Reduction and Preparation of a Phthalimide Derivative from a Furo-heliangolide

Departamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, Universidade de São Paulo, Av. Bandeirantes 3900, 14040-901 – Ribeirão Preto – SP, Brazil
*
Author to whom correspondence should be addressed.
Molecules 2000, 5(6), 908-915; https://doi.org/10.3390/50600908
Submission received: 18 February 2000
/
Accepted: 20 June 2000
/
Published: 27 June 2000
Abstract
:Goyazensolide, a biologically active natural product classified as a furoheliangolide, was transformed in a phthalimide derivative that showed an enhanced biological activity against Tripanosoma cruzi. The complex natural product was also reduced with high stereoselectivity by hydrogen/Wilkinson’s catalyst; an epimer of this product was obtained in the reduction with hydrogen/Pd-C.
Introduction
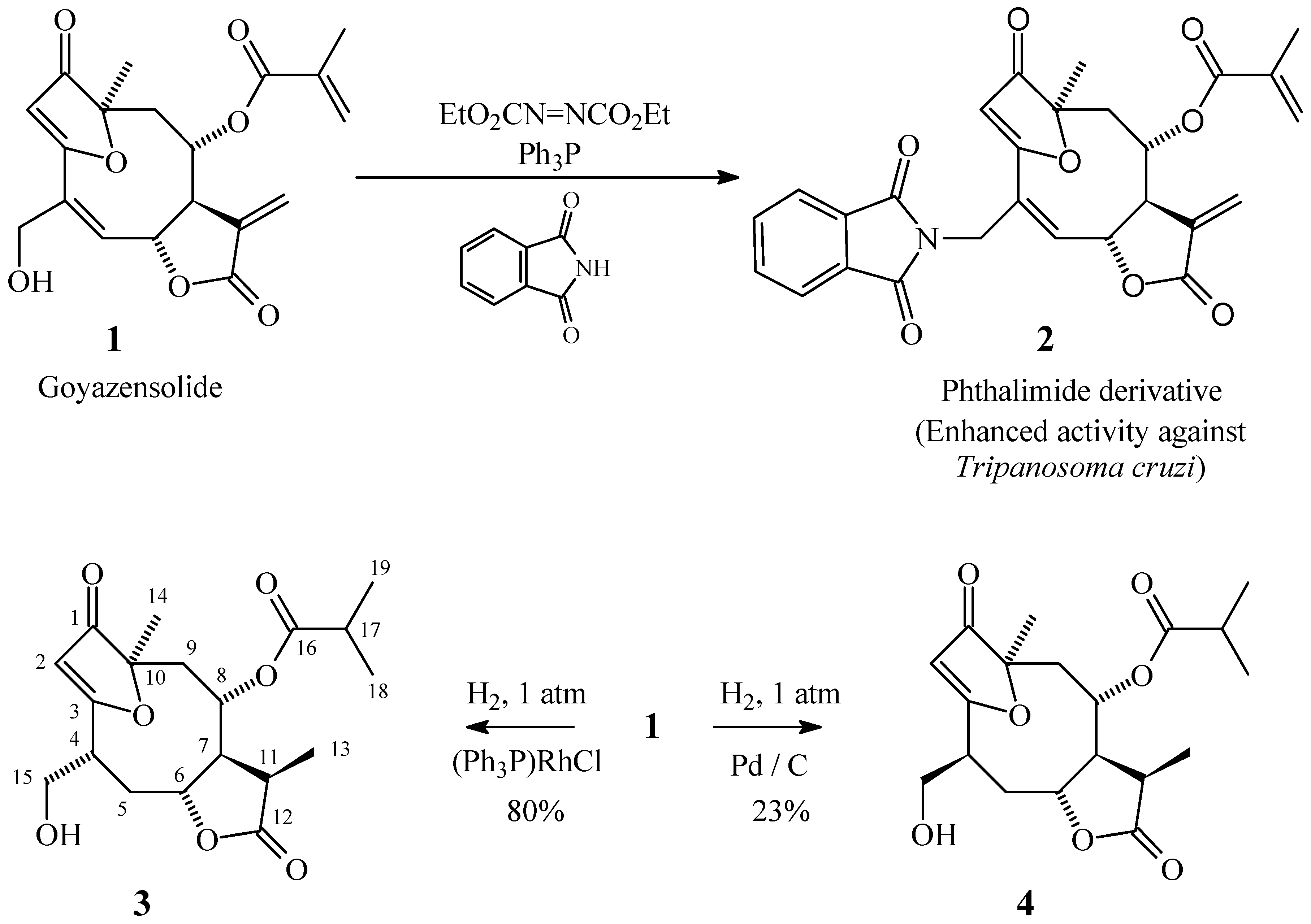
Goyazensolide is a natural product isolated from a Brazilian plant, Eremanthus goyazensis (Compositae, tribe Vernoniae), collected in Minas Gerais State [1,2]. It is a furo-heliangolide possessing schistosomicidal and cytotoxic properties and can usually be isolated in larger amounts than many other biologically active products of similar structure found on the same plant or in other plants of the same species. Seeking methods for the preparation of the more scarce natural products or biologically active derivatives from the relatively abundant goyazensolide, we have recently carried out some chemical transformations of this product. To this extent we have succeeded in preparing a phthalimide derivative 2 with enhanced biological activity against Tripanosoma cruzi and in reducing 1 with high stereoselectivity by H2/Wilkinson’s catalyst, thus obtaining compound 3. Reduction of 1 with H2/Pd/C produced a complex mixture from which 4, an epimer of 3, could be isolated.
Scheme 1.

Results and Discussion
Hydrogenation of goyazensolide (1) (20.0 mg) at room temperature for 2 h, with Pd / C as catalyst, produced a complex mixture that was separated by column chromatography into three fractions: F1 (10.8 mg), F2 (2.8 mg) and F3 (4.7 mg). Only the third fraction, F3, was formed by a pure product (4, thus obtained in 23% yield); the other two fractions were still mixtures of at least 4 compounds each, which led us to the decision that it would be worthless to attempt the isolation of other pure products, as they would be obtained in too low yield. Nevertheless, 1H NMR analysis of these fractions gave us some interesting information: 1) The less polar fraction F1 (obtained in higher yield) didn’t show, in its 1H NMR spectrum, the signals corresponding to the –CH2OH group, strongly suggesting that it consisted mainly of hydrogenolysis products; 2) Most (virtually all) of the components of all fractions showed the signal corresponding to the vinylic hydrogen of the furanone ring, leading to the conclusion that the double bond in the furanone ring is the most resistant (to this hydrogenation process) of all double bonds in the molecule of goyazensolide.
In sharp contrast, when goyazensolide was hydrogenated with Wilkinson’s catalyst, only one product was obtained in high yield. After the first column chromatography, to remove most of the impurities formed from the catalyst, a quantitative yield of a crude product containing essentially only 3 and some aromatic impurities was obtained. On further purification it gave 80% of pure 3.
Compounds 3 and 4 were submitted to a careful analysis mainly by 1H and 13C NMR spectra. Through the further use of techniques such as COSY, Jres, double irradiation, etc., we were able to assign all chemical shifts and to measure all H / H coupling constants. The results, comparing compounds 3 and 4, are shown in Table 1, Table 2 and Table 3.

{kind=link}
| H | 3 | 4 | δ(4) - δ(3) |
|---|---|---|---|
| 5α | 2.14 | 2.44 | 0.30 |
| 4 | 3.33 | 3.10 | -0.23 |
| 5β | 2.35 | 2.14 | -0.21 |
| 7 | 3.06 | 3.26 | 0.20 |
| 2 | 5.70 | 5.89 | 0.19 |
| 15a | 3.85 | 3.99 | 0.14 |
| 15b | 3.80 | 3.93 | 0.13 |
| 8 | 4.93 | 5.01 | 0.08 |
| 9β | 2.07 | 2.10 | 0.03 |
| 6 | 4.23 | 4.26 | 0.03 |
| 9α | 2.20 | 2.18 | -0.02 |
| 17 | 2.53 | 2.55 | 0.02 |
| 18 | 1.18 | 1.20 | 0.02 |
| 19 | 1.17 | 1.19 | 0.02 |
| 11 | 2.88 | 2.89 | 0.01 |
| 13 | 1.11 | 1.12 | 0.01 |
| 14 | 1.48 | 1.49 | 0.01 |
| J | 3 | 4 | J(4) – J(3) |
|---|---|---|---|
| 4,5β | 7.0 | 3.4 | -3.6 |
| 8,9α | 6.8 | 5.3 | -1.5 |
| 6,7 | 5.5 | 4.1 | -1.4 |
| 4,5α | 10.0 | 8.7 | -1.3 |
| 4,15a | 7.9 | 6.7 | -1.2 |
| 2,4 | 0.0 | 1.1 | 1.1 |
| 5α,6 | 8.6 | 9.7 | 1.1 |
| 7,11 | 8.3 | 9.3 | 1.0 |
| 5β,6 | 0.8 | 1.4 | 0.6 |
| 9α,9β | 15.0 | 15.5 | 0.5 |
| 8,9β | 2.5 | 3.0 | 0.5 |
| 7,8 | 9.3 | 9.8 | 0.5 |
| 17,18 | 7.2 | 6.8 | -0.4 |
| 5α,5β | 14.0 | 14.3 | 0.3 |
| 15a,15b | 10.7 | 10.5 | -0.2 |
| 4,15b | 5.9 | 5.8 | -0.1 |
| 11,13 | 7.4 | 7.5 | 0.1 |
| 3 | 4 | |
|---|---|---|
| C-1 | 205.5 | 205.9 |
| C-2 | 105.9 | 102.8 |
| C-3 | 189.0 | 189.0 |
| C-4 | 42.5 | 39.6 |
| C-5 | 35.2 | 34.5 |
| C-6 | 79.5 | 78.1 |
| C-7 | 51.9 | 49.8 |
| C-8 | 70.4 | 71.1 |
| C-9 | 40.4 | 37.4 |
| C-10 | 89.3 | 88.2 |
| C-11 | 38.4 | 37.3 |
| C-12 | 177.7 | 177.8 |
| C-13 | 10.8 | 10.3 |
| C-14 | 19.9 | 19.9 |
| C-15 | 63.4 | 62.0 |
| C-16 | 175.3 | 175.4 |
| C-17 | 34.2 | 34.2 |
| C-18 | 18.6 | 18.7 |
| C-19 | 18.8 | 18.7 |
The data on Table 1 and Table 2 are sorted according to decreasing absolute values of the difference between data for compound 4 and for compound 3 (last column). In this way it is easy to verify that, with a few exceptions, most of the larger differences (the values on the upper part of the tables) occur with hydrogens near to the chiral center C-4, the relative stereochemistry of which is the difference between isomers 3 and 4. These data are reasonably consistent with literature data [3] for natural products with structures similar to 3 and 4.
Although the general structure features of compounds 3 and 4 can be reasonably established by these data, the same can not be said about their relative stereochemistry. A few indications like the value of coupling constants J2,4 (the larger value for isomer 4 is in agreement with the expected values when considering the involved angles [4], according to Drieding molecular models), J4,5 and J7,11 were considered insufficient, and we have then performed some measures of the Nuclear Overhauser Effect (NOE) for both isomers. The results, summarized in Table 4, clearly and unambiguously establish the relative stereochemistry of compounds 3 and 4 as assigned in this paper.
| Irradiated Hydrogen | Enhanced Signals | |
|---|---|---|
| Compound 3 | Compound 4 | |
| 5α | 5β, 7, 15, 6 | 5β, 4, 7, 6 (small) |
| (+9α+9β in 3) | (8, through 9α and 9β) | |
| 5β | 5α, 4, 15, 6 | 5α, 6, 15 (small) |
| (+9α+9β in 4) | (8, through 9α and 9β) | |
| 6 | 5α, 5β, 13, 7 | 5β, 13 (small) |
| 7 | 11, 5α [8, 6 small] | 11, 5α |
| 13 | 11, 8, 6 | 8, 11, 6 (small) |
| (+18+19) | (17, through 18 and 19) | (17, through 18 and 19) |
| 11 | 7, 13 | 7, 13 |
| 4 | Not effected | 15, 5α |
The stereochemical outcome of these hydrogenation reactions is not surprising, particularly in the case of the chiral center C-11. Examining a 3-D model (or a stereoscopic projection such as the one found in reference 2) of goyazensolide we can see that, contrary to the first impression given by the planar representation, the methacrylate ester group is above the methylene of the lactone ring, blocking up the β-face of the double bond, thus forcing the approach of the catalysts (and the hydrogen) from the α- face. Around the center C-4 there is not a so strong sterical hindrance; the goyazensolide molecule, however, is rather bent downward and favors the approach of a bulk reagent such as the Wilkinson’s catalyst from the β-face. We cannot draw many conclusions about the Pd / C catalyst because a number of unidentified products were formed in this reduction. However, the absence of 3 among the products suggests that, when Pd / C approaches goyazensolide from the β-face, hydrogenolysis products are rather formed; also, as this catalyst is not as much sensitive to sterical hindrance, a possible complexation with the oxygen of the furanone ring could account for the formation of a representative amount of isomer 4.
Several phthalimide derivatives are known to be active against Tripanosoma cruzi [5]; as Mitsunobu [6,7] has described a general method for transforming alcohols into phthalimide derivatives under reasonably mild conditions, we decided to apply this method to goyazensolide. The reaction is very simple, consisting only in mixing the reagents in THF and stirring at room temperature for several hours; separation and purification of the product was not as easy because some excess reagents and by-products have Rf values similar to the Rf of the product, rendering the purification a rather laborious process.
The biological activity of derivative 2 against Tripanosoma cruzi (Y strain) was evaluated and compared to the activity of goyazensolide by Professor Sérgio de Albuquerque. The results [8] show that the derivative is considerably more active than goyazensolide.
Experimental
General
NMR spectra were measured using a Bruker DPX-300 (300 MHz 1H NMR and 75 MHz 13C NMR) instrument; deuterochloroform was used as solvent and tetramethylsilane as the internal standard. IR spectra were measured with a Perkin-Elmer 1600-FT or Nicolet 5ZDX spectrometers. TLC was performed on precoated silica gel 60 F254 plates (0.25 mm thick, Merck), and 70-230 mesh silica gel 60 (Merck) was used for column chromatography.
Preparation of Compound 2
Ethyl azodicarboxylate [9,10] (15 mg, 0.088 mmol), phthalimide (13 mg, 0.091 mmol), triphenylphosphine (23 mg, 0.089 mmol), goyazensolide (1) (32 mg, 0.088 mmol) and dry THF (3 mL) were mixed and stirred at room temperature, checking the progress of the reaction by TLC. After 23 h, as no further consumption of 1 seemed to be taking place, new portions of reagents and solvent were added (same amounts as above, except that no goyazensolide (1) was added). Stirring was continued for an additonal 18 h period at room temperature. Column chromatography of the crude reaction product using 9:1 ethyl acetate / hexane as eluent produced 67 mg of material containing the desired product. This was again chromatographed with a different eluent (1:1 ethyl acetate / hexane) thus yielding pure 2 (26 mg, 60%).
1H NMR (300 MHz, CDCl3) δ 7.90 and 7.78 (AA’BB’ system) (J1 5.4 Hz, J2 3.0 Hz), 6.22 (m, 1 H, ΣJ = 5 Hz), 6.20 (d, 1H, J 3.0 Hz), 5.99 (m, 1H, ΣJ = 6 Hz), 5.80 (s, 1H), 5.53 (m, 1H, ΣJ = 6 Hz), 5.43 (d, 1H, J 3.0 Hz), 5.29 (m, 1H, ΣJ = 12 Hz), 4.60 (dt, 1H, J1 15.8 Hz, J2 2.0 Hz, J3 2.0 Hz), 4.54 (dt, 1H, J1 15.8 Hz, J2 2.0 Hz, J3 2.0 Hz), 4.49 (dt, 1H, J1 11.7 Hz, J2 2.5 Hz, J3 2.0 Hz), 3.72 (m, 1H, ΣJ = 14 Hz), 2.44 (dd, 1H, J1 14.0 Hz, J2 11.7 Hz), 2.27 (dd, 1H, J1 14.0 Hz, J2 2.0 Hz), 1.81 (br. s, 3H), 1.43 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 204.6 (C=O); 183.4 (C), 168.6 (C=O), 167.4 (C=O), 166.8 (C=O), 137.6 (CH), 135.4 (C), 134.5 (CH), 133.0 (C), 131.7 (C), 129.3 (C), 126.6 (CH2), 124.7 (CH2), 123.7 (CH), 106.6 (CH), 89.9 (C), 81.4 (CH), 73.3 (CH), 50.9 (CH), 43.9 (CH), 39.6 (CH2), 20.4 (CH3), 18.0 (CH3); IR (liquid film) νmax 1772, 1714, 1588, 1424, 1393, 1289, 1138, 916, 756, 722cm−1.
Preparation of Compound 3
A solution of goyazensolide (1) (164 mg, 0.46 m mol) and Wilkinson’s catalyst [(Ph3P)3RhCl] [11,12,13] in dry toluene (24 mL) was bubbled with nitrogen for 15 minutes to remove dissolved oxygen. Then the nitrogen atmosphere inside the system was changed to a hydrogen atmosphere (1 atm) and the reaction mixture was stirred at room temperature under hydrogen for 7.5 h. After filtration through silica gel, the solvent was removed under vacuum. The residue was chromatographed in a column of silica gel using 8:2 ethyl acetate / hexane as eluent, yielding 180 mg of a crude product containing considerable amount of aromatic impurities, but no starting material. This product was again chromatographed with the same eluent, but with higher proportion of silica gel (60 g silica gel for 1 g of crude product), separating the pure fractions and submitting the other fractions to a new chromatography. Yield of pure 3 134 mg (80%).
1H NMR (300 MHz, CDCl3) δ 5.70 (s, 1H), 4.93 (ddd, 1H, J1 9.3 Hz, J2 6.8 Hz, J3 2.5 Hz), 4.23 (ddd, 1H, J1 8.6 Hz, J2 5.5 Hz, J3 0.8 Hz), 3.85 (dd, 1H, J1 10.7 Hz, J2 7.9 Hz), 3.80 (dd, 1H, J1 10.7 Hz, J2 5.9 Hz), 3.33 (m, 1H, ΣJ = 30.5 Hz), 3.06 (dt, 1H, J1 9.3 Hz, J2 8.3 Hz, J3 5.5 Hz), 2.88 (dq, 1H, J1 8.3 Hz, J2 7.4 Hz), 2.53 (hep, 1H, J 7.2 Hz), 2.35 (ddd, 1H, J1 14.0 Hz, J2 7.0 Hz, J3 0.8 Hz), 2.20 (dd, 1H, J1 15.0 Hz, J2 6.8 Hz), 2.14 (ddd, 1H, J1 14.0 Hz, J2 10.0 Hz, J3 8.6 Hz), 2.07 (dd, 1H, J1 15.0 Hz, J2 2.5 Hz), 1.48 (s, 3H), 1.18 (d, 3H, J 7.2 Hz), 1.17 (d, 3H, J 7.2 Hz), 1.11 (d, 3H, J 7.4 Hz); 13C NMR (75 MHz, CDCl3) δ 205.5 (C=O), 189.0 (C), 177.7 (C=O), 175.3 (C=O), 105.9 (CH), 89.3 (C), 79.5 (CH), 70.4 (CH), 63.4 (CH2), 51.9 (CH), 42.5 (CH), 40.4 (CH2), 38.4 (CH), 35.2 (CH2), 34.2 (CH), 19.9 (CH3), 18.8 (CH3), 18.6 (CH3), 10.8 (CH3); IR (liquid film) νmax 3437, 1773, 1732, 1697, 1603, 1463, 1385, 1355, 1194, 1133, 1061, 756 cm−1.
Preparation of Compound 4
A mixture of 95% ethanol (2 mL) and 5% Pd / C (1 mg) was stirred under hydrogen atmosphere (1 atm) for 2 h. A solution of goyazensolide (1) (20 mg, 0.056 m mol) in ethanol (1.5 mL) was then added, and stirring under hydrogen atmosphere was continued until no further comsumption of hydrogen could be detected (2 h). The reaction mixture was filtered through Celite® and the solvent was removed under vacuum. The crude product was chromatographed in a silica gel column using 8:2 ethyl acetate / hexane as eluent. Three fractions were separated: F1 (10.8 mg), F2 (2.8 mg) and F3 (4.7 mg); the first two fractions were mixtures of at least 4 products each (by 1H NMR), but fraction F3 was pure compound 4, yield 4.7 mg (23%).
1H NMR (300 MHz, CDCl3) δ 5.89 (d, 1H, J 0.6 Hz); 5.01 (ddd, 1H, J1 9.8 Hz, J2 5.3 Hz, J3 3.0 Hz), 4.26 (ddd, 1H, J1 9.7 Hz, J2 4.1 Hz, J3 1.4 Hz), 3.99 (dd, 1H, J1 10.5 Hz, J2 6.7 Hz), 3.93 (dd, 1H, J1 10.5 Hz, J2 5.8 Hz), 3.26 (dt, 1H, J1 9.8 Hz, J2 9.3 Hz, J3 4.1 Hz), 3.10 (m, 1H, ΣJ = 23.4 Hz), 2.89 (dq, 1H, J1 9.3 Hz, J2 7.5 Hz), 2.55 (hep, 1H, J 6.8 Hz), 2.44 (ddd, 1H, J1 14.3 Hz, J2 9.8 Hz, J3 8.4 Hz), 2.18 (dd, 1H, J1 15.5 Hz, J2 5.3 Hz), 2.14 (ddd, 1H, J1 14.3 Hz, J2 3.6 Hz, J3 2.0 Hz), 2.10 (dd, 1H, J1 15.5 Hz, J2 3.0 Hz), 1.49 (s, 3H), 1.20 (d, 3H, J 6.8 Hz), 1.19 (d, 3H, J 6.8 Hz), 1.12 (d, 3H, J 7.5 Hz); 13C NMR (75 MHz, CDCl3) δ 205.9, (C=O), 189.0 (C), 175.4 (C=O), 157.8 (C=O), 102.8 (CH), 88.2 (C), 78.1 (CH), 71.1 (CH), 62.0 (CH2), 49.8 (CH), 39.6 (CH), 37.4 (CH2), 37.3 (CH), 34.5 (CH2), 34.2 (CH), 19.9 (CH3), 18.7 (CH3), 10.3 (CH3); IR (liquid film) νmax 3462, 1770, 1732, 1706, 1592, 1460, 1384, 1360, 1193, 1134, 1065, 810, 733 cm−1.
| Compound | % lysis | |||
| 100 | 250 | 500 μg / mL | ||
| Negative control | 0.0 | 0.0 | 0.0 | |
| Goyazensolide (1) | 9.9 | 12.9 | 22.4 | |
| Phthalimide derivative (2) | 50.0 | 52.9 | 61.5 | |
| *positive control gentian violet 250 μg / mL | ||||
Acknowledgements
The authors thank the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and the Coordenadoria de Aperfeiçoamento de Pessoal do Ensino Superior (CAPES) for financial support. We are grateful to Professor Walter Vichnewski and to Professor João Luis Callegari Lopes, as well as to other members of their research group, for supplying the samples of goyazensolide used in this work, and to Professor Sérgio de Albuquerque for performing the tests of biological activity.
References and Notes
- Vichnewski, W.; Takahashi, A. M.; Nasi, A. M. T. T.; Gonçalves, D. C. R. G.; Dias, D. A.; Lopes, J. N. C.; Goedken, V. L.; Gutiérrez, A. B.; Herz, W. Phytochemistry 1989, 28, 1441.
- Herz, W.; Goedken, V. L. J. Org. Chem. 1982, 47, 2798. [CrossRef]
- Cf. compounds 2a and 9a of reference 1. To make some data match better we had to assume that hydrogens 5α and 5β for compound 9a are inverted in reference [1].
- Jackman, L. M.; Sternhell, S. Applications of Nuclear Magnetic Resonance Spectroscopy in Organic Chemistry, 2nd Edition; Pergamon Press: Oxford, 1978; p. 316. [Google Scholar]
- Albuquerque, S.; Nasi, A. M. T. T.; Ribeiro, R. D.; Lopes, R. A.; Carraro, A. A.; Rodrigues, E. R. Mem. Inst. Oswaldo Cruz 1991, 86 Suppl.I, 234.
- Mitsunobu, O. Synthesis 1981, 1.
- Mitsunobu, O.; Wada, M.; Sano, T. J. Amer. Chem. Soc. 1972, 94, 679. [CrossRef]
- The results of biological activity measurements are summarized in the table below:
- Rabjohn, N. Organic Syntheses; Horning, E. C., Ed.; John Wiley & Sons: New York, 1963; Coll. Vol. III, p. 375. [Google Scholar]
- Kauer, J. C. Organic Syntheses; Rabjohn, N., Ed.; John Wiley & Sons: New York, 1967; Coll. Vol. IV, p. 411. [Google Scholar]
- Harwood, L. M.; Moody, C. J. Experimental Organic Chemistry: Principles and Practice; Blackwell Scientific Publications: Oxford, 1989; pp. 511–513. [Google Scholar]
- March, J. Advanced Organic Chemistry, 4th Edition; John Wiley & Sons: New York, 1992; p. 771. [Google Scholar]
- Ireland, R. E.; Bey, P. Organic Syntheses; Noland, W. E., Ed.; John Wiley & Sons: New York, 1988; Coll. Vol. VI, p. 459. [Google Scholar]
- Sample Availability: Not available.
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Da Silva, G.V.J.; Heleno, V.C.G.; Constantino, M.G. Reduction and Preparation of a Phthalimide Derivative from a Furo-heliangolide. Molecules 2000, 5, 908-915. https://doi.org/10.3390/50600908
AMA Style
Da Silva GVJ, Heleno VCG, Constantino MG. Reduction and Preparation of a Phthalimide Derivative from a Furo-heliangolide. Molecules. 2000; 5(6):908-915. https://doi.org/10.3390/50600908
Chicago/Turabian StyleDa Silva, Gil Valdo José, Vladimir Constantino Gomes Heleno, and Mauricio Gomes Constantino. 2000. "Reduction and Preparation of a Phthalimide Derivative from a Furo-heliangolide" Molecules 5, no. 6: 908-915. https://doi.org/10.3390/50600908