Synthesis of Polycyclic Ring Systems Using Transition Metal Catalyzed Cyclizations of Diazo Alkynyl Ketones

Department of Chemistry, Emory University, Atlanta, Georgia 30322, USA
Molecules 2001, 6(1), 1-12; https://doi.org/10.3390/60100001
Submission received: 3 November 2000
/
Accepted: 6 November 2000
/
Published: 22 December 2000
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
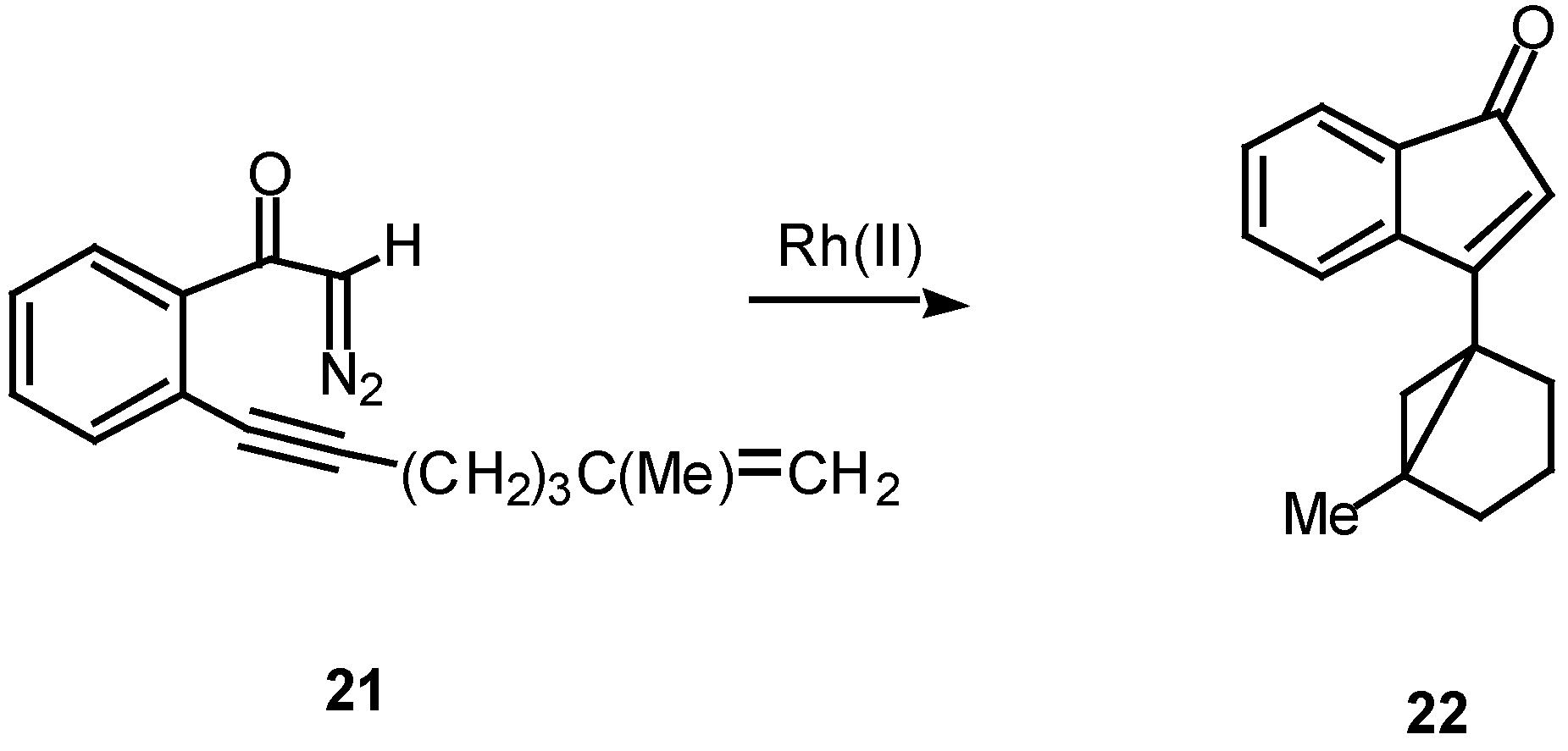{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The rhodium(II)-catalyzed reaction of α-diazo ketones bearing tethered alkyne units represents a new and useful method for the construction of a variety of substituted cyclopentenones. The process proceeds by addition of the rhodium-stabilized carbenoid onto the acetylenic π-bond to give a vinyl carbenoid intermediate. The resulting rhodium complex undergoes a wide assortment of reactions including cyclopropanation, 1,2-hydrogen migration, CH-insertion, addition to tethered alkynes and ylide formation. When 2-alkynyl-2-diazo-3-oxobutanoates were treated with a Rh(II)-catalyst, furo[3,4-c]furans were formed in excellent yield.
Keywords:
rhodium; catalyst; diazo; ketone; alkyne; cyclization; CH-insertion; ylide; furo[3,4-c]furansIntroduction
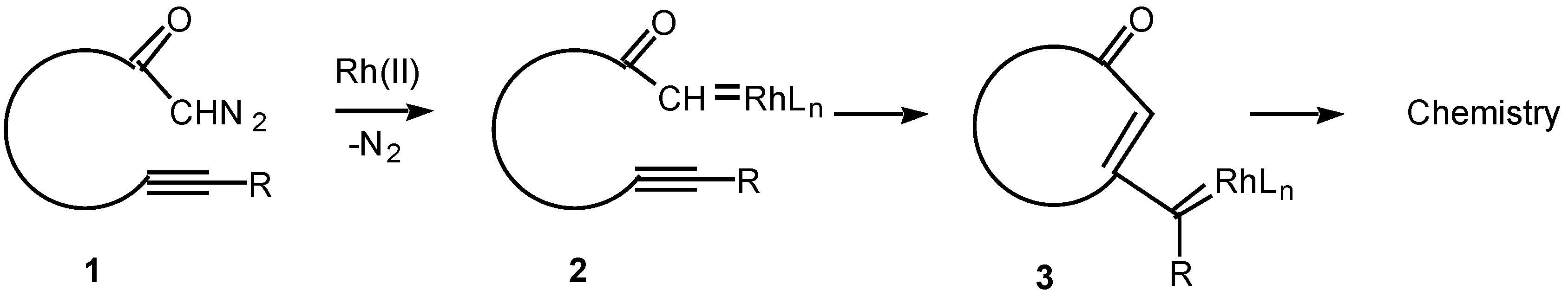
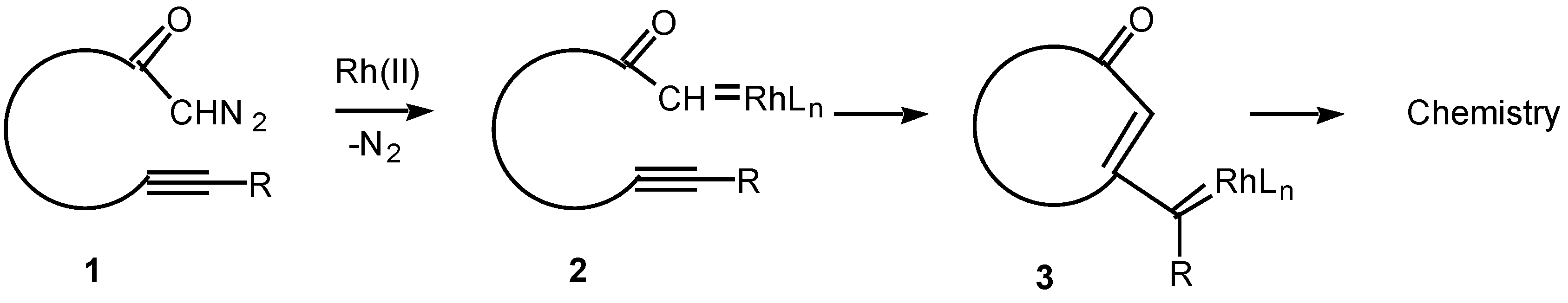
The chemistry of transition metal carbene complexes has been a subject of intense activity over the past two decades [1]. Current interest in this field stems from the role of metal carbenes in alkene metathesis [2], in alkene and alkyne polymerization [3], in cyclopropanation chemistry [4], and as intermediates in an impressive array of synthetic methodology [5,6]. The intramolecular reactions of metal carbene complexes derived from α-diazo carbonyl compounds have been extensively studied from both a mechanistic and synthetic viewpoint [7]. Rhodium(II) carboxylates are particularly effective catalysts for the decomposition of diazo compounds and many chemical syntheses are based on this methodology [8]. Among the more synthetically useful processes of the resulting carbenoid intermediates are intramolecular C-H insertion [9], cyclopropanation [10], and ylide generation [11]. In contrast to these processes, the corresponding reaction of alkynes with metal carbenes has been far less studied. Only in recent years has some attention been focused on the intramolecular cyclization reactions of α-diazo ketones containing tethered alkynes (i.e., 1) in the presence of transition metal catalysts. The overall reaction observed is believed to proceed via an initial decomposition of the α-diazo ketone to generate a rhodium carbenoid intermediate 2. Attack on the carbenoid carbon by the tethered alkyne generates a new intermediate (3) in which carbene-like character has been transferred to the beta-carbon of the alkyne. The intermediate vinyl carbenoid may then react further in either an intramolecular or intermolecular fashion to give novel products. This article describes some of our work in this area.
Scheme 1.
General Mechanistic Considerations
Scheme 2.

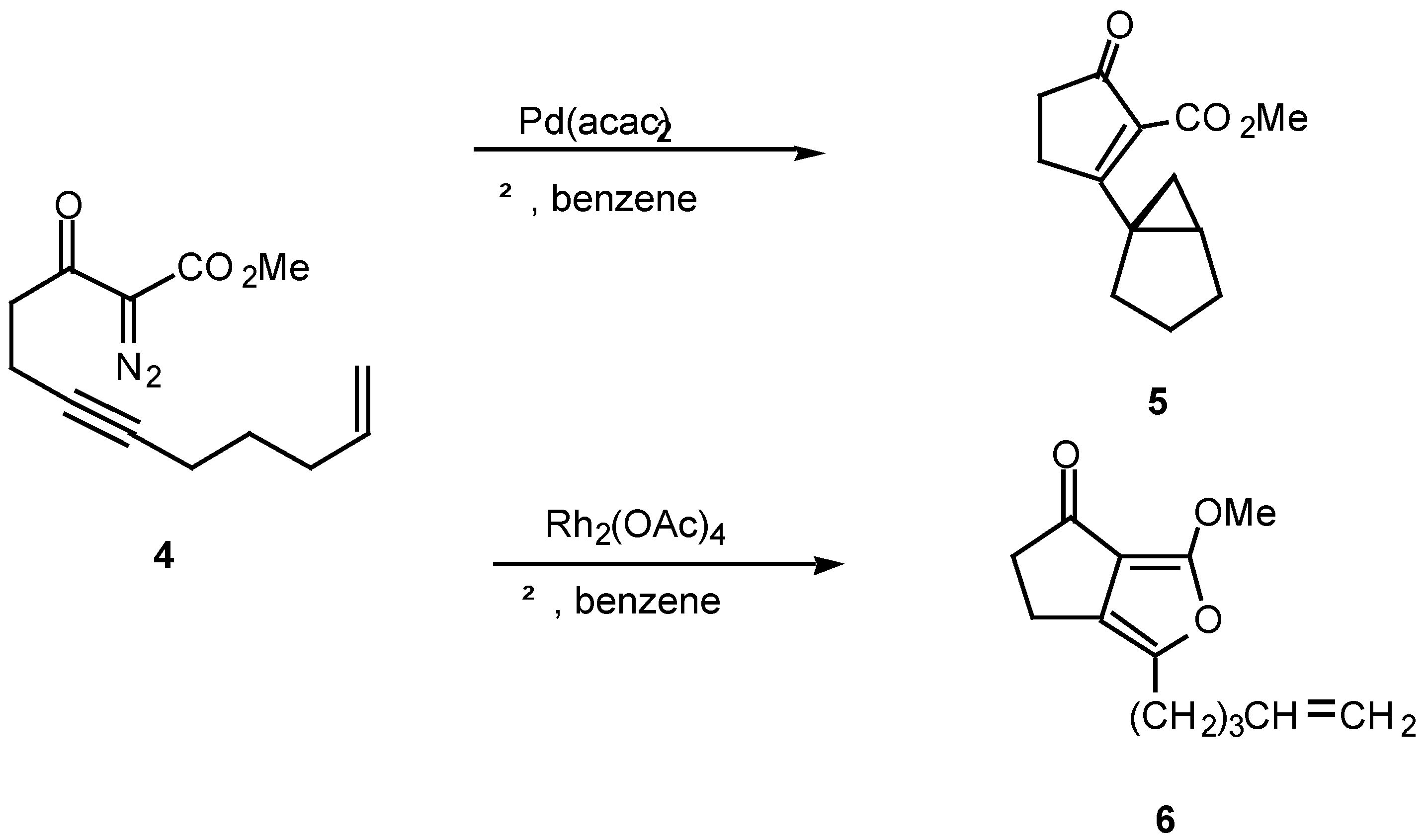
The mechanism of the diazo ketone-alkyne cyclization reaction has been the subject of some study over the past several years [12,13,14]. For example, treatment of ketone, α-diazo ketoester 4 with catalytic palladium(II) acetoacetonate produced cyclopropane 5 in 78% yield, while the reaction with rhodium(II) acetate provided furan 6 in 56% yield. Furan 6 arises from a 1,5-electrocyclization of the initially produced vinyl carbenoid intermediate onto the adjacent carbonyl group (vide infra).
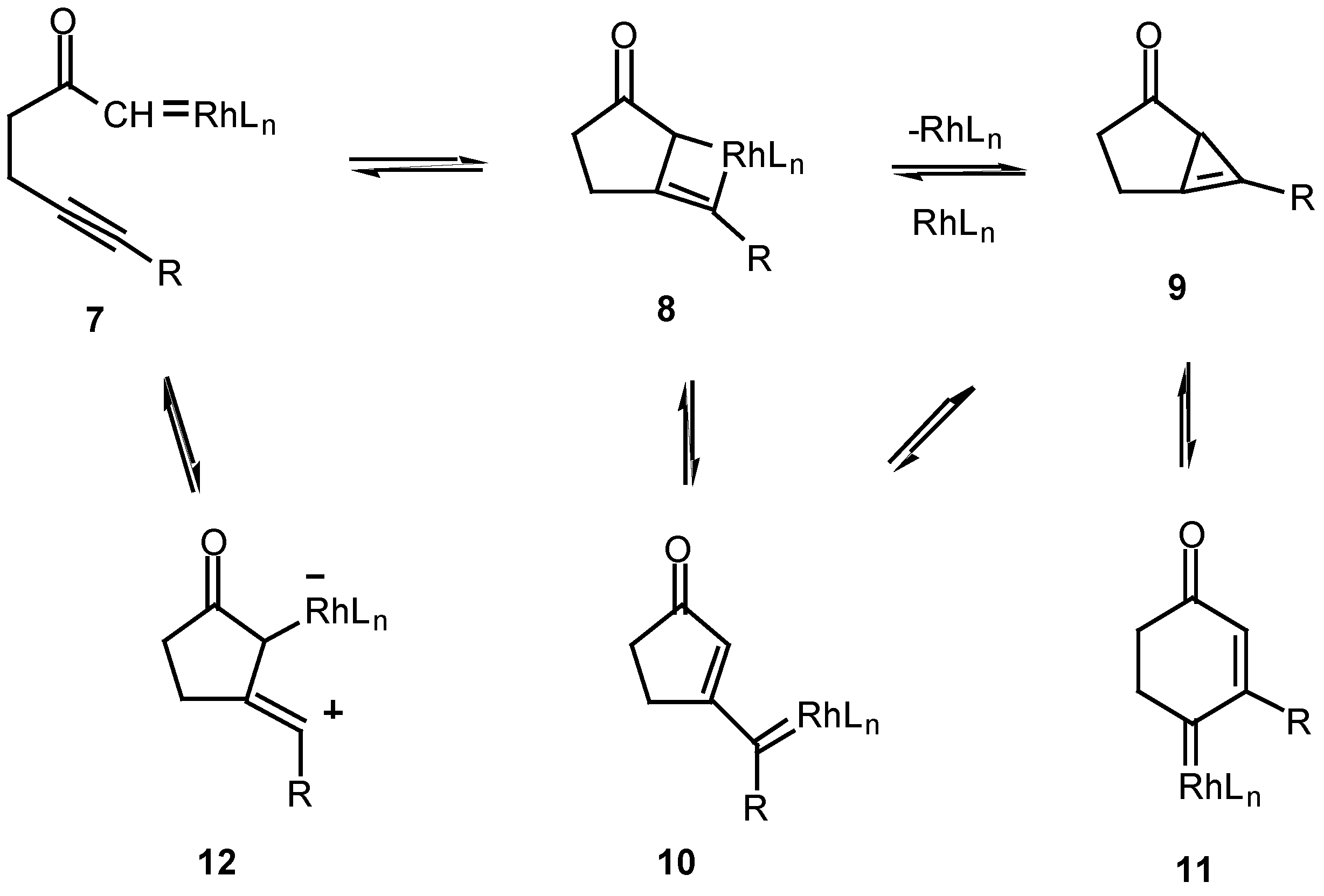
The fact that the chemistry of 4 is catalyst dependent suggests that a metalated species is involved in the product-determining step [12]. One possible mechanism to explain the products involves the initial decomposition of the α-diazo moiety to give the metal carbenoid 7. In the next step, the rhodium metal migrates from the original diazo carbon to the alkynyl carbon via a metathesis reaction and ultimately produces metallocyclobutene 8. This intermediate could then ring open to furnish the vinyl carbenoid 10 which goes on to afford the observed products. Another possible variation would be formation of the highly strained cyclopropene 9. This intermediate could then be rapidly converted into the 5-exo vinyl carbenoid 10 or the 6-endo carbenoid 11, both of which can undergo further chemistry. This pathway has precedent from the known metal catalyzed ring opening of cyclopropenes to vinyl carbenes [14].
Scheme 3.
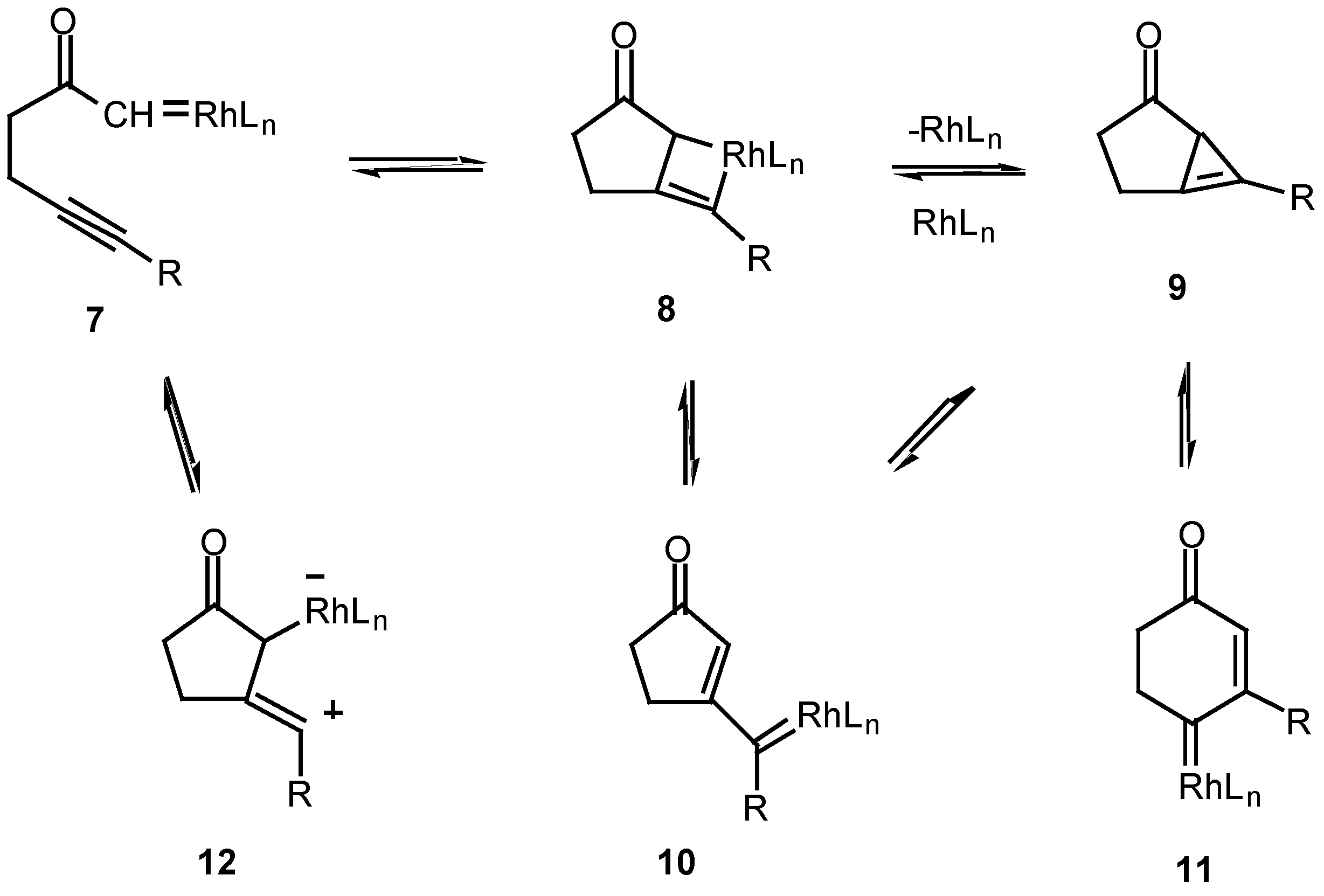
Scheme 4.
Scheme 5.
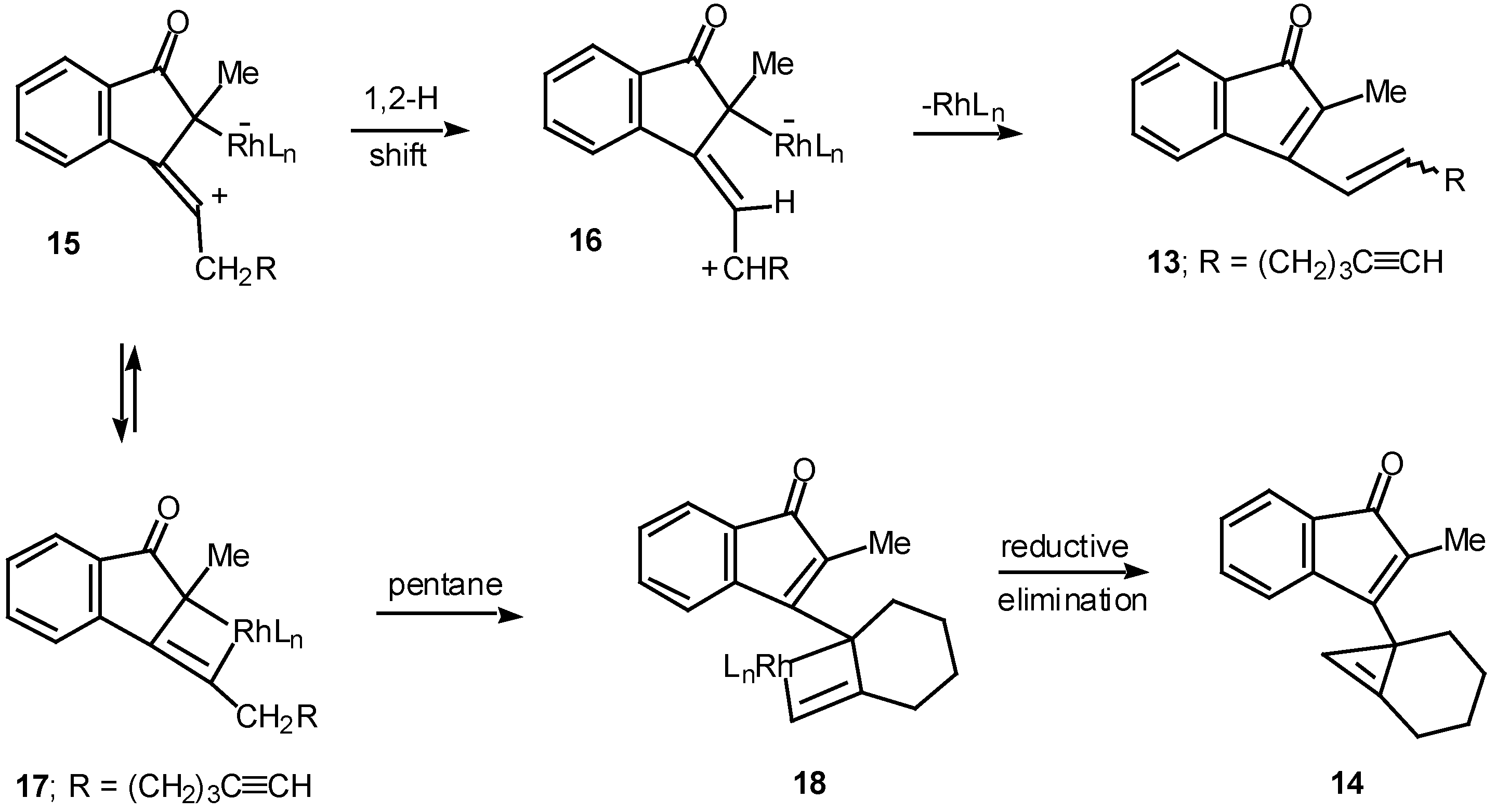
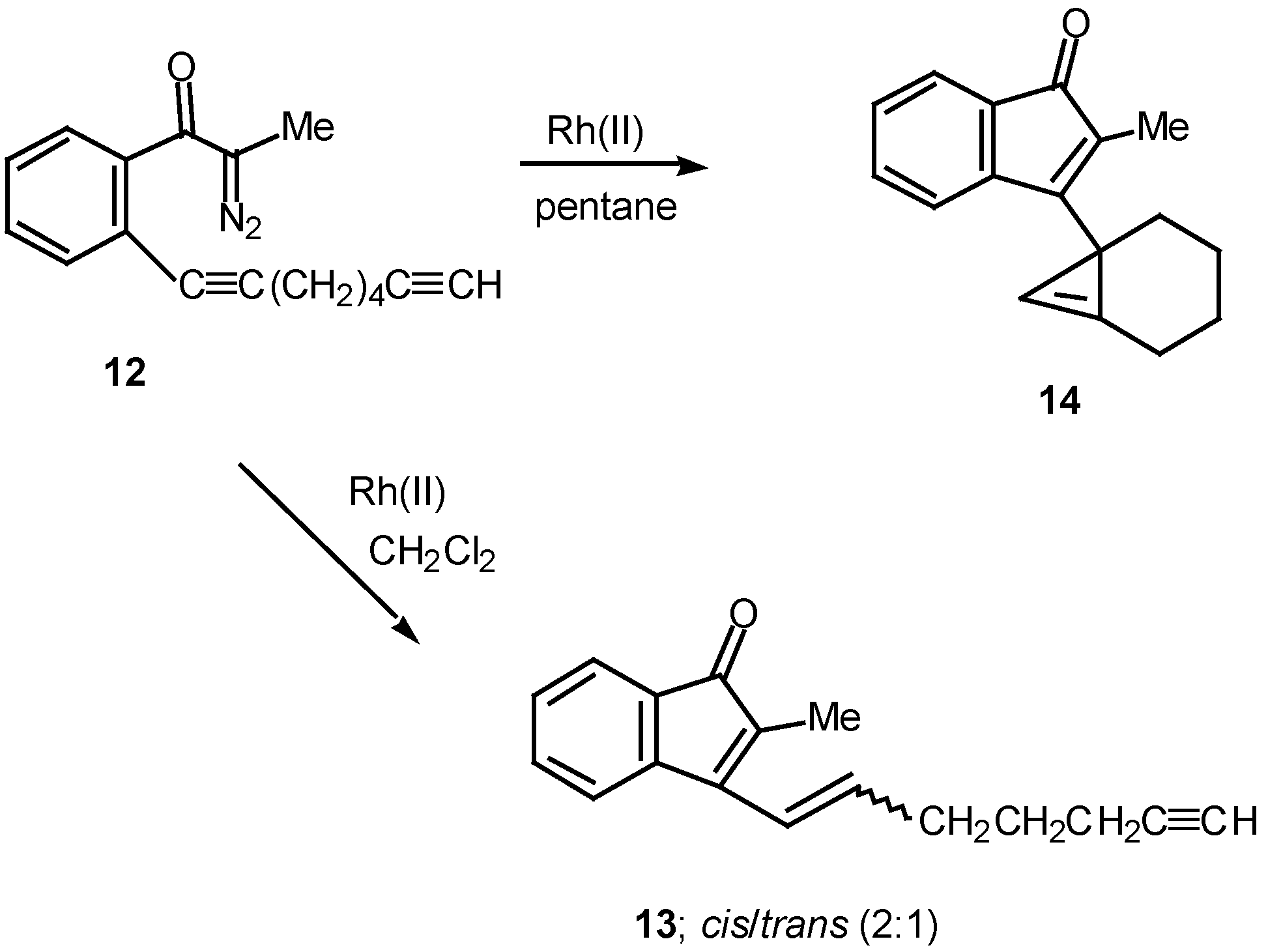
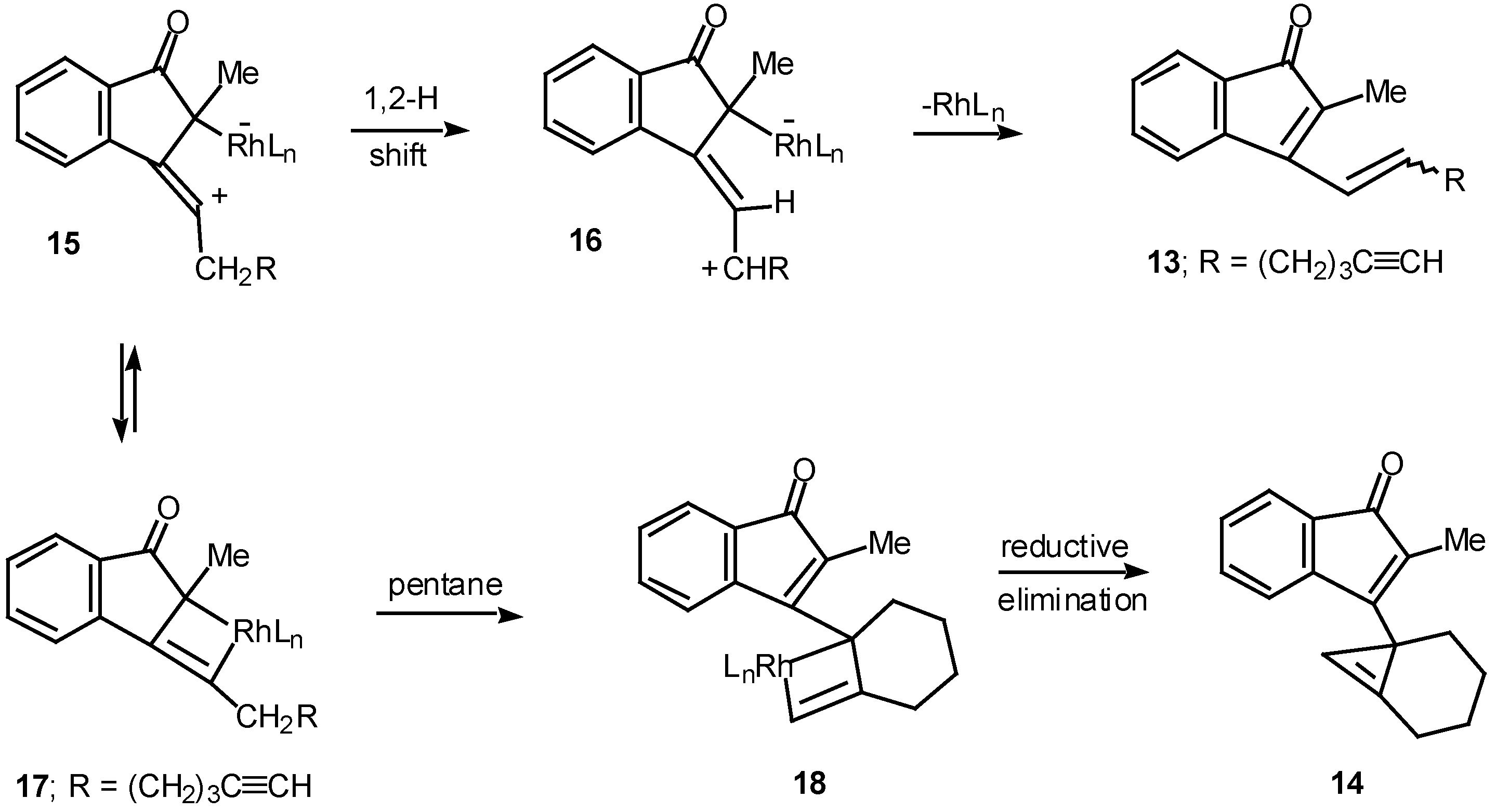
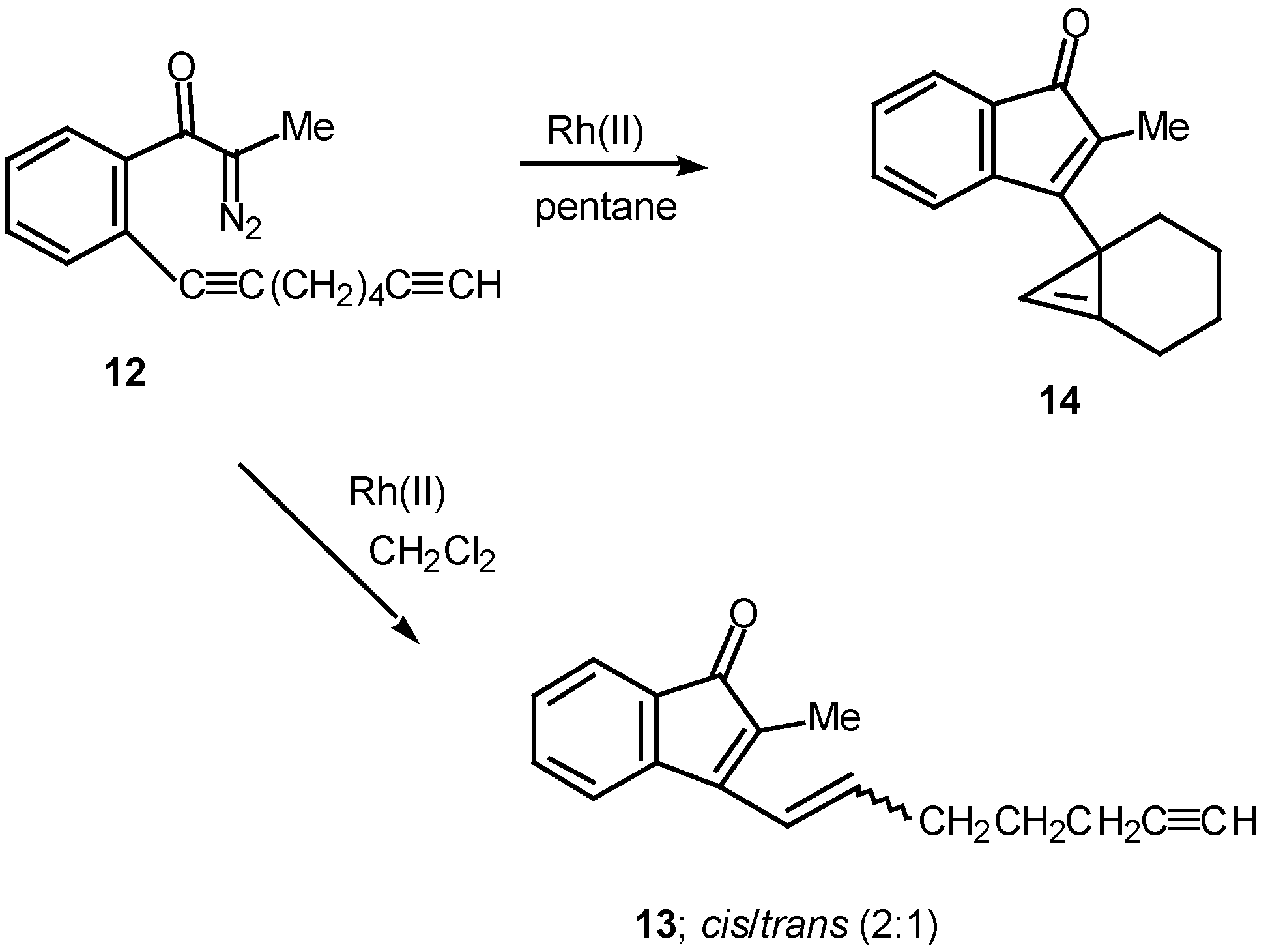
Results in our laboratory showed that the reaction mechanism is markedly dependent on the solvent employed in these Rh(II)-catalyzed insertion processes. Thus, treatment of 12 with a catalytic amount of rhodium(II) acetate resulted in a 2:1-mixture of the cis and trans-alkenyl substituted indenones 13 (85% combined yield). No signs of cyclopropene 14 (<2%) could be detected in the crude reaction mixture by NMR spectroscopy. Interestingly, when pentane was used as the solvent, cyclopropene 14 (80%) was the exclusive product. The degree of chemoselectivity that was achieved in this reaction by simply changing the solvent from dichloromethane to pentane is most remarkable [17]. A reasonable explanation that nicely accounts for the formation of indenone 13 involves stepwise cyclization of the initially formed keto carbenoid to give 15 (Scheme 5). A 1,2-hydrogen shift results in the formation of allylic cation 16 and this is followed by collapse to 13 and regeneration of the rhodium catalyst. The intermediates involved in the formation of 13 are dipolar, which would explain why the formation of 13 is strongly inhibited in nonpolar solvents. When pentane is used as the solvent, metal migration occurs via the metallocyclobutene intermediates 17 and 18 so as to avoid charge buildup [17]. Thus, it would appear as though the reaction mechanism of these alkyne cyclizations is markedly dependent on the nature of the solvent used.
Cyclopropenation
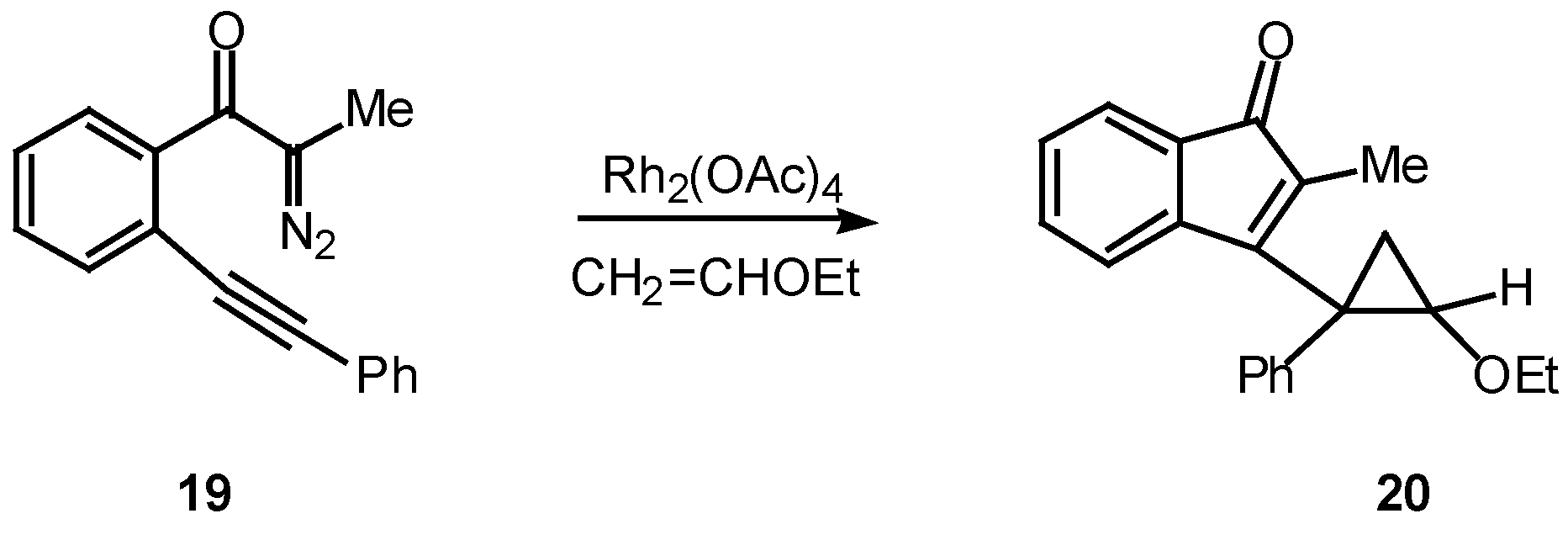
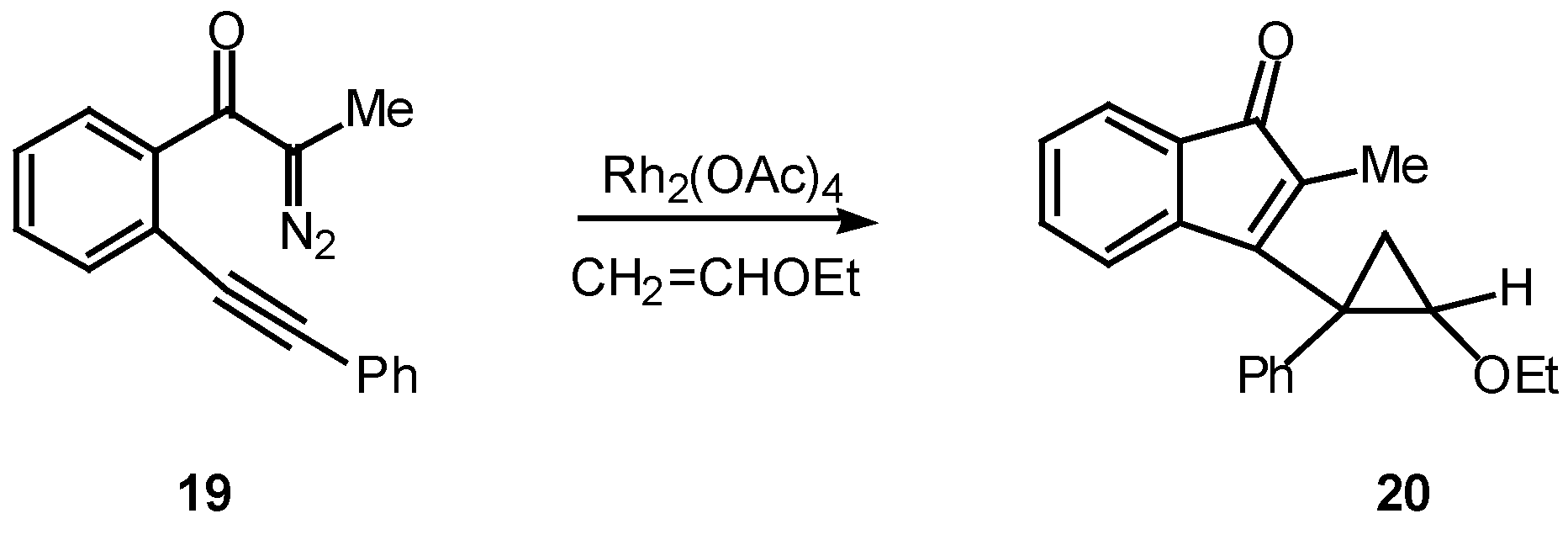
Many of the earlier systems studied involved trapping the vinyl carbenoid intermediate as a cyclopropane via reaction with external or tethered alkenes [12,18,19]. What is most interesting about this reaction is the formation of three new rings in one step from an acyclic precursor. A typical example is outlined in Scheme 6. Treatment of 19 with a catalytic quantity of rhodium(II) acetate in the presence of two equiv of vinyl ether afforded cyclopropane 20 in 91% yield [18].
Scheme 6.
In the case of intramolecular trapping with tethered alkenes, two basic structural variations are possible. These will depend on the point of attachment of the alkenyl group and each variation will lead to very different cyclization products. In type I systems, the alkenyl group is tethered onto the alkynyl carbon atom and this is illustrated with α-diazo ketone 21. Treatment of 21 with a catalytic quantity of rhodium(II) acetate gave indenone 22 in 60% yield.16
Scheme 7.
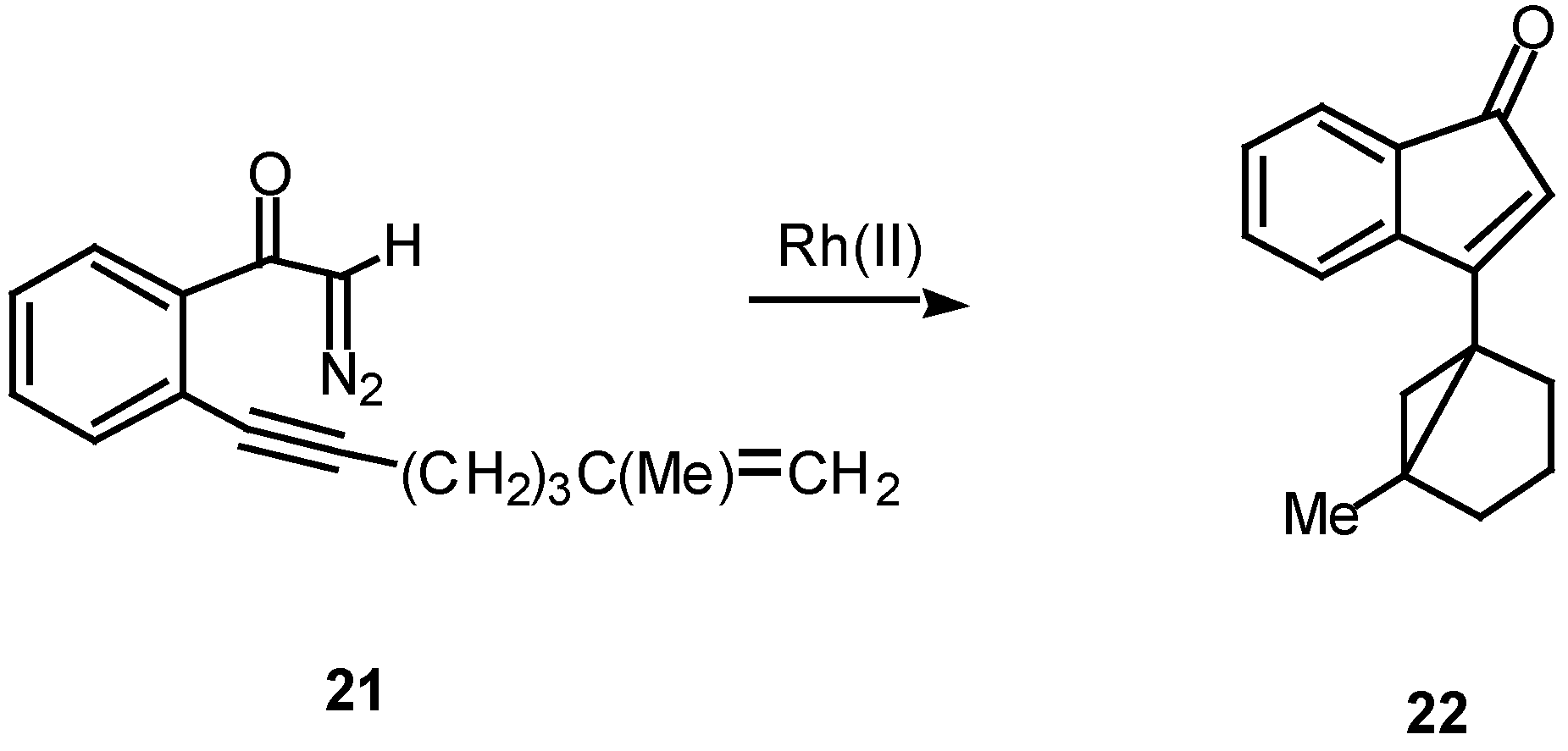
Scheme 8.
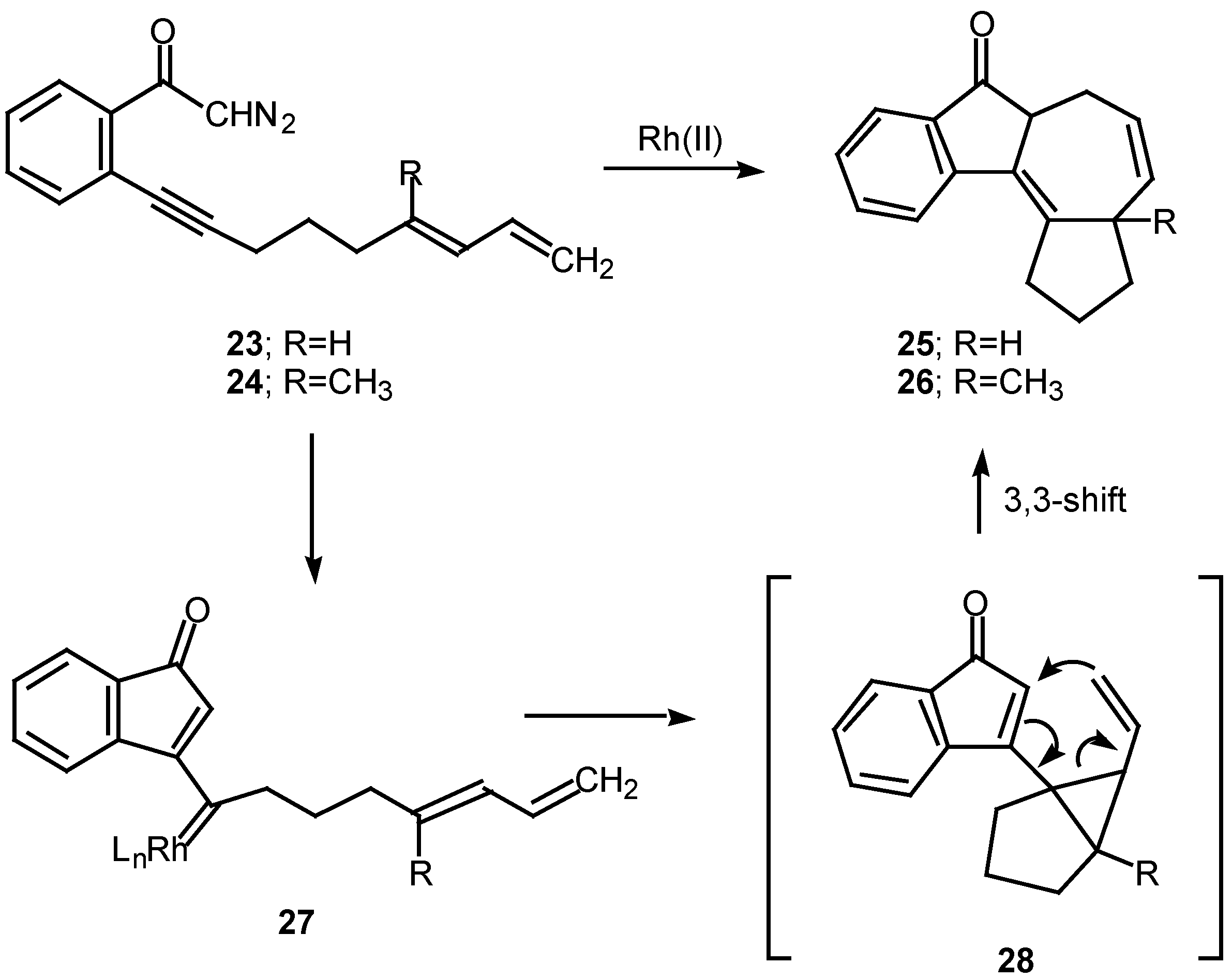
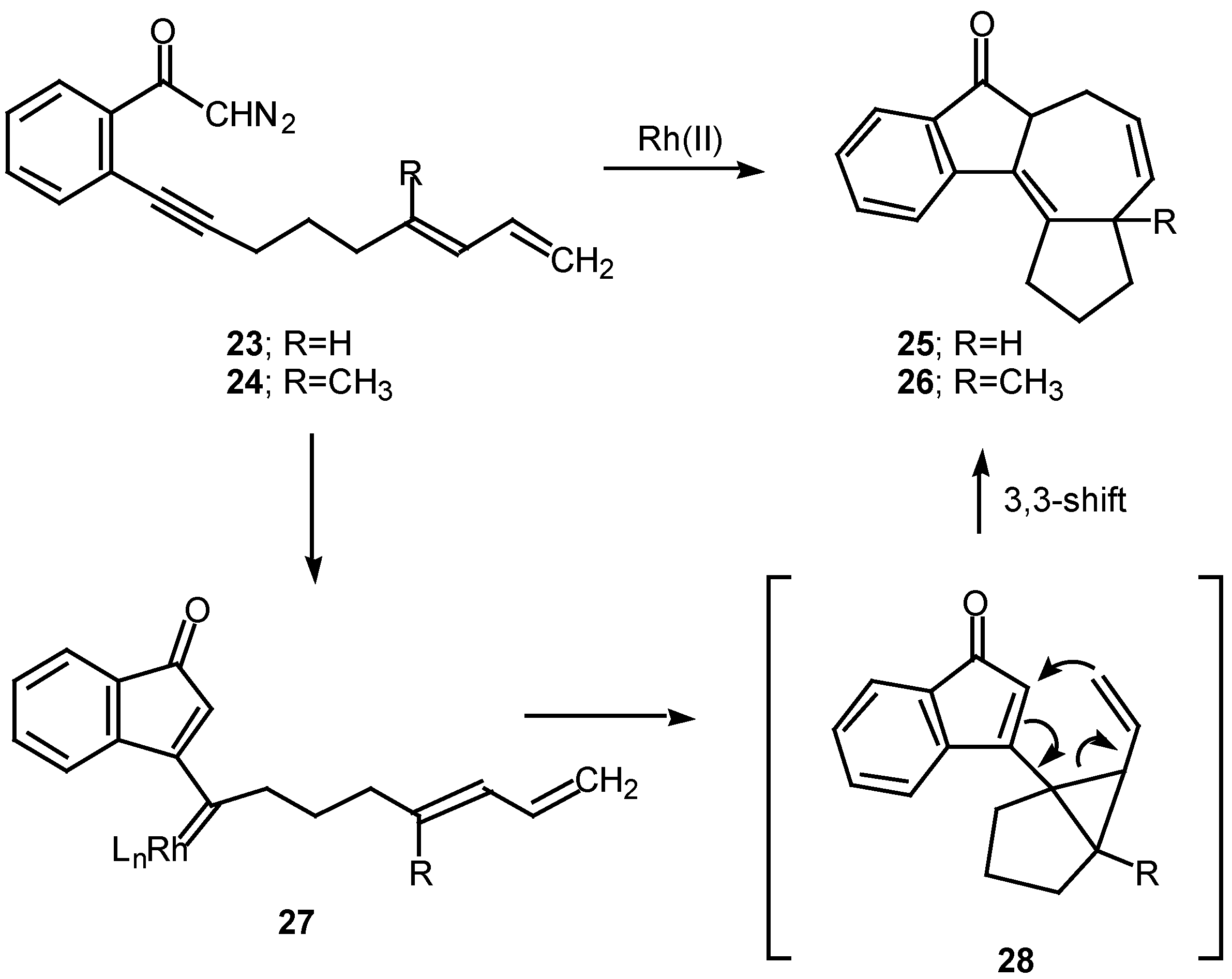
A number of experiments designed to probe the scope and generality of the intramolecular alkyne cyclopropanation reaction were carried out in an effort to exploit this tandem sequence as a synthetic method. Initial efforts focused on the rhodium(II) catalyzed reaction of o-(6,8-nonadien-1-ynyl)-α-diazoacetophenone 23. Treatment of 23 with a catalytic quantity of rhodium(II) mandelate gave cycloheptadiene 25 in 58% yield. In a similar manner, treating the closely related diazo ketone 24 (R=CH3) with rhodium(II) mandelate also gave cyclopent[g]azulenone 26 [13]. The formation of the fused cycloheptadienes 25 and 26 can be rationalized by assuming that the reaction proceeds through the divinylcyclopropane intermediate 28. When the internal double bond of the diene possesses the E-geometry, intramolecular cyclopropanation gives rise to a cis-divinyl cyclopropane, which rapidly undergoes a Cope rearrangement under the conditions used [13]. It should be noted that intramolecular cyclopropanation of dienes by simple carbenoids followed by rearrangement of the resulting vinylcyclopropane has been effectively in several elegant syntheses. The overall process is also closely related to work by Davies who developed a synthesis of fused seven membered carbocycles based on a formal intramolecular [3+4]-cycloaddition of vinyl carbenoids with dienes [20].
Cyclization of vinyl carbenoids to produce cyclopropenes is another common reaction that is often encountered with these systems [21]. For example, treatment of α-diazo ketone 29 with a catalytic quantity of rhodium(II) acetate afforded cyclopropene 30 in 95% yield.
Scheme 9.

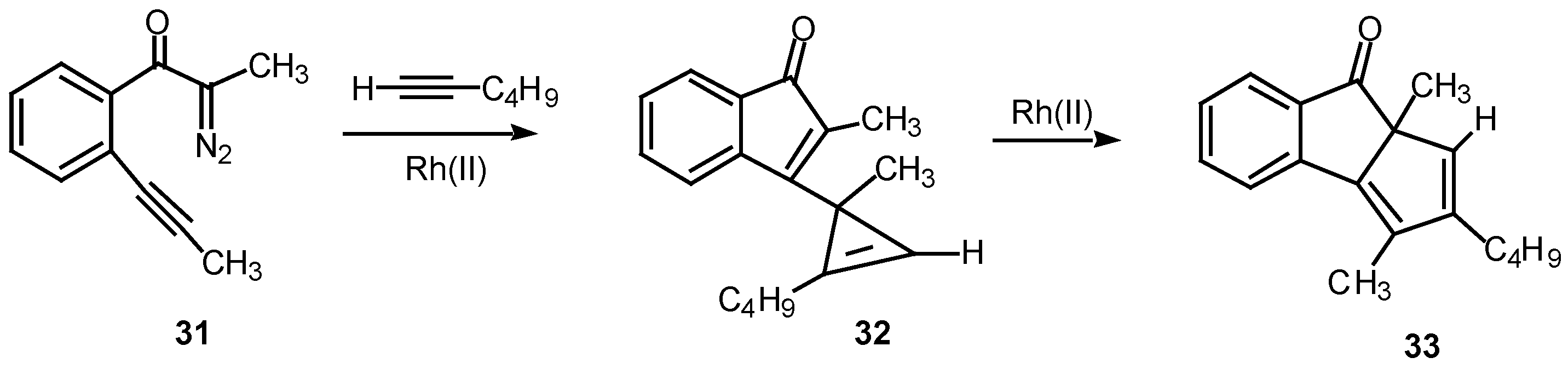
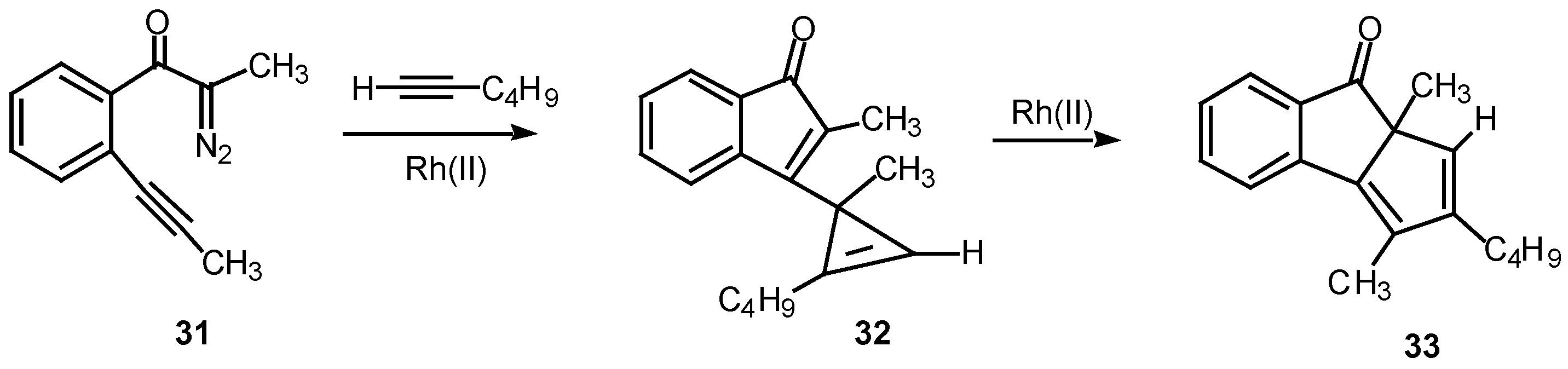
A double internal/external alkyne cyclization of acetylenic α-diazo ketone 31 with 1-hexyne was also studied in our laboratory. Stirring this mixture in the presence of rhodium(II) acetate at 25°C for 1 h afforded the novel cyclopentadiene derivative 33 in 81% yield. Control experiments established that the initial product that was first formed was indenone 32. This product is the result of the vinyl carbenoid adding across the acetylenic π-bond of 1-hexyne. When the reaction was carried out for only 10 min at 25°C, indenone 32 could be isolated in 85% yield. Further stirring of 32 with rhodium(II) acetate induced a subsequent rearrangement and ultimately produced 33 in 92% yield [22].
Scheme 10.
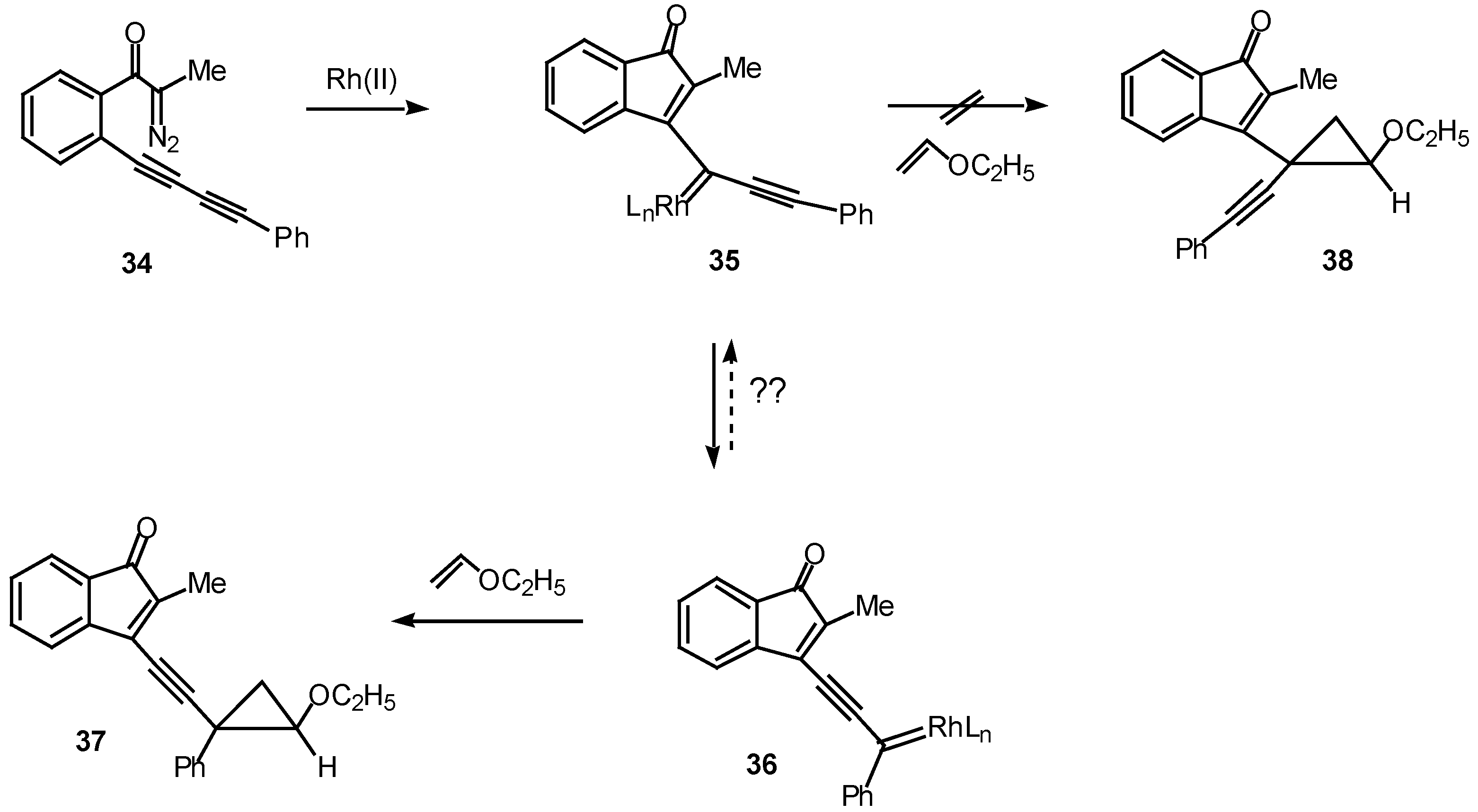
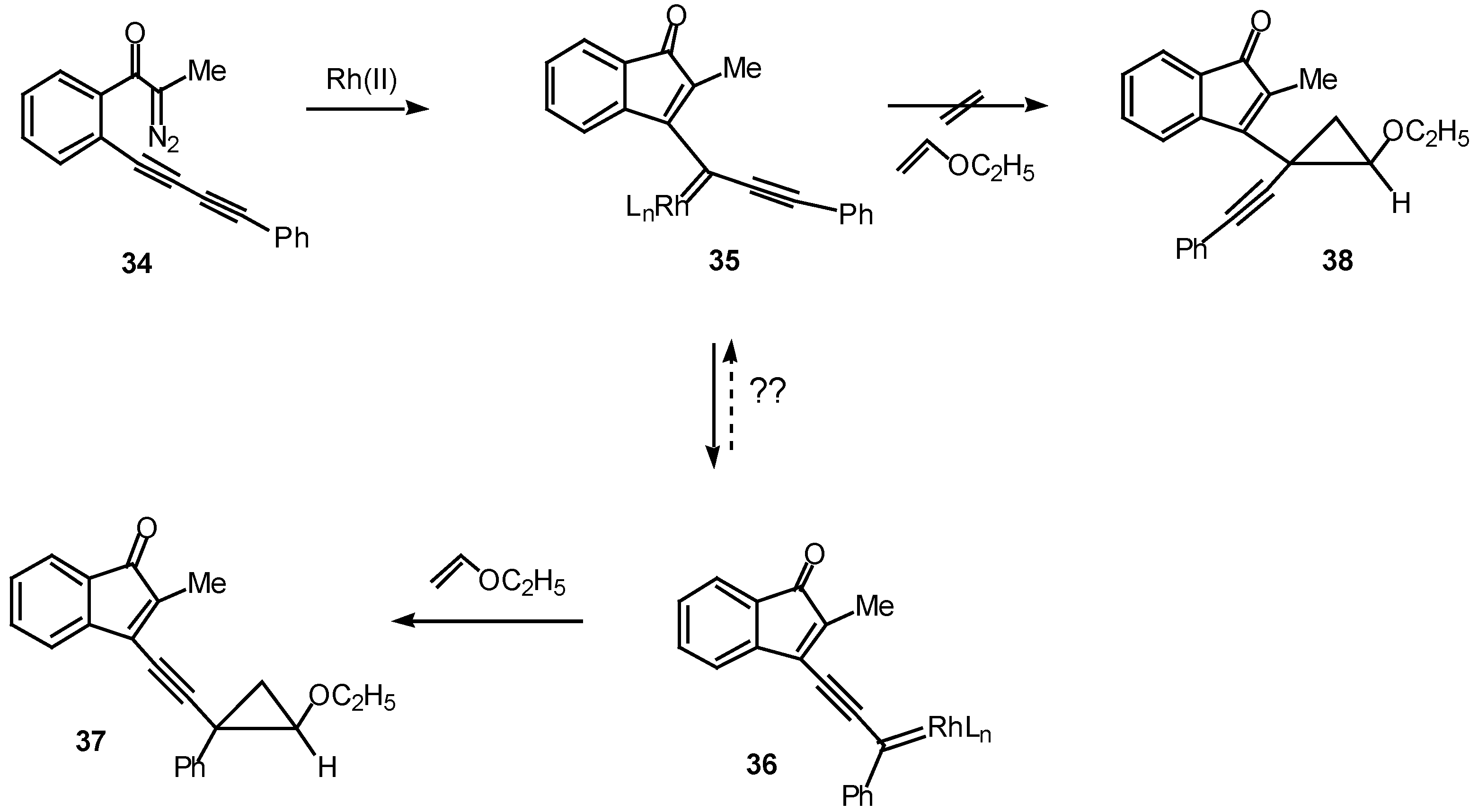
The ease with which these systems undergo the rhodium(II) catalyzed cyclization to give cyclopropenyl substituted indenones suggested that a similar transformation might occur with diacetylenic systems [23]. Such a study was carried out using diazo ketone 34. A critical issue is whether the cyclization will occur to give products derived from the fully rearranged carbenoid 36 or from the initially formed carbenoid 35. In fact, treatment of 34 with a catalytic quantity of rhodium(II) acetate at 25°C in the presence of ethyl vinyl ether afforded cyclopropane 37 with notable efficiency (90% chemical yield) and selectivity (>95% isomeric purity). No signs of the isomeric cyclopropane 38 could be detected in the crude reaction mixture [24]. The exclusive formation of cyclopropane 37 was attributed to a slower rate of trapping of vinyl carbenoid 35 by ethyl vinyl ether, perhaps as a consequence of a more congested transition state. Another possibility is that the equilibrium between the two carbenoids lies completely in favor of the more stable phenyl substituted isomer 36.
Scheme 11.
Ylide Formation and Subsequent Rearrangements
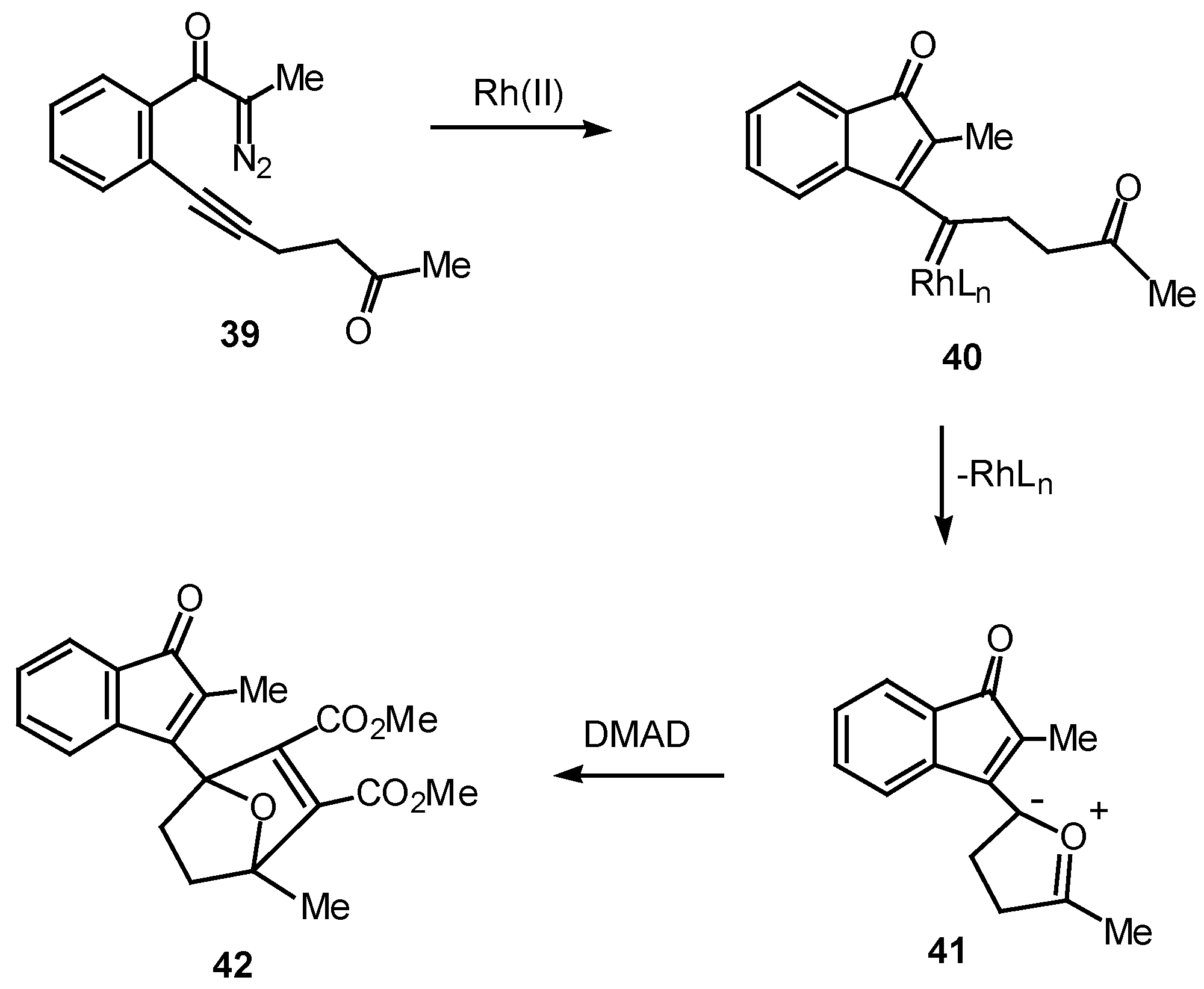
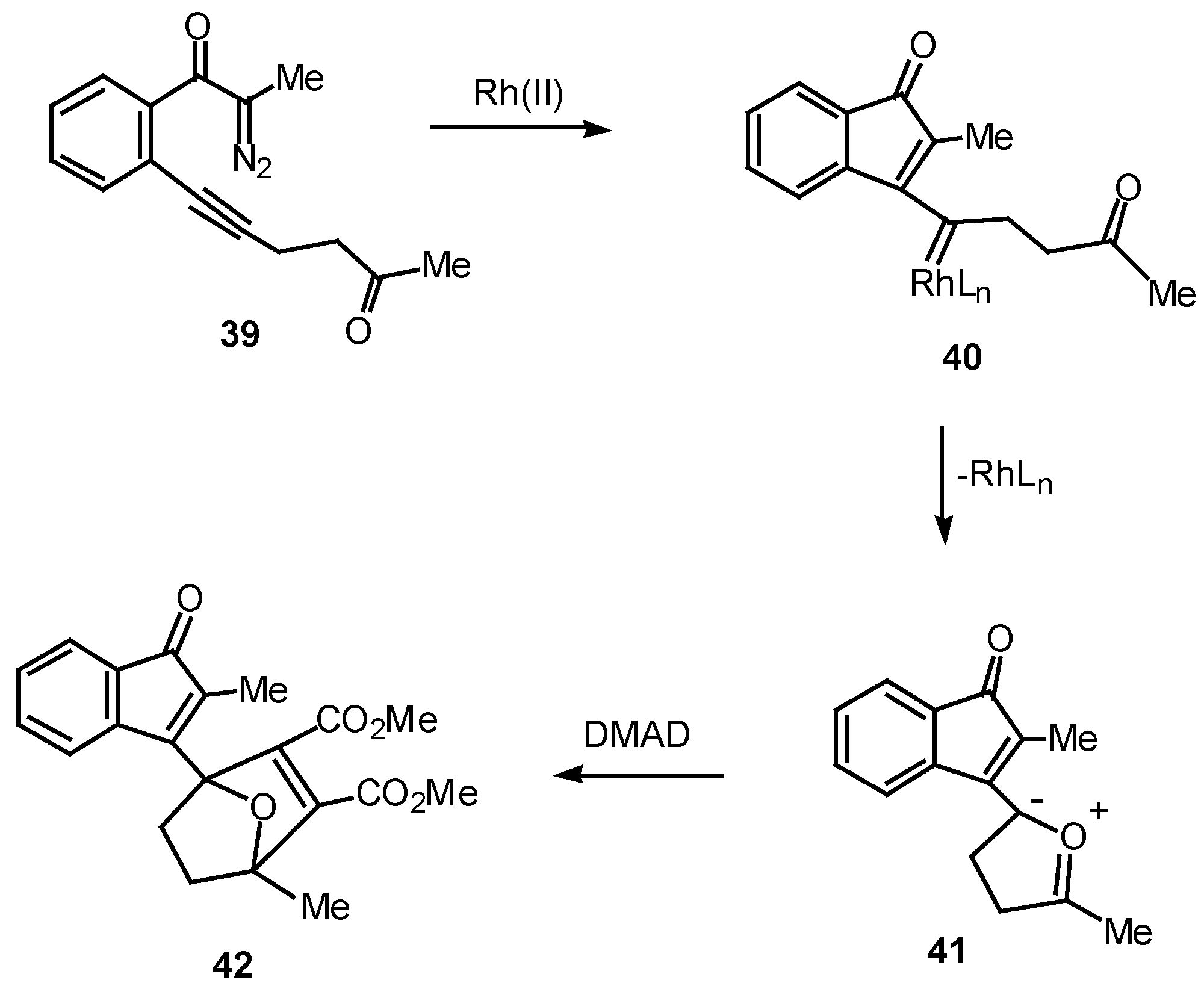
Over the past several years, our group has studied the Rh(II)-induced α-diazo ketone cyclization onto a neighboring carbonyl group followed by dipolar-cycloaddition of the resulting carbonyl ylide dipole as a method for generating oxapolycyclic ring systems [11]. The ease with which α-diazo ketones containing tethered carbonyl groups undergo this tandem process suggested that a similar sequence could also occur with a vinylogous keto carbenoid. In order to test this possibility, the Rh(II) catalyzed behavior of diazo ketone 39 was studied. Treatment of 39 with a catalytic amount of rhodium(II) octanoate in the presence of 1 equiv of dimethyl acetylenedicarboxylate afforded cycloadduct 42 in 97% yield. This result can be accounted for in terms of the intermediacy of vinyl carbenoid 40 which cyclizes onto the oxygen atom of the neighboring carbonyl group to give the resonance-stabilized dipole 41. Dipolar cycloaddition of 41 across the activated π-bond of DMAD affords cycloadduct 42 [25].
Scheme 12.
Diazoester Cyclizations
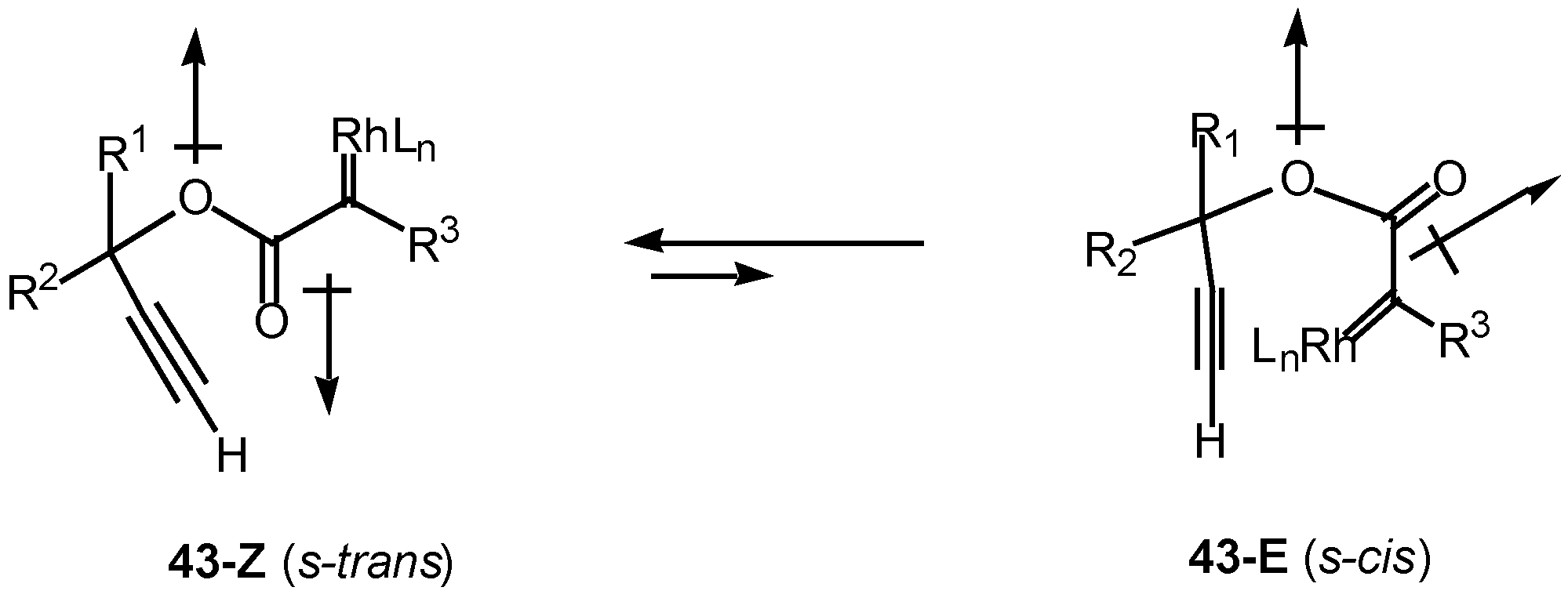
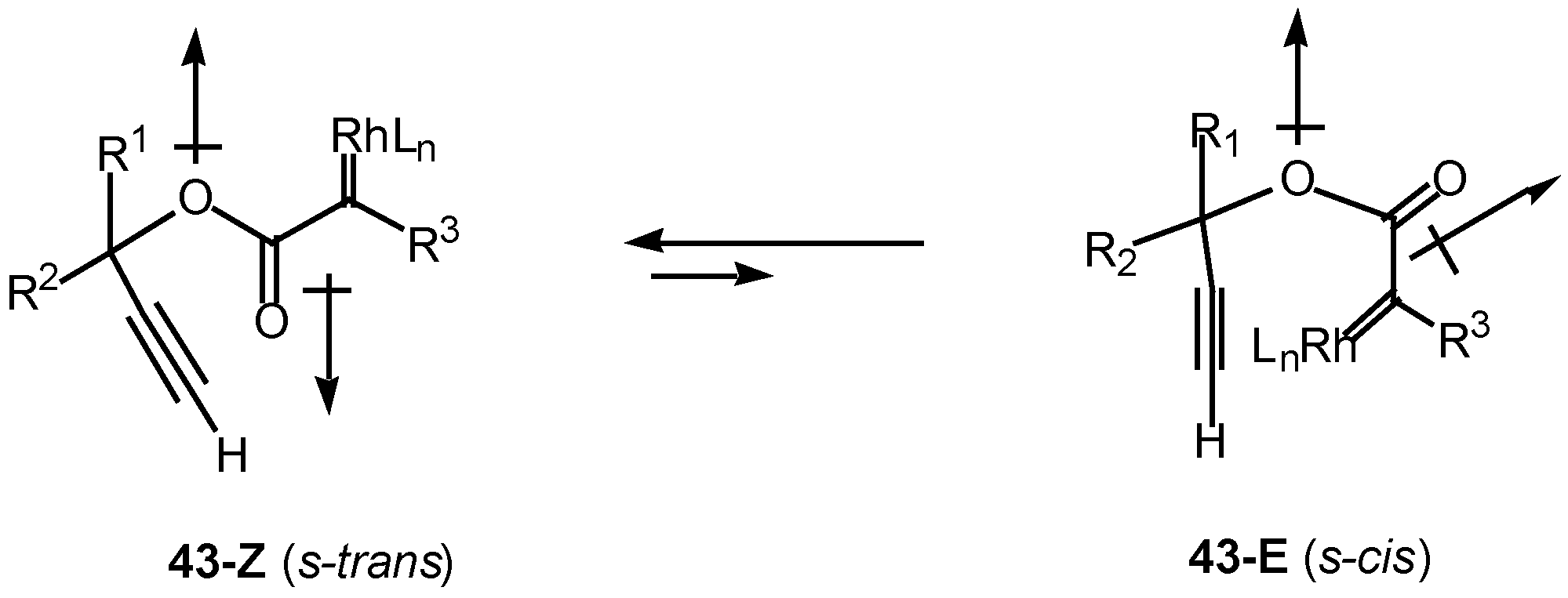
Introduction of a heteroatom α to the diazo carbonyl group may further complicate the cyclization chemistry. It is well known that esters exist primarily in the Z or s-trans (i.e., 43-Z) conformation about the carbonyl π-bond (Scheme 13). Esters are more stable in this conformation for several reasons, one of which is to minimize the overall dipole effect. In this orientation, intramolecular cyclization of the rhodium carbenoid onto the alkyne π-bond cannot occur. In order for cyclization to take place, there must be rotation about the ester bond to generate the E or s-cis conformer 43-E, which can then achieve the necessary geometry to allow the cyclization to proceed [26].
Scheme 13.
Scheme 14.
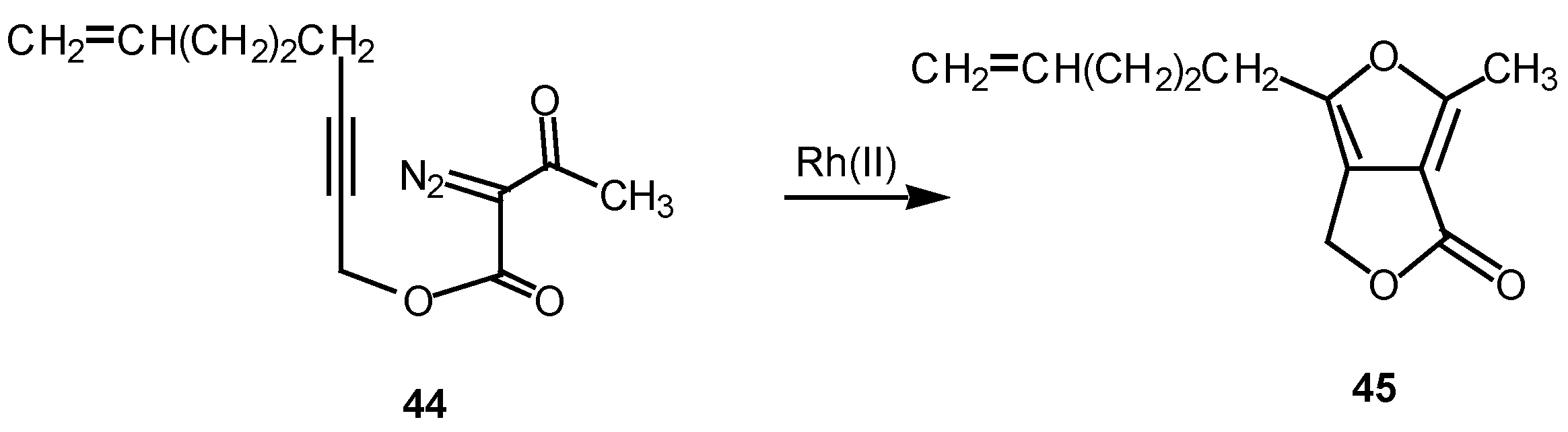
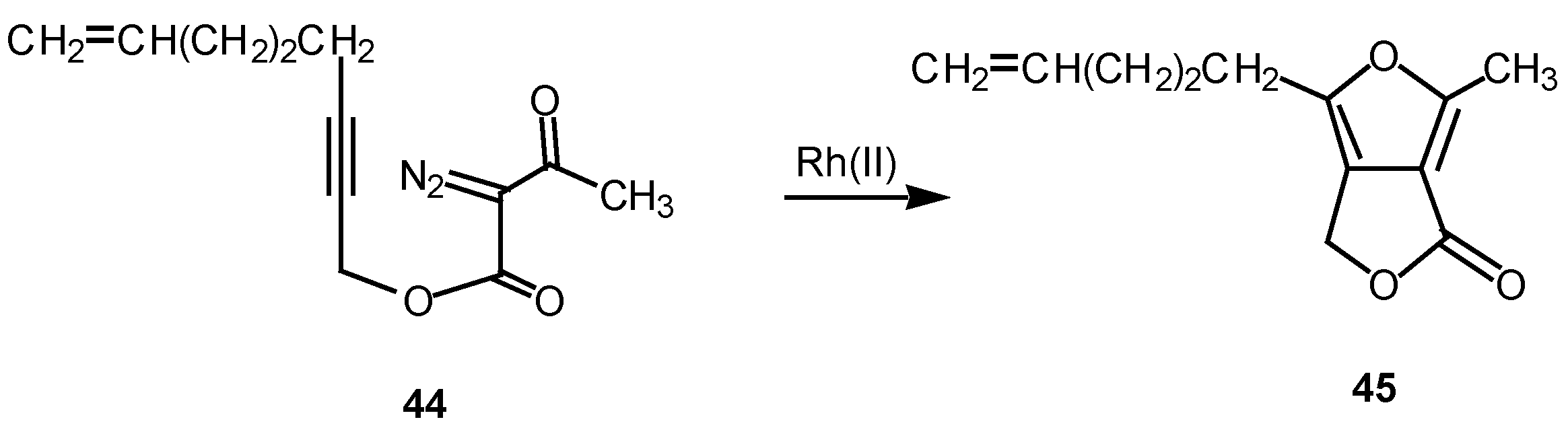
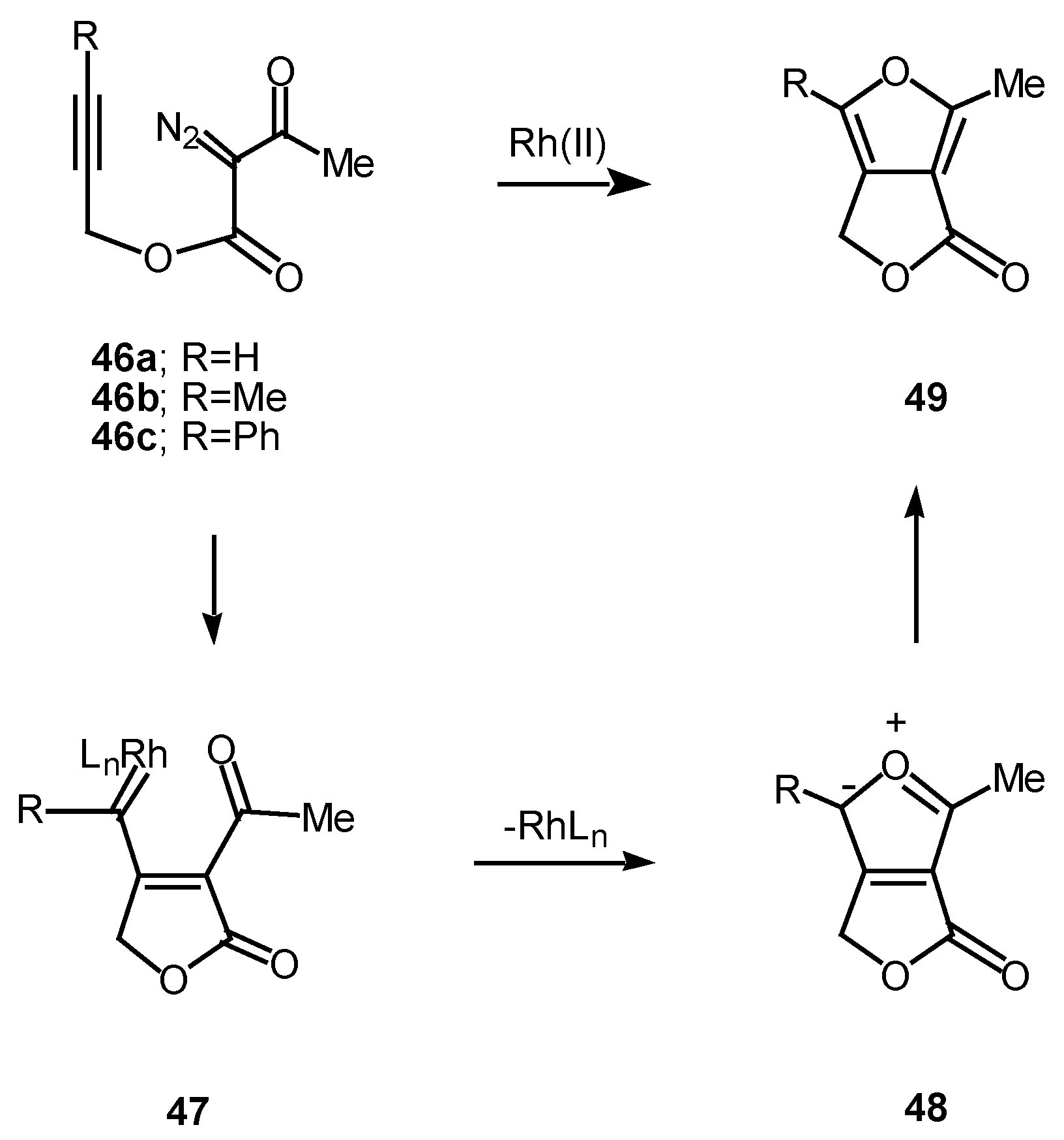
We have found that cyclization of the distabilized diazo ketoester 44 with rhodium(II) octanoate furnished furan 45 in 77% yield [27]. This transformation proceeds by addition of the rhodium-stabilized carbenoid onto the acetylenic π-bond to produce an electrophilic vinyl carbenoid intermediate (i.e., 47) which is subsequently attacked by the adjacent carbonyl oxygen bond. The resulting dipole 48 undergoes subsequent collapses to give furan 49 [28]. 6π-Electrocyclization reactions to produce five membered rings are well precedented transformations in heterocyclic chemistry [29]. Related furan cyclizations have also been observed in ortho constrained systems [30].
Scheme 15.
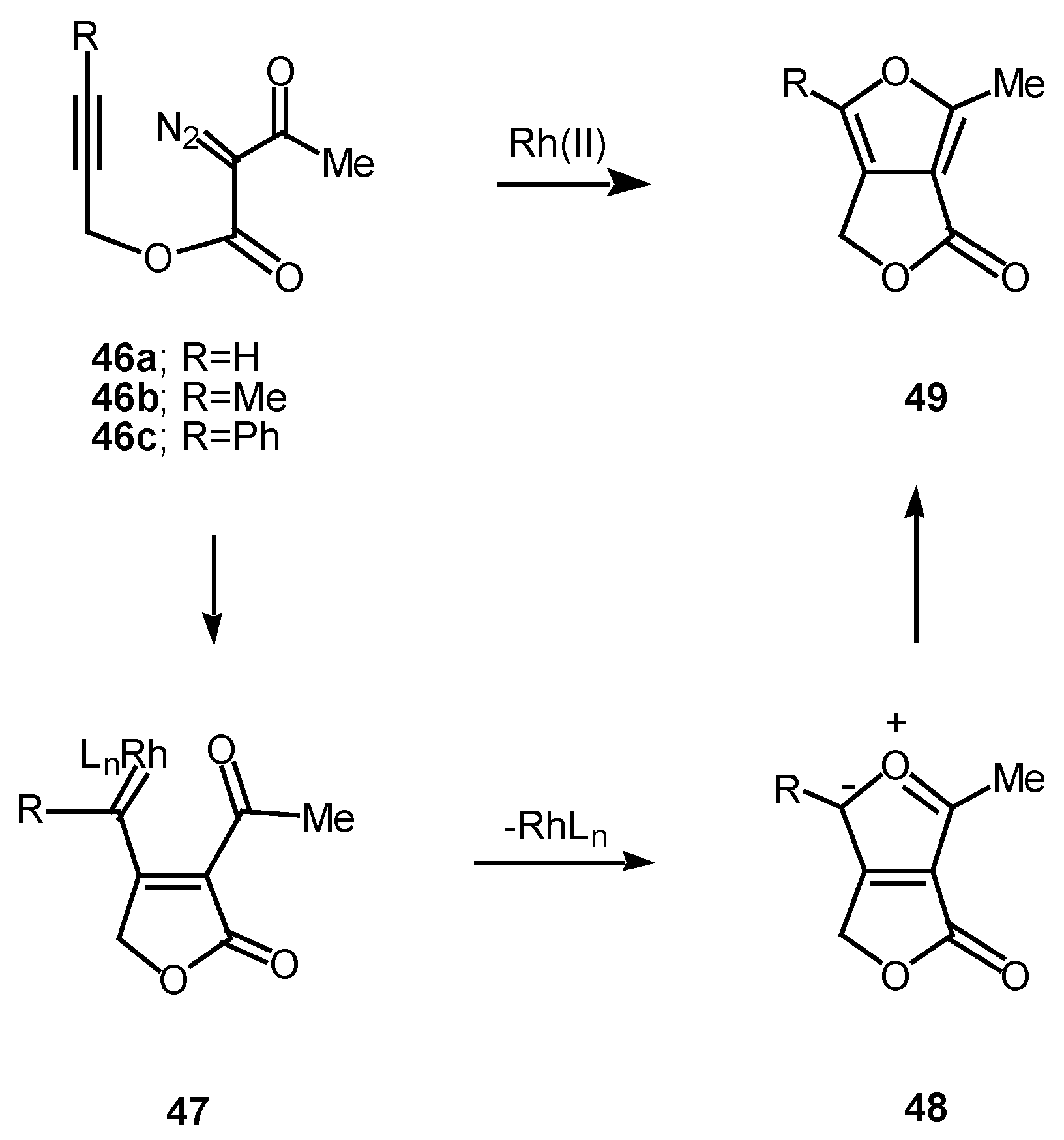
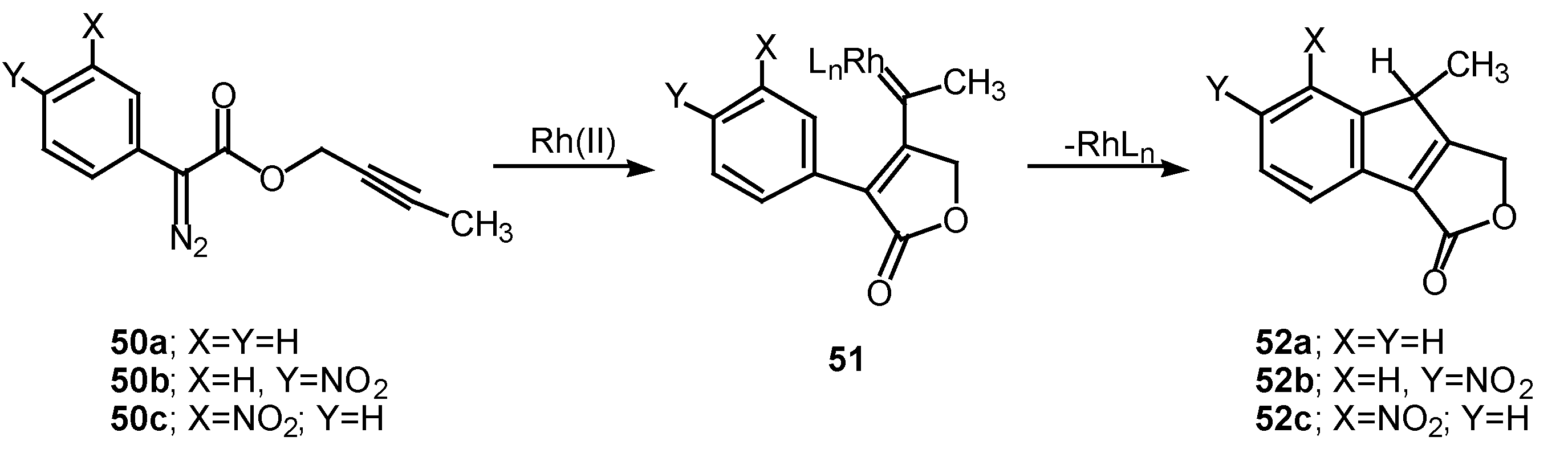
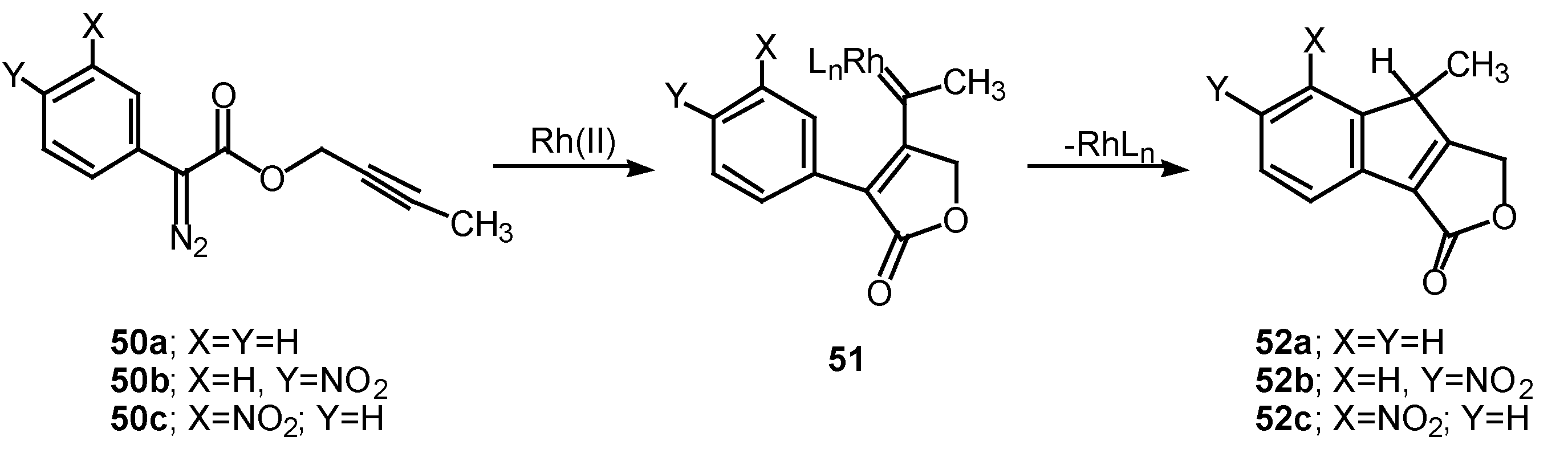
The 1,5-electrocyclization process involved in furan formation has also been utilized to produce indeno[1,2-c]furans such as 52a-c in 45-60% yield. Treatment of the starting α-diazo esters 50a-c with rhodium(II) catalysts gave indenes 52a-c via an electrocyclization of the transient vinyl carbenoid 51 [26]. There seemed to be little effect displayed by the nature of the substituent groups on the aromatic ring as indeno[1,2-c]furans 52b and 52c were isolated as the exclusive products. The fact that the insertion reactions occurs ortho to the nitro group (i.e., 50c to 52c) rather than producing a mixture of ortho and para isomers, suggests that subtle factors play a role in this process as well.
Scheme 16.
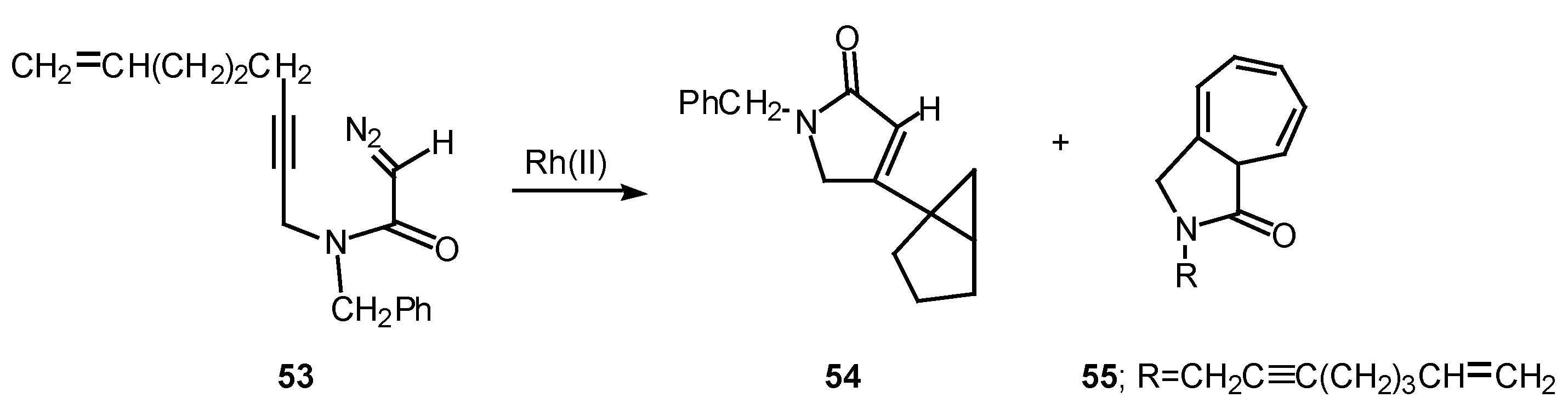
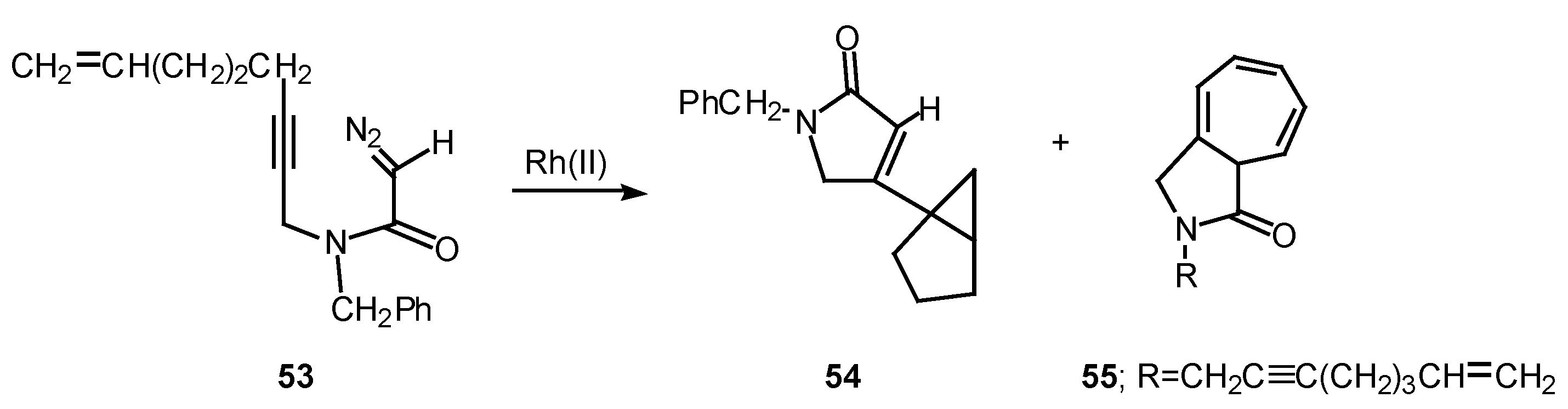
Rotamer population can play a significant role in determining the chemoselectivity of rhodium(II) catalyzed reactions of α-diazo amide systems containing tethered alkynes. The reaction of diazo amide 53 with rhodium(II) octanoate was found to undergo attack on the π-system of the acetylenic tether to give a transient vinyl carbenoid. The next step involved an internal cyclopropanation reaction to produce 54 in 41% yield. Cycloheptatriene 55, which is derived by insertion of the carbenoid into the N-benzyl substituent, was also isolated from this reaction in 33% yield. Rotamer populations nicely account for the behavior of this system.
Scheme 17.
Amide rotamers generally interconvert in solution with lifetimes of 10-1-10-2 sec. The geometry of a typical amide C-N bond will be fixed during the entire lifetime of the acyl rhodium carbenoid intermediate. Assuming that both amide rotamers are equally reactive toward π-addition, the relative amounts of compounds 54 and 55 that are formed are determined by the equilibrium concentration of the starting rotamers.
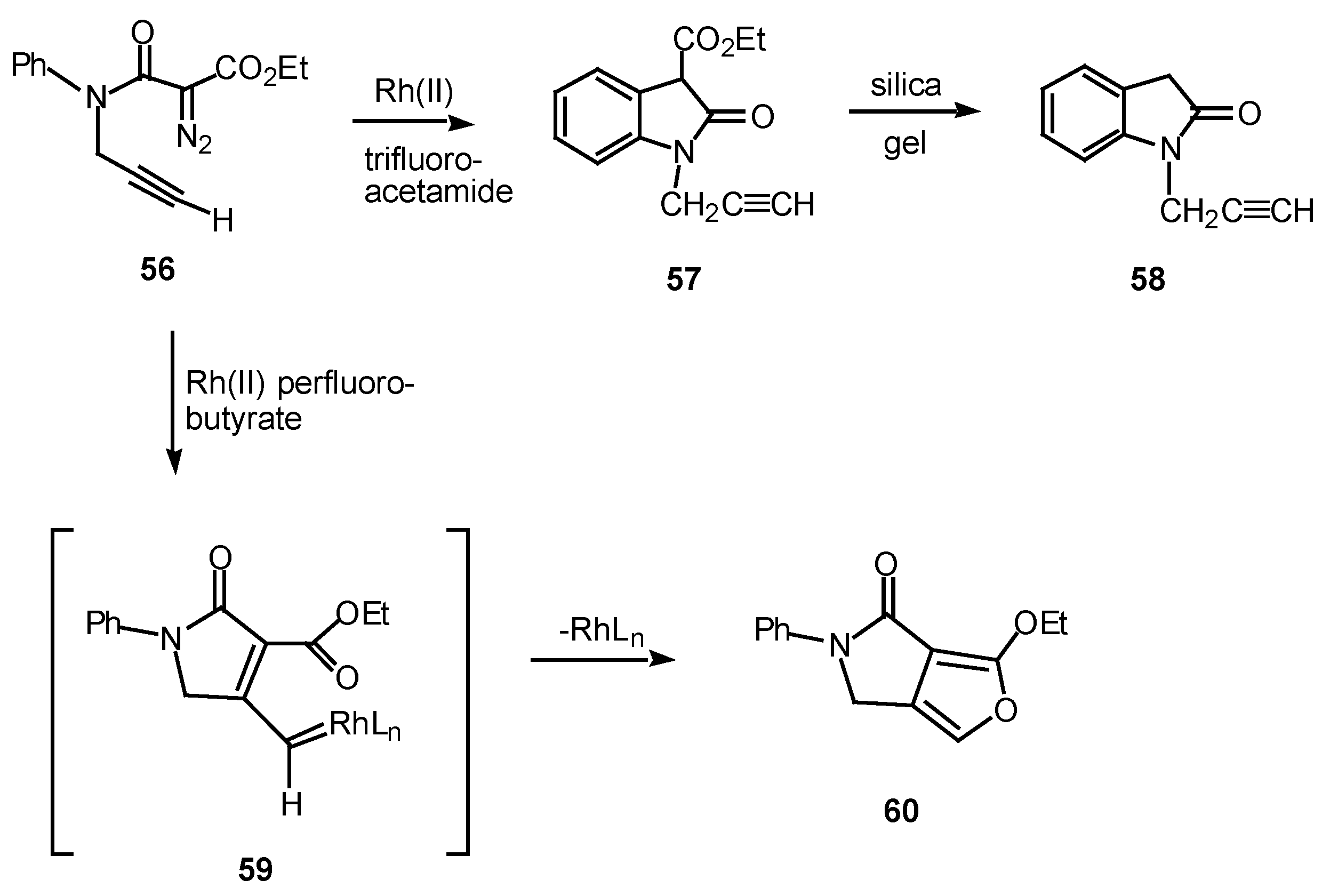
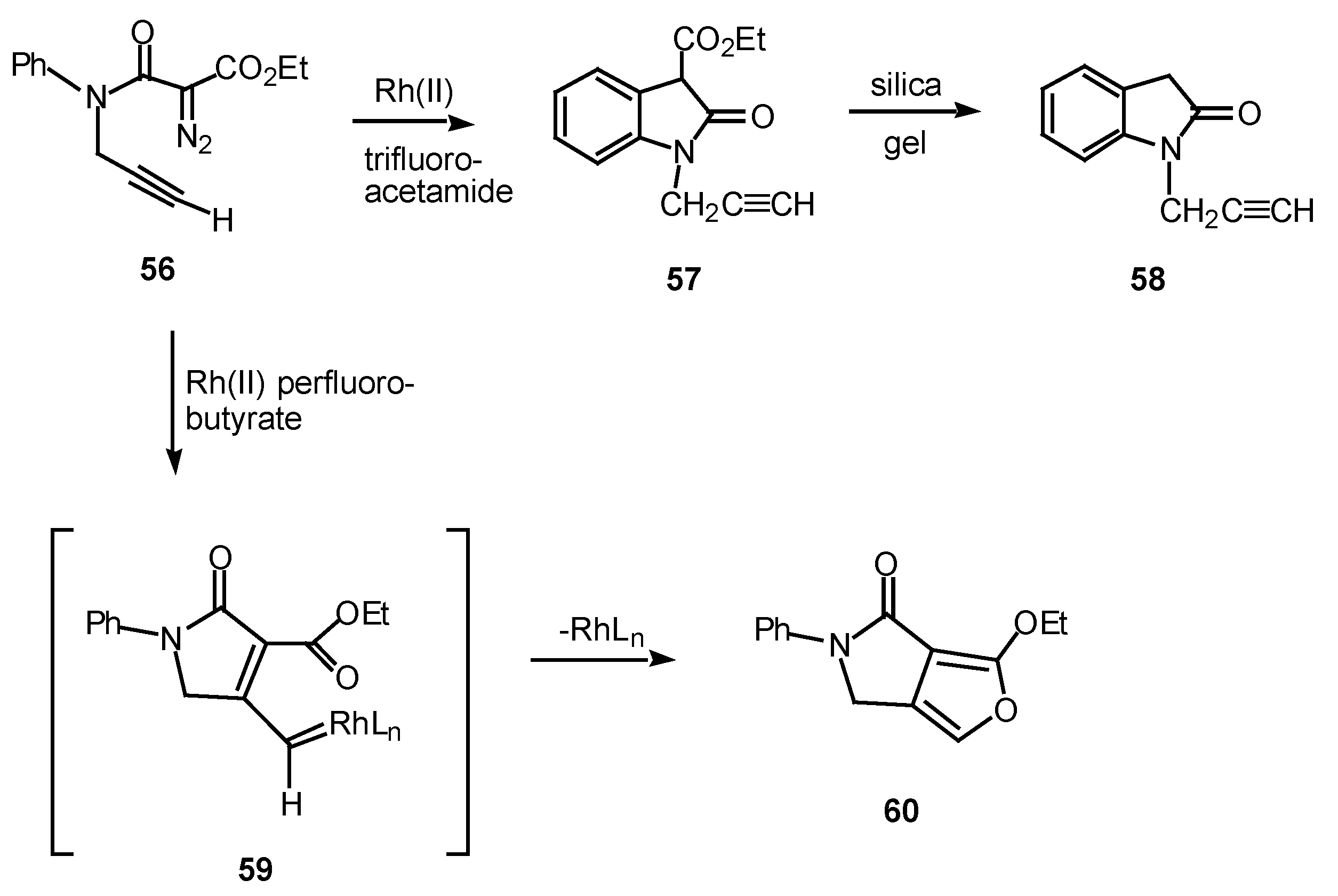
In one study, the mode of cyclization of a distabilized α-diazo amide was varied by changing the ligands on the rhodium catalyst. Reaction of 56 with rhodium(II) trifluoroacetamide in benzene at 25°C provided oxindole 57 in 87% yield. On the other hand, when rhodium(II) perfluorobutyrate was used as a catalyst, fluropyrrolone 60 was formed in 98% yield [31], in line with previous observations [32].
Scheme 18.
Conclusions
It is clear from the above discussion that the reaction of α-diazo carbonyl compounds with tethered alkynes is both a mechanistically complex and synthetically useful process. Four major factors dictate the mode of reaction of the initially formed rhodium carbenoid species. The electronics about the carbenoid center is perhaps the most important factor. Conformation of the molecule is also quite impactful. The geometrical orientation can be influenced by both the nature of the carbenoid stabilizing group (amide vs. ester vs. ketone), and by substitution on the carbonyl group. Steric factors appear to influence the process in subtle ways. Finally, the polarity of the solvent used in these reactions has also been shown to influence both the mechanism and chemoselectivity of the reaction. These factors can be exploited and manipulated in many ways to generate a wide variety of interesting products. Application of the methodology to the synthesis of natural products is still relatively unexplored.
Acknowledgments
We thank the National Science Foundation for generous support of this work. We also acknowledge the contributions of the graduate and postdoctoral students who participated in this research area. Their names are given in the literature references.
References
- Collman, J.P.; Hegedus, L.S.; Norton, J.R.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry; Mill Valley, CA, 1987. [Google Scholar] Doyle, M.P. Acc. Chem. Res. 1986, 19, 348. [PubMed]
- Grubbs, R.H. Comprehensive Organometallic Chemistry; Wilkinson, G., Stone, F.G.A., Able, E.W., Eds.; Pergamon: New York, 1982; Vol. 8, p. 499. [Google Scholar]
- Dragutan, V.; Balaban, A.T.; Doimonie, M. Olefin Metathesis and Ring-Opening Polymerization of Cyclo-Olefins, 2nd ed.; Wiley-Interscience: New York, 1985. [Google Scholar]
- Brookhart, M.; Studabaker, W.B. Chem. Rev. 1987, 87, 411.
- Casey, C.P. Reactive Intermediates; Jones, M., Moss, R.A., Eds.; Wiley: New York, 1981; p. 135. [Google Scholar]
- Dötz, K.H. Angew. Chem., Int. Ed. Engl. 1984, 23, 587. [CrossRef]
- Padwa, A.; Krumpe, K.E. Tetrahedron 1992, 48, 5385.
- Adams, J.; Spero, D.M. Tetrahedron 1991, 47, 1765.
- Taber, D.F. Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon Press: New York; Vol IV, 1991; p. 1046. [Google Scholar]
- Maas, G. Top Curr. Chem. 1987, 137, 77.
- Padwa, A.; Hornbuckle, S.F. Chem. Rev. 1991, 91, 263.
- Hoye, T.R.; Dinsmore, C.J.; Johnson, D.S.; Korkowski, P.F. J. Org. Chem. 1990, 55, 4518. [CrossRef]
- Padwa, A.; Krumpe, K.E.; Gareau, Y.; Chiacchio, U. J. Org. Chem. 1991, 56, 2523. [CrossRef]
- Padwa, A.; Xu, S.L. J. Am. Chem. Soc. 1992, 114, 5881. Padwa, A.; Kassir, J.M.; Xu, S.L. J. Org. Chem. 1991, 56, 6971. Padwa, A.; Carter, S.P.; Nimmesgern, H.; Stull, P. J. Am. Chem. Soc. 1998, 110, 2894. Padwa, A.; Fryxell, G.E.; Zhi, L. J. Org. Chem. 1988, 53, 2875.
- Hoye, T.R.; Dinsmore, C.J. J. Am Chem. Soc. 1991, 113, 4343. [CrossRef]
- Padwa, A.; Krumpe, K.E.; Zhi, L. Tetrahedron Lett. 1989, 30, 2633.
- Padwa, A.; Austin, J.A.; Xu, S.L. J. Org. Chem. 1992, 57, 1330.
- Padwa, A.; Austin, D.J.; Chiacchio, U.; Kassir, J.M.; Rescifina, A.; Xu, S.L. Tetrahedron Lett. 1991, 32, 5923.
- Mueller, P.H.; Kassir, J.M.; Semones, M.A.; Weingarten, M.D.; Padwa, A. Tetrahedron Lett. 1993, 34, 4285.
- Davies, H.M.L.; McAfee, M.J.; Oldenburg, C.E.M.J. J. Org. Chem. 1989, 54, 930. Davies, H.M.L.; Oldenburg, C.E.M.; McAfee, M.J.; Nordahl, J.G.; Henretta, J.P.; Romines, K.R. Tetrahedron Lett. 1988, 975.
- Deem, M.L. Synthesis 1982, 701.
- Padwa, A.; Kassir, J.M.; Xu, S.L. J. Org. Chem. 1997, 62, 1642.
- Padwa, A.; Gareau, Y.; Xu, S.L. Tetrahedron Lett. 1991, 32, 983.
- Padwa, A.; Austin, D.J.; Gareau, Y.; Kassir, J.M.; Xu, S.L. J. Am Chem. Soc. 1993, 115, 2637.
- Kassir, J.M.; Semones, M.A.; Weingarten, M.D.; Padwa, A. Tetrahedron Lett. 1993, 34, 7853.
- Padwa, A.; Kinder, F.R. J. Org. Chem. 1993, 58, 21.
- Padwa, A.; Dean, D.C.; Fairfax, D.J.; Xu, S.L. J. Org. Chem. 1993, 58, 4646.
- Padwa, A.; Kinder, F.R. Tetrahedron Lett. 1990, 31, 6835.
- Taylor, E.C.; Turchi, I.J. Chem. Rev. 1979, 79, 181. Huisgen, R. Angew. Chem., Int. Ed. Engl. 1980, 19, 947.
- Padwa, A.; Fairfax, D.J.; Austin, D.J.; Xu, S.L. J. Chem. Soc., Perkin Trans. I 1992, 2837.
- Brown, D.S.; Elliott, M.C.; Moody, C.J.; Mowlem, T.J.; Marino, J.P.; Padwa, A. J. Org. Chem. 1994, 59, 2447. [CrossRef]
- Wee, A.G.H.; Liu, B.; Zhang, L. J. Org. Chem. 1992, 57, 4404. Liu, B.; Wee, A.G.H. Heterocycles 1993, 36, 445.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Padwa, A. Synthesis of Polycyclic Ring Systems Using Transition Metal Catalyzed Cyclizations of Diazo Alkynyl Ketones. Molecules 2001, 6, 1-12. https://doi.org/10.3390/60100001
AMA Style
Padwa A. Synthesis of Polycyclic Ring Systems Using Transition Metal Catalyzed Cyclizations of Diazo Alkynyl Ketones. Molecules. 2001; 6(1):1-12. https://doi.org/10.3390/60100001
Chicago/Turabian StylePadwa, Albert. 2001. "Synthesis of Polycyclic Ring Systems Using Transition Metal Catalyzed Cyclizations of Diazo Alkynyl Ketones" Molecules 6, no. 1: 1-12. https://doi.org/10.3390/60100001