Three-Dimensional Quantitative Structural Activity Relationship (3D-QSAR) Studies of Some 1,5-Diarylpyrazoles: Analogue Based Design of Selective Cyclooxygenase-2 Inhibitors

Abstract
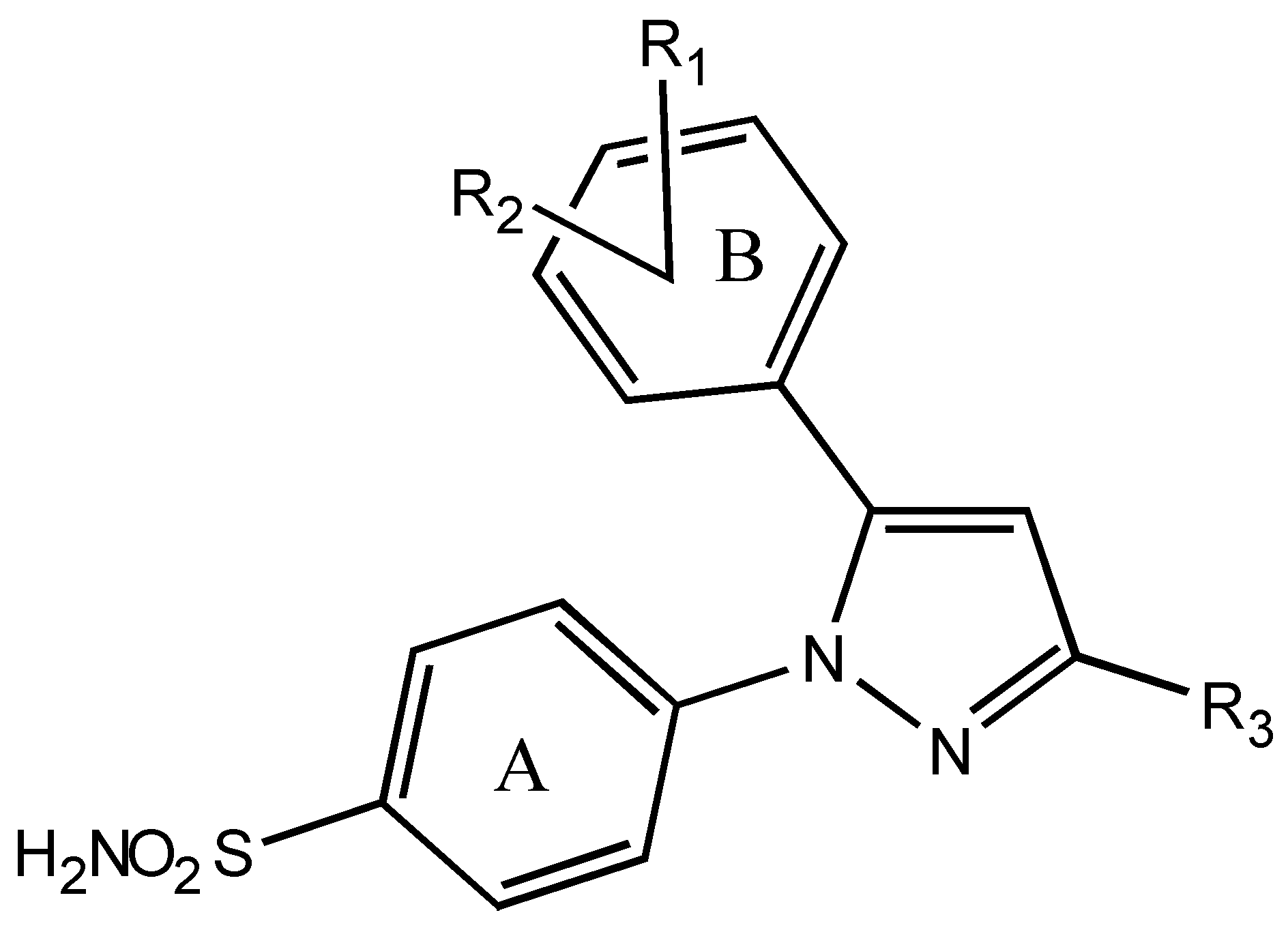
:Introduction
Computational Methods
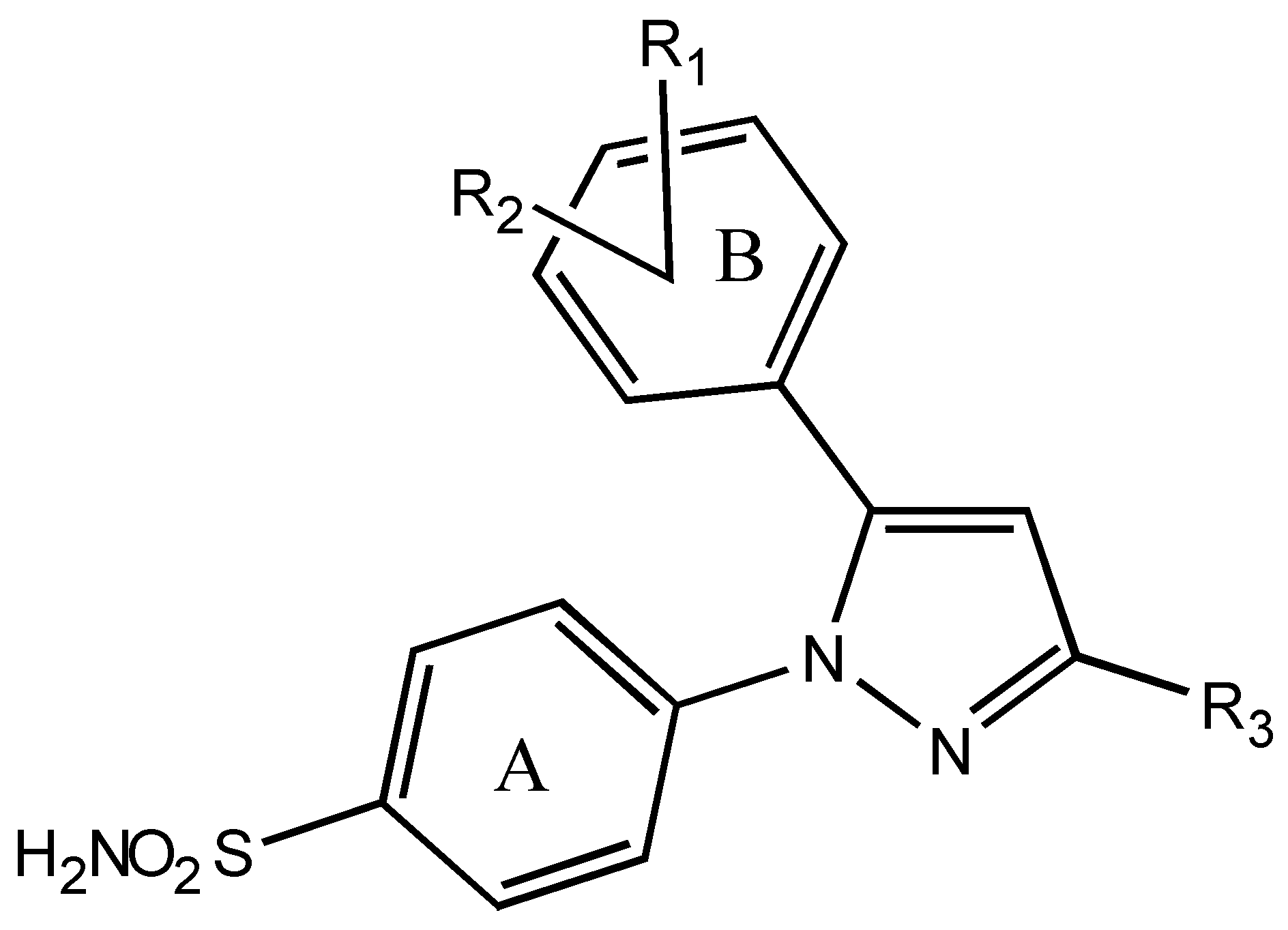
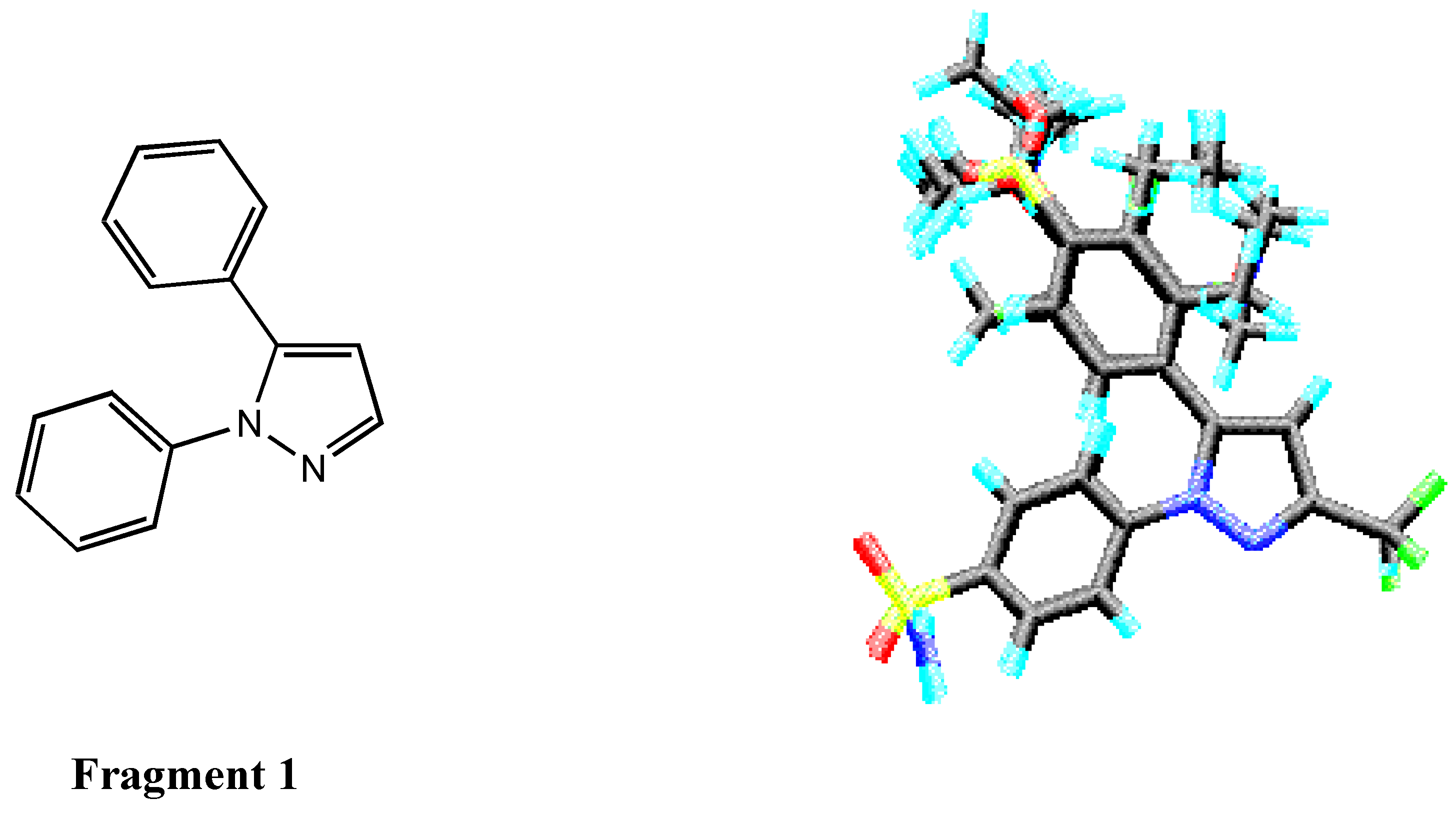
Molecular 3D Structure Building
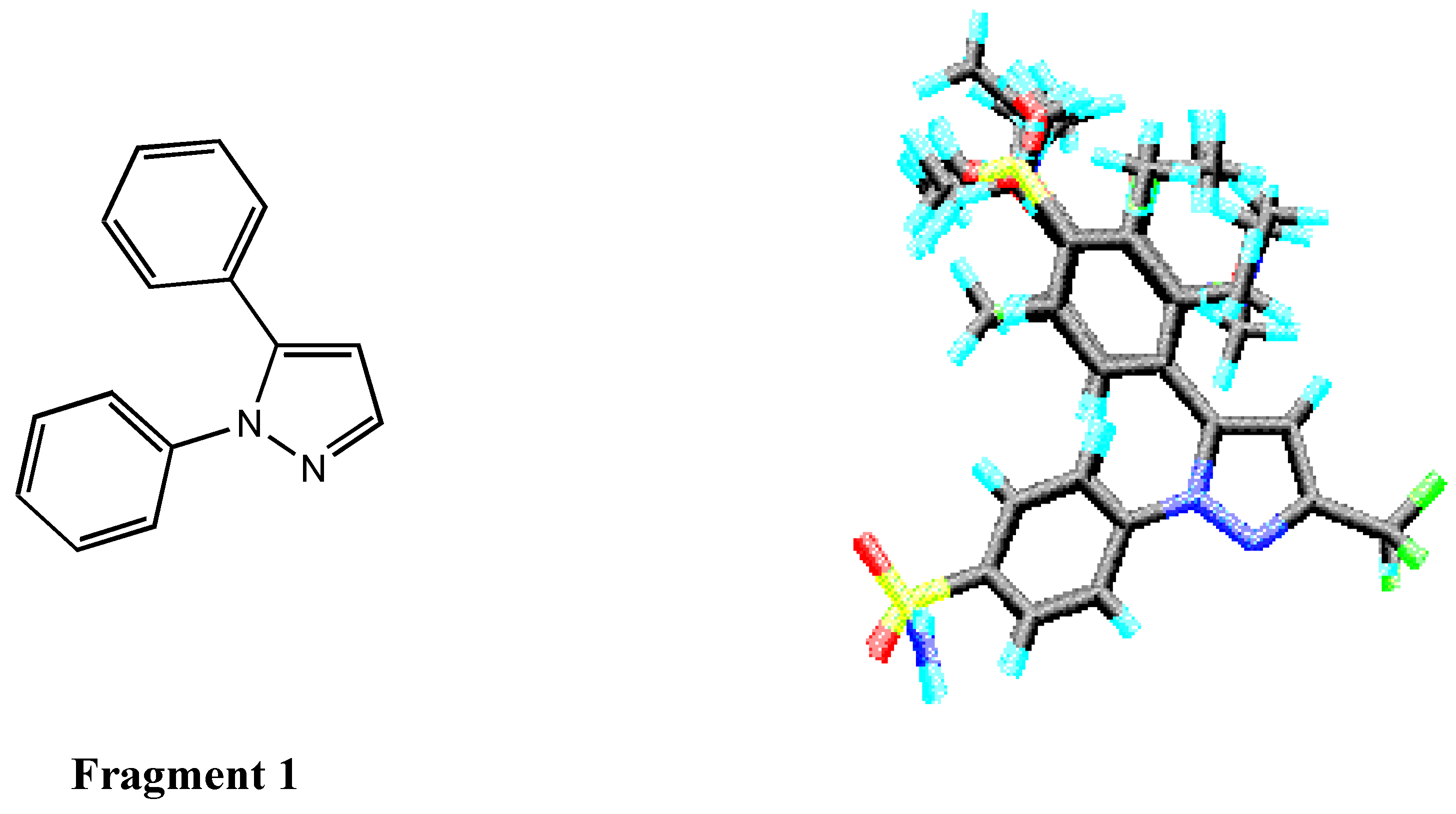
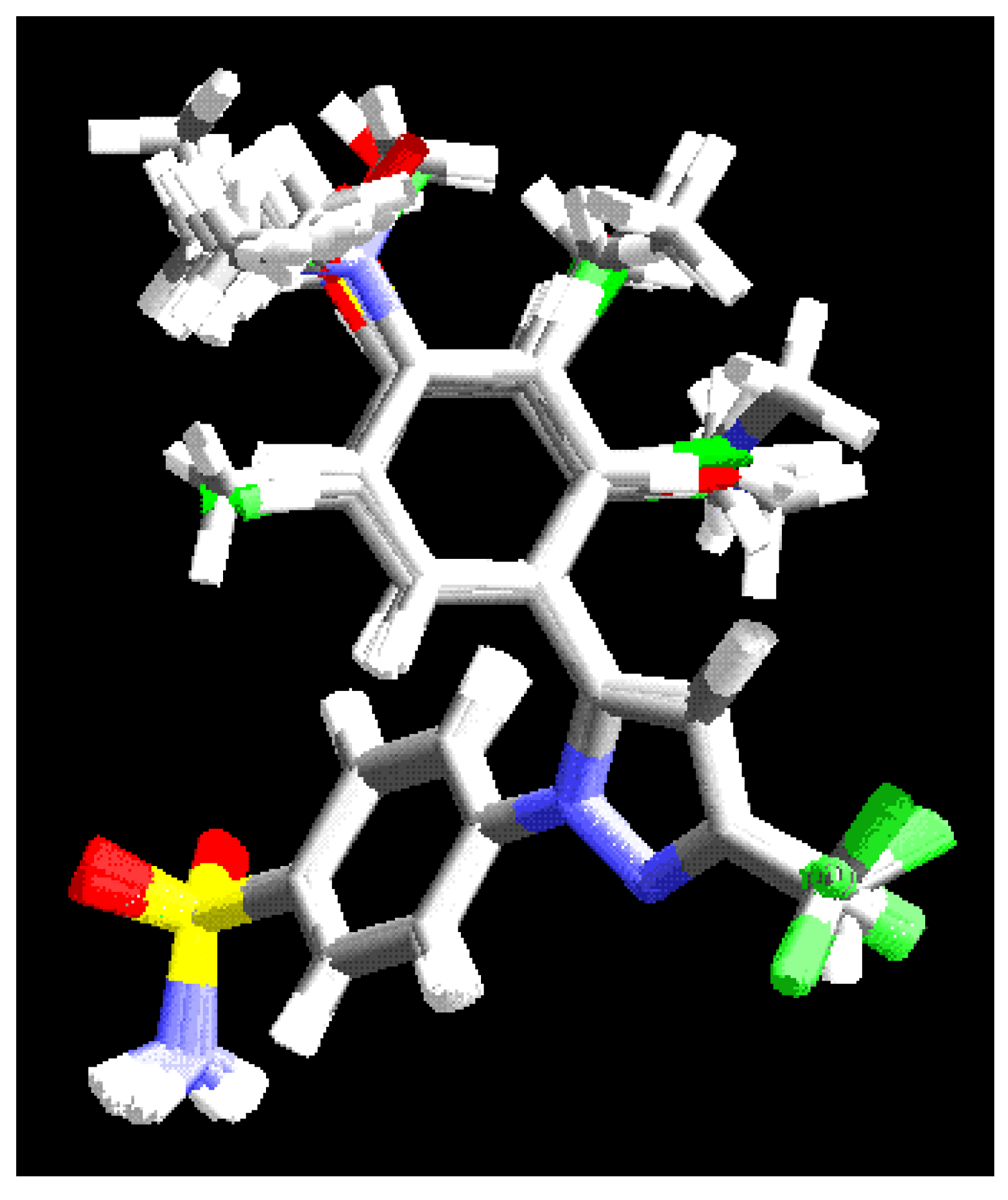
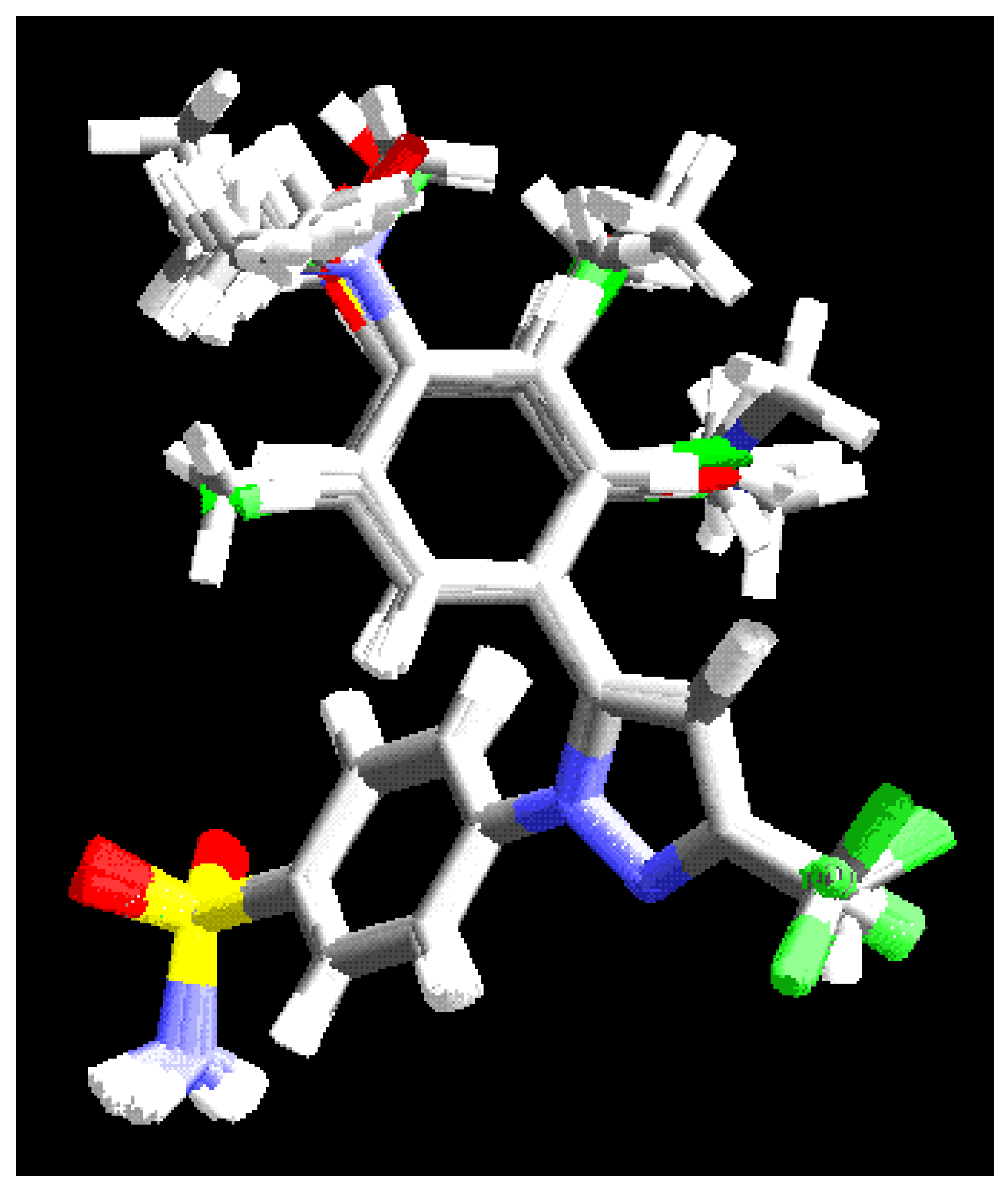
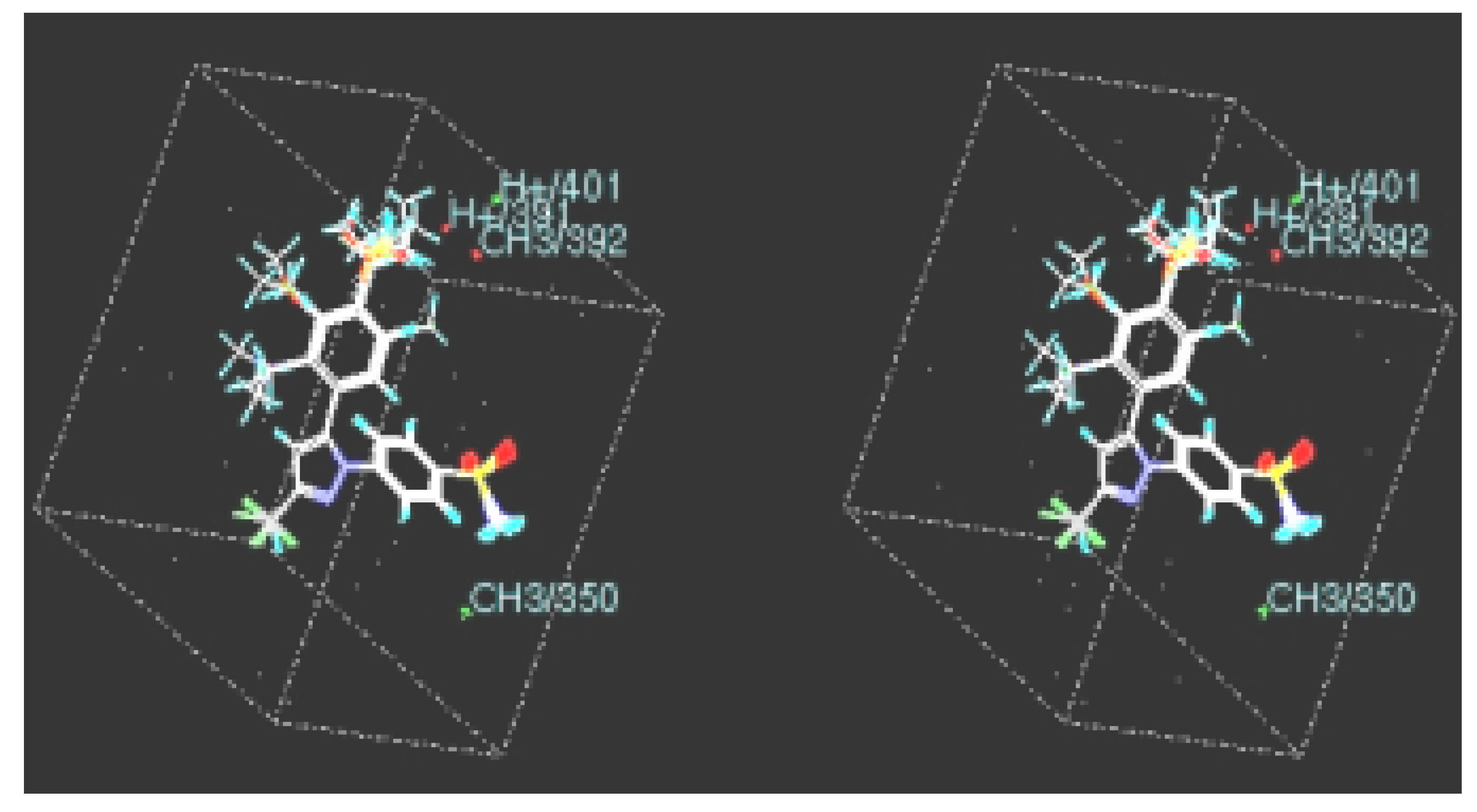
Alignment
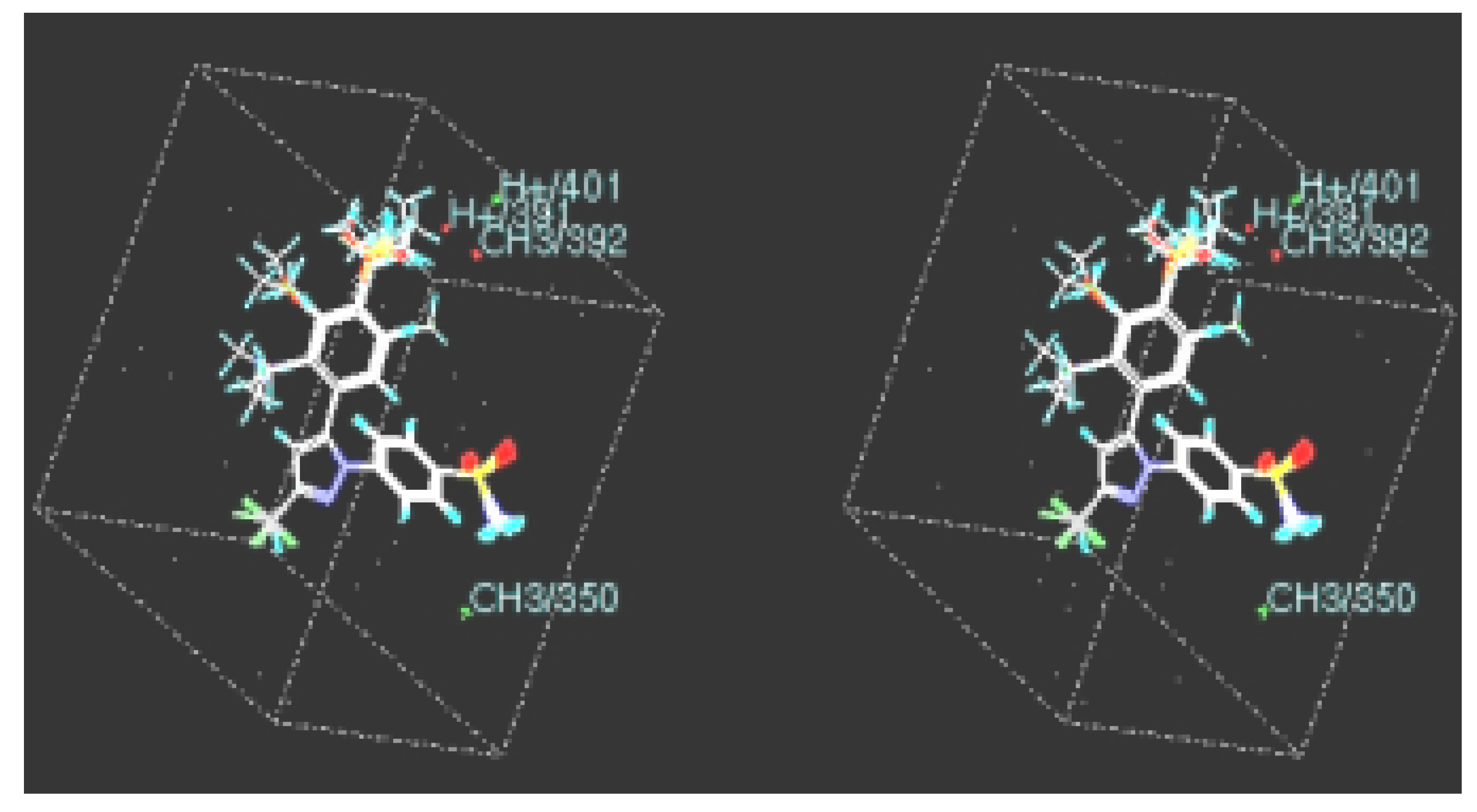
CoMFA
MFA
RSA
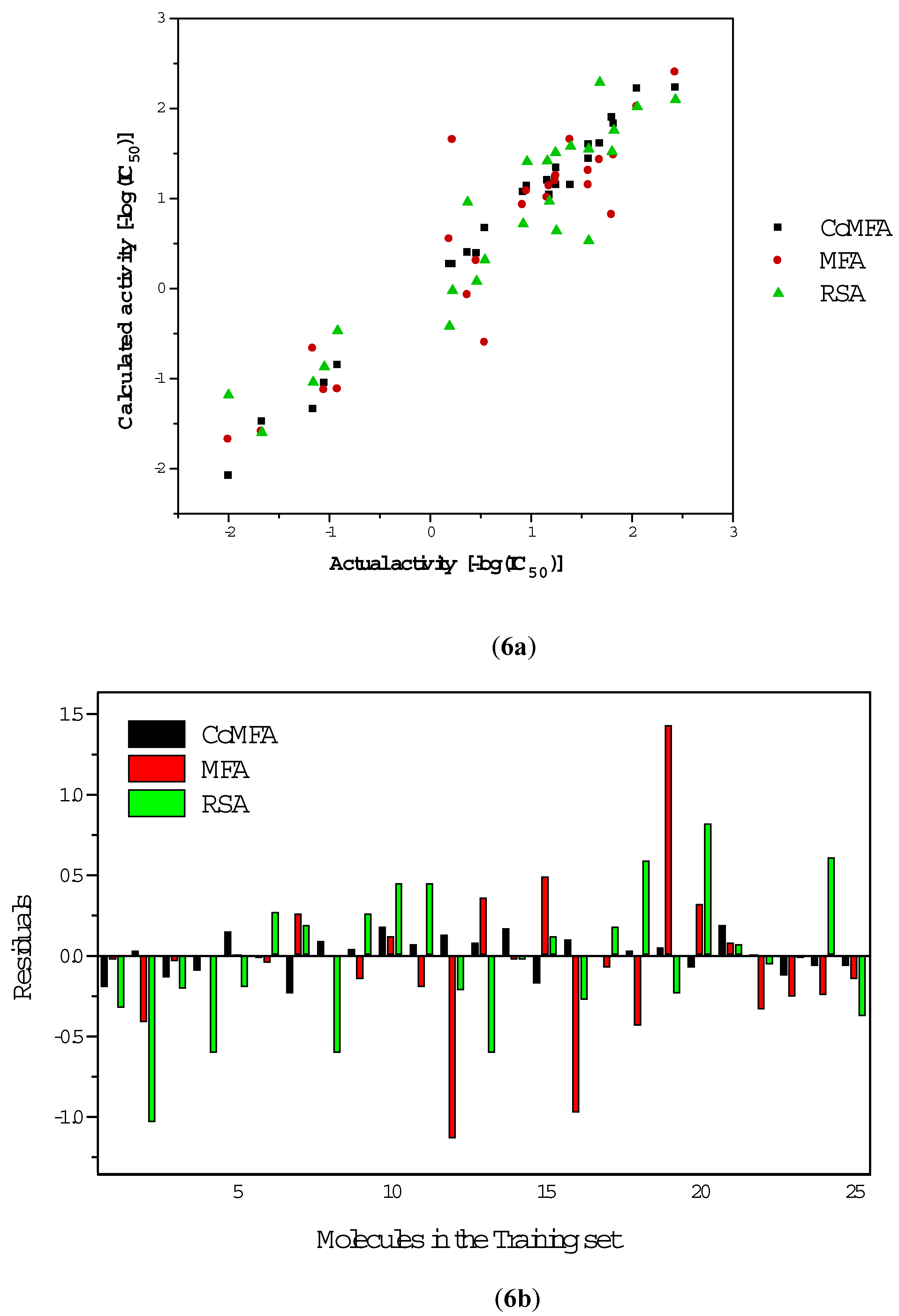
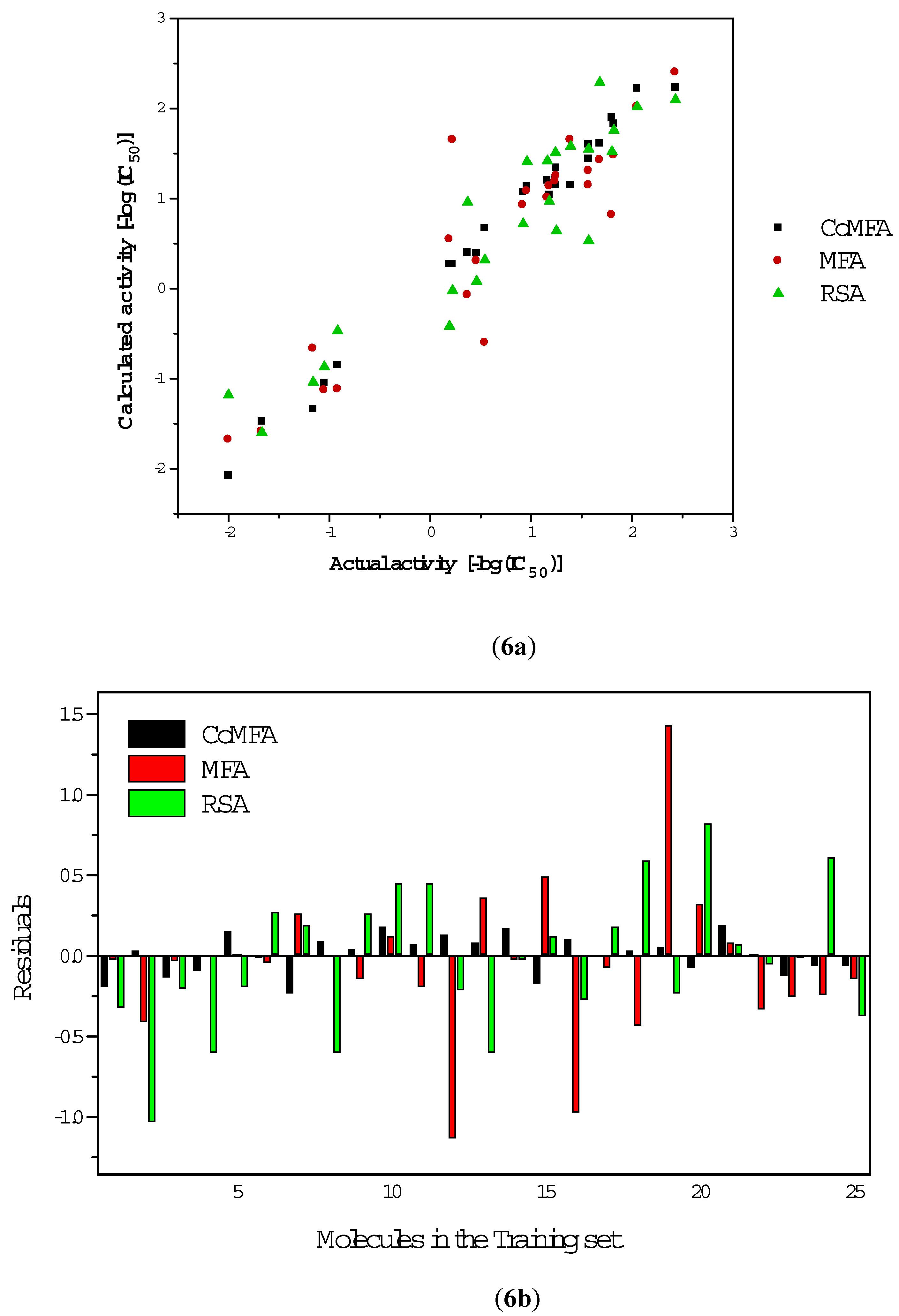
Results and Discussion
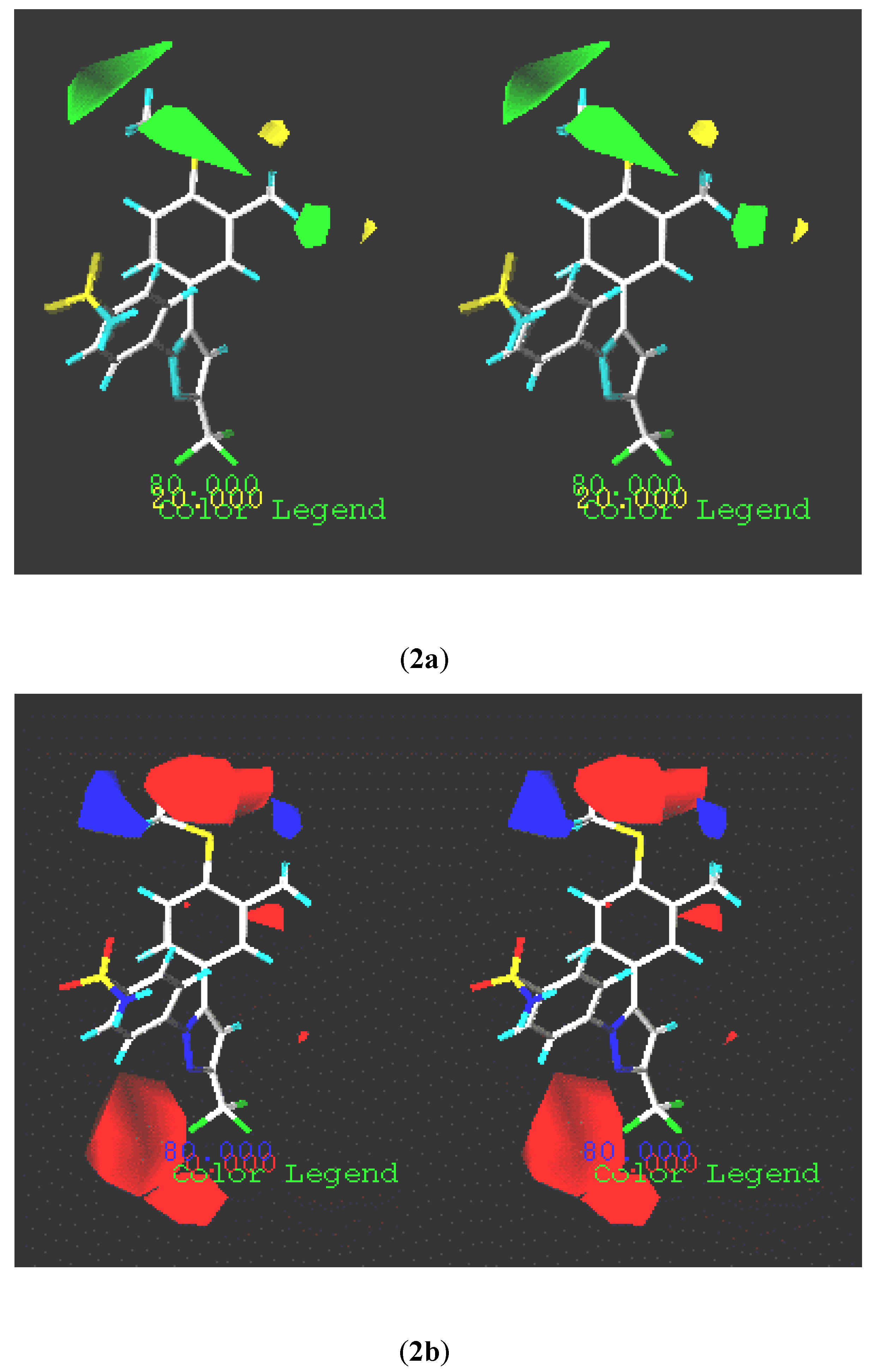
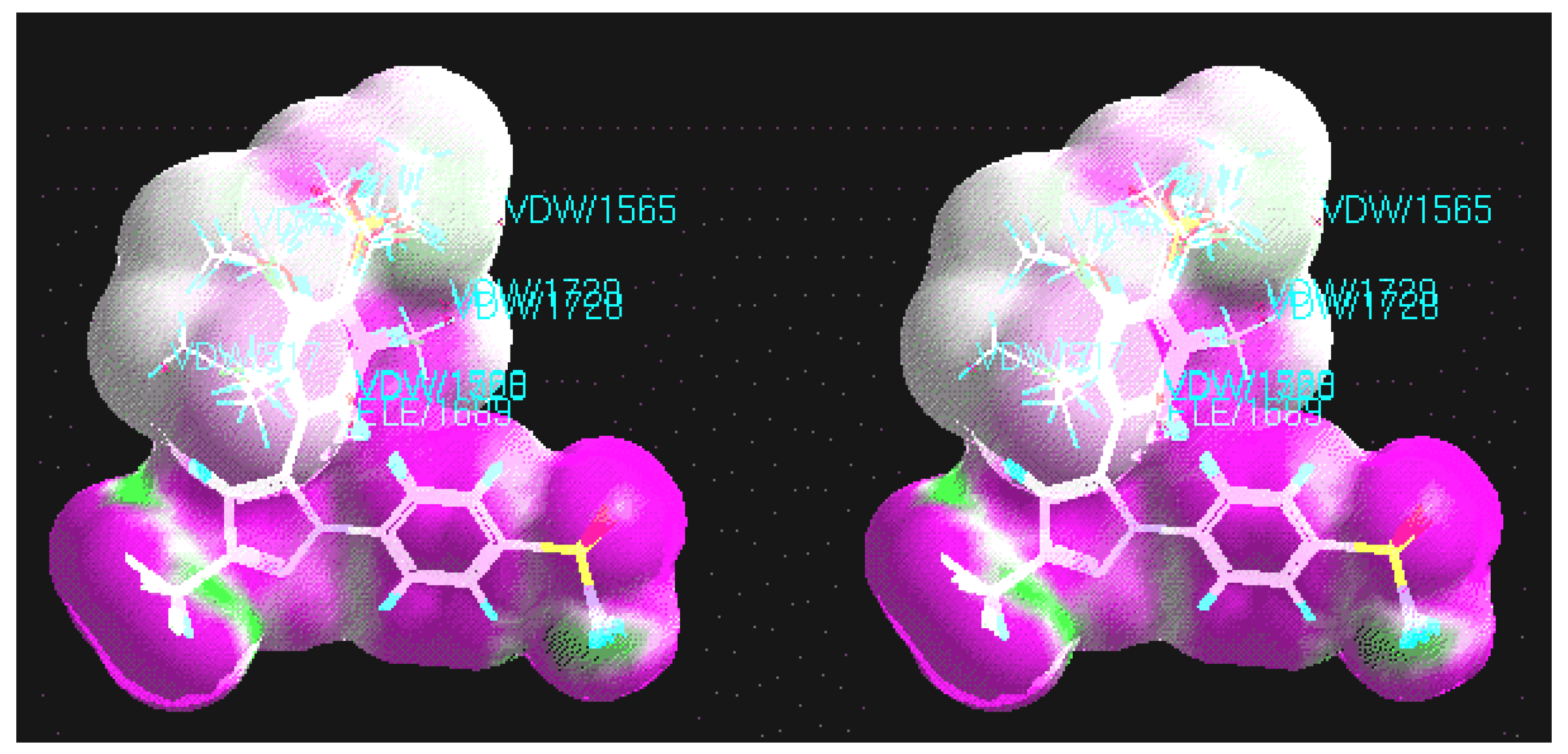
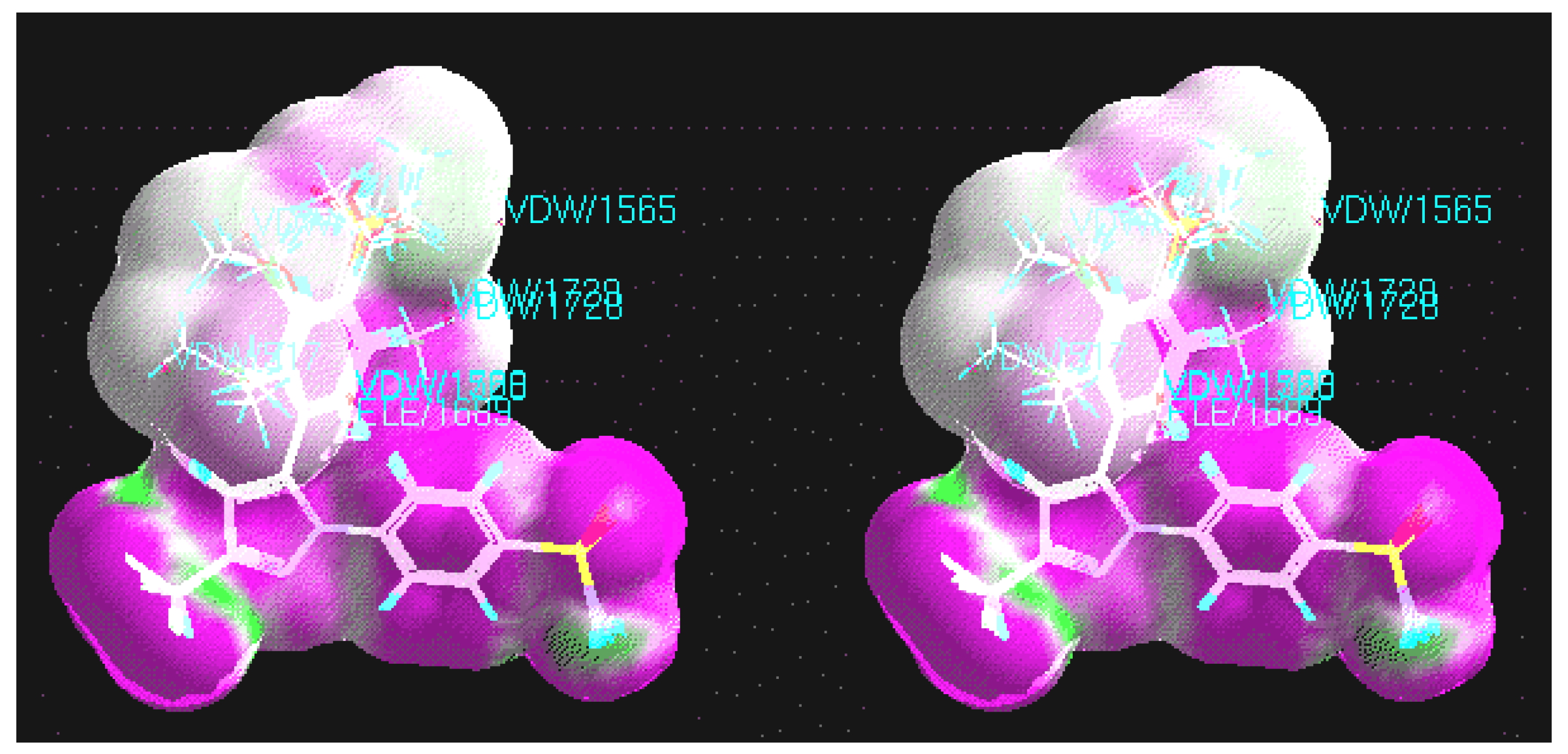
CoMFA
CoMFA coefficient contour maps
MFA
RSA
Conclusions
Acknowledgements:
References and Notes
- Picot, D.; Loll, P. J.; Garavito, R. M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 1994, 367, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Kurumbail, R. G.; Stevens, A. M.; Gierse, J. K.; McDonald, J. J.; Stegeman, R. A.; Pak; Gildehaus, D.; Miyashiro, J. M.; Penning, T. D.; Seibert, K.; Isakson, P. C.; Stallings, W. C. Structural basis for selective inhibition of cyclooxygenase -2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A. S.; Crews, B. C.; Rowlinson, S. W.; Garner, C.; Seibert, K.; Marnett, L. J. Asprin-like molecules that covalently inactivete cyclooxygenase-2. Science 1998, 280, 1268–1270. [Google Scholar] [CrossRef] [PubMed]
- Penning, T. D.; Talley, J. J.; Bertenshaw, S. R.; Carter, J. S.; Collins, P. W.; Docter, S.; Graneto, M. J.; Lee, L. F.; Malecha, J. W.; Miyashiro, J. M.; Rogers, R. S.; Rogier, D.; Yu, S. S.; Anderson, G. D.; Burton, E. G.; Cogburn, J. N.; Gregory, S. A.; Koboldt, C. M.; Perkins, W. E.; Seibert, K.; Veenhuizen, A. W.; Zhang, Y. Y.; Isakson, P. C. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluromethyl)-1H-pyrazol-1-yl]benzensulfonamide (SC-58635, Celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef] [PubMed]
- SYBYL 6.5 Molecular Modeling Software, Tripos Associates Inc.: S. Hanley Road. St. Louis, MO 63144-2913, 1699.
- Stewart, J. J. P. MOPAC; A semiemperical molecular orbital program. J. Comput.-Aided Mol. Des. 1990, 4, 1–105. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M. J. S.; Zoebish, E. G.; Healy, E. F.; Stewart, J. J. P. AM1 A New General Purpose Quantum Mechanical Molecular Model. J. Am. Chem. Soc. 1985, 107, 3902. [Google Scholar] [CrossRef]
- Reddy, G. O.; Dev, R. V.; Rekha, K. S.; Vyas, K.; Mohanti, S. B.; Kumar, P. R. Acta Crystallogr. 1999, C55, IUC9900161.
- Cramer, R. D., III; Patterson, D E.; Bunce, J. D. Comparative Molecular Field Analysis (CoMFA). 1 Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar]
- Cerius2 A program suite for molecular modelling activities, Molecular Simulations Inc.: Scranton Road, San Diego, CA 92121-3752, USA.
- Rohrbaugh, R. H.; Jurs, P. C. Descriptions of molecular shape applied in studies of structure/activity and structure/property relationships. Analytica Chimica Acta 1987, 199, 99–109. [Google Scholar] [CrossRef]
- Rogers, D.; Hopfinger, A. J. Application of genetic function approximation to quantitative structure-activity relationships and quantitative structure property relationships. J. Chem. Inf. Comput. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Stanton, D.T.; Jurs, P.C. Development and use of charge partial surface area structural descriptors in computer-assisted quantitative structure-property relationship studies. Anal. Chem. 1990, 62, 2323–29. [Google Scholar] [CrossRef]
- Sample Availability: Not available.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. No. | R1, R2 | R3 | Activity-(logIC50) | Calculated activity | ||
| CoMFA | MFA | RSA | ||||
| 1 | 3-CH3-4-SCH3 | CF3 | 2.43 | 2.23 | 2.40 | 2.10 |
| 2 | 3-Cl-4-NHCH3 | CF3 | 1.57 | 1.60 | 1.15 | 0.53 |
| 3 | 3-Cl-4-OCH3-5-CH3 | CF3 | 1.18 | 1.04 | 1.14 | 0.97 |
| 4 | 2,4-di-Cl | CF3 | 1.25 | 1.15 | 1.25 | 0.64 |
| 5 | 2,4-di-CH3 | CF3 | 0.92 | 1.07 | 0.93 | 0.72 |
| 6 | 2-F | CF3 | 1.24 | 1.22 | 1.19 | 1.51 |
| 7 | 4-F | CF3 | 1.39 | 1.15 | 1.65 | 1.58 |
| 8 | 2-Cl | CF3 | 1.25 | 1.34 | 1.25 | 0.64 |
| 9 | 2-Me | CF3 | 1.16 | 1.20 | 1.01 | 1.42 |
| 10 | 3-Me | CF3 | 0.96 | 1.14 | 1.08 | 1.41 |
| 11 | 4-CF3 | CF3 | -0.92 | -0.85 | -1.12 | -0.47 |
| 12 | 2-OMe | CF3 | 0.54 | 0.67 | -0.60 | 0.32 |
| 13 | 4-OEt | CF3 | 0.19 | 0.27 | 0.55 | -0.42 |
| 14 | 4-SMe | CF3 | 2.05 | 2.22 | 2.02 | 2.02 |
| 15 | 2-NMe2 | CF3 | -1.16 | -1.34 | -0.67 | -1.04 |
| 16 | 4-NHMe | CF3 | 1.80 | 1.90 | 0.82 | 1.52 |
| 17 | 4-CO2H | CF3 | -1.05 | -1.05 | -1.13 | -0.87 |
| 18 | 3-C2H5-4-OCH3 | CF3 | 0.37 | 0.40 | -0.07 | 0.96 |
| 19 | 3,4-di-OCH3 | CF3 | 0.22 | 0.27 | 1.65 | -0.02 |
| 20 | 4-SO2Me | CF3 | -2.00 | -2.08 | -1.68 | -1.18 |
| 21 | 4-CO2H | CHF2 | -1.67 | -1.48 | -1.59 | -1.60 |
| 22 | 4-OMe | CHF2 | 1.82 | 1.83 | 1.48 | 1.76 |
| 23 | 3-Cl-4-OCH3 | CHF2 | 1.57 | 1.44 | 1.31 | 1.55 |
| 24 | 3,5-di-Cl-4-OCH3 | CHF2 | 1.68 | 1.61 | 1.43 | 2.29 |
| 25 | 3,5-di-F-4-OCH3 | CHF2 | 0.46 | 0.39 | 0.31 | 0.08 |
| Comp. No. | R1, R2 | R3 | Activity-(logIC50) | Predicted Activitya | ||
| CoMFA | MFA | RSA | ||||
| 26 | 4-Cl | CF3 | 2.00 | 1.04 | 1.84 | 1.25 |
| 27 | 4-Me | CF3 | 1.40 | 0.79 | 1.11 | 1.24 |
| 28 | 4-NO2 | CF3 | -0.42 | -0.77 | -1.72 | 0.33 |
| 29 | 4-NMe2 | CF3 | 2.33 | 2.32 | -0.70 | 0.89 |
| 30 | 3-F-4-OCH3 | CHF2 | 1.30 | 1.40 | 1.54 | 1.32 |
| CoMFA | MFAc | RSAd | |
| r 2 cv | 0.66 | 0.73 | 0.77 |
| ar 2 | 0.99 (0.62 and 0.37) | 0.86 | 0.90 |
| No. of components | 6 | 4 | 4 |
| PRESSb | 0.4 | 9.7 | 7.756 |
| Standard deviation | 0.13 | 0.43 | 0.4 |
© 2000 by MDPI (http://www.mdpi.org).
Share and Cite
Desiraju, G.R.; Gopalakrishnan, B.; Jetti, R.K.R.; Raveendra, D.; Sarma, J.A.R.P.; Subramanya, H.S. Three-Dimensional Quantitative Structural Activity Relationship (3D-QSAR) Studies of Some 1,5-Diarylpyrazoles: Analogue Based Design of Selective Cyclooxygenase-2 Inhibitors. Molecules 2000, 5, 945-955. https://doi.org/10.3390/50700945
Desiraju GR, Gopalakrishnan B, Jetti RKR, Raveendra D, Sarma JARP, Subramanya HS. Three-Dimensional Quantitative Structural Activity Relationship (3D-QSAR) Studies of Some 1,5-Diarylpyrazoles: Analogue Based Design of Selective Cyclooxygenase-2 Inhibitors. Molecules. 2000; 5(7):945-955. https://doi.org/10.3390/50700945
Chicago/Turabian StyleDesiraju, Gautam R., Bulusu Gopalakrishnan, Ram K. R. Jetti, Dayam Raveendra, Jagarlapudi A. R. P. Sarma, and Hosahalli S. Subramanya. 2000. "Three-Dimensional Quantitative Structural Activity Relationship (3D-QSAR) Studies of Some 1,5-Diarylpyrazoles: Analogue Based Design of Selective Cyclooxygenase-2 Inhibitors" Molecules 5, no. 7: 945-955. https://doi.org/10.3390/50700945