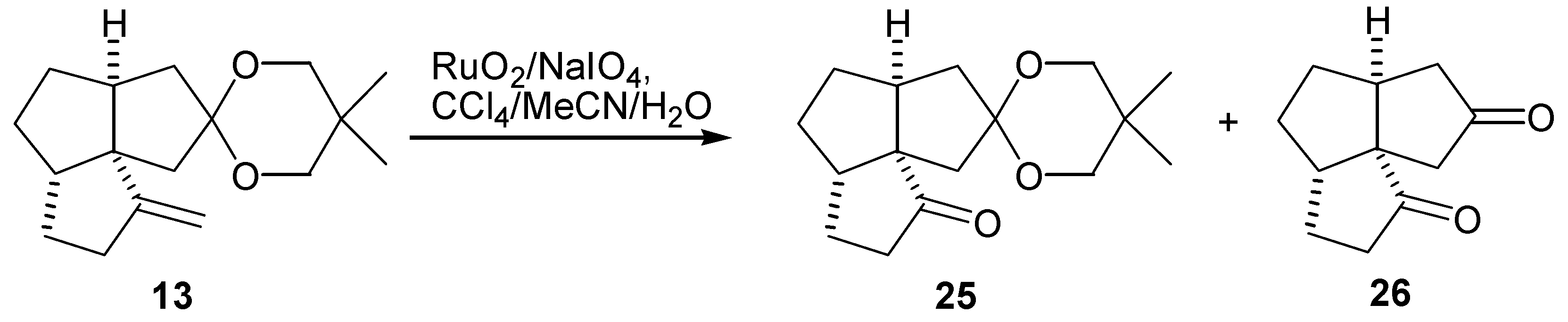
The keto ketal (25) was synthesized by a ruthenium tetroxide oxidation of the ketal alkene (13), according to Sharpless' procedure [33]. When the reaction was carried out at room temperature for 1 hr the keto ketal (25) was obtained in 79% yield, accompanied by 3% (as determined by GLC analysis of the crude products) of the diketone (26). However, when the reaction time was extended to 24 hr, the only product was the diketone (26), obtained in 71% yield (Scheme 4). It was possible to completely suppress hydrolysis of the ketal group with a phosphate buffer (pH 7.2). However, the use of a buffer decreased the solubility of sodium periodate and the work-up was complicated by presence of a large amount of precipitated salts.
Preparation of the Diketone (26)
The keto ketal (25) was deketalized by treatment with a 1:1 mixture of 5% hydrochloric acid and acetone to provide the diketone (26) in 93% yield. It is interesting that compound (26) was relatively unstable. Even when stored in a freezer (-4°C) under an argon atmosphere, the diketone (26) quickly darkened, and turned black in a few days. Thus, either the diketone (26) was used immediately after preparation or it was freshly distilled before use.
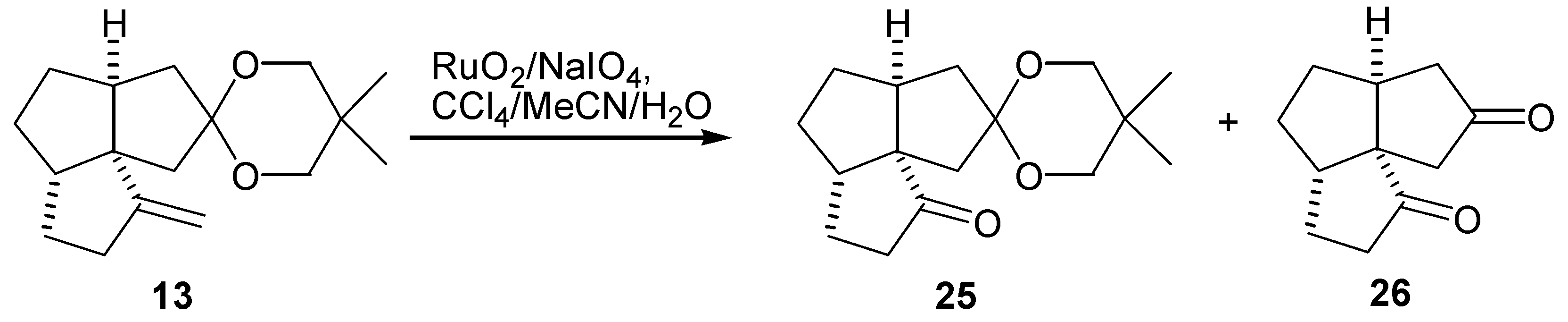
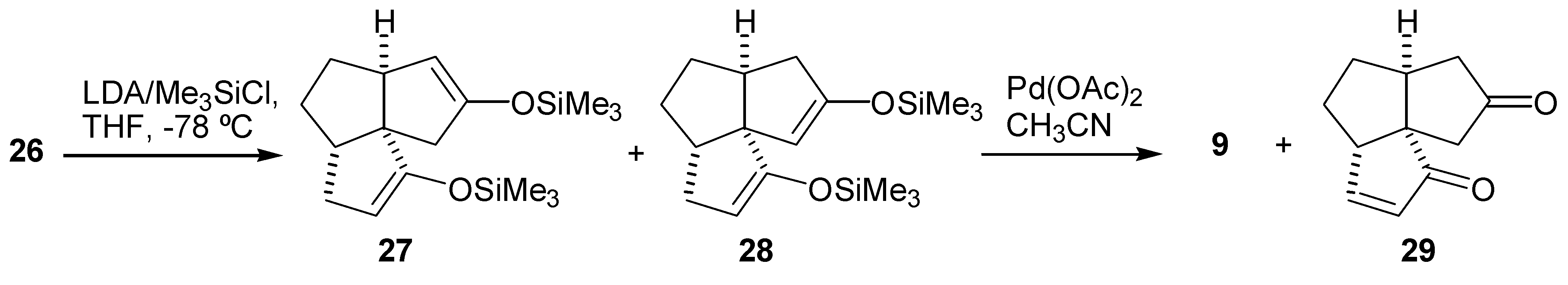
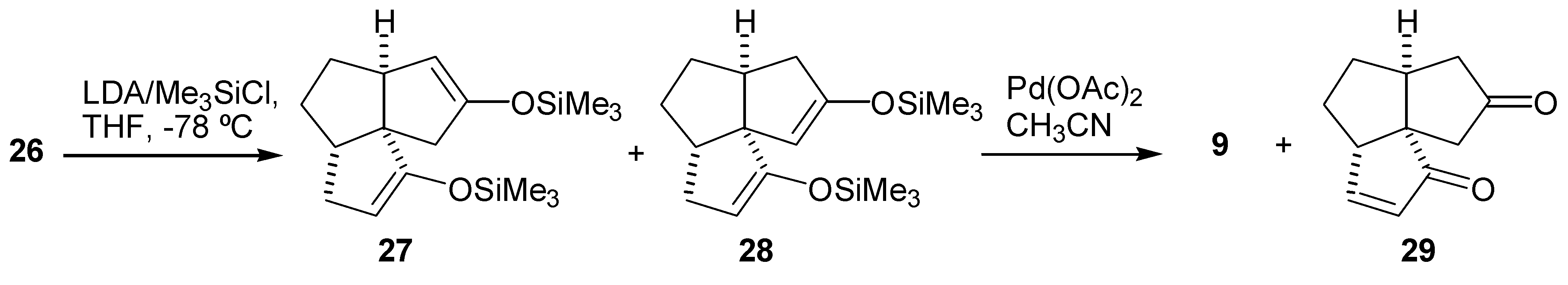
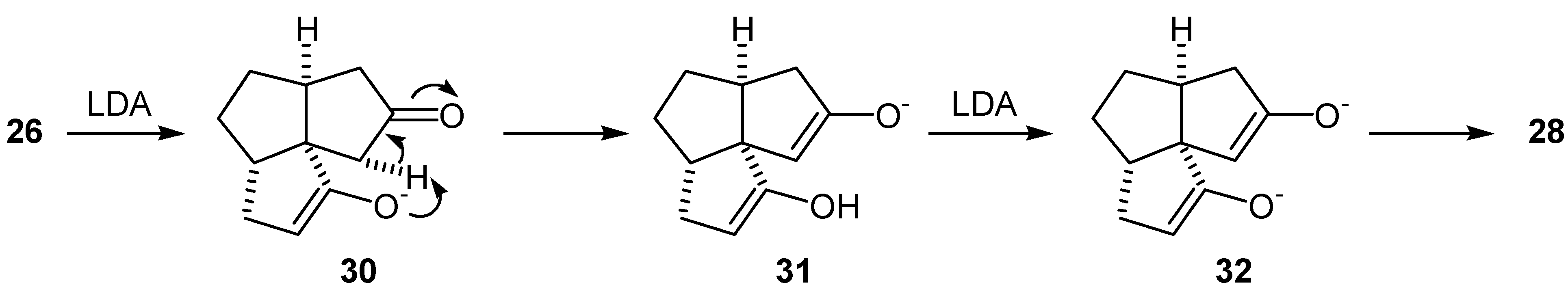
Initial synthetic plan called for a conversion of the diketone (26) into the dienedione (9) following Saegusa procedure [19]. Thus, the diketone (26) was converted to a mixture of the bisenol silyl ethers (27) and (28) upon treatment with LDA-TMSCl according to the procedure of Corey et al [34]. A GLC analysis of the crude product indicated that the two bisenol silyl ethers were formed in a ratio of about 1:1. Oxidation of this 1:1 mixture of (27) and (28) with palladium acetate gave the dienedione (9) and enedione (29) in 20% and 10% yield, respectively (Scheme 5). A variety of more hindered bases were used instead of LDA in an attempt to achieve a regioselective formation of the bisenol silyl ether (27). These included both lithium and potassium bis(trimethylsilyl)amides, lithium tetramethylpiperidide and lithium bis(phenyldimethylsilyl)amide. The reactions were carried out in THF at -78°C, in an attempt to prepare the kinetically favored bisenol silyl ether. Alternatively, preparation of the bisenol silyl ether (27) under thermodynamic conditions, by treatment of the diketone (26) with TMSBr-Et3N in DMF [35], or TMSI-Et3N in CH2Cl2 was attempted [18]. In all cases, the results were similar in that the ratio of regioisomers (27) and (28) was close to 1:1, as determined by GLC analysis. The subsequent oxidations with palladium acetate produced consistently the dienedione (9) and the enedione (29) in ≈ 2:1 ratio and in a low yield. Formation of a relatively large amount of the bisenol silyl ether (28) may be rationalized by proposing that the base first abstracts the least hindered C-3 proton to give the lithium enolate (30) (Scheme 6). This enolate may then, in an intramolecular reaction, abstract the C-11 proton and produce the undesired enolate (31). Deprotonation of (31) with a second equivalent of base, followed by treatment with trimethylsilyl chloride, would produce (28). If this rationale is correct then the use of a more hindered base is of no benefit as it would only increase the difference in reactivity between the C-9 and C-3 protons, and favor the pathway described in Scheme 6. Therefore, it was decided to execute the oxidation steps sequentially.
Preparation of the Tricyclic Enedione (29)
The tricyclic keto ketal (25) was converted to the corresponding enol silyl ether by treatment with trimethylsilyl iodide and triethylamine in methylene chloride. Oxidation of the enol silyl ether with palladium acetate in acetonitrile [19] gave the enone ketal (14) in 74% overall yield (Scheme 1). Acidmediated deketalization of the tricyclic enone ketal (14) provided the enedione (29) in 93% yield.
Preparation of the Tricyclic Dienedione (9)
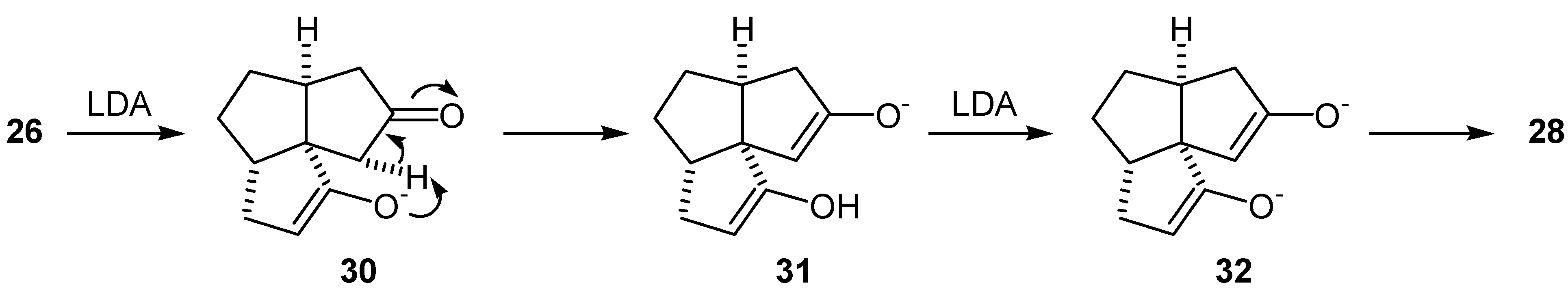
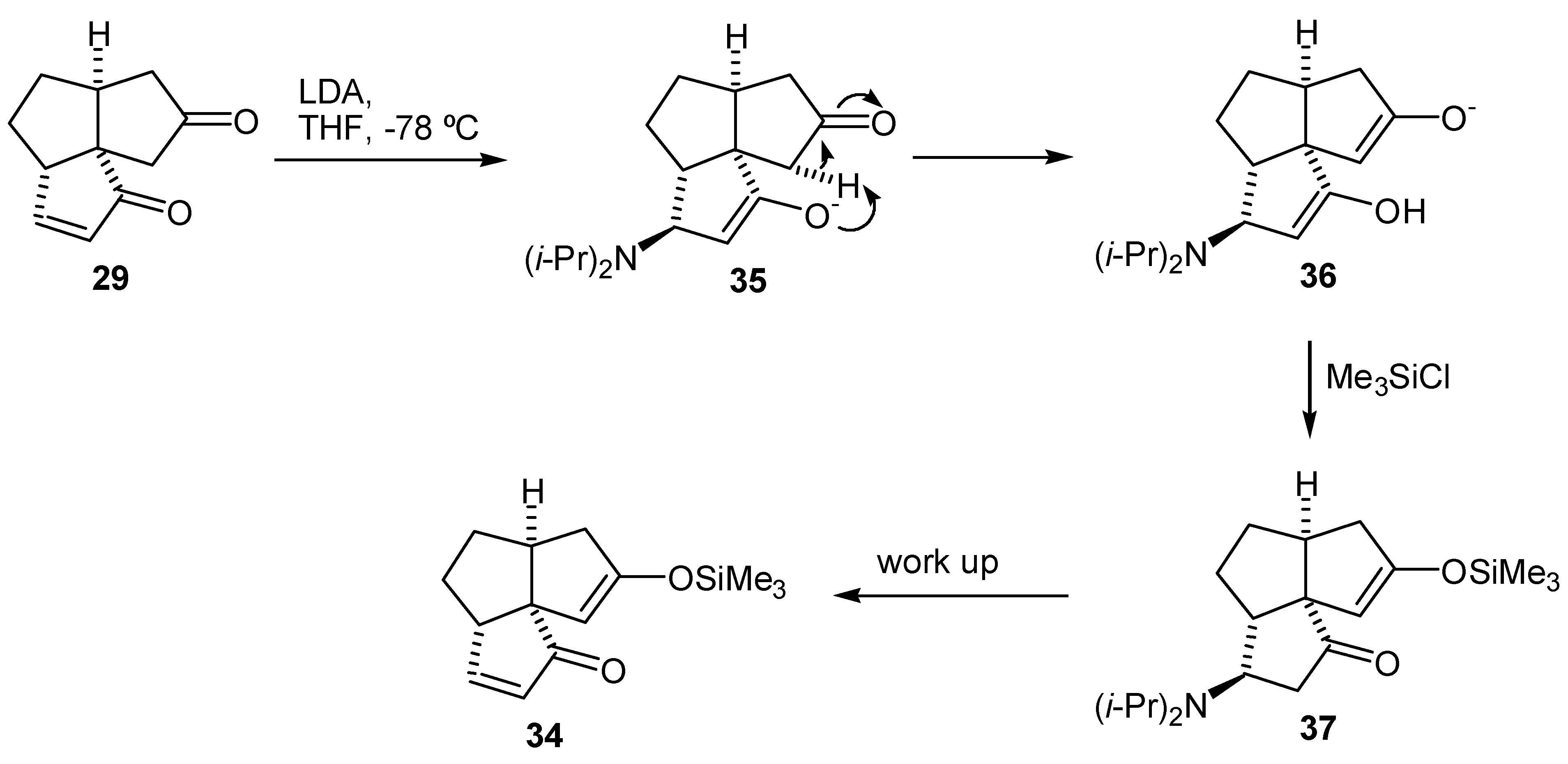
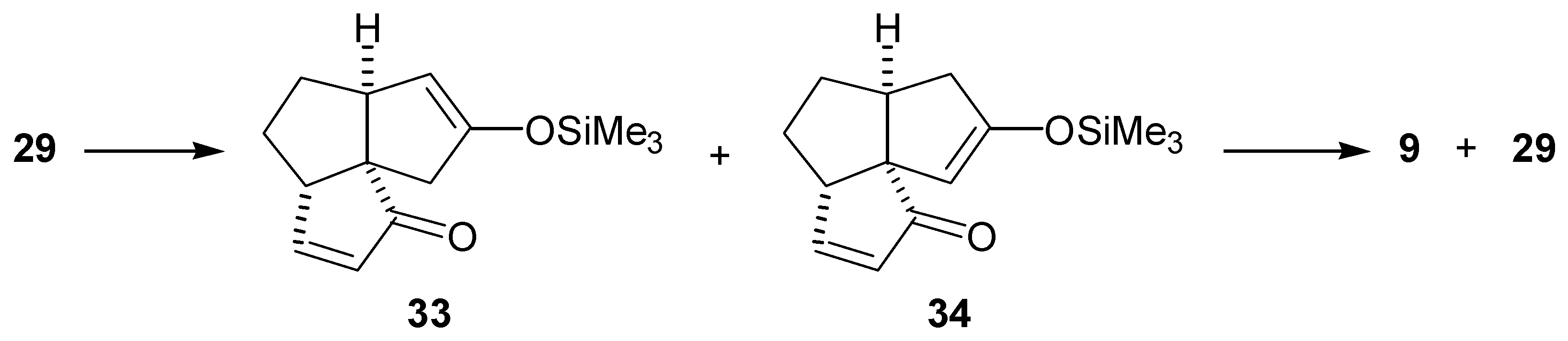
The enedione (29) was treated with a variety of bases (LDA, lithium and potassium bis(trimethylsilyl)amides and lithium bis(phenyldimethylsilyl)amide) in THF at -78°C. In all cases, GLC analysis of the crude mixture indicated that two two regioisomeric enol silyl ethers (33) and (34) were formed in a 1:1 ratio (Scheme 7). Subsequent oxidation of this mixture of enol silyl ethers (33) and (34) with palladium acetate met with no more success than the previous attempts to oxidize bisenol silyl ethers (27) and (28). The material balances in the conversions of the diketone (26) or the enedione (29) to the dienedione (9) were very low. As the loss of material may be due to the complexation of the product to the palladium(0), reactions were conducted under higher dilution (5-30 mmol/L of the substrate) than normal (250 mmol/L of the substrate). Under such conditions, it was necessary to extend the reaction time from 2 to 24 hr. Oxidation of the mixture of enol silyl ethers (33) and (34) under higher dilution conditions provided only a moderate improvement and a 28% overall yield of the dienedione (9) was achieved after reoxidation of the recovered enedione (29).
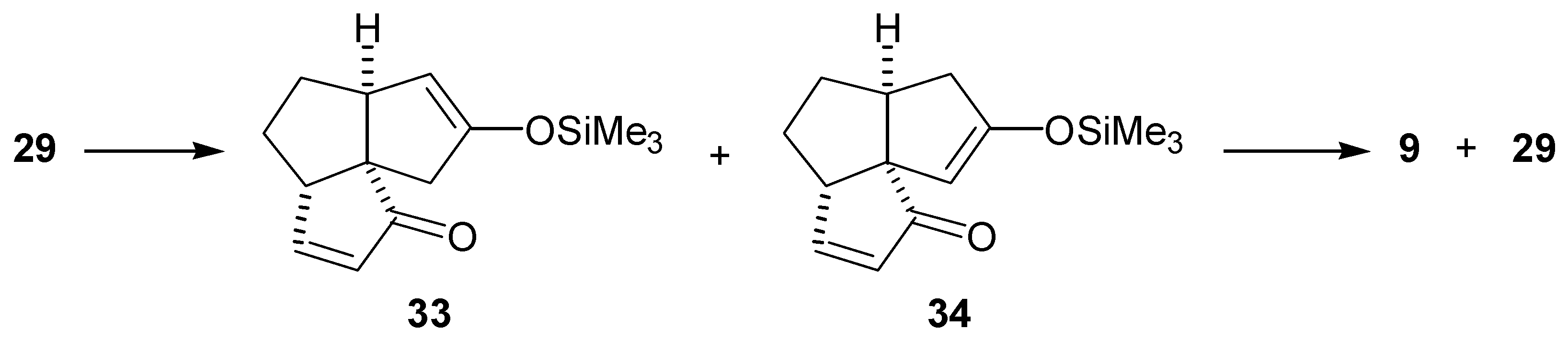
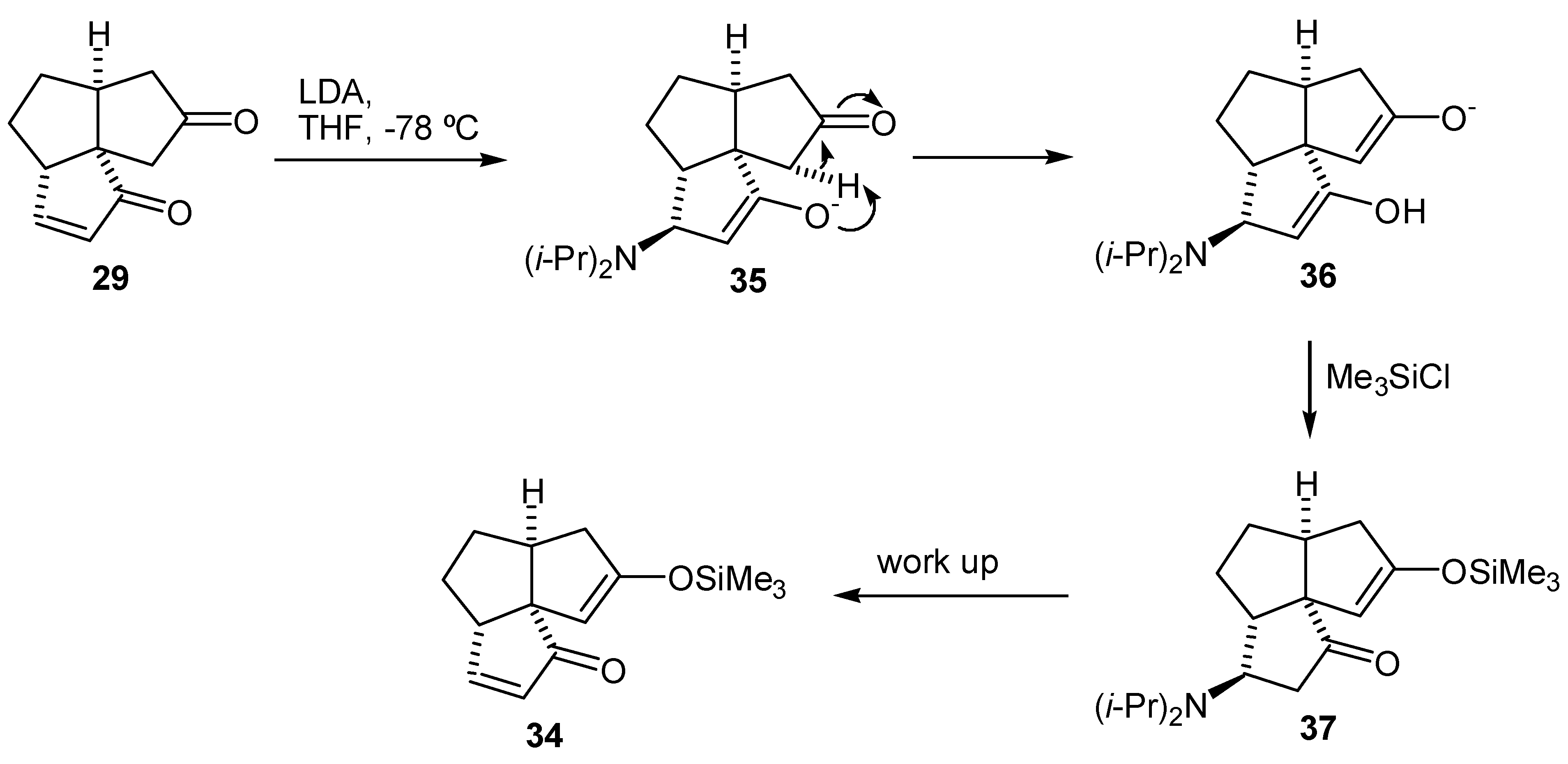
It was later found from a study on the alkylation of the dienedione (9) that the 1,4-addition of amide bases to the A-ring enone functionality occurs with ease (vide infra). Thus, it is likely that the reason for the lack of selectivity in formation of the enol silyl ether (33) was that the intermediate enolate (35) obtained after the 1,4-addition of LDA, abstracted a C-11 proton. Treatment of the intermediate (36) with trimethylsilyl chloride, followed by warming up of the reaction mixture resulted in the undesired enol silyl ether (34) (Scheme 8). The results from the alkylation of dienedione (9) indicated that it might be possible to prevent 1,4-addition of a base to the enone function of (29) by use of lithium tetramethylpiperidide. Thus, treatment of the enedione (29) with 1.1 equivalent of lithium tetramethylpiperidide at -78°C, followed by an excess of trimethylsilyl chloride provided predominantly the enol silyl ether (33) (97% by GLC analysis of the crude product). Treatment of the enedione (29) with a larger excess of lithium tetramethylpiperidide significantly reduced selectivity of the formation of (33). Subsequent oxidation of (33) with palladium acetate, in DMF, provided the dienedione (9). Application of this sequence on 8.2 mg of the enedione (29) gave the dienedione (9) in 46% yield. Unfortunately, this development occurred toward the end of the project, after the study on the preparation of the dienedione (9) was concluded. For this reason, only a limited quantity of the enedione (29) was available for reexamination of the Saegusa procedure and, therefore, this route was not fully optimized.
In addition to the enol silyl ether/palladium acetate method, other procedures involving palladium(II) oxidation of the diketone (26) or enedione (29)were examined. [36,37,38]. Either a mixture of products was obtained, none of which was the desired dienedione (9), or the dienedione (9) was obtained in very low yield (<10%).
In the late-1970s and early-1980s, Barton's group reported the use of benzeneseleninic acid anhydride (BSA) in oxidation of ketones to enones [39,40,41]. An interesting feature of this reaction is that the α-phenylselenoxy ketone is formed directly and, if appropriately positioned, undergoes synelimination in situ. Furthermore, the formation of the α-phenylselenoxy ketone is reversible allowing for the equilibration of intermediates that cannot eliminate. The drawback of this method is that benzeneseleninic anhydride and selenium-containing reaction products are powerful oxidants that may react further with the newly formed enone, thus diminishing its yield. After extensive research, optimal conditions for preparation of the dienedione (9), and cyclopentenones in general, were found [42]. The reaction was allowed to proceed until conversion of approximately 50% of the starting enedione (29) (by GLC analysis of the reaction mixture) had been achieved. The dienedione (9) and the recovered enedione (29) were obtained in yields of 38% and 43%, respectively. Recycling the enedione (29) twice provided the dienedione (9) in 59% overall yield.
The dienedione (9) allows for introduction of substituents at any ring A or ring C carbon atom. Ketone functionality, which allows functionalization of ring B carbon atoms, was removed earlier in the synthesis. In order to explore synthetic utility of dienedione (9), a brief study on introduction of silphiperfolane C-11 methyl group was performed.
Alkylation of the Dienedione (9)
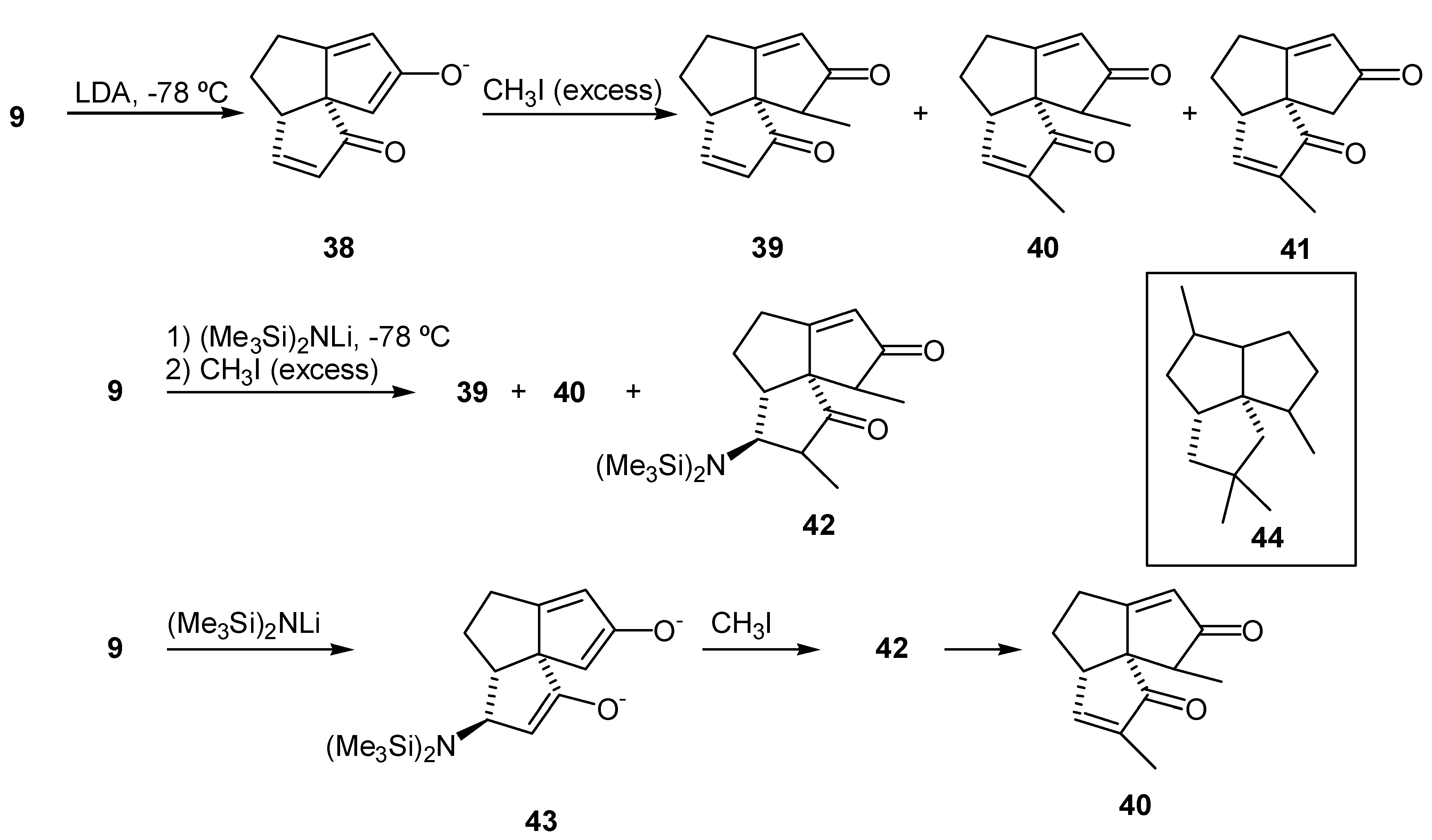
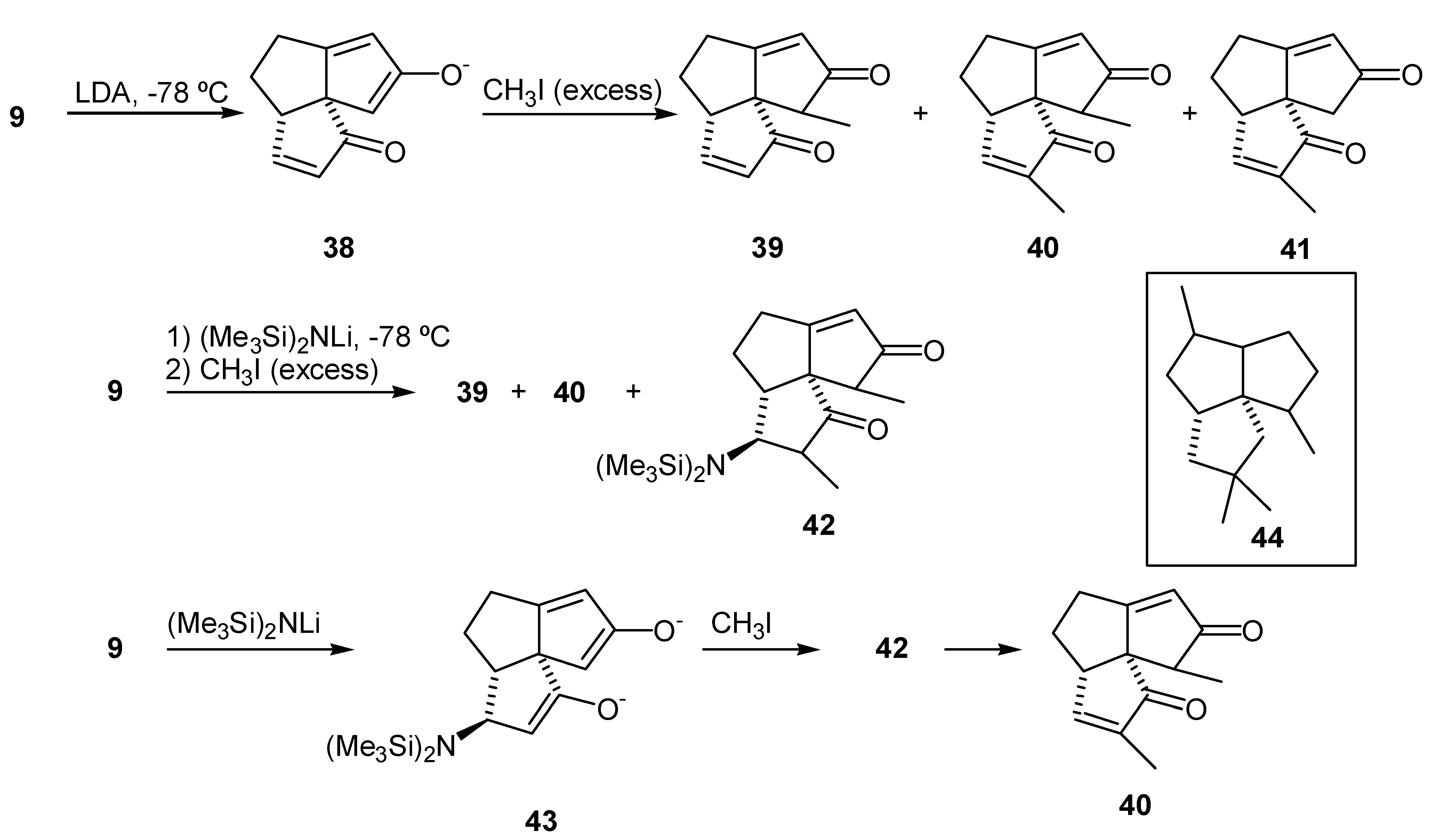
Treatment of (9) with LDA was expected to provide the "kinetic" enolate (38) (Scheme 9) by removal of the kinetically most acidic proton at C-11 (subergorgic acid numbering). Treatment of the enolate (38) with iodomethane was expected to provide the alkylated product (39). However, alkylation of the dienedione (9) with LDA-MeI was rather capricious. Mixtures of mono- and dialkylated products, some recovered dienedione (9), as well as other unidentified products, which proved to be very difficult to separate from the 11-methyl dienedione (39), were observed in the crude product mixtures. The isolated byproducts (40) and (41) were identified on the basis of their 1H NMR spectra (CDCl3, 200 MHz). The 3,11-dimethyl dienedione (40) exhibited resonances due to Me-12 (δ 1.08, d, 3H), Me-13 (δ 1.81, s, 3H), H-5 (δ 3.28, br d, 1H), H-9 (δ 5.94, d, 1H) and H-4 (δ 7.33, d, 1H). The 3-methyl dienedione (41) exhibited resonances due to Me-12 (δ 1.82, s, 3H), H-11 α or β (δ 2.36, d, 1H), H-11 α or β (δ 2.61, d, 1H), H-5 (δ 3.18, m, 1H), H-9 (δ 6.00, d, 1H) and H-4 (δ 7.35, d, 1H). It is interesting that functionalization of C-3 was unusually facile. That fact may be exploited in a synthesis of triquinane molecules with pentalene skeleton (44) which has a gem-dimethyl substituents at C-3. In an attempt to improve selectivity of the alkylation a more bulky base, lithium bis(trimethylsilyl)amide, was employed. The mixture of products thus obtained was separated by radial chromatography and the components were characterized. A new product was isolated along with the 11-methyl dienedione (39), and the 3,11-dimethyl dienedione (40). This new product was identified as the 4-bis(trimethylsilyl)amino-3,11-dimethyl dienedione (42). The 1H NMR spectrum (CDCl3, 200MHz) of compound (42) exhibited a signal at δ 0.25 (d, 18 H) due to the protons in trimethylsilyl groups. The resonances due to two methyl groups were overlapping at δ 1.05 (m, 6H, Me-12 and Me-13), and the olefinic proton exhibited a resonance at δ 5.98 (d, 1H, H-9). Therefore, the products (40) and (42) are a result of 1,4-addition of the base to the A-ring enone of (9) (Scheme 9), followed by subsequent trapping of the intermediate enolate (43) with iodomethane. This was an unexpected outcome, as lithium bases do not usually add in a 1,4-fashion to enones in THF at -78°C [43]. Compound (42) was moderately stable with a half-life of approximately 24 hr in deuteriochloroform solution at room temperature. This material eliminated hexamethyldisilazane to provide the 3,11-dimethyl dienedione (40).
Lithium tetramethylpiperidide was used next as a non-nucleophilic base [44]. 11-Methyl dienedione (39) was obtained in acceptable yield (53%) when 1.1 equivalent of base was used. Surprisingly, when a larger excess of base was employed, a significant amount of dialkylated product (40) was obtained. With these results in hand we reexamined the formation of enol silyl ethers from the enedione (29) (vide supra), and the subsequent palladium acetate oxidation. NOE difference experiments performed in order to determine the configuration of the methyl group were inconclusive. Molecule (39) has three sp2 carbons in each of the A and C rings. This flattens the molecule and changes the orientation of the protons of interest and the distances between them, as compared to a saturated triquinane ring system. Thus, usual NOE experiments that were successfully applied in determination of the stereochemistry of C-11 methyl group in other silphiperfolanes [16,45,46] did not provide a conclusive result in this case.
Conclusion
The work described herein represents a new approach to the construction of the silphiperfolane carbon skeleton. The key step of the overall synthetic sequence was the one pot conversion of the enone (11) to the triquinane (12) involving a copper(I)-catalyzed reaction of the enone ketal (11) with the bifunctional reagent (7) followed by addition of DMPU. The compound (9) was thus prepared in 11 steps and an overall yield of 11% from the known keto ketal (10) (Scheme 1). The employed methodology allowed for an efficient construction of a highly functionalized triquinane skeleton in general and subergorgic acid skeleton in particular. Therefore this appears to be a viable general route towards the total synthesis of subergorgic acid and its analogues.
Experimental
General
Infrared (IR) spectra were recorded on liquid films (sodium chloride plates) or on potassium bromide discs, by means of a Perkin-Elmer model 1710 Fourier transform spectrophotometer (internal calibration). Proton nuclear magnetic resonance (1H NMR) spectra were recorded on deuteriochloroform solutions (unless otherwise stated) using Varian model XL-300 or Bruker models AC-200 or WH-400 spectrometers. Signal positions were measured relative to those of tetramethylsilane (δ=0). Carbon nuclear magnetic resonance (13C NMR) spectra and the attached proton test (APT) experiments were recorded on deuteriochloroform solutions at 75.3 MHz using the Varian spectrometer noted above, or the Bruker models AC-200 (50.5 MHz) or AMX-500 (125.8 MHz). Signal positions are given relative to deuteriochloroform (δ=77.0). Signals with negative intensities in the attached proton test (APT) experiments are indicated by (-ve) following the chemical shift. Nuclear Overhauser enhancement (NOE) difference experiments were recorded on a Bruker model WD-400 spectrometer. 1H-1H Homonuclear correlation (COSY) experiments were recorded on Bruker models AC-200 or WD-400 spectrometers. Low resolution and high resolution mass spectra were recorded with a Kratos/AEI MS 50 (70 eV) mass spectrometer. All compounds subjected to high resolution mass measurements were homogeneous by GLC and/or TLC analysis. Microanalysis were performed on a Carlo Erba CHN elemental analyzer (Model 1106) in the microanalytical laboratory at the University of British Columbia. Gas-liquid chromatography (GLC) analyses were performed on Hewlett-Packard models 5880 or 5890 capillary gas chromatographs, employing 25 m × 0.21 mm fused silica columns coated with cross-linked SE-54 and equipped with flame ionization detectors. Conventional (drip) and flash column chromatography [49] were performed on 230-400 mesh silica gel (grade 60). In addition to column chromatography, separations were carried out on a centrifugally accelerated, radial, thinlayer chromatograph (Chromatotron, Model 7924) using 1, 2 or 4 mm silica gel plates (grade 60, E. Merck 7749). Distillation temperatures (uncorrected) were recorded as air-bath temperatures required for short-path bulb-to-bulb (Kugelrohr) distillation. Melting points were measured on a Fisher-Johns apparatus and are uncorrected. All reactions were carried out under an atmosphere of dry argon using flame dried glassware unless stated otherwise. Solvents and reagents were purified and dried using established procedures. THF and diethyl ether were distilled from sodium benzophenone ketyl radical anion. Carbon tetrachloride was distilled from P2O5. Acetonitrile, diisopropylamine, triethylamine, HMPA, DMPU, benzene, trimethylsilyl chloride, chloroform and dichloromethane were distilled from calcium hydride. Iodomethane was passed through a short column of flame dried basic alumina (activity I) before use. Solutions of methyllithium (LiBr complex) in diethyl ether, n-butyllithium in hexane and t-butyllithium in pentane were obtained from Aldrich Chemical Co. Inc. and were standardized using the method of Kofron and Baclawski [50]. Magnesium bromide-etherate complex was prepared by the reaction of freshly distilled 1,2-dibromoethane with magnesium turnings (flame dried under ar-gon atmosphere) in dry ether, with subsequent removal of ether under vacuum (0.1 mmHg) at room temperature. Cuprous bromide-dimethyl sulfide complex was prepared by the method of Wuts [51]. Lithium diisopropylamide (LDA) and other lithium dialkylamides were prepared by the addition of a solution of methyllithium (1.0 equiv.) in diethyl ether to a solution of the appropriate amine (1.1 equiv.) in dry THF at -78°C. The resulting solution was then stirred at 0°C for 5 min before use. Potassium hydride was obtained as 35% suspension in mineral oil from the Aldrich Chemical Company, Inc. and was rinsed free of oil (with dry THF) and dried under stream of argon before use. Aqueous NH4Cl-NH4OH (pH 8) was prepared by the addition of 50 mL of aqueous ammonium hydroxide (58%) to 1L of saturated aqueous ammonium chloride. All other reagents were commercially available and were utilized without purification unless stated otherwise.
Enone Ketal (11)
To a cold (-78°C), stirred solution (argon atmosphere) of the keto ketal (10) (1.80 g, 8.0 mmol) in dry CH2Cl2 (100 mL), were added Et3N (3.44 mL, 24 mmol) and Me3SiI (2.35 mL, 16 mmol). After the mixture had been stirred at -78°C for 30 min, saturated aqueous NaHCO3 (10 mL) was added, and the mixture was allowed to warm to room temperature. The phases were separated, and the aqueous phase was extracted with Et2O (3 × 10 mL). The organic extracts were combined and dried over anhydrous MgSO4. Evaporation of the solvent gave the enol silyl ether (2.19 g, 92%), as a colorless oil. The product was used without further purification. The product was characterized with a sample obtained by distillation (120°C/0.1 mm Hg).
IR (neat): 2957, 1645, 1114 cm-1. 1H NMR (400 MHz, CDCl3) δ: 0.12 (s, 9H, Me3Si-), 0.90 (s, 6H, Me-12 and Me-13), 1.49 (m, 2H), 1.94 (dd, 1H, J=4.2, 2.4 Hz), 2.18-2.32 (m, 2H), 2.36-2.62 (m, 2H), 3.03 (m, 1H, H-1), 3.38 (s, 2H, ketal CH2), 3.42 (s, 2H, ketal CH2), 4.55 (d, 1H, H-2, J=1.8 Hz). HRMS. for C16H28O3Si: calcd. 296.1807, found: 296.1804.
To a solution of Pd(OAc)2 (1.59 g, 7.1 mmol) in 90 mL of dry acetonitrile at rt was added a solution of 2.09 g (7.1 mmol) of the crude enol silyl ether in dry acetonitrile (10 mL). After the mixture had been stirred at rt for 2 hr, it was filtered through a short column of Florisil. The column was washed with Et2O and the combined filtrate was concentrated under reduced pressure. A GLC analysis of the residual oil showed that the enone (11) and ketone (10) were present in a ratio of 94:6. Flash chromatography (4 cm × 25 cm silica gel column, 2:1 petroleum ether/EtOAc) of the residual material gave 1.29 g (72%) of enone (11) as a white solid. Recrystallization (hexane) provided long white needles that exhibited m.p. 84-86°C.
IR (KBr): 2959, 1704, 1635, 1103 cm-1. 1H NMR (400 MHz, CDCl3) δ: 0.94, 1.01 (s, s, 3H each, Me-12 and Me-13), 1.40 (dd, 1H, H-6 α or β, J=6.2, 6.2 Hz), 2.08 (dd, 1H, H-4 α or β, J=9.0, 1.7 Hz), 2.60 (dd, 1H, H-4α or β, J=9.0, 3.0 Hz), 2.65 (dd, 1H, H-6α or β, J=6.2, 4.0 Hz), 2.87 (d, 1H, H-8α or β, J=9.0 Hz), 2.99 (d, 1H, H-8α or β, J=9.0 Hz), 3.12 (m, 1H, H-5), 3.47 (dd, 2H, ketal CH2, J=5.0 Hz), 3.52 (dd, 2H, ketal CH2, J=5.0 Hz), 5.89 (s, 1H, H-2). 13C NMR (75 MHz, CDCl3) δ: 22.36 (C-12), 22.40 (C-13), 30.08 (C-10), 38.15 (C-4), 42.05 (C-6 or C-8), 42.09 (C-6 or C-8), 43.42 (C-5), 71.65 (C-9), 72.55 (C-11), 109.01 (C-7), 125.76 (C-2), 185.62 (C-1), 209.66 (C-3). HRMS for C13H18O3: calcd. 222.1256, found: 222.1255. Anal. calcd. for C13H18O3: C, 70.25; H, 8.16, found: C, 70.36; H, 8.30.
Tricyclic Ketone (12)
To a cold (-78°C), stirred solution of the freshly distilled 4-chloro-2-trimethylstannyl-1-butene (13) (1.09 g, 4.28 mmol) in 100 mL of dry THF was added a solution of MeLi in Et2O (2.85 mL, 1.5 M, 4.28 mmol). After the solution had been stirred at -78°C for 15 min, solid MgBr2·Et2O (1.10 g, 4.28 mmol) was added in one portion. After stirring at -78°C had been continued for another 10 min period, solid CuBr·Me2S (102 mg, 0.43 mmol) was added in one portion, followed by a solution of the enone (11) (678 mg, 3.05 mmol) in dry THF (5 mL). The mixture was stirred at -78°C for 10 min and dry DMPU (0.74 ml, 6.1 mmol) was added. The solution was stirred for an additional 15 min at -78°C and was allowed to warm up to rt. After stirring for 3 hr at rt, aqueous NH4Cl-NH4OH (pH 8) and Et2O (50 mL) were added and the mixture was filtered through a short column of Florisil. The column was washed with Et2O and the combined filtrate was concentrated under reduced pressure. Flash chromatography (2 cm × 15 cm silica gel column, 3:1 petroleum ether/Et2O) of the residual material, gave 683 mg (81%) of the tricyclic ketone (12) as a white solid. Recrystallization (methanol) provided white cubic crystals that exhibited m.p. 59-61°C.
IR (KBr): 2927, 1741, 1651, 1119 cm-1. 1H NMR (400 MHz, CDCl3) δ: 0.90, 0.93 (s, s, 3H each, Me-16 and Me-17), 1.80-1.96 (m, 3H, H-4 α and β, H-9 α or β), 2.19 (d, 1H, H-11 α or β, J=13.9 Hz), 2.32 (d, 1 H, H-11 α or β, J=13.9 Hz), 2.18-2.42 (m, 4H, H-3 α and β, H-7 α or β, H-9 α or β), 2.47 (dd, 1H, H-7 α or β, J=18.5, 9.0 Hz), 2.57 (m, 1H, H-8), 2.61 (m, 1H, H-5), 3.42 (d, 1H, ketal CH2, J=11.2 Hz), 3.46 (d, 1H, ketal CH2, J=11.2 Hz), 3.50 (s, 2H, ketal CH2), 4.98 (t, 1H, H-12, J=1.8 Hz), 5.01 (t, 1H, H-12, J=1.8 Hz). 13C NMR (75 MHz, CDCl3) δ: 22.39 (C-16, -ve), 22.43 (C-17, -ve), 27.59 (C-4), 30.03 (C-14), 33.74 (C-3), 42.06 (C-7), 44.25 (C-8, -ve), 45.02 (C-9 or C-11), 47.73 (C-9 or C-11), 58.80 (C-1), 60.21 (C-5, -ve), 71.55 (C-13), 72.41 (C-15), 105.37 (C-12), 108.76 (C-10), 159.79 (C-2), 221.90 (C-6). HRMS calcd. for C17H24O3: 276.1725, found: 276.1725. Anal. calcd. for C17H24O3: C, 73.88; H, 8.75, found: C, 74.04; H, 8.69.
LiAlH4 Reduction of the Ketone (12)
To a cold (0°C), stirred suspension of LiAlH4 (30 mg, 0.75 mmol) in 20 mL of dry THF was added a solution of the ketone (12) (158 mg, 0.57 mmol) in 2 mL of dry THF. After the mixture had been stirred at 0°C for 30 min, solid Na2SO4·10H2O was added in one portion. The slurry thus formed was filtered through a sintered glass funnel and the collected material was rinsed with Et2O. The combined filtrates were dried (MgSO4) and concentrated under reduced pressure to provide a crude product as a white solid. Recrystallization (pentane) provided 155 mg (97%) of the alcohol (18) as white cubic crystals that exhibited m.p. 80-81°C.
IR (KBr): 3296, 2956, 1647, 1111 cm-1. 1H NMR (400 MHz, CDCl3) δ: 0.91, 0.99 (s, s, 3H each, Me-16 and Me-17), 1.37 (m, 1H, H-4 α), 1.67 (dt, 1H, H-7 α or β, J=13.3, 4.9 Hz), 1.85 (m, 1H, H-4 β), 2.06 (d, 1H, H-11 α or β, J=14 Hz), 2.14 (m, 1H, H-7 α or β), 2.20 (m, 1H, H-5), 2.12-2.42 (m, 5H, H-3 α and β, H-8, H-9 α and β), 2.45 (d, 1H, H-11 α or β, J=14 Hz), 3.06 (br, 1H, -OH, exchanges with D2O), 3.44 (s, 2H, ketal CH2), 3.51 (s, 2H, ketal CH2), 3.84 (br, 1H, H-6), 4.84 (s, 1H, H-12), 4.86 (s, 1H, H-12). 13C NMR (100 MHz, CDCl3) δ: 22.43 (C-16, -ve), 22.59 (C-17, -ve), 28.35 (C-4), 30.02 (C-14), 33.44 (C-7), 41.76 (C-9 or C-11), 41.96 (C-9 or C-11), 48.20 (C-3), 48.32 (C-8, -ve), 61.56 (C-1), 61.87 (C-5, -ve), 71.40 (C-13), 72.63 (C-15), 79.07 (C-6, -ve), 104.07 (C-10), 110.45 (C-12), 162.76 (C-2). HRMS calcd. for C17H26O3: 278.1882, found: 278.1884.
L-Selectride® Reduction of the Ketone (12)
To a cold (-78°C), stirred solution of L-Selectride® (Aldrich) (400 μL of a 1.0 M solution in THF, 0.4 mmol) in 5 mL of dry THF was added a solution of the ketone (12) (55 mg, 0.20 mmol) in 2 mL of dry THF. After the solution had been stirred at -78°C for 3 hr, a 5% solution of KOH in MeOH (30 µL) was added followed by 60 µL of H2O2 (30% wt. solution in water). The reaction mixture was filtered through a short column of Florisil, and eluted with Et2O. The combined filtrates were dried (MgSO4) and concentrated under reduced pressure. Flash chromatography (1 cm × 6 cm silica gel column, 3:1 petroleum ether/ether) of the crude material produced 51 mg (92%) of the alcohol (19) as a white solid. Recrystallization (pentane) provided white needles that exhibited m.p. 80-82°C.
IR (KBr): 3213, 2956, 1655, 1109 cm-1. 1H NMR (400 MHz, CDCl3) δ: 0.89, 0.98 (s, s, 3H each, Me-16 and Me-17), 1.33 (br s, 1H, -OH, exchanges with D2O), 1.61 (m, 1H, H-4 α or β), 1.68 (m, 1H, H-4 α or β), 1.80 (m, 3H, H-7 α and β, H-9 α or β), 2.07 (d, 1H, H-11 α or β, J=13.8 Hz), 2.17 (d, 1H, H-9 α or β, J=12.1 Hz), 2.19 (d, 1H, H-11 α or β, J=13.8 Hz), 2.33-2.47 (m, 4H, H-3 α and β, H-5, H-8), 3.44 (dd, 4H, ketal CH2's, J=11.5 Hz), 4.43 (q, 1H, H-6, J=6.6 Hz), 4.81 (s, 1H, H-12), 4.88 (s, 1H, H-12). 13C NMR (100 MHz, CDCl3) δ: 22.45 (C-16, -ve), 22.63 (C-17, -ve), 23.83 (C-4), 29.95 (C-14), 35.06 (C-7), 40.11 (C-9 or C-11), 41.81 (C-9 or C-11), 46.73 (C-8, -ve), 48.13 (C-3), 57.46 (C-5, -ve), 60.67 (C-1), 72.05 (C-13 and C-15), 74.86 (C-6, -ve), 103.57 (C-12), 109.34 (C-10), 163.34 (C-2). HRMS calcd. for C17H26O3: 278.1882, found: 278.1883.
Phenyl Thionocarbonate (21)
To a stirred solution of the alcohol (19) (45.3 mg, 0.16 mmol) in 8 mL of dry acetonitrile were added DMAP (80 mg, 0.65 mmol) and phenyl chlorothionocarbonate (33 µL, 0.24 mmol). After the reaction mixture had been stirred for 24 hr at rt, it was concentrated under reduced pressure. The residue was partitioned between water (2 mL) and EtOAc (5 mL), and the phases were separated. The aqueous layer was extracted with EtOAc (2 × 2 mL). The combined organic extracts were washed with 0.1 M HCl, followed by aqueous NaHCO3 and brine. The organic extracts were dried (MgSO4) and concentrated under reduced pressure. Two sequential flash chromatographies (1 cm × 15 cm silica gel column, 3:1 hexanes/Et2O; 1 cm × 12 cm silica gel column, 5:1 hexanes/Et2O) of the residue thus obtained, provided 51 mg (76%) of the phenyl thionocarbonate (21) as a white solid that exhibited m.p. 89-91°C (decomposes).
IR (KBr): 2955, 1775, 1737, 1650, 1594, 1491, 1112 cm-1. 1H NMR (400 MHz, CDCl3) δ: 0.91, 0.98 (s, s, 3H each, Me-16 and Me-17), 1.61 (m, 1H, H-4 α), 1.71 (m, 1H, H-4 β), 1.90 (dd, 1H, H-9 β, J=13.4, 5.5 Hz), 2.07-2.15 (m, 2H, H-7 α and β), 2.19 (m, 3H, H-9 α, H-11 α and β), 2.31-2.48 (m, 3H, H-3 α and β, H-8), 2.79 (q, 1H, H-5, J=7.4 Hz), 3.46 (m, 4H, ketal CH2's), 4.86 (s, 1H, H-12), 4.90 (s, 1H, H-12), 5.81 (q, 1H, H-6, J=7.4 Hz), 7.07 (m, 2H, Ph), 7.26 (m, 1H, Ph), 7.38 (m, 2H, Ph). 13C NMR (75 MHz, CDCl3) δ: 22.47 (C-16, -ve), 22.61 (C-17, -ve), 25.05, 30.03 (C-14), 34.81, 36.30, 41.44, 46.46 (C-5 or C-8, -ve), 48.45, 54.77 (C-5 or C-8, -ve), 60.76, 71.83 (C-13), 72.32 (C-15), 87.37 (C-6, -ve), 104.22, 109.03, 121.87, 121.91 (-ve), 126.40 (-ve), 129.44 (-ve), 153.31, 162.39, 194.64 (C-18). HRMS calcd. for C24H30SO4: 414.1865, found: 414.1860.
Phenyl Thionocarbonate (20)
Using the previously described procedure, the alcohol (18) (1.49 g, 5.36 mmol) was converted to the corresponding phenyl thionocarbonate (20) in 1.57 g (71%) isolated yield. The white solid thus obtained exhibited m.p. 83-86°C (decomposes).
IR (KBr): 2953, 1784, 1652, 1592, 1491, 1123 cm-1. 1H NMR (400 MHz, CDCl3) δ: 0.93, 1.02 (s, s, 3H each, Me-16 and Me-17), 1.60 (m, 1H, H-4 α), 1.87 (m, 1H, H-4 β), 1.93 (m, 1H, H-7 β), 2.07 (m, 1H, H-9 α or β), 2.20 (m, 3H, H-11 α and β, H-8), 2.32-2.50 (m, 4H, H-3 α and β, H-7 α, H-9 α or β), 2.69 (m, 1H, H-5), 3.49 (m, 4H, ketal CH2's), 4.91 (m, 1H, H-12), 4.98 (s, 1H, H-12), 5.32 (dd, 1H, H-6, J=11.3, 5.7 Hz), 7.10 (m, 2H, Ph), 7.28 (1H, Ph), 7.40 (m, 2H, Ph). 13C NMR (75 MHz, CDCl3) δ: 22.50 (C-16, -ve), 22.64 (C-17, -ve), 27.48, 30.06 (C-14), 33.24, 36.70, 41.06, 47.96 (C-3), 48.02 (C-8, -ve), 58.22 (C-5, -ve), 60.42 (C-1), 71.97 (C-13), 72.10 (C-15), 90.53 (C-6, -ve), 105.31 (C-12), 109.59 (C-10), 122.00 (Ph, -ve), 126.43 (Ph, -ve), 129.47 (Ph, -ve), 153.34 (Ph), 161.71 (C-2), 194.81 (C-18). HRMS calcd. for C24H30SO4: 414.1864, found: 414.1864.
Alkene (13) from Phenyl Thionocarbonate (20)
To a stirred solution of the phenyl thionocarbonate (20) (772 mg, 1.87 mmol) in dry benzene (50 mL) were added tributyltin hydride (1.0 mL, 3.73 mmol) and solid AIBN (15 mg, 0.1 mmol). After the reaction mixture had been heated to reflux for 2 hr, the solution was cooled to rt and concentrated under reduced pressure. Two consecutive column chromatographies (3 cm × 15 cm silica gel columns, eluting the first column with petroleum ether, and the second column with 5:1 petroleum ether/Et2O) of the material thus obtained gave a colorless oil. The crude product thus obtained was distilled (80°C/0.1 mm Hg) to provide 425 mg (87%) of the alkene (13).
IR (neat): 2954, 1728, 1650, 1123 cm-1. 1H NMR (400 MHz, CDCl3) δ: 0.91, 1.02 (s, s, 3H each, Me-16 and Me-17), 1.28 (m, 1H), 1.37 (m, 1H), 1.46 (m, 1H), 1.69-1.82 (m, 3H), 1.88 (m, 1H), 2.02 (d, 1H, H-11 β, J=14.0 Hz), 2.16 (ddd, 1H, H-9 α, J=13.5, 8.7, 1.8 Hz), 2.25 (d, 1H, H-11 α, J=14.0, 1.8 Hz), 2.24-2.42 (m, 4H, H-3 α and β, H-9 β), 3.45 (s, 2H, ketal CH2), 3.46 (d, 1H, ketal CH2, J=11.0 Hz), 3.52 (d, 1H, ketal CH2, J=11.0 Hz), 4.83 (br s, 1H, H-12), 4.91 (br s, 1H, H-12). 13C NMR (75 MHz, CDCl3) δ: 22.47 (C-16, -ve), 22.68 (C-17, -ve), 29.66, 30.02 (C-14), 30.92, 31.40, 34.35 (C-9 or C-11), 47.98 (C-3), 50.80 (C-8, -ve), 53.69 (C-5, -ve), 61.84 (C-1), 71.89 (C-13), 72.13 (C-15), 104.07 (C-12), 109.61 (C-10),163.42 (C-2). HRMS calcd. for C17H26O2: 262.1932, found: 262.1930.
Alkene (13) from Phenyl Thionocarbonate (21)
Using the previously described procedure, the phenyl thionocarbonate (21) (193.4 mg) was converted to the alkene (13) in 102.2 mg (83%) yield. The spectral properties of this material were identical with those described for the compound (13) obtained previously.
Keto Ketal (25)
To a stirred solution of the alkene (13) (101 mg, 0.39 mmol), in 6 mL of CCl4, 6 mL of acetonitrile and 9 mL of water were added solid NaIO4·H2O (329 mg, 1.54 mmol) and solid RuO2·xH2O (5.5 mg, 0.04 mmol). After the reaction mixture had been stirred at rt for 1 hr, the phases were separated and the aqueous layer was extracted with 2 mL CCl4. Isopropanol (0.5 mL) was added to the combined organic extracts and stirring was continued at rt for another 1 hr period. The slurry was filtered through a column of Florisil (2 cm × 10 cm, eluting with Et2O (50 mL)). The combined filtrates were concentrated under reduced pressure. Flash chromatography (1 cm × 12 cm silica gel column, 3:1 petroleum ether/Et2O) and distillation (100°C/0.1 mmHg) of the crude product produced 79 mg (79%) of the keto ketal (25) as a colorless oil.
IR (neat): 2952, 1732, 1114 cm-1. 1H NMR (400 MHz, CDCl3) δ: 0.92, 0.93 (s, s, 3H each, Me-15 and Me-16), 1.38 (m, 1H, H-7 β), 1.54 (m, 1H, H-9 β), 1.64 (m, 1H, H-4 α or β), 1.87 (m, 1H, H-9 α), 1.93-2.07 (m, 5H, H-11 α or β, H-4 α or β, H-6 α and β, H-7 α), 2.16 (m, 1H, H-11 α or β), 2.23 (m, 1H, H-3 α or β), 2.39 (m, 1H, H-3 α or β), 2.50 (m, 1H, H-8), 2.55 (m, 1H, H-5), 3.48 (m, 4H, ketal CH2's). 13C NMR (75 MHz, CDCl3) δ: 22.52 (C-15, -ve), 22.54 (C-16, -ve), 24.00, 29.94 (C-13), 32.48, 33.02, 36.58 (C-3), 39.95 (C-9 or C-11), 42.68 (C-9 or C-11), 47.68 (C-5 or C-8, -ve), 49.35 (C-5 or C-8, -ve), 64.74 (C-1), 71.45 (C-12), 72.67 (C-14), 109.84 (C-10), 224.47 (C-2). HRMS calcd. for C16H24O3: 264.1726, found: 264.1728. Anal. calcd. for C16H24O3: C 72.69%; H 9.15%, found: C 72.43%; H 9.04%.
Diketone (26)
A solution of the keto ketal (25) (46 mg, 0.17 mmol) in a mixture of acetone and 5% aqueous HCl (1:1, 10 mL) was stirred at rt for 20 min. Diethyl ether (5 mL) was added and the phases were separated. The aqueous layer was extracted with Et2O (3 × 3 mL). The combined organic extracts were washed with water, aqueous NaHCO3, brine, and dried (MgSO4). The mixture was concentrated under reduced pressure, and the residual material distilled (90°C/0.1 mmHg) give 28.9 mg (93%) of the diketone (26) as colorless oil.
IR (neat): 2955, 1741 cm-1. 1H NMR (400 MHz, CDCl3) δ: 1.48 (dd, 1H, J=12.0, 6.0 Hz), 1.66 (m, 2H), 2.02-2.18 (m, 5H), 2.32 (ddd, 1H, J=18.5, 9.0, 6.0 Hz), 2.50 (ddd, 1H, J=16.5, 9.0, 0.5 Hz), 2.56 (m, 1H), 2.65 (dd, 1H, H-11 α or β, J=18.5, 1.4 Hz), 2.68 (m, 1H), 2.72 (dd, 1H, H-11 α or β, J=18.5, 1.5 Hz). 13C NMR (75 MHz, CDCl3) δ: 25.21, 32.41, 33.40, 37.12 (C-3), 44.05 (C-9 or C-11), 45.77 (C-8, -ve), 46.26 (C-9 or C-11), 49.01 (C-5, -ve), 62.87 (C-1), 217.08 (C-10), 222.01 C-2). HRMS calcd. for C11H14O2: 178.0994, found: 178.0994.
Enone Ketal (14)
To a cold (-78°C), stirred solution of the keto ketal (25) (360 mg, 1.36 mmol) in 100 mL of dry CH2Cl2 were added freshly distilled Et3N (568 µL, 4.08 mmol) and Me3SiI (387 µL, 2.7 mmol). After the mixture had been stirred at -78°C for 2 hr, a saturated aqueous NaHCO3 solution (20 mL) was added. The phases were separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were dried (MgSO4) and concentrated under reduced pressure to gave the enol silyl ether. IR (neat): 2953, 1645, 1113 cm-1. This product was used without further purification.
To a stirred solution of Pd(OAc)2 (305 mg, 1.36 mmol) in 100 mL of dry acetonitrile was added a solution of the crude enol silyl ether in dry acetonitrile (5 mL). After the mixture had been stirred at rt for 2 hr, it was filtered through a short column of Florisil and eluted with Et2O (50 mL). Concentration of the filtrate under reduced pressure, followed by a flash chromatography (2 cm × 12 silica gel column, 10:1 petroleum ether/EtOAc) of the residual material, gave 264.2 mg (74%) of the enone ketal (14) as a white solid. Recrystalization (hexane) provided material that exhibited m.p. 87-88°C.
IR (KBr): 2949, 1697, 1584, 1122 cm-1. 1H NMR (400 MHz, CDCl3) δ: 0.95, 1.01 (s, s, 3H each, H- 15 and H-16), 1.52 (m, 3H), 1.79 (ddd, 1H, H-9 α or β, J=14.0, 7.0, 1.5 Hz), 2.11 (d, 1H, H-11 α or β, J=14.0 Hz), 2.15 (m, 1H), 2.26 (ddd, 1H, H-9 α or β, J=14.0, 10.0, 1.0 Hz), 2.43 (d, 1H, H-11 α or β, J=14.0 Hz), 2.54 (m, 1H, H-8), 3.08 (br d, 1H, H-5, J=9.0 Hz), 3.50 (s, 2H, ketal CH2), 3.47 (d, 1H, ketal CH2, J=11.5 Hz), 3.56 (d, 1H, ketal CH2, J=11.5 Hz), 6.14 (dd, 1H, H-3, J=5.5, 2.0 Hz), 7.43 (dd, 1H, H-4, J=5.5, 2.5 Hz). 13C NMR (75 MHz, CDCl2) δ: 22.42 (C-15, -ve), 22.56 (C-16, -ve), 27.05 (C-6 or C-7), 29.71 (C-6 or C-7), 30.00 (C-13), 40.94 (C-9 or C-11), 41.51 (C-9 or C-11), 46.35 (C-8, -ve), 55.26 (C-5, -ve), 63.29 (C-1), 71.97 (C-12), 72.38 (C-14), 109.27 (C-10), 133.17 (C-3, -ve), 166.39 (C-4, -ve), 212.83 (C-2). HRMS calcd. for C16H22O3: 262.1571, found: 262.1570. Anal. calcd. for C16H22O3: C 73.25%; H 8.45%, found: C 73.06%; H 8.38%.
Enedione (29)
A solution of the enone ketal (14) (26.4 mg, 0.1 mmol) in a mixture of acetone and 5% aqueous HCl (1:1, 4 mL) was stirred at rt overnight. The reaction mixture was diluted with Et2O (2 mL). The phases were separated and the aqueous layer extracted with Et2O (3 × 3 mL). The combined extracts were washed with water, aqueous NaHCO3, brine, and dried (MgSO4). The mixture was concentrated under reduced pressure to gave 16.4 mg (93%) of the enedione (29) as a white solid. Recrystalization (hexane) provided white needles that exhibited m.p. 82-83°C.
IR (KBr): 2929, 1739, 1702, 1584 cm-1. 1H NMR (400 MHz, CDCl3) δ: 1.70 (m, 3H, H-7 α and β, H-6 α), 2.05 (m, 2H, H-6 β, H-9 α or β), 2.33 (d, 1H, H-11 α or β, J=18.7 Hz), 2.70 (m, 2H, H-8 and H-9 α or β), 2.86 (d, 1H, H-11 α or β, J=18.7, 1.1 Hz), 3.19 (br d, 1H, H-5, J=9.0 Hz), 6.24 (dd, 1H, H-3, J=5.7, 1.7 Hz), 7.55 (dd, 1H, H-4, J=5.7, 2.6 Hz). 13C NMR (75 MHz, CDCl3) δ: 27.04 (C-6 or C-7), 29.82 (C-6 or C-7), 42.70 (C-9), 44.11 (C-8, -ve), 46.39 (C-11), 54.47 (C-5, -ve), 61.57 (C-1), 133.28 (C-3, -ve), 166.06 (C-4, -ve), 212.00 (C-2), 217.50 (C-10). HRMS calcd. for C11H12O2: 176.0837, found: 176.0839.
Dienedione (9)
To a stirred solution of the enedione (29) (94.5 mg, 0.54 mmol) in dry benzene (20 mL) heated to reflux, was added solid benzeneseleninic anhydride (272 mg, 0.76 mmol) in small portions (every 5-7 min.) over a period of 3 hr and 15 min. After the reaction mixture had been stirred at 80°C for a further 15 min, it was cooled and concentrated to a small volume under reduced pressure. Drip column chromatography (TLC grade silica gel without binder [52], 10 g, (2.5 cm × 5 cm column), elution first with hexane until PhSeSePh was eluted, then with Et2O until the enedione (29) was eluted, and finally with EtOAc to elute the dienedione (9)) of the residual material, gave 35.2 mg (38%) of the dienedione (9) as a white solid that exhibited m.p. 125-128°C, and 40.5 mg (43%) of the recovered enedione (29) as a white solid. The overall yield of the dienedione (9) was 54.8 mg (59%) after recycling twice the enedione (29).
IR (KBr): 2926, 1713, 1630, 1585 cm-1. 1H NMR (400 MHz, CDCl3) δ: 2.10 (m, 2H, H-6 β and H-7 β), 2.27 (m, 1H, H-6 α), 2.39 (d, 1H, H-11 α or β, J=16.8 Hz), 2.59 (m, 1H, H-7 α), 2.63 (d, 1H, H-11 α or β, J=16.8 Hz), 3.33 (ddd, 1H, H-5, J=3.0, 1.3, 1.2 Hz), 6.02 (d, 1H, H-9, J=1.9 Hz), 6.32 (dd, 1H, H-3, J=5.6, 1.3 Hz), 7.78 (dd, 1H, H-4, J=5.6, 3.0 Hz). 13C NMR (75 MHz, CDCl3) δ: 25.77 (C-6 or C-7), 29.85 (C-6 or C-7), 46.36 (C-11), 48.34 (C-5, -ve), 67.42 (C-1), 125.65 (C-9, -ve), 132.80 (C-3, -ve), 166.45 (C-4, -ve), 182.81 (C-8), 207.65 (C-2 or C-10), 208.35 (C-2 or C-10). HRMS calcd. for C13H18O3: 174.0681, found: 174.0681.
11-Methyl Dienedione (39)
To a cold (-78°C), stirred solution of lithium tetramethylpiperidide (0.046 mmol) in 3 mL of dry THF, was added a solution of the dienedione (9) (7.3 mg, 0.042 mmol) in 2 mL of dry THF. After the solution had been stirred at -78°C for 10 min, freshly distilled CH3I (103 μL, 2.1 mmol) was added. The reaction mixture was stirred at -78°C for an another 10 min period and 1 mL of aqueous NaHCO3 and 1 mL Et2O were added. The mixture was allowed to warm to rt, the phases were separated and the aqueous layer was extracted with CH2Cl2 (5 × 1 mL). The combined organic extracts were washed with brine, dried (MgSO4) and concentrated under reduced pressure. The residual material was dissolved in 0.5 mL of CH2Cl2 and purified on silica gel (chromatotron, 1 mm plate, 9:9:2 hexane/ CH2Cl2/Et2O) to give 4.2 mg (53%) of the 11-methyl dienedione (39) as a white solid that exhibited m.p. 134-135°C.
IR (KBr): 2922, 1710, 1632, 1584 cm-1. 1H NMR (400 MHz, CDCl ) δ: 1.10 (d, 3H, H-12, J=8.0 Hz), 1.97 (ddd, 1H, H-6 β, J=13.0, 9.5, 6.5 Hz), 2.09 (m, 1H, H-7 β), 2.27 (m, 1H, H-7 α, shows cou-pling with H-9 J=2.0 Hz), 2.60 (dd, 1H, H-6 α, J=13.0, 6.5 Hz), 2.60 (q, 1H, H-11, J=8.0 Hz), 3.42 (ddd, 1H, H-5, J=9.5, 3.0, 1.5 Hz), 5.96 (d, 1H, H-9, J=2.0 Hz), 6.27 (dd, 1H, H-3, J=5.8, 1.5 Hz), 7.74 (dd, 1H, H-4, J=5.8, 3.0 Hz). 13C NMR (125 MHz, CDCl3) δ: 15.74 (C-12, -ve), 25.88 (C-6 or C-7), 30.11 (C-6 or C-7), 44.22 (C-5 or C-8, -ve), 50.46 (C-5 or C-8, -ve), 69.61 (C-1), 123.3 (C-9, -ve), 132.68 (C-3, -ve), 166.45 (C-4, -ve), 182.06 (C-8), 207.98 (C-2 or C-10), 212.63 (C-2 or C-10). HRMS calcd. for C12H12O2: 188.0838, found: 188.0841.
Acknowledgement
The work described herein was done at the Department of Chemistry, University of British Columbia, Vancouver, BC, Canada. I thank NSERC of Canada for financial support and the University of British Columbia for the University Graduate Fellowship. Thanks are also due to Edward Piers for the use of his laboratory facilities and helpful discussions in the course of this project.
References and Notes
- Paquette, L. A. The Development of Polyquinane Chemistry. Top. Curr. Chem. 1979, 79, 41–165. [Google Scholar]
- Paquette, L. A. Recent Synthetic Developments in Polyquinane Chemsitry. Top. Curr. Chem. 1984, 119, 1–160. [Google Scholar]
- Paquette, L. A.; Doherty, A. M. Polyquinane Chemistry. Reactivity and Structure Concepts in Or- ganic Chemistry 1987, 26, 1–225. [Google Scholar]
- Mehta, G.; Srikrishna, A. Synthesis of Polyquinane Natural Products: An Update. Chem. Rev. 1997, 97, 671–719. [Google Scholar] [CrossRef] [PubMed]
- Groweiss, A.; Fenical, W.; Cun-heng, H.; Clardy, J.; Zhongde, W.; Zhongnian, Y.; Kanghou, L. Subergorgic Acid, A Novel Tricyclopentanoid Cardiotoxin from the Pacific Gorgonian Coral Subergorgia Suberosa. Tetrahedron Lett. 1985, 26, 2379–2382. [Google Scholar] [CrossRef]
- Parameswaran, P. S.; Naik, C. G.; Kamat, S. Y.; Puar, M. S.; Das, P.; Hegde, V. R. Studies on the Secondary Metabolites from the Indian Gorgonian Subergorgia suberosa: Isolation and Characterization of Four Analogues of the Cardiotoxin Subergorgic Acid. J. Nat. Prod. 1998, 61, 832–834 and 1074. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-H.; Jiao, K.-F.; Ji, Q.-E.; Song, H.-Q. Studies on the Relationship between the MO Indices and Anticholinesterase Activity of Subergorgin. Journal of Molecular Structure (Teochem) 1989, 188, 167–174. [Google Scholar] [CrossRef]
- Tan, X.; Ye, H.; Zeng, L.; Cui, Z.; He, S. An Organophosphorus (Soman) Antidote from Gorgonian Coral Subergorgia Suberosa. Zhongguo Haiyang Yaowu 1990, 9, 11–12, (Chemical Abstracts 1991, 115: 35564g). [Google Scholar]
- Trost, B. M. Some Aspects of Organosulfur-Mediated Synthetic Methods. Acc. Chem. Res. 1978, 453–461. [Google Scholar] [CrossRef]
- Piers, E.; Karunaratne, V. Organitin-Based Bifunctional Reagents: 4-chloro-2-lithio-1-butene and Related Substances. Methylenecyclopentane Annulations. Total Synthesis of (±)-Δ9(12)-Capnellene. Tetrahedron 1989, 45, 1089–1104. [Google Scholar] [CrossRef]
- Piers, E.; Karunaratne, V. Bifunctional Reagents in organic Synthesis. Total Synthesis of the Ses- quiterpenoids (±)-Pentalene and (±)-9-epi-Pentalene. Can. J. Chem. 1989, 67, 160–164. [Google Scholar] [CrossRef]
- Piers, E.; Reanud, J. Annulations via Bifunctional Reagents. Total Synthesis of (±)-Methyl Canta- brenonate and (±)-Methyl Epoxycantabrenonate. Synthesis 1992, 74–82. [Google Scholar] [CrossRef]
- Iwata, C.; Takemoto, Y.; Doi, M.; Imanishi, T. Stereoselective Total Synthesis of (±)-Subergorgic Acid, A New Type of Angular Triquinane Sesquiterpene. J. Org. Chem. 1988, 53, 1623–1628. [Google Scholar] [CrossRef]
- Wender, P.; deLong, M. Synthetic Studies on Arene-Olefin Cycloadditions. XII. Total Synthesis of (±)-Subergorgic Acid. Tetrahedron Lett. 1990, 31, 5429–5432. [Google Scholar] [CrossRef]
- Crimmins, M.; Dudek, C.; Cheung, A. W.-H. A Fragmentation-Rearrangement Sequence of Cyclobutylcarbinyl Radicals. Tetrahedron Lett. 1992, 33, 181–184. [Google Scholar] [CrossRef]
- Paquette, L. A.; Meister, P. G.; Friedrich, D.; Sauer, D. R. Enantioselective Total Synthesis of (-)- Subergorgic Acid. J. Am. Chem. Soc. 1993, 115, 49–56. [Google Scholar] [CrossRef]
- Piers, E.; Moss, N. Thermal Rearrangement of Divinylcyclopropane Systems. A New Formal Total Synthesis of (±)-Quadrone. Tetrahedron Lett. 1985, 26, 2735–2738. [Google Scholar] [CrossRef]
- Miller, R. D.; McKaen, D. R. The Facile Silylation of Aldehydes and Ketones using Trimethylsilyl Iodide: An Exceptionally simple Procedure for the Generation of Thermodinamically Equilibrated Trimethylsilylenol Ethers. Synthesis 1979, 730–732. [Google Scholar] [CrossRef]
- Ito, Y.; Hirao, T.; Saegusa, T. Synthesis of α,β-Unsaturated Carbonyl Compounds by Palla-dium(II)-Catalyzed Dehydrosilylation of Silyl Enol Ethers. J. Org. Chem. 1978, 43, 1011–1013. [Google Scholar] [CrossRef]
- Piers, E.; Chong, J. M. Regioselective Addition of Trimethylstannylcopper-Dimethyl sulphide to 1-Alkynes: Synthesis of ω-Substituted 2-(Trimethylstannyl)-1-Alkenes. J. Chem. Soc. Chem. Commun. 1983, 934–935. [Google Scholar]
- Piers, E.; Karunaratne, V. 4-Chloro-2-lithio-1-butene, a Novel Donor-Acceptor Conjuctive Reagent. J. Org. Chem. 1983, 48, 1774–1776. [Google Scholar] [CrossRef]
- Taylor, R. J. K. Organocopper Conjugate Addition-Enolate Trapping Reactions. Synthesis 1985, 364–392. [Google Scholar] [CrossRef]
- Robins, M. J.; Wilson, J. S. Smooth and Efficient Deoxygentation of Secondary Alcohols. A General Procedure for the Conversion of Ribonucleosides to 2’-Deoxynucleosides. J. Am. Chem. Soc. 1981, 103, 932–933. [Google Scholar] [CrossRef]
- Robins, M. J.; Wilson, J. S.; Hansske, F. Nucleic Acids Related Compounds. 42. A General Procedure for the Efficient Deoxygenation of Secondary Alcohols. Regiospecific and Stereoselective Conversion of Ribonucleosides to 2’-Deoxynucleosides. J. Am. Chem. Soc. 1983, 105, 4059–4065. [Google Scholar]
- Story, B.-A. The Total Synthesis of (±)-β-Panasinsene. Ph. D. Thesis, University of British Columbia, 1991; pp. 87–92. [Google Scholar]
- Brendel, J.; Weyerstahl, P. Total Synthesis of (±)-Silphiperfol-5-en-3-ol. Tetrahedron Lett. 1989, 30, 2371–2374. [Google Scholar]
- Cram, D. J.; Abd Elhafez, F. A. Studies in Stereochemistry. X. The Rule of “Steric Control of Asymmetric Induction” in the Synthesis of Acyclic Systems. J. Am. Chem. Soc. 1952, 74, 5828–5835. [Google Scholar] [CrossRef]
- Chérest, M.; Felkin, H.; Prudent, N. Torsional Strain Involving Partial Bonds. The Stereochemistry of the Lithium Aluminium Hydride Reduction of Some Simple Open-Chain Ketones. Tetrahe- dron Lett. 1968, 2199–2204. [Google Scholar]
- Wipke, W. T.; Gund, P. Simulation and Evaluation of Chemical Synthesis. Congestion: a Conformation-Dependent Function of Steric Environment at a Reaction Center. Application with Torsional Terms to Stereoselectivity of Nucleophilic Additions to Ketones. J. Am. Chem. Soc. 1976, 98, 8107–8118. [Google Scholar] [CrossRef]
- Wu, Y.-D.; Houk, K. N. Electronic and Conformational Effects of π-Facial Stereoselectivity in Nucleophilic Additions to Carbonyl Compounds. J. Am. Chem. Soc. 1987, 109, 908–910. [Google Scholar]
- Wu, Y.-D.; Houk, K. N.; Trost, B. M. Origin of Enhanced Axial Attack by Sterically Undemanding Nucleophiles on Cyclohexanones. J. Am. Chem. Soc. 1987, 109, 5560–5561. [Google Scholar] [CrossRef]
- Wu, Y.-D.; Houk, K. N. Effect of Torsional Strain and Electrostatic Interactions on the Stereochemistry of Nucleophilic Additions to Cyclohexanone and Related Systems. Angew. Chem. Int. Ed. Engl. 1992, 31, 1019–1021. [Google Scholar] [CrossRef]
- Carlsen, P. H. J.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. A Greatly Improved Procedure for Ruthenium Tetraoxide Catalyzed Oxidations of Organic Compounds. J. Org. Chem. 1981, 46, 3936–3938. [Google Scholar] [CrossRef]
- Corey, E. J.; Gross, A. W. Highly Selective, Kinetically Controlled Enolate Formation using Lithium Dialkylamides in the Presence of Trimethylchlorosilane. Tetrahedron Lett. 1984, 25, 495–498. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, M. A.; Iqbal, J. An Efficient Regio and Stereoselective Synthesis of Silyl Enol Ethers. Synth. Commun. 1988, 18, 1679–1683. [Google Scholar] [CrossRef]
- Tsuji, J.; Minami, I.; Shimizu, I. A Novel Palladium-Catalyzed Preparative Method of α,β-Unsaturated Ketones and Aldehydes from Saturated Ketones and Aldehydes via Their Silyl Enol Ethers. Tetrahedron Lett. 1983, 24, 5635–5638. [Google Scholar] [CrossRef]
- Minami, I.; Takahashi, K.; Shimizu, I.; Tsuji, J. New Synthetic Methods for α,β-Unsaturated Ketones, Aldehydes, Esters and Lactones by Palladium-Catalyzed reactions of Silyl Enol Acetates with Allyl Carbonates. Tetrahedron 1986, 42, 2971–2977. [Google Scholar] [CrossRef]
- Mincone, E.; Ortaggi, G.; Sirna, A. The Regioselective α,β-Dehydrogenation of Ketosteroids by Palladium(II) Chloride. Synthesis 1977, 773–774. [Google Scholar] [CrossRef]
- Barton, D. H. R.; Lester, D. J.; Ley, S. V. Dehydrogenation of Steriodal Ketones using Benzeneseleninic Anhydride. J. Chem. Soc. Chem. Commun. 1978, 130–131. [Google Scholar] [CrossRef]
- Barton, D. H. R.; Lester, D. J.; Ley, S. V. Dehydrogenation of Steriodal and Triterpenoid Ketones using Benzeneseleninic Anhydride. J. Chem. Soc. Perkin Trans I 1980, 2209–2212. [Google Scholar] [CrossRef]
- Barton, D. H. R.; Morzycki, J. W.; Motherwell, W. B.; Ley, S. V. Oxygen Atom Transfer from Iodylbenzene to Diphenyl Diselenides – A Convenient Method for Dehydrogenation of Steroidal 3-Ketones. J. Chem. Soc. Chem. Commun. 1981, 1044–1045. [Google Scholar] [CrossRef]
- Dragojlovic, V. Preparation of Cyclopentenones by Benzeneseleninic Anhydride Oxidation of Cyclopentanones. J. Chem. Res. (S) 1999, 256–257. [Google Scholar] [CrossRef]
- Lee, R. A.; McAndrews, C.; Patel, K. M.; Reusch, W. Methylation of Kinetically Generated Dienolate Anions Derived from α,β-Unsaturated Ketones. Tetrahedron Lett. 1973, 965–968. [Google Scholar] [CrossRef]
- Olofson, R. A.; Dougherty, C. M. Lithium 2,2,6,6-Tetramethylpiperidide and Related, Strong, Proton-Specific Bases. Evaluation in Synthesis. J. Am. Chem. Soc. 1973, 95, 582–584. [Google Scholar] [CrossRef]
- Bohlmann, F.; Jakupovic, J. Neue Sesquiterpen-Kohlenwasserstoffe mit Anomalen Kohlenstoffgerüst aus Silphium-Arten. Phytochemistry 1980, 19, 259–265. [Google Scholar] (the spectra have been erroneously reversed Wender, P.; Singh, S. K. Synthetic Studies on Arene-Olefin Cycloadditions-VIII. Total Synthesis of (±)-Silphiperfol-6-ene, (±)-7αH-Silphiperfol-5-ene and (±)-7βH-Silphiperfol-5- ene. Tetrahedron Lett. 1985, 26, 5987–5990. [Google Scholar] ).
- Jakupovic, J.; Zdero, C.; Paredes, L.; Bohlmann, F. Sesquiterpene Glycosides and Other Constituents from Osteospermum Species. Phytochemistry 1988, 27, 2881–2886. [Google Scholar] [CrossRef]
- Knudsen, M. J.; Shore, N. E. Synthesis of the Angularly Fused Triquinane Skeleton via Intramolecular Organometallic Cyclization. J. Org. Chem. 1984, 49, 5025–5026. [Google Scholar] [CrossRef]
- Huguet, J.; Karpf, M.; Dreiding, A. S. Synthesis of a Stereoisomer of Ptychanolide. Tetrahedron Lett. 1983, 24, 4177–4180. [Google Scholar] [CrossRef]
- Still, W. C.; Kahn, M.; Mitra, A. Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution. J. Org. Chem. 1978, 43, 2923–2925. [Google Scholar] [CrossRef]
- Kofron, W. G.; Baclawski, L. M. A Convenient method for Estimation of Alkyllithium Concentrations. J. Org. Chem. 1976, 41, 1879–1880. [Google Scholar] [CrossRef]
- Wuts, P. G. M. An Expedient Procedure for the Purification of the CuBr℘CH SCH Complex. Synth. Commun. 1981, 11, 139–140. [Google Scholar] [CrossRef]
- Sigma Chemical Co., S-6628 Silica-Gel for Thin Layer Chromatography, Type H, Size 10-40 μ, binder: none.
- Samples Availability: Not available.
Figure 1.

Scheme 1.
Scheme 2.
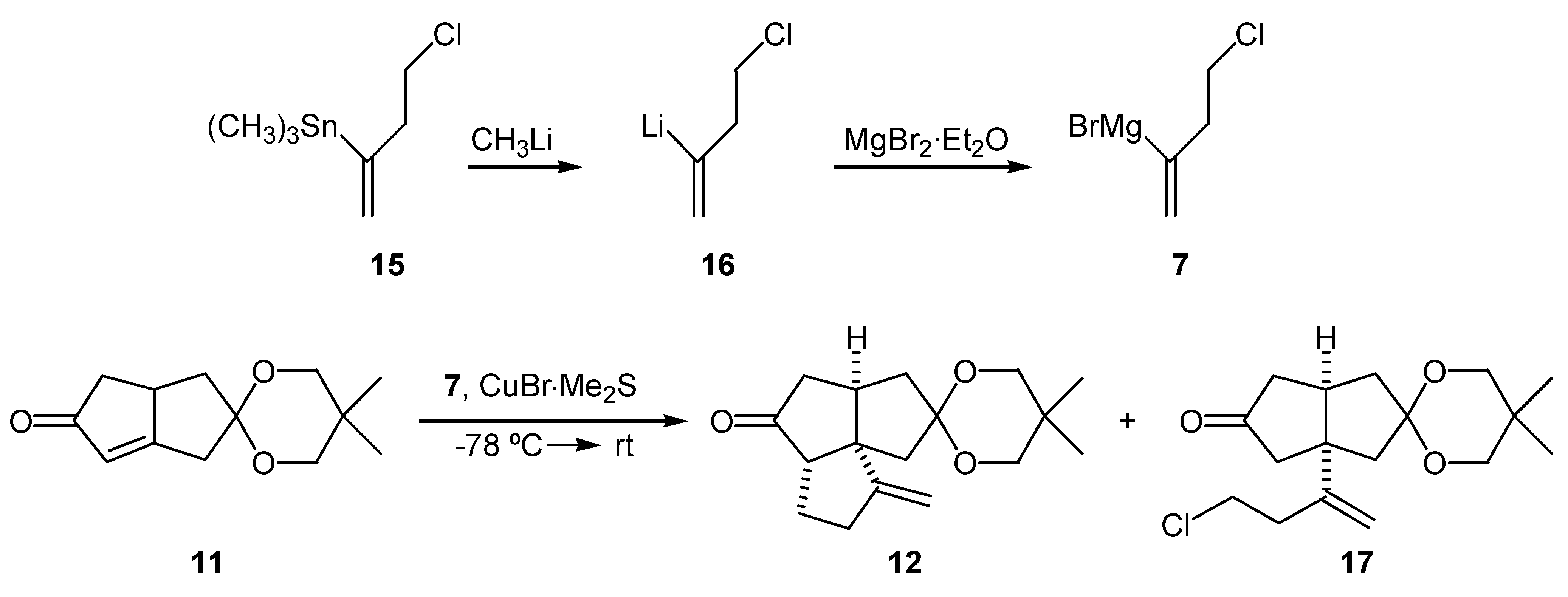
Figure 2.
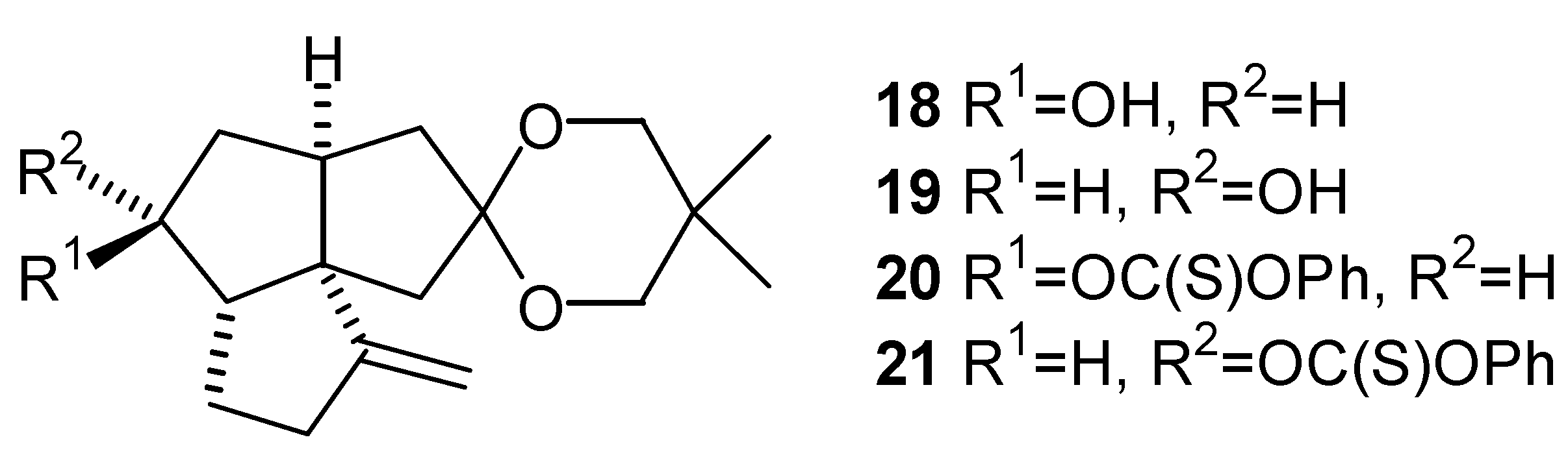
Scheme 3.
Scheme 4.
Scheme 5.
Scheme 6.
Scheme 7.
Scheme 8.
Scheme 9.

{kind=link}
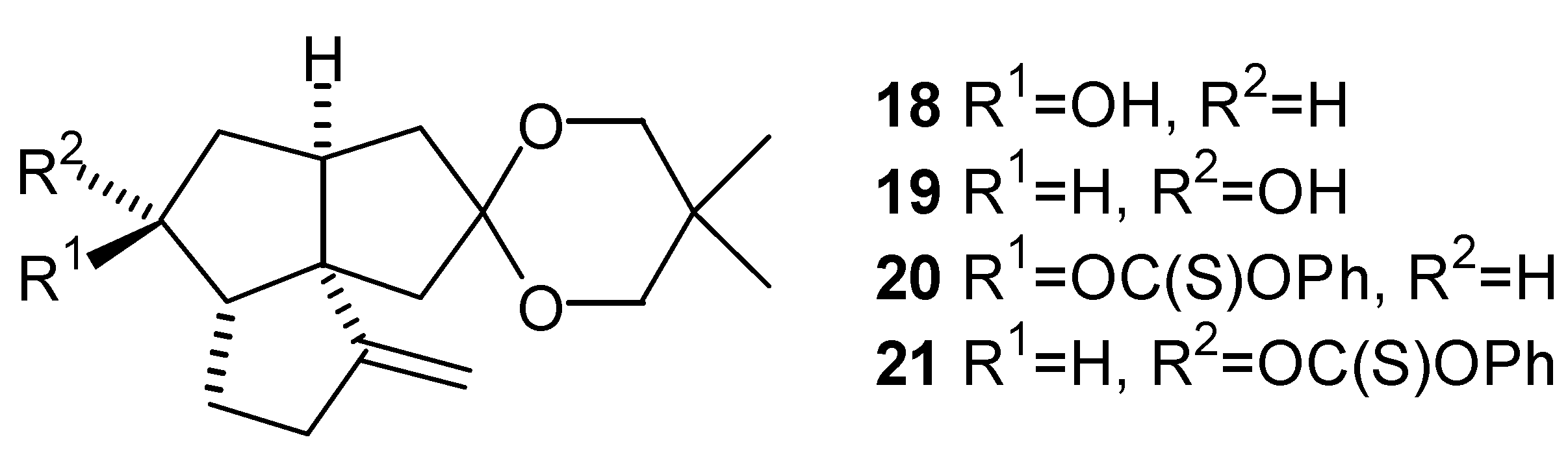{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Additive | (12)/(17)a | yield of (12)(%) |
| (1) | (none) | 50/50 | 74b |
| (2) | TMEDA (4 eq.) | 90/10 | 75 |
| (3) | HMPA (2 eq.) | 95/5 | 80 |
| (4) | DMPU (2 eq.) | 95/5 | 81 |
GLC ratio; b) Isolated yield of (12) after treatment of (12)/(17) mixture with KH/THF.
| Entry | Reducing Agent | (18)/(19)a | Yield (%) |
| (1) | LiAlH4 | >98/<2 | 97b |
| (2) | NaBH4 | 75/25 | 97c |
| (3) | DIBAL-H | 20/80 | 96c |
| (4) | L-Selectride® | 4/96 | 92b |
a) 1H NMR ratio. b) Isolated yield of the major epimer. c) Isolated yield of the mixture of alcohols.
| Proton | δ | COSY correlationsb | NOEsb,c |
| phenylthionocarbonate (20) | |||
| H-4 α | 1.60 | H-4 β, H-5 | 1.87 (H-4 β) 5.32 (H-6) |
| H-4 β | 1.87 | H-4 α, H-5 | 1.60 (H-4 α) 2.69 (H-5) |
| H-5 | 2.69 | H-4 α & β, H-6 | 1.87 (H-4 β) 2.20 (H-11 β) 5.32 (H-6) |
| H-6 | 5.32 | H-5, H-7 α and β | 2.32-2.50 (H-3 α & H-7 α) 2.69 (H-5) |
| phenyl thionocarbonate (21)a | |||
| H-4 α | 1.61 | H-5 | |
| H-4 β | 1.71 | H-5 | 1.61 (H-4 α) 2.79 (H-5) |
| H-5 | 2.79 | H-4 α & β, H-6 | 1.71 (H-4 β) 2.19 (H-11 β) 5.81 (H-6) |
| H-6 | 5.81 | H-5, H-7 α & β | 1.90 (H-9 β) 2.07-2.15 (H-7 α & β) 2.79 (H-5) |
| H-9 α | 2.19 | H-9 β | |
| H-9 β | 1.90 | H-9 α | 2.19 (H-9 α) 5.81 (H-6) |
a) Subergorgic acid numbering. b) Only those COSY correlations and NOE data that could be unambigously assigned and were crucial to the determination of the stereochemistry of (20) and (21) are reported. c) Enhanced resonances attributed to the proton(s) resulting from the irradiation at δ corresponding to the proton column.
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Dragojlovic, V. Synthesis of a Highly Functionalized Triquinane: Studies Towards a Total Synthesis of Subergorgic Acid and Its Analogues. Molecules 2000, 5, 674-698. https://doi.org/10.3390/50400674
AMA Style
Dragojlovic V. Synthesis of a Highly Functionalized Triquinane: Studies Towards a Total Synthesis of Subergorgic Acid and Its Analogues. Molecules. 2000; 5(4):674-698. https://doi.org/10.3390/50400674
Chicago/Turabian StyleDragojlovic, Veljko. 2000. "Synthesis of a Highly Functionalized Triquinane: Studies Towards a Total Synthesis of Subergorgic Acid and Its Analogues" Molecules 5, no. 4: 674-698. https://doi.org/10.3390/50400674